
A Practical Laboratory-Scale Synthesis of All Eight Stereoisomeric Forms of Terpene Linalool Oxide


1
Consiglio Nazionale delle Ricerche (C.N.R.) Istituto di Scienze e Tecnologie Chimiche, Via Mancinelli 7, 20131 Milano, Italy
2
Dipartimento di Scienze Farmaceutiche, Università degli Studi di Milano, 20133 Milano, Italy
*
Author to whom correspondence should be addressed.
Chemistry 2021, 3(4), 1247-1257; https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry3040090
Submission received: 17 September 2021
/
Revised: 11 October 2021
/
Accepted: 12 October 2021
/
Published: 16 October 2021
(This article belongs to the Special Issue Flavors and Fragrances: Biology, Chemistry and Biotechnology)
Abstract
:In this work, we describe a user-friendly procedure for the preparation of all the isomeric forms of the terpene linalool oxide. The presented method is based on the transformation of the linalool enantiomers into the corresponding diastereoisomeric mixtures of the two furanoid oxides and two pyranoid oxides. Taking advantage of the different steric hindrance of the hydroxyl functional groups, the pyranoid forms were separated as a diastereoisomeric mixtures of their benzoate esters. Conversely, the cis- and trans-furanoid isomers were transformed in the corresponding acetates, which were directly separated by chromatography. The hydrolysis of the latter esters afforded cis- and trans-furanoid linalool oxides whereas the same reaction performed on the benzoates mixture afforded a separable mixture of cis- and trans-pyranoid linalool oxide. Overall, the method features, as a unique mandatory requirement, the availability of both linalool enantiomers, and can be conveniently performed from a milligram to a multigram scale.
1. Introduction
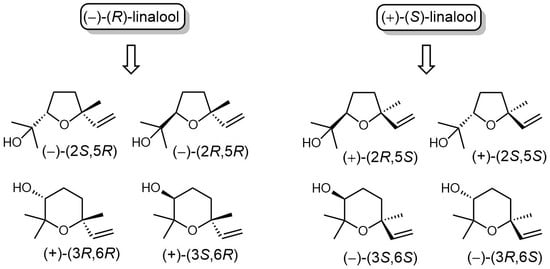
All eight ether stereoisomers with general structure 1 and 2 (Figure 1) are naturally occurring monoterpenes, collectively called linalool oxides [1]. They comprise the two tetrahydrofuran diastereoisomers 1a and 1b (cis and trans furanoid linalool oxides isomers) or tetrahydropyran derivatives 2a and 2b (cis and trans pyranoid linalool oxides isomers). Most likely, the biosynthesis of these compounds proceeds through the regioselective mono-epoxidation of the linalool trisubstituted double bond, followed by the intramolecular cyclization of the obtained epoxy-alcohol [2]. Due to the natural occurrence of both (R)- and (S)-linalool [3] as precursors, this mechanism justifies the formation of both furanoid and pyranoid isomers, as well as the presence in nature of all their possible stereoisomeric forms. Despite the differences in structure, the aromas of the different forms of linalool oxide have a strong family resemblance [4]. The furan-based linalool oxide has a floral character, somewhat reminiscent of lavender, but this is dominated by the profile of black tea. The pyran-based linalool oxide is more obviously floral, with less resemblance to tea. All these monoterpenes are important flavours and are thought to give a relevant contribute to the scent of different foods and beverages of vegetal origin such as grape [5], papaya [2,6], wine [7], and tea [8] where they are often present as glycosides derivatives [9,10] or less frequently as acetate [11]. In addition, these monoterpene isomers are important chiral building blocks for the synthesis of different bioactive natural products [12,13,14,15,16,17].
As we are involved in a number of studies focused on the stereoselective synthesis of flavors, fragrances and sesquiterpenes structurally related to the linalool and linalool oxides [18,19,20,21], we became interested in developing a synthetic method for the preparation of all the linalool oxide stereoisomers. Although a number of chemical [12,14,22,23,24,25,26,27,28,29,30] and chemoenzymatic syntheses [31,32,33,34,35] of enantioenriched linalool oxides have been previously reported, only a few [22,23,24] are suitable for the stereoselective preparation of all the isomeric forms of these monoterpenes. Among the latter three approaches, Klein et Al. [22] described the most direct one. Accordingly, a given linalool enantiomer was treated with peracetic acid to afford a diastereoisomeric mixture of the enantiopure furanoid and pyranoid linalool oxides. Epoxidation and cyclization steps occurred consecutively, in one pot. The first reaction was highly regioselective and the diastereoisomeric mixture of the trisubstituted epoxides was formed preferentially. Then, the acid-catalyzed cyclization step provided furanoid and pyranoid oxides in a 82:18 ratio.
Even if the above procedure appears very simple and provides all the isomers at once, their chromatographic separation is troublesome. Indeed, the linalool oxide isomers required separation by means of a two steps procedure based on fractional distillation. Accordingly, distillation of the crude reaction mixture allowed the separation of pyranoid oxides whereas furanoid oxides had to be converted into the corresponding acetates before fractionation. Moreover, as reported by Klein et al., their procedure was effective on a large scale only, i.e., starting from five moles of linalool. On the contrary, this technique is not conveniently performable at laboratory scale where only few grams of the pure linalool oxide stereoisomers are usually required either as synthetic building blocks or as analytic standards.
Hence, in this work, we describe a user-friendly procedure for the preparation of the linalool oxide isomers starting from linalool enantiomers. Our proposed method allows the separation of all linalool oxide forms from their crude mixtures available by Klein’s process. To accomplish our method, we take advantage of two additional key steps. The first step consisted of the chemoselective benzoylation of the secondary alcohol functional group of the pyranoid oxides 2a and 2b only followed by the chromatographic separation of these esters from the unreacted furanoid oxides. The second step is the transformation of the furanoid isomers 1a and 1b into their corresponding acetate esters, which are separable by chromatography. Finally, the removal of the acetate functional group afforded cis- and trans-furanoid linalool oxides whereas the hydrolysis of the benzoate esters afforded a mixture of cis- and trans-pyranoid linalool oxide, which were separated by chromatography. Since the cyclization step did not involve racemization [22], the linalool oxide isomers obtained through our procedure possess the same enantiomeric purity of the starting linalool, namely 97% ee for (R)-linalool and 50% ee for (S)-linalool.
2. Materials and Methods
2.1. Materials and General Methods
All moisture and air sensitive reactions were carried out using dry solvents and under a static atmosphere of nitrogen.
All solvents and reagents were of commercial quality.
(−)-(R)-Linalool, extracted from Cinnamomun canphora (L.) and possessing 98% chemical purity by GC and 97% ee by chiral GC, [α= −20.9 (neat), lot MKBR2739V, was purchased from Sigma-Aldrich (St. Louis, MO, USA).
Coriander oil, [α = +8.3 (neat) was purchased from Sigma-Aldrich (lot MKCC6867) and was used as source of (+)-(S)-linalool (91% chemical purity by GC-MS analysis and 50% ee by chiral GC).
2.2. Analytical Methods and Characterization of the Chemical Compounds
1H and 13C-NMR spectra and DEPT experiments (Supplementary Materials): CDCl3 solutions at rt using a Bruker-AC-400 spectrometer (Billerica, MA, USA) at 400, 100 and 100 MHz, respectively; 13C spectra are proton decoupled; chemical shifts in ppm rel to internal SiMe4 (=0 ppm).
TLC: Merk silica gel 60 F254 plates (Merck Millipore, Milan, Italy). Column chromatography: silica gel.
Melting points were measured on a Reichert apparatus, equipped with a Reichert microscope, and are uncorrected.
Optical rotations were measured on a Jasco-DIP-181 digital polarimeter (Jasco, Tokyo, Japan).
Mass spectra were recorded on a Bruker ESQUIRE 3000 PLUS spectrometer (Electrospray Ionization (ESI) detector, Billerica, MA, USA or by GC-MS analyses.
GC-MS analyses: HP-6890 gas chromatograph equipped with a 5973 mass detector, using a HP-5MS column (30 m × 0.25 mm, 0.25 μm film thickness; Hewlett Packard, Palo Alto, CA, USA) with the following temp. program: 60° (1 min), then 6°/min to 150° (held 1 min), then 12°/min to 280° (held 5 min); carrier gas: He; constant flow 1ml/min; split ratio: 1/30; tR given in minutes.
tR(linalool) 9.19, tR(1a) 8.88, tR(1b) 8.50, tR(2a) 11.14, tR(2b) 10.99, tR(3a) 23.90, tR(3b) 23.79, tR(4a) 13.13, tR(4b) 12.71.
2.3. Procedure for the Preparation of the Linalool Oxide Isomers (2S,5R)-1a, (2R,5R)-1b, (3R,6R)-2a and (3S,6R)-2b
2.3.1. Preparation of the Mixture of the Linalool Oxide Isomers 1a, 1b, 2a and 2b from (R)-Linalool
m-Chloroperbenzoic acid (77% w/w, 40 g, 178 mmol) was added portion-wise to a mechanically stirred solution of (−)-(R)-linalool (25 g, 162 mmol, 98% purity, ee > 95%) in CH2Cl2 (150 mL) at 0 °C. The reaction was stirred 2 hours at 0°C, overnight and then was treated with PTSA (1 g, 5.2 mmol) and quenched by the addition of aq. Na2S2O5 (10% w/w, 60 mL). The reaction was stirred for one further hour and then was cooled to 0 °C. The m-chlorobenzoic acid formed was removed by filtration and the liquid phase was concentrated under reduced pressure. The residue was dissolved in diethyl ether (250 mL) and was washed in turn with, aq. NaOH (5% w/v) and brine. Then, the organic phase was dried (Na2SO4) and the solvent was evaporated. The obtained oil was roughly purified by filtration on an alumina column (activated, basic, Brockmann I) eluting with hexane/diethyl ether, to afford the crude mixture of the linalool oxide isomers (26.9 g, 1a = 39%, 1b = 42%, 2a = 10%, 2b = 9%).
2.3.2. Procedure for the Separation of the Linalool Oxide Isomers
The above-described mixture of the linalool oxide isomers was dissolved in a mixture of dry CH2Cl2 (30 mL) and dry pyridine (5 mL). The resulting solution was cooled to 0 °C and benzoyl chloride (5.4 g, 38.4 mmol) was added dropwise under stirring. The reaction was stirred at rt. for 4 hours, then was poured in crushed ice, was extracted with ethyl acetate and the organic phase was washed in turn with, aq. NaHCO3 (saturated solution) and brine. The organic phase was dried (Na2SO4), was concentrated under reduced pressure and the residue was chromatographed using n-hexane/AcOEt (95:5–7:3) as eluent to afford the benzoate esters (6.9 g, 25.2 mmol, pyranoid/furanoid ratio 96:4) and the mixture of the furanoid linalool oxide diastereoisomers (19.6 g, 115 mmol).
The benzoate esters were dissolved in methanol (40 mL) and were heated under reflux with a solution of NaOH (3 g, 75 mmol) in methanol (30 mL). After complete hydrolysis of the esters (TLC monitoring, 3 h), the main part of the solvent was removed under reduced pressure and the reaction was quenched by the addition of crushed ice followed by the extraction with diethyl ether (3 × 70 mL). The combined organic phases were dried (Na2SO4), were concentrated under reduced pressure and the residue was chromatographed using n-hexane/AcOEt (9:1–7:3) as eluent to afford a small amount of furanoid isomers (0.1 g, 0.6 mmol) followed by the pyranose isomers. The latter compounds were eluted in the following order:
cis-linalool oxide (+)-2a (first eluted isomer): 1.65 g (9.7 mmol)
mixture of cis/trans-linalool oxide: 0.95 g (5.6 mmol)
trans-linalool oxide (+)-2b (second eluted isomer): 1.25 g (7.3 mmol)
pyranoid cis-linalool oxide (+)-2a = (3R,6R)-2,2,6-trimethyl-6-vinyltetrahydro-2H-pyran-3-ol
colorless oil which solidified on standing. Mp 95–96 °C; 98% of diastereoisomeric purity and 98% of chemical purity by GC. [α = +3.6 (c 2.3, CH2Cl2), Lit. [25]: [α = +1.9 (c 0.5, CH2Cl2)
1H NMR (400 MHz, CDCl3) δ 5.97 (dd, J = 17.8, 11.1 Hz, 1H), 4.99 (d, J = 17.8 Hz, 1H), 4.98 (d, J = 11.1 Hz, 1H), 3.49–3.38 (m, 1H), 2.12 (dt, J = 13.6, 3.7 Hz, 1H), 1.79–1.51 (m, 3H), 1.42–1.31 (m, 1H), 1.25 (s, 3H), 1.17 (s, 3H), 1.16 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 146.3 (CH), 110.6 (CH2), 75.9 (C), 74.9 (CH), 73.4 (C), 32.5 (CH2), 31.6 (Me), 29.5 (Me), 25.7 (CH2), 20.8 (Me). GC-MS m/z (rel intensity) 155 (M+−Me, 6), 143 (2), 137 (4), 125 (2), 119 (1) 109 (3), 102 (4), 94 (76), 79 (23), 68 (100), 59 (67), 43 (32).
pyranoid trans-linalool oxide (+)-2b = (3S,6R)-2,2,6-trimethyl-6-vinyltetrahydro-2H-pyran-3-ol.
Colorless oil. 93% of diastereoisomeric purity and 96% of chemical purity by GC. [α = +11.4 (c 2.4, CH2Cl2), Lit. [25]: [α = +11.0 (c 0.5, CH2Cl2)
1H NMR (400 MHz, CDCl3) δ 5.94 (dd, J = 17.8, 11.0 Hz, 1H), 5.03 (d, J = 17.8 Hz, 1H), 4.96 (d, J = 11.0 Hz, 1H), 3.42 (br s, 1H), 2.03–1.92 (m, 1H), 1.85–1.66 (m, 4H), 1.24 (s, 3H), 1.22 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 146.8 (CH), 110.4 (CH2), 75.2 (C), 73.5 (C), 71.2 (CH), 30.7 (Me), 27.6 (CH2), 27.2 (Me), 26.3 (Me), 24.2 (CH2). GC-MS m/z (rel intensity) 155 (M+−Me, 10), 143 (3), 137 (4), 125 (2), 119 (1) 109 (4), 102 (4), 94 (74), 79 (21), 68 (100), 59 (61), 43 (30)
The diastereoisomeric mixture of furanoid linalool oxides 1a and 1b (19.6 g, 115 mmol) was heated at reflux, under nitrogen, with acetic anhydride (20 mL, 212 mmol) and anhydrous NaOAc (2 g, 24 mmol). The reaction was stirred until complete transformation of the starting alcohol isomers (3 h, TLC monitoring), then was cooled, and was quenched by addition of water (100 mL). The obtained mixture was extracted with diethyl ether (3 × 100 mL) and the combined organic phases were washed in turn with water, aq. NaHCO3 (saturated solution) and brine. The organic phase was dried (Na2SO4), concentrated under reduced pressure and the residue was chromatographed using n-hexane/Et2O (99:1–8:2) as eluent to afford the following fractions:
furanoid trans-linalool oxide acetate (−)-4b (first eluted isomers, Rf = 0.23 in hexane/diethyl ether 9:1): 5.3 g (25 mmol)
mixture of cis/trans-linalool oxide acetate: 11.8 g (55.7 mmol)
furanoid cis-linalool oxide acetate (+)-4a (second eluted isomers, Rf = 0.19 in hexane/diethyl ether 9:1): 4.4 g (20.7 mmol)
Furanoid cis-linalool oxide acetate (+)-4a = 2-((2S,5R)-5-methyl-5-vinyltetrahydrofuran-2-yl)propan-2-yl acetate. Colorless oil; [α = +0.3 (c 2.8, CHCl3), Lit. [15]: [α = −0.3 (neat)
1H NMR (400 MHz, CDCl3) δ 5.97 (dd, J = 17.5, 10.8 Hz, 1H), 5.22 (dd, J = 17.5, 1.6 Hz, 1H), 4.98 (dd, J = 10.8, 1.6 Hz, 1H), 4.13–4.05 (m, 1H), 2.00–1.71 (m, 4H), 1.98 (s, 3H), 1.49 (s, 3H), 1.48 (s, 3H), 1.31 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 170.4 (C), 144.2 (CH), 111.3 (CH2), 84.1 (CH), 83.1 (C), 82.9 (C), 37.7 (CH2), 26.6 (CH2), 25.9 (Me), 22.5 (Me), 22.4 (Me), 21.8 (Me). GC-MS m/z (rel intensity) 197 (M+−Me, 7), 185 (1), 169 (2), 152 (M+−AcOH, 29), 143 (3), 137 (27), 119 (6), 111 (100), 101 (9), 93 (63), 81 (31), 67 (32), 55 (60)
Furanoid trans-linalool oxide acetate (−)-4b = 2-((2R,5R)-5-methyl-5-vinyltetrahydrofuran-2-yl)propan-2-yl acetate. Colorless oil; [α = −8.2 (c 2.9, CHCl3), Lit. [15]: [α = −5.0 (neat)
1H NMR (400 MHz, CDCl3) δ 5.86 (dd, J = 17.2, 10.6 Hz, 1H), 5.18 (dd, J = 17.2, 1.2 Hz, 1H), 4.99 (dd, J = 10.6, 1.2 Hz, 1H), 4.07 (t, J = 6.5 Hz, 1H), 1.99 (s, 3H), 1.94–1.79 (m, 3H), 1.77–1.67 (m, 1H), 1.48 (s, 6H), 1.32 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 170.4 (C), 143.6 (CH), 111.3 (CH2), 83.9 (CH), 83.3 (C), 83.0 (C), 37.1 (CH2), 26.6 (Me), 26.4 (CH2), 22.4 (Me), 22.4 (Me), 21.5 (Me). GC-MS m/z (rel intensity) 197 (M+−Me, 2), 185 (2), 169 (3), 152 (M+−AcOH, 26), 143 (3), 137 (21), 119 (5), 111 (100), 101 (8), 93 (56), 81 (29), 67 (33), 55 (49)
A sample of the ester (+)-4a or (−)-4b (1 g, 4.7 mmol) was dissolved in dry diethyl ether (30 mL) and treated under stirring with LiAlH4 (0.2 g, 5.3 mmol). As soon as the starting ester was no longer detectable by TLC analysis (1 hour), the reaction was diluted with diethyl ether (70 mL), was quenched with crushed ice and was acidify with aq. HCl (3% w/v). The separated organic phase was dried (Na2SO4), concentrated under reduced pressure and the residue was purified by bulb-to-bulb distillation to afford linalool oxide (−)-1a (0.71 g, 88% yield) or linalool oxide (−)-1b (0.73 g, 91% yield), respectively.
Furanoid cis-linalool oxide (−)-1a = 2-((2S,5R)-5-methyl-5-vinyltetrahydrofuran-2-yl)propan-2-ol. Colorless oil; [α = −4.1 (c 2.1, CHCl3), Lit. [13]: [α = −3.8 (c 0.72, CHCl3)
1H NMR (400 MHz, CDCl3) δ 5.98 (dd, J = 17.5, 10.8 Hz, 1H), 5.19 (dd, J = 17.5, 1.3 Hz, 1H), 5.00 (dd, J = 10.8, 1.3 Hz, 1H), 3.85 (t, J = 7.1, 1H), 2.12–2.04 (m, 1H), 1.96–1.72 (m, 4H), 1.31 (s, 3H), 1.22 (s, 3H), 1.12 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 144.3 (CH), 111.5 (CH2), 85.5 (CH), 82.7 (C), 71.2 (C), 37.9 (CH2), 27.3 (Me), 26.4 (CH2), 26.0 (Me), 24.3 (Me). GC-MS m/z (rel intensity) 155 (M+−Me, 11), 137 (9), 125 (1), 119 (2), 111 (43), 102 (3), 94 (62), 81 (24), 68 (36), 59 (100)
Furanoid trans-linalool oxide (−)-1b = 2-((2R,5R)-5-methyl-5-vinyltetrahydrofuran-2-yl)propan-2-ol. Colorless oil; [α = −6.5 (c 2.4, CHCl3), Lit. [13] for the (2S,5S) enantiomer: [α = +5.7 (c 0.95, CHCl3)
1H NMR (400 MHz, CDCl3) δ 5.86 (dd, J = 17.4, 10.8 Hz, 1H), 5.17 (dd, J = 17.4, 1.5 Hz, 1H), 4.98 (dd, J = 10.8, 1.5 Hz, 1H), 3.78 (t, J = 7.1, 1H), 2.16 (s, 1H), 1.93–1.77 (m, 3H), 1.75–1.66 (m, 1H), 1.30 (s, 3H), 1.21 (s, 3H), 1.12 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 143.7 (CH), 111.3 (CH2), 85.5 (CH), 83.0 (C), 71.1 (C), 37.4 (CH2), 27.1 (Me), 26.8 (Me), 26.3 (CH2), 24.1 (Me). GC-MS m/z (rel intensity) 155 (M+−Me, 6), 137 (7), 125 (2), 111 (39), 102 (3), 94 (55), 81 (23), 68 (34), 59 (100)
2.4. Procedure for the Preparation of the Linalool Oxide Isomers (2R,5S)-1a, (2S,5S)-1b, (3S,6S)-2a and (3R,6S)-2b
The procedure described above (paragraph 2.3) was repeated using (S)-linalool as starting material (obtained by rectification of coriander oil, 50% e.e.). The obtained linalool oxide isomers (2R,5S)-1a, (2S,5S)-1b, (3S,6S)-2a and (3R,6S)-2b showed the same analytical data of their corresponding enantiomers with the exception of the sign and magnitude of their optical rotation values. Below are reported the measured optical rotation values:
(2R,5S)-1a: [α = +1.8 (c 2.1, CHCl3)
(2S,5S)-1b: [α = +3.0 (c 2.4, CHCl3)
(3S,6S)-2a: [α = −1.5 (c 2.3, CH2Cl2)
(3R,6S)-2b: [α = −6.3 (c 2.3, CH2Cl2)
3. Results and Discussion
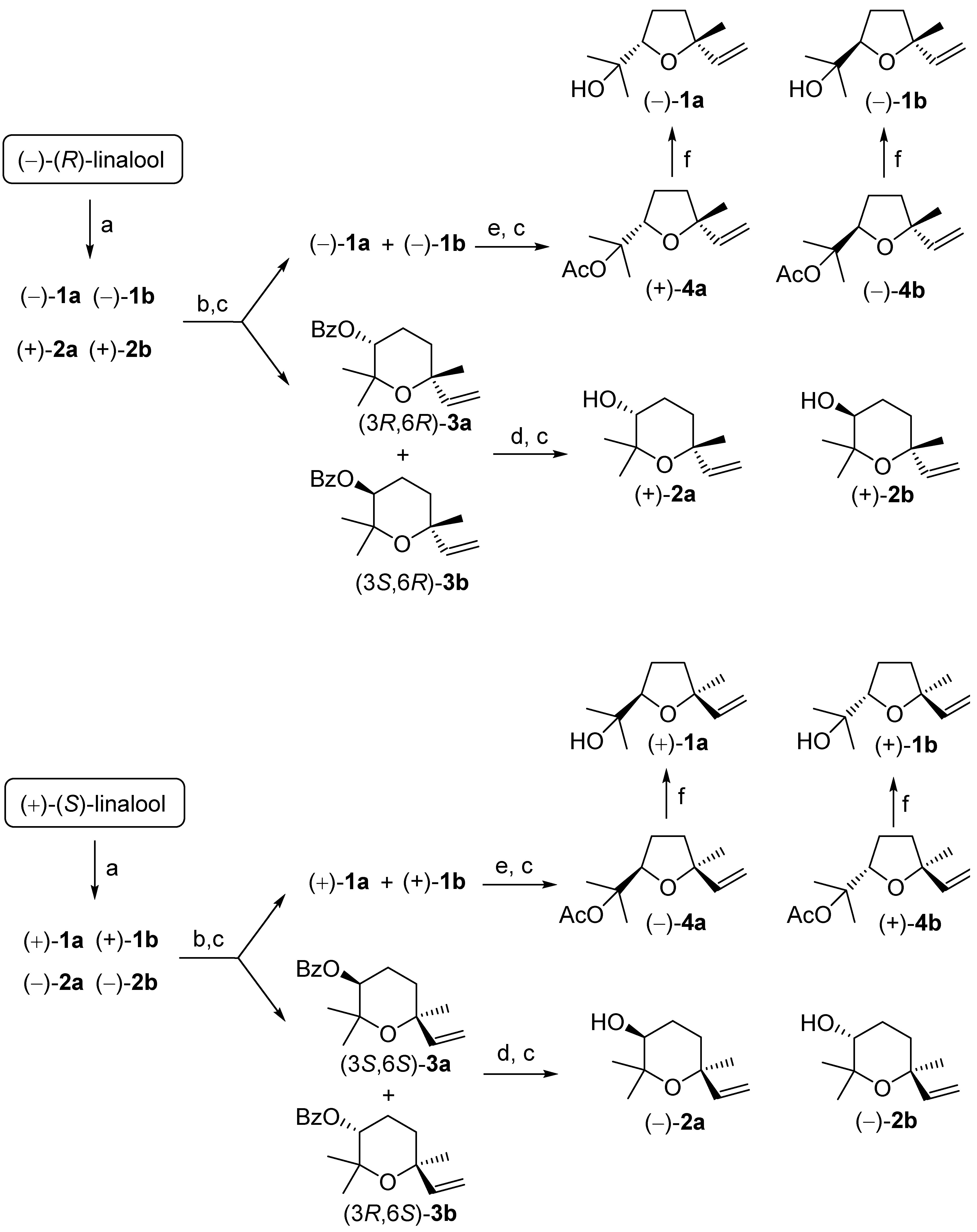
The first step of our procedure followed closely the process reported by Klein et al. [22]. The only difference lies in the use of m-chloroperbenzoic acid (MCPBA) as reagent of choice for linalool epoxidation instead of peracetic acid.
Accordingly, the reaction of (R)-linalool (Scheme 1) with an equimolar amount of MCPBA at 0 °C, afforded the corresponding mono-epoxide derivative. The reaction was highly regioselective since only the trisubstituted double bond was converted in the corresponding epoxide. The latter compound was not isolated because of its high instability to acid environment. In fact, the cyclization reaction started during the epoxidation reaction and proceeded rapidly with the work-up procedure, making unnecessary the epoxide isolation. Therefore, the completion of the cyclization step was warranted by adding to the reaction mixture a catalytic amount of p-toluenesulfonic acid (PTSA) as soon as the starting linalool was completely transformed. The obtained crude linalool oxides mixture consisted of a combination of furanoid linalool oxide isomers (81% of the mixture, of which 1a = 39% and 1b = 42%) and pyranoid isomers (19% of the mixture, of which 2a = 10% and 2b = 9%).
The chromatographic separation of these isomers is very complex. Furanoid linalool oxides 1a and 1b show the same retention factor (Rf) and are not separable.
On the contrary, pyranoid linalool oxides have different Rf but although the cis-isomer 2a is separable from the trans-isomer 2b, their partial co-elution with furanoid oxide isomers result in the difficult isolation of the isomeric pure forms. Overall, the purification of the linalool oxide isomers by direct chromatographic separation is not a feasible procedure.
Therefore, we investigated the possibility of derivatize selectively the oxides 2a and 2b in presence of the oxides 1a and 1b. The obtained new compounds, possibly possessing Rf very different from the starting alcohols, should be easier separable. Then, the removal of the derivatizing moiety should give oxides 2a and 2b free of the furanoid isomers. Following this reasoning, we took advantage of the different steric hindrance of the hydroxyl functional groups present in the linalool oxide framework.
Thus, the secondary alcohol functional group of the pyranoid isomers we expected to be transformed into the corresponding esters easier than the tertiary alcohol present in the furanoid oxides. To this end, we selected benzoyl chloride (BzCl) as derivatizing agent. Indeed, the obtained benzoate esters were stable, easily separable from the linalool oxides mixture and straightforwardly hydrolysable by alkali treatment. Accordingly, we tested the benzoylation reaction using different experimental conditions and employing the crude linalool oxides mixture as substrate (Table 1). It is worth noting that only the 19% of the latter mixture had to be esterified. Therefore, the reaction should proceed with high chemoselectivity.
The addition of an excess of BzCl (three equivalents in relation to the starting amount of pyranoid linalool oxide) and of a catalytic amount of dimethylaminopyridine (DMAP) at room temperature, lead to the complete esterification of the pyranoid oxides although with the concomitant formation of a considerable quantity of furanoid esters (entry 1). Since DMAP strongly catalyzes the acylation reactions, we further ran the reaction in the same experimental conditions but without any DMAP addition. Indeed, with this experiment, we observed that the pyranoid oxides were completely esterified whereas the furanoid esters were yet formed, although in minor amount (entry 2).
In order to increase further the selectivity of this transformation, we reduced both the equivalent of BzCl (1.2) and the temperature (0 °C). These new experimental conditions (entry 3) allowed the formation of the pyranoid benzoate ester in high yield (96% of conversion) and with very high chemoselectivity (the formed esters contained only 3% of furanoid isomers).
Taking advantage of the latter successful derivatization procedure, we checked its applicability for the large-scale separation of the linalool oxide isomers (150 mmol scale). Accordingly, (−)-(R)-linalool was treated with mCPBA and was transformed in the mixture of the linalool oxide isomers (−)-1a, (−)-1b, (+)-2a and (+)-2b (Scheme 1). The following reaction with BzCl, using the previously selected experimental conditions, afforded an equimolar mixture of the benzoate (3R,6R)-3a and (3S,6R)-3b, which was easily separated from the furanoid isomers (−)-1a, (−)-1b by chromatography. Hence, the purified esters were hydrolyzed using NaOH in methanol and the obtained linalool oxides were chromatographed to afford cis-pyranoid isomer (+)-2a (first eluted isomer) and trans-pyranoid isomers (+)-2b (second eluted isomer).
As mentioned before, the diastereoisomers (−)-1a and (−)-1b could not be separated by chromatography because they possess the same retention factor (Rf). This problem has been previously resolved through transformation of the cis- and trans-furanoid oxides mixture in the corresponding acetate esters followed by fractional distillation [15,22]. In our work, we observed that these diastereomerically related acetates, 4a and 4b (Scheme 1) can be, however, separated in pure stereoisomeric form by chromatographic separation.
Accordingly, we treated the mixture of compounds (−)-1a and (−)-1b with acetic anhydride in the presence of sodium acetate to give the expected acetates (+)-4a and (−)-4b in almost quantitative yield. The subsequent chromatographic separation afforded isomers (−)-4b (first eluted) and (+)-4a (second eluted), both in diastereoisomerically pure form. Finally, the cleavage of the acetate functional group was performed by reduction using LiAlH4, to give pure trans- and cis-furanoid linalool oxides (−)-1b and (−)-1a, respectively.
Overall, we obtained the pyranoid isomers (3R,6R)-2a, (3S,6R)-2b and the furanoid isomers (2S,5R)-1a, (2R,5R)-1b in isomerically pure forms. Since our synthesis starts from (R)-linalool, all these compounds possess configuration R in position 6 (for tetrahydropyrane ring) or position 5 (for tetrahydrofuran ring) of their framework. In order to prepare the lacking four linalool oxides enantiomers, we applied the same reaction sequence starting from (S)-linalool. The linalool enantiomers are available from many plant essential oils where they are present in different enantiomeric purity. Differently from (R)-linalool, which occurred in rosewood and in ho leaf in almost enantiopure form, the most relevant source of (S)-linalool is coriander oil, where the enantiomeric excess of the terpene is rather modest (40–60%).
Thus, we purified commercially available coriander oil and the obtained (S)-linalool, possessing about 50% ee. was employed for the synthesis of the linalool oxide isomers. Accordingly, the application of our procedure afforded the pyranoid isomers (3S,6S)-(−)-2a, (3R,6S)-(−)-2b and the furanoid isomers (2R,5S)-(+)-1a, (2S,5S)-(+)-1b, which showed optical rotation values of opposite sign and inferior magnitude of the corresponding enantiomeric forms.
In conclusion, we present a user-friendly procedure for the preparation of all the isomeric forms of the terpene linalool oxide. The method features, as a unique mandatory requirement, the availability of both linalool enantiomers and can be conveniently performed from a milligram to a multigram scale. The linalool oxide isomers obtained through our procedure possess the same enantiomeric purity of the starting linalool and could be employed both as chiral building blocks for stereoselective synthesis and as reference standards in F&F formulation and essential oils analysis.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/chemistry3040090/s1. Study of the benzoylation of the crude mixture of the linalool oxide isomers obtained from linalool; Figure S1: 1H-NMR of the cis linalool oxide (pyranoid); Figure S2: 13C-NMR of the cis linalool oxide (pyranoid); Figure S3: DEPT experiment of the cis linalool oxide (pyranoid); Figure S4: 1H-NMR of the trans linalool oxide (pyranoid); Figure S5: 13C-NMR of the trans linalool oxide (pyranoid); Figure S6: DEPT experiment of the trans linalool oxide (pyranoid); Figure S7: 1H-NMR of the cis linalool oxide (furanoid); Figure S8: 13C-NMR of the cis linalool oxide (furanoid); Figure S9: DEPT experiment of the cis linalool oxide (furanoid); Figure S10: 1H-NMR of the trans linalool oxide (furanoid); Figure S11: 13C-NMR of the trans linalool oxide (furanoid); Figure S12: DEPT experiment of the trans linalool oxide (furanoid); Figure S13: 1H-NMR of the cis linalool oxide acetate (furanoid); Figure S14: 13C-NMR of the cis linalool oxide acetate (furanoid); Figure S15: DEPT experiment of the cis linalool oxide acetate (furanoid); Figure S16: 1H-NMR of the trans linalool oxide acetate (furanoid); Figure S17: 13C-NMR of the trans linalool oxide acetate (furanoid); Figure S18: DEPT experiment of the trans linalool oxide acetate (furanoid).
Author Contributions
Conceptualization, methodology and validation, S.S.; investigation and data curation, S.S., D.D.S. and S.P.; writing, S.S.; review and editing, S.S. and D.D.S.; funding acquisition, S.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Regione Lombardia within the project POR-FESR 2014-2020 n° 228775 VIPCAT (Value Added Innovative Protocols for Catalytic Transformations).
Data Availability Statement
Data is contained within the article or supplementary material.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bauer, K.; Garbe, D.; Surburg, H. Common Fragrance and Flavor Materials, 4th ed.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2001; p. 145. [Google Scholar]
- Winterhalter, P.; Katzenberger, D.; Schreier, P. 6,7-Epoxy-linalool and related oxygenated terpenoids from Carica papaya fruit. Phytochemistry 1986, 25, 1347–1350. [Google Scholar] [CrossRef]
- Aprotosoaie, A.C.; Hăncianu, M.; Costache, I.-I.; Miron, A. Linalool: A review on a key odorant molecule with valuable biological properties. Flavour Fragr. J. 2014, 29, 193–219. [Google Scholar] [CrossRef]
- Wright, J. Linalool oxide. Perfum. Flavorist 2013, 38, 18–19. [Google Scholar]
- Stevens, K.L.; Bomben, J.; Lee, A.; McFadden, W.H. Volatiles from grapes. Muscat of Alexandria. J. Agric. Food Chem. 1966, 14, 249–252. [Google Scholar] [CrossRef]
- Flath, R.A.; Forrey, R.R. Volatile components of papaya (Carica papaya L., Solo variety). J. Agric. Food Chem. 1977, 25, 103–109. [Google Scholar] [CrossRef]
- Williams, P.J.; Strauss, C.R.; Wilson, B. New linalool derivatives in muscat of Alexandria grapes and wines. Phytochemistry 1980, 19, 1137–1139. [Google Scholar] [CrossRef]
- Wang, D.; Ando, K.; Morita, K.; Kubota, K.; Kobayashi, A. Optical isomers of linalool and linalool oxides in tea aroma. Biosci. Biotech. Biochem. 1994, 58, 2050–2053. [Google Scholar] [CrossRef] [Green Version]
- Moon, J.-H.; Watanabe, N.; Ijima, Y.; Yagi, A.; Sakata, K. Cis- and trans-linalool 3,7-oxides and methyl salicylate glycosides and (Z)-3-hexenyl β-D-glucopyranoside as aroma precursors from tea leaves for oolong tea. Biosci. Biotech. Biochem. 1996, 60, 1815–1819. [Google Scholar] [CrossRef]
- Jiang, L.; Kojima, H.; Yamada, K.; Kobayashi, A.; Kubota, K. Isolation of some glycosides as aroma precursors in young leaves of japanese pepper (Xanthoxylum piperitum DC.). J. Agric. Food Chem. 2001, 49, 5888–5894. [Google Scholar] [CrossRef] [PubMed]
- Chanotiya, C.S.; Sammal, S.S.; Mathela, C.S. Composition of a new chemotype of Tanacetum nubigenum. Indian J. Chem. B 2005, 44, 1922–1926. [Google Scholar] [CrossRef]
- Gajula, S.; Madhu, M.; Chintakrinda, S.K.; Yadav, J.S.; Mohapatra, D.K. A protecting-group-free synthesis of arbusculone, andirolactone, pinnatolide, ipomolactone, cyclocapitelline and isocyclocapitelline. Tetrahedron Lett. 2018, 59, 4172–4175. [Google Scholar] [CrossRef]
- Daub, M.E.; Prudhomme, J.; Ben Mamoun, C.; Le Roch, K.G.; Vanderwal, C.D. Antimalarial properties of simplified kalihinol analogues. ACS Med. Chem. Lett. 2017, 8, 355–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartung, J.; Drees, S.; Geiss, B.; Schmidt, P. Vanadium(V)-catalyzed oxidation of (3R)-linalool—The selective formation of furanoid linalool oxides and their conversion into isocyclocapitelline derivatives. Synlett 2003, 223–226. [Google Scholar] [CrossRef]
- Mahboobi, S.; Koller, M.; Schollmeyer, D. Synthesis of the racemates of the β-carboline alkaloid chrysotricine and its diastereomer. Monatshefte Chem. 2000, 131, 383–392. [Google Scholar] [CrossRef]
- Díez, D.; Moro, R.F.; Marcos, I.S.; Sánchez López, J.; Urones, J.G. Synthesis of an analog of usneoidol E. Part II. Synlett 2000, 794–796. [Google Scholar] [CrossRef]
- Urones, J.G.; Díez, D.; Marcos, I.S.; Basabe, P.; Lithgow, A.M.; Moro, R.F.; Garrido, N.M.; Escarcena, R. The use of acyclic monoterpenes in the preparation of β-pyrones: Synthesis of the right-hand fragment of usneoidone E. Tetrahedron 1995, 51, 3691–3704. [Google Scholar] [CrossRef]
- Serra, S.; De Simeis, D. A study on the lipase-catalysed acylation of 6,7-dihydroxy-linalool. Nat. Prod. Commun. 2016, 11, 1217–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serra, S.; De Simeis, D.; Brenna, E. Lipase mediated resolution of cis- and trans-linalool oxide (pyranoid). J. Mol. Catal. B: Enzym. 2016, 133, S420–S425. [Google Scholar] [CrossRef]
- Serra, S.; De Simeis, D. Two complementary synthetic approaches to the enantiomeric forms of the chiral building block (2,6,6-trimethyltetrahydro-2H-pyran-2-yl)methanol: Application to the stereospecific preparation of the natural flavor linaloyl oxide. Catalysts 2018, 8, 362. [Google Scholar] [CrossRef] [Green Version]
- Serra, S. Bisabolane sesquiterpenes: Synthesis of (R)-(+)-sydowic acid and (R)-(+)curcumene ether. Synlett 2000, 2000, 890–892. [Google Scholar] [CrossRef]
- Klein, E.; Farnow, H.; Rojahn, W. Die chemie der linalool-oxide. Justus Liebigs Ann. Chem. 1964, 675, 73–82. [Google Scholar] [CrossRef]
- Méou, A.; Bouanah, N.; Archelas, A.; Zhang, X.M.; Guglielmetti, R.; Furstoss, R. Synthesis of all four stereoisomers of enantiomerically pure cis- and trans-linalyl oxides. Synthesis 1990, 1990, 752–753. [Google Scholar] [CrossRef]
- Méou, A.; Bouanah, N.; Archelas, A.; Zhang, X.M.; Guglielmetti, R.; Furstoss, R. Synthesis of all four stereoisomers of enantiomerically pure tetrahydro-2,2,6-trimethyl-6-vinyl-2H-pyran-3-ols. Synthesis 1991, 1991, 681–682. [Google Scholar] [CrossRef]
- Vidari, G.; Di Rosa, A.; Zanoni, G.; Bicchi, C. Enantioselective synthesis of each stereoisomer of the pyranoid linalool oxides: The linalool route. Tetrahedron Asymmetry 1999, 10, 3547–3557. [Google Scholar] [CrossRef]
- Vidari, G.; Di Rosa, A.; Castronovo, F.; Zanoni, G. Enantioselective synthesis of each stereoisomer of the pyranoid linalool oxides: The geraniol route. Tetrahedron Asymmetry 2000, 11, 981–989. [Google Scholar] [CrossRef]
- Duan, S.; Moeller, K.D. Anodic coupling reactions: Probing the stereochemistry of tetrahydrofuran formation. A short, convenient synthesis of linalool oxide. Org. Lett. 2001, 3, 2685–2688. [Google Scholar] [CrossRef]
- Volz, F.; Wadman, S.H.; Hoffmann-Röder, A.; Krause, N. Gold catalysis in stereoselective natural product synthesis: (+)-linalool oxide, (−)-isocyclocapitelline, and (−)-isochrysotricine. Tetrahedron 2009, 65, 1902–1910. [Google Scholar] [CrossRef]
- Wan, K.K.; Litz, J.P.; Vosburg, D.A. Two-step, stereoselective synthesis of linalyl oxides by asymmetric allylic o-alkylation. Tetrahedron Asymmetry 2010, 21, 2425–2428. [Google Scholar] [CrossRef]
- Al Hazmi, A.M.; Sheikh, N.S.; Bataille, C.J.R.; Al-Hadedi, A.A.M.; Watkin, S.V.; Luker, T.J.; Camp, N.P.; Brown, R.C.D. Trans-2-tritylcyclohexanol as a chiral auxiliary in permanganate-mediated oxidative cyclization of 2-methylenehept-5-enoates: Application to the synthesis of trans-(+)-linalool oxide. Org. Lett. 2014, 16, 5104–5107. [Google Scholar] [CrossRef]
- David, L.; Veschambre, H. Preparation d’oxydes de linalol par bioconversion. Tetrahedron Lett. 1984, 25, 543–546. [Google Scholar] [CrossRef]
- Miyazawa, M.; Yokote, K.; Kameoka, H. Resolution of racemic linalool oxide-pyranoid by microbial esterification. Tetrahedron Asymmetry 1995, 6, 1067–1068. [Google Scholar] [CrossRef]
- Demyttenaere, J.C.R.; Adams, A.; Vanoverschelde, J.; De Kimpe, N. Biotransformation of (S)-(+)-linalool by Aspergillus niger: An investigation of the culture conditions. J. Agric. Food Chem. 2001, 49, 5895–5901. [Google Scholar] [CrossRef] [PubMed]
- Mirata, M.-A.; Wüst, M.; Mosandl, A.; Schrader, J. Fungal biotransformation of (±)-linalool. J. Agric. Food Chem. 2008, 56, 3287–3296. [Google Scholar] [CrossRef] [PubMed]
- Van Lint, M.J.; Gümüs, A.; Ruijter, E.; Faber, K.; Orru, R.V.A.; Hall, M. Enantioselective bio-hydrolysis of geranyl-derived rac-epoxides: A chemoenzymatic route to trans-furanoid linalool oxide. Adv. Synth. Catal. 2019, 361, 813–825. [Google Scholar] [CrossRef]
Figure 1.
The eight linalool oxide isomers and their postulated biosynthesis from linalool enantiomers.
Figure 1.
The eight linalool oxide isomers and their postulated biosynthesis from linalool enantiomers.

Scheme 1.
Experimental procedure for the preparation of all the isomeric forms of linalool oxide starting from (R)-linalool and (S)-linalool. Reagents and conditions: (a) mCPBA, CH2Cl2, 0 °C, PTSA cat.; (b) BzCl, Py, CH2Cl2, 0 °C; (c) chromatographic separation; (d) NaOH/MeOH, reflux, 3 h; (e) NaOAc, Ac2O, reflux, 3 h; (f) LiAlH4, dry Et2O, r.t. 1 h.
Scheme 1.
Experimental procedure for the preparation of all the isomeric forms of linalool oxide starting from (R)-linalool and (S)-linalool. Reagents and conditions: (a) mCPBA, CH2Cl2, 0 °C, PTSA cat.; (b) BzCl, Py, CH2Cl2, 0 °C; (c) chromatographic separation; (d) NaOH/MeOH, reflux, 3 h; (e) NaOAc, Ac2O, reflux, 3 h; (f) LiAlH4, dry Et2O, r.t. 1 h.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Study of the chemoselective benzoylation of the crude mixture of the linalool oxide isomers obtained from linalool.
Table 1.
Study of the chemoselective benzoylation of the crude mixture of the linalool oxide isomers obtained from linalool.
| Entry | Experimental Conditions 1 | Conversion of Linalool Oxide (Pyranoid) % 4 | Benzoate Ester (Pyranoid) % 2,4 | Benzoate Ester (Furanoid) % 3,4 | Benzoate Esters Ratio (Pyranoid/Furanoid) |
|---|---|---|---|---|---|
| 1 | BzCl (3 eq.); DMAP cat.; rt; 4 h | >99 | 55 | 45 | 1.2 |
| 2 | BzCl (3 eq.); rt; 4 h | >99 | 73 | 27 | 2.7 |
| 3 | BzCl (1.2 eq.); from 0 °C to rt; 4 h | 96 | 97 | 3 | 32.3 |
1 Reaction conditions: Each experiment was performed using 10 mMol of a mixture of linalool oxide isomers (81:19 ratio of furanoid/pyranoid isomers). The alcohols mixture, in dry CH2Cl2 (4 mL) was treated under stirring with 1 mL of pyridine followed by the addition of BzCl, according to the experimental conditions indicated in the table. The molar equivalents of BzCl were calculated in relation to the starting amount of pyranoid linalool oxide. 2 Percentage of the pyranoid benzoate esters in relation to the sum of all the linalool oxide benzoate ester isomers. 3 Percentage of the furanoid benzoate esters in relation to the sum of all the linalool oxide benzoate ester isomers. 4 Compounds concentrations were determined by GC-MS analysis.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Serra, S.; De Simeis, D.; Papili, S. A Practical Laboratory-Scale Synthesis of All Eight Stereoisomeric Forms of Terpene Linalool Oxide. Chemistry 2021, 3, 1247-1257. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry3040090
AMA Style
Serra S, De Simeis D, Papili S. A Practical Laboratory-Scale Synthesis of All Eight Stereoisomeric Forms of Terpene Linalool Oxide. Chemistry. 2021; 3(4):1247-1257. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry3040090
Chicago/Turabian StyleSerra, Stefano, Davide De Simeis, and Sara Papili. 2021. "A Practical Laboratory-Scale Synthesis of All Eight Stereoisomeric Forms of Terpene Linalool Oxide" Chemistry 3, no. 4: 1247-1257. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry3040090