
When Stereochemistry Raised Its Ugly Head in Coordination Chemistry—An Appreciation of Howard Flack


Department of Chemistry, University of Basel, BPR 1096, Mattenstrasse 24a, CH-4058 Basel, Switzerland
*
Author to whom correspondence should be addressed.
Chemistry 2020, 2(3), 759-776; https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry2030049
Submission received: 24 August 2020
/
Revised: 3 September 2020
/
Accepted: 8 September 2020
/
Published: 12 September 2020
(This article belongs to the Special Issue Memorial Issue Dedicated to Dr. Howard D. Flack: The Man behind the Flack Parameter)
Abstract
:Chiral compounds have played an important role in the development of coordination chemistry. Unlike organic chemistry, where mechanistic rules allowed the establishment of absolute configurations for numerous compounds once a single absolute determination had been made, coordination compounds are more complex. This article discusses the development of crystallographic methods and the interplay with coordination chemistry. Most importantly, the development of the Flack parameter is identified as providing a routine method for determining the absolute configuration of coordination compounds.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
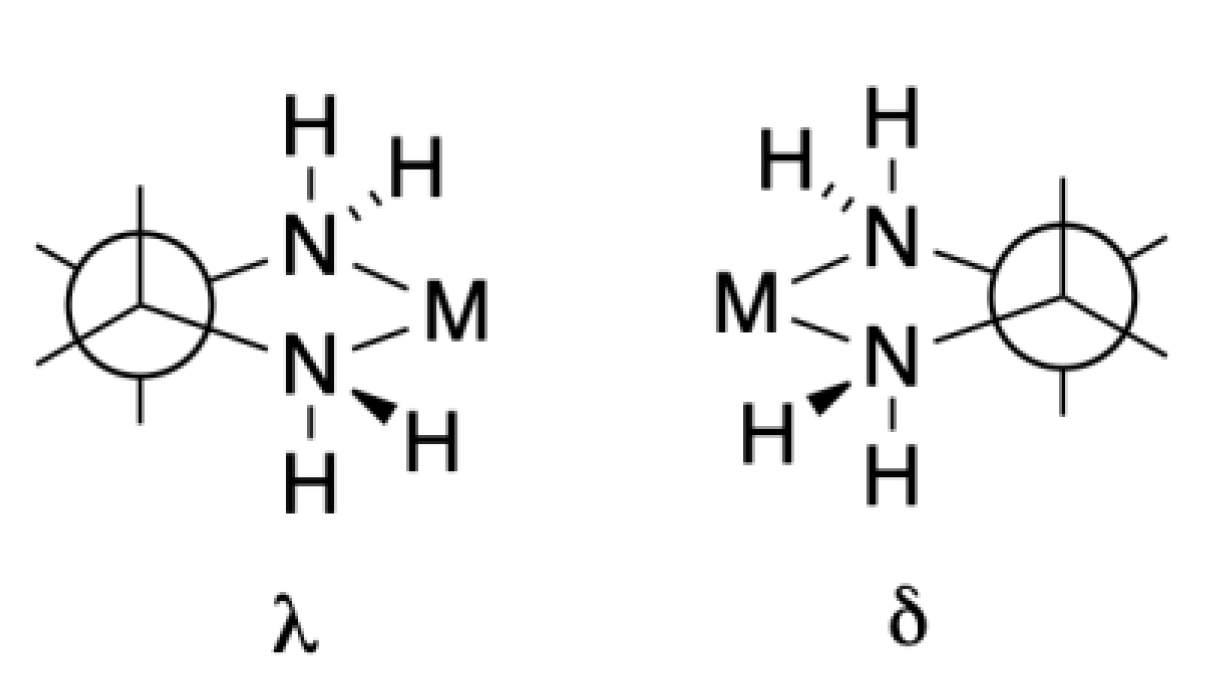{kind=link}
1. Introduction
When the authors first studied chemistry, the accepted mantra was that crystallography could not be used routinely to determine the absolute configuration of a compound. The basis for this assertion was subsequently shown to be incorrect, but routine determination of the absolute configuration of a compound by crystallography remained in the realms of the relatively exotic. In 1966, a comprehensive listing identified 54 organic structures, the absolute configurations of which had been determined by crystallographic methods [1]. Over the next few years, more determinations were reported, with an additional 40, 39 and 133 absolute structures identified in 1968 [2], 1969 [3] and 1970 [4], respectively. For organic compounds, classical correlation methods relying on real or virtual chemical transformation were still used to relate the configuration to one of the known absolute configurations. Indirect methods of determining the absolute configuration relied upon esoteric spectroscopic methods or empirical correlations such as the octant rule [5]. In 1983, the situation was changed by a paradigm-shifting publication by Howard Flack [6].
This article is not a detailed account of the crystallographic background, but rather takes the opportunity to survey the impact of crystallographic methods on the investigation of coordination compounds and to appreciate the broader contributions of Howard Flack. Nevertheless, a short introduction to the crystallographic complexities is included. Optical activity and subsequently chirality have had a profound influence on the development of coordination chemistry, from the time of Werner onwards.
2. Chirality
2.1. Through a Glass, Darkly
From the earliest documented history, mirrors have excited and fascinated Mankind. In the New Testament of the Christian Bible, the phrase “For now we see through a glass, darkly” refers to a poorly discerned image in a mirror. Mirrors, images in mirrors, and mirror images influenced Western art and literature from the early Renaissance to modern times [7]. The 19th Century C.E. imagination was energized and inspired by mirror images and their relationship to “reality”. One of the best known literary works on this theme is the 1871 work Through the Looking-Glass, and What Alice Found There by Lewis Carroll (Figure 1) [8].
By the end of the 19th Century C.E., chemists were familiar with the fact that some compounds exhibited the phenomenon of optical activity. Jacobus Henricus van’t Hoff [9,10] and Joseph Achille LeBel [11] had independently explained the phenomenon of optical activity as being a consequence of four groups attached to a carbon atom being oriented in space in the form of a tetrahedron. In his paper, “A suggestion looking to the extension into space of the structural formulas at present used in chemistry and a note upon the relation between the optical activity and the chemical constitution of organic compounds”, van’t Hoff used the word asymmetric to describe a carbon atom with four different groups attached. Both van’t Hoff and LeBel showed that when four different groups were bonded in a tetrahedral arrangement about a carbon atom, the object and its mirror image were non-superposable. Although it is critical to distinguish between the observable phenomenon (optical activity) and its origin (dissymmetry), it was slowly becoming clear by the end of the 19th Century, that optical activity should be regarded as arising from a molecular dissymmetry rather than an asymmetry associated with a single atomic centre [12,13,14,15,16,17,18].
Although most people “knew” what a mirror image was and understood, on some level at least, the consequent left–right inversion, it fell to William Thomson (1824–1907), better known under his later title of Lord Kelvin to make a scientific definition that brought clarity to the concept of mirror images. Kelvin is well known for his works in physical chemistry and thermodynamics, indeed he is commemorated in the S.I. unit of temperature, the kelvin. Nevertheless, for us his critical contribution, which was not widely recognized or adopted at the time, is the introduction of the terms chiral and chirality; “I call any geometrical figure, or group of points, chiral, and say it has chirality, if its image in a plane mirror, ideally realized, cannot be brought to coincide with itself. Two equal and similar right hands are homochirally similar. Equal and similar right and left hands are heterochirally similar or ‘allochirally’ similar (but heterochirally is better). These are also called ‘enantiomorphs,’ after a usage introduced, I believe, by German writers. Any chiral object and its image in a plane mirror are heterochirally similar” [19]. The word ‘chiral’ is derived from the Greek word for hand, χειρ. Kelvin’s dismissal of enantiomorphs is a little disingenuous. Not only was this description being used by the United Kingdom chemical community in the 1890s [20,21], but also had a long tradition in the German literature [22,23,24] being introduced by Naumann in 1856 [25] and treated at length in Schoenflies’ 1891 text on crystallography [26]. The IUPAC has subsequently made recommendations on the basic terminology of stereochemistry [27] and Gal has published extensively on the early etymology of stereochemical terms [28,29,30,31,32].
As mentioned above, Kelvin’s proposal was widely ignored by the community and according to a Scifinder search, the first subsequent mention of the word chiral in the chemical literature occurred in the early 1920s [33,34] and with no further mention up to 1950, when Raman used the term in the context of quartz crystals [35]. The rehabilitation of the word seems to stem from letters to Nature in the late 1950s [36,37]. It is interesting to note that in the first two publications introducing the Cahn–Ingold–Prelog system, the words chiral and chirality are not used [38,39] and only in the third paper do they appear extensively, with the comment “This useful word [chirality] was brought to our attention by Professor K. Mislow, who referred us to Webster’s Dictionary (2nd Edition), where chiral is defined as Of, or pertaining to the hand, specifically turning the plane of polarisation of light to either hand”[40].
2.2. Some Definitions
In introducing the concept of chirality, Kelvin made the link between molecular dissymmetry and the experimental observable of optical activity. It cannot be stressed strongly enough that chirality is a property of an object as a whole rather than something associated with a portion of that object, such as an “asymmetric atom”. The term chirality equates exactly with the term dissymmetry (dissymmétrie) originally used by Pasteur [28,29,41,42,43]. Note that dissymmetric and asymmetric are not equivalent—asymmetric means without symmetry whereas dissymmetric is more specific and means “lacking improper symmetry elements”. In simpler language, dissymmetric means chiral. The IUPAC definition of asymmetric is very clear [44]:
"Lacking all symmetry elements (other than the trivial one of a one-fold axis of symmetry), i.e., belonging to the symmetry point group C1. The term has been used loosely (and incorrectly) to describe the absence of a rotation–reflection axis (alternating axis) in a molecule, i.e., as meaning chiral, and this usage persists in the traditional terms asymmetric carbon atom, asymmetric synthesis, asymmetric induction, etc".
If an object is chiral, then the object and its mirror image are not identical and cannot be superposed. The IUPAC uses the word superpose rather than the more familiar English word superimpose.
The concepts of chirality and symmetry are closely related. This article is not intended as a primer in group theory, and the requirements for chirality are presented without clarification of the symmetry operations that are involved. The reader is referred to standard texts on group theory for further education and elucidation [45].
For a molecule to be chiral, it must have no symmetry elements of the second kind, such as a mirror plane, a centre of inversion or a rotation–reflection axis. The presence of any one of these symmetry elements precludes chirality. It is not sufficient to only look for a mirror plane—even if no mirror plane is present, the presence of an inversion centre will ensure that an object is not chiral. This reinforces the primary criterion for chirality—that of non-superposability of the object on its mirror image. If an object is superposable on its mirror image, it is described as being achiral. Such objects possess a mirror plane, a centre of inversion, or a rotation–reflection axis. And remember, nothing can surpass a good quality model or diagram to determine whether a molecule is chiral. The IUPAC definition of chiral is concise and precise [44]:
"The geometric property of a rigid object (or spatial arrangement of points or atoms) of being non-superposable on its mirror image; such an object has no symmetry elements of the second kind (a mirror plane, σ = S1, a centre of inversion, i = S2, a rotation-reflection axis, S2n). If the object is superposable on its mirror image the object is described as being achiral."
3. The Importance of Chirality in Coordination Chemistry
3.1. It all Began with Werner
The observation of different optical forms of chiral coordination compounds played a critical role in the development and acceptance of Werner’s coordination theory and the establishment of the octahedral geometry of six-coordinate metal complexes. Werner reported the resolution of salts of cis-[CoCl(en)2(NH3)]2+ [46], cis-[CoBr(en)2(NH3)]2+ [46], cis-[Co(en)2(NO2)2]+ [47], cis-[CoCl(en)2(NO2)]+ [48], cis-[CoCl2(en)2]+ [49], [Co(en)3]3+ [50], [Rh(en)3]3+ [51] and the preparation by ligand exchange reactions of resolved precursors of optically active salts or by seeding of cis-[Co(en)2(H2O)(NH3)]3+ [46], cis-[Co(CO3)(en)2]+ [52], cis-[Co(C2O4)(en)2]+ [53,54], cis-[CoBrCl(en)2]+ [55], [(en)2Co(NH2)(NO2)Co(en)2]4+ [56], [(en)2Co(NH2)(O2)Co(en)2]4+ [57] and, finally the “all-inorganic” compound [Co{(OH)2Co(NH3)4}3]6+ [58]. For determining the optical rotation, Werner utilized an F. Schmidt and Haensch polarimeter [59], and originally used the d and l descriptors to describe the compounds with positive and negative rotation of 656.3 nm wavelength linearly polarized light, respectively. He subsequently reported optical rotatory dispersion (ORD) spectra which are simply plots of the variation in optical rotation of linearly polarized light with wavelength [60]. The development of the successful resolution methods is described in the doctoral thesis of Victor King [61]. A number of excellent surveys of this aspect of Werner’s work have been published [62,63,64].
3.2. Non-Crystallographic Approaches to Determining the Absolute Configuration of Metal Complexes
Although Werner had successfully obtained the large selection of optically pure compounds described above, they were only defined by a relative configuration. Subsequent workers adopted the practice of using the (+)d or (−)d notation to identify the clockwise or anticlockwise rotation at the sodium D-line (589 nm). The situation regarding absolute configuration was worse than that with organic compounds, as only few transformations between optically active species were known and there was no understanding of rules for retention or inversion at octahedral centres. This meant that it was not possible to use mechanistic principles to correlate the relative configuration in a series of complexes. An example of the complexity of the situation is seen in a classical publication from John Bailar Jr. in which the the reaction of (−)d-cis-[CoCl2(en)2]Cl with liquid ammonia at 196 K gave (−)d-cis-[Co(en)2(NH3)2]Cl (−32°), whereas reaction with a saturated solution of ammonia in methanol at 298 K gave (+)d-cis-[Co(en)2(NH3)2]Cl (+31°) [65].
In the 1930s, Matthieu attempted with only poor success to correlate the relative (or indeed absolute) configuration of complexes [66,67,68,69,70,71,72,73,74,75,76,77,78,79] with the sign of the Cotton effects that they exhibited [80,81,82]. If a compound under investigation has an absorption band in the region of the optical rotatory dispersion spectrum, an anomalous dispersion effect is seen. This is called the Cotton effect and refers to the change in sign of the optical rotatory dispersion close to an absorption band. Close to the region where light is absorbed, the magnitude of the optical rotation varies rapidly with wavelength, passes through zero at the absorption maximum and continues varies with wavelength but with an opposite sign.
The first successful attempts at determining the absolute configuration of octahedral transition metal complexes were made by Werner Kuhn in the 1930s [83,84]. Kuhn used a coupled oscillator model to calculate the sign of the Cotton effect. Kuhn assigned the absolute configuration of Δ to the (−)d-[Co(C2O4)3]3– anion [83] and the (−)d-[Co(en)3]3+ cation (Figure 2) [84].
4. A Brief History of Crystallography
4.1. X-rays—The Early Days
The availability of routine X-ray crystallographic methods for the determination of solid state structures has had a profound effect on the way in which coordination (and other) chemists characterize their compounds. It is necessary to add a small caveat to curb the enthusiasm of the chemist—typically a single crystal structure provides information about the arrangement of atoms, molecules and ions within the crystal studied but, in the absence of other methods, gives no information about the bulk material.
X-rays were discovered in 1895 by Wilhelm Conrad Roentgen when he identified a new type of radiation from a Crookes tube [85,86]. Laue demonstrated that the wavelengths of X-rays were commensurate with the spacings between atoms, ions and molecules in crystals and that, therefore, an optical diffraction was to be expected [87,88,89]. This critical work by Laue was paradigm shifting for chemistry—suddenly, atoms became objects which could be detected and quantified in terms of size and spatial position. By 1913, the Braggs had built their first single-crystal X-ray spectrometer with a gold leaf electroscope as the detector and the crystal could be rotated in the X-ray beam.
4.2. From X-rays to Chemical Crystallography
Chemical crystallography began when William Henry Bragg and his son William Lawrence Bragg showed that the diffraction data could be solved by ad hoc trial-and-error methods to yield models of the spatial arrangement of atoms and ions in crystalline solids such as NaCl, KCl, KBr, KI, ZnS and diamond [97,98,99,100,101,102,103,104,105].
4.3. Initial Approaches to the Phase Problem
X-ray detectors measure the intensity of radiation but not the phase of that radiation. Any phase change due to scattering of radiation is almost the same for all atoms. The underlying physics of X-ray diffraction was developed by Darwin and Bragg from 1914 onwards [106,107,108]. The diffraction data correspond to the amplitude of the Fourier transform of the unit cell electron density in the unit cell. Friedel pairs of diffraction spots are Bragg reflections which are reflected through the origin and related by Friedel’s law, which states that they have equal amplitudes and opposite phases [109].
The electron density can be obtained by Fourier synthesis if the phase is known. The introduction of Fourier analyses [110], and subsequent elaboration by Patterson with his eponymous function, started the transition of crystallography to become a more routine technique and also provided the methods that could be used for phase evaluation of the diffraction data [111,112]. The Patterson method recognizes that although the phase information is needed to locate the peaks in electron density within a unit cell, the magnitudes of the structure factors contain information about the spacings. The Patterson map shows peaks at all positions corresponding to an interatomic vector rather than the position of the atoms. Patterson methods can only be used for relatively small molecules (<50 atoms) and are not appropriate for the ab initio determination of absolute configuration.
The next development was the study of series of compounds which only differed by the replacement of one atom in the structure. The method relied upon the replaced atom being on a special position and having a different scattering factor. Within these constraints, the effect is the modification of the structure factors by values dependent on the phase of the reflection [113]. One of the earliest applications was to the alums, and diffraction data from the compounds AB(SO4)2•12H2O (A = NH4, K, Rb, Cs, Tl; B = Cr, Al) were used to determine the structures using this isomorphous replacement method [114]. Probably the most spectacular early success used the related heavy atom method in an isomorphous series for solving and refining the centrosymmetric phthalocyanine complexes [M(pc)] (M = Ni, Pt) (Figure 3) and consequently, the correct phasing of H2Pc [115,116,117]. One of the early applications to a chiral molecule was reported in the structure of cholesteryl iodide. In this compound, the iodine is not on a special position and the step-wise work-flow is worth rehearsing to demonstrate the skill and tenacity of the early crystallographers: (1) measure the intensities of all reflections, (2) locate the heavy atoms from the Patterson map, (3) calculate the phases from the heavy atom contributions, (4) use chemical knowledge to choose correct distances and angles, and (5) recalculate phases on all the atoms and repeat the Fourier summations.
4.4. The Bijvoet Method
Most crystallographers accepted Friedel’s law that the Bragg pairs had equal amplitudes and opposite phases and considered that X-ray methods could not be used directly for the determination of absolute configuration without chemical modification or isomorphous replacement. However, a number of physicists and crystallographers remained open-minded. If the X-ray radiation has a wavelength close to the absorption edge of an atom in the compound, a small phase change occurs in the scattered X-rays from these atoms, phenomenologically similar to the Cotton effect. As a consequence, the diffraction pattern is no longer centrosymmetric but has pairs of spots with unequal intensities. This effect was first reported in studies of zinc blende (ZnS) using W Lβ (1.2447–1.3017 Å) [118] or Au Lα1 (0.8638 Å) [119] radiation, which are close to the Zn K edge (1.2837 Å). However, these results were forgotten by the community for over a decade.
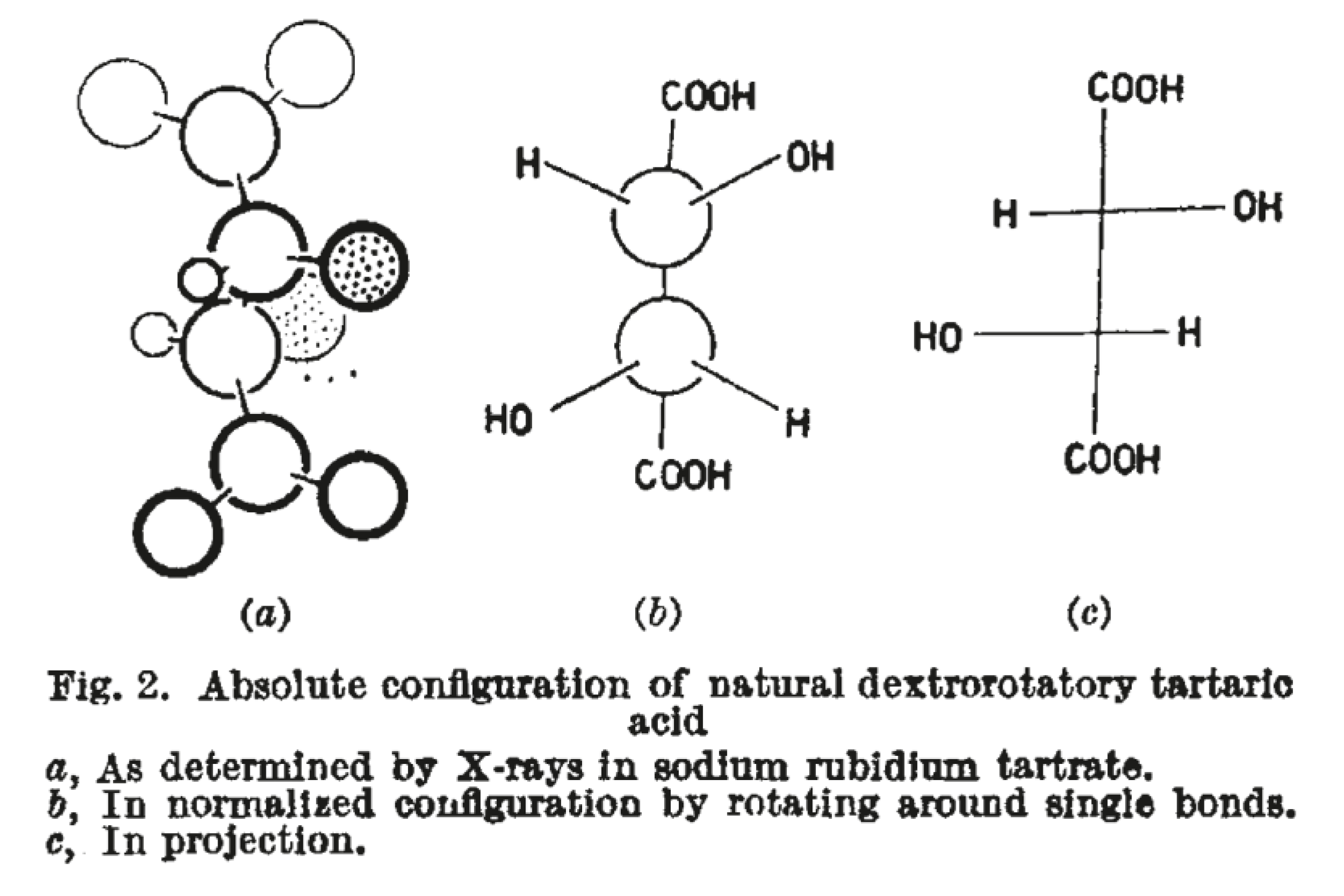
At the end of the 1940s, the Dutch crystallographer Johannes Bijvoet developed a general isomorphous replacement method for non-centrosymmetric structures and successfully solved the structure of strychnine using the sulfate and selenate salts [120,121,122,123,124,125]. However, the most significant contribution of Bijvoet was in rediscovering that X-ray analysis could determine absolute configurations using the anomalous scattering of X-rays of a wavelength close to an X-ray absorption of an atom in the compound. This effect was observed in sodium rubidium tartrate (Figure 4) using Zr Kα radiation, which is close to the K-edge of rubidium, and in the early 1950′s, Bijvoet published a series of landmark papers, commencing in 1951 with “Determination of absolute configuration of optically active compounds by means of X-rays” [95,126,127,128].
A further simplification was introduced by Mathieson who, making use of the known absolute configurations that were available from the Bijvoet method, proposed the use of diastereoisomers in which one of the stereochemical centres was absolutely defined. This avoided the need to use X-ray radiation of a wavelength close to an absorption edge in the compound and utilized the heavy atom method with auxiliaries of known absolute configuration such as (R) or (S)-chloroiodoacetate [129].
In parallel, direct methods were being developed to permit the extraction of the phase information from the crystallographic structure factors and crystallography entered its modern phase [130,131]. Direct methods estimate and then test and select the initial phases and were initially introduced for centrosymmetric space groups and subsequently extended to non-centrosymmetric space groups [132,133].
5. Chirality and Crystallography
Chirality, crystallography and symmetry are related in intimate and subtle ways that lead to fascination and frustration. The nuances and manifestations of these relationships delighted and occupied Howard Flack through much of his career. In this section, we rehearse a few of the consequences of these relationships for coordination compounds.
5.1. Absolutism
Chemists tend to talk of absolute configuration meaning the spatial arrangement of the atoms of in a chiral molecular entity and denoted by a stereochemical descriptor such as R/S, P/M, D/L or Δ/Λ. In contrast, crystallographers use the term absolute structure, introduced in 1984 by Peter Jones [134], to describe spatial arrangement of atoms in a non-centrosymmetric crystal [135]. The term absolute structure refers specifically to the crystalline state, whereas the broader term absolute configuration can be applied to any phase or to solutions.
5.2. Chiral Space Groups and Chiral Molecules
There are 230 three-dimensional space groups. Of these 22 are chiral space groups comprising 11 enantiomorphic pairs (P41-P43: P4122-P4322: P41212-P43212: P31-P32: P3121-P3221: P3112-P3212: P61-P65: P6122-P6522: P62-P64: P6222-P6422: P4132-P4332). It would be tempting to expect that a chiral molecule would crystallize in a chiral space group, but that would be too simple, and would not take the infinite perversity of nature into account. In Section 2.2, we stated that for an object to be chiral, it must possess no symmetry elements of the second kind. Of the 230 space groups, there are 65 (including the 22 chiral groups) which only possess operations of the first kind (rotations, rotation–translations, and translations) and these 65 are known collectively as the Sohncke groups [136]. Enantiopure chiral molecules must crystallize in one of these 65 Sohncke space groups.
Returning to the perversity of nature, achiral molecules can also occur in any of the 230 space groups—the packing of achiral objects may be in a chiral or an achiral manner. A systematic survey of achiral molecules in non-centrosymmetric space groups was reported in 2005 [137]. Similarly, a crystal composed of equal numbers of the two enantiomers of a molecule (a racemate) may occur in any of the 230 space groups, although examples in the Sohncke set are very rare.
5.3. The Flack Parameter
By the early 1980s, the ratios of crystallographic R or Rw values for structures refined with alternative absolute configurations were being used to assign absolute configurations and absolute structures, although the statistical methods were debated [138,139]. In 1983, Flack published a paper entitled “On Enantiomorph-Polarity Estimation” which revolutionized the field of structure determination of chiral molecules and structures [6]. Rogers had introduced a parameter η which he proposed as a good method for distinguishing between refinements of structures with opposite configurations. Flack pointed out an inherent problem with the use of η and introduced a new parameter that he called x and defined in terms of the structure factor for reflection h in Equation (1).
If the experimental data and the model used for refinement have the same chirality, the value of x is 0, and if they have opposite configurations, then x has a value of 1. A value of 0.5 indicates a racemic crystal with equal amounts of both enantiomer. This single publication revolutionized the determination of absolute structures. Flack also recognized that there was no need to formally solve the structures in both configurations and in a note added in proof to the original paper, he notes that “The refinement of … x has now been added as a permanent feature in our implementation of the X-RAY76 system … x is varied automatically in the final stages of refinement with a non-centrosymmetric structure.” The direct refinement of the Flack parameter together with other structural parameters is now routine. To date, this paper has been cited 11,161 times. The parameter x rapidly, and universally, became known as the Flack Parameter. To date, the Flack parameter does not yet belong to the data indexed by the Cambridge Structural Database (CSD) [140,141].
5.4. Some Musings on Racemates, Spontaneous Resolution and Other Complexities
Although the introduction of the Flack parameter transformed the mechanics and quantification of the determination of absolute configurations, it is our opinion that a second paper by Flack is equally important. The 2003 article entitled “Chiral and Achiral Crystal Structures” provides the clearest and most comprehensive compilation of the language of chirality and crystallography that we know. He commences by identifying three origins of chirality in crystal structures: (i) the molecular components (ii) the crystal structure itself and (iii) the symmetry group of the structure. The really valuable part of the publication concerns the quantification of a descriptor (racemic, racemate) that is often used loosely or incorrectly by the chemical community [142]. These descriptors simply refer to equimolar amounts of opposite enantiomers with no restriction to phase. If the two enantiomers are not present in equal amounts, the correct description is an enantiomeric mixture.
Particularly important is the identification of a racemic conglomerate, obtained when the crystallization of a solution of a racemic compound results in spontaneous resolution and the generation of equal numbers (strictly equal weights) of enantiopure crystals each only containing components with only one chirality.
He then identified the term ordered racemic crystal structure or racemic structure to describe a crystal containing an ordered array of equal numbers of the different enantiomers. The term anomalous racemate had been used to describe ordered crystals in which the ratio of the two enantiomers was not 1:1, and Flack proposed a new description of M:N mixed enantiomeric crystal structure or M:N enantiomeric structure, where M:N is the ratio of the two enantiomers present. He further proposed the term disordered mixed enantiomeric structure to replace pseudoracemate in describing crystals in which each position can be occupied by a molecule of either configuration. This publication is recommended to anyone who wishes to think more deeply about the consequences of chirality in the solid state.
6. Chiral Coordination Compounds
Although chirality has been so important in the development of coordination chemistry, it is surprising how few monographs or comprehensive reviews exist. The standard works on stereochemistry concentrate on organic compounds [143]. For coordination chemistry, the bibles are von Zelewsky’s 1996 work “Stereochemistry of Coordination Compounds” [144] and Hawkins earlier work “Absolute Configuration of Metal Complexes” [145]. A more organometallic-oriented, but extremely useful and relevant, presentation is found in the 2008 book “Chirality in Transition Metal Chemistry” by Amouri and Gruselle [146]. Two earlier reviews complement these to provide an overview of the period at which crystallography was starting to provide information about absolute stereochemistry and a volume of the ACS Symposium Series from 1980 that was dedicated to the “Stereochemistry of Optically Active Transition Metal Complexes” provides an excellent overview of the state of the art at that time [147,148,149]. Volume 12 of Topics in Stereochemistry was entitled “Topics in Inorganic and Organometallic Stereochemistry”and for the real afficionados, the chapter “Conformational Analysis and Steric Effects in Metal Chelates” provides a masterly overview of conformational effects within chelate rings and is both a tour de force and a challenge [150].
6.1. The First Absolute Determination
The first determinations of the absolute configuration of metal complexes using the Bijvoet method was reported in 1955 by Yoshihiko Saito who studied Λ- and Δ-[Co(en)3]Cl3·0.5NaCl [151]. In a 1974 review, Saito surveyed the literature up to 1972 and reported that, in the intervening 23 years, the absolute configuration of salts of only an additional 53 metal cations had been determined through structural characterization [148].
The list of compounds characterized provides a snap-shot of the contemporary coordination chemistry. The first class included those which required the “pure” Bijvoet anomalous dispersion approach for compounds such as [ML3]n+ (L = chelating bidentate ligand) or cis-[ML2X2]n+ (L = chelating bidentate ligand, X = monodentate ligand). The structural elucidation of the diamines into related polyamines with polymethylene spacers resulted in new classes of chiral complexes in which the chirality arises from the dissymmetric arrangement of the ligand donor atoms about the metal centre (see the general references at the beginning of this section). These latter compounds can be seen as the direct progenitors of macrocyclic chemistry and the grandparents of supramolecular chemistry. Interesting as these compounds are, we return to our main theme when we consider the remaining types of compounds characterized.
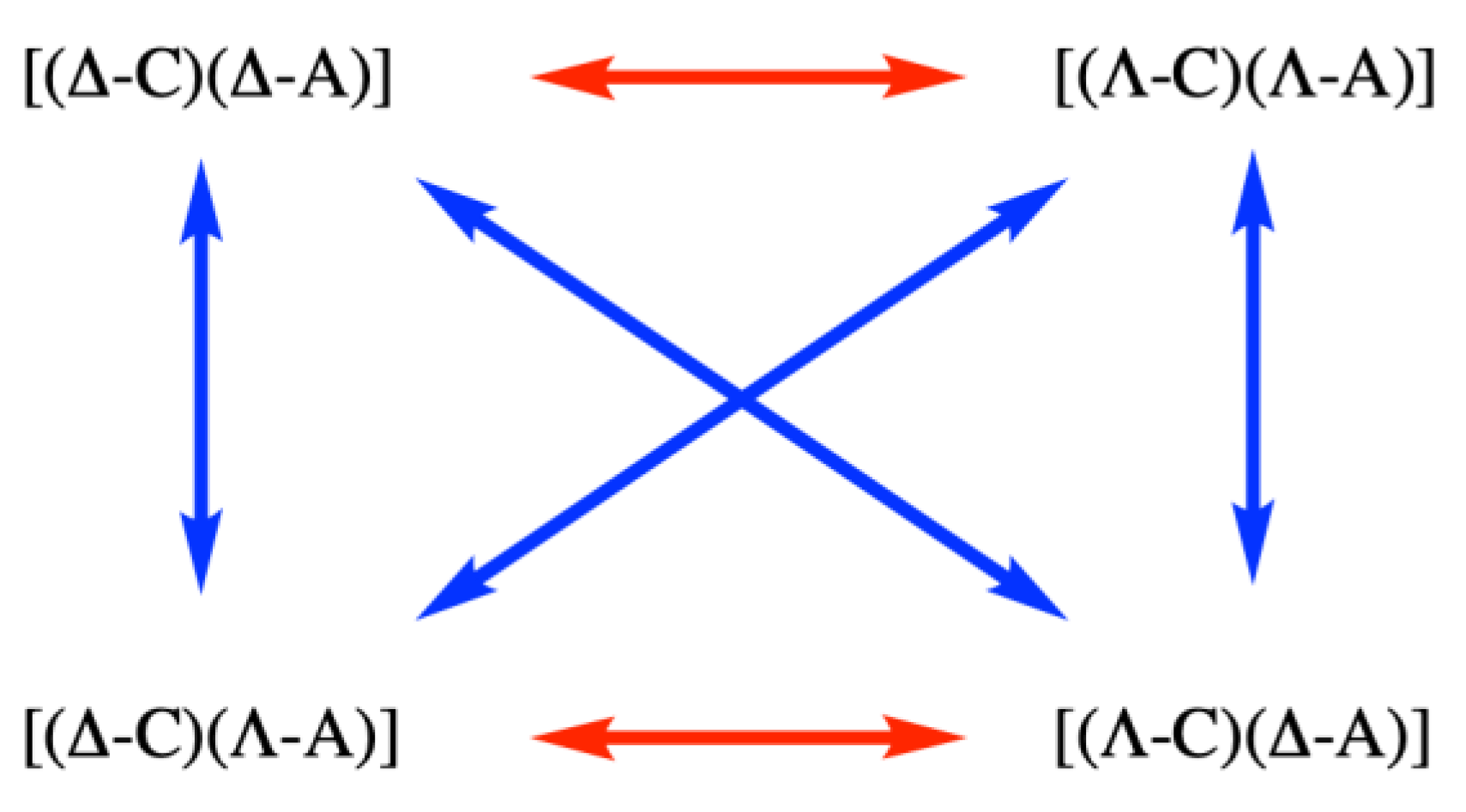
We start with a small digression into the consequences of multiple stereogenic centres in a compound. The classical method of resolving a chiral coordination compound, for example a cation C+, is to form salts with a chiral anion A–. Four possible compounds can be formed, of which [(Δ-C)(Δ-A)] and [(Λ-C)(Λ-A)] form a pair of enantiomers as do [(Δ-C)(Λ-A)] and [(Λ-C)(Δ-A)] (Figure 5). All other relationships between the combinations are as diastereoisomers. Enantiomers have identical physical properties (solubility, melting point, NMR spectra) and only differ in their interactions with other chiral agents (for example polarized light). On the other hand, diastereoisomers differ in physical properties as the spatial interactions between, for example, (Δ-C) with (Λ-A) and (Δ-A) will be different (think about putting your left foot into a left shoe and into a right shoe—the thermodynamics of the pairing are different). A typical resolution method might involve treating a racemic mixture of cations (Λ-C) and (Δ-C) with (Λ-A) and hoping that the solubility of the diastereoisomeric salts [(Δ-C)(Λ-A)] and [(Δ-C)(Δ-A)] might be sufficiently large that one selectively precipitates or crystallizes. Salts of this type provided the next class that were studied extensively in this first period of determining the absolute configuration of coordination compounds. Very typically, chiral organic anions, or anions containing coordinated chiral organic ligands, were used for the precipitation. After Bijvoet established the absolute configuration of tartrate, known chemical transformations allowed correlation to a large number of other “simple” chiral organic compounds. As a consequence, the absolute configuration of the organic component of the anion was generally known when the structure of the diastereoisomeric salt [(C)(A)] was determined and the configuration at the cation followed from the correct assignment of the (known) configuration to the anion.
The final class of compounds studied belonged to the next level of structural development in which the ligands themselves are chiral. Consider propane-1,2-diamine (H2NCH2C*H(Me)NH2, pn) in which C2 (indicated with an asterisk) is a stereogenic centre; as a result the ligand pn is typically encountered as one of the two pure enantiomers R-pn or S-pn, as the racemate containing equal amounts of R-pn and S-pn or the enantiomeric mixture x(R-pn):1–x(S-pn). We now consider the formation of the octahedral complex [M(pn)3]n+.
Starting with enantiomerically pure R-pn or S-pn, four compounds could be obtained: Δ-[M(S-pn)3]n+ and Λ-[M(R-pn)3]n+ (a pair of enantiomers) and a second pair of enantiomers Λ-[M(S-pn)3]n+ and Δ-[M(R-pn)3]n+. All other relationships are diastereoisomeric. Thus, reaction with R-pn will give two chemically distinct diastereoisomers Λ-[M(R-pn)3]n+ and Δ-[M(R-pn)3]n+.
The more perverse readers will now ask what happens with the racemic ligand? In this case, in addition to the four homoleptic complexes already mentioned, we now have the possibility of the heteroleptic complexes Δ-[M(S-pn)2(R-pn)]n+ and Λ-[M(R-pn)2(S-pn)]n+ (a pair of enantiomers) as well as Δ-[M(R-pn)2(S-pn)]n+ and Λ-[M(S-pn)2(R-pn)]n+ (a second pair of enantiomers). But it gets worse! The two nitrogen donor atoms of each pn ligand are not chemically equivalent as one is attached to the stereogenic C2 and the other to C1. As a consequence, there are also the complexes with a facial or meridional arrangement of the C*-NH2 donors! Once again, this is not the place to follow the fascinating stereochemistry of “simple” systems like this.
We make one further foray into the world of stereochemical complexity revealed in these early crystallographic studies. Our discussion commenced with the definition of the Δ- and Λ-configuration using [M(bpy)3]n+ complexes as an example (Figure 2). Why did we not start with Werner’s [M(en)3]n+ compounds? Whereas the chelate ring in bpy complexes is planar, that in en complexes is non-planar and chiral. The configuration is denoted by the descriptor δ or λ as defined in Figure 6. Although individual δ or λ chelate rings are enantiomeric, the situation is different in [M(en)3]n+ when we have the possibilities of Δ-[M(δ-en)3]n+ and Λ-[M(λ-en)3]n+ (a pair of enantiomers), Λ-[M(δ-en)3]n+ and Δ-[M(λ-en)3]n+ (a second pair of enantiomers) and Δ-[M(δ-en)2(λ-en)]n+ and Λ-[M(λ-en)2(δ-en)]n+ (a third pair of enantiomers) as well as Δ-[M(λ-en)2(δ-en)]n+ and Λ-[M(δ-en)2(λ-en)]n+ (a fourth pair of enantiomers), with each pair of enantiomers having a different thermodynamic stability. The early crystallographic studies confirmed that the favoured crystal forms were typically Λ-[M(δ-en)3]n+ and Δ-[M(λ-en)3]n+.
6.2. The Influence of Flack
It is in the area of dissymmetric metal complexes that we see the real influence of Flack and his parameter. If a metal complex crystallizes in one of the Sohncke space groups, most crystallographic software will automatically generate the Flack parameter (hopefully close to 0 or 1) to indicate whether the choice of absolute configuration is correct. A Flack parameter value close to 0.5 is an indication that something is amiss, typically an inversion twin. What is remarkable is how often the chemist, as opposed to the crystallographer, does not comment on the absolute configuration of the complex. There are many examples of spontaneous resolution of tris(chelate)metal complexes in the CSD which are not specifically identified in their associated publications.
Today, we have a veritable wealth of chiral systems and different types of chirality that Werner and Pasteur could only have dreamed of. The Flack parameter finds widespread use in the study of novel chiral systems such as helicates, cyclic helicates and knotted systems exhibiting topological chirality and has been used to establish the absolute configuration of molecular trefoil knots assembled in self-sorting processes [152]. The use of the Flack parameter to establish the asymmetric crystallization of coordination networks and metal-organic frameworks is an interesting new development [153,154,155].
And what of the future? Routine inclusion of the Flack parameter in CIF files should be encouraged, if only to stimulate further discussion of chirality aspects within the inorganic and coordination chemistry communities! The merits and demerits of determining the Flack parameter during the refinement or post-refinement are currently being discussed [156]. Finally, data-mining activities would be dramatically improved by the inclusion of the Flack parameter in the standard data in Crystal Structure Databases. In the course of writing this article, we became aware of the difficulty of answering simple questions such as “How many chiral coordination compounds have been structurally characterized?” and “How many coordination compounds exhibit asymmetric crystallization?”.
7. Concluding Remarks
This article is an attempt to express our thanks to Howard Flack for providing a tool that made our lives as coordination chemists more interesting and rewarding and, at the same time, to place this innovation of the Flack parameter in its historical perspective.
Author Contributions
This article was conceived and written jointly by E.C.C. and C.E.H. All authors have read and agreed to the published version of the manuscript.
Funding
This work received no external funding.
Acknowledgments
As always, we give our thanks to the various library and abstracting services which have aided us in identifying and sourcing material.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Allen, F.H.; Rogers, D. A reference list of organic structures whose absolute configurations have been determined by X-ray fluorescence. Chem. Commun. 1966, 838–841. [Google Scholar] [CrossRef]
- Allen, F.H.; Neidle, S.; Rogers, D. A reference list of organic structures whose absolute configurations have been determined by X-ray methods. Part 2. Chem. Commun. 1968, 308–310. [Google Scholar] [CrossRef]
- Allen, F.H.; Neidle, S.; Rogers, D. A reference list of organic structures whose absolute configurations have been determined by X-ray methods. Part 3. J. Chem. Soc. D 1969, 452–454. [Google Scholar] [CrossRef]
- Neidle, S.; Rogers, D.; Allen, F.H. A reference list of organic structures whose absolute configurations have been determined by X-ray methods. Part IV. J. Chem. Soc. C 1970, 2340. [Google Scholar] [CrossRef]
- Lightner, D.A. Chapter 5 Determination of Absolute Configuration by Cd. Applications of the Octant Rule and the Exciton Chirality Rule. In Analytical Applications of Circular Dichroism; Purdie, N., Brittain, H.G., Eds.; Elsevier: Amsterdam, The Netherlands, 1994; pp. 131–174. [Google Scholar]
- Flack, H.D. On enantiomorph-polarity estimation. Acta Crystallogr. Sect. A Found. Crystallogr. 1983, 39, 876–881. [Google Scholar] [CrossRef]
- Yiu, Y. The Mirror and Painting in Early Renaissance Texts. Early Sci. Med. 2005, 10, 187–210. [Google Scholar] [CrossRef]
- Carroll, L. Through the Looking Glass, and What Alice Found There; Macmillan and Co.: London, UK, 1871. [Google Scholar]
- van’t Hoff, J.H. Sur les Formules de Structure dans L’Espace. Arch. Neerl. Sci. Exactes Nat. 1874, 9, 445. [Google Scholar]
- van’t Hoff, J.H. Voorstel Tot Uitbreiding Der Tegenwoordige in De Scheikunde Gebruikte Structuurformules in De Ruimte, Benevens Een Daarmee Samenhangende Opmerking Omtrent Het Verband Tusschen Optisch Actief Vermogen En Chemische Constitutie Van Organische Verbindingen; Greven: Utrecht, The Netherlands, 1874. [Google Scholar]
- Le Bel, J.A. Sur les relations qui existent entre les formules atomiques des corps organiques et le pouvoir rotatoire de leurs dissolutions. Bull. Soc. Chim. Fr. 1874, 22, 337–347. [Google Scholar]
- Riddell, F.; Robinson, M.J. J.H. van’t Hoff and J.A. Le Bel — their historical context. Tetrahedron 1974, 30, 2001–2007. [Google Scholar] [CrossRef]
- Ramberg, P.J. Chemical Structure, Spatial Arrangement; Routledge: London, UK, 1988. [Google Scholar]
- Drayer, D.E. The Early History of Stereochemistry: From the Discovery of Molecular Asymmetry and the First Resolution of a Racemate By Pasteur to the Asymmetrical Chiral Carbon of Van’t Hoff and Le Bel. In Drug Stereochemistry: Analytical Methods and Pharmacology, 3rd ed.; Jóźwiak, K., Lough, W.J., Wainer, I.W., Eds.; Informa Healthcare: London, UK, 2012; pp. 1–16. [Google Scholar]
- Drayer, D.E. The Early History of Stereochemistry: From the Discovery of Molecular Asymmetry and the First Resolution of a Racemate by Pasteur to the Asymmetrical Chiral Carbon of Van’t Hoff and Le Bel. Clin. Res. Regulat. Aff. 2001, 18, 181–203. [Google Scholar] [CrossRef]
- Meijer, E.W.; van’t Hoff, J.H. Hundred Years of Impact on Stereochemistry in The Netherlands. Angew. Chem. Int. Ed. 2001, 40, 3783–3789. [Google Scholar] [CrossRef]
- Cintas, P. On the Origin of Tetrahedral Carbon: A Case for Philosophy of Chemistry? Found. Chem. 2002, 4, 149–161. [Google Scholar] [CrossRef]
- Ochiai, H. Philosophical Foundations of Stereochemistry. HYLE 2015, 21, 1–18. [Google Scholar]
- Kelvin, W.T. The Molecular Tactics of a Crystal; Clarendon Press: Oxford, UK, 1894. [Google Scholar]
- Purdie, T. Resolution of lactic acid into its optically active components. J. Chem. Soc. Trans. 1893, 63, 1143–1157. [Google Scholar] [CrossRef] [Green Version]
- Kipping, F.S.; Pope, W.J. Studies of the terpenes and allied compounds. The sulphonic derivatives of camphor. Part I. J. Chem. Soc. Trans. 1893, 63, 548–604. [Google Scholar] [CrossRef] [Green Version]
- Einhorn, A. Ueber die Beziehungen des Cocaïns zum Atropin. Ber. Dtsch. Chem. Ges. 1890, 23, 1338–1344. [Google Scholar] [CrossRef] [Green Version]
- Zelinsky, N. Ueber die Stereoisomerie der Dimethyldioxyglutarsäuren. Ber. Dtsch. Chem. Ges. 1891, 24, 4006–4017. [Google Scholar] [CrossRef] [Green Version]
- Gal, J. Carl Friedrich Naumann and the introduction of enantio terminology: A review and analysis on the 150th anniversary. Chirality 2007, 19, 89–98. [Google Scholar] [CrossRef]
- Naumann, C.F. Elemente Der Theoretischen Krystallographie; W. Engelmann: Leipzig, Germany, 1856. [Google Scholar]
- Schoenflies, A. Krystallsysteme Und Krystallstructur; Teubner: Leipzig, Germany, 1891. [Google Scholar]
- Moss, G.P. Basic terminology of stereochemistry (IUPAC Recommendations 1996). Pure Appl. Chem. 1996, 68, 2193–2222. [Google Scholar] [CrossRef]
- Gal, J. Louis Pasteur, Chemical Linguist: Founding the Language of Stereochemistry. Helv. Chim. Acta 2019, 102, e1900098. [Google Scholar] [CrossRef]
- Gal, J. Louis Pasteur, language, and molecular chirality. I. Background and Dissymmetry. Chirality 2011, 23, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Gal, J. Stereochemical vocabulary for structures that are chiral but not asymmetric: History, analysis, and proposal for a rational terminology. Chirality 2011, 23, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Gal, J. Molecular Chirality in Chemistry and Biology: Historical Milestones. Helv. Chim. Acta 2013, 96, 1617–1657. [Google Scholar] [CrossRef]
- Gal, J. Molecular Chirality: Language, History, and Significance. In Differentiation of Enantiomers I; Schurig, V., Ed.; Springer: Cham, Switzerland, 2013; pp. 1–20. [Google Scholar]
- Larmor, J. On Electro-crystalline properties as conditioned by atomic lattices. Proc. R. Soc. Lond. Ser. A 1921, 99, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Larmor, J. The structural significance of optical rotatory quality. Rep. Br. Ass. Advmt. Sci. 1922, 351–352. [Google Scholar]
- Raman, C.V. Crystals of quartz with iridescent faces. Proc. Indian Acad. Sci. Sect. A 1950, 31A, 275–279. [Google Scholar] [CrossRef]
- Whyte, L.L. Chirality. Nature 1957, 180, 513. [Google Scholar] [CrossRef]
- Whyte, L.L. Chirality. Nature 1958, 182, 198. [Google Scholar] [CrossRef]
- Cahn, R.S.; Ingold, C.K. Specification of configuration about quadricovalent asymmetric atoms. J. Chem. Soc. 1951, 612. [Google Scholar] [CrossRef]
- Cahn, R.S.; Ingold, C.K.; Prelog, V. The specification of asymmetric configuration in organic chemistry. Experientia 1956, 12, 81–94. [Google Scholar] [CrossRef]
- Cahn, R.S.; Ingold, C.; Prelog, V. Specification of Molecular Chirality. Angew. Chem. Int. Ed. Engl. 1966, 5, 385–415. [Google Scholar] [CrossRef]
- Gal, J. The discovery of biological enantioselectivity: Louis Pasteur and the fermentation of tartaric acid, 1857--a review and analysis 150 yr later. Chirality 2008, 20, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Gal, J. When did Louis Pasteur present his memoir on the discovery of molecular chirality to the Académie des sciences? Analysis of a discrepancy. Chirality 2008, 20, 1072–1084. [Google Scholar] [CrossRef] [PubMed]
- Gal, J. In defense of Louis Pasteur: Critique of Gerald Geison’s deconstruction of Pasteur’s discovery of molecular chirality. Chirality 2019, 31, 261–282. [Google Scholar] [CrossRef]
- Mcnaught, A.D.; Wilkinson, A.; IUPAC. Compendium of Chemical Terminology, 2nd ed.; (the “Gold Book”); Blackwell Scientific Publications: Oxford, UK, 1997; Online Version (2019-) Created By S.J. Chalk; ISBN 0-9678550-9-8. [Google Scholar] [CrossRef]
- Cotton, F.A. Chemical Applications of Group Theory, 3rd; Wiley: New York, NY, USA, 1990. [Google Scholar]
- Werner, A. Zur Kenntnis des asymmetrischen Kobaltatoms. I. Ber. Dtsch. Chem. Ges. 1911, 44, 1887–1898. [Google Scholar] [CrossRef] [Green Version]
- Werner, A. Zur Kenntnis des asymmetrischen Kobaltatoms. II. Ber. Dtsch. Chem. Ges. 1911, 44, 2445–2455. [Google Scholar] [CrossRef]
- Werner, A. Zur Kenntnis des asymmetrischen Kobaltatoms. III. Ber. Dtsch. Chem. Ges. 1911, 44, 3272–3278. [Google Scholar] [CrossRef] [Green Version]
- Werner, A. Zur Kenntnis des asymmetrischen Kobaltatoms. IV. Ber. Dtsch. Chem. Ges. 1911, 44, 3279–3284. [Google Scholar] [CrossRef] [Green Version]
- Werner, A. Zur Kenntnis des asymmetrischen Kobaltatoms. V. Ber. Dtsch. Chem. Ges. 1912, 45, 121–130. [Google Scholar] [CrossRef] [Green Version]
- Werner, A. Über Spiegelbildisomerie bei Rhodium-Verbindungen. I. Ber. Dtsch. Chem. Ges. 1912, 45, 1228–1236. [Google Scholar] [CrossRef] [Green Version]
- Werner, A.; McCutcheon, T.P. Zur Kenntnis des asymmetrischen Kobaltatoms. VI. Ber. Dtsch. Chem. Ges. 1912, 45, 3281–3287. [Google Scholar] [CrossRef]
- Werner, A.; Shibata, Y. Zur Kenntnis des asymmetrischen Kobaltatoms. VII. Ber. Dtsch. Chem. Ges. 1912, 45, 3287–3293. [Google Scholar] [CrossRef] [Green Version]
- Werner, A. Zur Kenntnis des asymmetrischen Kobaltatoms XI. Über Oxalo-diäthylendiamin-kobaltisalze und eine neue Spaltungsmethode für racemische anorganische Verbindungen. Ber. Dtsch. Chem. Ges. 1914, 47, 2171–2182. [Google Scholar] [CrossRef]
- Werner, A.; Tschernoff, G. Zur Kenntnis des asymmetrischen Kobaltatoms. VIII. Ber. Dtsch. Chem. Ges. 1912, 45, 3294–3301. [Google Scholar] [CrossRef] [Green Version]
- Werner, A. Zur Kenntnis des asymmetrischen Kobaltatoms IX. Ber. Dtsch. Chem. Ges. 1913, 46, 3674–3683. [Google Scholar] [CrossRef]
- Werner, A. Zur Kenntnis des asymmetrischen Kobaltatoms X. Ber. Dtsch. Chem. Ges. 1914, 47, 1961–1979. [Google Scholar] [CrossRef] [Green Version]
- Werner, A. Zur Kenntnis des asymmetrischen Kobaltatoms XII. Über optische Aktivität bei kohlenstofffreien Verbindungen. Ber. Dtsch. Chem. Ges. 1914, 47, 3087–3094. [Google Scholar] [CrossRef] [Green Version]
- Jensen, W.B. Polarimeters; Oesper Museum Booklets on the History of Chemical Apparatus, University of Cincinnati: Cincinnati, OH, USA, 2014. [Google Scholar]
- Hargreaves, M.K. Optical Rotatory Dispersion: Its Nature and Origin. Nature 1962, 195, 560–566. [Google Scholar] [CrossRef]
- King, V.L. Über Spaltungsmethoden Und Ihre Anwendung Auf Komplexe Metall-Ammoniakverbindungen. Ph.D. Dissertation, University of Zürich, Zürich, Switzerland, 1912. [Google Scholar]
- Kauffman, G.B. The Discovery of Optically Active Coordination Compounds: A Milestone in Stereochemistry. Isis 1975, 66, 38–62. [Google Scholar] [CrossRef]
- Kauffman, G.B. A Stereochemical Achievement of the First Order: Alfred Werner’s Resolution of Cobalt Complexes, 85 Years Later. Bull. Hist. Chem. 1997, 20, 50–59. [Google Scholar]
- Ernst, K.H.; Berke, H. Optical activity and Alfred Werner’s coordination chemistry. Chirality 2011, 23, 187–189. [Google Scholar] [CrossRef] [PubMed]
- Bailar, J.C.; Haslam, J.H.; Jones, E.M. The Stereochemistry of Complex Inorganic Molecules. III. The Reaction of Ammonia with levo-Dichlorodiethylenediaminocobaltic Chloride. J. Am. Chem. Soc. 1936, 58, 2226–2227. [Google Scholar] [CrossRef]
- Mathieu, J.-P. Activité optique et solubilité de quelques cobaltammines. C. R. Hebd. Seances Acad. Sci. 1934, 199, 278–280. [Google Scholar]
- Mathieu, J.-P. Configuration de quelques complexes hexacoordinés optiquement actifs. C. R. Hebd. Seances Acad. Sci. 1934, 198, 1598–1600. [Google Scholar]
- Mathieu, J.-P. Absorption, activité optique et configuration de complexes minéraux. C. R. Hebd. Seances Acad. Sci. 1935, 201, 1183–1184. [Google Scholar]
- Mathieu, J.-P. Werner complexes; optical activity and the configuration of ions of the type MA3. J. Chim. Phys. 1936, 33, 78–96. [Google Scholar] [CrossRef]
- Mathieu, J.-P. Recherches expérimentales sur le dichroisme circulaire et sur quelques applications physico-chimiques de ce phénomène. Ann. Phys. 1935, 11, 371–460. [Google Scholar] [CrossRef]
- Mathieu, J.-P. Recherches sur les complexes de Werner activité optique et configuration des ions du type MeA3. J. Chim. Phys. 1936, 3, 476–498. [Google Scholar] [CrossRef]
- Mathieu, J.-P. The Werner complexes. Optical activity and configuration of the ions containing the groups M en2 and M ox2. Bull. Soc. Chim. Fr. Mem. 1936, 3, 476–498. [Google Scholar]
- Mathieu, J.-P. The Werner complexes. Absorption of the hexacoördinates of cobalt and chromium in aqueous solution. Bull. Soc. Chim. Fr. Mem. 1936, 3, 463–475. [Google Scholar]
- Mathieu, J.-P. Experiments on the complexes of Werner. Substitution in the optically active complex chlorides. Bull. Soc. Chim. Fr. Mem. 1937, 4, 687–700. [Google Scholar]
- Mathieu, J.-P. Recent views on the stereochemistry of complex inorganic compounds. Bull. Soc. Chim. Fr.; Mem. 1938, 5, 725–805. [Google Scholar]
- Mathieu, J.-P. The Werner complexes-absorption and optical activity of cobalt compounds with a double nucleus. Bull. Soc. Chim. Fr. Mem. 1938, 5, 105–113. [Google Scholar]
- Mathieu, J.-P. Researches on Werner complexes. Cobaltammines containing optically active amino acids. Bull. Soc. Chim. Fr. Mem. 1939, 6, 873–882. [Google Scholar]
- Mathieu, J.-P. Researches on Werner complexes. Optical activity and configuration of platinum. IV. Triethylenediamine ion. Bull. Soc. Chim. Fr. Mem. 1939, 6, 1258–1259. [Google Scholar]
- Mathieu, J.-P. Activité Optique Naturelle. In Handbuch der Physik/Encyclopedia of Physics; Spektroskopie II/Spectroscopy II; Madelung, O., Ed.; Springer: Berlin/Heidelberg, Germany, 1957; pp. 333–432. [Google Scholar]
- Cotton, A. Recherches sur l’absorption et la Dispersion de la lumière par les Milieux Doués du Pouvoir Rotatoire. Ann. Chim. Phys. Sér. 7 1896, 8, 347–432. [Google Scholar] [CrossRef]
- Cotton, A. Absorption inégale des rayons circulaires droit et gauche dans certains corps actifs. C. R. Hebd. Seances Acad. Sci. 1895, 120, 989–991. [Google Scholar]
- Cotton, A. Dispersion rotatoire anomale des corps absorbants. C. R. Hebd. Seances Acad. Sci. 1895, 120, 1044–1046. [Google Scholar]
- Kuhn, W.; Bein, K. Konfiguration Und Optische Drehung Bei Anorganischen Komplexverbindungen, Z. Anorg. Allg. Chem. 1934, 216, 321–348. [Google Scholar] [CrossRef]
- Kuhn, W. Das Problem der absoluten Konfiguration optisch aktiver Stoffe. Sci. Nat. 1938, 26, 289–296. [Google Scholar] [CrossRef]
- Röntgen, W.C. Ueber eine neue Art von Strahlen. Ann. Phys. 1898, 300, 12–17. [Google Scholar] [CrossRef] [Green Version]
- Röntgen, W.C. Ueber eine neue Art von Strahlen (Vorläufige Mittheilung). Sonderabbdruck Sitz. Würzburger Physik.Medic. Ges. 1895, 1896, 2–20. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, W.K.; Laue, M. Interferenz -Erscheinungen bei Röntgenstrahlen (mit 5 Tafeln). Sitz. Bayer. Akad. Wiss. 1912, 303–322. [Google Scholar]
- Laue, M. Eine quantitative Prüfung der Theorie für die Interferenz-Erscheinungen bei Röntgenstrahlen. Sitz. Bayer. Akad. Wiss. 1912, 363–373. [Google Scholar]
- Eckert, M. Max von Laue and the discovery of X-ray diffraction in 1912. Ann. Phys. 2012, 524, A83–A85. [Google Scholar] [CrossRef]
- Hendrickson, W.A. Evolution of diffraction methods for solving crystal structures. Acta Crystallogr. Sect. A Found. Crystallogr. 2013, 69, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Ewald, P.P. Fifty Years of X-Ray Diffraction; Oosthoek’s Uitgevers Mij. N.V.: Utrecht, The Netherlands, 1962; ISBN 978-1-4615-9961-6. [Google Scholar]
- Bragg, W.L. The Development of X-Ray Analysis; G. Bell: London, UK, 1975; ISBN 0486673162. [Google Scholar]
- Bacon, G.E. X-Ray and Neutron Diffraction, 1st ed.; Ter Haar, D., Ed.; Pergamon: Oxford, UK, 1966; ISBN 0080119999. [Google Scholar]
- Hildebrandt, G. The Discovery of the Diffraction of X-rays in Crystals—A Historical Review. Cryst. Res. Technol. 1993, 28, 747–766. [Google Scholar] [CrossRef]
- Bijvoet, J.M.; Bernal, J.D.; Patterson, A.L. Forty years of X-ray diffraction. Nature 1952, 169, 949–950. [Google Scholar] [CrossRef]
- Bijvoet, J.M.; Burgers, W.G.; Hägg, G. (Eds.) Early Papers on Diffraction of X-Rays By Crystals; Oosthoek’s Uitgevers Mij. N.V.: Utrecht, The Netherlands, 1969; Volume 1, ISBN 978-1-4615-6880-3. [Google Scholar]
- Bragg, H.B.; Bragg, W.L. The structure of the diamond. Proc. R. Soc. Lond. Ser. A 1913, 89, 277–291. [Google Scholar] [CrossRef]
- Bragg, W.L.; Bragg, W.L. The Structure of the Diamond. Nature 1913, 91, 557. [Google Scholar] [CrossRef]
- Bragg, W.L. The Specular Reflection of X-rays. Nature 1912, 90, 410. [Google Scholar] [CrossRef] [Green Version]
- Bragg, W.L.; James, R.W.; Bosanquet, C.H. The intensity of reflexion of X-rays by rock-salt.—Part II. Philos. Mag. (1798–1977) 1921, 42, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Bragg, W.L.; James, R.W.; Bosanquet, C.H. The intensity of reflexion of X-rays by rock-salt. Philos. Mag. (1798–1977) 1921, 41, 309–337. [Google Scholar] [CrossRef] [Green Version]
- Bragg, W.L.; James, R.W.; Bosanquet, C.H. The distribution of electrons around the nucleus in the sodium and chlorine atoms. Philos. Mag. (1798–1977) 1922, 44, 433–449. [Google Scholar] [CrossRef]
- Bragg, W.L. The Diffraction of Short Electromagnetic Waves by a Crystal. Proc. Camb. Philos. Soc. 1913, 17, 43–57. [Google Scholar]
- Bragg, W.L. The structure of some crystals as indicated by their diffraction of X-rays. Proc. R. Soc. Lond. Ser. A 1913, 89, 248–277. [Google Scholar] [CrossRef] [Green Version]
- Bragg, W.L. The analysis of crystals by the X-ray spectrometer. Proc. R. Soc. Lond. Ser. A 1914, 89, 468–489. [Google Scholar] [CrossRef] [Green Version]
- Darwin, C.G. LXXVIII. The theory of X-ray reflexion. Part II. Philos. Mag. (1798–1977) 1914, 27, 675–690. [Google Scholar] [CrossRef] [Green Version]
- Darwin, C.G. XXXIV. The theory of X-ray reflexion. Philos. Mag. (1798–1977) 1914, 27, 315–333. [Google Scholar] [CrossRef] [Green Version]
- Bragg, W.L.; Darwin, C.G.; James, R.W. LXXXI. The intensity of reflexion of X-rays by crystals. Philos. Mag. (1798–1977) 1926, 1, 897–922. [Google Scholar] [CrossRef]
- Friedel, G. Sur les symétries cristallines que peut révéler la diffraction des rayons X. C.R. Acad. Sci. Paris 1913, 157, 1533–1536. [Google Scholar]
- Bragg, W.L. The determination of parameters in crystal structures by means of Fourier series. Proc. R. Soc. Lond. Ser. A 1929, 123, 537–559. [Google Scholar] [CrossRef] [Green Version]
- Patterson, A.L. A Fourier Series Method for the Determination of the Components of Interatomic Distances in Crystals. Phys. Rev. 1934, 46, 372–376. [Google Scholar] [CrossRef]
- Patterson, A.L. A Direct Method for the Determination of the Components of Interatomic Distances in Crystals. Z. Kristallogr. Kristallgeom. Kristallphys. Kristallchem. 1935, 90, 517–542. [Google Scholar] [CrossRef]
- Robertson, J.M. X-ray analysis and application of Fourier series methods to molecular structures. Rep. Prog. Phys. 1937, 4, 332–367. [Google Scholar] [CrossRef]
- Cork, J.M. The crystal structure of some of the alums. Philos. Mag. (1798–1977) 1927, 4, 688–698. [Google Scholar] [CrossRef]
- Robertson, J.M. An X-ray study of the phthalocyanines. Part II. Quantitative structure determination of the metal-free compound. J. Chem. Soc. 1936, 1195–1209. [Google Scholar] [CrossRef]
- Robertson, J.M.; Woodward, I. An X-ray study of the phthalocyanines. Part III. Quantitative structure determination of nickel phthalocyanine. J. Chem. Soc. 1937, 219–230. [Google Scholar] [CrossRef]
- Robertson, J.M.; Woodward, I. An X-ray study of the phthalocyanines. Part IV. Direct quantitative analysis of the platinum compound. J. Chem. Soc. 1940, 36–48. [Google Scholar] [CrossRef]
- Nishikawa, S.; Matukawa, K. Hemihedry of Zincblende and X-Ray Reflexion. Proc. Imp. Acad. Jpn. 1928, 4, 96–97. [Google Scholar] [CrossRef]
- Coster, D.; Knol, K.S.; Prins, J.A. Unterschiede in der Intensität der Röntgenstrahlen-reflexion an den beiden 111-Flächen der Zinkblende. Z. Phys. 1930, 63, 345–369. [Google Scholar] [CrossRef]
- Bokhoven, C.; Schoone, J.C.; Bijvoet, J.M. The Fourier synthesis of the crystal structure of strychnine sulphate pentahydrate. Acta Crystallogr. 1951, 4, 275–280. [Google Scholar] [CrossRef]
- Bijvoet, J.M. Phase determination in direct Fourier synthesis of crystal structure. Proc. Sect. Sci. K. Ned. Akad. Wet. 1949, 52, 313–314. [Google Scholar]
- Bokhoven, C.; Schoone, J.C.; Bijvoet, J.M. On the crystal structure of strychnine sulphate and selenate. III. [001] projection. Proc. Sect. Sci. K. Ned. Akad. Wet. 1949, 52, 120–121. [Google Scholar]
- Bokhoven, C.; Schoone, J.C.; Bijvoet, J.M. On the crystal structure of strychnine sulphate and selenate. I. Cell dimensions and spacegroup. Proc. Sect. Sci. K. Ned. Akad. Wet. 1947, 50, 825. [Google Scholar]
- Bokhoven, C.; Schoone, J.C.; Bijvoet, J.M. On the crystal structure of strychnine sulphate and selenate. II. [010] projection and structure formula. Proc. Sect. Sci. K. Ned. Akad. Wet. 1948, 51, 990. [Google Scholar]
- Groenewege, M.P.; Peerdeman, A.F. Johannes Martin Bijvoet. 23 January 1892–4 March 1980. Biogr. Mem. Fellows R. Soc. 1983, 29, 26–41. [Google Scholar] [CrossRef]
- Bijvoet, J.M.; van Bommel, A.J.; Peerdeman, A.F. Determination of absolute configuration of optically active compounds by means of X-rays. Proc. Sect. Sci. K. Ned. Akad. Wet. 1951, 54, 16–19. [Google Scholar] [CrossRef]
- Bijvoet, J.M.; Peerdeman, A.F.; van Bommel, A.J. Determination of the Absolute Configuration of Optically Active Compounds by Means of X-Rays. Nature 1951, 168, 271–272. [Google Scholar] [CrossRef]
- Bijvoet, J.M.; Peerdeman, A.F.; van Bommel, A.J. Structure of Optically Active Compounds in the Solid State. Nature 1954, 173, 888–891. [Google Scholar] [CrossRef]
- Mathieson, A.M. The determination of absolute configuration by the use of an internal reference asymmetric centre. Acta Crystallogr. 1956, 9, 317. [Google Scholar] [CrossRef]
- Karle, J.; Hauptman, H. The phases and magnitudes of the structure factors. Acta Crystallogr. 1950, 3, 181–187. [Google Scholar] [CrossRef]
- Karle, J.; Hauptman, H. A theory of phase determination for the four types of non-centrosymmetric space groups 1P222, 2P22, 3P12, 3P22. Acta Crystallogr. 1956, 9, 635–651. [Google Scholar] [CrossRef]
- Woolfson, M.M. Direct methods in crystallography. Rep. Prog. Phys. 1971, 34, 369–434. [Google Scholar] [CrossRef]
- Ladd, M.F.C.; Palmer, R.A. (Eds.) Theory and Practice of Direct Methods in Crystallography; Springer: New York, NY, USA, 1980. [Google Scholar]
- Jones, P.G. The determination of absolute structure. I. Some experiences with the Rogers η refinement. Acta Crystallogr. Sect. A Found. Crystallogr. 1984, 40, 660–662. [Google Scholar] [CrossRef]
- Flack, H.D. Absolute-structure determination: Past, present and future. Chimia 2014, 68, 26–30. [Google Scholar] [CrossRef]
- Sohncke, L. Entwickelung Einer Theorie Der Krystallstruktur; B.G. Teubner: Leipzig, Germany, 1879. [Google Scholar]
- Pidcock, E. Achiral molecules in non-centrosymmetric space groups. Chem. Commun. 2005, 3457–3459. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, W.C. Significance tests on the crystallographic R factor. Acta Crystallogr. 1965, 18, 502–510. [Google Scholar] [CrossRef]
- Rogers, D. On the application of Hamilton’s ratio test to the assignment of absolute configuration and an alternative test. Acta Crystallogr. Sect. A Cryst. Phys. Diffr. Theor. Gen. Crystallogr. 1981, 37, 734–741. [Google Scholar] [CrossRef]
- CCDC the Cambridge Structural Database (CSD). Available online: https://www.ccdc.cam.ac.uk (accessed on 20 July 2020).
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Flack, H. Chiral and Achiral Crystal Structures. Helv. Chim. Acta 2003, 86, 905–921. [Google Scholar] [CrossRef]
- Eliel, E.L.; Wilen, S.H.; Mander, L.N. Stereochemistry of Organic Compounds; Wiley-Interscience: New York, NY, USA, 1994. [Google Scholar]
- von Zelewsky, A. Stereochemistry of Coordination Compounds; John Wiley & Sons Inc.: Chichester, UK, 1996. [Google Scholar]
- Hawkins, C.J. Absolute Configuration of Metal Complexes; Wiley-Interscience: New York, NY, USA, 1971. [Google Scholar]
- Amouri, H.; Gruselle, M. Chirality in Transition Metal Chemistry; John Wiley & Sons Inc.: Chichester, UK, 2008. [Google Scholar]
- Gillard, R.D.; Mitchell, P.R. The Absolute Configuration of Transition Metal Complexes. In Structure and Bonding; Springer: Berlin/Heidelberg, Germany, 1970; Volume 7, pp. 46–86. [Google Scholar]
- Saito, Y. Absolute configurations of metal complexes determined by X-ray analysis. Coord. Chem. Rev. 1974, 13, 305–337. [Google Scholar] [CrossRef]
- Saito, Y. Absolute Configuration of Transition Metal Complexes. In ACS Symposium Series: Stereochemistry of Optically Active Transition Metal Compounds; American Chemical Society: Washington, DC, USA, 1980; pp. 13–42. [Google Scholar]
- Buckingham, D.A.; Sargeson, A.M. Conformational Analysis and Steric Effects in Metal Chelates. In Topics in Stereochemistry; Allinger, N.L., Eliel, E.L., Eds.; Wiley: Chichester, UK, 1971; Volume 6, pp. 219–277. [Google Scholar]
- Saito, Y.; Nakatsu, K.; Shiro, M.; Kuroya, H. Determination of the absolute configuration of optically active complex ion, [Coen3]3+, by means of X-rays. Acta Crystallogr. 1955, 8, 729–730. [Google Scholar] [CrossRef]
- Zhong, J.; Zhang, L.; August, D.P.; Whitehead, G.F.S.; Leigh, D.A. Self-Sorting Assembly of Molecular Trefoil Knots of Single Handedness. J. Am. Chem. Soc. 2019, 141, 14249–14256. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, S.; Nieto, R.A.; Wu, T.; Feng, P.; Bu, X. A tale of three carboxylates: Cooperative asymmetric crystallization of a three-dimensional microporous framework from achiral precursors. Angew. Chem. Int. Ed. Engl. 2010, 49, 1267–1270. [Google Scholar] [CrossRef]
- Zheng, W.; Wei, Y.; Xiao, X.; Wu, K. Spontaneous asymmetric crystallization of a quartz-type framework from achiral precursors. Dalton Trans. 2012, 41, 3138–3140. [Google Scholar] [CrossRef]
- Yuan, S.; Deng, Y.-K.; Xuan, W.-M.; Wang, X.-P.; Wang, S.-N.; Dou, J.-M.; Sun, D. Spontaneous chiral resolution of a 3D (3,12)-connected MOF with an unprecedented ttt topology consisting of cubic [Cd4(μ3-OH)4] clusters and propeller-like ligands. Cryst. Eng. Comm. 2014, 16, 3829–3833. [Google Scholar] [CrossRef]
- Watkin, D.J.; Cooper, R.I. Why direct and post-refinement determinations of absolute structure may give different results. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 661–683. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Mankind has long been fascinated by objects and their mirror images. In this illustration by John Tenniel from Through the Looking-Glass, and What Alice Found There we see the subtley different reality in the enantiomeric world. (Public domain image. Source https://en.wikipedia.org/wiki/Through_the_Looking-Glass#/media/File:Aliceroom3.jpg).
Figure 1.
Mankind has long been fascinated by objects and their mirror images. In this illustration by John Tenniel from Through the Looking-Glass, and What Alice Found There we see the subtley different reality in the enantiomeric world. (Public domain image. Source https://en.wikipedia.org/wiki/Through_the_Looking-Glass#/media/File:Aliceroom3.jpg).

Figure 2.
(a) IUPAC recommends the notation Δ and Λ for denoting the absolute configuration of octahedral metal complexes. The Δ (pale blue) and Λ (pink) enantiomers of an [M(bpy)3]n+ complex (bpy = 2,2′-bipyridine) are presented here. (b) The absolute configuration proposed by Kuhn and Bein for (−)d-[Co(C2O4)3]3− and a representation using the same colour coding as in (a) to show the Δ configuration (only the carbon and the coordinated oxygen atoms of the oxalate ligands are shown) [83]. Figure 2b ©1934 Kuhn, W.; Bein, K. Z. Anorg. Allg. Chem. 1934, 216, 321–348 reproduced with kind permission of John Wiley and Sons.
Figure 2.
(a) IUPAC recommends the notation Δ and Λ for denoting the absolute configuration of octahedral metal complexes. The Δ (pale blue) and Λ (pink) enantiomers of an [M(bpy)3]n+ complex (bpy = 2,2′-bipyridine) are presented here. (b) The absolute configuration proposed by Kuhn and Bein for (−)d-[Co(C2O4)3]3− and a representation using the same colour coding as in (a) to show the Δ configuration (only the carbon and the coordinated oxygen atoms of the oxalate ligands are shown) [83]. Figure 2b ©1934 Kuhn, W.; Bein, K. Z. Anorg. Allg. Chem. 1934, 216, 321–348 reproduced with kind permission of John Wiley and Sons.

Figure 3.
The projection of the nickel phthalocyanine molecule presented by Robertson in his pioneering work on the crystallography of phthalocyanines [116]. ©1937 Robertson, J.M.; Woodward, I. J. Chem. Soc. 1937, 219–230 reproduced with kind permission of The Royal Society of Chemistry.
Figure 3.
The projection of the nickel phthalocyanine molecule presented by Robertson in his pioneering work on the crystallography of phthalocyanines [116]. ©1937 Robertson, J.M.; Woodward, I. J. Chem. Soc. 1937, 219–230 reproduced with kind permission of The Royal Society of Chemistry.

Figure 4.
The absolute configuration of tartaric acid as depicted by Bijvoet after the determination of the structure of sodium rubidium tartrate. [116,127]. ©1951 Bijvoet, J.M.; Peerdeman, A.F.; van Bommel, A.J. Nature 1951, 168, 271–272, reproduced with kind permission of Springer Nature. The absolute configuration confirmed the random choice selected by Fischer.
Figure 4.
The absolute configuration of tartaric acid as depicted by Bijvoet after the determination of the structure of sodium rubidium tartrate. [116,127]. ©1951 Bijvoet, J.M.; Peerdeman, A.F.; van Bommel, A.J. Nature 1951, 168, 271–272, reproduced with kind permission of Springer Nature. The absolute configuration confirmed the random choice selected by Fischer.

Figure 5.
The four possible combinations of a chiral cation and a chiral anion: [(Δ-C)(Δ-A)] and [(Λ-C)(Λ-A)] form a pair of enantiomers (denoted by a red double-headed arrow) as do [(Δ-C)(Λ-A)] and [(Λ-C)(Δ-A)]; all other relationships are as diastereoisomers (denoted by a blue double-headed arrow).
Figure 5.
The four possible combinations of a chiral cation and a chiral anion: [(Δ-C)(Δ-A)] and [(Λ-C)(Λ-A)] form a pair of enantiomers (denoted by a red double-headed arrow) as do [(Δ-C)(Λ-A)] and [(Λ-C)(Δ-A)]; all other relationships are as diastereoisomers (denoted by a blue double-headed arrow).

Figure 6.
The two enantiomeric forms of a five-membered chelate ring involving an en-like ligand are denoted by the descriptors δ or λ.
Figure 6.
The two enantiomeric forms of a five-membered chelate ring involving an en-like ligand are denoted by the descriptors δ or λ.
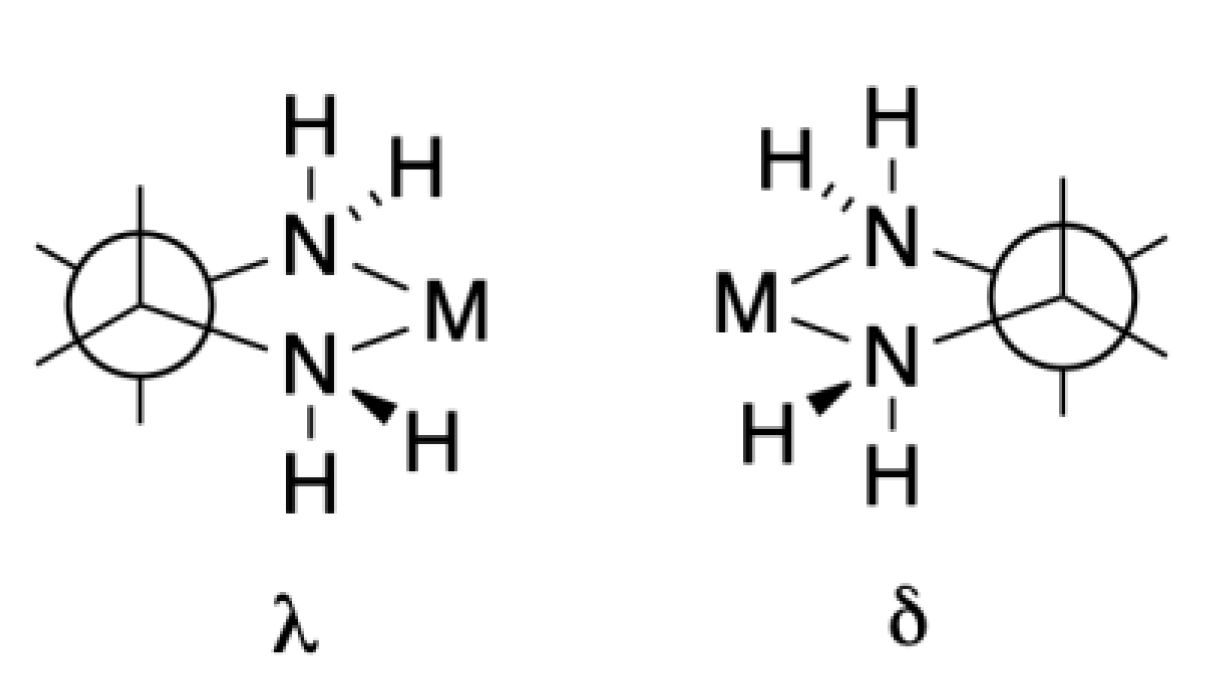
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Constable, E.C.; Housecroft, C.E. When Stereochemistry Raised Its Ugly Head in Coordination Chemistry—An Appreciation of Howard Flack. Chemistry 2020, 2, 759-776. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry2030049
AMA Style
Constable EC, Housecroft CE. When Stereochemistry Raised Its Ugly Head in Coordination Chemistry—An Appreciation of Howard Flack. Chemistry. 2020; 2(3):759-776. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry2030049
Chicago/Turabian StyleConstable, Edwin C., and Catherine E. Housecroft. 2020. "When Stereochemistry Raised Its Ugly Head in Coordination Chemistry—An Appreciation of Howard Flack" Chemistry 2, no. 3: 759-776. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry2030049