
Efficient Electrocatalytic Approach to Spiro[Furo[3,2-b]pyran-2,5′-pyrimidine] Scaffold as Inhibitor of Aldose Reductase

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Experimental Section
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schneider, P.; Schneider, G. A Computational Method for Unveiling the Target Promiscuity of Pharmacologically Active Compounds. Angew. Chem. Int. Ed. 2017, 56, 7971–7974. [Google Scholar] [CrossRef] [Green Version]
- Evans, B.E.; Rittle, K.E.; Bock, M.G.; DiPardo, R.M.; Freidinger, R.M.; Whitter, W.L.; Lundell, C.F.; Veber, D.F.; Anderson, P.S.; Chang, R.S.L.; et al. Methods for drug discovery: Development of potent, selective, orally effective cholecystokinin antagonists. J. Med. Chem. 1988, 31, 2235–2246. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.-F.; Wang, W. (Eds.) Catalytic Cascade Reactions; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014. [Google Scholar]
- Hammerich, O.; Speiser, B. (Eds.) Organic Electrochemistry: Revised and Expanded, 5th ed.; CRS Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Yan, M.; Kawamata, Y.; Baran, P.S. Synthetic Organic Electrochemistry: Calling All Engineers. Angew. Chem. Int. Ed. 2018, 57, 4149–4155. [Google Scholar] [CrossRef] [PubMed]
- Elinson, M.N.; Makhova, I.V.; Nikishin, G.I. Electrochemical Synthesis of the Aryl α-Ketoesters from Acetophenones Mediated by KI. Angew. Chem. Int. Ed. 1988, 27, 1716–1717. [Google Scholar]
- Frankle, R.; Little, D. Redox catalysis in organic electrosynthesis: Basic principles and recent developments. Chem. Soc. Rev. 2014, 43, 2492–2521. [Google Scholar]
- Ogibin, Y.N.; Elinson, M.N.; Nikishin, G.I. Mediator oxidation systems in organic electrosynthesis. Russ. Chem. Rev. 2009, 78, 89–114. [Google Scholar] [CrossRef]
- Tang, H.-T.; Jia, J.-S.; Pan, Y.-M. Halogen mediated electrochemical organic synthesis. Org. Biomol. Chem. 2020, 18, 5315–5333. [Google Scholar] [CrossRef] [PubMed]
- Karkas, M.D. Electrochemical strategies for C-H functionalization and C-N bond formation. Chem. Soc. Rev. 2018, 47, 5786–5865. [Google Scholar] [CrossRef] [Green Version]
- Elinson, M.N.; Vereshchagin, A.N.; Ryzhkov, F.V. Catalysis of Cascade and Multicomponent Reactions of Carbonyl Compounds and C-H Acids by Electricity. Chem. Rec. 2016, 16, 1950–1964. [Google Scholar] [CrossRef]
- Vereshchagin, A.N.; Elinson, M.N.; Zaimovskaya, T.A.; Nikishin, G.I. Electrocatalytic cascade multicomponent assembling: Stereoselective one-pot synthesis of the substituted 3-azabicyclo[3.1.0]hexane-1-carboxylate system from aldehyde, malononitrile, malonate and methanol. Tetrahedron 2008, 64, 9766–9770. [Google Scholar] [CrossRef]
- Elinson, M.N.; Feducovich, S.K.; Starikova, Z.A.; Vereshchagin, A.N.; Nikishin, G.I. Stereoselective electrocatalytic transformation of arylidenemalononitriles and malononitrile into (1R,5S,6R)*-6-aryl-2-amino-4,4-dialkoxy-1,5-dicyano-3-azabicyclo-[3.1.0]hex-2-enes. Tetrahedron 2004, 60, 11743–11749. [Google Scholar] [CrossRef]
- Elinson, M.N.; Dorofeeva, E.O.; Vereshchagin, A.N.; Nikishin, G.I. Electrochemical synthesis of cyclopropanes. Russ. Chem. Rev. 2015, 84, 485–497. [Google Scholar] [CrossRef]
- Elinson, M.N.; Feducovich, S.K.; Vereshchagin, A.N.; Gorbunov, S.V.; Belyakov, P.A.; Nikishin, G.I. Electrocatalytic multicomponent cyclization of an aldehyde, malononitrile and a malonate into 3-substituted-2,2-dicyanocyclopropane-1,1-dicarbo-xylate—the first one-pot synthesis of a cyclopropane ring from three different molecules. Tetrahedron Lett. 2006, 47, 9129–9133. [Google Scholar] [CrossRef]
- Elinson, M.N.; Feducovich, S.K.; Bushuev, S.G.; Zakharenkov, A.A.; Pashchenko, D.V.; Nikishin, G.I. Electrochemical transformation of malonate and alkylidenemalonates into 3-substituted cyclopropane-1,1,2,2-tetracarboxylates. Mendeleev Commun. 1998, 8, 15–17. [Google Scholar] [CrossRef]
- Elinson, M.N.; Vereshchagin, A.N.; Ryzkov, F.V. Electrochemical Synthesis of Heterocycles via Cascade Reactions. Curr. Org. Chem. 2017, 21, 1427–1439. [Google Scholar] [CrossRef]
- Taylor, A.P.; Robinson, R.P.; Fobian, Y.M.; Blamore, D.C.; Jones, L.H.; Fadeyi, O. Modern advances in heterocyclic chemistry in drug discovery. Org. Biomol. Chem. 2016, 14, 6611–6637. [Google Scholar] [CrossRef] [PubMed]
- Brunton, L.L.; Chabner, B.A.; Knollmann, B.C.; Keith, L.P. Goodman & Gilman’s the Pharmacological Basis of Therapeutics, 12th ed.; The McGraweHill Companies: New York, NY, USA, 2011. [Google Scholar]
- Naguib, F.N.M.; Levesque, D.L.; Eng-Chi, W.; Panzica, R.P.; El Kouni, M.H. Phenylselenenyl- and phenylthio-substituted pyrimidines as inhibitors of dihydrouracil dehydrogenase and uridine phosphorylase. Biochem. Pharmacol. 1993, 46, 1273–1283. [Google Scholar] [CrossRef]
- Grams, F.; Brandstetter, H.; D’Alo, S.; Geppert, D.; Krell, H.-W.; Leinert, H.; Livi, V.; Menta, E.; Oliva, A.; Zimmermann, G. Pyrimidine-2,4,6-Triones: A New Effective and Selective Class of Matrix Metalloproteinase Inhibitors. Biol. Chem. 2001, 382, 1277–1285. [Google Scholar] [CrossRef]
- Maquoi, E.; Sounni, N.E.; Devy, L.; Olivier, F.; Frankenne, F.; Krell, H.-W.; Grams, F.; Foidart, J.-M.; Noël, A. Anti-Invasive, Antitumoral, and Antiangiogenic Efficacy of a Pyrimidine-2,4,6-trione Derivative, an Orally Active and Selective Matrix Metalloproteinases Inhibitor. Clin. Cancer Res. 2004, 10, 4038–4047. [Google Scholar] [CrossRef] [Green Version]
- Bentley, R. From miso, sake and shoyu to cosmetics: A century of science for kojic acid. Nat. Prod. Rep. 2006, 23, 1046–1062. [Google Scholar] [CrossRef]
- Chang, T.-S. An Updated Review of Tyrosinase Inhibitors. Int. J. Mol. Sci. 2009, 10, 2440–2475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, B.V.S.; Reddy, M.R.; Madan, C.; Kumar, K.P.; Rao, M.S. Indium(III) chloride catalyzed three-component coupling reaction: A novel synthesis of 2-substituted aryl(indolyl)kojic acid derivatives as potent antifungal and antibacterial agents. Bioorg. Med. Chem. Lett. 2010, 20, 7507–7511. [Google Scholar] [CrossRef] [PubMed]
- Rho, H.S.; Ahn, S.M.; Yoo, D.S.; Kim, M.K.; Cho, D.H.; Cho, J.I. Kojyl thioether derivatives having both tyrosinase inhibitory and anti-inflammatory properties. Bioorg. Med. Chem. Lett. 2010, 20, 6569–6571. [Google Scholar] [CrossRef] [PubMed]
- Aytemir, M.D.; Özçelik, B. A study of cytotoxicity of novel chlorokojic acid derivatives with their antimicrobial and antiviral activities. Eur. J. Med. Chem. 2010, 45, 4089–4095. [Google Scholar] [CrossRef]
- Tanaka, R.; Tsujii, H.; Yamada, T.; Kajimoto, T.; Amano, F.; Hasegawa, J.; Hamashima, Y.; Node, M.K.; Katoh, K.; Takebe, Y. Novel 3α-methoxyserrat-14-en-21β-ol (PJ-1) and 3β-methoxyserrat-14-en-21β-ol (PJ-2)-curcumin, kojic acid, quercetin, and baicalein conjugates as HIV agent. Bioorg. Med. Chem. 2009, 17, 5238–5246. [Google Scholar] [CrossRef]
- Lucas, A.S.D.; Gonçalves, L.M.; Carvalho, L.A.R.; Correia, H.F.; Da Costa, E.M.R.; Guedes, R.A.; Moreira, R.; Guedes, R.C. Optimization of O3-Acyl Kojic Acid Derivatives as Potent and Selective Human Neutrophil Elastase Inhibitor. J. Med. Chem. 2013, 56, 9802–9806. [Google Scholar] [CrossRef]
- Zheng, Y.; Tice, C.M.; Singh, S.B. The use of spirocyclic scaffolds in drug discovery. Bioorg. Med. Chem. Lett. 2014, 24, 3673–3682. [Google Scholar] [CrossRef] [Green Version]
- King, S.B.; Statford, E.S.; Craig, C.R.; Fifer, E.K. Synthesis and Pharmacological Evaluation of Spiro-Analogues of 5-Benzyl-5-ethyl Barbituric Acid. Pharm. Res. 1995, 12, 1240–1243. [Google Scholar] [CrossRef]
- Galati, E.M.; Monforte, M.T.; Miceli, N.; Ranerill, E. Anticonvulsant and sedative effects of some 5-substituted bromopyrazolinic spirobarbiturates. Farmaco 2001, 56, 459–461. [Google Scholar] [CrossRef]
- Kim, S.-H.; Pudzianowski, A.T.; Leavitt, K.J.; Barbosa, J.; McDonnell, P.A.; Metzler, W.J.; Rankin, B.M.; Liu, R.; Vaccaro, W.; Pitts, W. Structure-based design of potent and selective inhibitors of collagenase-3 (MMP-13). Bioorg. Med. Chem. Lett. 2005, 15, 1101–1106. [Google Scholar] [CrossRef]
- Fraser, W.; Suckling, C.J.; Wood, H.C.S. Latent inhibitors. Part 7. Inhibition of dihydro-orotate dehydrogenase by spirocyclopropanobarbiturates. J. Chem. Soc. Perkin Trans. 1990, 1, 3137–3144. [Google Scholar] [CrossRef]
- Duan, J.; Jiang, B.; Chen, L.; Lu, Z.; Barbosa, J.; Pitts, W.J. Barbituric acid derivatives as inhibitors of the TNF-alpha converting enzyme (TACE) and/or matrix metalloproteinases. U.S. Patent Application No. 0229084, 7 July 2003. [Google Scholar]
- Elinson, M.N.; Dorofeeva, E.O.; Vereshchagin, A.N.; Nasybullin, F.R.; Egorov, M.P. Electrocatalytic stereoselective transformation of aldehydes and two molecules of pyrazolin-5-one into (R*,R*)-bis(spiro-2,4-dihydro-3H-pyrazol-3-one)cyclopropa-nes. Cat. Sci. Technol. 2015, 5, 2384–2387. [Google Scholar] [CrossRef]
- Vereshchagin, A.N.; Elinson, M.N.; Dorofeeva, E.O.; Nasybullin, R.F.; Bushmarinov, I.S.; Goloveshkin, A.S.; Egorov, M.P. Electrocatalytic cyclization of 3-(5-hydroxy-3-methylpyrazol-4-yl)-3-arylpropionitriles: ‘one-pot’ simple fast and efficient way to substituted spirocyclopropylpyrazolones. Electrochim. Acta 2015, 165, 116–121. [Google Scholar] [CrossRef]
- Elinson, M.N.; Feducovich, S.K.; Starikova, Z.A.; Vereshchagin, A.N.; Gorbunov, S.V.; Nikishin, G.I. Stereoselective electrocatalytic transformation of arylidene- or alkylidenemalononitriles and malonate into alkyl (1R,5R,6R)*-6-substituted-5-cyano-4,4-dialkoxy-2-oxo-3-azabicyclo[3.1.0]hexane-1-carboxylates. Tetrahedron Lett. 2005, 46, 6389–6393. [Google Scholar] [CrossRef]
- Nikishin, G.I.; Elinson, M.N.; Lizunova, T.L.; Ugrak, B.I. Electrochemical transformation of malononitrile and ketones into 3,3-disubstituted-1,1,2,2-tetracyanocyclopropanes. Tetrahedron Lett. 1991, 32, 2655–2656. [Google Scholar] [CrossRef]
- Nikishin, G.I.; Elinson, M.N.; Fedukovich, S.K. Electrochemical dehydrotrimerization of dimethyl malonate to the hexamethyl ester of cyclopropanehexacarboxylic acid. Russ. Chem. Bull. 1986, 35, 1749. [Google Scholar] [CrossRef]
- Bruker. APEX-III; Bruker AXS Inc.: Madison, WI, USA, 2019. [Google Scholar]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Cryst. 2015, 48, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Cryst. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Elinson, M.N.; Ryzhkova, Y.E.; Krymov, S.K.; Vereshchagin, A.N.; Goloveshkin, A.S.; Egorov, M.P. Electrochemically induced multicomponent ‘one-pot’ assembling benzaldehydes, N,N′-dimethylbarbituric acid, and kojic acid. Monatsh. Chem. 2020, 151, 567–573. [Google Scholar] [CrossRef]
- Vereshchagin, A.N.; Elinson, M.N.; Dorofeeva, E.O.; Stepanov, N.O.; Zaimovskaya, T.A.; Nikishin, G.I. Electrocatalytic and chemical methods in MHIRC reactions: The first example of the multicomponent assembly of medicinally relevant spirocyclopropylbarbiturates from three different molecules. Tetrahedron 2013, 69, 1945–1952. [Google Scholar] [CrossRef]
- Elinson, M.N.; Fedukovich, S.K.; Vereshchagin, A.N.; Dorofeev, A.S.; Dmitriev, D.V.; Nikishin, G.I. Electrocatalytic transformation of malononitrile and cycloalkylidenemalononitriles into spirobicyclic and spirotricyclic compounds containing 1,1,2,2-tetracyanocyclopropane fragment. Russ. Chem. Bull. 2003, 52, 2235–2240. [Google Scholar] [CrossRef]
- Elinson, M.N.; Lizunova, T.L.; Dekaprilevich, M.O.; Struchkov, Y.T.; Nikishin, G.I. Electrochemical cyclotrimerization of cyanoacetic ester into trans-1,2,3-tricyanocyclopropane-1,2,3-tricarboxylate. Mendeleev Commun. 1993, 3, 192–193. [Google Scholar] [CrossRef]
- Petrash, J.M. All in the family: Aldose reductase and closely related aldo-keto reductases. Cell. Mol. Life Sci. C. 2004, 61, 737–749. [Google Scholar] [CrossRef]
- Schemmel, K.E.; Padiyara, R.S.; D’Souza, J.J. Aldose reductase inhibitors in the treatment of diabetic peripheral neuropathy: A review. J. Diabetes Complicat. 2010, 24, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Steuber, H.; Zentgraf, M.; La Motta, C.; Sartini, S.; Heine, A.; Klebe, G. Evidence for a novel binding site conformer of aldose reductase in ligand-bound state. J. Mol. Biol. 2007, 369, 186–197. [Google Scholar] [CrossRef]
- Ballante, F. Protein-Ligand Docking in Drug Design: Performance Assessment and Binding-Pose Selection. In Rational Drug Design: Methods and Protocols; Mavromoustakos, T., Kellici, T.F., Eds.; Springer: New York, NY, USA, 2018; pp. 67–88. [Google Scholar]
- Lead Finder, Version 1909 Build 2; BioMolTech®: Toronto, ON, Canada; Available online: http://www.cresset-group.com/lead-finder/ (accessed on 24 May 2021).
- Flare, Version 3.0; Cresset®: Litlington, UK; Available online: http://www.cresset-group.com/flare/ (accessed on 24 May 2021).
- Cheeseright, T.; Mackey, M.; Rose, S.; Vinter, A. Molecular Field Extrema as Descriptors of Biological Activity: Definition and Validation. J. Chem. Inf. Model. 2006, 46, 665–676. [Google Scholar] [CrossRef]
- Bauer, M.R.; Mackey, M.D. Electrostatic Complementarity as a Fast and Effective Tool to Optimize Binding and Selectivity of Protein–Ligand Complexes. J. Med. Chem. 2019, 62, 3036–3050. [Google Scholar] [CrossRef]
- Kuhn, M.; Firth-Clark, S.; Tosco, P.; Mey, A.S.J.S.; Mackey, M.; Michel, J. Assessment of Binding Affinity via Alchemical Free-Energy Calculations. J. Chem. Inf. Model. 2020, 60, 3120–3130. [Google Scholar] [CrossRef]
- Stroganov, O.V.; Novikov, F.N.; Stroylov, V.S.; Kulkov, V.; Chilov, G.G. Lead finder: An approach to improve accuracy of protein-ligand docking, binding energy estimation, and virtual screening. J. Chem. Inf. Model. 2008, 48, 2371–2385. [Google Scholar] [CrossRef]
- Howard, E.I.; Sanishvili, R.; Cachau, R.E.; Mitschler, A.; Chevrier, B.; Barth, P.; Lamour, V.; Van Zandt, M.; Sibley, E.; Bon, C.; et al. Ultrahigh resolution drug design I: Details of interactions in human aldose reductase-inhibitor complex at 0.66 A. Proteins 2004, 55, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Urzhumtsev, A.; Tête-Favier, F.; Mitschler, A.; Barbanton, J.; Barth, P.; Urzhumtseva, L.; Biellmann, J.-F.; Podjarny, A.D.; Moras, D. A ‘specificity’ pocket inferred from the crystal structures of the complexes of aldose reductase with the pharmaceutically important inhibitors tolrestat and sorbinil. Structure 1997, 5, 601–612. [Google Scholar] [CrossRef] [Green Version]
- Steuber, H.; Zentgraf, M.; Podjarny, A.; Heine, A.; Klebe, G. High-resolution Crystal Structure of Aldose Reductase Complexed with the Novel Sulfonyl-pyridazinone Inhibitor Exhibiting an Alternative Active Site Anchoring Group. J. Mol. Biol. 2006, 356, 45–56. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
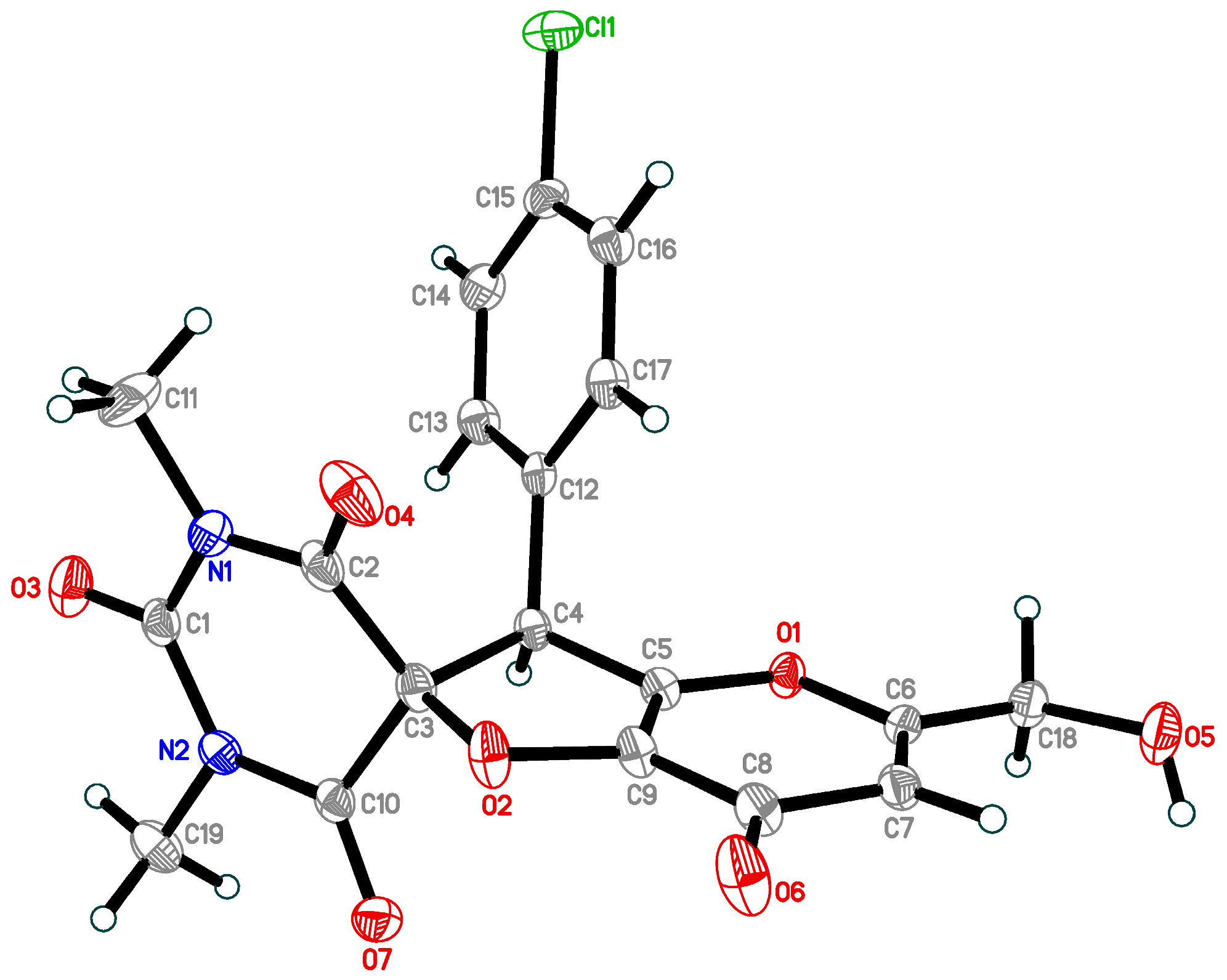{kind=link}
{kind=link}
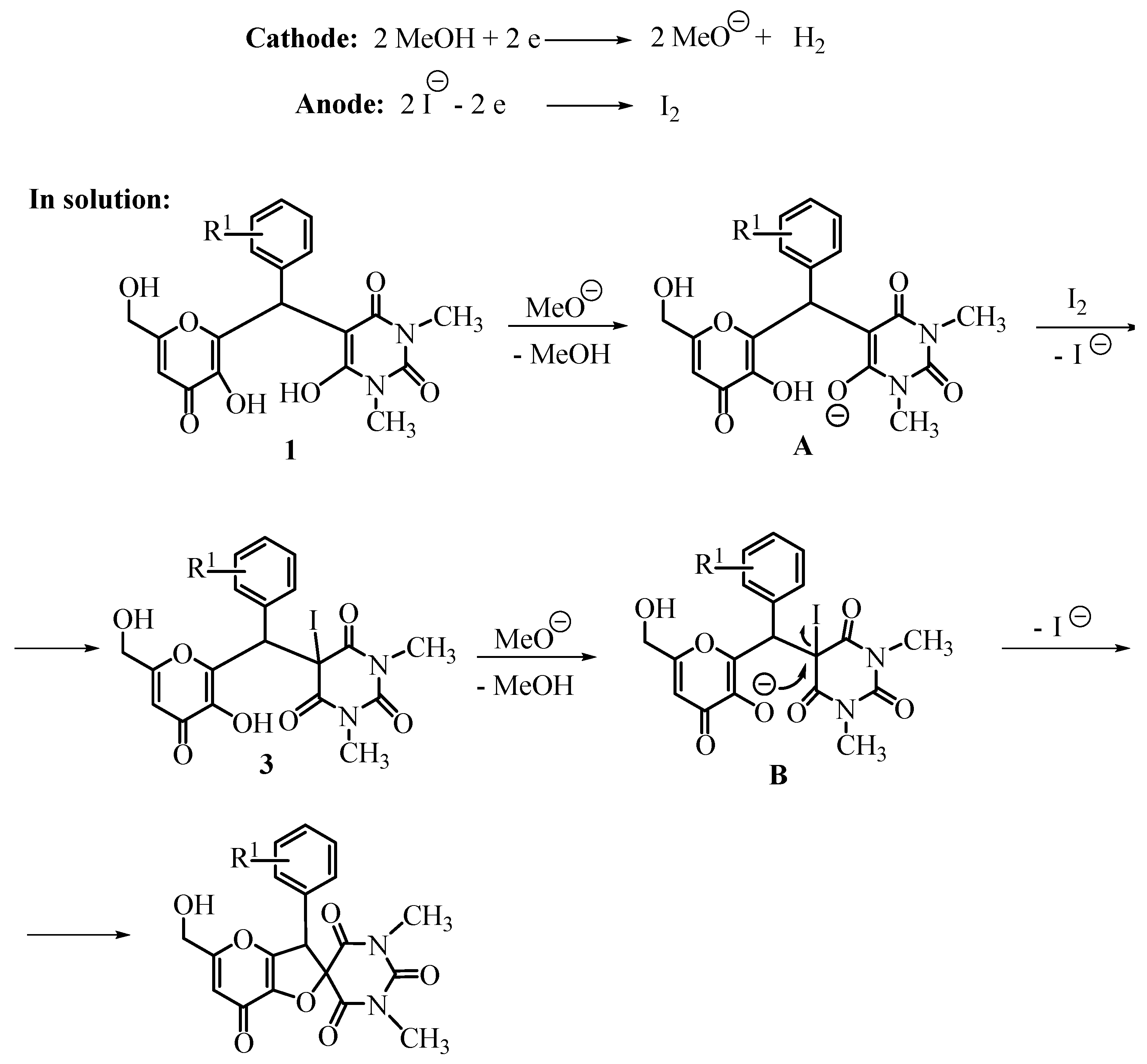{kind=link}
| Entry | Solvent | Mediator | Time (min) | Electricity (F/mol) | Yield of 2a (%) |
|---|---|---|---|---|---|
| 1 | MeOH | LiBr | 64 | 2.0 | 59 |
| 2 | MeOH | NaBr | 64 | 2.0 | 65 |
| 3 | MeOH | KBr | 64 | 2.0 | 62 |
| 4 | MeOH | LiI | 64 | 2.0 | 63 |
| 5 | MeOH | NaI | 64 | 2.0 | 70 |
| 6 | MeOH | KI | 64 | 2.0 | 66 |
| 7 | MeOH | NH4I | 64 | 2.0 | 53 |
| 8 | EtOH | NaI | 64 | 2.0 | 64 |
| 9 | n-PrOH | NaI | 64 | 2.0 | 60 |
| 10 | MeOH | NaI | 70 | 2.2 | 74 |
| 11 | MeOH | NaI | 77 | 2.4 | 77 |
| 12 | MeOH | NaI | 83 | 2.6 | 80 |
| 13 | MeOH | NaI | 90 | 2.8 | 82 |
| 14 | MeOH | NaI | 96 | 3.0 | 78 |
 |
| Compound | LFdG (kcal/mol) | LFVS | Interaction | Residues | Remark |
|---|---|---|---|---|---|
| 2a | −8.426 | −9.520 |  | Trp20 Tyr209 Ser210 | Formed an interaction with the key residue Trp20. Formed additional interactions with Tyr209 and Ser210 (which are binding residues in the reference ligand) [51]. |
| 2b | −8.301 | −9.381 |  | Tyr20 Asn160 Tyr209 | Formed an interaction with the key residue Trp20. Formed additional interactions with Asn160 and Cys298. |
| 2c | −8.269 | −9.951 |  | Trp20 Trp111 Asn160 Tyr209 Ser210 Cys298 | Formed an interaction with the key residue Trp20. Formed additional interactions with Ser210 (which is a binding residue in the reference ligand), Trp111, Asn160, Tyr209, and Cys298. |
| 2d | −7.874 | −8.951 |  | Ser210 | The only interaction revealed was with Ser210. |
| 2e | −9.124 | −10.496 |  | Tyr20 Asn160 Tyr209 Ser210 | Formed an interaction with the key residue Trp20. Formed an additional interaction to Ser110 (which is a binding residue in the reference ligand). The selection of the best pose was performed manually. |
| 2f | −8.424 | −9.04 |  | Trp20 Leu300 | Formed an interaction with the key residue Trp20. Formed an additional binding with Leu300. |
| 2g | −9.588 | −10.799 |  | Trp20 Tyr48 Trp111 Asn160 Tyr209 | Formed an interaction with the key residue Trp20. Formed an additional interaction with Tyr48 (which is a binding residue in the reference ligand), Trp111, Asn160, and Tyr209. |
| 2h | −6.399 | −7.686 |  | Trp20 Ser210 Ser214 | Formed an interaction with the key residue Trp20. Formed additional interactions with Ser210 and Ser214 (which are binding residues in the reference ligand). |
| 2i | −9.238 | −10.634 |  | Trp20 Val47 Tyr48 | Formed an interaction with the key residue Trp20. Formed an additional interaction with Val47 and Tyr48 (which are binding residues in the reference ligand) [51]. |
| Co-crystallized ligand | −8.545 | −10.089 |  | Trp20 Tyr48 His110 Ser210 Ser214 | The reference co-crystallized ligand [51]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elinson, M.N.; Vereshchagin, A.N.; Ryzhkova, Y.E.; Ryzhkov, F.V.; Fakhrutdinov, A.N.; Egorov, M.P. Efficient Electrocatalytic Approach to Spiro[Furo[3,2-b]pyran-2,5′-pyrimidine] Scaffold as Inhibitor of Aldose Reductase. Electrochem 2021, 2, 295-310. https://0-doi-org.brum.beds.ac.uk/10.3390/electrochem2020021
Elinson MN, Vereshchagin AN, Ryzhkova YE, Ryzhkov FV, Fakhrutdinov AN, Egorov MP. Efficient Electrocatalytic Approach to Spiro[Furo[3,2-b]pyran-2,5′-pyrimidine] Scaffold as Inhibitor of Aldose Reductase. Electrochem. 2021; 2(2):295-310. https://0-doi-org.brum.beds.ac.uk/10.3390/electrochem2020021
Chicago/Turabian StyleElinson, Michail N., Anatoly N. Vereshchagin, Yuliya E. Ryzhkova, Fedor V. Ryzhkov, Artem N. Fakhrutdinov, and Mikhail P. Egorov. 2021. "Efficient Electrocatalytic Approach to Spiro[Furo[3,2-b]pyran-2,5′-pyrimidine] Scaffold as Inhibitor of Aldose Reductase" Electrochem 2, no. 2: 295-310. https://0-doi-org.brum.beds.ac.uk/10.3390/electrochem2020021