1. Introduction
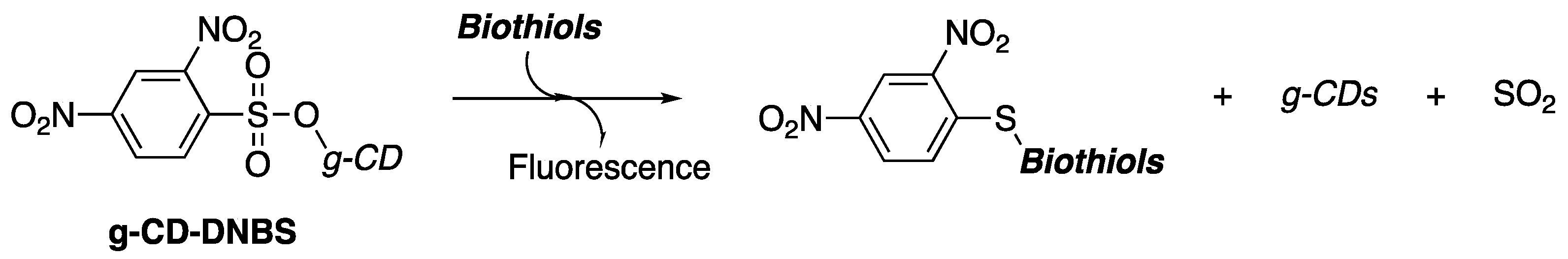
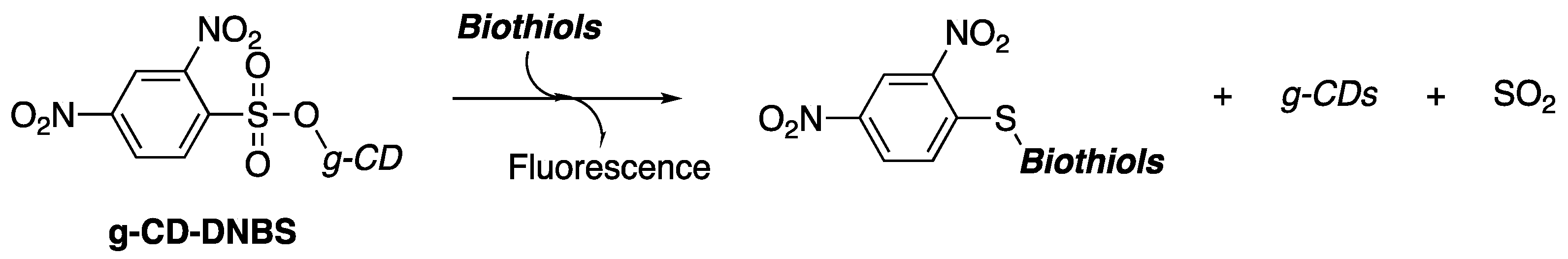
A reliable method for producing arylsulfonates involves the nucleophilic substitution reaction of alcohols and sulfonyl halides. This reaction is highly efficient in creating the sulfonate ester, a synthetically important electrophile in organic chemistry due to its high chemical reactivity [
1]. This property has been utilized in the detection and fluorescent imaging of biologically important compounds. In particular, recent research has shown that 2,4-dinitrobenzenesulfonate-functionilized carbon dots (g-CD-DNBS) are significant regarding the detection and fluorescence imaging of biothiols (
Scheme 1) [
2].
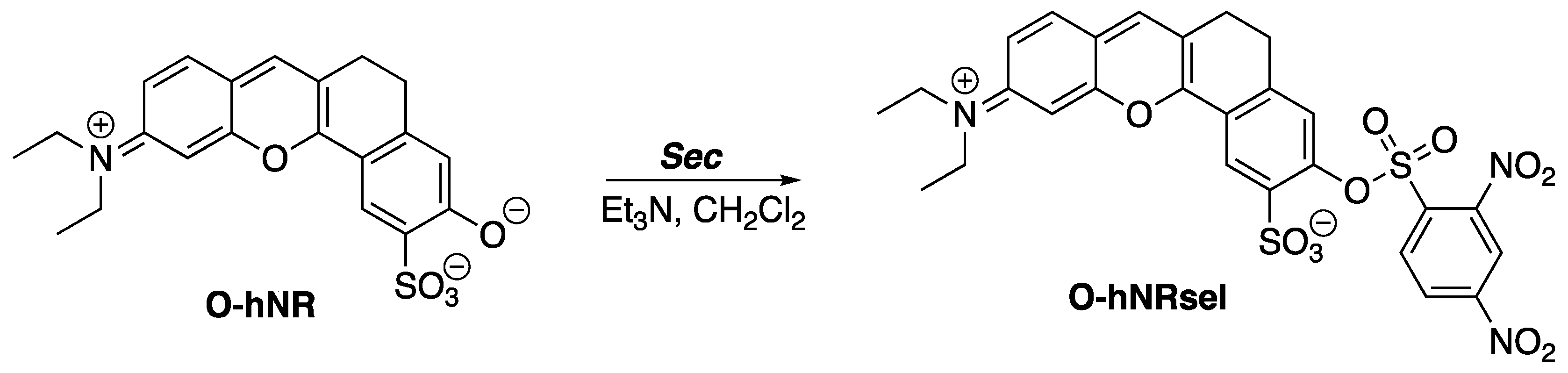
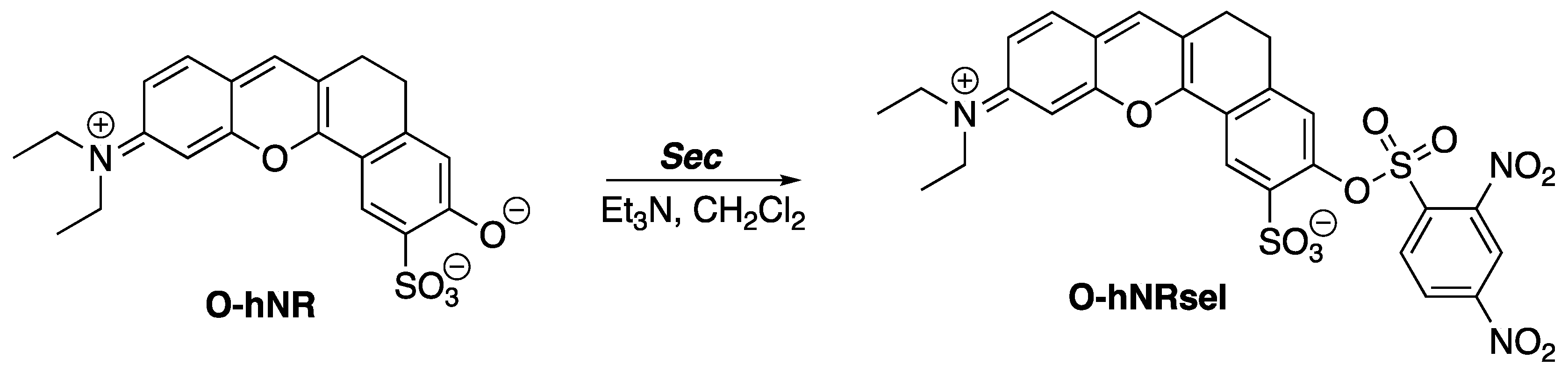
The 2,4-dinitrobenzenesulfonyl (DNBS) group resembles the x-substituted 2,4-dinitrophenyl-4′-phenylbenzenesulfonate compounds. Similarly, this resemblance is found in the fluorescent probe O-hNRSel. O-hNRSel was developed for the detection and imaging of selenocysteine (Sec) in living cells (
Scheme 2) [
3]. Sec is a selenium-containing amino acid that resides in proteins of organisms and viruses. Selenium is linked to several health benefits, including the prevention of cancer and cardiovascular diseases [
4]. The role of Sec in human health underlines the importance of Sec fluorescence probes.
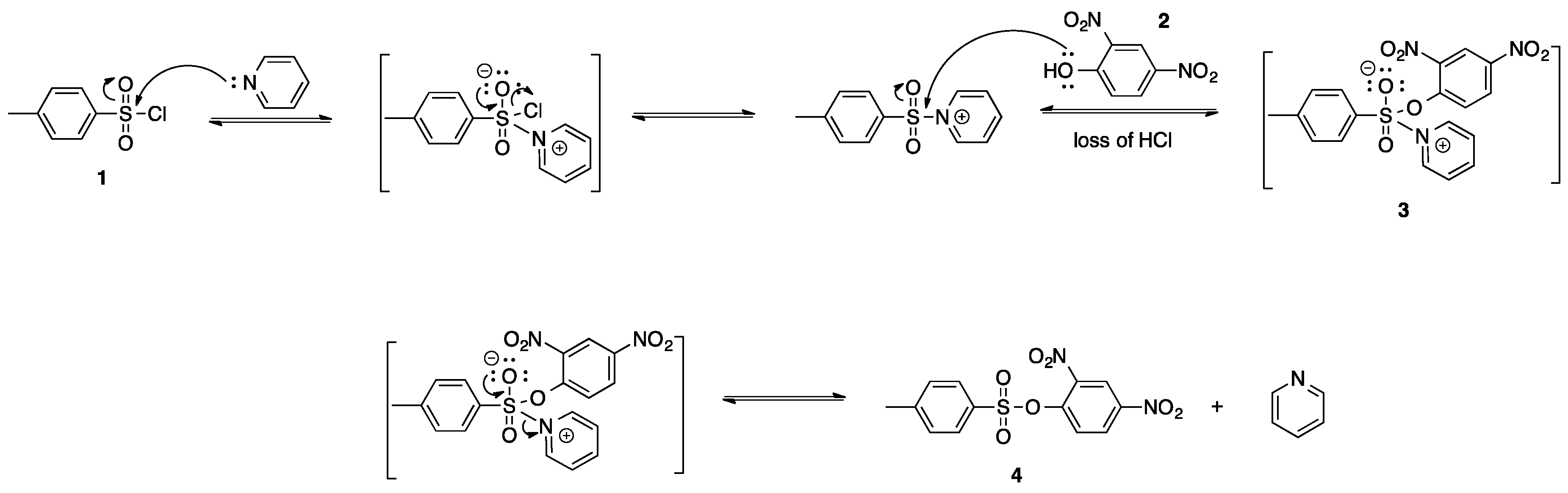
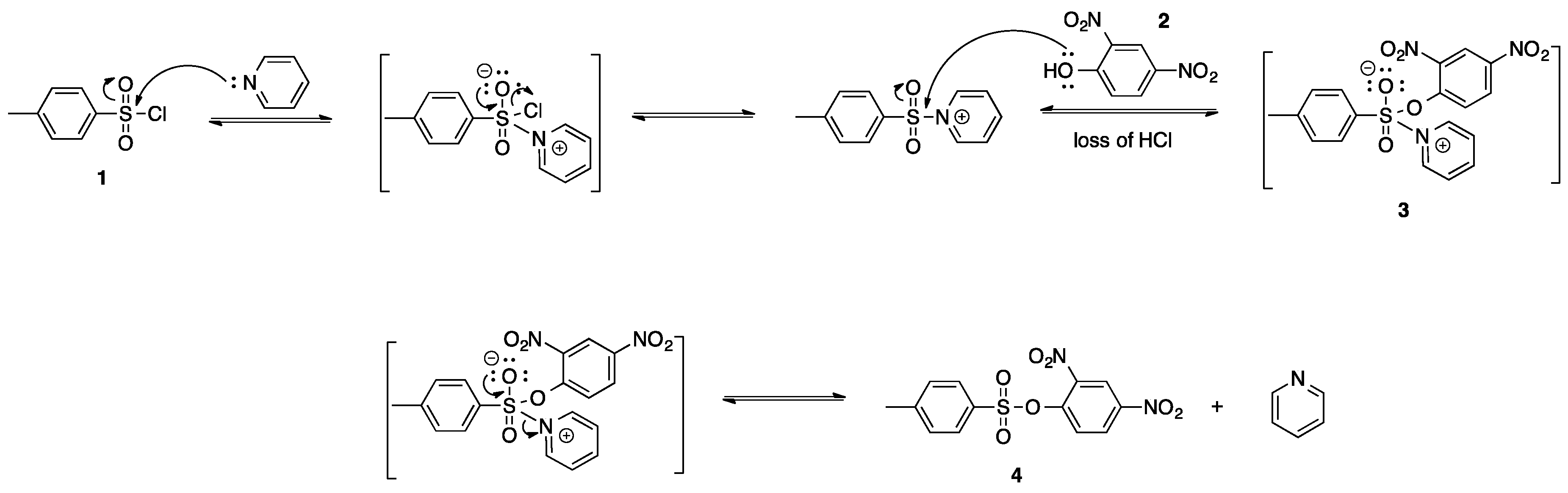
A facile synthesis of sulfonates is necessary to produce these biologically significant compounds. A review of the current literature suggests the use of sulfonic acids or sulfonyl halides for synthesizing sulfonates [
5,
6]. The compounds synthesized hereafter were done so through the sulfonylation of 2,4-dinitrophenol with sulfonyl chloride in the presence of a base. Basic conditions allow for the neutralization of hydrochloric acid. The presence of hydrochloric acid lowers the pH of the reaction, resulting in poor deprotonation of the intermediary structure. Preliminary experiments for the synthesis of 2,4-dinitrophenyl-4-methylbenzenesulfonate involved the treatment of 4-methylbenzenesulfonyl chloride (
1) with 2,4-dinitrophenol (
2) in the presence of pyridine and dichloromethane. This reaction was done under N
2 atmosphere to avoid hydrolysis of 4-methylbenzenesulfonyl chloride. The production of the insoluble pyridinium salt, the pyridine adduct of 2,4-dinitrophenyl 4-methylbenzenesulfonate (
3), was evidence of an unsuccessful reaction. Compound
3 is insoluble in dichloromethane and pulled out of the solution as a white precipitate. The proposed mechanism for the formation of the pyridinium salt (
3) in the presence of pyridine is found in
Scheme 3. The desired sulfonate (
4) was not formed as a result of compound
3 being insoluble in solution. A review of the literature supports the theory that a pyridinium salt was formed [
7].
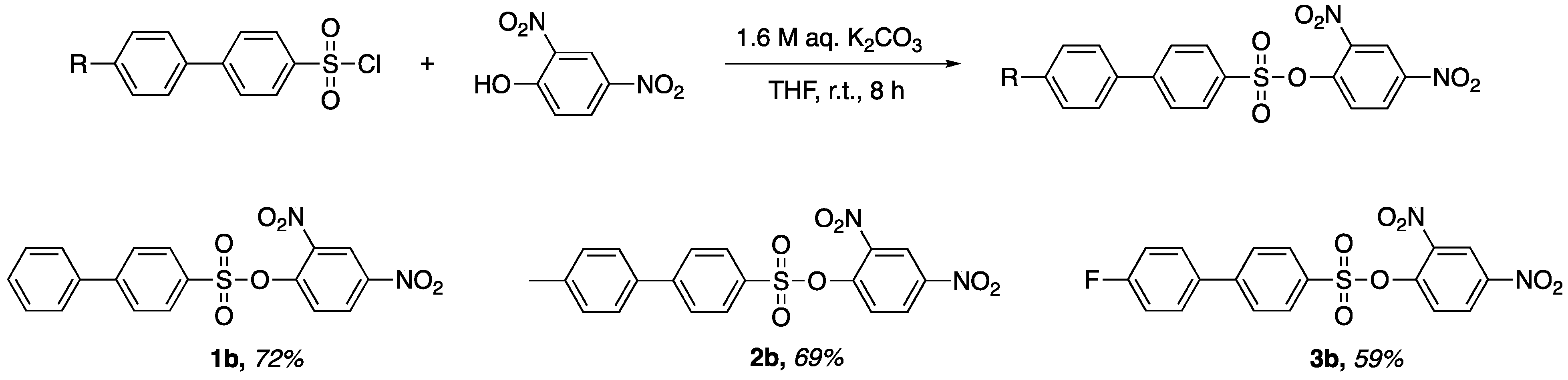
A new synthetic method was proposed and applied to the synthesis of three x-substituted 2,4-dinitrophenyl-4′-phenylbenzenesulfonates. The resulting products were characterized by crystallographic and spectroscopic means. Crystallographic characterization offers insight into the structural features and inter- and intramolecular interactions of molecules. Herein, we report the facile synthesis and characterization of x-substituted 2,4-dinitrophenyl-4′-phenylbenzenesulfonates.
2. Experimental
The reagents used in the synthesis of x-substituted 2,4-dinitrophenyl-4′-phenylbenzenesulfonates were obtained from commercial sources and used without further purification. Thin-layer chromatography (TLC) was used to track reaction progress and obtain Rf values for the reactions.
Preparation of 2,4-Dinitrophenyl 4-methylbenzenesulfonate in the presence of N,N-diisopropylethylamine (4). 2,4-dinitrophenol (2.025 g, 11.0 mmol) was dissolved in a flask containing 11 mL of chilled dichloromethane. 4-Methylbenzenesulfonyl chloride (2.090 g, 11.0 mmol) was added dropwise to the stirred solution. This was followed by the dropwise addition of N,N-diisopropylethylamine (3.8 mL, 21.8 mmol). The stirred solution was left at room temperate for 24 h under N2 atmosphere. After 24 h, the mixture was diluted with 15 mL of dichloromethane and transferred to a separatory funnel. The organic layer was washed with water (3 × 10 mL). The aqueous layers were combined and back-extracted with 10 mL of dichloromethane. All organic layers were combined, washed with brine (10 mL), and dried over anhydrous sodium sulfate. Evaporation of solvent yielded a solid, yellow residue as a crude product. Purification via column chromatography with dichloromethane as the solvent yielded a yellow powder (1.712 g, 46%). M.p. 196–202 °C. Rf = 0.74 (CH2Cl2). 1H-NMR (400 MHz, DMSO-d6) δ 8.79 (d, J = 2.8 Hz, 1H), 8.54 (dd, J = 9.1, 2.8 Hz, 1H), 7.78–7.71 (m, 2H), 7.54 (d, J = 9.1 Hz, 1H), 7.49 (m, 2H), 2.42 (s, 3H). HRMS (ESI): cald. For C13H10N2NaO7S [M + Na]+ 361.0100; found 361.0110.
Preparation of pyridinium adduct of 2,4-Dinitrophenyl 4-methylbenzenesulfonate in the presence of pyridine (3). 2,4-dinitrophenol (2.016 g, 11.0 mmol) was dissolved in a flask containing 11 mL of chilled dichloromethane. 4-Methylbenzenesulfonyl chloride (2.088 g, 10.9 mmol) was added dropwise to the stirred solution. This was followed by the dropwise addition of pyridine (1.80 mL, 21.8 mmol). The stirred solution was left at room temperate for 24 h under N2 atmosphere. After reaction completion, an insoluble precipitate was isolated via vacuum filtration giving a fine, white powder. Recrystallization in 2:1 acetone/water, followed by trituration with hexanes afforded the product as small, translucent crystalline sheets (1.875 g, 41% yield). M.p. 240–248 °C. Rf = 0.70 (80:20 ACN/H2O w/1 mL NH4OH). 1H-NMR (400 MHz, DMSO-d6) δ 9.40–9.33 (m, 2H), 9.05 (d, J = 2.5 Hz, 1H), 8.96–8.82 (m, 2H), 8.50–8.35 (m, 3H), 7.43–7.36 (m, 2H), 7.06 (d, J = 7.8 Hz, 2H), 2.24 (s, 3H).
Preparation of 2,4-Dinitrophenyl 4-methylbenzenesulfonate in the presence of triethylamine (4). 2,4-dinitrophenol (1.999 g, 10.9 mmol) was dissolved in a flask containing 11 mL of chilled dichloromethane. 4-Methylbenzenesulfonyl chloride (2.076 g, 10.9 mmol) was added dropwise to the stirred solution. This was followed by the dropwise addition of triethylamine (3.0 mL, 21.9 mmol). The stirred solution was left at room temperate for 24 h under N2 atmosphere. After 24 h, the mixture was diluted with 15 mL of dichloromethane and transferred to a separatory funnel. The organic layer was washed with water (3 × 10 mL). The aqueous layers were combined and back-extracted with 10 mL of dichloromethane. All organic layers were combined, washed with brine (10 mL), and dried over anhydrous sodium sulfate. The resulting solution was evaporated to afford a solid, yellow residue. The crude product was recrystallized in 2:1 DCM/ethyl acetate to afford large, pale-yellow, translucent crystals (1.768, 50%).
Preparation of 2,4-Dinitrophenyl 4-methylbenzenesulfonate in the presence of aqueous sodium hydroxide (4). 2,4-dinitrophenol (1.021 g, 5.25 mmol) was dissolved in a flask containing 10 mL of THF. 4-Methylbenzenesulfonyl chloride (1.052 g, 5.25 mmol) was then added to the flask, followed by the dropwise addition of 1 M NaOH (10 mL, 10.5 mmol). The solution was stirred at room temperate for 6 h. After reaction completion, a yellow precipitate was isolated via vacuum filtration to afford a yellow powder. The crude product was recrystallized in ethanol to afford large, pale-yellow, translucent crystals (1.204 g, 66%).
Preparation of 2,4-Dinitrophenyl 2,4,6-trimethylbenzenesulfonate (
Table 1,
entry 1). 2,4-dinitrophenol (0.842 g, 4.57 mmol) and 2,4,6-trimethylbenzenesulfonyl chloride (1.002 g, 4.58 mmol) were added to a flask containing 10 mL of tetrahydrofuran. Of aqueous potassium carbonate 0.915 M (10 mL, 9.15 mmol) was added dropwise to the stirred solution. The solution was then stirred at room temperate for 6 h. After reaction completion, a yellow precipitate was isolated via vacuum filtration to afford a yellow powder. The crude product was recrystallized in ethanol to afford large, pale-yellow, translucent crystals (1.470 g, 88%). M.p. 128–132 °C. R
f = 0.86 (CH
2Cl
2).
1H-NMR (400 MHz, Chloroform-
d) δ 8.75 (d,
J = 2.8 Hz, 1H), 8.44 (dd,
J = 9.0, 2.8 Hz, 1H), 7.58 (d,
J = 9.0 Hz, 1H), 7.03 (s, 2H), 2.57 (s, 6H), 2.35 (s, 3H). HRMS (ESI): cald. For C
15H
14N
2NaO
7S [M + Na]
+ 389.0400; found 389.0410.
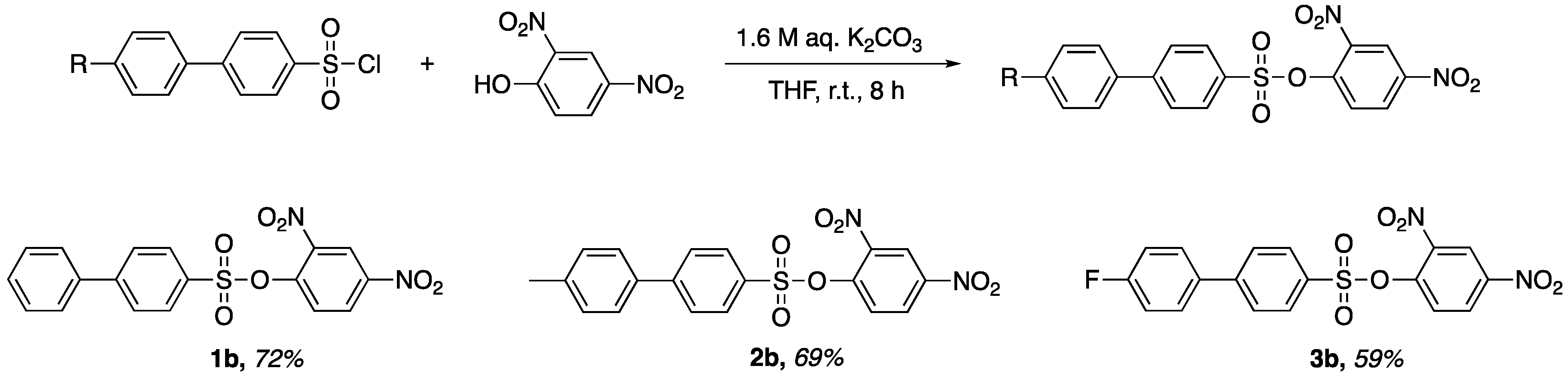
Preparation of 2,4-Dinitrophenyl-4′-phenylbenzenesulfonate (1b). 2,4-dinitrophenol (0.729 g, 3.96 mmol) and biphenyl-4-sulfonyl chloride (1.005 g, 3.96 mmol) were added to a flask containing 10 mL of tetrahydrofuran. Of aqueous potassium carbonate 1.6 M (5.0 mL, 7.92 mmol) was added dropwise to the stirred solution. The stirred solution was left at room temperate for 8 h. After reaction completion, a yellow precipitate was isolated via vacuum filtration to afford a yellow powder. The crude product was recrystallized in a small amount of dichloromethane to afford pale-yellow translucent crystals (1.136 g, 72% yield). M.p. 137–140 °C, Rf = 0.69 (CH2Cl2). 1H-NMR (400 MHz, Chloroform-d) δ 8.78 (d, J = 2.7 Hz, 1H), 8.50 (dd, J = 9.0, 2.7 Hz, 1H), 8.01–7.96 (m, 2H), 7.82–7.75 (m, 3H), 7.64–7.60 (m, 2H), 7.53–7.43 (m, 3H). HRMS (ESI): cald. For C18H12N2NaO7S [M + Na]+ 423.0300; found 423.3700.
Preparation of 2,4-Dinitrophenyl-4′-(4-methylphenyl)-benzenesulfonate (2b). 2,4-dinitrophenol (0.692 g, 3.75 mmol) and 4′-methylbiphenyl-4-sulfonyl chloride (1.001 g, 3.75 mmol) were added to a flask containing 10 mL of tetrahydrofuran. Of aqueous potassium carbonate 1.6 M (5.0 mL, 7.92 mmol) was added dropwise to the stirred solution. The stirred solution was left at room temperate for 8 h. After reaction completion, a yellow precipitate was isolated via vacuum filtration to afford a yellow powder. The crude product was recrystallized in a small amount of dichloromethane to afford pale-yellow translucent crystals (1.075 g, 69% yield). M.p. 152–155 °C, Rf = 0.66 (CH2Cl2). 1H NMR (400 MHz, Chloroform-d) δ 8.78 (d, J = 2.7 Hz, 1H), 8.50 (dd, J = 9.0, 2.7 Hz, 1H), 7.99–7.93 (m, 2H), 7.81–7.74 (m, 3H), 7.56–7.49 (m, 2H), 7.30 (d, J = 7.9 Hz, 2H), 2.42 (s, 3H). HRMS (ESI): cald. For C19H14N2NaO7S [M + Na]+ 437.0400; found 437.3800.
Preparation of 2,4-Dinitrophenyl-4′-(4-flourophenyl)-benzenesulfonate (3b). 2,4-dinitrophenol (0.680 g, 3.69 mmol) and 4′-flourobiphenyl-4-sulfonyl chloride (1.006 g, 3.69 mmol) were added to a flask containing 10 mL of tetrahydrofuran. Of aqueous potassium carbonate 1.6 M (5.0 mL, 7.92 mmol) was added dropwise to the stirred solution. The stirred solution was left at room temperate for 8 h. After reaction completion, a yellow precipitate was isolated via vacuum filtration to afford a yellow powder. The crude product was recrystallized in a small amount of dichloromethane to afford pale-yellow translucent crystals (0.917 g, 59% yield). M.p. 140–143 °C, Rf = 0.64 (CH2Cl2). 1H-NMR (400 MHz, Chloroform-d) δ 8.78 (d, J = 2.7 Hz, 1H), 8.51 (dd, J = 9.0, 2.7 Hz, 1H), 8.02–7.94 (m, 2H), 7.82–7.71 (m, 3H), 7.65–7.55 (m, 2H), 7.24–7.14 (m, 2H). HRMS (ESI): cald. For C18H11FN2NaO7S [M + Na]+ 441.0200; found 441.3500.
Spectroscopic and Crystallographic Characterization
1H-NMR spectra (400 MHz) were recorded on a JEOL ECZ400 spectrometer using a DMSO-
d6 or Chloroform-
d solvent. Chemical shifts are reported in parts per million (ppm, δ) relative to the residual solvent peak, and coupling constants (J) are reported in Hertz (Hz). Results were analyzed and figures were created with the use of MestReNov [
8]. The spectra of all compounds synthesized can be found in
Figures S1–S8. Spectra were obtained from 16 scans.
The data yielded from the crystallographic characterization of x-substituted 2,4-dinitrophenyl-4′-phenylbenzenesulfonates can be found in
Tables S1–S15. Molecular structures of 2,4-dinitrophenyl-4′-phenylbenzenesulfonate (
1b), 2,4-dinitrophenyl-4′-(4-methylphenyl)-benzenesulfonate (
2b), and 2,4-dinitrophenyl-4′-(4-flourophenyl)-benzenesulfonate (
3b) can be found in
Figures S9–S11. X-ray diffraction was carried out on a Bruker APEXII CCD diffractometer with Mo
Kα radiation. The software used for data collection is as follows: Data collection:
APEX2 [
9]; cell refinement:
SAINT [
10]; data reduction:
SAINT [
10]; program used to solve structure: ShelXT [
11]; program used to refine structure:
OLEX2 [
12,
13]; program used to generate figures: Mercury [
14,
15,
16,
17,
18]; and absorbance correction:
SADABS [
19]. Interactive links for the structures of x-substituted 2,4-dinitrophenyl-4′-phenylbenzenesulfonates are found in the following figures: 2,4-dinitrophenyl-4′-phenylbenzenesulfonate (
1b),
Figure S9; 2,4-dinitrophenyl-4′-(4-methylphenyl)-benzenesulfonate (
2b),
Figure S10; 2,4-dinitrophenyl-4′-(4-flourophenyl)-benzenesulfonate (
3b), and
Figure S11.
3. Results and Discussion
In our work towards developing a facile synthesis of sulfonates, various reaction conditions were investigated. A variety of solvent and base combinations were used to ascertain their effects on the yield and reaction time of sulfonate derivatives, the results of which can be found in
Table 1. Entries are presented in order of decreasing yield. Successful formation of the desired product occurred in all cases, except entry 8 where dichloromethane and pyridine were used. This led to the development of a new synthetic method using an aqueous base and the water-miscible solvent, tetrahydrofuran. This single-phase solvent system defeats the need for a phase transfer catalyst and can support a wider range of starting materials. The use of an aqueous base and tetrahydrofuran resulted in higher yields, less environmental impact, and shorter reaction times. Additionally, the desired sulfonate product was isolated directly from the reaction mixture with good purity. An initial concern with this new synthetic method was the hydrolysis of sulfonyl chloride due to the presence of water. However, results show the rate of hydrolysis has little effect on yield.
A comparison of entries 2, 3, and 5 revealed the electronic and steric effects from biphenyl sulfonyl chloride derivatives and their implications in the synthesis of x-substituted 2,4-dinitrophenyl-4′-phenylbenzenesulfonates. The structure and yield of these sulfonates are shown in
Figure 1. The highest yielding x-substituted 2,4-dinitrophenyl-4′-phenylbenzenesulfonate was compound
1b, which has no substituent. Compounds
2b and
3b, which contain electron-donating and electron-withdrawing groups, respectively, were slightly lower in yield. In general, the reaction yield may be affected by electronic factors, steric factors or a combination of both. Although it is not clear to us which factor is responsible for the difference in yields, one can imagine a combination of electronic factors and steric factors to be responsible. However, the difference in yields was not statistically significant enough for us to draw clear conclusions on what exactly accounts for the difference in reaction yields.
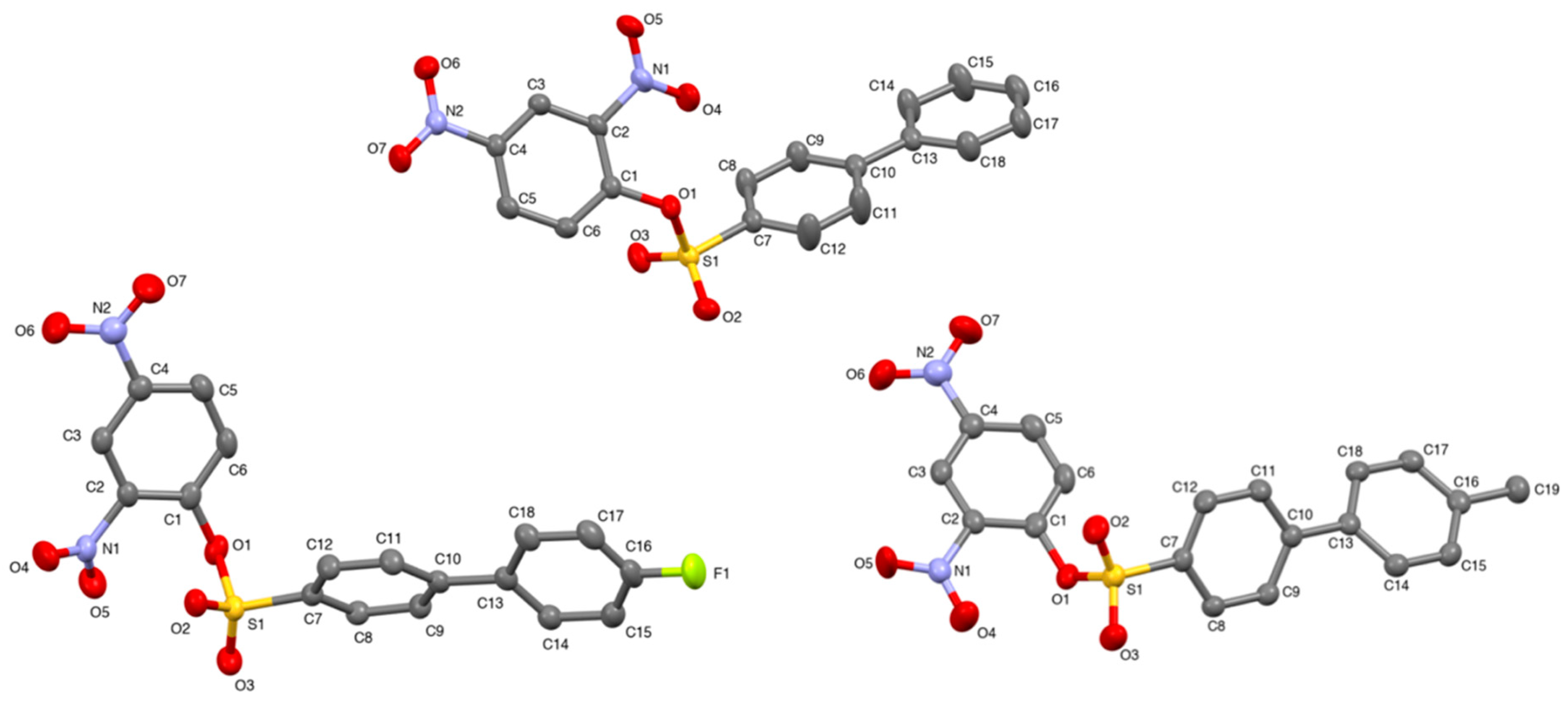
Crystal structures of the x-substituted 2,4-dinitrophenyl-4′-phenylbenzenesulfonates were obtained by single crystal X-ray diffraction. Pertinent data such as bond angles, bond lengths, torsion angles, and other crystallographic parameters can be found in the supplementary material. The asymmetric units of 2,4-dinitrophenyl-4′-phenylbenzenesulfonate (
1b), 2,4-dinitrophenyl-4′-(4-methylphenyl)-benzenesulfonate (
2b), and 2,4-dinitrophenyl-4′-(4-flourophenyl)-benzenesulfonate (
3b) can be found in
Figure 2.
The x-substituted 2,4-dinitrophenyl-4′-phenylbenzenesulfonates exhibited a two-fold screw axis (−x, 1/2 + y, 1/2 − z), and glide plane geometry (x, 1/2 − y, 1/2 + z) with an inversion center (−x, −y, −z). Screw axis and glide plane geometries are indicative of efficient packing. All three sulfonate structures exhibited a monoclinic system (
P2
1/
c space group). The central sulfur atom, S1, is a slightly distorted tetrahedron according to the τ
4 descriptor for four-fold coordination [
20]. The aryl groups of the sulfonates were oriented gauche about the S1–O1 bond with the following C7–S1–O1–C1 torsion angles: 131.6 (1)°,
1b; −94.0 (1)°,
2b; −92.7 (1)°, and
3b (
Table 2). The S1 = O2 and S1 = O3 bond lengths were in good agreement with known values. The S1–C7 and S1–O1 bond lengths were 1.751 (2) Å and 1.626 (1) Å for compound
1b; 1.745 (2) Å and 1.619 (1) Å for compound
2b; and 1.743 (2) Å and 1.623 (1) Å for compound
3b, respectively. The S1–O1–C1 bond angles for compounds
1b,
2b, and
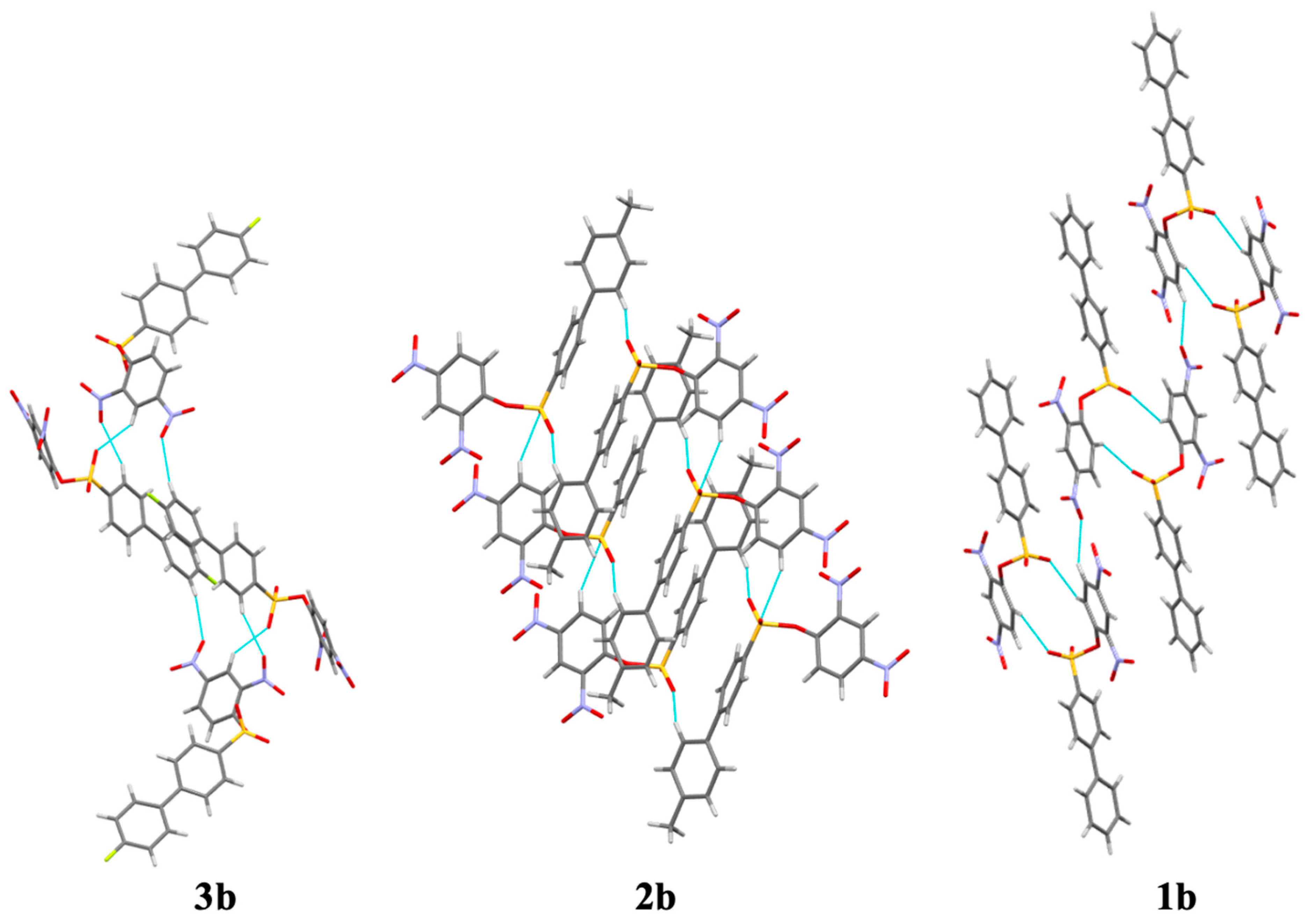
3b were 120.5 (1)°, 120.4 (1)°, and 119.2 (1)°, respectively. Molecules were liked by π–π interactions, C–H···O hydrogen bonds, C–H···π interactions, and, in the case of compound
3b, C–H···F hydrogen bonds (
Figure 3). Hydrogen atoms bonded to carbon atoms were placed in calculated positions and refined as riding: C–H = 0.95–1.00 Å with a fixed
Uiso(H) = 1.2
Ueq(C) for all C–H groups.
The extent of hydrogen bonding varied significantly in the sulfonate derivatives. In the case of compound 2b, only two C–H··· O hydrogen bonds were observed (
Table 3). A greater number of hydrogen bonds in compounds
1b and
3b lend credence to a lattice dependent on hydrogen bond contacts for efficient packing. The F1···H17 bond in compound
3b had a bond length of 2.450 Å, the shortest and therefore strongest hydrogen bond of the sulfonate compounds.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
