Manipulating the Conformation of 3,2′:6′,3″-Terpyridine in [Cu2(μ-OAc)4(3,2′:6′,3″-tpy)]n 1D-Polymers

 , , and
, , and
Abstract
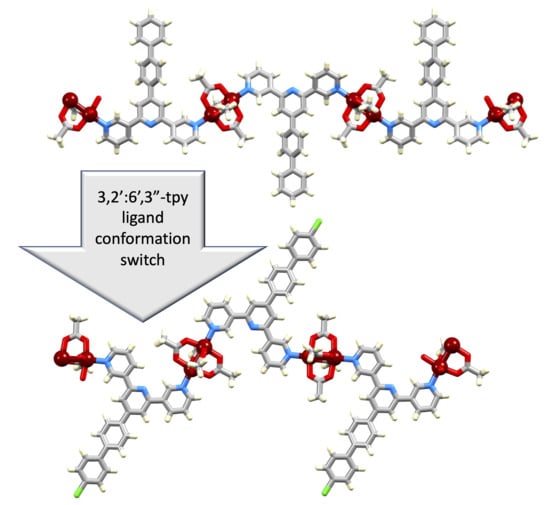
:
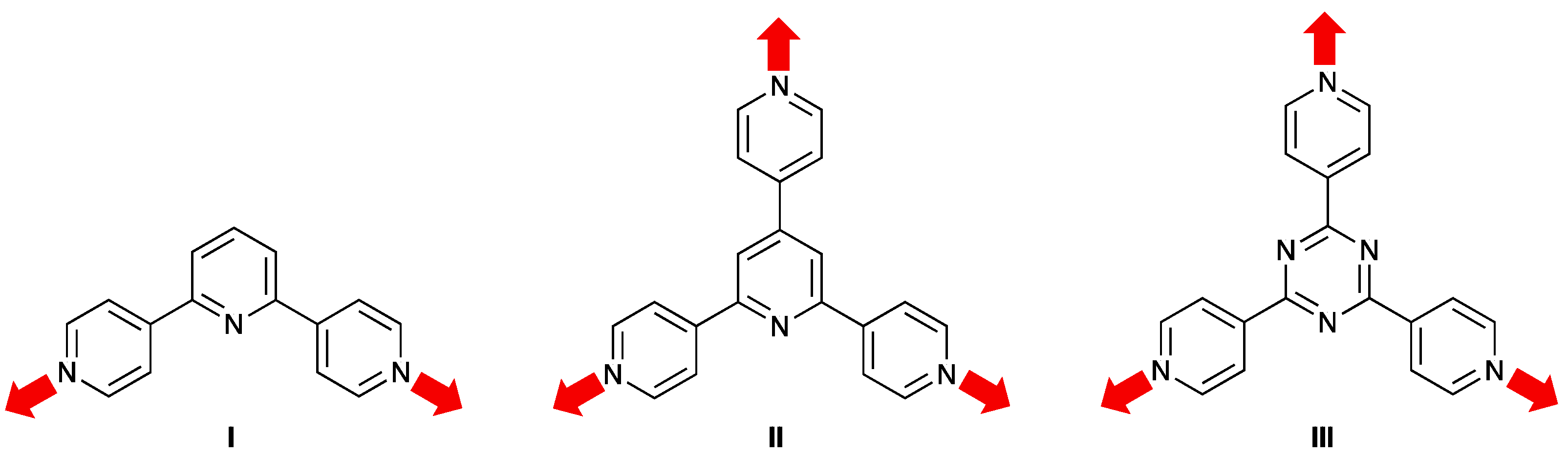
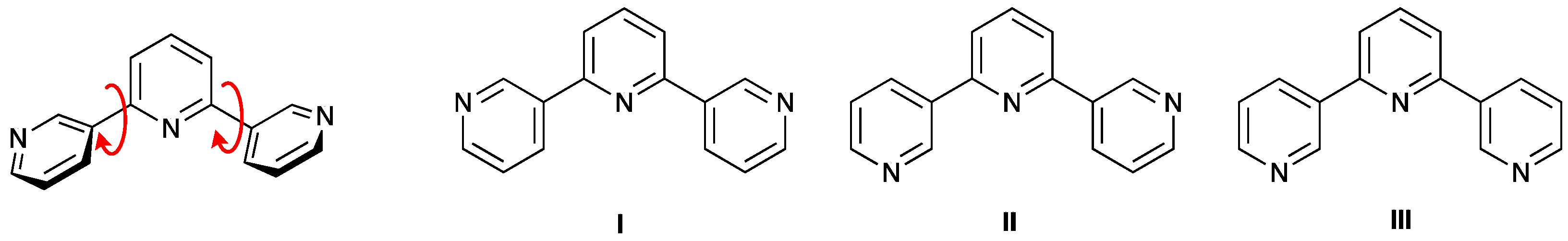
1. Introduction
2. Materials and Methods
2.1. General
2.2. Compound 1
2.3. Compound 2
2.4. Compound 3
2.5. Compound 4
2.6. Compound 5
2.7. Crystal Growth of [Cu2(μ-OAc)4(1)]n and Preparative Scale Reaction
2.8. Crystal Growth of [Cu2(μ-OAc)4(2)]n and Preparative Scale Reaction
2.9. Crystal Growth of [Cu2(μ-OAc)4(3)]n and Preparative Scale Reaction
2.10. Crystal Growth of [Cu2(μ-OAc)4(4)]n and Preparative Scale Reaction
2.11. Crystal Growth of [Cu2(μ-OAc)4(5)]n·nMeOH and Preparative Scale Reaction
2.12. Crystallography
2.13. [Cu2(μ-OAc)4(1)]n
2.14. [Cu2(μ-OAc)4(2)]n
2.15. [Cu2(μ-OAc)4(3)]n
2.16. [Cu2(μ-OAc)4(4)]n
2.17. [Cu2(μ-OAc)4(5)]n·nMeOH
2.18. Density Functional Theory (DFT) Calculations
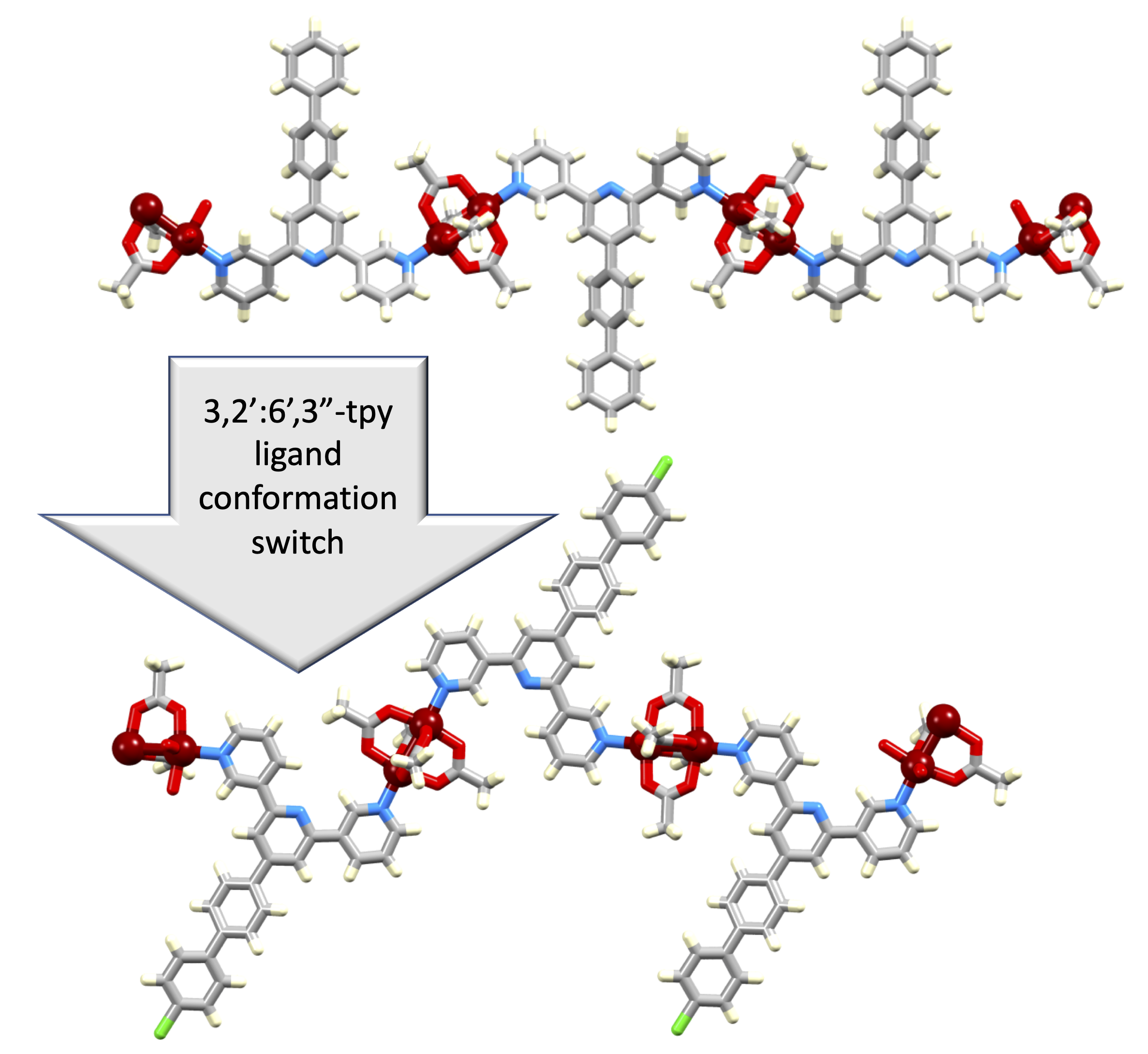
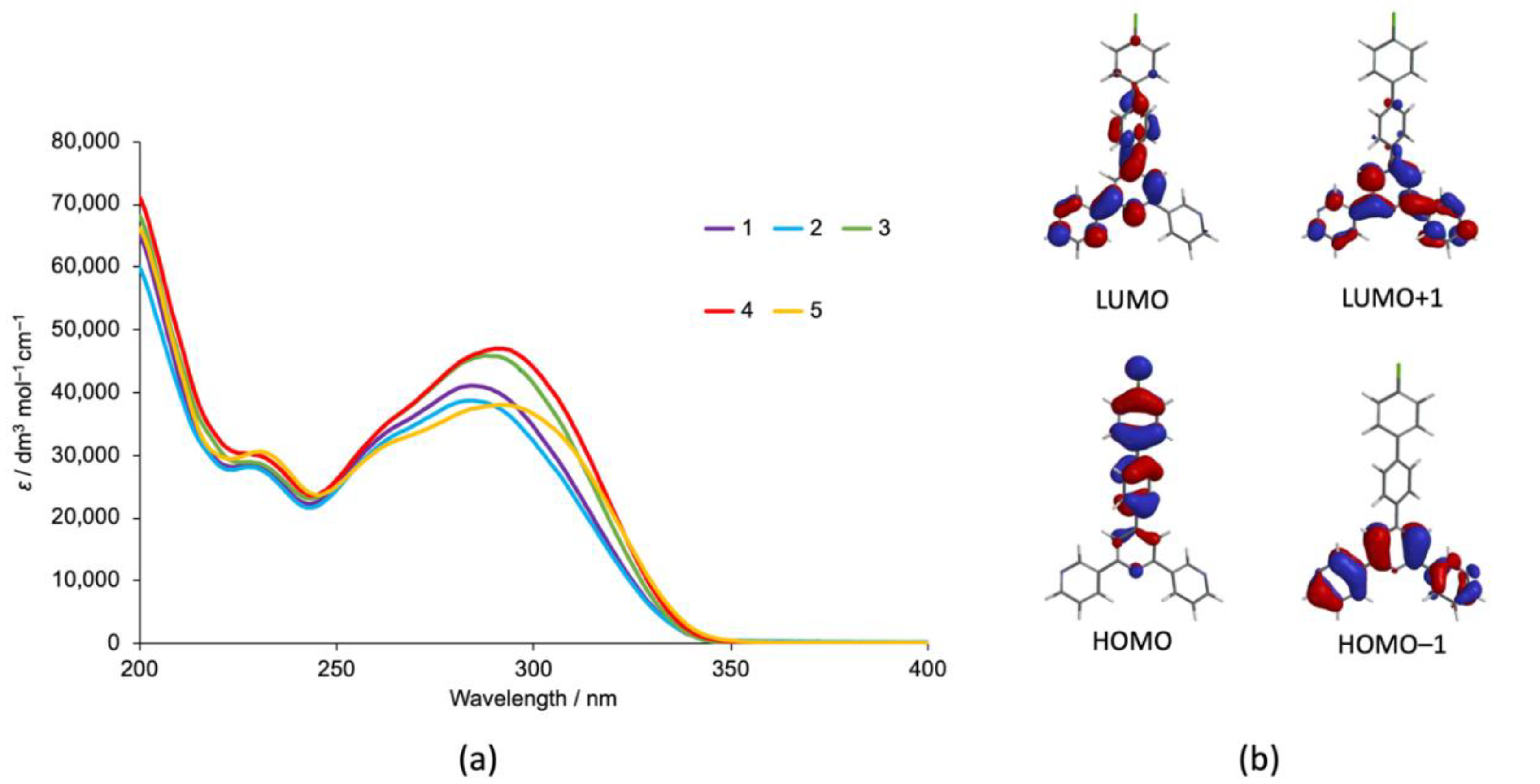
3. Results and Discussion
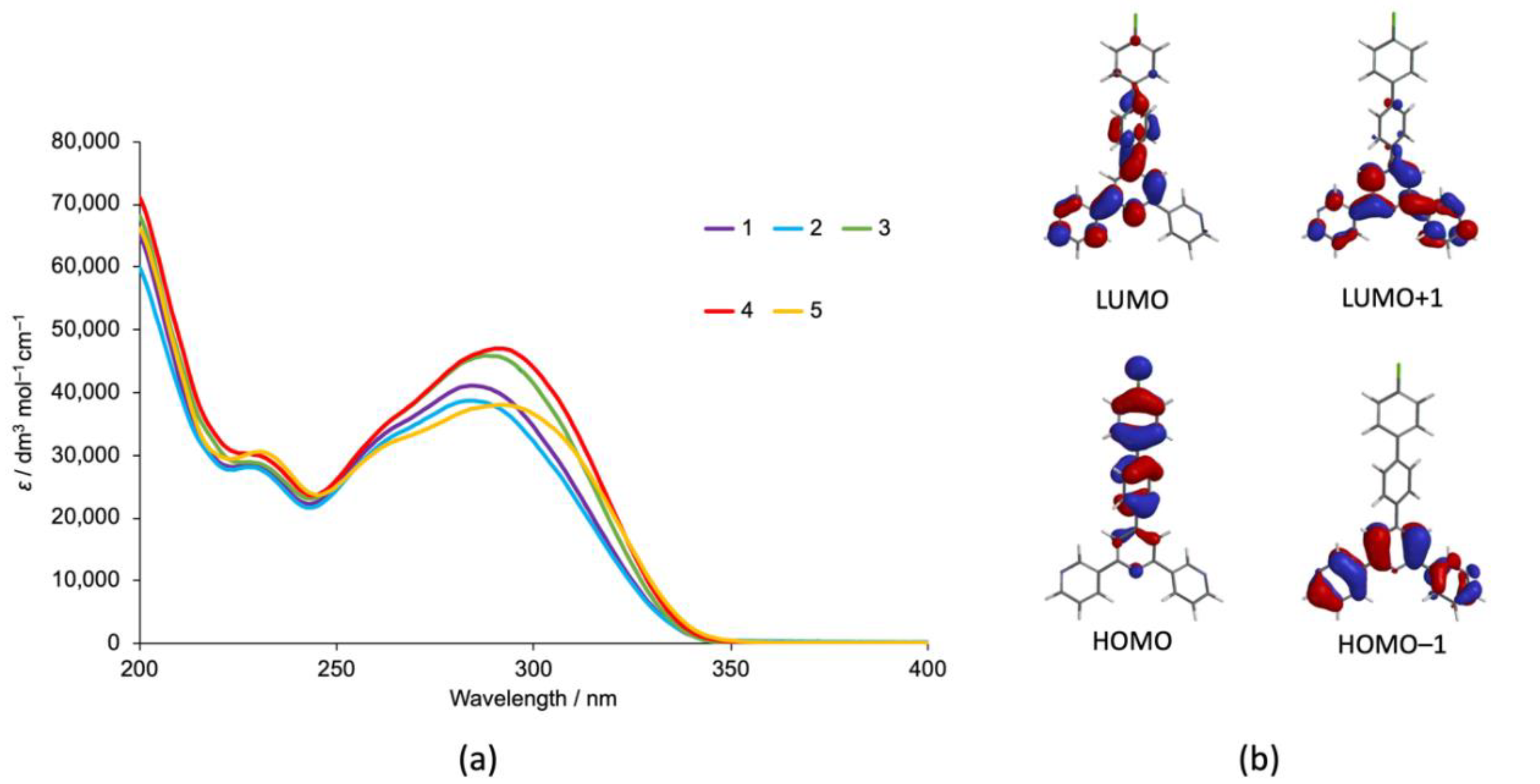
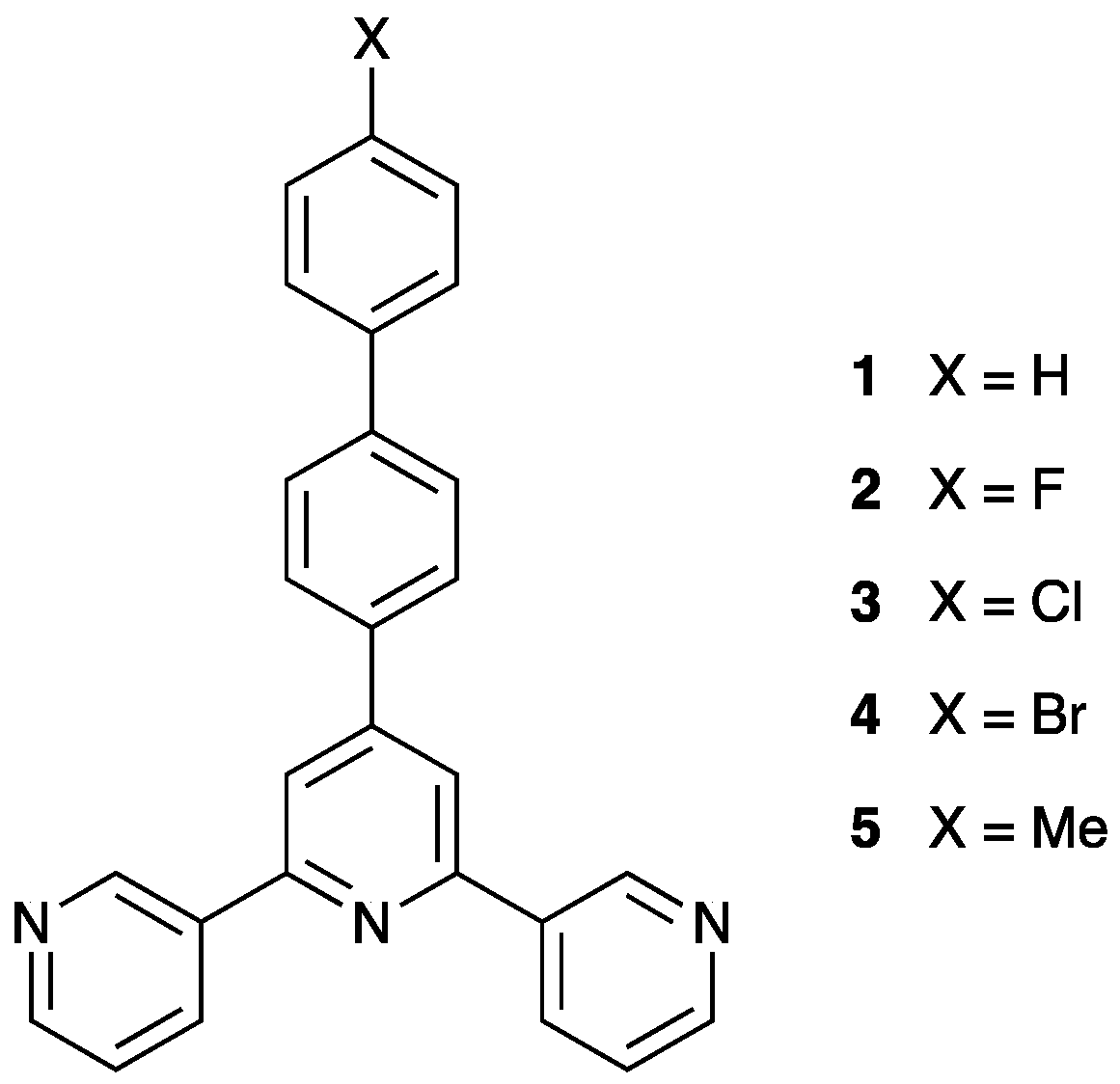
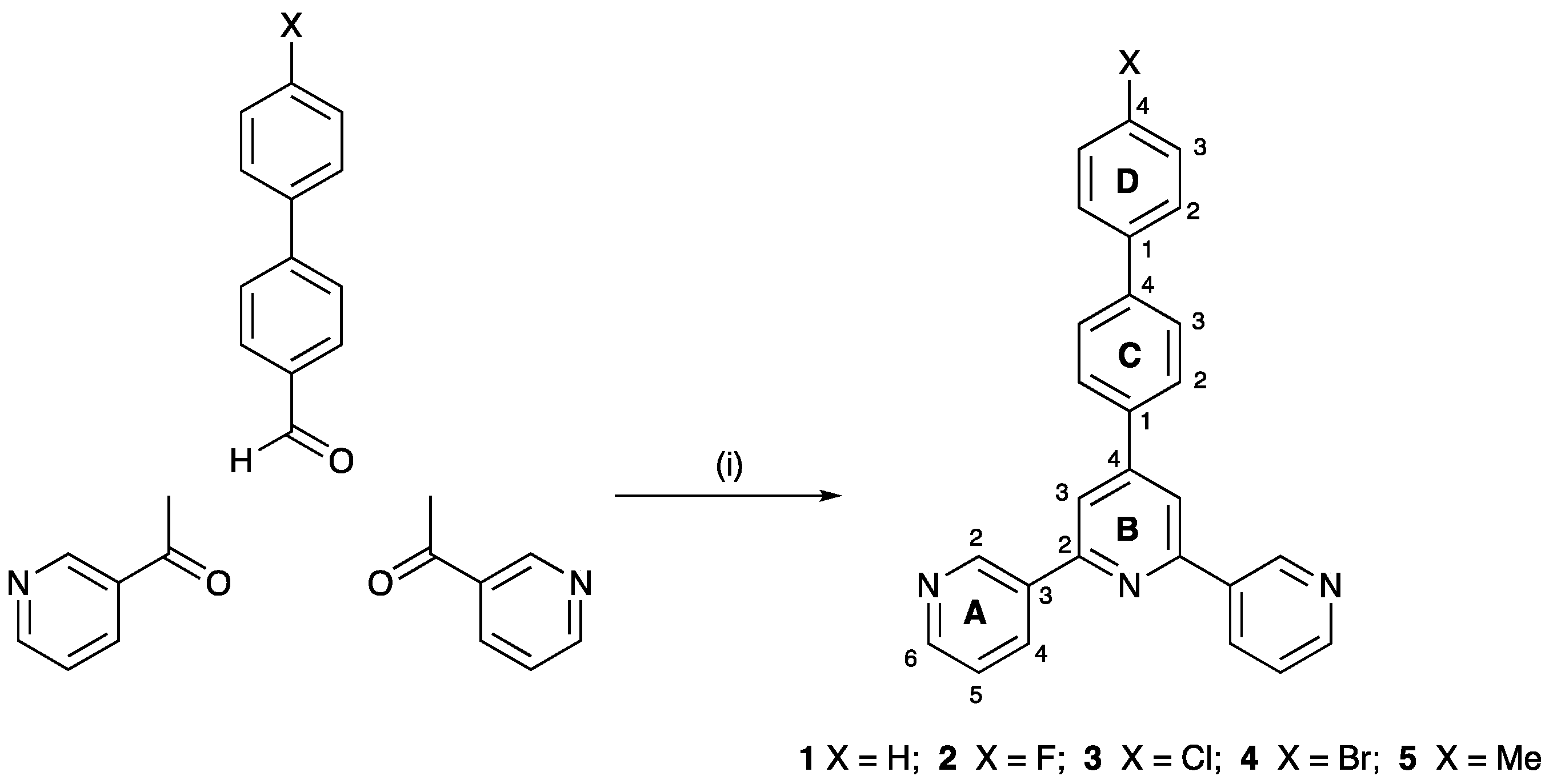
3.1. Ligand Synthesis and Characterization
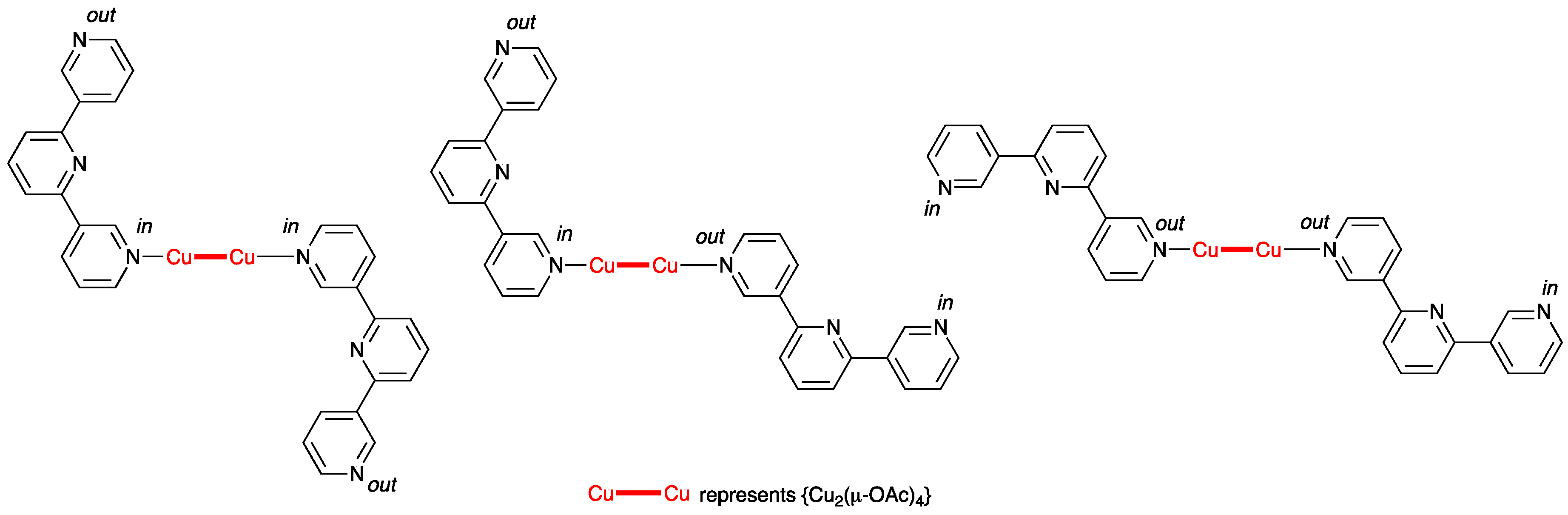
3.2. Reactions of Copper(II) Acetate and Ligands 1–5
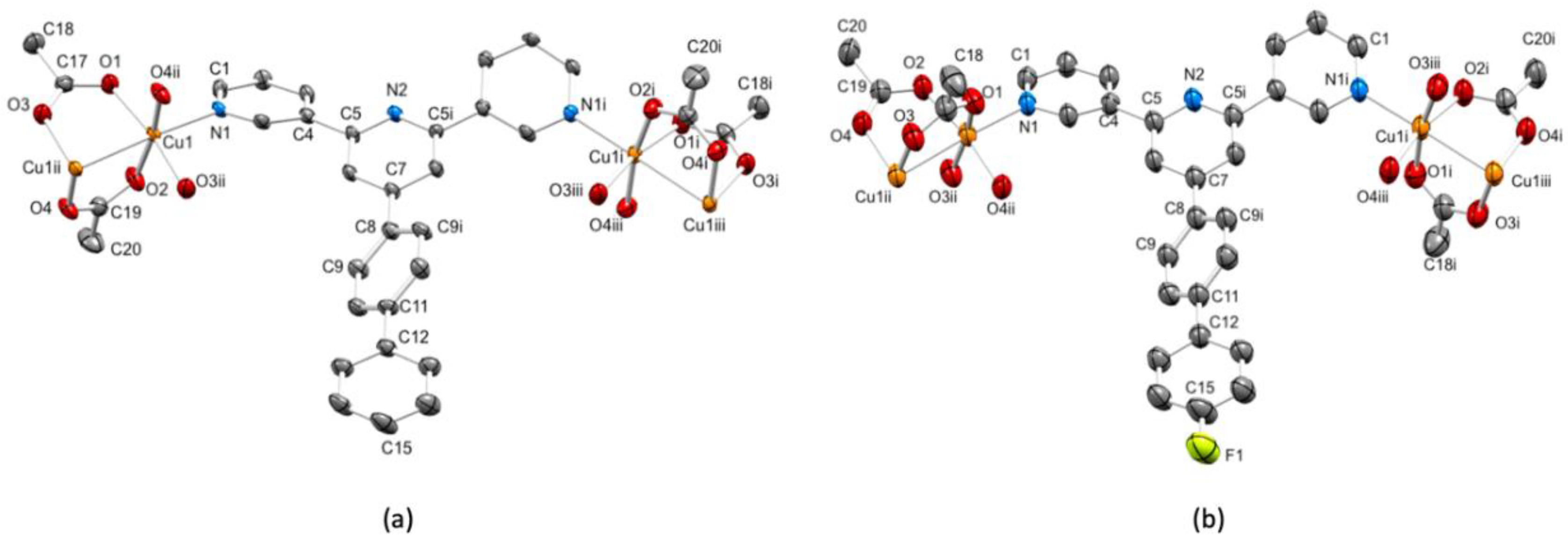
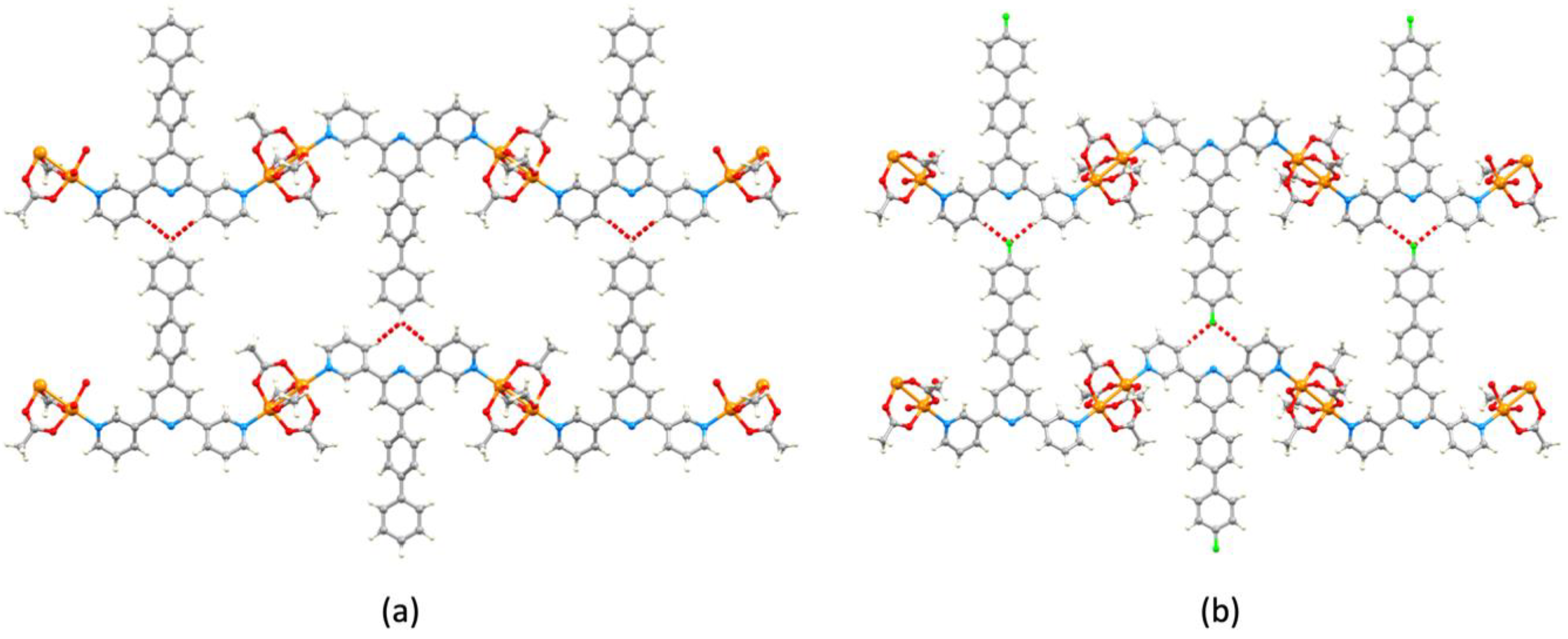
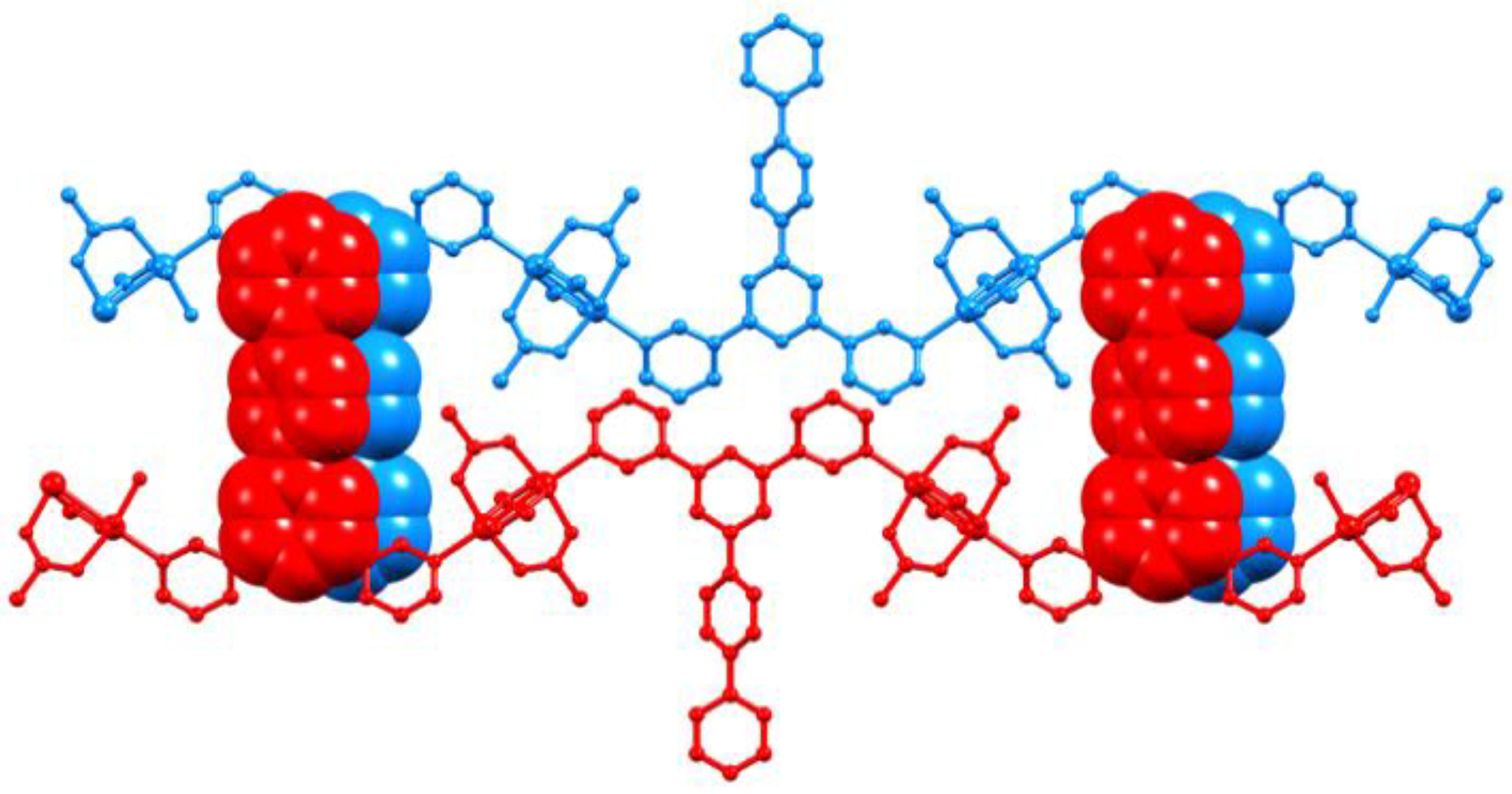
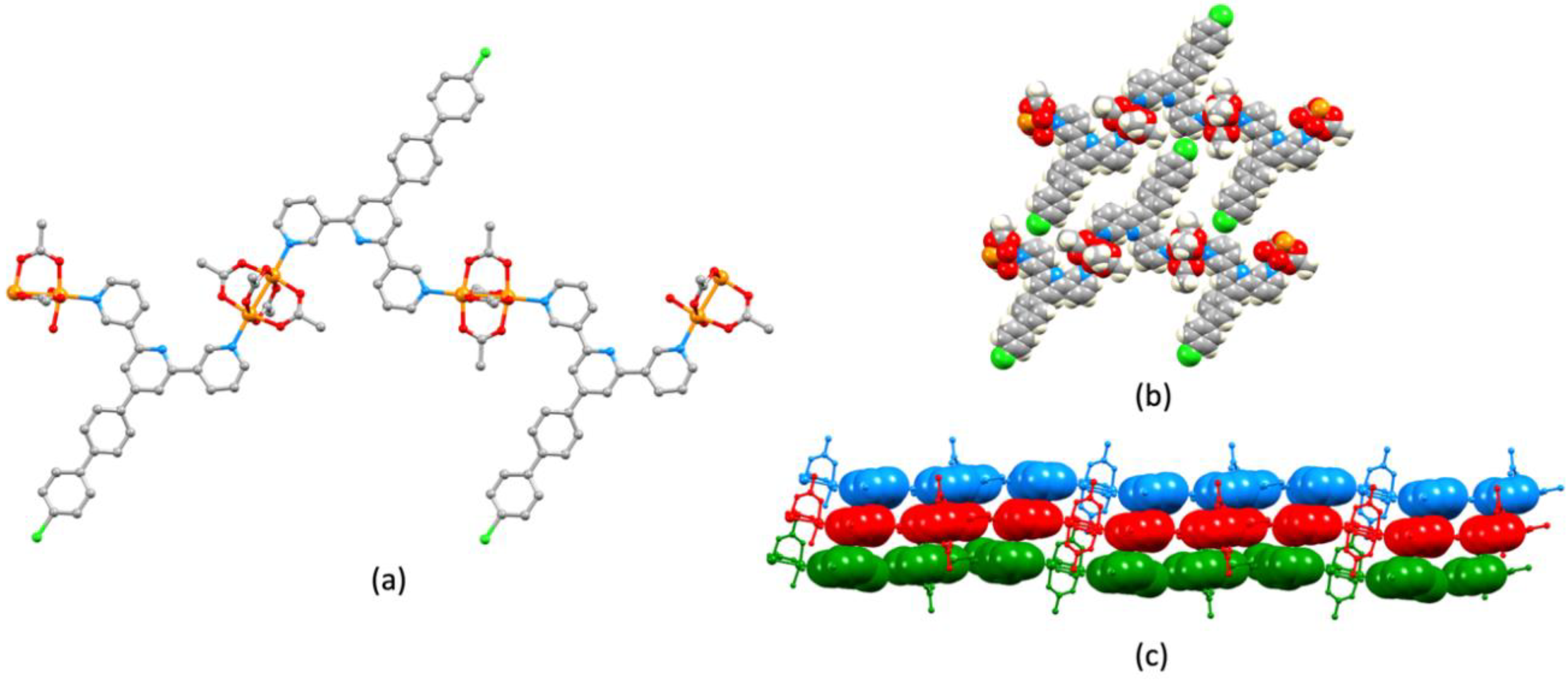
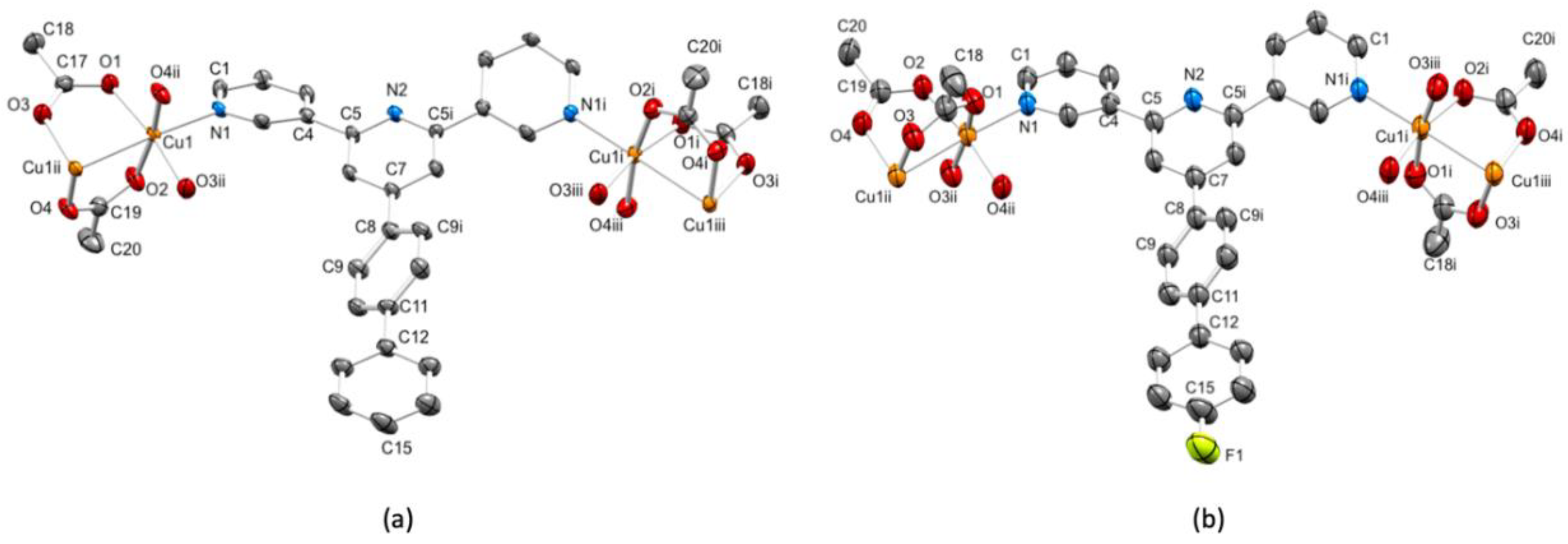
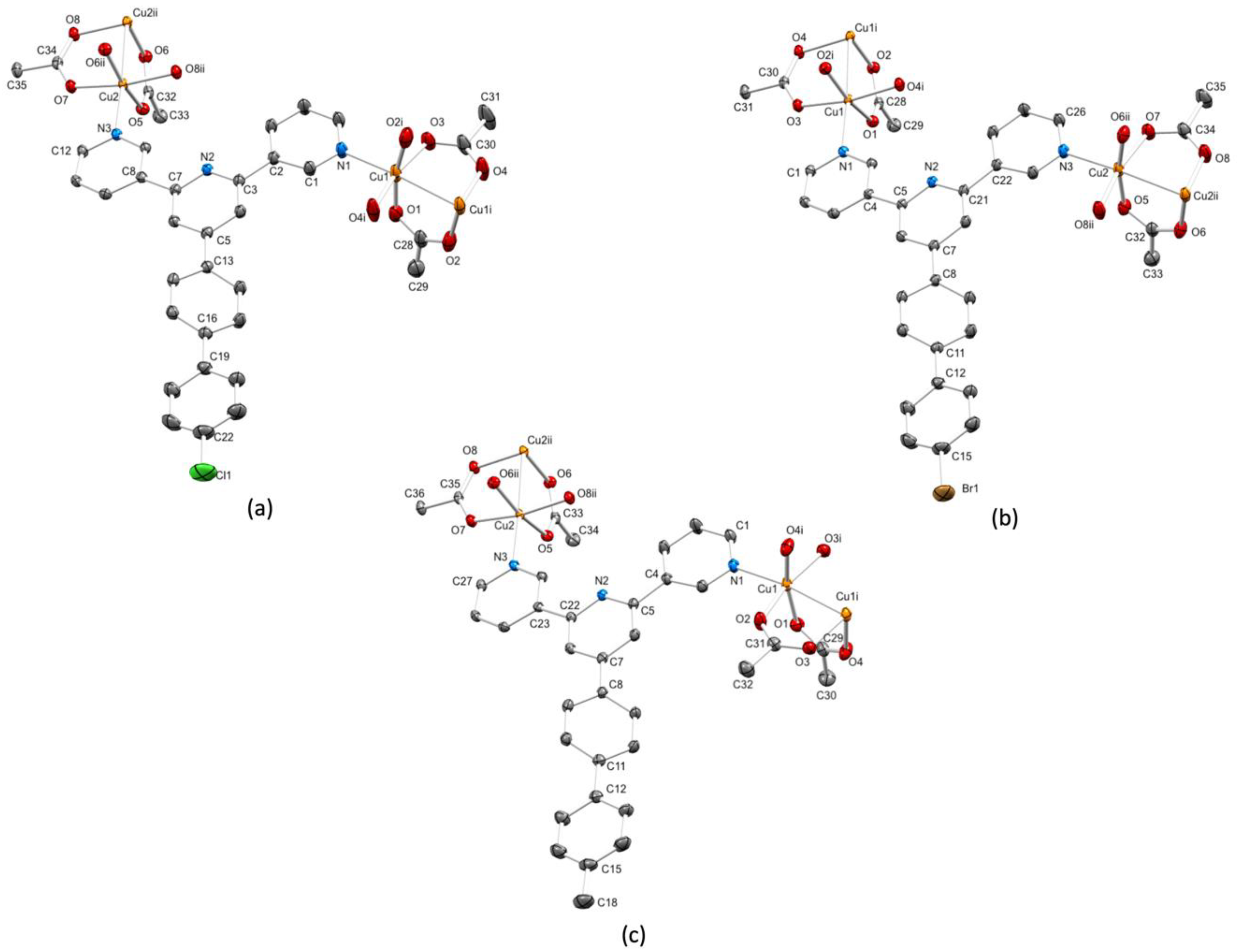
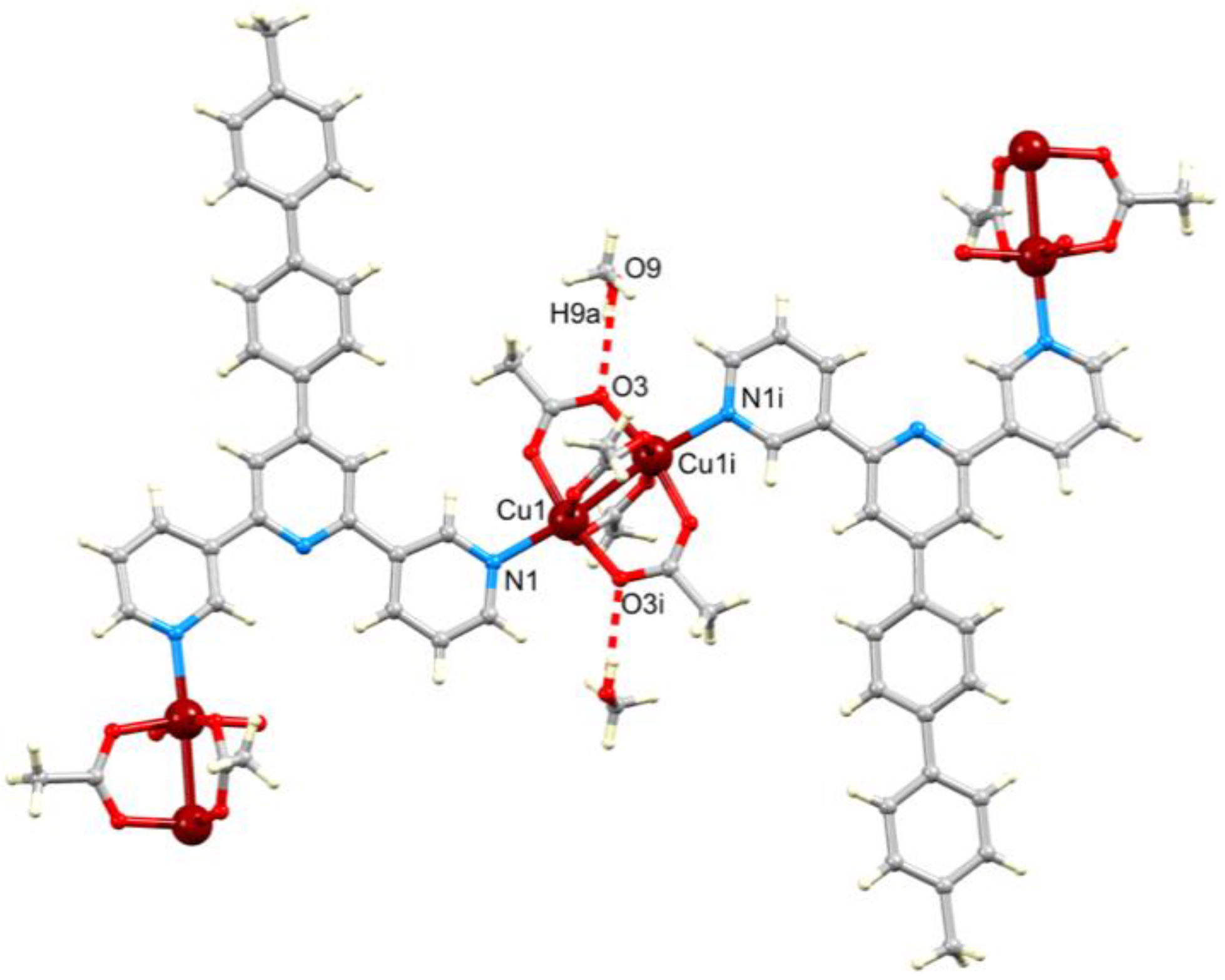
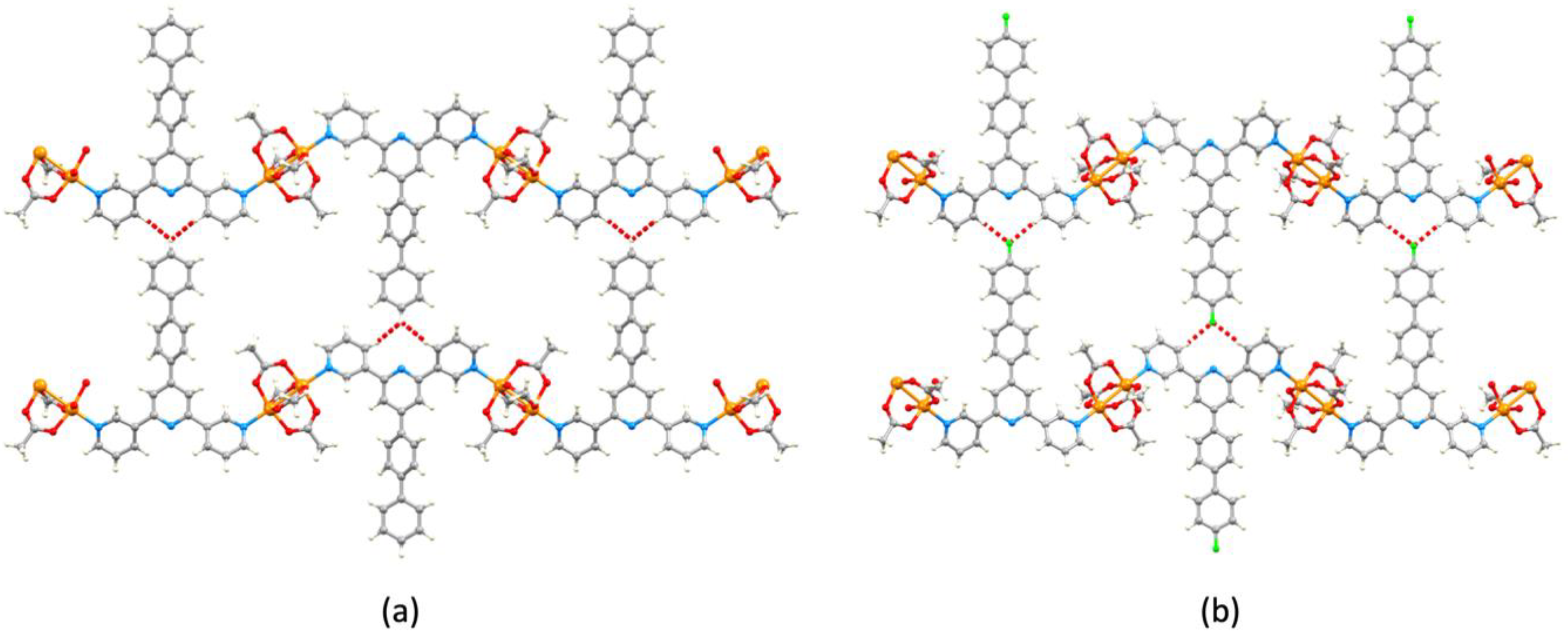
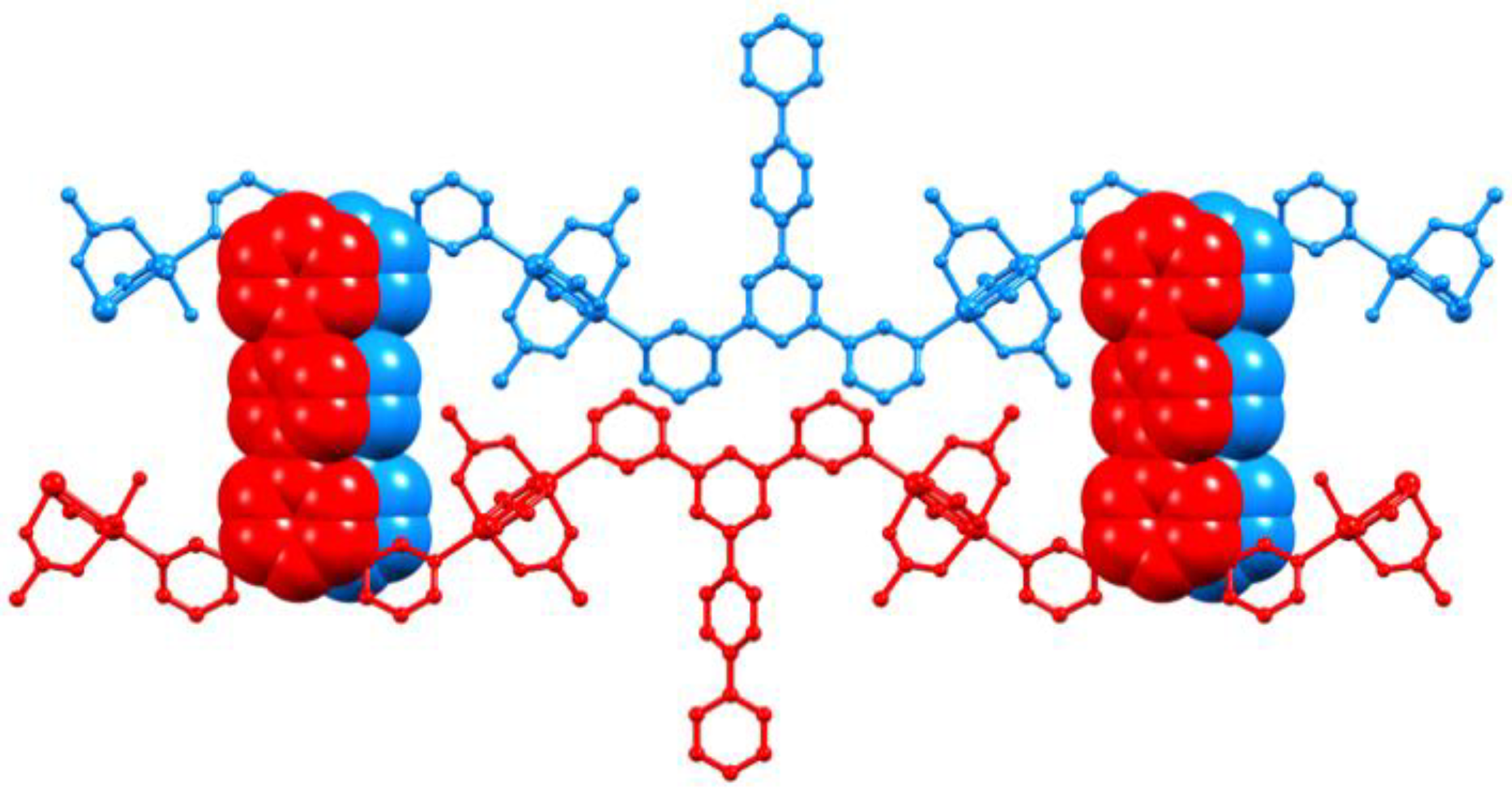
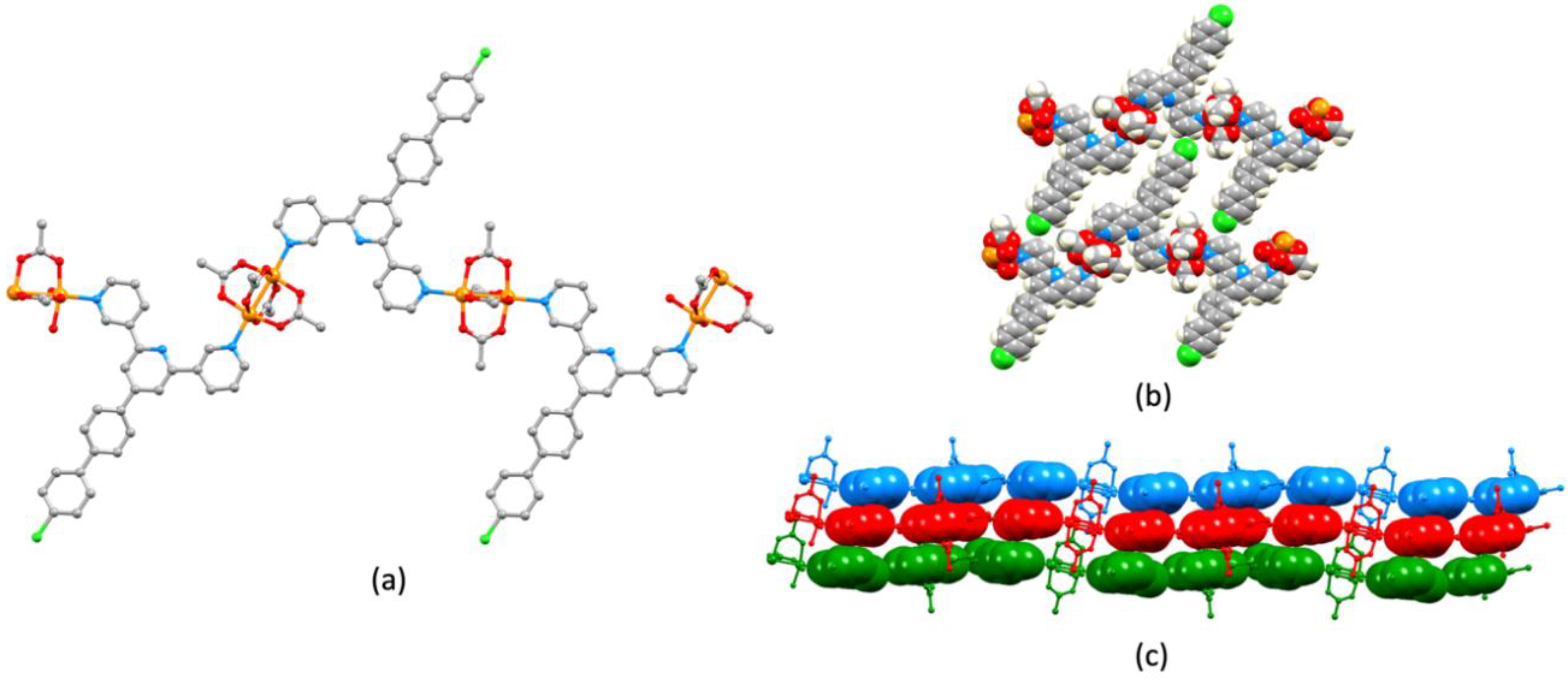
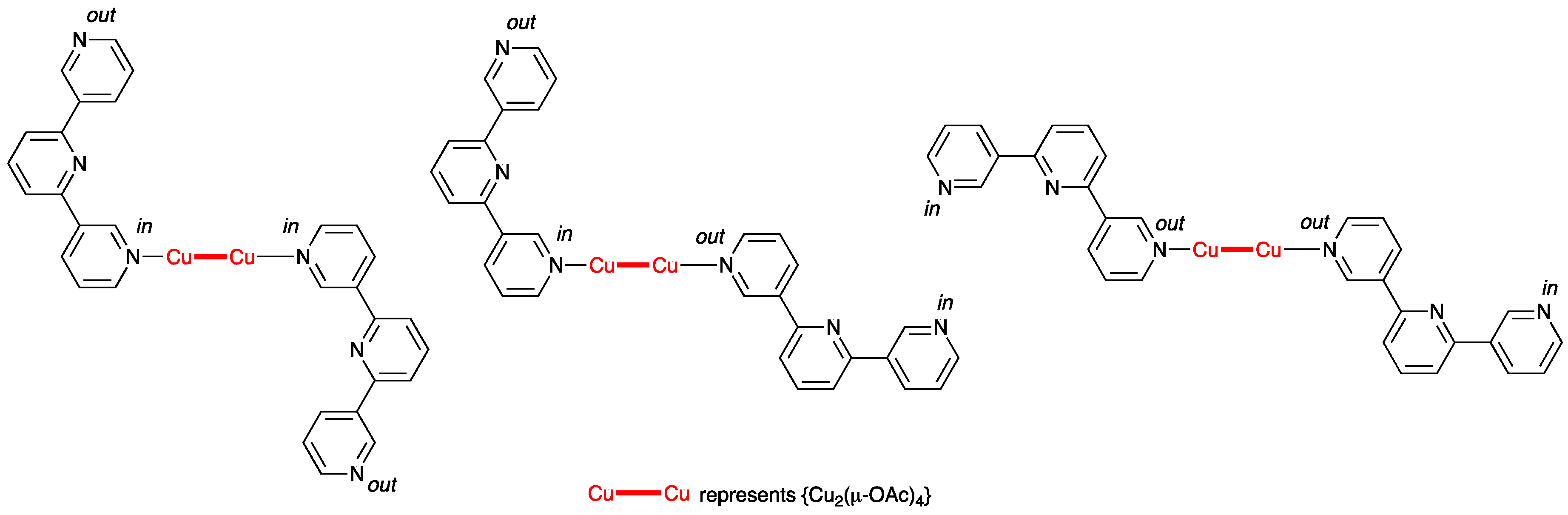
3.3. Single Crystal Structures
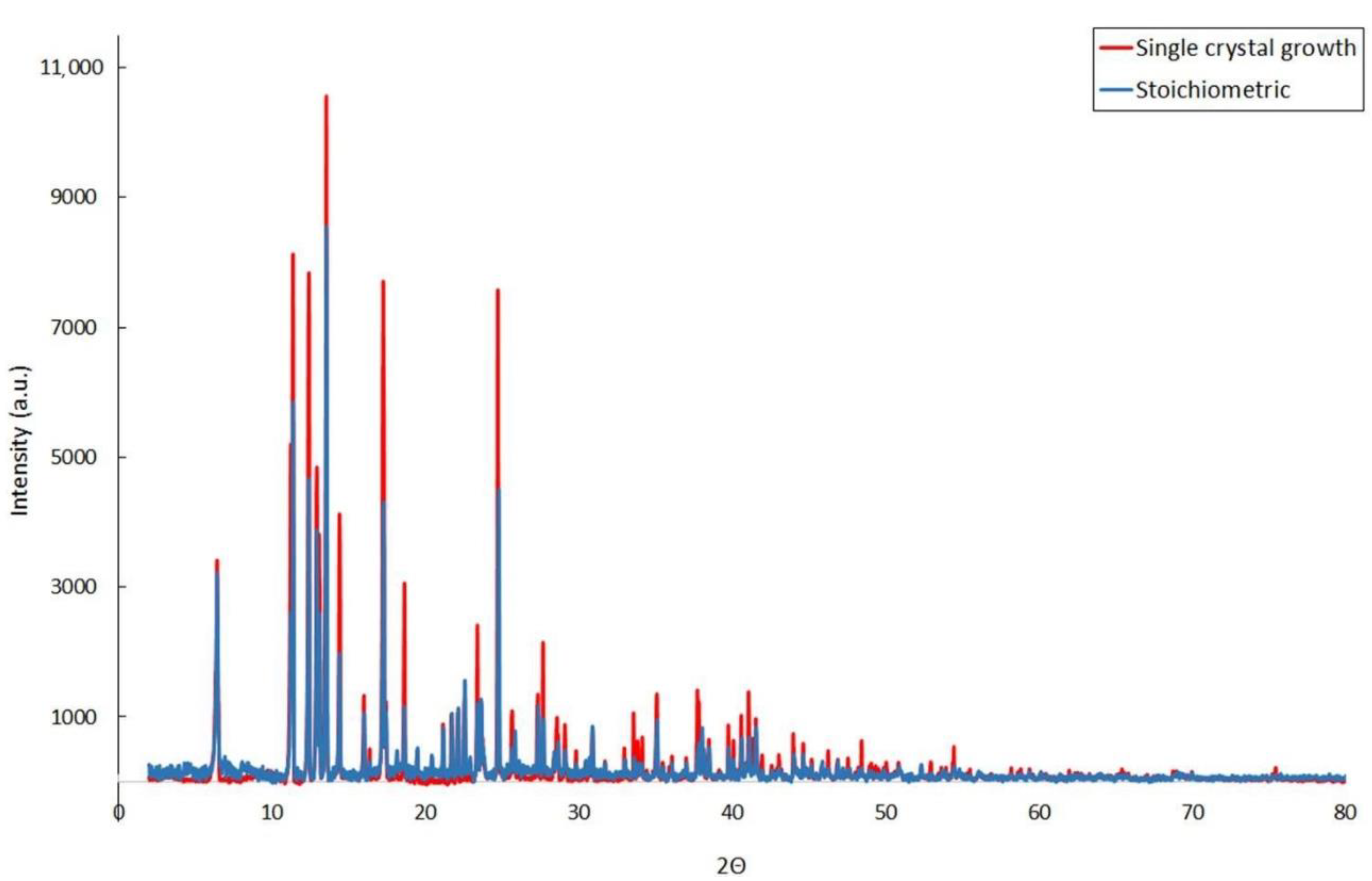
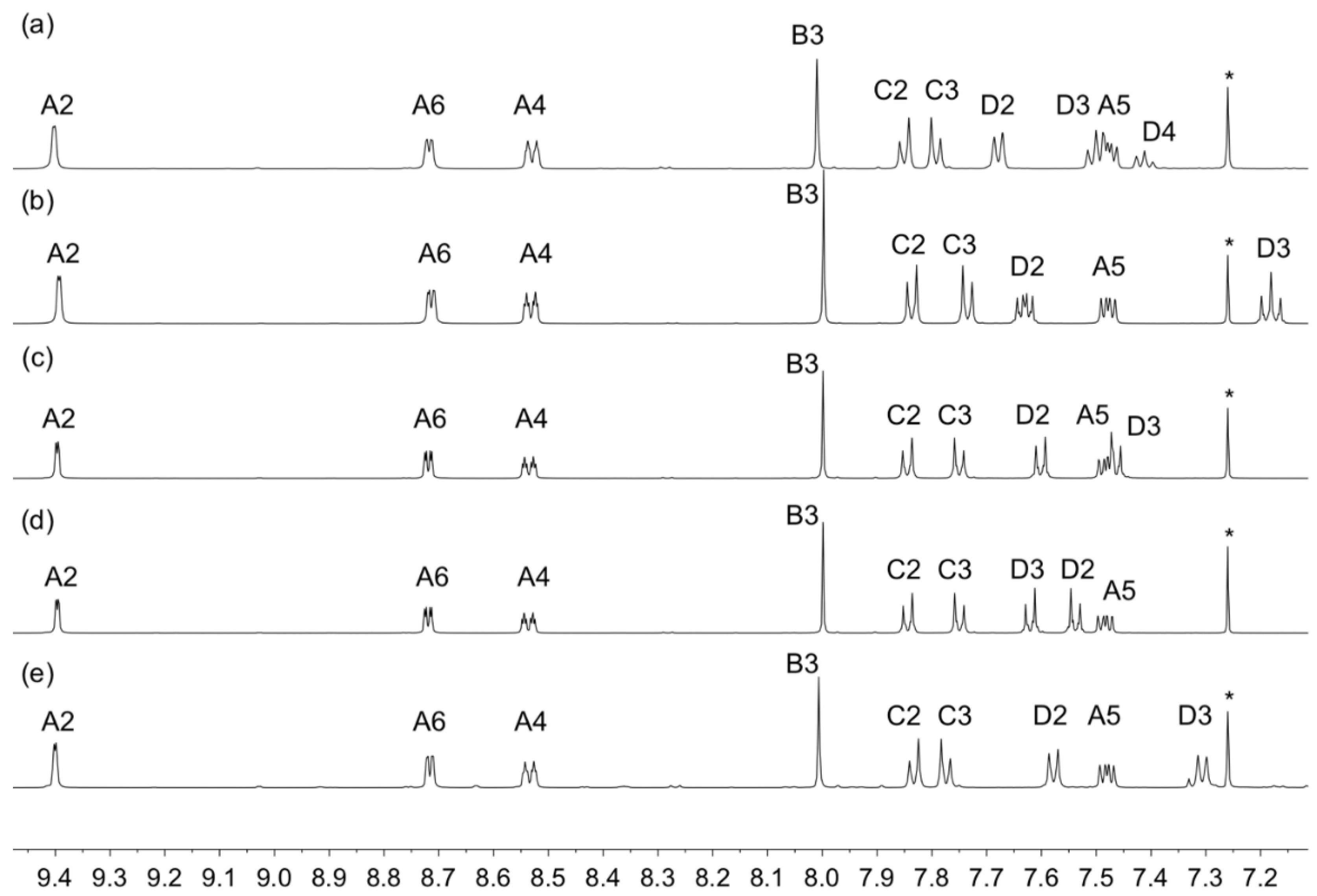
3.4. Characterization by PXRD
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Constable, E.C. The Coordination Chemistry of 2,2′:6′,2″-Terpyridine and Higher Oligopyridines. Adv. Inorg. Chem. Radiochem. 1986, 30, 69–121. [Google Scholar] [CrossRef]
- Constable, E.C. 2,2′:6′,2″-Terpyridines: From chemical obscurity to common supramolecular motifs. Chem. Soc. Rev. 2007, 36, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Schubert, U.S.; Hofmeier, H.; Newkome, G.R. Modern Terpyridine Chemistry; Wiley–VCH: Weinheim, Germany, 2006; ISBN 978-3-527-31475-1. [Google Scholar]
- Wei, C.; He, Y.; Shi, X.; Song, Z. Terpyridine-metal complexes: Applications in catalysis and supramolecular chemistry. Coord. Chem. Rev. 2019, 385, 1–19. [Google Scholar] [CrossRef]
- Schubert, U.S.; Winter, A.; Newkome, G.R. Terpyridine-Based Materials: For Catalytic, Optoelectronic and Life Science Applications; Wiley–VCH: Weinheim, Germany, 2011; ISBN 9783527330386. [Google Scholar]
- Constable, E.C. Expanded ligands: An assembly principle for supramolecular chemistry. Coord. Chem. Rev. 2008, 252, 842–855. [Google Scholar] [CrossRef]
- Chakraborty, S.; Newkome, G.R. Terpyridine-based metallosupramolecular constructs: Tailored monomers to precise 2D-motifs and 3D-metallocages. Chem. Soc. Rev. 2018, 47, 3991–4016. [Google Scholar] [CrossRef] [PubMed]
- Housecroft, C.E.; Constable, E.C. The Terpyridine Isomer Game: From Chelate to Coordination Network Building Block. Chem. Commun. 2020, 56, 10786–10794. [Google Scholar] [CrossRef]
- Kröhnke, F. Syntheses Using Pyridinium Salts. Angew. Chem. Int. Ed. 1963, 2, 225. [Google Scholar] [CrossRef]
- Wang, J.; Hanan, G.S. A facile route to sterically hindered and non-hindered 4′-aryl-2,2′:6′,2″-terpyridines. Synlett 2005, 1251–1254. [Google Scholar] [CrossRef]
- Sato, S.; Murase, T.; Fujita, M. Self-Assembly of Coordination Cages and Spheres. In Supramolecular Chemistry: From Molecules to Nanomaterials; Gale, P.A., Steed, J.W., Eds.; Wiley-Blackwell: Oxford, UK, 2012. [Google Scholar] [CrossRef]
- Rocco, D.; Prescimone, A.; Constable, E.C.; Housecroft, C.E. Switching the conformation of 3,2′:6′,3″-tpy domains in 4′-(4-n-alkyloxyphenyl)-3,2′:6′,3″-terpyridines. Molecules 2020, 25, 3162. [Google Scholar] [CrossRef]
- Rocco, D.; Manfroni, G.; Prescimone, A.; Klein, Y.M.; Gawryluk, D.J.; Constable, E.C.; Housecroft, C.E. Single and double-stranded 1D-coordination polymers with 4′-(4-alkyloxyphenyl)-3,2′:6′,3″-terpyridines and {Cu2(μ-OAc)4} or {Cu4(μ3-OH)2(μ-OAc)2(μ3-OAc)2(AcO-κO)2} motifs. Polymers 2020, 12, 318. [Google Scholar] [CrossRef] [Green Version]
- Rocco, D.; Prescimone, A.; Constable, E.C.; Housecroft, C.E. Straight versus branched chain substituents in 4′-(butoxyphenyl)-3,2′:6′,3″-terpyridines: Effects on (4,4) coordination network assemblies. Polymers 2020, 12, 1823. [Google Scholar] [CrossRef] [PubMed]
- Rocco, D.; Prescimone, A.; Constable, E.C.; Housecroft, C.E. Directing 2D-coordination networks: Combined effects of a conformationally flexible 3,2′:6′,3″-terpyridine and chain length variation in 4′-(4-n-alkyloxyphenyl) substituents. Molecules 2020, 25, 1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granifo, J.; Gavino, R.; Freire, E.; Baggio, R. The new sulphur-containing ligand 4′-(4-methylthiophenyl)-3,2′:6′,3″-terpyridine (L1) and the supramolecular structure of the dinuclear complex [Zn2(μ-L1)(acac)4] (acac = acetylacetonato): The key role of non-covalent S…O contacts and C–H…S hydrogen bonds. J. Mol. Struct. 2011, 1006, 684–691. [Google Scholar] [CrossRef]
- Henling, L.M.; Marsh, R.E. Some more space-group corrections. Acta Crystallogr. 2014, C70, 834–836. [Google Scholar] [CrossRef] [Green Version]
- Granifo, J.; Vargas, M.; Garland, M.T.; Ibanez, A.; Gavino, R.; Baggio, R. The novel ligand 4′-phenyl-3,2′:6′,3″-terpyridine (L) and the supramolecular structure of the dinuclear complex [Zn2(μ-L)(acac)4]·H2O (acac = acetylacetonato). Inorg. Chem. Comm. 2008, 11, 1388–1391. [Google Scholar] [CrossRef]
- Klein, Y.M.; Lanzilotto, A.; Prescimone, A.; Krämer, K.W.; Decurtins, S.; Liu, S.-X.; Constable, E.C.; Housecroft, C.E. Coordination behaviour of 1-(3,2′:6′,3″-terpyridin-4′-yl)ferrocene: Structure and magnetic and electrochemical properties of a tetracopper dimetallomacrocycle. Polyhedron 2017, 129, 71–76. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Tan, J.; Su, J.; Zhang, J.; Zhang, S.; Wu, J.; Tian, Y. Syntheses, crystal structures and third-order nonlinear optical properties of two series of Zn(II) complexes using the thiophene-based terpyridine ligands. Dyes Pigments 2016, 130, 216–225. [Google Scholar] [CrossRef]
- Zhang, L.; Li, C.-J.; He, J.-E.; Chen, Y.-Y.; Zheng, S.-R.; Fan, J.; Zhang, W.-G. Construction of New Coordination Polymers from 4′-(2,4-disulfophenyl)-3,2′:6′,3″-terpyridine: Polymorphism, pH-dependent syntheses, structures, and properties. J. Solid State Chem. 2016, 233, 244–254. [Google Scholar] [CrossRef]
- Cheng, Y.; Yang, M.-L.; Hu, H.-M.; Xu, B.; Wang, X.; Xue, G. Syntheses, structures and luminescence for zinc coordination polymers based on a multifunctional 4′-(3-carboxyphenyl)-3,2′:6′,3″-terpyridine ligand. J. Solid State Chem. 2016, 239, 121–130. [Google Scholar] [CrossRef]
- Wang, T.-T.; Zhang, J.-L.; Hu, H.-M.; Cheng, Y.; Xue, L.-L.; Wang, X.; Wang, B.-Z. Syntheses, structures and luminescent properties of Zn/Cd coordination polymers based on 4′-(2-carboxyphenyl)-3,2′:6′,3″-terpyridine. Polyhedron 2018, 151, 43–50. [Google Scholar] [CrossRef]
- Lufei, X.; Yupeng, T. CSD Refcode VUKMOF. In CSD Communication CCDC 1407410; The Cambridge Crystallographic Data Centre (CCDC): Cambridge, UK, 2015. [Google Scholar]
- Liu, C.; Ding, Y.-B.; Shi, X.-H.; Zhang, D.; Hu, M.-H.; Yin, Y.-G.; Li, D. Interpenetrating Metal−Organic Frameworks Assembled from Polypyridine Ligands and Cyanocuprate Catenations. Cryst. Growth Des. 2009, 9, 1275–1277. [Google Scholar] [CrossRef]
- Li, L.; Zhang, Y.Z.; Yang, C.; Liu, E.; Golen, J.A.; Zhang, G. One-dimensional copper(II) coordination polymers built on 4′-substituted 4,2′:6′,4″- and 3,2′:6′,3″-terpyridines: Syntheses, structures and catalytic properties. Polyhedron 2016, 105, 115–122. [Google Scholar] [CrossRef]
- Köberl, M.; Cokoja, M.; Herrmann, W.A.; Kühn, F.E. From molecules to materials: Molecular paddle-wheel synthons of macromolecules, cage compounds and metal-organic frameworks. Dalton Trans. 2011, 40, 6834–6859. [Google Scholar] [CrossRef] [PubMed]
- Constable, E.C.; Housecroft, C.E.; Neuburger, M.; Schönle, J.; Vujovic, S.; Zampese, J.A. Molecular recognition between 4′-(4-biphenylyl)-4,2′:6′,4″-terpyridine domains in the assembly of d9 and d10 metal ion-containing one-dimensional coordination polymers. Polyhedron 2013, 60, 120–129. [Google Scholar] [CrossRef]
- Constable, E.C.; Housecroft, C.E.; Vujovic, S.; Zampese, J.A.; Crochet, A.; Batten, S.R. Do perfluoroarene… arene and C–H...F interactions make a difference to 4,2′:6′,4”-terpyridine-based coordination polymers? CrystEngComm 2013, 15, 10068–10078. [Google Scholar] [CrossRef] [Green Version]
- Software for the Integration of CCD Detector System Bruker Analytical X-ray Systems, Bruker axs, Madison, WI (after 2013).
- Sheldrick, G.M. ShelXT-Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. Olex2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with ShelXL. Acta Cryst. 2015, C27, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Palatinus, L.; Chapuis, G. Superflip-A Computer Program for the Solution of Crystal Structures by Charge Flipping in Arbitrary Dimensions. J. Appl. Cryst. 2007, 40, 786–790. [Google Scholar] [CrossRef] [Green Version]
- Palatinus, L.; Prathapa, S.J.; van Smaalen, S. EDMA: A Computer Program for Topological Analysis of Discrete Electron Densities. J. Appl. Cryst. 2012, 45, 575–580. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeBail, A.; Duroy, H.; Fourquet, J.L. Ab-initio structure determination of LiSbWO6 by X-ray powder diffraction. Mater. Res. Bull. 1988, 23, 447–452. [Google Scholar] [CrossRef]
- Pawley, G.S. Unit-cell refinement from powder diffraction scans. J. Appl. Cryst. 1981, 14, 357–361. [Google Scholar] [CrossRef]
- Rodríguez-Carvajal, J. Recent Advances in Magnetic Structure Determination by Neutron Powder Diffraction. Physica B 1993, 192, 55–69. [Google Scholar] [CrossRef]
- Roisnel, T.; Rodríguez-Carvajal, J. WinPLOTR: A Windows tool for powder diffraction patterns analysis Materials Science Forum. In Proceedings of the Seventh European Powder Diffraction Conference (EPDIC 7), Barcelona, Spain, 20–23 May 2000; pp. 118–123. [Google Scholar]
- Spartan ’18 Version 1.4.4; Wavefunction Inc.: Irvine, CA, USA, 2019.
- Bienz, S.; Bigler, L.; Fox, T.; Meier, H. Spektroskopische Methoden in der Organischen Chemie, 9th ed.; Thieme: Stuttgart, Germany, 2016; ISBN 9783135761091. [Google Scholar]
- Janiak, C. A critical account on π–π stacking in metal complexes with aromatic nitrogen-containing ligands. J. Chem. Soc. Dalton Trans. 2000, 3885–3896. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cu–O/Å | Cu–N/Å | Cu…Cu/Å | |
|---|---|---|---|
| [Cu2(μ-OAc)4(1)]n | 1.954(4), 1.975(4), 1.990(4), 1.961(4) | 2.157(4) | 2.6051(13) |
| [Cu2(μ-OAc)4(2)]n | 1.953(5), 1.994(5), 1.979(5), 1.959(5) | 2.168(5) | 2.6149(17) |
| [Cu2(μ-OAc)4(3)]n | 1.9760(18), 1.9894(18), 1.9759(18), 1.9789(18), 1.979(3), 1.970(2), 1.966(3), 1.971(3) | 2.167(2), 2.151(2) | 2.6292(8), 2.6319(7) |
| [Cu2(μ-OAc)4(4)]n | 1.973(2), 1.983(2), 1.976(2), 1.979(2), 1.981(3), 1.971(2), 1.972(3), 1.974(3) | 2.153(2), 2.158(3) | 2.6352(8), 2.6235(9) |
| [Cu2(μ-OAc)4(5)]n·nMeOH | 1.9885(15), 1.9787(15), 1.9773(15), 1.9756(15), 1.9887(18), 1.9700(18), 1.9809(19), 1.9644(19) | 2.1510(18), 2.1813(19) | 2.6551(6), 2.6331(6) |
| py–py/o | pyN2–Phenylene/o | Phenylene–phenyl/o | |
|---|---|---|---|
| [Cu2(μ-OAc)4(1)]n | 25.1 | 38.3 | 41.7 |
| [Cu2(μ-OAc)4(2)]n | 21.1 | 39.8 | 38.5 |
| [Cu2(μ-OAc)4(3)]n | 8.0, 4.5 | 24.0 | 27.2 |
| [Cu2(μ-OAc)4(4)]n | 8.1, 4.0 | 22.9 | 26.5 |
| [Cu2(μ-OAc)4(5)]n·nMeOH | 6.5, 4.0 | 27.8 | 28.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rocco, D.; Novak, S.; Prescimone, A.; Constable, E.C.; Housecroft, C.E. Manipulating the Conformation of 3,2′:6′,3″-Terpyridine in [Cu2(μ-OAc)4(3,2′:6′,3″-tpy)]n 1D-Polymers. Chemistry 2021, 3, 182-198. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry3010015
Rocco D, Novak S, Prescimone A, Constable EC, Housecroft CE. Manipulating the Conformation of 3,2′:6′,3″-Terpyridine in [Cu2(μ-OAc)4(3,2′:6′,3″-tpy)]n 1D-Polymers. Chemistry. 2021; 3(1):182-198. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry3010015
Chicago/Turabian StyleRocco, Dalila, Samantha Novak, Alessandro Prescimone, Edwin C. Constable, and Catherine E. Housecroft. 2021. "Manipulating the Conformation of 3,2′:6′,3″-Terpyridine in [Cu2(μ-OAc)4(3,2′:6′,3″-tpy)]n 1D-Polymers" Chemistry 3, no. 1: 182-198. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry3010015