Synthesis and Photostability of Cyclooctatetraene-Substituted Free Base Porphyrins

 , , , ,
, , , ,
Abstract
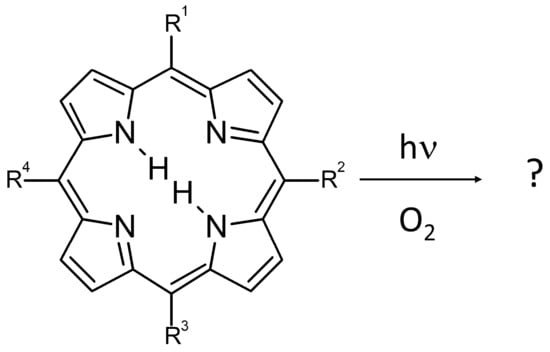
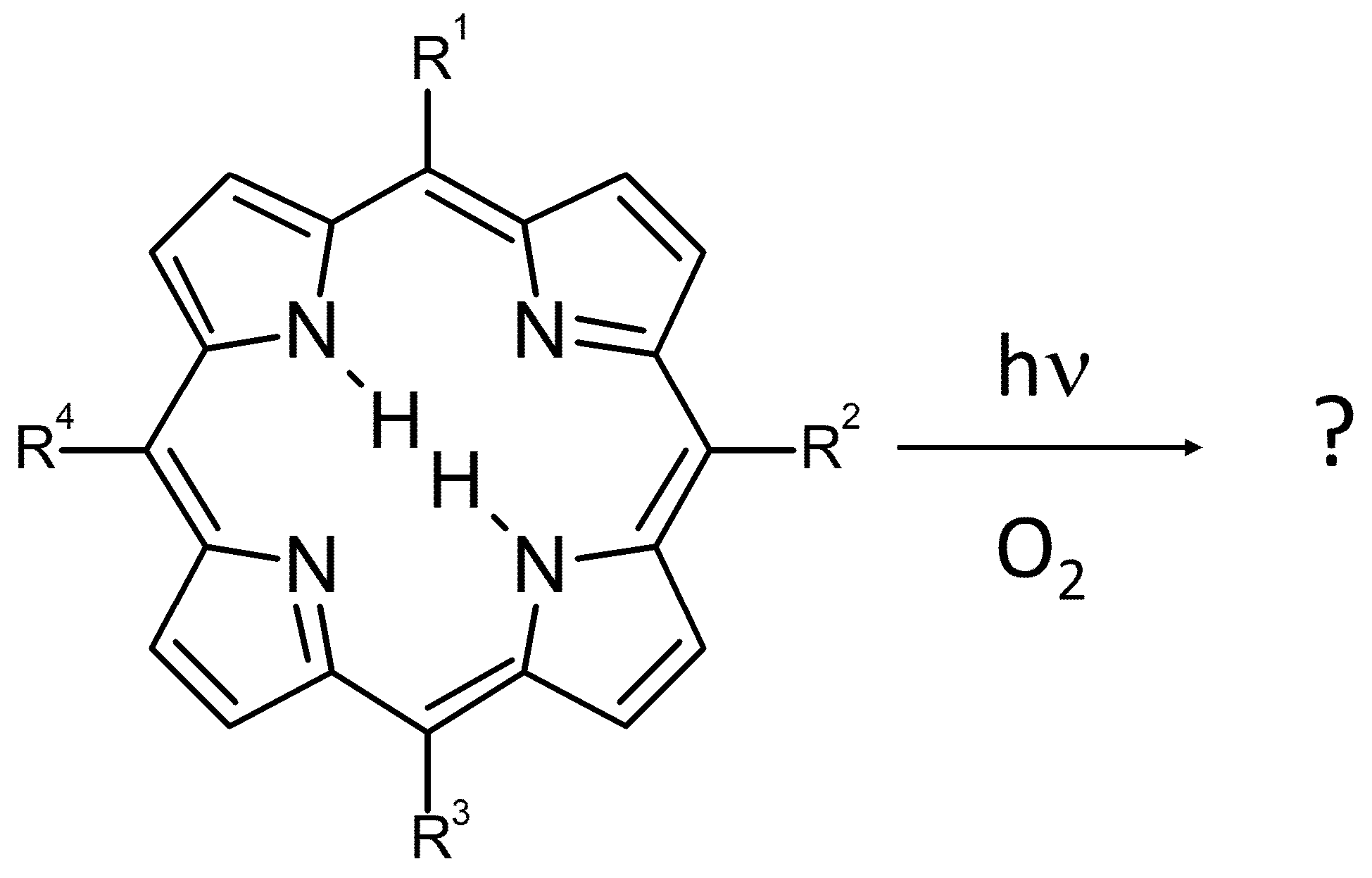
:
1. Introduction
2. Materials and Methods
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Demchenko, A.P. Photobleaching of organic fluorophores: Quantitative characterization, mechanisms, protection. Methods Appl. Fluoresc. 2020, 8, 022001. [Google Scholar] [CrossRef] [PubMed]
- Ha, T.; Tinnefeld, P. Photophysics of Fluorescent Probes for Single-Molecule Biophysics and Super-Resolution Imaging. Ann. Rev. Phys. Chem. 2012, 63, 595–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bregnhøj, M.; Prete, M.; Turkovic, V.; Petersen, A.U.; Nielsen, M.B.; Madsen, M.; Ogilby, P.R. Oxygen-dependent photophysics and photochemistry of prototypical compounds for organic photovoltaics: Inhibiting degradation initiated by singlet oxygen at a molecular level. Methods Appl. Fluoresc. 2019, 8, 014001. [Google Scholar] [CrossRef]
- Grabenhorst, L.; Trofymchuk, K.; Steiner, F.; Glembockyte, V.; Tinnefeld, P. Fluorophore photostability and saturation in the hotspot of DNA origami nanoantennas. Methods Appl. Fluoresc. 2020, 8, 024003. [Google Scholar] [CrossRef]
- Widengren, J.; Chmyrov, A.; Eggeling, C.; Löfdahl, P.-Å.; Seidel, C.A.M. Strategies to Improve Photostabilities in Ultrasensitive Fluorescence Spectroscopy. J. Phys. Chem. A 2007, 111, 429–440. [Google Scholar] [CrossRef]
- Liphardt, B.; Lüttke, W. Laser-Dyes. I. Bifluorophoric Laser-Dyes for Increase of the Efficiency of Dye-Lasers. Liebigs Ann. Chem. 1981, 1118–1138. [Google Scholar] [CrossRef]
- Liphardt, B.; Liphardt, B.; Lüttke, W. Laser dyes with intramolecular triplet quenching. Opt. Commun. 1981, 38, 207–210. [Google Scholar] [CrossRef]
- Schäfer, F.P.; Zhang, F.G.; Jethwa, J. Intramolecular TT-energy transfer in bifluorophoric laser dyes. Appl. Phys. B 1982, 28, 37–41. [Google Scholar] [CrossRef]
- Altman, R.B.; Terry, D.S.; Zhou, Z.; Zheng, Q.; Geggier, P.; Kolster, R.A.; Zhao, Y.; Javitch, J.A.; Warren, J.D.; Blanchard, S.C. Cyanine fluorophore derivatives with enhanced photostability. Nat. Methods 2012, 9, 68–71. [Google Scholar] [CrossRef] [Green Version]
- Tinnefeld, P.; Cordes, T. ‘Self-healing’ dyes: Intramolecular stabilization of organic fluorophores. Nat. Methods 2012, 9, 426–427. [Google Scholar] [CrossRef] [Green Version]
- Altman, R.B.; Zheng, Q.; Zhou, Z.; Terry, D.S.; Warren, J.D.; Blanchard, S.C. Enhanced photostability of cyanine fluorophores across the visible spectrum. Nat. Methods 2012, 9, 428–429. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Jockusch, S.; Zhou, Z.; Altman, R.B.; Warren, J.D.; Turro, N.J.; Blanchard, S.C. On the Mechanisms of Cyanine Fluorophore Photostabilization. J. Phys. Chem. Lett. 2012, 3, 2200–2203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Velde, J.H.M.; Ploetz, E.; Hiermaier, M.; Oelerich, J.; de Vries, J.W.; Roelfes, G.; Cordes, T. Mechanism of Intramolecular Photostabilization in Self-Healing Cyanine Fluorophores. ChemPhysChem 2013, 14, 4084–4093. [Google Scholar] [CrossRef] [PubMed]
- Van der Velde, J.H.M.; Oelerich, J.; Huang, J.Y.; Smit, J.H.; Hiermaier, M.; Ploetz, E.; Herrmann, A.; Roelfes, G.; Cordes, T. The Power of Two: Covalent Coupling of Photostabilizers for Fluorescence Applications. J. Phys. Chem. Lett. 2014, 5, 3792–3798. [Google Scholar] [CrossRef]
- Zheng, Q.; Juette, M.F.; Jockusch, S.; Wasserman, M.R.; Zhou, Z.; Altman, R.B.; Blanchard, S.C. Ultra-stable organic fluorophores for single-molecule research. Chem. Soc. Rev. 2014, 43, 1044–1056. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Q.; Jockusch, S.; Zhou, Z.; Blanchard, S.C. The Contribution of Reactive Oxygen Species to the Photobleaching of Organic Fluorophores. Photochem. Photobiol. 2014, 90, 448–454. [Google Scholar] [CrossRef] [Green Version]
- Juette, M.F.; Terry, D.S.; Wasserman, M.R.; Zhou, Z.; Altman, R.B.; Zheng, Q.; Blanchard, S.C. The bright future of single-molecule fluorescence imaging. Curr. Opin. Chem. Biol. 2014, 20, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Van der Velde, J.H.M.; Uusitalo, J.J.; Ugen, L.J.; Warszawik, E.M.; Herrmann, A.; Marrink, S.J.; Cordes, T. Intramolecular photostabilization via triplet-state quenching: Design principles to make organic fluorophores “self-healing”. Faraday Discuss. 2015, 184, 221–235. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Q.S.; Jockusch, S.; Rodriguez-Calero, G.G.; Zhou, Z.; Zhao, H.; Altman, R.B.; Abruna, H.D.; Blanchard, S.C. Intra-molecular triplet energy transfer is a general approach to improve organic fluorophore photostability. Photochem. Photobiol. Sci. 2016, 15, 196–203. [Google Scholar] [CrossRef] [Green Version]
- Van der Velde, J.H.M.; Oelerich, J.; Huang, J.; Smit, J.H.; Aminian Jazi, A.; Galiani, S.; Kolmakov, K.; Gouridis, G.; Eggeling, C.; Herrmann, A.; et al. A simple and versatile design concept for fluorophore derivatives with intramolecular photostabilization. Nature Comm. 2016, 7, 10144. [Google Scholar] [CrossRef]
- Zheng, Q.; Jockusch, S.; Zhou, Z.; Altman, R.B.; Zhao, H.; Asher, W.; Holsey, M.; Mathiasen, S.; Geggier, P.; Javitch, J.A.; et al. Electronic tuning of self-healing fluorophores for live-cell and single-molecule imaging. Chem. Sci. 2017, 8, 755–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Q.; Lavis, L.D. Development of photostable fluorophores for molecular imaging. Curr. Opin. Chem. Biol. 2017, 39, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Minoshima, M.; Kikuchi, K. Photostable and photoswitching fluorescent dyes for super-resolution imaging. JBIC J. Biol. Inorg. Chem. 2017, 22, 639–652. [Google Scholar] [CrossRef] [PubMed]
- Van der Velde, J.H.M.; Smit, J.H.; Hebisch, E.; Punter, M.; Cordes, T. Self-healing dyes for super-resolution fluorescence microscopy. J. Phys. D Appl. Phys. 2018, 52, 034001. [Google Scholar] [CrossRef]
- Glembockyte, V.; Wieneke, R.; Gatterdam, K.; Gidi, Y.; Tampé, R.; Cosa, G. Tris-N-Nitrilotriacetic Acid Fluorophore as a Self-Healing Dye for Single-Molecule Fluorescence Imaging. J. Am. Chem. Soc. 2018, 140, 11006–11012. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Liu, L.; Jing, T.; Ruan, Z.; Yuan, P.; Yan, L. Self-Healing Organic Fluorophore of Cyanine-Conjugated Amphiphilic Polypeptide for Near-Infrared Photostable Bioimaging. ACS Appl. Mater. Interfaces 2018, 10, 14517–14530. [Google Scholar] [CrossRef]
- Gong, W.; Das, P.; Samanta, S.; Xiong, J.; Pan, W.; Gu, Z.; Zhang, J.; Qu, J.; Yang, Z. Redefining the photo-stability of common fluorophores with triplet state quenchers: Mechanistic insights and recent updates. Chem. Comm. 2019, 55, 8695–8704. [Google Scholar] [CrossRef]
- Smit, J.H.; van der Velde, J.H.M.; Huang, J.; Trauschke, V.; Henrikus, S.S.; Chen, S.; Eleftheriadis, N.; Warszawik, E.M.; Herrmann, A.; Cordes, T. On the impact of competing intra- and intermolecular triplet-state quenching on photobleaching and photoswitching kinetics of organic fluorophores. Phys. Chem. Chem. Phys. 2019, 21, 3721–3733. [Google Scholar] [CrossRef] [Green Version]
- Isselstein, M.; Zhang, L.; Glembockyte, V.; Brix, O.; Cosa, G.; Tinnefeld, P.; Cordes, T. Self-Healing Dyes—Keeping the Promise? J. Phys. Chem. Lett. 2020, 11, 4462–4480. [Google Scholar] [CrossRef]
- Pati, A.K.; El Bakouri, O.; Jockusch, S.; Zhou, Z.; Altman, R.B.; Fitzgerald, G.A.; Asher, W.B.; Terry, D.S.; Borgia, A.; Holsey, M.D.; et al. Tuning the Baird aromatic triplet-state energy of cyclooctatetraene to maximize the self-healing mechanism in organic fluorophores. Proc. Natl. Acad. Sci. USA 2020, 117, 24305–24315. [Google Scholar] [CrossRef]
- Yang, Z.; Li, L.; Ling, J.; Liu, T.; Huang, X.; Ying, Y.; Zhao, Y.; Zhao, Y.; Lei, K.; Chen, L.; et al. Cyclooctatetraene-conjugated cyanine mitochondrial probes minimize phototoxicity in fluorescence and nanoscopic imaging. Chem. Sci. 2020, 11, 8506–8516. [Google Scholar] [CrossRef]
- Ostapko, J.; Gorski, A.; Buczyńska, J.; Golec, B.; Nawara, K.; Kharchenko, A.; Listkowski, A.; Ceborska, M.; Pietrzak, M.; Waluk, J. Towards More Photostable, Brighter, and Less Phototoxic Chromophores: Synthesis and Properties of Porphyrins Functionalized with Cyclooctatetraene. Chem. Eur. J. 2020, 70, 16666–16675. [Google Scholar] [CrossRef] [PubMed]
- Pineiro, M.; Carvalho, A.L.; Pereira, M.M.; Gonsalves, A.M.A.R.; Arnaut, L.G.; Formosinho, S.J. Photoacoustic measurements of porphyrin triplet-state quantum yields and singlet-oxygen efficiencies. Chem. Eur. J. 1998, 11, 2299–2307. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, R.; Tanielian, C.; Dunsbach, R.; Wolff, C. Phenalenone, a universal reference compound for the determination of quantum yields of singlet oxygen O2 (1Δg) sensitization. J. Photochem. Photobiol. A Chem. 1994, 79, 11–17. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Cavaleiro, J.A.S.; Görner, H.; Lacerda, P.S.S.; MacDonald, J.G.; Mark, G.; Neves, M.G.P.M.S.; Nohr, R.S.; Schuchmann, H.P.; van Sonntag, C.; Tome, A.C. Singlet oxygen formation and photostability of meso-tetraarylporphyrin derivatives and their copper complexes. J. Photochem. Photobiol. A Chem. 2001, 144, 131–140. [Google Scholar] [CrossRef]
- Cavaleiro, J.A.S.; Hewlins, M.J.E.; Jackson, A.H.; Neves, G.P.M.S. Structures of the Ring-Opened Oxidation Products from meso-Tetraphenylporphyrins. J. Chem. Soc. Chem. Comm. 1986, 142–144. [Google Scholar] [CrossRef]
- Cavaleiro, J.A.S.; Neves, M.G.P.S.; Hewlins, M.J.E.; Jackson, A.H. The Photooxidation of Meso-Tetraphenylporphyrins. J. Chem. Soc. Perkin Trans. 1 1990, 7, 1937–1943. [Google Scholar] [CrossRef]
- Smith, K.M.; Brown, S.B.; Troxler, R.F.; Lai, J.J. Mechanism of photo-oxygenation of meso-tetraphenylporphyrin metal complexes. Tetrahedron Lett. 1980, 21, 2763–2766. [Google Scholar] [CrossRef]
- Smith, K.M.; Brown, S.B.; Troxler, R.F.; Lai, J.J. Photooxygenation of meso-tetraphenylporphyrin complexes. Photochem. Photobiol. 1982, 36, 147–152. [Google Scholar] [CrossRef]
- Wojaczyński, J.; Popiel, M.; Szterenberg, L.; Latos-Grażyński, L. Common Origin, Common Fate: Regular Porphyrin and N-Confused Porphyrin Yield an Identical Tetrapyrrolic Degradation Product. J. Org. Chem. 2011, 76, 9956–9961. [Google Scholar] [CrossRef]
- Silva, A.M.S.; Neves, M.G.P.M.S.; Martins, R.R.L.; Cavaleiro, J.A.S.; Boschi, T.; Tagliatesta, P. Photo-oxygenation of meso-tetraphenylporphyrin derivatives: The influence of the substitution pattern and characterization of the reaction products. J. Porphyr. Phthalocyanines 1998, 2, 45–51. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
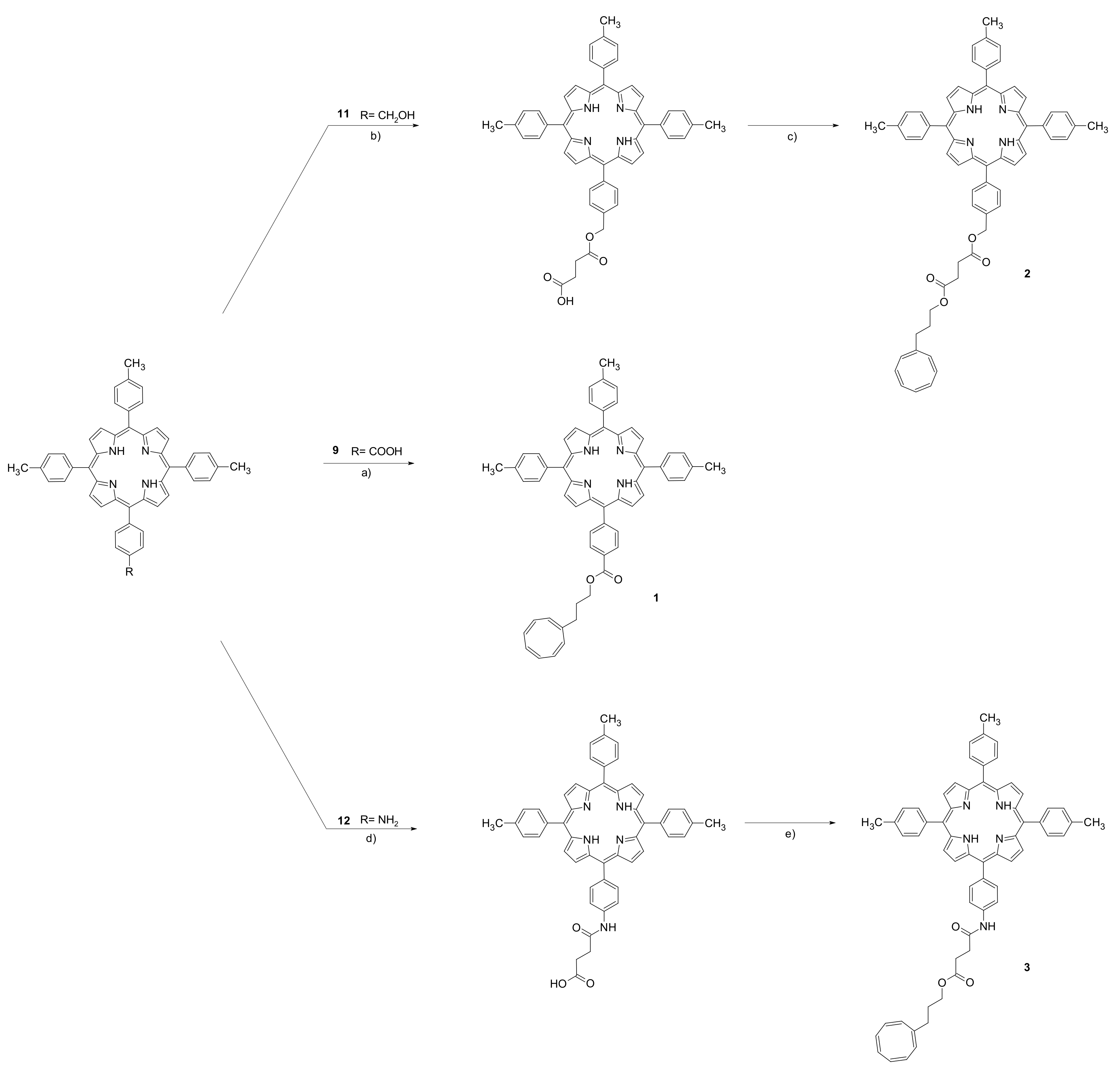{kind=link}
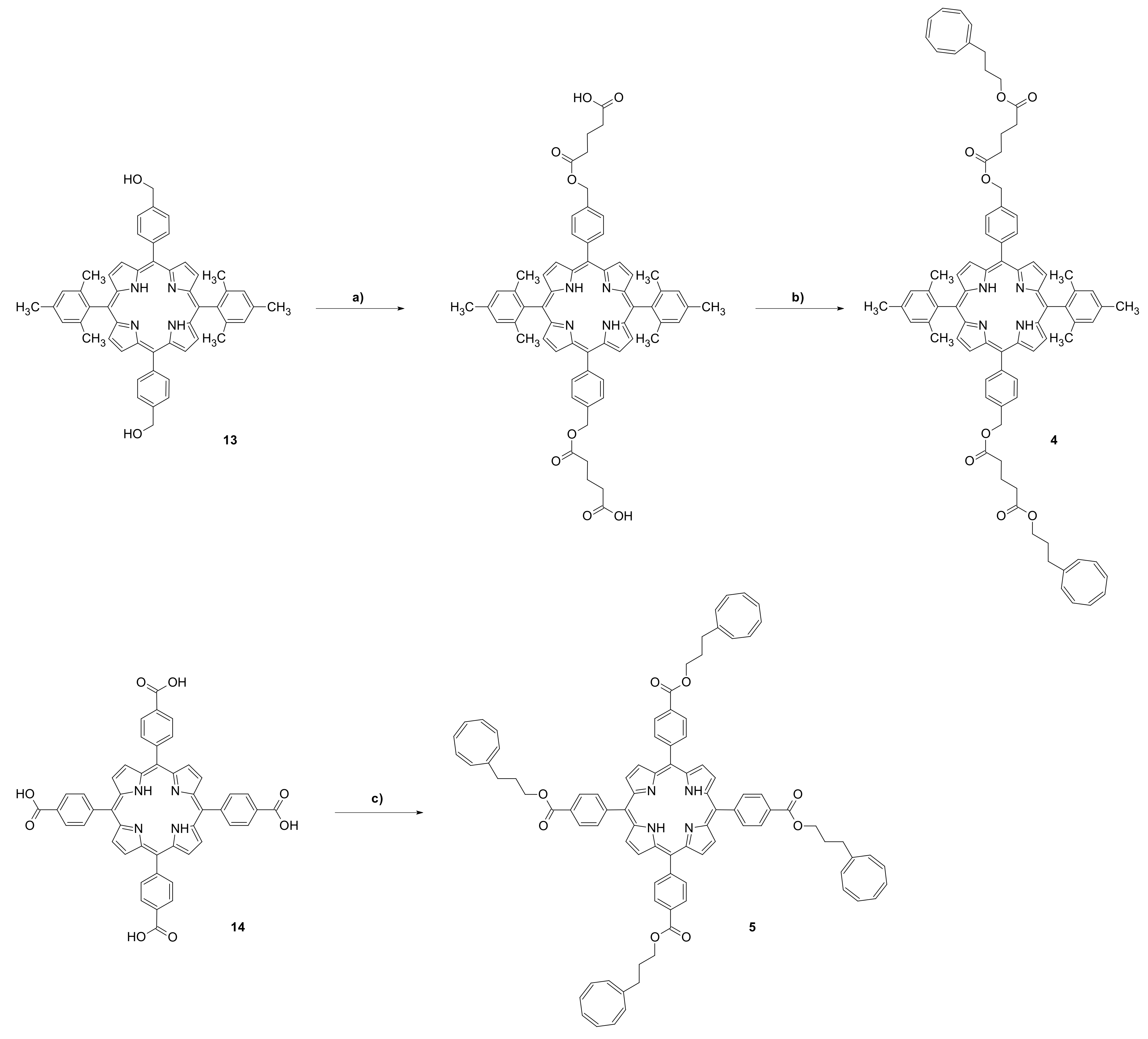{kind=link}
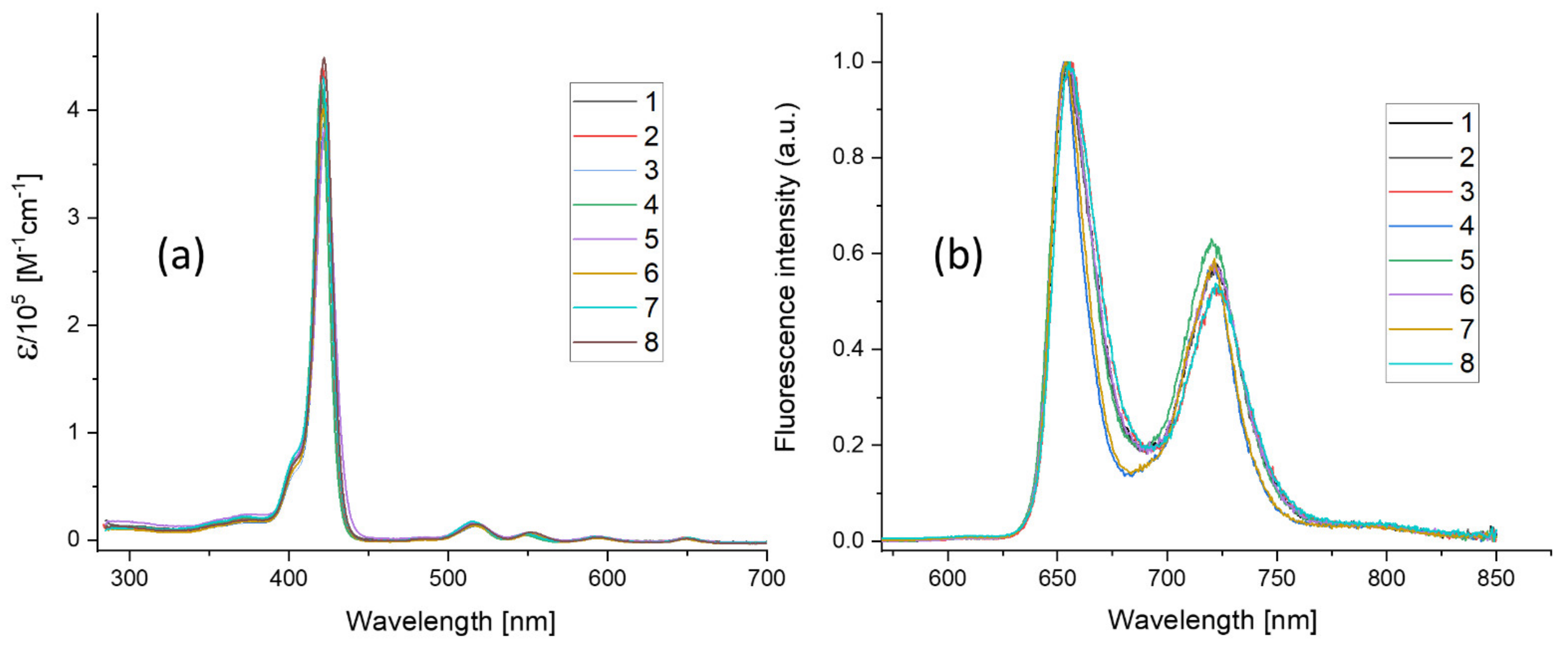
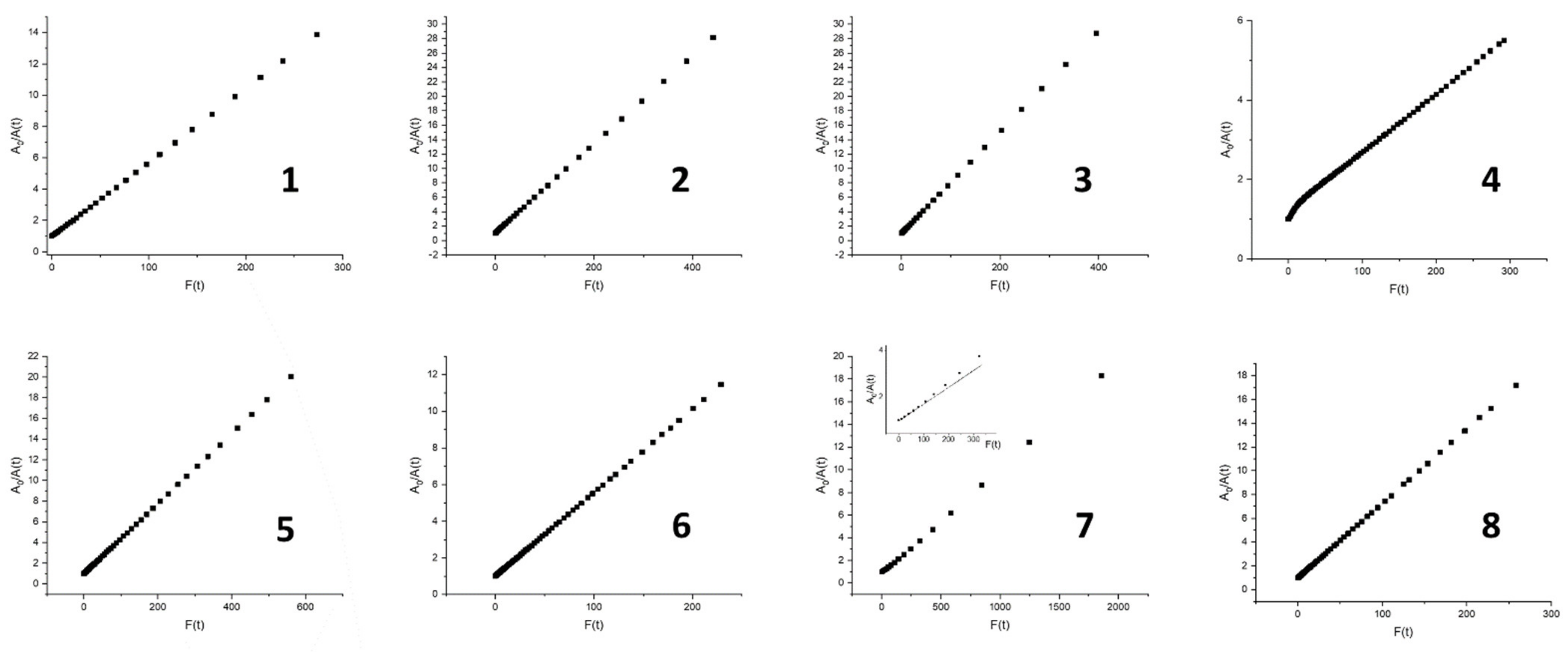
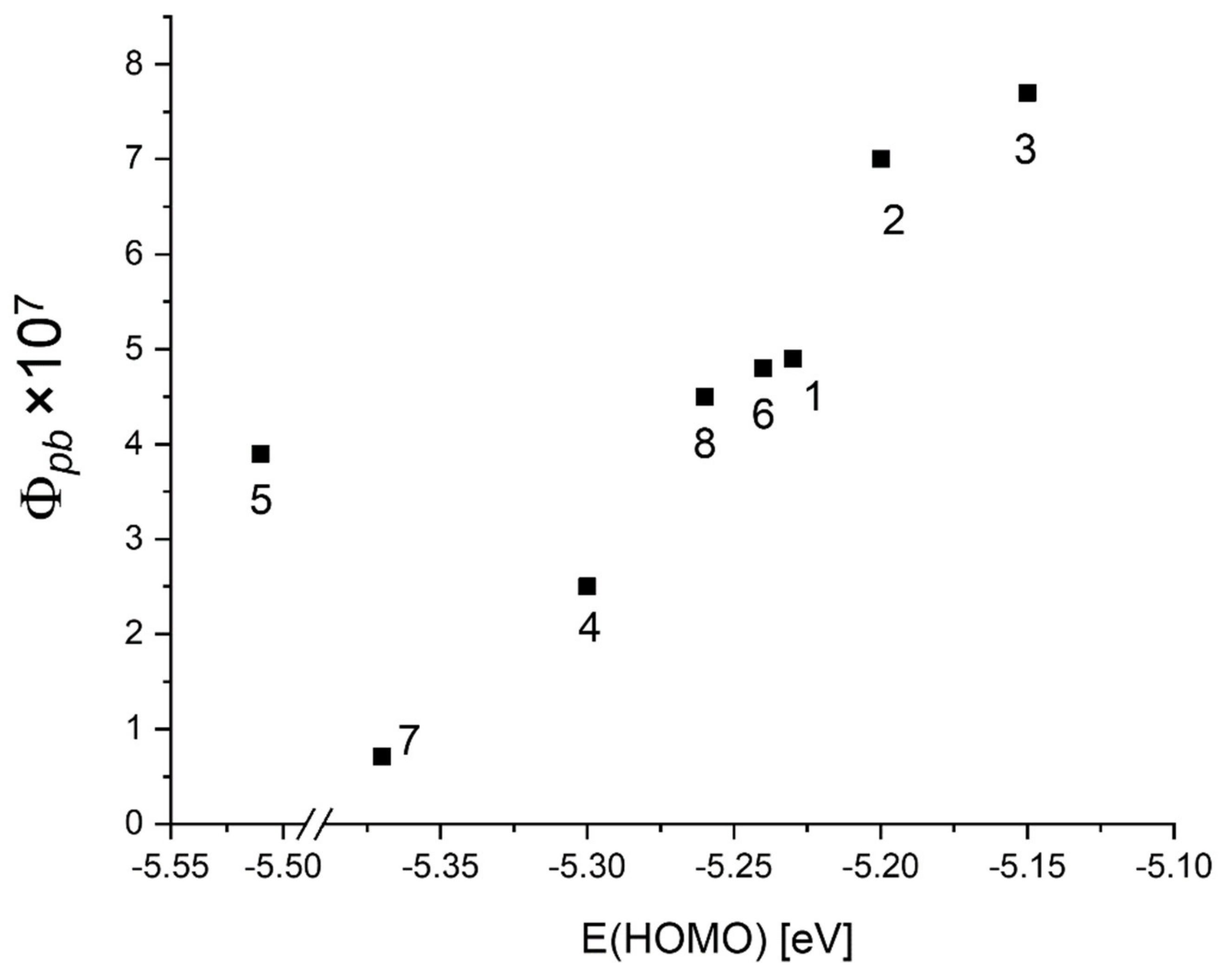
| Abs. Maxima [nm] | Abs. Coeff [M−1 cm−1] | Φfl | S1 Lifetime [ns] | T1 Lifetime Aerated Degassed [ns] [μs] | ΦΔ | Φpb | |
|---|---|---|---|---|---|---|---|
| 1 | 421 516 551.5 593.5 649.5 | 416,860 17,558 9056 5016 3901 | 0.12 | 9.7 | 293 458 | 0.78 | 4.9 × 10−7 |
| 2 | 420.5 516 551 592.5 648.5 | 440,270 17,921 9077 5035 4014 | 0.12 | 9.6 | 297 14.7 | 0.7 | 7.0 × 10−7 |
| 3 | 421.5 517 552 594 649.5 | 389,104 15,590 8793 4486 3872 | 0.12 | 9.5 | 298 112 | 0.75 | 7.7 × 10−7 |
| 4 | 420 514.5 548 591.5 650 | 426,346 18,376 7350 5052 4380 | 0.13 | 10.3 | 386 9.3 | 0.72 | 2.5 × 10−7 |
| 5 | 422 516 550.5 593 649 | 383,662 15,931 7171 4281 2949 | 0.13 | 10.2 | 329 257 | 0.66 | 3.9 × 10−7 |
| 6 | 421 516 551 593.5 649.5 | 401,207 16,709 8627 4795 3735 | 0.12 | 9.6 | 297 645 | 0.78 | 4.8 × 10−7 |
| 7 | 420.5 515 548.5 592.5 650.5 | 431,582 20,127 8252 5515 4621 | 0.14 | 10.4 | 509 1070 | 0.78 | 7.1 × 10−8 |
| 8 | 421.5 517 552.5 593.5 650.5 | 449,392 15,922 8839 4545 3849 | 0.13 | 9.5 | 294 820 | 0.79 | 4.5 × 10−7 |
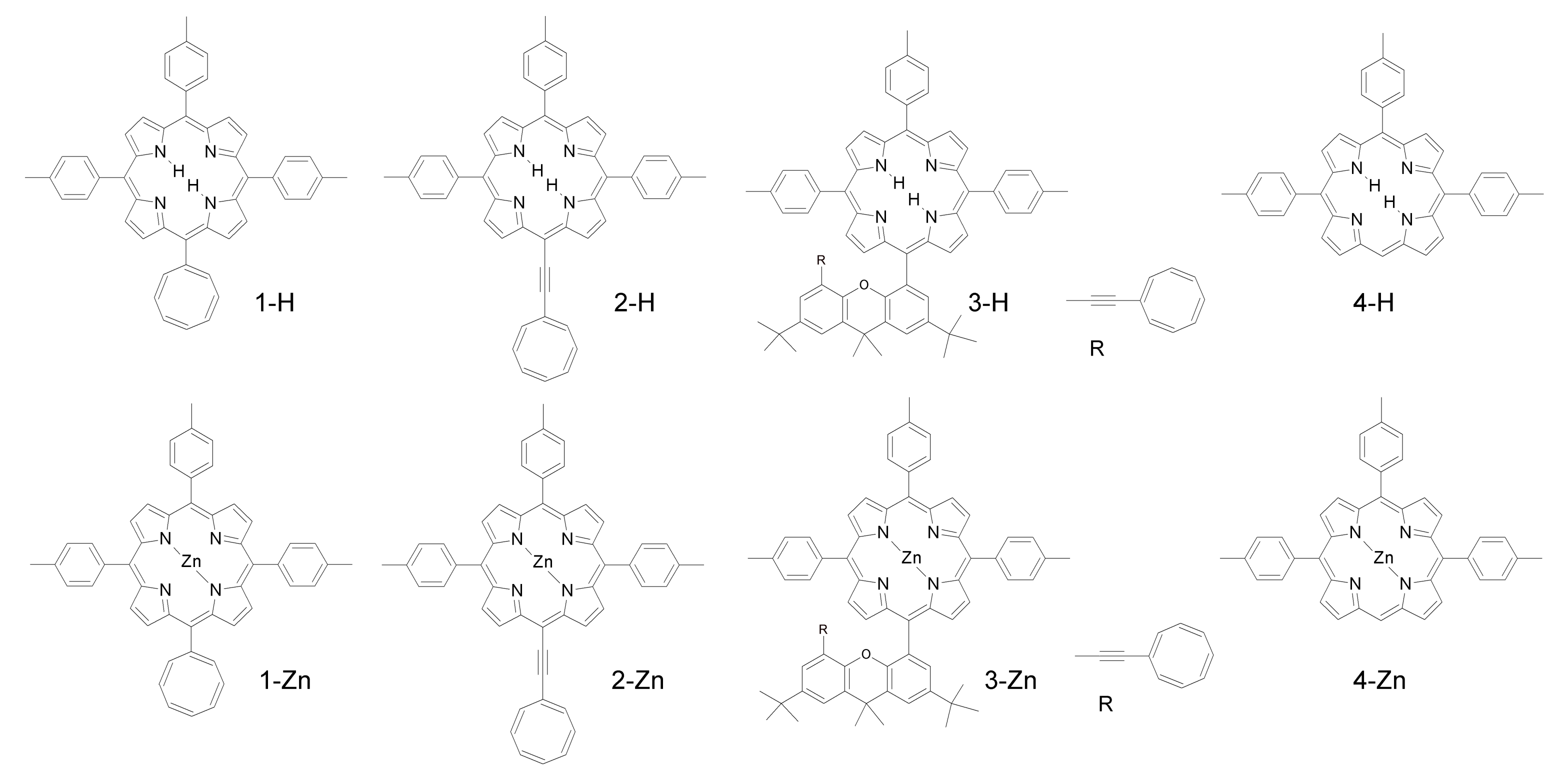
| Compound | kintr [106 s−1] | kq[O2] [106 s−1] |
|---|---|---|
| 1-H | 5.6 | 3.8 |
| 2-H | 5.4 | 7.6 |
| 3-H | 5.2 | 6.8 |
| 4-H | 0.0048 | 4 |
| 1-Zn | 5.6 | 4.1 |
| 2-Zn | 5.2 | 7.8 |
| 3-Zn | 17 | 4 |
| 4-Zn | 0.0038 | 2.8 |
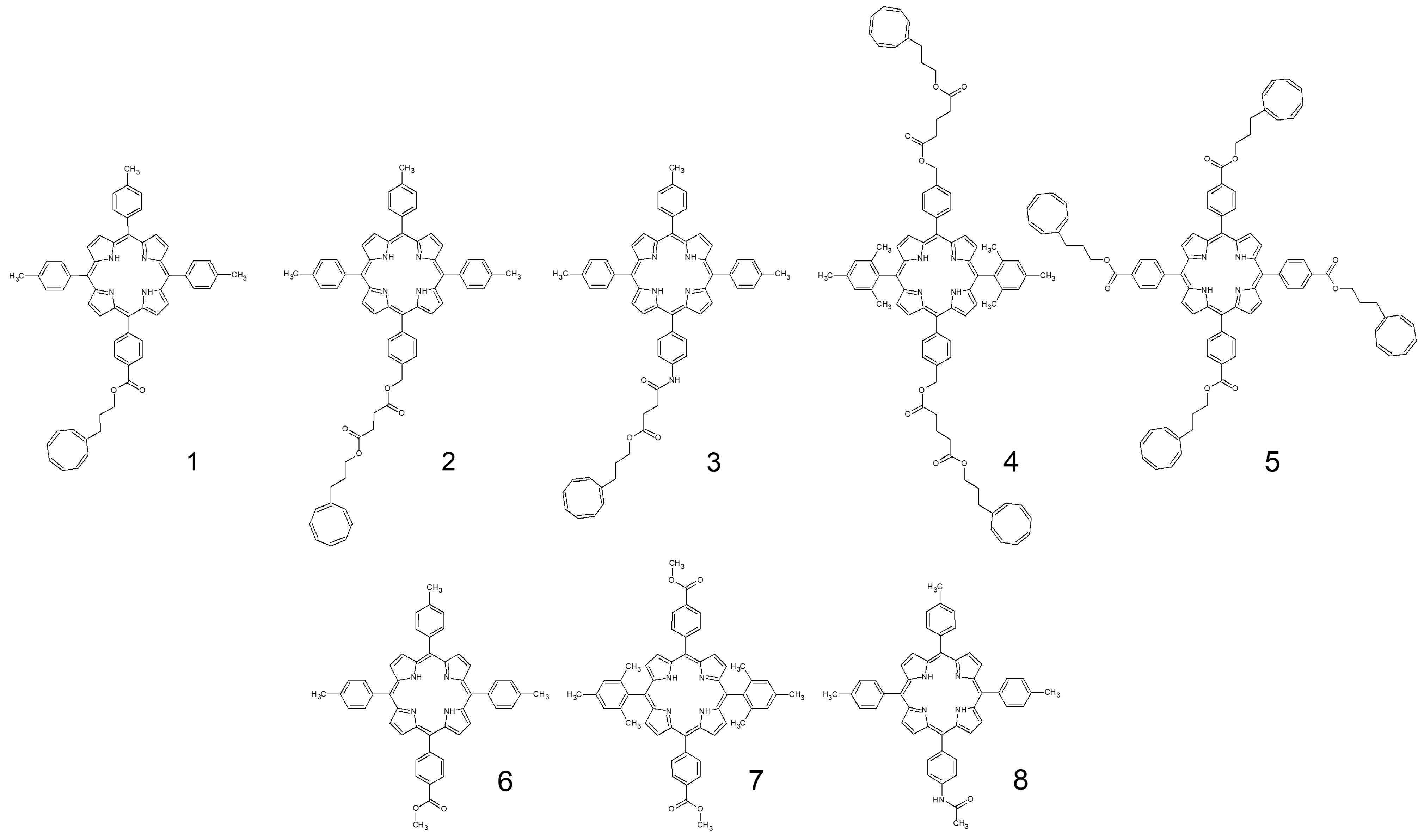
| 1 | 0.0022 | 3.4 |
| 2 | 0.068 | 3.4 |
| 3 | 0.0089 | 3.4 |
| 4 | 0.11 | 2.5 |
| 5 | 0.0039 | 3 |
| 6 | 0.0016 | 3.4 |
| 7 | 0.00093 | 2 |
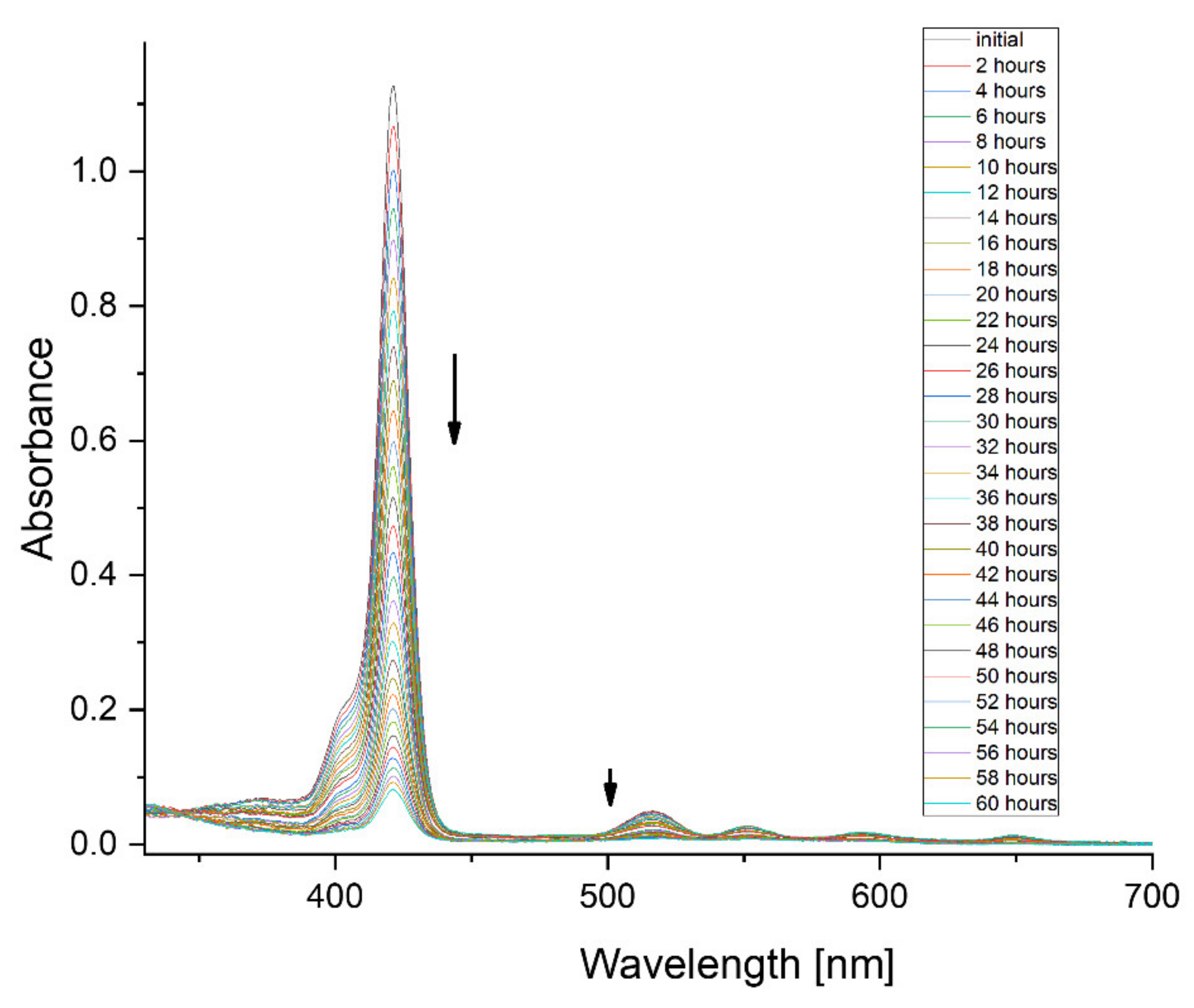
| 8 | 0.0012 | 3.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buczyńska, J.; Gajewska, A.; Gorski, A.; Golec, B.; Nawara, K.; Rybakiewicz, R.; Waluk, J. Synthesis and Photostability of Cyclooctatetraene-Substituted Free Base Porphyrins. Chemistry 2021, 3, 104-115. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry3010008
Buczyńska J, Gajewska A, Gorski A, Golec B, Nawara K, Rybakiewicz R, Waluk J. Synthesis and Photostability of Cyclooctatetraene-Substituted Free Base Porphyrins. Chemistry. 2021; 3(1):104-115. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry3010008
Chicago/Turabian StyleBuczyńska, Joanna, Agnieszka Gajewska, Aleksander Gorski, Barbara Golec, Krzysztof Nawara, Renata Rybakiewicz, and Jacek Waluk. 2021. "Synthesis and Photostability of Cyclooctatetraene-Substituted Free Base Porphyrins" Chemistry 3, no. 1: 104-115. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry3010008