
The B2 Structural Motif as a Tool for Modulating Ring Currents in Monocyclic Li Clusters


Faculty of Science, University of Kragujevac, Radoja Domanovića 12, 34000 Kragujevac, Serbia
*
Author to whom correspondence should be addressed.
Chemistry 2021, 3(3), 1063-1073; https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry3030077
Submission received: 28 July 2021
/
Revised: 3 September 2021
/
Accepted: 4 September 2021
/
Published: 14 September 2021
(This article belongs to the Special Issue Current Density and Spectroscopy—A Themed Issue in Honor of Professor Riccardo Zanasi on the Occasion of His 70th Birthday)
Abstract
:Magnetically induced current densities, calculated at the M06-2X/def2-TZVP level using the diamagnetic-zero version of the continuous transformation of origin of current density (CTOCD-DZ) method, were employed to study the aromaticity in and . It was found that the rings in and remarkably resemble the monocyclic and clusters. Unlike the parent and systems that sustain negligibly weak global current density circulation, the and clusters exhibit a strong diatropic current density. The present work demonstrates how structural modifications introduced by the unit can be used for modulating the current density in cyclic Li-based clusters.
1. Introduction
Cyclic electron delocalization is among the most intriguing phenomena in chemistry. If present in a molecule, cyclic electron delocalization determines rather unique structural, magnetic, thermodynamic, and reactivity-based properties [1,2,3]. All these features coming due to cyclic electron delocalization are usually referred to as aromaticity. Understanding, defining, and evaluating the magnetic aspects of molecular aromaticity have been in focus of chemists for almost a whole century [4,5]. Already, London [6] recognized that an aromatic molecule sustains an induced ring current as a response to the applied external magnetic field perpendicular to the molecular plane. Nowadays, it is widely accepted that the specific behavior of molecules in the presence of the external magnetic field can be used for qualitative and quantitative assessment of aromaticity [4,5,7,8]. The magnetically induced current density is the most important concept in the evaluation of the magnetic aspect of aromaticity. Based on the current densities, one can measure both global and local aromaticity [8]. In addition, other indices of magnetic aromaticity, including the nucleus-independent chemical shift (NICS) [9], as one of the most employed aromaticity indices, can be obtained from the induced current density [4,10]. On the other hand, there is no unique way to reconstruct current density maps from NICS values [11].
One of the most important challenges in the computation of magnetically induced current densities and related molecular magnetic response properties is how to treat the so-called gauge origin problem [8]. The gauge origin problem refers to the dependence of the calculated magnetic properties on the translation of the origin of the coordinate system. The gauge origin problem can be generally eliminated either by employing a basis set with an explicit magnetic field dependence or by an explicit treatment of the gauge origin dependence [8]. The first group of methods are based on perturbation-dependent basis functions, such as gauge-including atomic orbitals (GIAO) [6]. Calculation of the induced current density by means of the GIAO can be performed by means of the gauge-including magnetically induced current (GIMIC) method [8,12]. Within the second approach, the most employed methods are based on the continuous transformations of origin of current density (CTOCD) technique [13,14]. In this approach, the gauge origin problem is resolved by using a different gauge origin for each point for which the current density is calculated. It should be noted that this method was originally introduced as the so-called continuous set of gauge transformation (CSGT) method and that the name CTOCD was suggested somewhat later [15,16,17]. Over almost a half of a century, Riccardo Zanasi contributed to the development of the CTOCD method and other approaches for calculation of the magnetic response properties, as well as in the software implementation of these theories [18,19]. In the present work, the diamagnetic zero (DZ) version of the CTOCD method was employed. In the CTOCD-DZ, also known as the ipsocentric method [20,21], the current density at each point is calculated by choosing itself as the origin of the vector potential. More details on the CTOCD method can be found in reviews [4,16] and elsewhere [22].
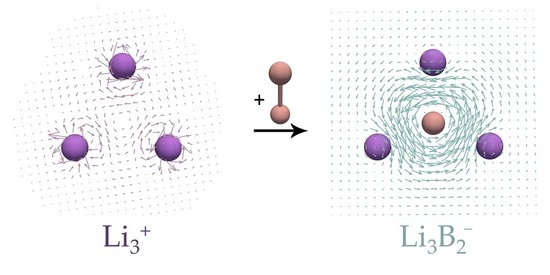
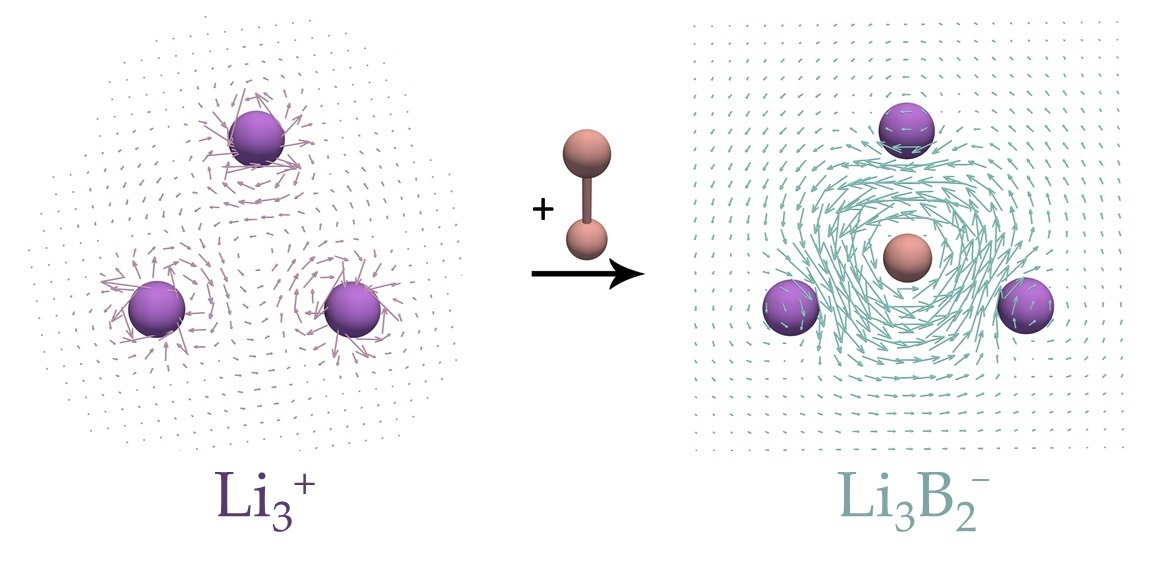
There are many cases showing that the NICS and current density results do not always agree [23,24,25,26,27]. One of the most-known examples is the monocyclic cluster. has two valence electrons, and according to the Hückel 4n + 2 rule, this system should be aromatic. Large negative NICS values wrongly predicted the aromatic character of [23] because this system does not sustain global ring currents, but rather local circulations around Li atoms. In what follows, the magnetically induced current densities in two recently characterized complexes [28], and , are examined and compared to those in the corresponding reference and systems. To the best of the authors’ knowledge, the target and clusters have not so far been experimentally observed. However, a closely related species, , which is isoelectronic to , has been recently experimentally characterized [29]. In what follows, it is shown how the presence of the unit can drastically change magnetic response properties of the parent ring.
2. Computational Methods
The molecular structures were fully optimized at the M06-2X/def2-TZVP level of theory by means of the Gaussian 09 program [30]. Frequency calculations confirmed that the obtained structures have no imaginary frequencies. The NBO analysis [31] was performed at the same level as implemented in Gaussian 09 program. Magnetically induced current densities were calculated at the M06-2X/def2-TZVP level using the CTOCD-DZ approach. These calculations were performed using our in-house Fortran code, which requires the data from the Gaussian 09 wfx-file (obtained with the keyword NMR = CSGT). The current densities were calculated for a grid of points belonging to the plane formed by Li atoms. The so-obtained current density maps were visualized using the Paraview program [32], utilizing the following convention: counterclockwise/clockwise circulations represent diatropic/paratropic current densities. The bond current strengths were obtained through the disc-based quadrature scheme [33], which employs numerical integration, as devised by Elhay and Kautsky [34]. The integration disc perpendicularly bisects the given bond, and the disc radius is the average of the covalent radii of the two bonded atoms.
The multicenter delocalization index (MCI) [3], which quantifies electron delocalization in a given ring, can be calculated using different partition schemes [35,36,37]. In this work, the natural atomic orbital (NAO) density matrices obtained from NBO analysis were used to calculate the MCI. In order to eliminate the ring-size dependence of the MCI, the nth root of the original index, denoted by , was employed [3]. These calculations were performed using our in-house FORTRAN program. The NICS values were calculated at the M06-2X/def2-TZVP level of theory through the gauge-including atomic orbital (GIAO) method [38,39]. The dissected canonical molecular orbital NICS (CMO-NICS) was calculated using the NBO 7.0 program [40].
3. Results and Discussion
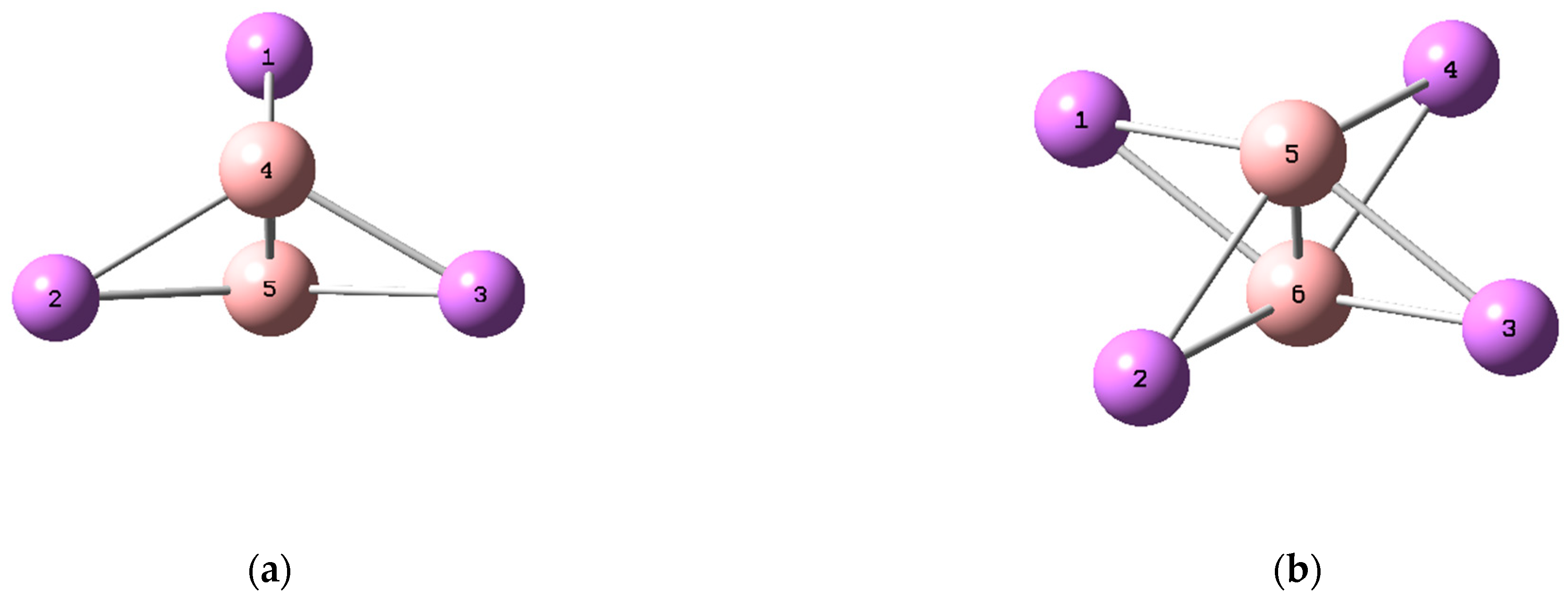
The optimized structures of and , having and symmetries, respectively, are presented in Figure 1. These structures can be described as inverse sandwich complexes, consisting of the ring sandwiched between two boron atoms. Both studied complexes are characterized by a short B−B distance: 1.53 and 1.52 Å in and , respectively. These B−B distances are shorter than those reported in the cluster, for which the respective B−B bond was characterized as triple [41]. It is worth noting that, according to the NBO analysis, the B−B bond in studied clusters is a triple bond: 1σ + 2π bonds.
It has previously been shown that the cluster exhibits the structure. It was found that the presence of the unit in elongates the Li−Li distances (being equal to 3.30 and 3.65 Å) with respect to that in monocyclic cluster (3.05 Å) optimized at the same level of theory. The analogous planar geometry of the bare complex does not correspond to minima on the potential energy surfaces. The global minimum of was found to have tetrahedral structure [42]. The strain in the ring of the complex is reduced in comparison to the ring in . Along these lines, it was observed that Li−Li distance in is 2.89 Å, which is significantly shorter than that in .
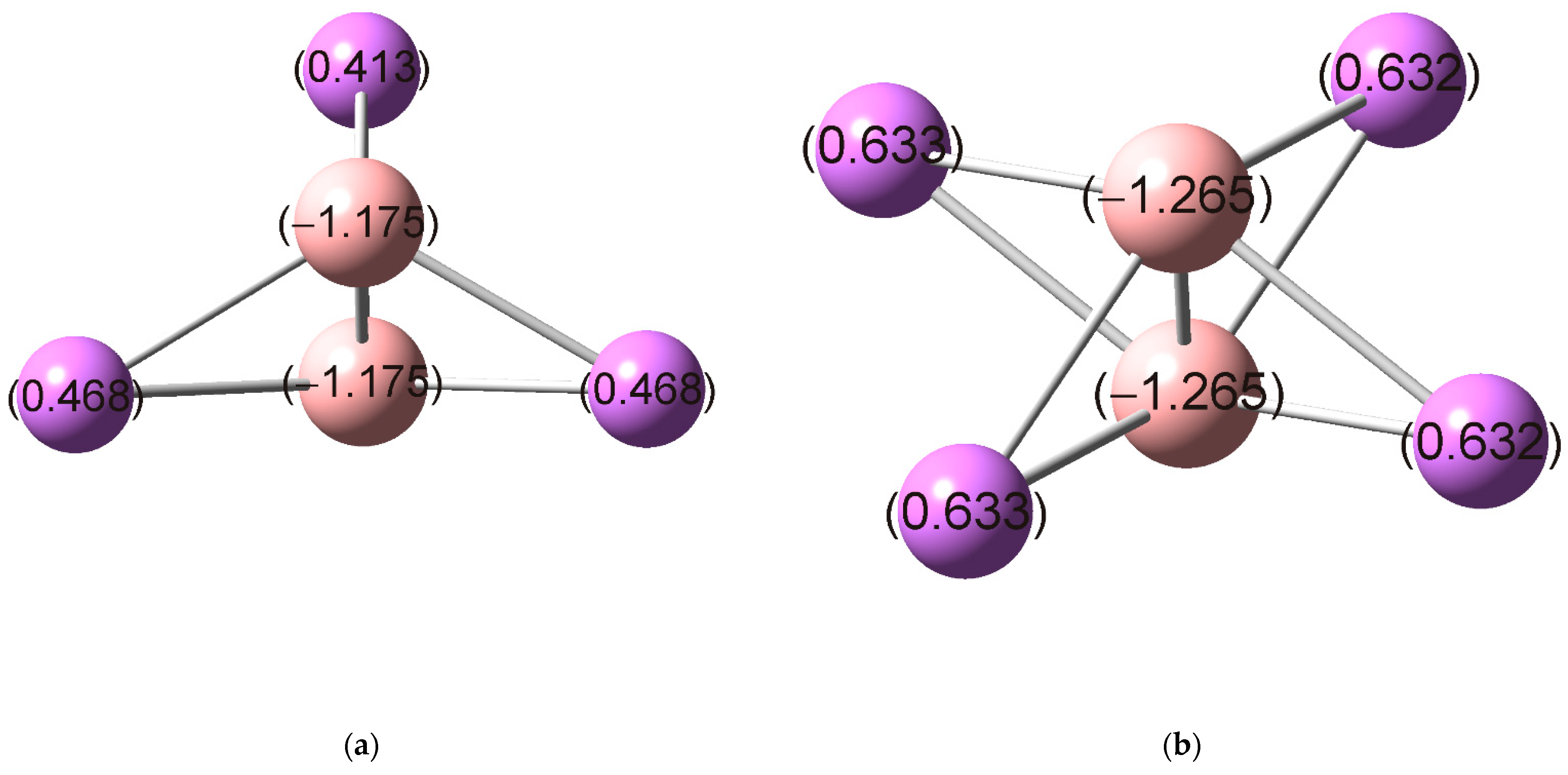
Natural atomic charges of the studied systems are shown in Figure 2. As can be seen, B atoms in and carry negative charges of −1.18 and −1.27, respectively. This reveals a significant charge transfer from Li atoms to the central unit. The obtained charges on B atoms are approximately equal to −1, and and can be viewed as charge-transfer complexes: and . The calculated dissociation energies (Table 1) showed a strong stabilizing electrostatic interaction between the and fragments in the studied clusters.
The obtained structural and electronic features of indicate that the ring in this complex bears a significant resemblance to the monocyclic cluster. In what follows, the nature of magnetically induced current densities in the complex is compared with that in the monocyclic cluster. As mentioned above, the global minimum of has a tetrahedral structure. Therefore, the current density induced in will be contrasted with the monocyclic cluster whose structure was extracted from the optimized geometry of the complex.
The calculated NICS and values of the studied systems are presented in Table 2. In order to verify these values, the diffuse functions were introduced through the ma-def2-TZVP basis set. It was revealed that the addition of the diffuse functions invokes only negligible changes in the aromaticity index values. The reference and systems are characterized by large and negative values, in agreement with the previous work [23]. The CMO σ- showed the values in these two systems are practically fully determined by the HOMO level contribution. It should be noted that the NICS(0) in and is calculated in, or very close, to the B–B bond critical point [43]. Therefore, the values cannot be used to compare the aromaticity of the target complexes and their reference systems. The values predict a high extent of electron delocalization in the reference and systems. Based on the values, the introduction of the fragment into the rings causes a reduction in the intensity of cyclic electron delocalization. This result goes in line with the calculated NBO-based Wiberg bond indices, which also suggest notably weaker Li–Li bonding interactions in and than in the reference bare Li-clusters.
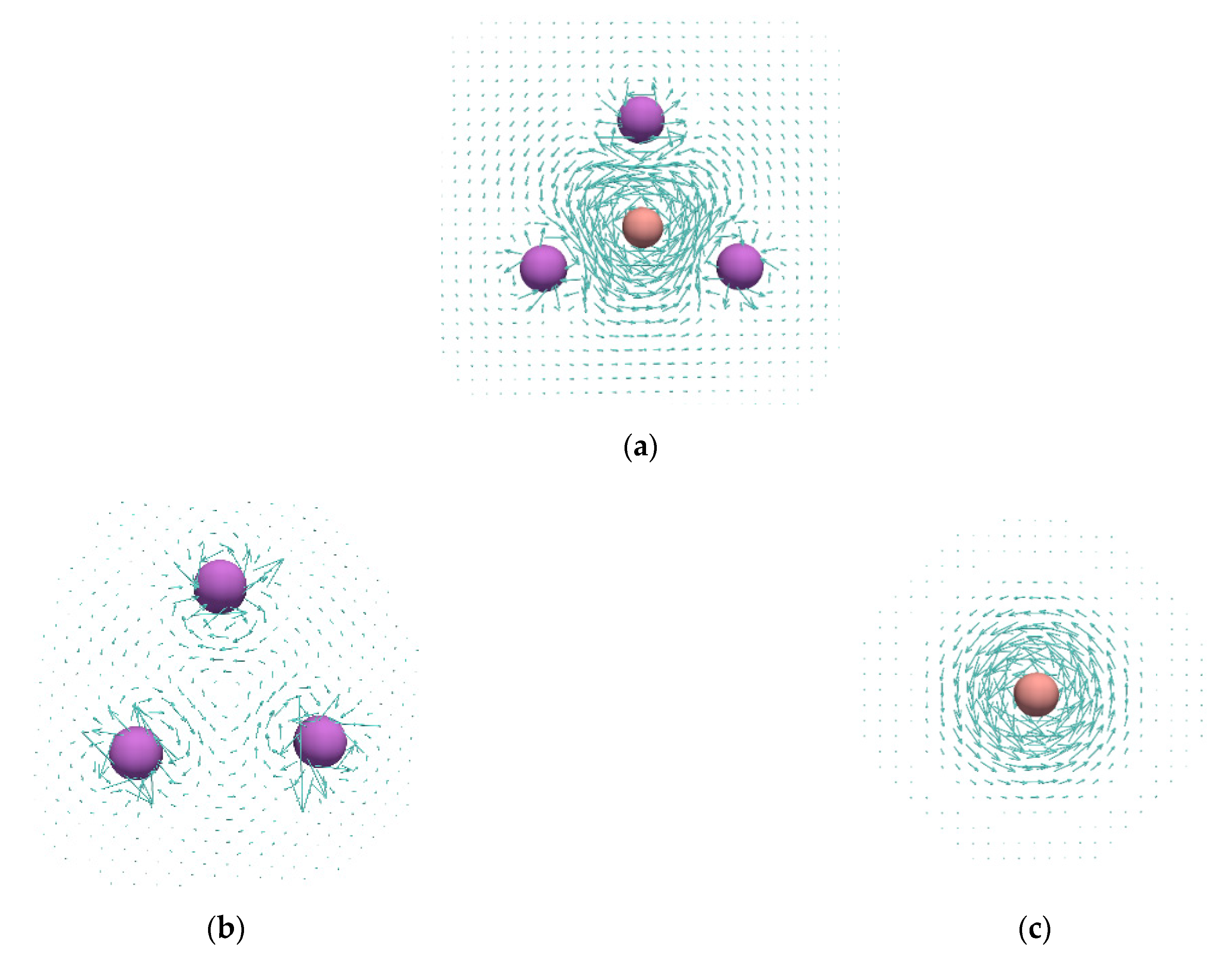
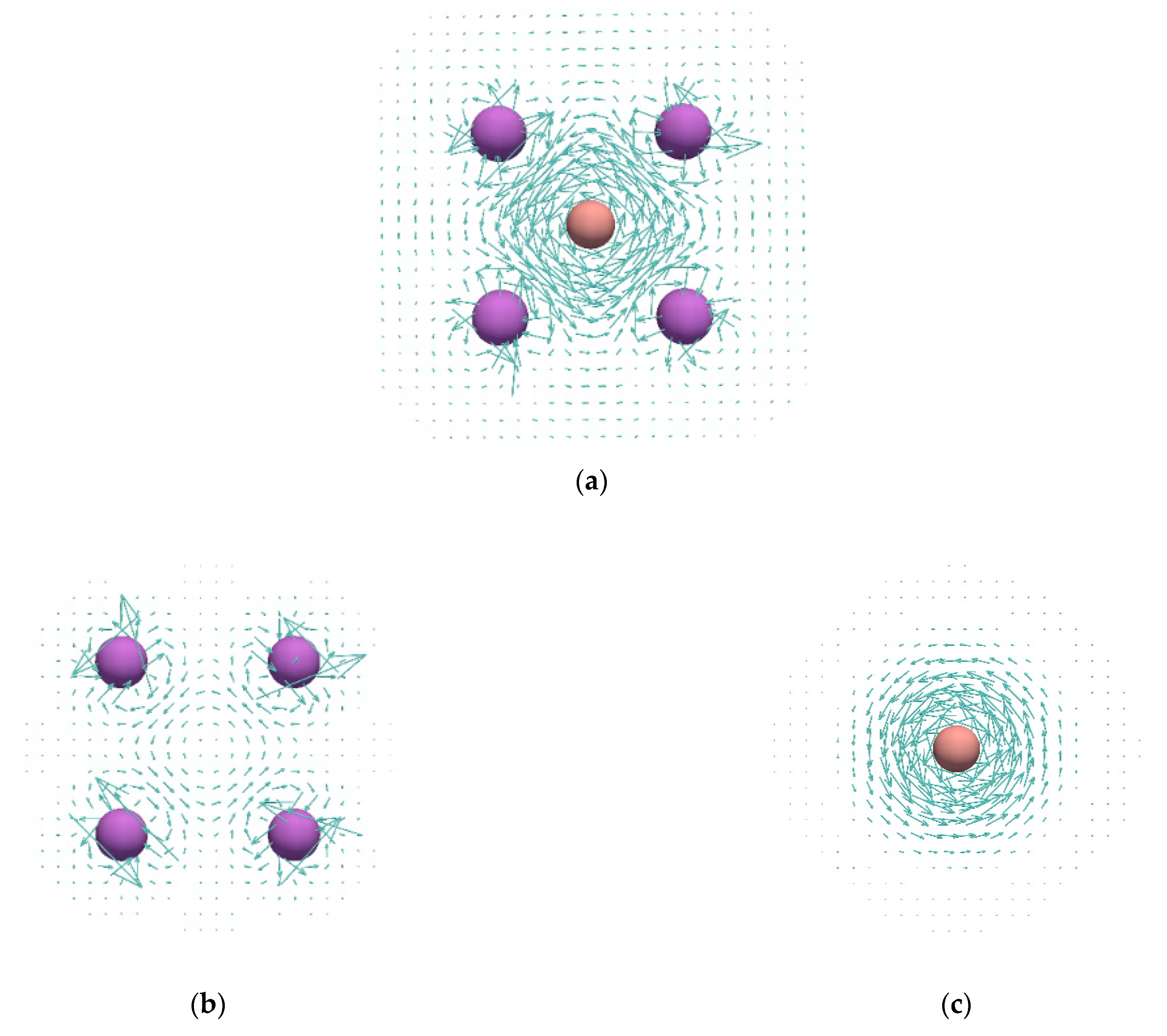
Current density maps of the studied and complexes, as well as those of the reference and systems, are shown in Figure 3 and Figure 4. In agreement with the previous study [23], it can be seen that the cluster shows no significant ring currents, but only local circulations around Li atoms. The same was found for the structure (Figure 4b). On the other hand, the and systems sustain rather strong diatropic ring currents. The most striking feature of the ring currents in / is that the strongest circulations occur inside the rings and around the B–B bonds. The obtained current densities in / resembles the current densities of π-electrons in the isolated . However, the current density maps from Figure 3 and Figure 4 clearly show that the Li atoms are substantially involved in the circulation occurring in the ring center. In addition, the isolated unit was found to be unstable relative to the dissociation product 2 (Table 1), which also confirms a strong stabilization of the fragment thorough the interaction with Li atoms in and . The obtained current densities completely oppose the conclusions derived from the MCI values.
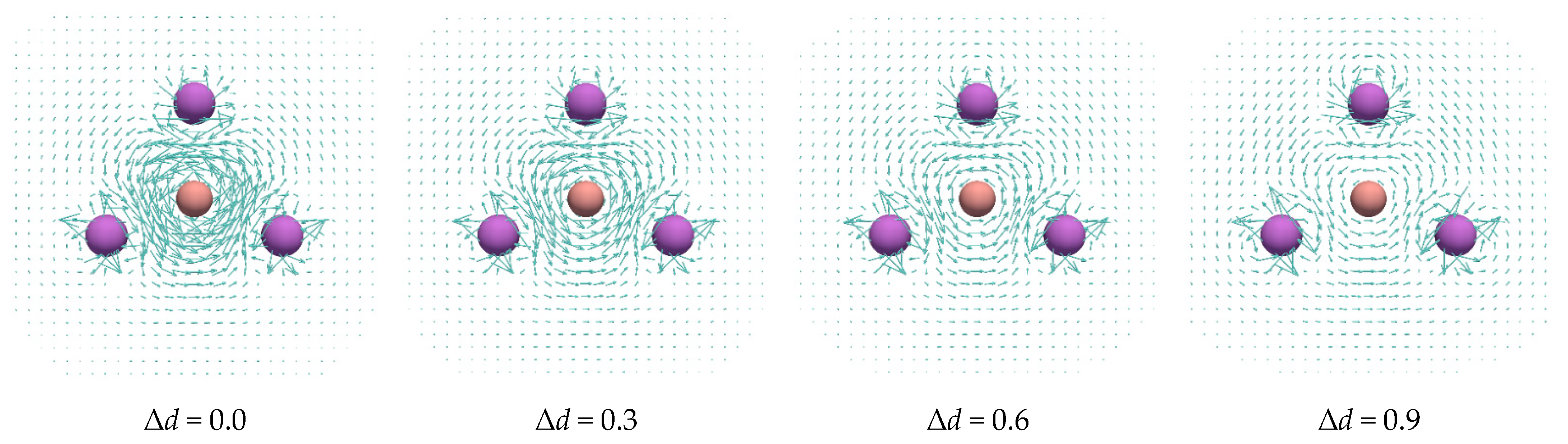
The nature of the current densities induced in the studied complexes was further analyzed by examining structures in which the B–B distances were simultaneously elongated by Δd value, while the Li-nuclei positions were kept frozen. The current density maps of the so-obtained geometries are displayed in Figure 5. As can be seen in and , increasing the B–B distances leads to a continuous weakening of the current density induced inside the Li-atom rings. Table 3 contains natural atomic charges of B atoms, as well as aromaticity indices of Li-atom rings obtained for the structures with the modified B–B distances. As can be seen, the charge of B atoms changes from around −1, at the equilibrium distance, to approximately −1/2 at the dissociation limit. In other words, the unit changes the charge from −2 to −1. While the values show the same trend as the current densities and NBO charges, the magnitudes do not change regularly with increasing distance between B atoms in the studied complexes. The also confirms that the B–B bond elongation reduces the intensity of cyclic delocalization in the two clusters. This implies that both and current density give the same prediction on the aromatic character of the systems obtained by the B–B bond elongation in the examined and clusters. The energetic data from Table 1 show that the dissociation of the studied complexes in and fragments is thermodynamically preferred over the dissociation in and fragments.
Current density maps are mainly used as a qualitative tool, and for quantitative purposes, the bond current strengths can be utilized. Table 4 presents bond current strengths for the studied molecules. The and complexes sustain strong diatropic current densities, with the respective bond current strengths of 8.81/8.57 and 8.11 nA T−1. For comparison, the C–C bond current strength for benzene at the same level of theory is 4.75 nA T−1. The obtained results show that the magnetic properties of the systems are substantially modulated with the introduction of the unit.
Within the ipsocentric method, total current density can be partitioned into molecular orbital contributions. The HOMO electrons in and contribute to a rather weak circulation that occurs in the very center of the ring (Figure 3d and Figure 4d). On the other hand, the quasi-degenerate HOMO-1 level in and the doubly degenerate HOMO-1 level in give the most important contribution to the total current density (Figure 3e and Figure 4e).
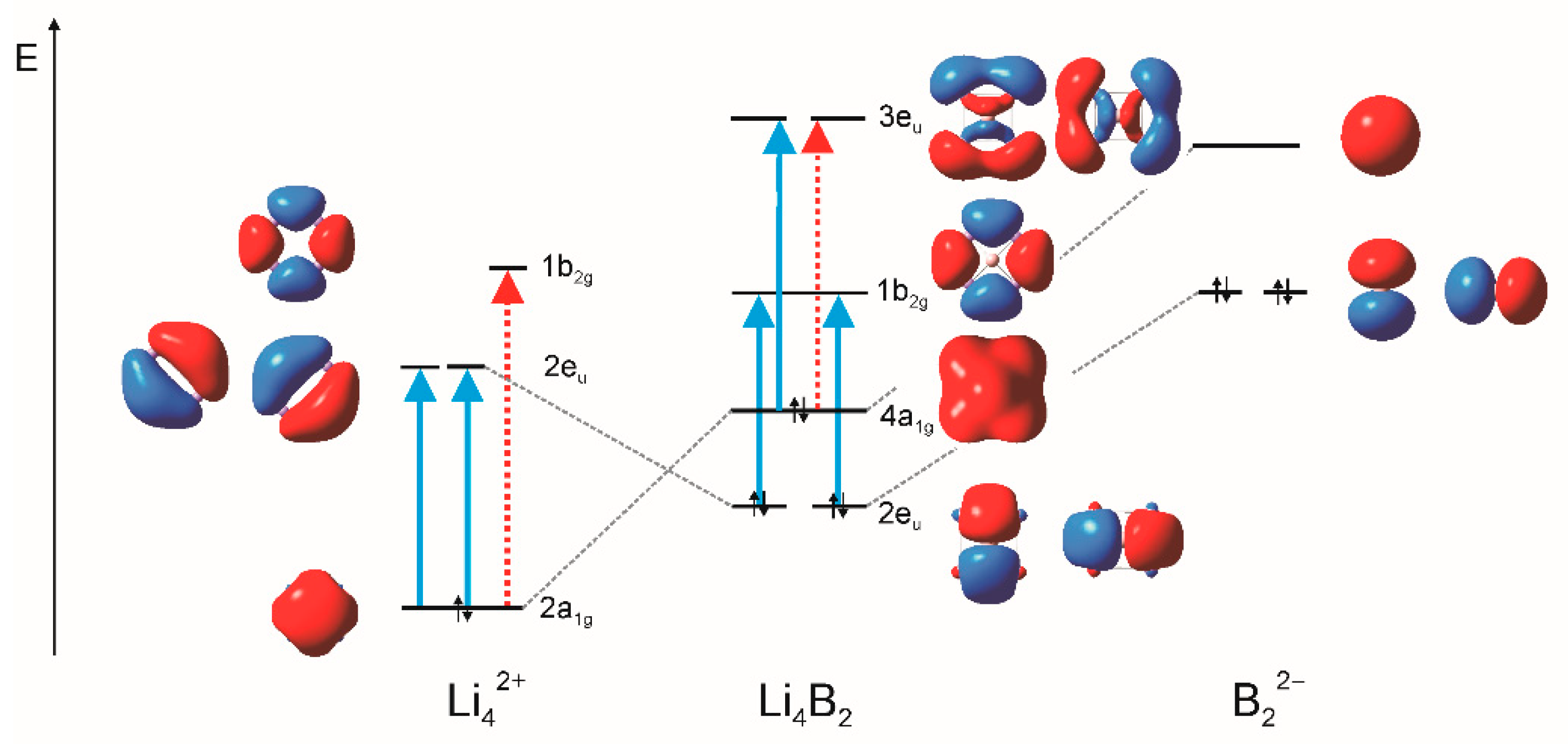
Relevant subsections of the orbital interaction diagrams of the and fragment valence orbitals are shown in Figure 6 and Figure 7. The HOMO () in is formed by mixing HOMO of and LUMO of the fragment. The quasi-degenerate HOMO-1 level, consisting of orbitals, comes as a result of the combination of LUMO of the fragment and the degenerate HOMO-1 level of the fragment. The same bonding situation was found in (Figure 7).
The CTOCD-DZ approach provides a unique interpretative capability, as the orbital current density contributions can be related to the virtual transitions between occupied and virtual orbitals. The virtual transitions must obey symmetry-based selection rules [20,21]. If the product of symmetries of the occupied and virtual orbitals contains the in-plane translation/rotation, then the given virtual excitation contributes to diatropic/paratropic current density. As already shown [23], the HOMO current density rather weakly contributes to the global currents in because the effect of the translational transition from HOMO to LUMO is annulled by the rotational transition to the virtual orbital (Figure 6). In , the HOMO electrons give small contribution to the global circulation. This is because the translational transition from HOMO to LUMO is accompanied by the rotational transition to the orbital. There are translational transitions from the quasi-doubly degenerate HOMO-1 level to the quasi-doubly degenerate virtual orbital level . These transitions are of the most importance for the global circulation in the cluster.
A very similar relation between the found magnetic properties and electronic structure can be found in the system. Figure 7 demonstrates that the HOMO electrons in induce weak currents because of the two supposed transitions: translational and rotational from HOMO ) to LUMO . This way, the HOMO current density contribution is quite small. On the other hand, the translational transition from the doubly degenerate HOMO-1 level to the virtual level gives rise to the strong currents induced by the HOMO-1 electrons.
4. Conclusions
The present paper shows how the unit can be used for the rational structural modifications of cyclic Li-based clusters, which can cause substantial changes in their aromatic characteristics. The magnetically induced current densities, as the most reliable magnetic criterion, revealed the aromatic character of and clusters. The orbital analysis showed that in both and , the main contribution to the global currents comes from the doubly degenerate HOMO-1 level, which can be deduced from the combination of the LUMO of the fragment and the doubly degenerate π-MO level of the fragment. Quite surprisingly, the MCI values predict a low level of electron delocalization in and clusters and, on the other hand, intensive electron delocalization in the reference systems. It is difficult to understand such a difference between the current-density- and electron-delocalization-based aromaticity predictions, and its rationalization will remain a task for the future.
Author Contributions
Conceptualization, S.R.; methodology, S.R.; software, S.R.; investigation, S.Đ.; writing—original draft preparation, S.Đ.; writing—review and editing, S.R.; visualization, S.Đ. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Serbian Ministry of Education, Science and Technological Development (Agreement No. 451-03-68/2020-14/200122).
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
The authors would like to thank Dušan Ćoćić and Ralph Puchta for helping us in calculations of CMO-NICS indices. We would like to thank Tim Clark and Wolfgang Hieringer for hosting these calculations at the CCC. The authors gratefully acknowledge the Regionales Rechenzentrum Erlangen (RRZE) for a generous allotment of computer time.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cyrański, M.K. Energetic Aspects of Cyclic π-Electron Delocalization: Evaluation of the Methods of Estimating Aromatic Stabilization Energies. Chem. Rev. 2005, 105, 3773–3811. [Google Scholar] [CrossRef] [PubMed]
- Gershoni-Poranne, R.; Stanger, A. Magnetic Criteria of Aromaticity. Chem. Soc. Rev. 2015, 44, 6597–6615. [Google Scholar] [CrossRef] [PubMed]
- Feixas, F.; Matito, E.; Poater, J.; Solà, M. Quantifying Aromaticity with Electron Delocalisation Measures. Chem. Soc. Rev. 2015, 44, 6434–6451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazzeretti, P. Ring Currents. Prog. Nucl. Magn. Reson. Spectrosc. 2000, 36, 1–88. [Google Scholar] [CrossRef]
- Gomes, J.A.N.F.; Mallion, R.B. Aromaticity and Ring Currents. Chem. Rev. 2001, 101, 1349–1384. [Google Scholar] [CrossRef] [PubMed]
- London, F. Théorie Quantique des Courants Interatomiques dans les Combinaisons Aromatiques. J. Phys. Radium 1937, 8, 397–409. [Google Scholar] [CrossRef] [Green Version]
- Geuenich, D.; Hess, K.; Köhler, F.; Herges, R. Anisotropy of the Induced Current Density (ACID), a General Method to Quantify and Visualize Electronic Delocalization. Chem. Rev. 2005, 105, 3758–3772. [Google Scholar] [CrossRef]
- Sundholm, D.; Fliegl, H.; Berger, R.J.F. Calculations of Magnetically Induced Current Densities: Theory and Applications. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2016, 6, 639–678. [Google Scholar] [CrossRef]
- Schleyer, P.v.R.; Maerker, C.; Dransfeld, A.; Jiao, H.; Hommes, N.J.v.R.E. Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. [Google Scholar] [CrossRef]
- Lazzeretti, P. Assessment of Aromaticity via Molecular Response Properties. Phys. Chem. Chem. Phys. 2004, 6, 217–223. [Google Scholar] [CrossRef]
- Van Damme, S.; Acke, G.; Havenith, R.W.A.; Bultinck, P. Can the Current Density Map Topology Be Extracted from the Nucleus Independent Chemical Shifts? Phys. Chem. Chem. Phys. 2016, 18, 11746–11755. [Google Scholar] [CrossRef]
- Jusélius, J.; Sundholm, D.; Gauss, J. Calculation of Current Densities Using Gauge-Including Atomic Orbitals. J. Chem. Phys. 2004, 121, 3952–3963. [Google Scholar] [CrossRef]
- Keith, T.A.; Bader, R.F.W. Calculation of Magnetic Response Properties Using a Continuous Set of Gauge Transformations. Chem. Phys. Lett. 1993, 210, 223–231. [Google Scholar] [CrossRef]
- Keith, T.A.; Bader, R.F.W. Topological Analysis of Magnetically Induced Molecular Current Distributions. J. Chem. Phys. 1993, 99, 3669–3682. [Google Scholar] [CrossRef]
- Lazzeretti, P.; Malagoli, M.; Zanasi, R. Computational Approach to Molecular Magnetic Properties by Continuous Transformation of the Origin of the Current Density. Chem. Phys. Lett. 1994, 220, 299–304. [Google Scholar] [CrossRef]
- Lazzeretti, P. Methods of Continuous Translation of the Origin of the Current Density Revisited. Theor. Chem. Acc. 2012, 131, 1222. [Google Scholar] [CrossRef]
- Zanasi, R. Coupled Hartree-Fock Calculations of Molecular Magnetic Properties Annihilating the Transverse Paramagnetic Current Density. J. Chem. Phys. 1996, 105, 1460–1469. [Google Scholar] [CrossRef]
- Lazzeretti, P.; Zanasi, R. SYSMO Package; University of Modena: Modena, Italy, 1980. [Google Scholar]
- Monaco, G.; Summa, F.F.; Zanasi, R. Program Package for the Calculation of Origin-Independent Electron Current Density and Derived Magnetic Properties in Molecular Systems. J. Chem. Inf. Model. 2021, 61, 270–283. [Google Scholar] [CrossRef]
- Steiner, E.; Fowler, P.W. Patterns of Ring Currents in Conjugated Molecules: A Few-Electron Model Based on Orbital Contributions. J. Phys. Chem. A 2001, 105, 9553–9562. [Google Scholar] [CrossRef]
- Steiner, E.; Fowler, P.W. Four- and Two-Electron Rules for Diatropic and Paratropic Ring Currents in Monocyclic π Systems. Chem. Commun. 2001, 2220–2221. [Google Scholar] [CrossRef]
- Soncini, A.; Teale, A.M.; Helgaker, T.; De Proft, F.; Tozer, D.J. Maps of Current Density Using Density-Functional Methods. J. Chem. Phys. 2008, 129, 74101. [Google Scholar] [CrossRef] [PubMed]
- Havenith, R.W.A.; De Proft, F.; Fowler, P.W.; Geerlings, P. σ-Aromaticity in H3+ and Li3+: Insights from Ring-Current Maps. Chem. Phys. Lett. 2005, 407, 391–396. [Google Scholar] [CrossRef]
- Badri, Z.; Pathak, S.; Fliegl, H.; Rashidi-Ranjbar, P.; Bast, R.; Marek, R.; Foroutan-Nejad, C.; Ruud, K. All-Metal Aromaticity: Revisiting the Ring Current Model among Transition Metal Clusters. J. Chem. Theory Comput. 2013, 9, 4789–4796. [Google Scholar] [CrossRef] [PubMed]
- Foroutan-Nejad, C. Is NICS a Reliable Aromaticity Index for Transition Metal Clusters? Theor. Chem. Acc. 2015, 134, 1–9. [Google Scholar] [CrossRef]
- Foroutan-Nejad, C.; Vícha, J.; Ghosh, A. Relativity or Aromaticity? A First-Principles Perspective of Chemical Shifts in Osmabenzene and Osmapentalene Derivatives. Phys. Chem. Chem. Phys. 2020, 22, 10863–10869. [Google Scholar] [CrossRef] [PubMed]
- Radenković, S.; Bultinck, P. Ring Currents in Polycyclic Sodium Clusters. J. Phys. Chem. A 2011, 115, 12493–12502. [Google Scholar] [CrossRef] [Green Version]
- Kalita, A.; Rohman, S.; Kashyap, C.; Ullah, S.; Baruah, I.; Marumder, L.; Das, K.; Guha, A. Boron-Boron Quadruple Bond in Li3B2− and Li4B2 Clusters. Phys. Chem. Chem. Phys. 2021. [Google Scholar] [CrossRef]
- Zhang, X.; Popov, I.A.; Lundell, K.A.; Wang, H.; Mu, C.; Wang, W.; Schnöckel, H.; Boldyrev, A.I.; Bowen, K.H. Realization of an Al≡Al Triple Bond in the Gas-Phase Na3Al2− Cluster via Double Electronic Transmutation. Angew. Chem. Int. Ed. 2018, 57, 14060–14064. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Foster, J.P.; Weinhold, F. Natural Hybrid Orbitals. J. Am. Chem. Soc. 1980, 102, 7211–7218. [Google Scholar] [CrossRef]
- Ayachit, U. The ParaView Guide: A Parallel Visualization Application; Kitware: New York, NY, USA, 2015; ISBN 978-1930934306. [Google Scholar]
- Irons, T.J.P.; Spence, L.; David, G.; Speake, B.T.; Helgaker, T.; Teale, A.M. Analyzing Magnetically Induced Currents in Molecular Systems Using Current-Density-Functional Theory. J. Phys. Chem. A 2020, 124, 1321–1333. [Google Scholar] [CrossRef]
- Elhay, S.; Kautsky, J. Algorithm 655: IQPACK: FORTRAN Subroutines for the Weights of Interpolatory Quadratures. ACM Trans. Math. Softw. 1987, 13, 399–415. [Google Scholar] [CrossRef]
- Bochicchio, R.; Ponec, R.; Torre, A.; Lain, L. Multicenter Bonding Within the AIM Theory. Theor. Chem. Acc. 2001, 105, 292–298. [Google Scholar] [CrossRef]
- Heyndrickx, W.; Salvador, P.; Bultinck, P.; Solà, M.; Matito, E. Performance of 3D-Space-Based Atoms-in-Molecules Methods for Electronic Delocalization Aromaticity Indices. J. Comput. Chem. 2011, 32, 386–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bultinck, P.; Rafat, M.; Ponec, R.; Van Gheluwe, B.; Carbó-Dorca, R.; Popelier, P. Electron Delocalization and Aromaticity in Linear Polyacenes: Atoms in Molecules Multicenter Delocalization Index. J. Phys. Chem. A 2006, 110, 7642–7648. [Google Scholar] [CrossRef] [PubMed]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient Implementation of the Gauge-Independent Atomic Orbital Method for NMR Chemical Shift Calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Cheeseman, J.R.; Trucks, G.W.; Keith, T.A.; Frisch, M.J. A Comparison of Models for Calculating Nuclear Magnetic Resonance Shielding Tensors. J. Chem. Phys. 1996, 104, 5497–5509. [Google Scholar] [CrossRef]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. NBO 7.0: New Vistas in Localized and Delocalized Cbonding Theory. J. Comput. Chem. 2019, 40, 2234–2241. [Google Scholar] [CrossRef]
- Ghorai, S.; Jemmis, E.D. A DFT Study on the Stabilization of the B≡B Triple Bond in a Metallaborocycle: Contrasting Electronic Structures of Boron and Carbon Analogues. Chem.–A Eur. J. 2017, 23, 9746–9751. [Google Scholar] [CrossRef]
- Alexandrova, A.; Boldyrev, A. σ-Aromaticity and σ-Antiaromaticity in Alkali Metal and Alkaline Earth Metal Small Clusters. J. Phys. Chem. A 2003, 107, 554–560. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules. Acc. Chem. Res. 1985, 18, 9–15. [Google Scholar] [CrossRef]
Figure 1.
Optimized structures of (a) and (b) obtained at the M06-2X/def2-TZVP level of theory.

Figure 2.
Natural atomic charges of (a) and (b) based on the natural bond orbital (NBO) analysis at the M06-2X/def2-TZVP level.
Figure 2.
Natural atomic charges of (a) and (b) based on the natural bond orbital (NBO) analysis at the M06-2X/def2-TZVP level.

Figure 3.
Current density maps calculated in the plane of the Li-atom ring: (a), (b), HOMO of (d), quasi-doubly degenerate HOMO-1 of (e); current densities plotted in the plane that perpendicularly bisects the B–B bond of the fragment extracted from the optimized geometry of (c).
Figure 3.
Current density maps calculated in the plane of the Li-atom ring: (a), (b), HOMO of (d), quasi-doubly degenerate HOMO-1 of (e); current densities plotted in the plane that perpendicularly bisects the B–B bond of the fragment extracted from the optimized geometry of (c).


Figure 4.
Current density maps calculated in the plane of the Li-atom ring: (a), (b), HOMO of (d), doubly degenerate HOMO-1 of (e); current densities plotted in the plane that perpendicularly bisects the B–B bond of the fragment extracted from the optimized geometry of (c).
Figure 4.
Current density maps calculated in the plane of the Li-atom ring: (a), (b), HOMO of (d), doubly degenerate HOMO-1 of (e); current densities plotted in the plane that perpendicularly bisects the B–B bond of the fragment extracted from the optimized geometry of (c).


Figure 5.
Current density maps of the structures in which, starting from the optimized geometries of (a) and (b), the B–B distances were elongated by Δd (in Å), while the Li-atom positions were kept unchanged.
Figure 5.
Current density maps of the structures in which, starting from the optimized geometries of (a) and (b), the B–B distances were elongated by Δd (in Å), while the Li-atom positions were kept unchanged.


Figure 6.
Orbital interaction diagram of obtained at the M06-2X/def2-TZVP level of theory. Full (blue) arrows represent the main translational transitions, and dashed (red) arrows represent the main rotational transitions.
Figure 6.
Orbital interaction diagram of obtained at the M06-2X/def2-TZVP level of theory. Full (blue) arrows represent the main translational transitions, and dashed (red) arrows represent the main rotational transitions.

Figure 7.
Orbital interaction diagram of obtained at the M06-2X/def2-TZVP level of theory. Full (blue) arrows represent the main translational transitions, and dashed (red) arrows represent the main rotational transitions.
Figure 7.
Orbital interaction diagram of obtained at the M06-2X/def2-TZVP level of theory. Full (blue) arrows represent the main translational transitions, and dashed (red) arrows represent the main rotational transitions.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Dissociation energies (DE, in kcal/mol) at the M06-2X/def2-TZVP level of theory.
| Dissociation Path | DE |
|---|---|
| 343.24 | |
| 569.61 | |
| 233.26 | |
| 358.96 | |
| −18.92 |
Table 2.
, , and CMO- values of the Li-atom rings in the studied systems. The results obtained with the ma-def2-TZVP basis set are given in the brackets.
Table 2.
, , and CMO- values of the Li-atom rings in the studied systems. The results obtained with the ma-def2-TZVP basis set are given in the brackets.
| Molecule | ||||
|---|---|---|---|---|
| 0.4214 (0.4012) | −54.62 (−53.58) | −45.48 | −6.2 | |
| 0.3176 (0.3081) | −46.21 (−46.50) | −36.17 | −6.64 | |
| 0.7631 (0.7631) | −9.00 (−9.06) | −11.47 | - | |
| 0.6575 (0.6575) | −9.59 (−9.73) | −11.08 | - |
Table 3.
Natural atomic charges of B atoms, , and values of the structures in which the B–B distances were simultaneously elongated by Δd value, while the Li-nuclei positions were kept unchanged.
Table 3.
Natural atomic charges of B atoms, , and values of the structures in which the B–B distances were simultaneously elongated by Δd value, while the Li-nuclei positions were kept unchanged.
| Δd | Charge of B | |||||
|---|---|---|---|---|---|---|
| 0.0 | −1.175 | −1.265 | −54.62 | −46.21 | 0.4213 | 0.3178 |
| 0.3 | −1.179 | −1.265 | −49.89 | −46.03 | 0.2581 | 0.3146 |
| 0.6 | −1.161 | −1.148 | −40.86 | −36.03 | 0.1613 | 0.1495 |
| 0.9 | −1.089 | −0.947 | −31.87 | −28.46 | 0.2728 | 0.2141 |
| 1.2 | −0.886 | −0.717 | −25.67 | −24.02 | 0.2571 | 0.1968 |
| 1.5 | −0.740 | −0.551 | −22.21 | −22.17 | 0.2444 | 0.1821 |
| 1.8 | −0.621 | −0.414 | −20.16 | −21.45 | 0.2315 | 0.1899 |
| 2.1 | −0.513 | −0.293 | −19.01 | −21.55 | 0.2237 | 0.2258 |
Table 4.
Bond current strengths (in nA T−1) for all symmetry-unique bonds.
| Bond | ||||
|---|---|---|---|---|
| Li1–Li2 | 2.28 | 1.59 | 8.81 | 8.11 |
| Li2–Li3 | 8.57 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Đorđević, S.; Radenković, S. The B2 Structural Motif as a Tool for Modulating Ring Currents in Monocyclic Li Clusters. Chemistry 2021, 3, 1063-1073. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry3030077
AMA Style
Đorđević S, Radenković S. The B2 Structural Motif as a Tool for Modulating Ring Currents in Monocyclic Li Clusters. Chemistry. 2021; 3(3):1063-1073. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry3030077
Chicago/Turabian StyleĐorđević, Slađana, and Slavko Radenković. 2021. "The B2 Structural Motif as a Tool for Modulating Ring Currents in Monocyclic Li Clusters" Chemistry 3, no. 3: 1063-1073. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry3030077