New Syntheses, Analytic Spin Hamiltonians, Structural and Computational Characterization for a Series of Tri-, Hexa- and Hepta-Nuclear Copper (II) Complexes with Prototypic Patterns

 , , and
, , and
Abstract
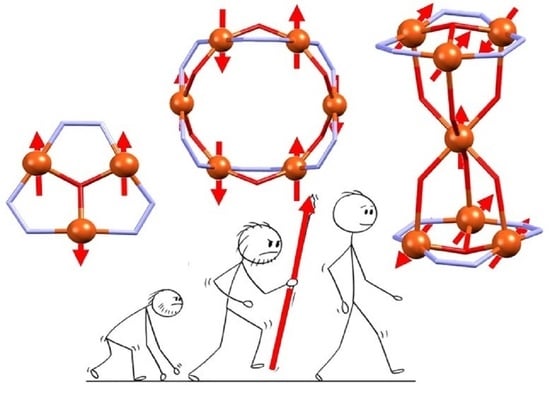
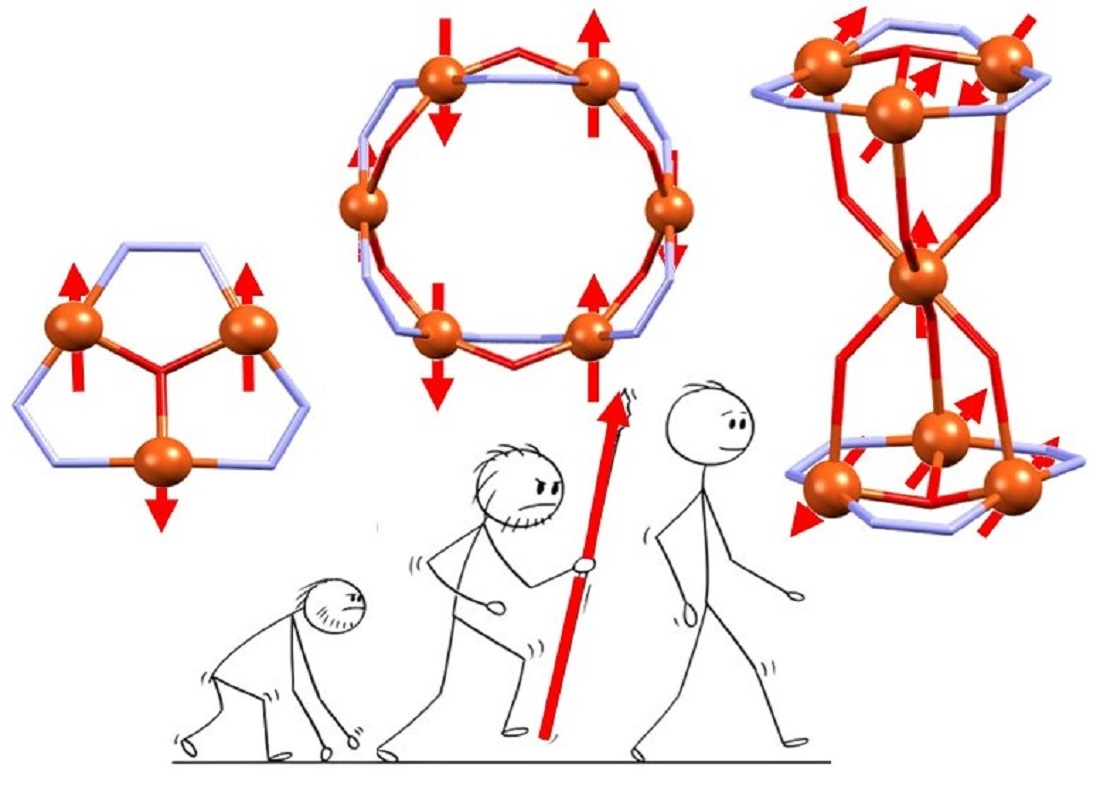
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
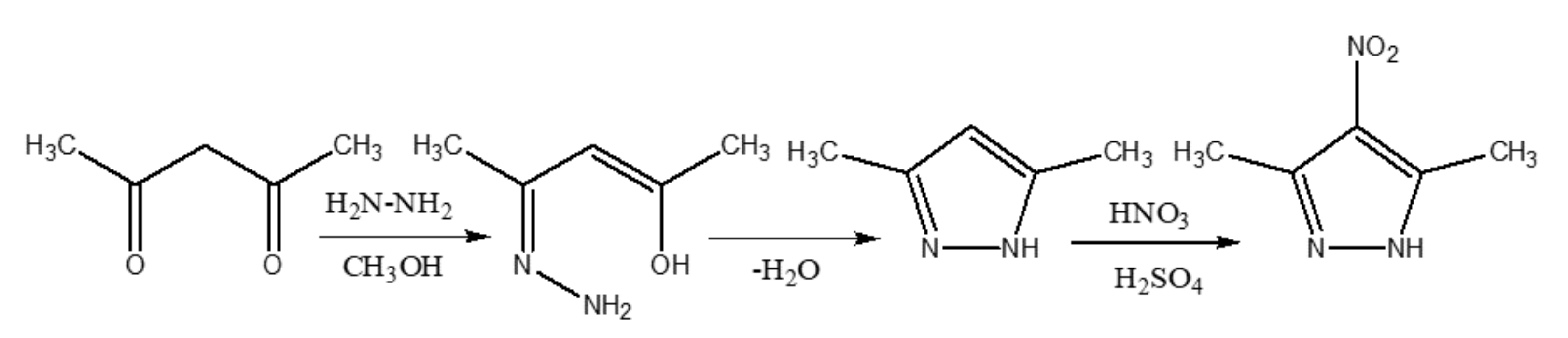
2.1. Materials and Physical Measurements
2.2. Synthesis
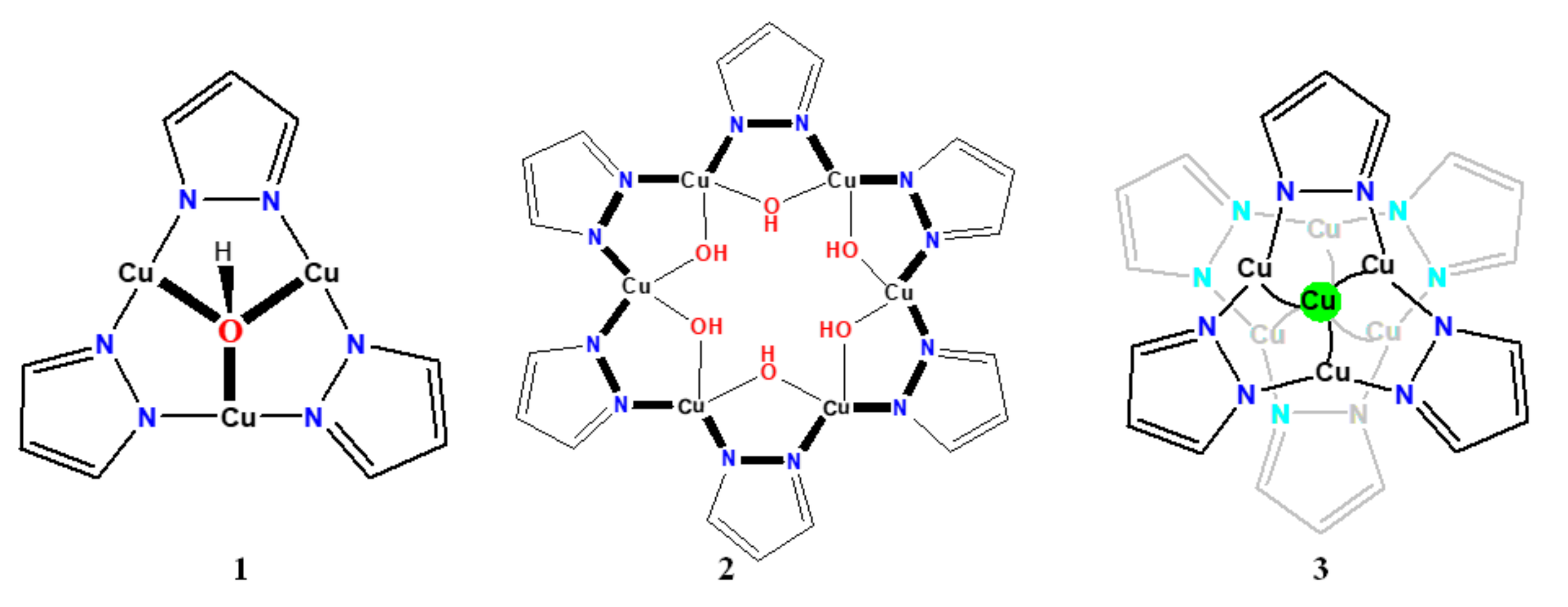
2.2.1. [Cu3(μ3-OH)(μ-dmnpz)3(OCOCH3)2(H2O)3]·H2O·EtOH (1)
2.2.2. [Cu6(μ-OH)6(μ-dmnpz)6(Py)6]·6Py·2EtOH (2)
2.2.3. [Cu7(μ-OH)2(μ-dmnpz)6(μ-OCOCH3)6(MeOH)6] (3)
2.3. X-ray Crystallography
2.4. Computational Methods
3. Results and Discussions
3.1. Structural Analysis from Crystallographic Data
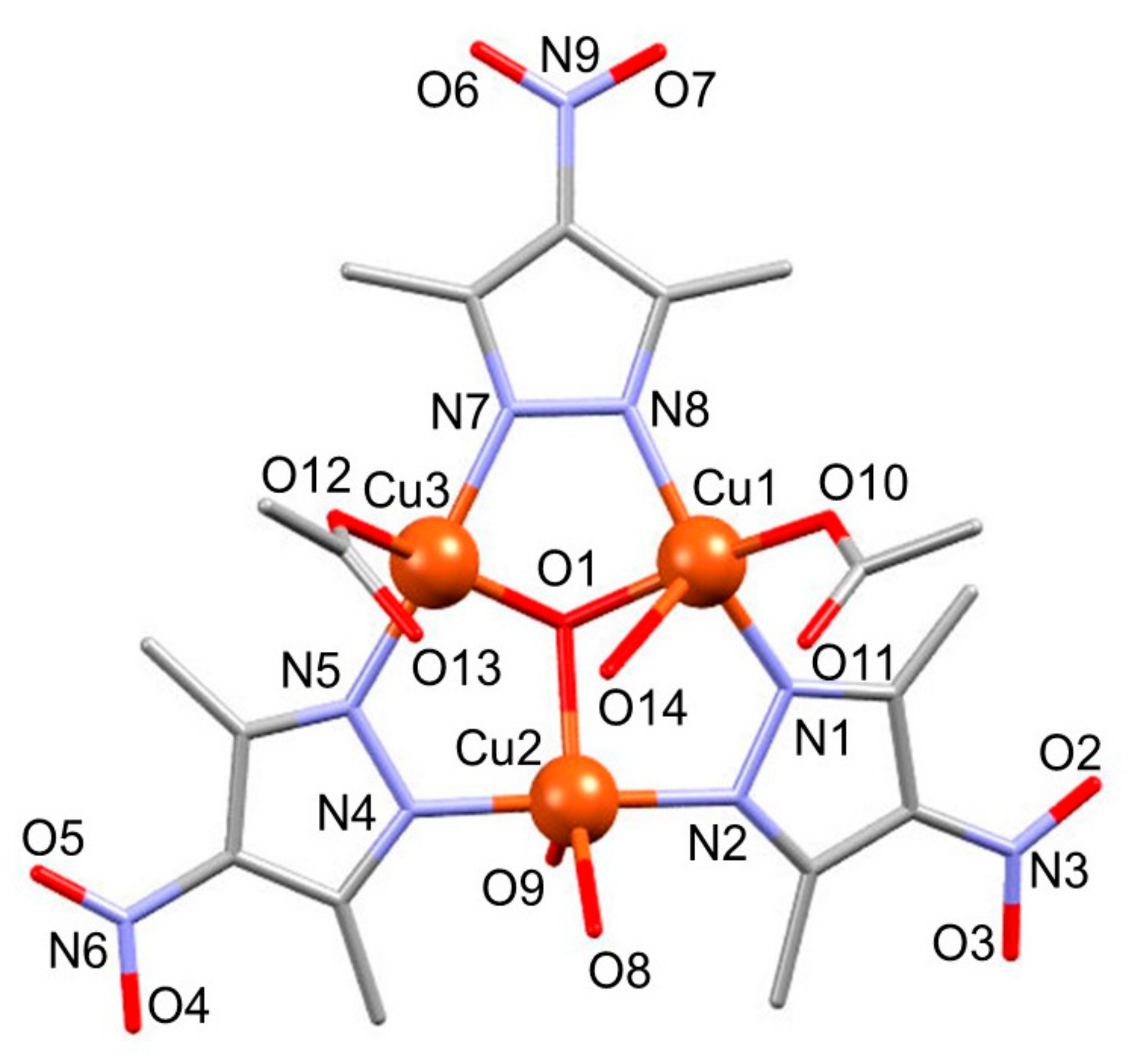
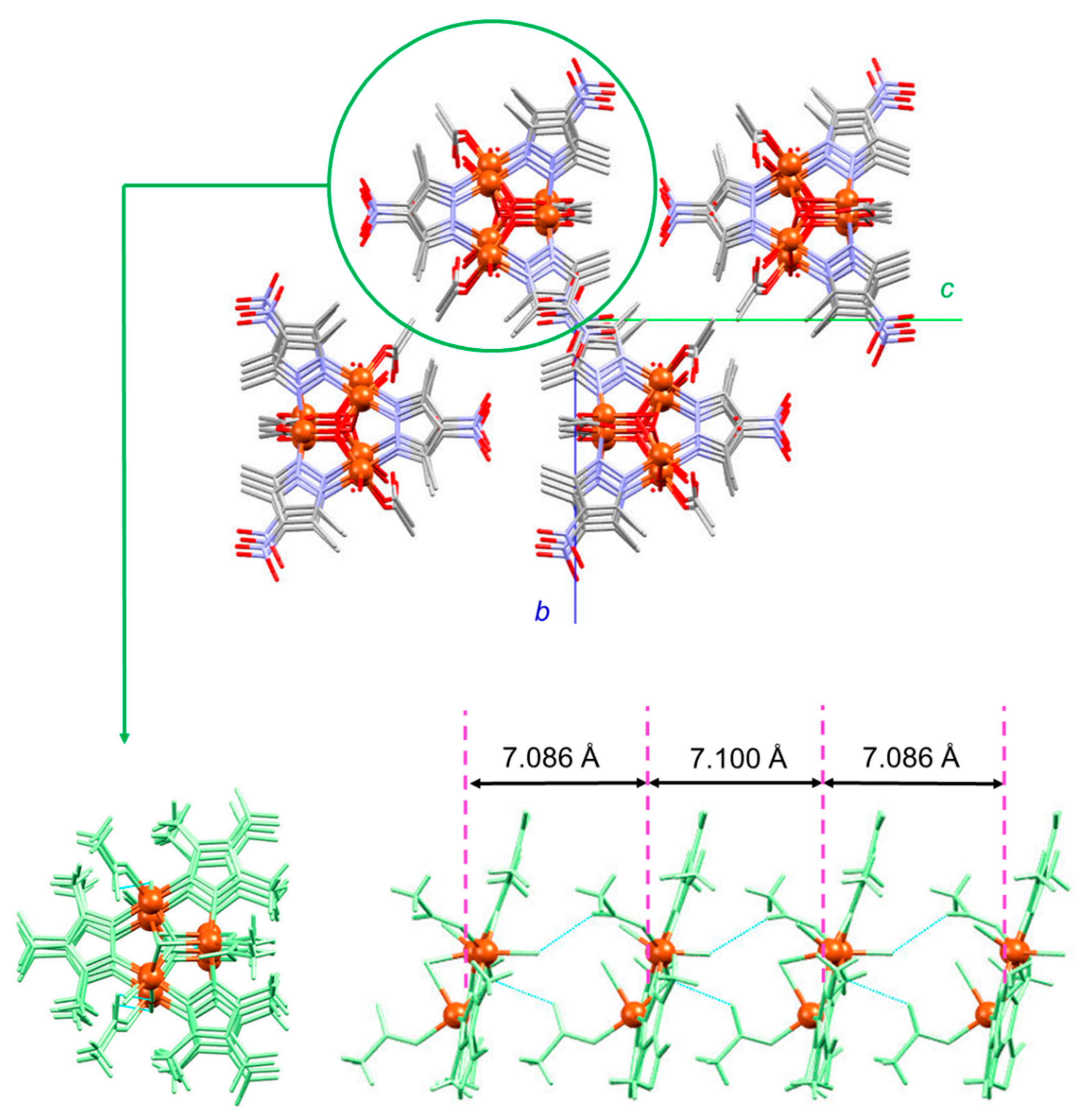
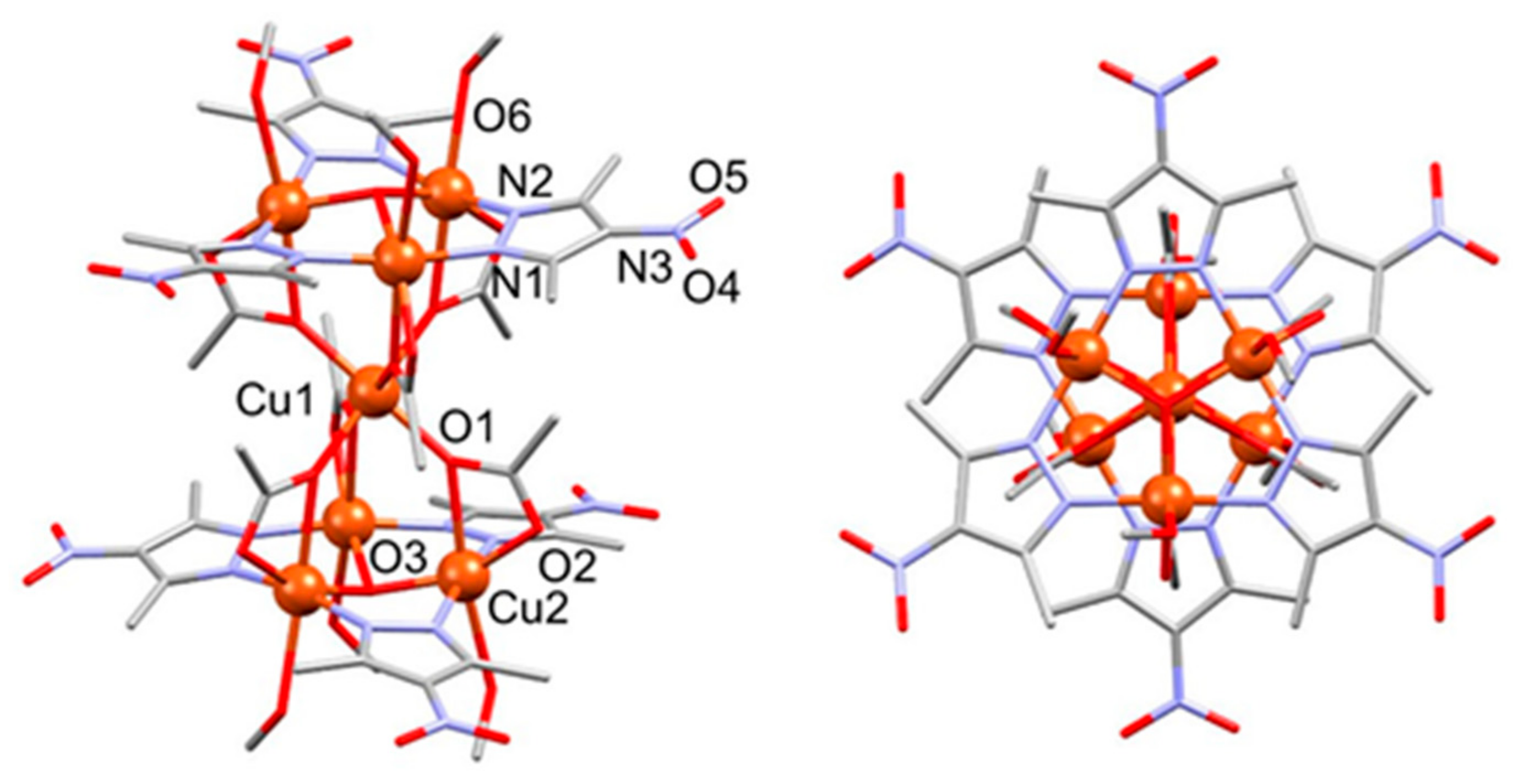
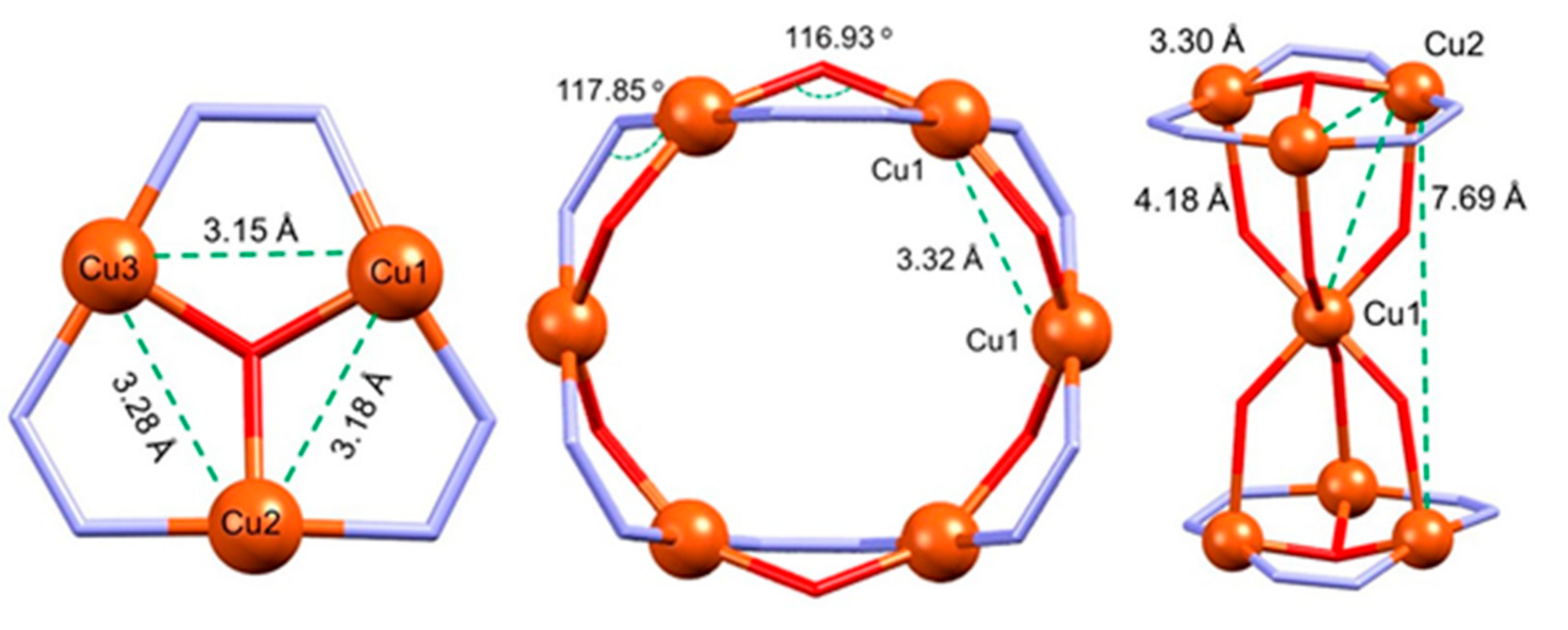
3.1.1. Structure of Compound 1
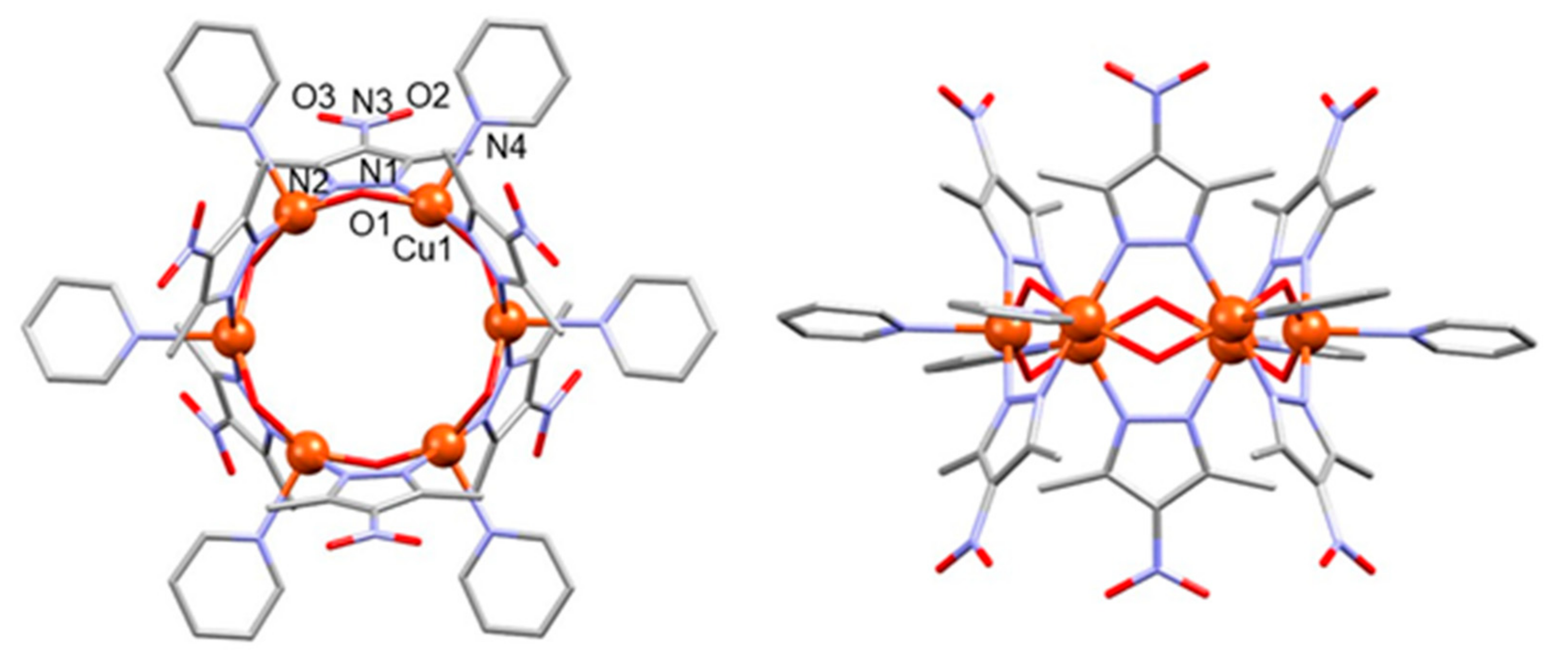
3.1.2. Structure of Compound 2
3.1.3. Structure of Compound 3
3.2. Spin Hamiltonians and Magnetic Data
3.2.1. Spin Hamiltonian and Magnetic Susceptibility Equations
3.2.2. The Magnetism of the Trinuclear Complex
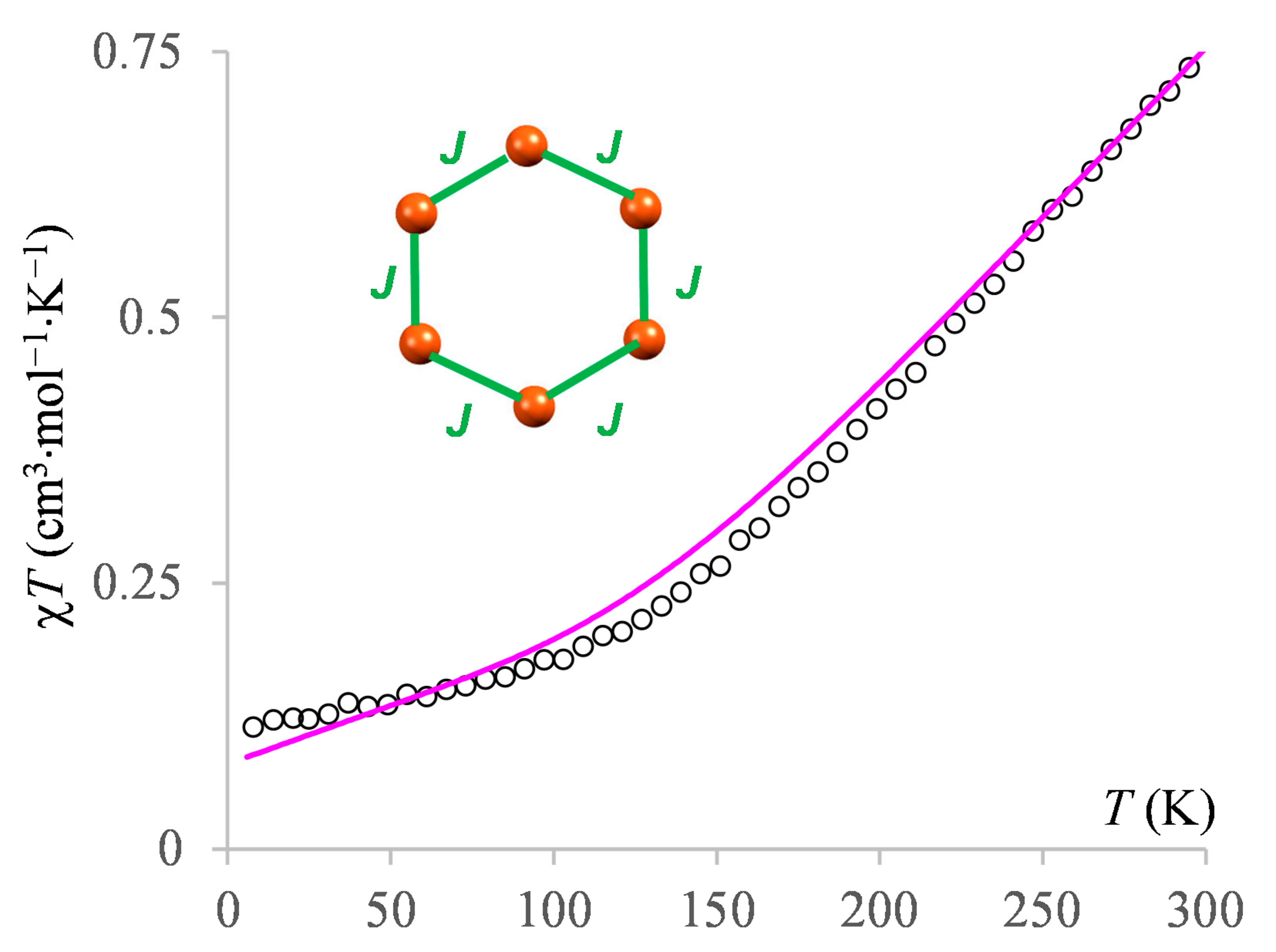
3.2.3. The Magnetism of the Hexanuclear Complex
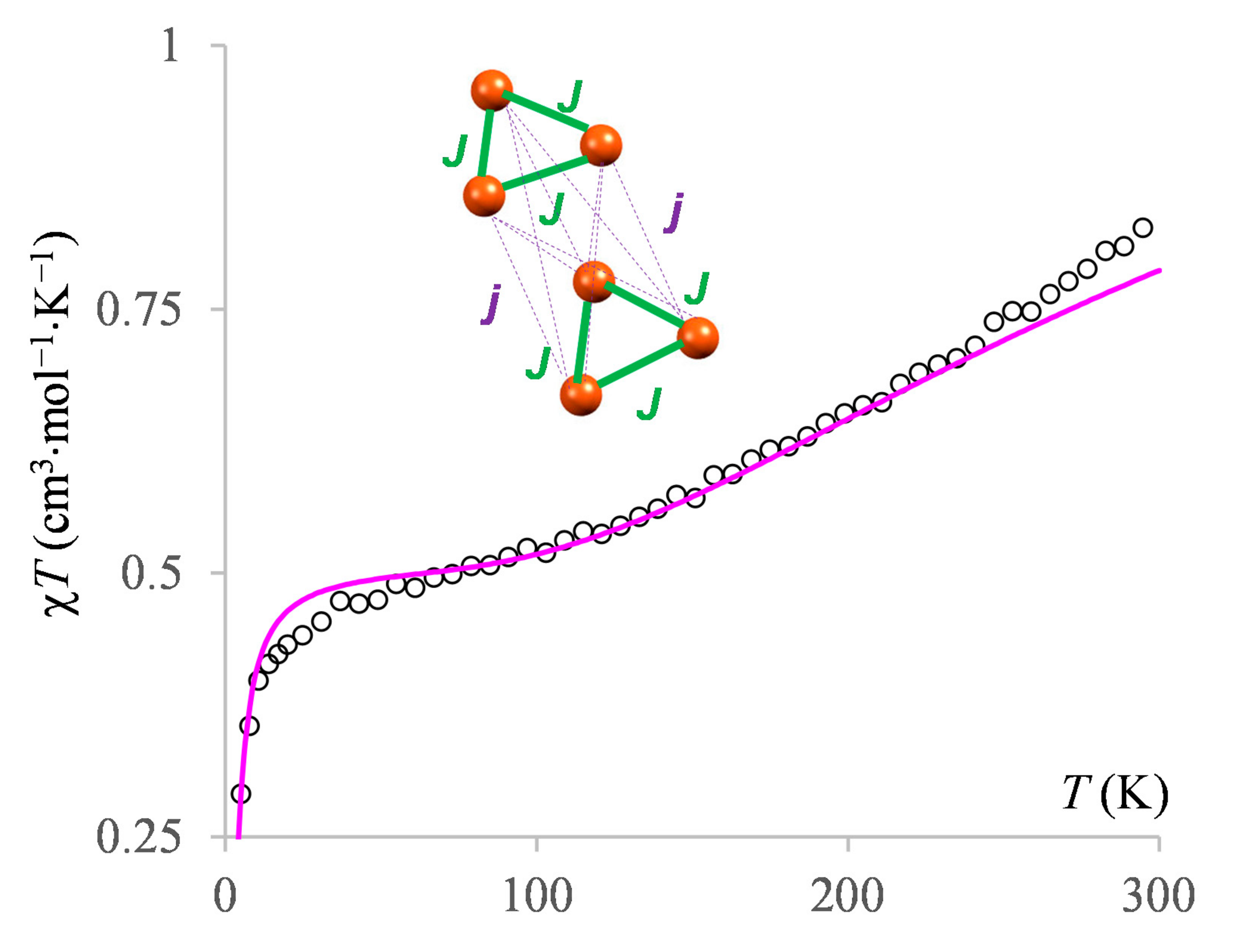
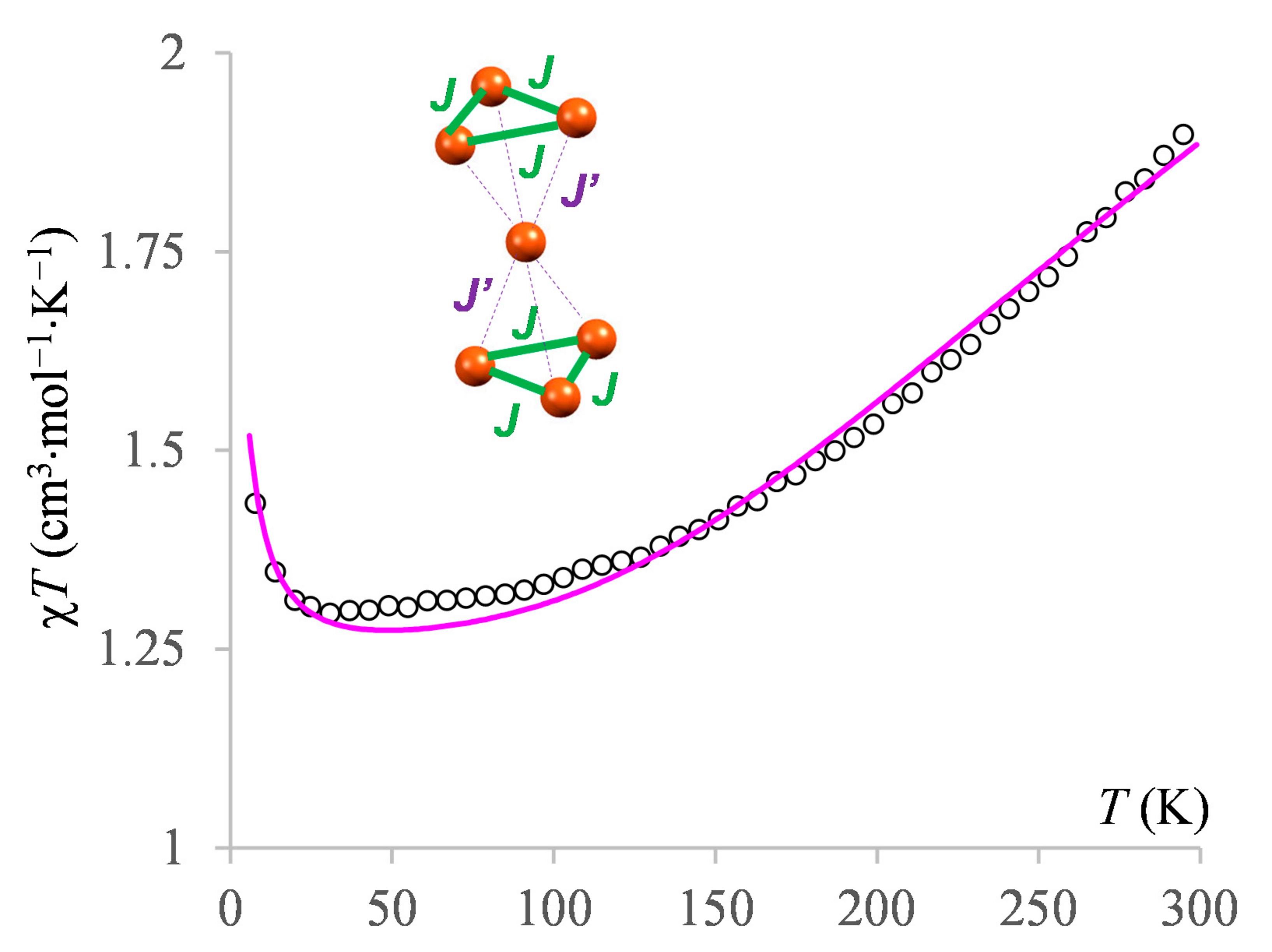
3.2.4. The Magnetism of the Heptanuclear Complex
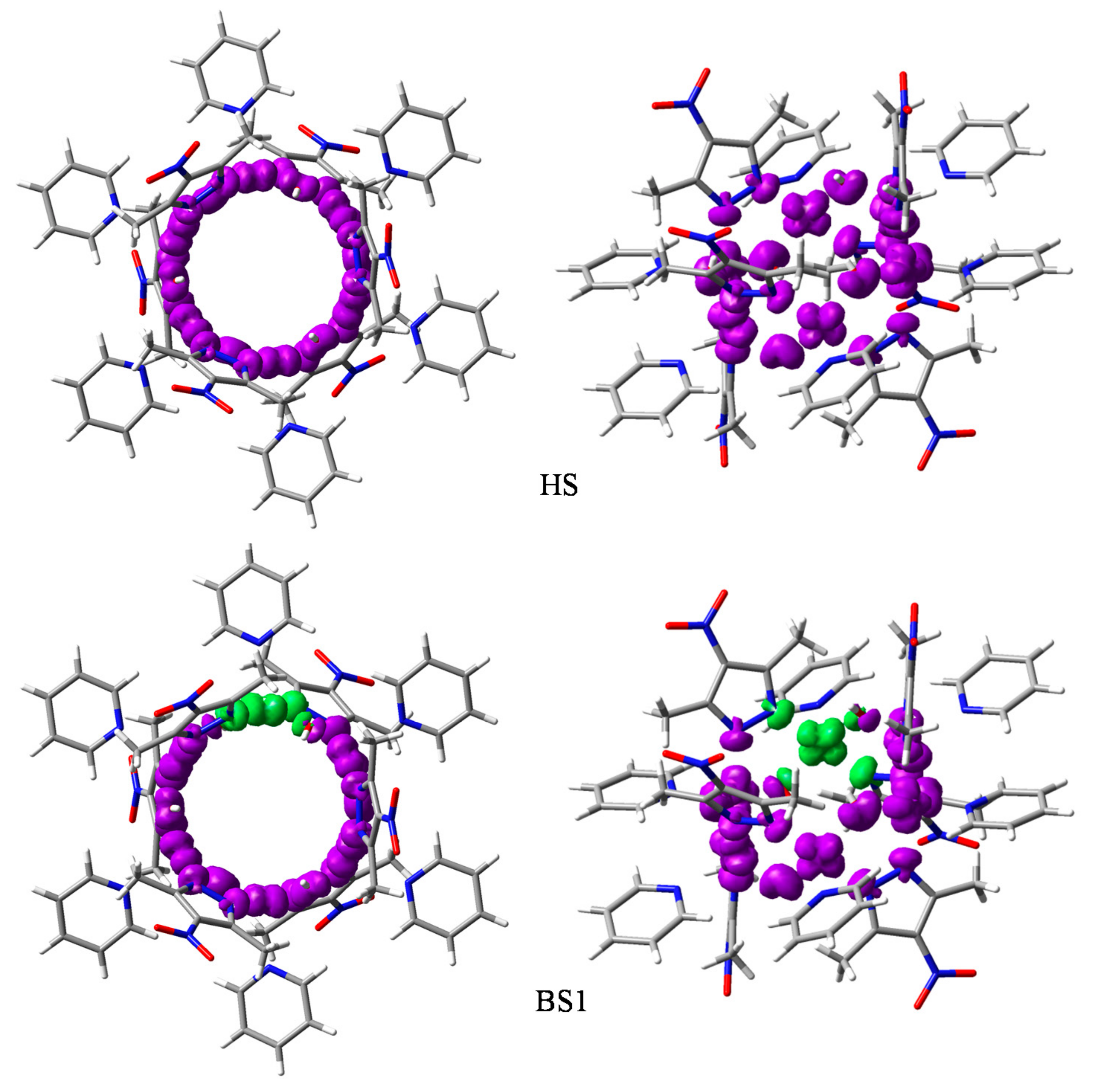
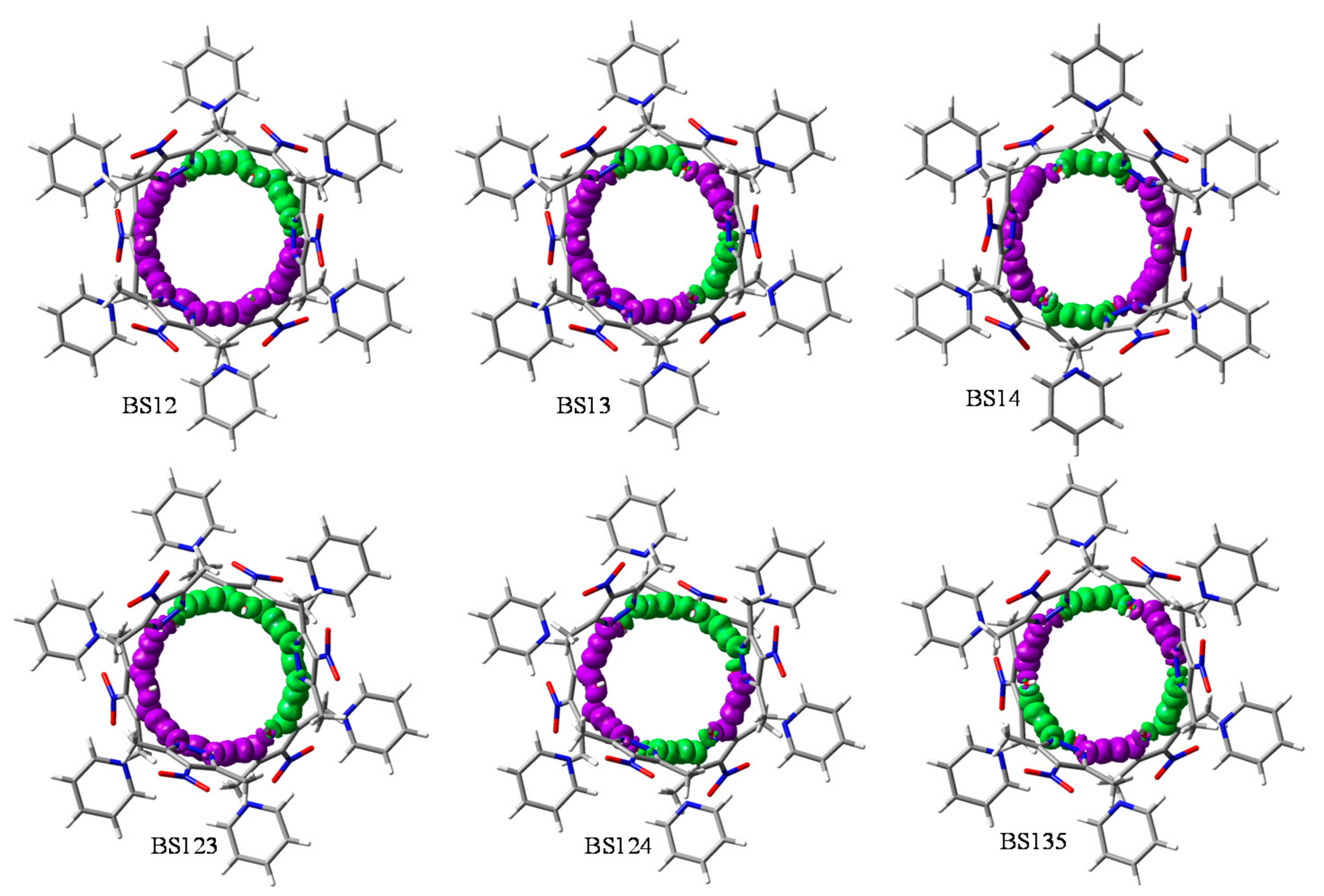
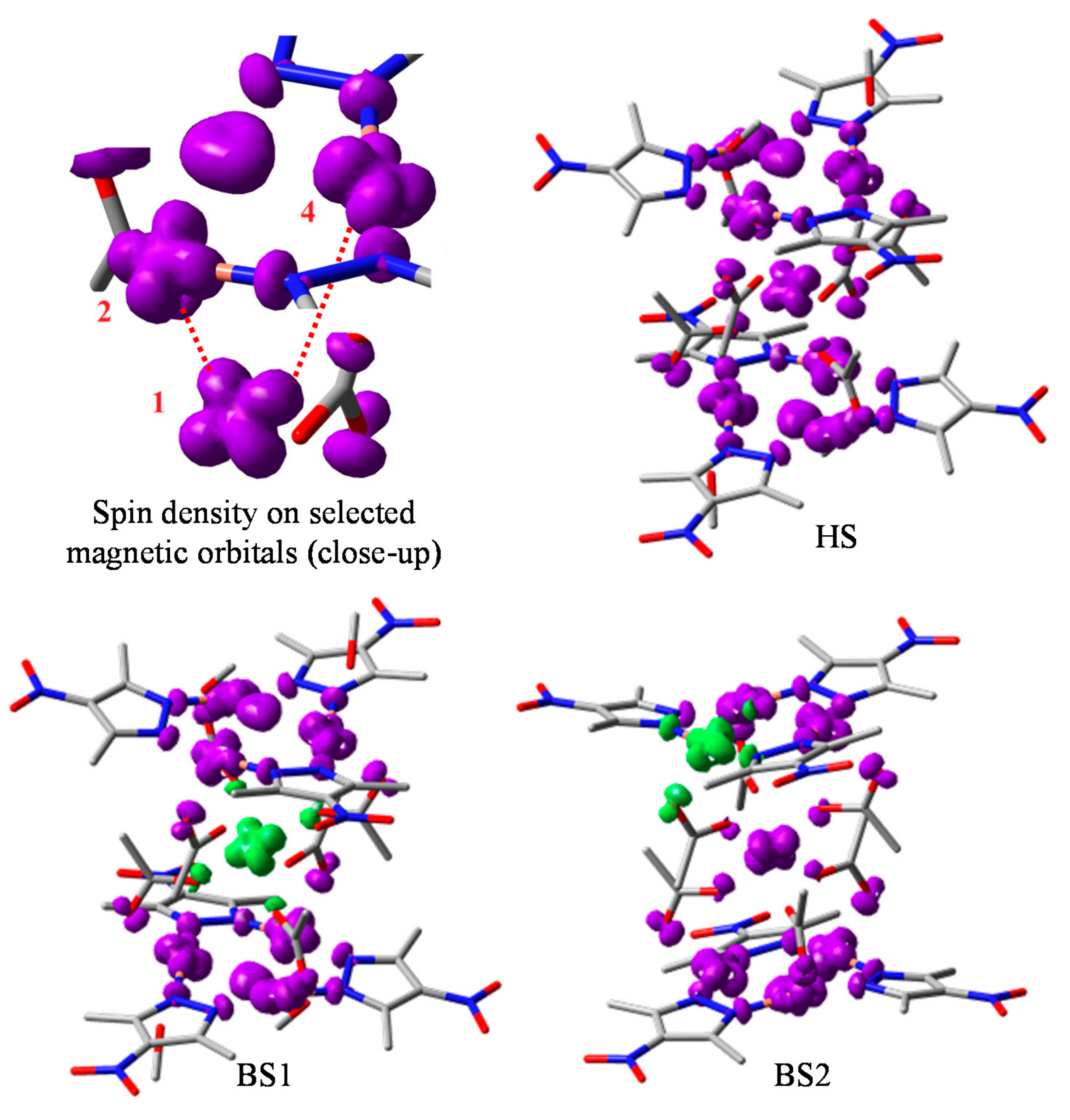
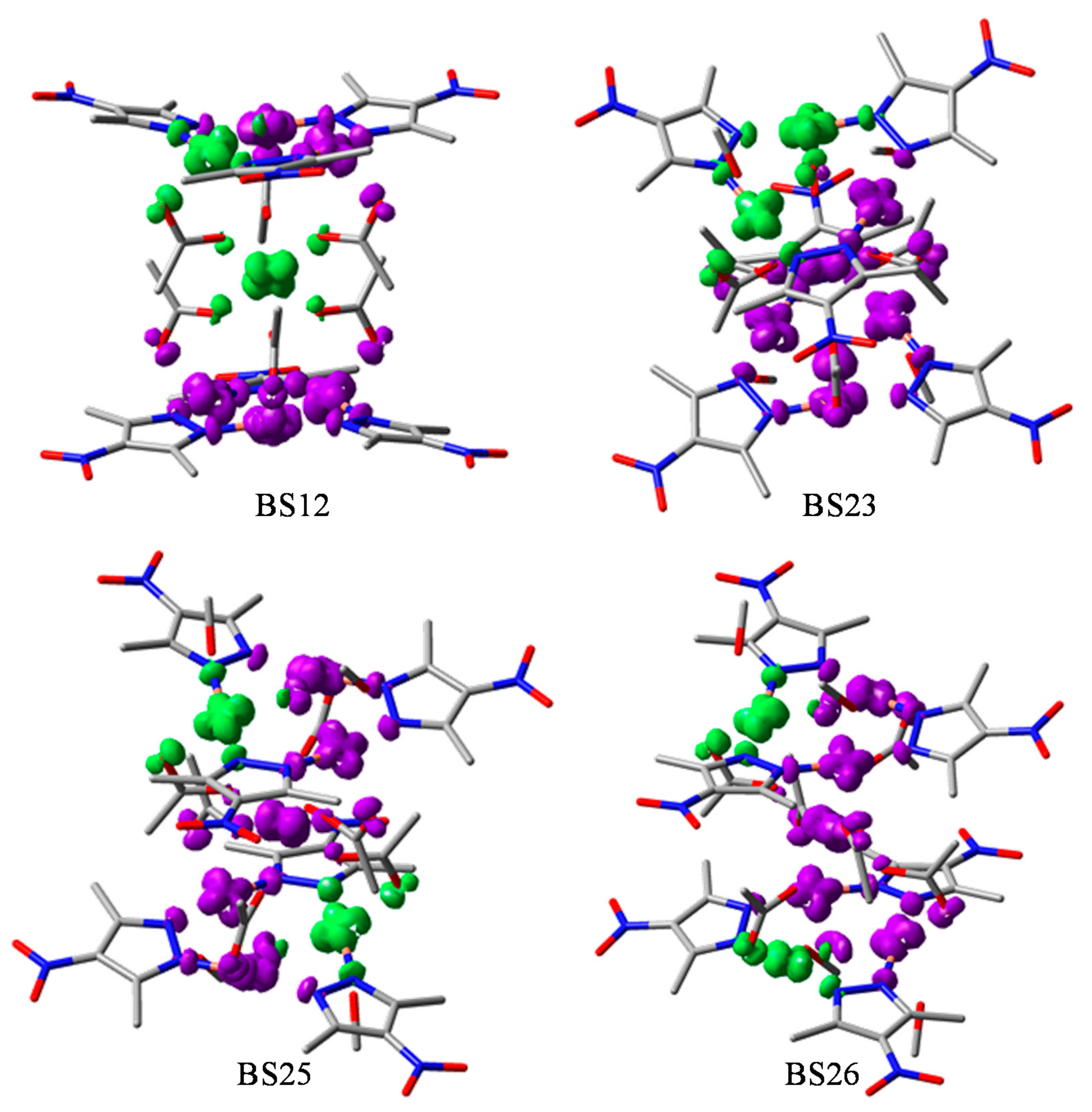
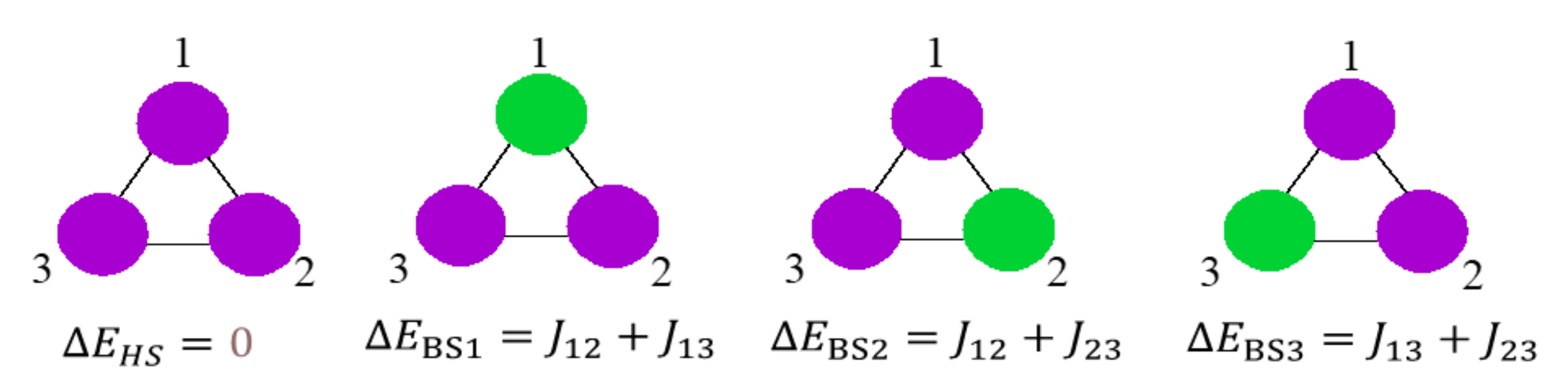
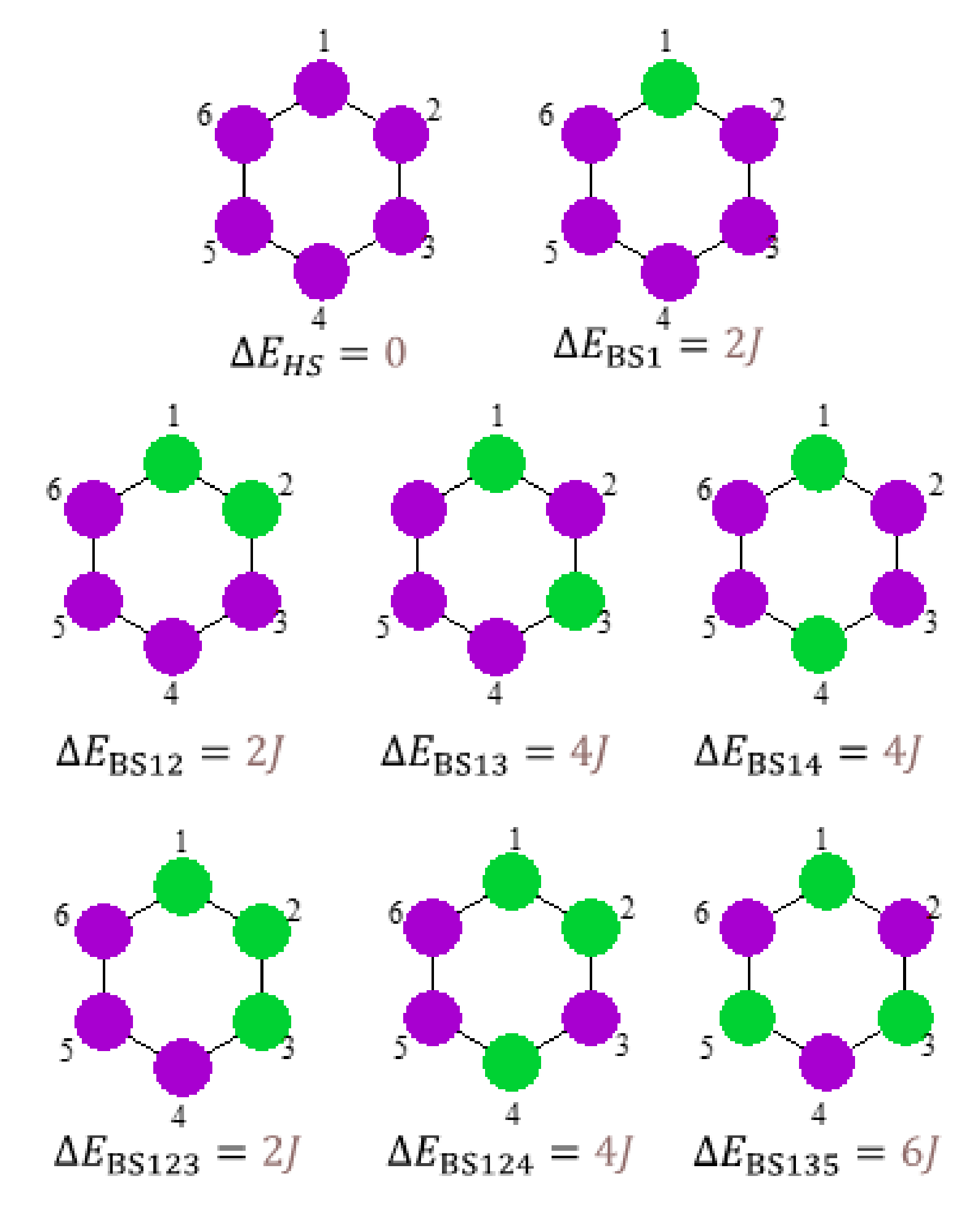
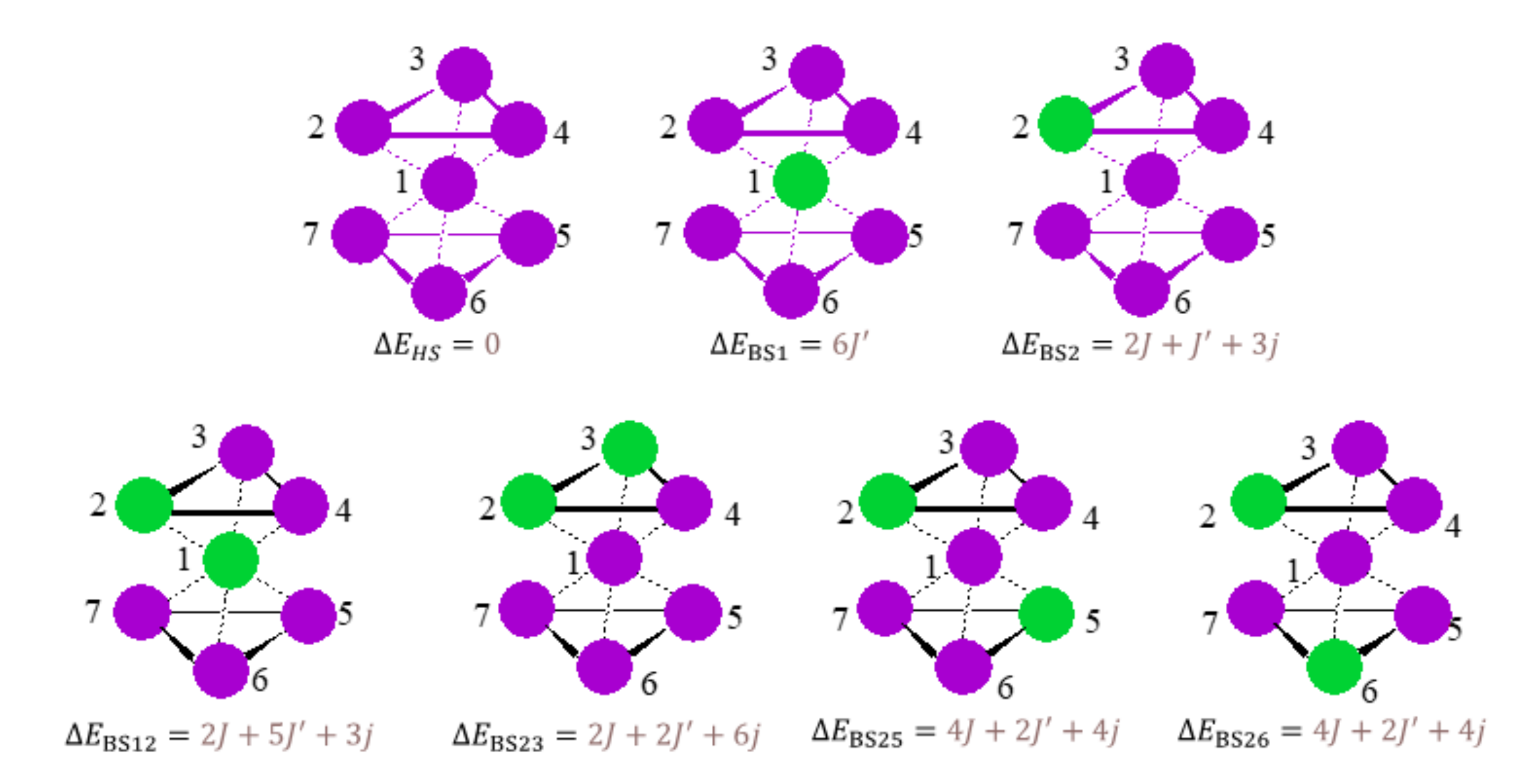
3.3. Broken Symmetry (BS) Calculations
3.3.1. Realization and Interpretation of BS Calculations
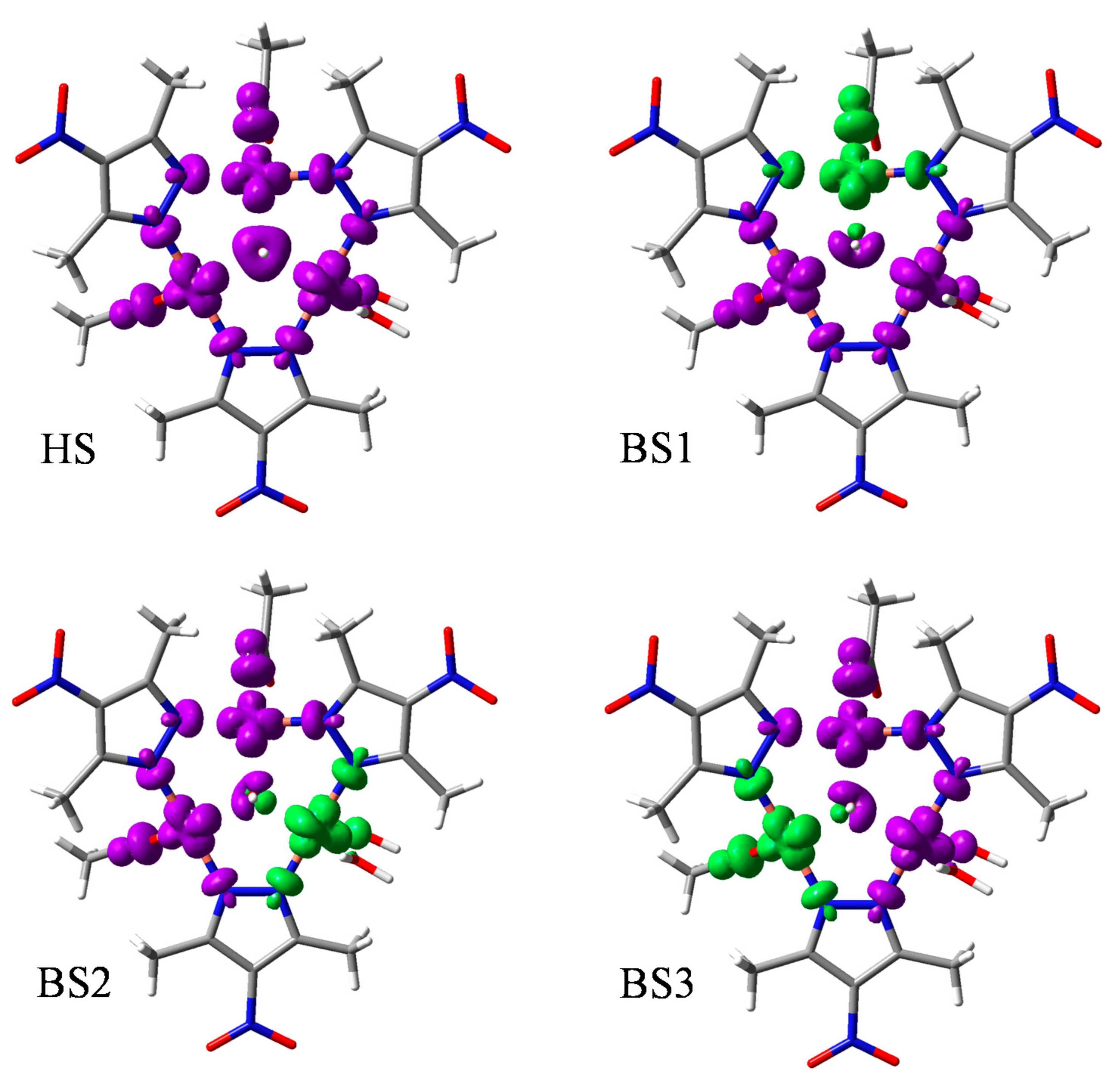
3.3.2. The BS Calculations of the Trinuclear Complex
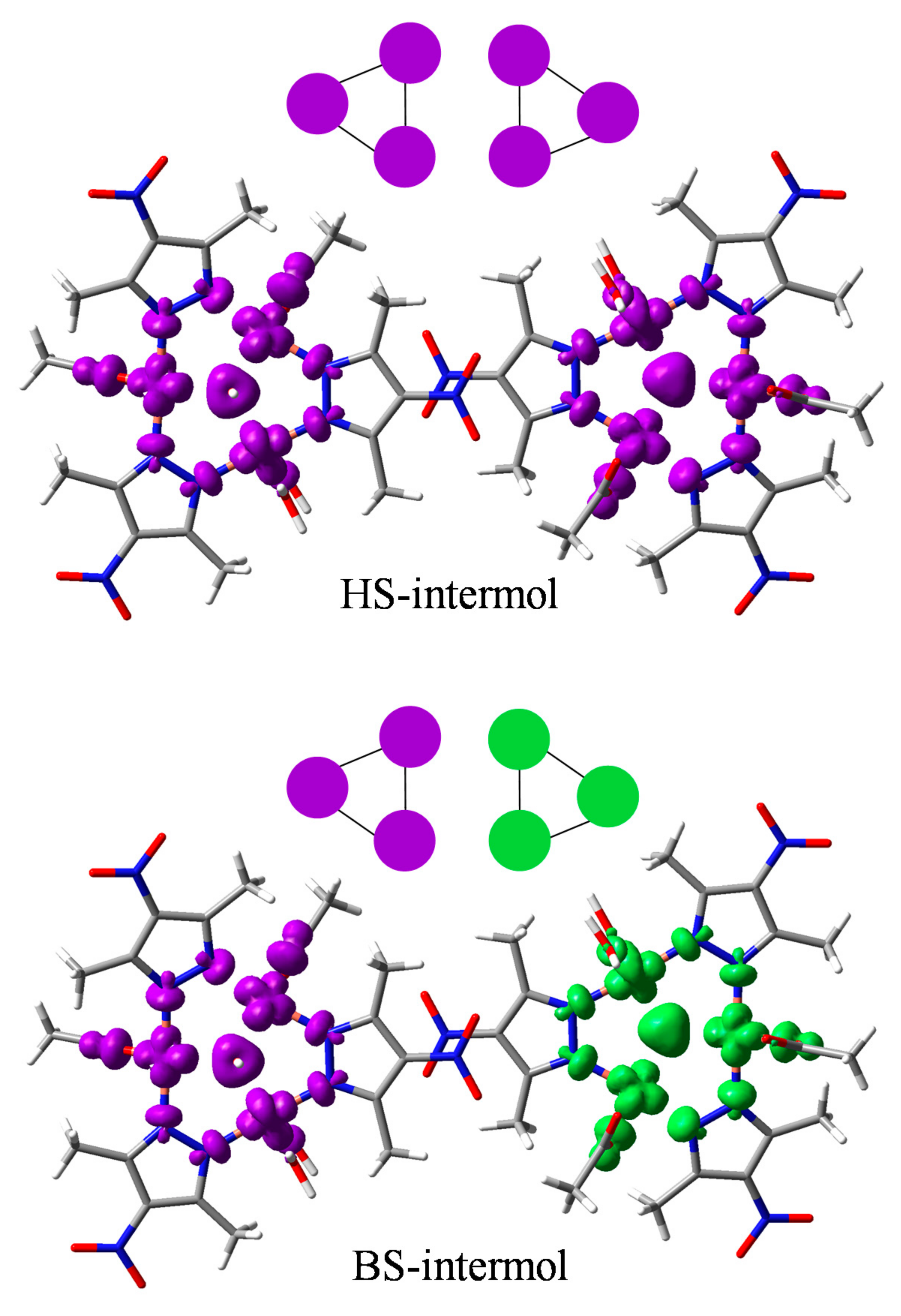
3.3.3. The BS Calculations of the Hexanuclear Complex
3.3.4. The BS Calculations of the Heptanuclear Complex
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Putz, M.V.; Cimpoesu, F.; Ferbinteanu, M. Structural Chemistry, Principles, Methods, and Case Studies; Springer: Cham, Switzerland, 2018; pp. 629–633. [Google Scholar] [CrossRef]
- Thompson, L.K. Polynuclear coordination complexes − from dinuclear to nonanuclear and beyond. Coord. Chem. Rev. 2002, 233–234, 193–206. [Google Scholar] [CrossRef]
- Halcrow, M.A. Pyrazoles and pyrazolides-flexible synthons in self-assembly. Dalton Trans. 2009, 2059–2073. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, R. Coordination chemistry with pyrazole-based chelating ligands: Molecular structural aspects. Coord. Chem. Rev. 2000, 203, 151–218. [Google Scholar] [CrossRef]
- Solomon, E.I.; Sundaram, U.M.; Machonkin, T.E. Multicopper Oxidases and Oxygenases. Chem. Rev. 1996, 96, 2563–2606. [Google Scholar] [CrossRef]
- Cole, A.P.; Root, D.E.; Mukherjee, P.; Solomon, E.I.; Stack, T.D. A trinuclear intermediate in the copper-mediated reduction of O2: Four electrons from three coppers. Science 1996, 273, 1848–1850. [Google Scholar] [CrossRef]
- Kaim, W.; Rall, J. Copper—A “Modern” Bioelement. Angew. Chem. Int. Ed. 1996, 35, 43–60. [Google Scholar] [CrossRef]
- Lee, S.K.; George, S.D.; Antholine, W.E.; Hedman, B.; Hodgson, K.O.; Solomon, E.I. Nature of the intermediate formed in the reduction of O2 to H2O at the trinuclear copper cluster active site in native laccase. J. Am. Chem. Soc. 2002, 124, 6180–6193. [Google Scholar] [CrossRef]
- Tsukerblat, B.S.; Kuyavskaya, B.Y.; Belinskii, M.I.; Ablov, A.V.; Novotortsev, V.M.; Kalinnikov, V.T. Antisymmetric Exchange in the Trinuclear Clusters of Copper (II). Theor. Chim. Acta 1975, 38, 131–138. [Google Scholar] [CrossRef]
- Ferrer, S.; Lloret, F.; Bertomeu, I.; Alzuet, G.; Borras, J.; Garcya-Granda, S.; Liu-Gonzalez, M.; Haasnoot, J.G. Cyclic Trinuclear and Chain of Cyclic Trinuclear Copper(II) Complexes Containing a Pyramidal Cu3O(H) Core. Crystal Structures and Magnetic Properties of [Cu3(μ3-OH)(aaat)3(H2O)3](NO3)2⋅H2O [aaat = 3-Acetylamino-5-amino-1,2,4-triazolate] and {[Cu3(μ3-OH)(aat)3(μ3-SO4)]⋅6H2O}n [aat = 3-Acetylamino-1,2,4-triazolate]: New Cases of Spin-Frustrated Systems. Inorg. Chem. 2002, 41, 5821–5830. [Google Scholar] [CrossRef]
- Rudra, I.; Wu, Q.; Voorhis, T.V. Predicting exchange coupling constants in frustrated molecular magnets using density functional theory. Inorg. Chem. 2007, 46, 10539–10548. [Google Scholar] [CrossRef]
- Wang, L.; Sun, Y.; Yu, Z.; Qi, Z.; Liu, C. Theoretical investigation on triagonal symmetry copper trimers: Magneto-structural correlation and spin frustration. J. Phys. Chem. A 2009, 113, 10534–10539. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.; Solomon, E.I. Ground-state electronic and magnetic properties of a μ3-Oxo-bridged trinuclear Cu(II) complex: Correlation to the native intermediate of the multicopper oxidases. Inorg. Chem. 2005, 44, 8076–8086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afrati, T.; Dendrinou-Samara, C.; Raptopoulou, C.; Terzis, A.; Tangoulis, V.; Tsipis, A.; Kessissoglou, D.P. Experimental and theoretical study of the antisymmetric magnetic behavior of copper inverse-9-metallacrown-3 compounds. Inorg. Chem. 2008, 47, 7545–7555. [Google Scholar] [CrossRef] [PubMed]
- Canon-Mancisidor, W.; Spodine, E.; Paredes-Garcia, V.; Venegas-Yazigi, D. Theoretical description of the magnetic properties of μ3-hydroxo bridged trinuclear copper(II) complexes. J. Mol. Model. 2013, 19, 2835–2844. [Google Scholar] [CrossRef]
- Angaroni, M.; Ardizzoia, G.A.; Beringhelli, T.; La Monica, G.; Gatteschi, D.; Masciocchi, N.; Moret, M. Oxidation reaction of [{Cu(Hpz)2Cl}2](Hpz = pyrazole): Synthesis of the trinuclear copper(II) hydroxo complexes [Cu3(OH)(pz)3(Hpz)2Cl2]·solv (solv = H2O or tetrahydrofuran). Formation, magnetic properties, and X-ray crystal structure of [Cu3(OH)(pz)3(py)2Cl2]·py (py = pyridine). J. Chem. Soc. Dalton Trans. 1990, 3305–3309. [Google Scholar] [CrossRef]
- Sakai, K.; Yamada, Y.; Tsubomura, T.; Yabuki, M.; Yamaguchi, M. Synthesis, Crystal Structure, and Solution Properties of a Hexacopper (II) Complex with Bridging Hydroxides, Pyrazolates, and Nitrates. Inorg. Chem. 1996, 35, 542–544. [Google Scholar] [CrossRef]
- Angaridis, P.A.; Baran, R.; Boča, R.; Cervantes-Lee, F.; Haase, W.; Mezei, G.; Raptis, R.G.; Werner, R. Synthesis and Structural Characterization of Trinuclear CuII − Pyrazolato Complexes Containing μ3-OH, μ3-O, and μ3-Cl Ligands. Magnetic Susceptibility Study of [PPN]2[(μ3-O)Cu3(μ-pz)3Cl3]. Inorg. Chem. 2002, 41, 2219–2228. [Google Scholar] [CrossRef]
- Mezei, G.; Baran, P.; Raptis, R.G. Anion Encapsulation by Neutral Supramolecular Assemblies of Cyclic CuII Complexes: A Series of Five Polymerization Isomers, [{cis-CuII(μ-OH)(μ-pz)}n], n=6, 8, 9, 12, and 14. Angew. Chem. Int. Ed. 2004, 43, 574–577. [Google Scholar] [CrossRef]
- Fernando, I.R.; Surmann, S.A.; Urech, A.A.; Poulsen, A.M.; Mezei, G. Selective total encapsulation of the sulfate anion by neutral nano-jars. Chem. Commun. 2012, 48, 6860–6862. [Google Scholar] [CrossRef]
- Canon-Mancisidor, W.; Gomez-Garcia, C.J.; Espallargas, G.M.; Vega, A.; Spodine, E.; Venegas-Yazigi, D.; Coronado, E. Structural re-arrangement in two hexanuclear CuII complexes: From a spin frustrated trigonal prism to a strongly coupled antiferromagnetic soluble ring complex with a porous tubular structure. Chem. Sci. 2014, 5, 324–332. [Google Scholar] [CrossRef]
- Henkelis, J.J.; Jones, L.F.; de Miranda, M.P.; Kilner, C.A.; Halcrow, M.A. Two Heptacopper(II) Disk Complexes with a [Cu7(μ3-OH)4(μ-OR)2]8+ Core. Inorg. Chem. 2010, 49, 11127–11132. [Google Scholar] [CrossRef]
- Mohamed, A.A.; Ricci, S.; Burini, A.; Galassi, R.; Santini, C.; Chiarella, G.M.; Melgarejo, D.Y.; Fackler, J.P., Jr. Halide and Nitrite Recognizing Hexanuclear Metallacycle Copper(II) Pyrazolates. Inorg.Chem. 2011, 50, 1014–1020. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-X.; Ren, Z.-G.; Liu, D.; Chen, Y.; Lang, J.-P.; Cheng, Z.-P.; Zhu, X.-L.; Abrahams, B.F. Single-crystal-to-single-crystal structural transformations of two sandwich-like Cu(II) pyrazolate complexes and their excellent catalytic performances in MMA polymerization. Chem. Commun. 2010, 46, 8430–8432. [Google Scholar] [CrossRef] [PubMed]
- Yakovleva, M.A.; Kushan, E.V.; Boltacheva, N.S.; Filyakova, V.I.; Nefedov, S.E. Deprotonation of pyrazole and its analogs by aqueous copper acetate in the presence of triethylamine. Russ. J. Inorg. Chem. 2012, 57, 181–192. [Google Scholar] [CrossRef]
- Di Nicola, C.; Garau, F.; Gazzano, M.; Guedes da Silva, M.F.C.; Lanza, A.; Monari, M.; Nestola, F.; Pandolfo, L.; Pettinari, C.; Pombeiro, A.J.L. New Coordination Polymers and Porous Supramolecular Metal Organic Network Based on the Trinuclear Triangular Secondary Building Unit [Cu3(μ3-OH)(μ-pz)3]2+ and 4,4′-Bypiridine. Cryst. Growth Des. 2012, 12, 2890–2901. [Google Scholar] [CrossRef]
- Di Nicola, C.; Garau, F.; Gazzano, M.; Lanza, A.; Monari, M.; Nestola, F.; Pandolfo, L.; Pettinari, C.; Zorzi, F. Reactions of a Coordination Polymer Based on the Triangular Cluster [Cu3(μ3-OH)(μ-pz)3]2+ with Strong Acids. Crystal Structure and Supramolecular Assemblies of New Mono-, Tri-, and Hexanuclear Complexes and Coordination Polymers. Cryst. Growth Des. 2010, 10, 3120–3131. [Google Scholar] [CrossRef]
- Zhou, Q.; Liu, Y.; Wang, R.; Fu, J.; Xu, J.; Lou, J. Synthesis, crystal structure and magnetic properties of a trinuclear Cu(II)-pyrazolate complex containing μ3-OH. J. Coord. Chem. 2009, 62, 311–318. [Google Scholar] [CrossRef]
- Zhou, J.H.; Liu, Z.; Li, Y.Z.; Song, Y.; Chen, X.T.; You, X.Z. Synthesis, structures and magnetic properties of two copper(II) complexes with pyrazole and pivalate ligands. J. Coord. Chem. 2006, 59, 147–156. [Google Scholar] [CrossRef]
- Davydenko, Y.M.; Demeshko, S.; Pavlenko, V.A.; Dechert, S.; Meyer, F.; Fritsky, I.O. Synthesis, Crystal Structure, Spectroscopic and Magnetically Study of Two Copper(II) Complexes with Pyrazole Ligand. Z. Anorg. Allg. Chem. 2013, 639, 1472–1476. [Google Scholar] [CrossRef]
- Bala, S.; Bhattacharya, S.; Goswami, A.; Adhikary, A.; Konar, S.; Mondal, R. Designing Functional Metal−Organic Frameworks by Imparting a Hexanuclear Copper-Based Secondary Building Unit Specific Properties: Structural Correlations with Magnetic and Photocatalytic Activity. Cryst. Growth Des. 2014, 14, 6391–6398. [Google Scholar] [CrossRef]
- Condello, F.; Garau, F.; Lanza, A.; Monari, M.; Nestola, F.; Pandolfo, L.; Pettinari, C. Synthesis and Structural Characterizations of New Coordination Polymers Generated by the Interaction Between the Trinuclear Triangular SBU [Cu3(µ3-OH)(µ-pz)3]2+ and 4,4′-Bipyridine. 3° Cryst. Growth Des. 2015, 15, 4854–4862. [Google Scholar] [CrossRef]
- Morgan, G.T.; Ackerman, I. CLII.—Substitution in the pyrazole series. Halogen derivatives of 3:5-dimethylpyrazole. J. Chem. Soc. Trans. 1923, 123, 1308–1318. [Google Scholar] [CrossRef]
- Hüttel, R.; Schäfer, O.; Jochum, P. Die Jodierung der Pyrazole. Justus Liebigs Ann. Chem. 1955, 593, 200–207. [Google Scholar] [CrossRef]
- Buchner, E.; Fritsch, M. VI. Darstellung und Derivate des freien Pyrazols. Justus Liebigs Ann. Chem. 1893, 273, 256–266. [Google Scholar] [CrossRef] [Green Version]
- Kahn, O. Molecular Magnetism; VCH Publishers: New York, NY, USA, 1993. [Google Scholar]
- Crystal Structure 4.2: Crystal Structure Analysis Package, Rigaku Corporation (2000–2015); Rigaku Corporation: Tokyo, Japan.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; Van De Streek, J. Mercury: Visualization and analysis of crystal Structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef] [Green Version]
- POVRAY, Version 3.5; Persistence of Vision Raytracer Pty. Ltd.: Williamstown, VIC, Australia, 2002.
- Koch, W.; Holthausen, M.C. A Chemist’s Guide to Density Functional Theory; VCH Publisher: Berlin, Germany, 2001. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General Atomic and Molecular Electronic Structure System. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Addison, A.W.; Rao, T.N.; Reedijk, J.; van Rijn, J.; Verschoor, G.C. Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen–sulphur donor ligands; the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane]copper(II) perchlorate. J. Chem. Soc. Dalton Trans. 1984, 7, 1349–1356. [Google Scholar] [CrossRef]
- Yang, L.; Powell, D.R.; Houser, R.P. Structural variation in copper(I) complexes with pyridylmethylamide ligands: Structural analysis with a new four-coordinate geometry index, τ4. Dalton Trans. 2007, 9, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Casarin, M.; Corvaja, C.; di Nicola, C.; Falcomer, D.; Franco, L.; Monari, M.; Pandolfo, L.; Pettinari, C.; Piccinelli, F.; Tagliatesta, P. Spontaneous Self-Assembly of an Unsymmetric Trinuclear Triangular Copper(II) Pyrazolate Complex, [Cu3(μ3-OH)(μ-pz)3(MeCOO)2(Hpz)] (Hpz = Pyrazole). Synthesis, Experimental and Theoretical Characterization, Reactivity, and Catalytic Activity. Inorg. Chem. 2004, 43, 5865–5876. [Google Scholar] [CrossRef] [PubMed]
- Heisenberg, W. Zur theorie des ferromagnetismus. Z. Phys. 1928, 49, 619–636. [Google Scholar] [CrossRef]
- van Vleck, J.H.; Sherman, A. The Quantum Theory of Valence. Rev. Mod. Phys. 1935, 7, 167–228. [Google Scholar] [CrossRef]
- Kambe, K. On the Paramagnetic Susceptibilities of Some Polynuclear Complex Salts. J. Phys. Soc. Jpn. 1950, 5, 48–51. [Google Scholar] [CrossRef]
- Sinn, E. Magnetic Exchange in Polynuclear Metal Complexes. Coord. Chem. Rev. 1970, 5, 313–347. [Google Scholar] [CrossRef]
- Bersuker, I.B. Modern aspects of the Jahn-Teller effect theory and applications to molecular problems. Chem. Rev. 2001, 101, 1067–1114. [Google Scholar] [CrossRef]
- Cage, B.; Cotton, F.A.; Dalal, N.S.; Hillard, E.A.; Rakvin, B.; Ramsey, C.M. Observation of Symmetry Lowering and Electron Localization in the Doublet-States of a Spin-Frustrated Equilateral Triangular Lattice: Cu3(O2C16H23)·1.2C6H12. J. Am. Chem. Soc. 2003, 125, 5270–5271. [Google Scholar] [CrossRef]
- Vreugdenhil, W.; Haasnoot, J.G.; Schoondergang, M.F.J. Reedijk, Spectroscopic and magnetic properties of transition metal(II) triflouromethanesulfonate compounds with bridging asymmetric 3,4-dialkyl substituted 1,2,4-triazole ligands. J. Inorg. Chim. Acta 1987, 130, 235–242. [Google Scholar] [CrossRef]
- Antolini, L.; Fabretti, A.C.; Gatteschi, D.; Giusti, A.; Sessoli, R. Synthesis, crystal and molecular structure, and magnetic properties of bis[tris(.mu.-3,5-diamino-1,2,4-triazole-N1,N2)tris(thiocyanato-N)nickel(II)]nickel(II) hexahydrate. Inorg. Chem. 1990, 29, 143–145. [Google Scholar] [CrossRef]
- Antolini, L.; Fabretti, A.C.; Gatteschi, D.; Giusti, A.; Sessoli, R. Synthesis, crystal and molecular structure, and magnetic properties of bis[(.mu.-3,5-diamino-1,2,4-triazole-N1,N2-bis(.mu.-3,5-diamino-1,2,4-triazolato-N1,N2)triaquacobalt(II))]cobalt(III) trichloride nonahydrate. Inorg. Chem. 1991, 30, 4858–4860. [Google Scholar] [CrossRef]
- Kolnaar, J.J.A.; van Dijk, G.; Kooijman, H.; Spek, A.L.; Ksenofontov, V.G.; Gütlich, P.; Haasnoot, J.G.; Reedijk, J. Synthesis, Structure, Magnetic Behavior, and Mössbauer Spectroscopy of Two New Iron(II) Spin-Transition Compounds with the Ligand 4-Isopropyl-1,2,4-triazole. X-ray Structure of [Fe3(4-isopropyl-1,2,4-triazole)6(H2O)6](tosylate)6·2H2O. Inorg. Chem. 1997, 36, 2433–2440. [Google Scholar] [CrossRef] [PubMed]
- Garcia, Y.; van Koningsbruggen, P.J.; Lapouyade, R.; Fournès, L.; Rabardel, L.; Kahn, O.; Ksenofonov, V.; Levchenko, G.P.; Gütlich, P. Influences of Temperature, Pressure, and Lattice Solvents on the Spin Transition Regime of the Polymeric Compound [Fe(hyetrz)3]A2·3H2O (hyetrz = 4-(2‘-hydroxyethyl)-1,2,4-triazole and A− = 3-nitrophenylsulfonate). Chem. Mater. 1998, 10, 2426–2433. [Google Scholar] [CrossRef]
- Roubeau, O.; Gomez, J.M.A.; Balskus, E.; Kolnaar, J.J.A.; Haasnoot, J.G.; Reedijk, J. Spin-transition behaviour in chains of FeII bridged by 4-substituted 1,2,4-triazoles carrying alkyl tails. New J. Chem. 2001, 25, 144–150. [Google Scholar] [CrossRef]
- Spielberg, E.T.; Fittipaldi, M.; Geibig, D.; Gatteschi, D.; Plass, W. Electronic and magnetic structure of a triacetylphlorogucinol-bridged C3-symmetric trinuclear copper complex: Magnetic characterization, ESR spectroscopy, and DFT calculations. Inorg. Chim. Acta 2010, 363, 4269–4276. [Google Scholar] [CrossRef]
- Shaik, S.; Hiberty, P.C. A Chemist’s Guide to Valence Bond. Theory; John Wiley & Sons: Hoboken, NJ, USA, 2007. [Google Scholar]
- Garciabach, M.A.; Blaise, P.; Malrieu, J.P. Dimerization of polyacetylene treated as a spin-Peierls distortion of the Heisenberg Hamiltonian. Phys. Rev. B Condens. Matter. 1992, 46, 15645–15651. [Google Scholar] [CrossRef]
- Mohamed, A.A.; Burini, A.; Galassi, R.; Paglialunga, D.; Galan-Mascaros, J.-R.; Dunbar, K.R.; Fackler, J.P., Jr. Self-assembly of a High-Nuclearity Chloride-Centered Copper(II) Cluster. Structure and Magnetic Properties of [Au(PPh3)2][trans-Cu6(μ-OH)6{μ-(3,5-CF3)2pz}6Cl]. Inorg. Chem. 2007, 46, 2348–2349. [Google Scholar] [CrossRef]
- Borrás-Almenar, J.J.; Clemente-Juan, J.M.; Coronado, E.; Tsukerblat, B.S. MAGPACK A Package to Calculate the Energy Levels, Bulk Magnetic Properties, and Inelastic Neutron Scattering Spectra of High Nuclearity Spin Clusters. J. Comput. Chem. 2001, 22, 985–991. [Google Scholar] [CrossRef]
- Rumer, G. Zum theorie der spinvalenz. Nachr. Ges. Wiss. Göttingen Math. Phys. Kl. 1932, 1932, 337–341. [Google Scholar]
- Cimpoesu, F.; Chihaia, V.; Stanica, N.; Hirao, K. The Spin Hamiltonian Effective Approach to the Vibronic Effects. Selected Cases. Adv. Quant. Chem. 2003, 44, 273–288. [Google Scholar] [CrossRef]
- Pauling, L.; Wheland, G.W. The Nature of the Chemical Bond. V. The Quantum-Mechanical Calculation of the Resonance Energy of Benzene and Naphthalene and the Hydrocarbon Free Radicals. J. Chem. Phys. 1933, 1, 362–374. [Google Scholar] [CrossRef] [Green Version]
- Ferbinteanu, M.; Buta, C.; Toader, A.M.; Cimpoesu, F. The spin coupling in the polyaromatic hydrocarbons and carbon-based materials. In Carbon-Related Materials-in Recognition of Nobel Lectures by Prof. Akira Suzuki in ICCE; Kaneko, S., Mele, P., Endo, T., Eds.; Springer: Cham, Switzerland, 2017; pp. 327–371. [Google Scholar] [CrossRef]
- Toader, A.M.; Buta, C.M.; Frecus, B.; Mischie, A.; Cimpoesu, F. Valence Bond Account of Triangular Polyaromatic Hydrocarbons with Spin: Combining Ab Initio and Phenomenological Approaches. J. Phys. Chem. C 2019, 123, 6869–6880. [Google Scholar] [CrossRef]
- Noodleman, L. Valence bond description of antiferromagnetic coupling in transition metal dimers. J. Chem. Phys. 1981, 74, 5737–5743. [Google Scholar] [CrossRef]
- Noodleman, L.; Davidson, E.R. Ligand spin polarization and antiferromagnetic coupling in transition metal dimers. Chem. Phys. 1986, 109, 131–143. [Google Scholar] [CrossRef]
- Ruiz, E.; Cano, J.; Alvarez, S.; Alemany, P. Broken symmetry approach to calculation of exchange coupling constants for homobinuclear and heterobinuclear transition metal complexes. J. Comp. Chem. 1999, 20, 1391–1400. [Google Scholar] [CrossRef]
- Bencini, A.; Totti, F.; Daul, C.A.; Doclo, K.; Fantucci, P.; Barone, V. Density Functional Calculations of Magnetic Exchange Interactions in Polynuclear Transition Metal Complexes. Inorg. Chem. 1997, 36, 5022–5030. [Google Scholar] [CrossRef]
- Shoji, M.; Koizumi, K.; Kitagawa, Y.; Kawakami, T.; Yamanaka, S.; Okumura, M.; Yamaguchi, K. A general algorithm for calculation of Heisenberg exchange integrals J. in multispin systems. Chem. Phys. Lett. 2006, 432, 343–347. [Google Scholar] [CrossRef]
- Ruiz, E. Theoretical study of the exchange coupling in large polynuclear transition metal complexes using DFT methods. In Principles and Applications of Density Functional Theory in Inorganic Chemistry II. Structure and Bonding; Springer: Berlin/Heidelberg, Germany, 2004; Volume 113, pp. 71–102. [Google Scholar] [CrossRef]
- Atitoaie, A.; Tanasa, R.; Enachescu, C. Size dependent thermal hysteresis in spincrossover nanoparticles reflected within a Monte Carlo based Ising-like model. J. Magn. Magn. Mater. 2012, 324, 1596–1600. [Google Scholar] [CrossRef]
- Varret, F.; Salunke, S.A.; Boukheddaden, K.; Bousseksou, A.; Codjovi, É.; Enachescu, C.; Linares, J. The Ising-like model applied to switchable inorganic solids: Discussion of the static properties. Comptes Rendus Chim. 2003, 6, 385–393. [Google Scholar] [CrossRef]
- Leininger, T.; Stoll, H.; Werner, H.-J.; Savin, A. Combining long-range configuration interaction with short-range density functionals. Chem. Phys. Lett. 1997, 275, 151–160. [Google Scholar] [CrossRef]
- Kohn, W.; Meir, Y.; Makarov, D.E. van der Waals Energies in Density Functional Theory. Phys. Rev. Lett. 1998, 80, 4153. [Google Scholar] [CrossRef] [Green Version]
- Iikura, H.; Tsuneda, T.; Yanai, T.; Hirao, K. A long-range correction scheme for generalized-gradient-approximation exchange functionals. J. Chem. Phys. 2001, 115, 3540–3544. [Google Scholar] [CrossRef]
- Tsuneda, T.; Kamiya, M.; Hirao, K. Regional self-interaction correction of density functional theory. J. Comput. Chem. 2003, 24, 1592–1598. [Google Scholar] [CrossRef]
- Grimme, S. Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 2004, 25, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Hay, P.J.; Thibeault, J.C.; Hoffmann, R. Orbital interactions in metal dimer complexes. J. Am. Chem. Soc. 1975, 97, 4884–4899. [Google Scholar] [CrossRef]
- Kahn, O.; Galy, J.; Journaux, Y.; Morgenstern-Badarau, I. Synthesis, crystal structure and molecular conformations, and magnetic properties of a copper-vanadyl (CuII-VOII) heterobinuclear complex: Interaction between orthogonal magnetic orbitals. J. Am. Chem. Soc. 1982, 104, 2165–2176. [Google Scholar] [CrossRef]
- Kahn, O. Magnetism of the Heteropolymetallic Systems. In Theoretical Approaches; Structure and Bonding; Springer: Berlin/Heidelberg, Germany, 1987; Volume 68. [Google Scholar] [CrossRef]
- Ruiz, E.; Alemany, P.; Alvarez, S.; Cano, J. Structural Modeling and Magneto− Structural Correlations for Hydroxo-Bridged Copper (II) Binuclear Complexes. Inorg. Chem. 1997, 36, 3683–3688. [Google Scholar] [CrossRef]



















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toader, A.M.; Buta, M.C.; Cimpoesu, F.; Toma, A.-I.; Zalaru, C.M.; Cinteza, L.O.; Ferbinteanu, M. New Syntheses, Analytic Spin Hamiltonians, Structural and Computational Characterization for a Series of Tri-, Hexa- and Hepta-Nuclear Copper (II) Complexes with Prototypic Patterns. Chemistry 2021, 3, 411-439. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry3010031
Toader AM, Buta MC, Cimpoesu F, Toma A-I, Zalaru CM, Cinteza LO, Ferbinteanu M. New Syntheses, Analytic Spin Hamiltonians, Structural and Computational Characterization for a Series of Tri-, Hexa- and Hepta-Nuclear Copper (II) Complexes with Prototypic Patterns. Chemistry. 2021; 3(1):411-439. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry3010031
Chicago/Turabian StyleToader, Ana Maria, Maria Cristina Buta, Fanica Cimpoesu, Andrei-Iulian Toma, Christina Marie Zalaru, Ludmila Otilia Cinteza, and Marilena Ferbinteanu. 2021. "New Syntheses, Analytic Spin Hamiltonians, Structural and Computational Characterization for a Series of Tri-, Hexa- and Hepta-Nuclear Copper (II) Complexes with Prototypic Patterns" Chemistry 3, no. 1: 411-439. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry3010031