Mechanisms Underlying Development of Taurine-Deficient Cardiomyopathy


1
Department of Pharmacology, College of Medicine, University of South Alabama, Mobile, AL 36688, USA
2
Faculty of Biotechnology, Fukui Prefectural University, Fukui 910-1195, Japan
3
Department of Pharmacy, School of Pharmacy, Hyogo University of Health Sciences, Kobe 650-8530, Japan
4
Department of Biochemistry and Molecular Medicine, The George Washington University, Washington, DC 20037, USA
*
Author to whom correspondence should be addressed.
†
Deceased: 20 September 2014.
Hearts 2020, 1(2), 86-98; https://0-doi-org.brum.beds.ac.uk/10.3390/hearts1020010
Submission received: 6 July 2020
/
Revised: 6 August 2020
/
Accepted: 7 August 2020
/
Published: 14 August 2020
(This article belongs to the Special Issue Nutrient Deficiency and Drug Induced Cardiac Injury and Dysfunction)
Abstract
:Taurine is a ubiquitous β-amino acid that plays an essential role in ensuring normal mitochondrial and myocardial function. In the mitochondria, taurine reacts with a tRNA forming a 5-taurinomethyluridine conjugate that primarily regulates the biosynthesis of the mitochondria encoded protein, ND6, which serves as a subunit of complex I of the respiratory chain. Impaired formation of the taurine conjugate reduces activity of complex I and plays a central role in the pathophysiology of the mitochondrial disease MELAS (myopathy, encephalopathy, lactic acidosis and stroke-like episodes). The restoration of mitochondrial levels of the taurine conjugate enhances electron flux through the respiratory chain, thereby preventing at least some of the symptoms of MELAS. Taurine therapy also diminishes the severity of congestive heart failure, an observation that led to its approval for the treatment of congestive heart failure in Japan. The review article discusses the role of defective calcium handling, reduced ATP generation, enhanced oxidative stress and apoptosis in the development of taurine-deficient cardiomyopathy. Some patients suffering from congestive heart failure are taurine-deficient, an observation supporting the hypothesis that low taurine levels contribute to the severity of heart failure. Thus, mishandling of taurine leads to mitochondrial dysfunction, which is involved in the development of both MELAS and congestive heart failure.
1. Introduction
Taurine (aminoethane sulfonate) is a ubiquitous, sulfur-containing β-amino acid, which largely exists as a free amino acid in excitable tissues such as the heart. Because little if any taurine is synthesized directly by the heart, maintenance of the large myocardial taurine pool relies upon uptake of the amino acid from the blood. The taurine transporter located on the cell membrane of the heart is a taurine-sodium cotransporter that utilizes the sodium gradient of the cardiomyocyte to drive the uptake of taurine against a concentration gradient. While cytosolic taurine levels of the heart range from 1 to 30 mM, plasma concentrations in most mammals vary from 44 to 224 μM, with the mean being 63 μM [1]. The uptake of taurine by the heart is generally a slow process but the rate of taurine turnover by the heart is an even slower event, which together ensures regulation of the intracellular taurine pool by blood taurine content [2].
The size of the intracellular taurine pool is species dependent, with levels varying from 1.8 mg/g wet wt. in the frog to 39.4 mg/g wet wt. in the mouse [3]. According to Kocsis et al. [3], taurine content of the heart is related to heart rate and myocardial workload, with higher rates of contraction found in animals whose hearts contain higher levels of taurine. Moreover, the endocardium, which contains higher levels of taurine than other regions of the left ventricle, is subjected to greater workloads than the areas of the left ventricle containing reduced taurine content. Also of interest is the observation that severe reductions in myocardial taurine content invariably lead to the development of cardiomyopathies [4,5,6].
Taurine is an essential nutrient in animals, such as the cat and fox, whose hepatic biosynthesis of taurine is low but the excretion of taurine as a bile acid conjugate is relatively high. Because the enzymes involved in the conjugation of bile acids in cats specifically utilize taurine as a substrate, elimination of taurine from the body is related to the composition of the bile conjugate. Thus, high levels of dietary taurine are required to avoid taurine deficiency in the cat. In contrast to the cat, taurine is a semi-essential nutrient in humans. Despite low levels of hepatic taurine biosynthesis, humans are capable of conjugating bile acids with either taurine or glycine, thereby preserving taurine for more important functions. Thus, humans are less dependent upon dietary sources of taurine than cats. In contrast to both cats and humans, rodents generate large amounts of taurine in the liver, eliminating the need for a dietary source of taurine to avoid taurine deficiency.
2. Animal Models of Taurine Deficiency
Two animal models of taurine deficiency have been used to study the properties of the taurine-deficient cardiomyopathy [5,6]. Cats are dependent upon a dietary source of taurine to maintain normal myocardial taurine content and ensure normal contractile function. According to Novotny et al. [7], the taurine-depleted cat heart exhibits defects in both systolic and diastolic function. During the initial phase of the cardiomyopathy, the left ventricle undergoes a significant elevation in diastolic compliance accompanied by systolic dysfunction, which together suggests remodeling of the heart leads to eccentric hypertrophy with increased ventricular diameter and wall thickness [8]. However, during later stages of the cardiomyopathy, cells die and the thickness of the ventricular wall decreases. In contrast to cats, mice do not develop a cardiomyopathy when maintained on a taurine-deficient diet. However, blocking myocardial taurine uptake leads to development of a taurine-deficient cardiomyopathy in rodents [6]. The taurine transporter knockout mouse (TauTKO) appears to undergo a unique pathway of remodeling [6]. While the nine-month old mouse heart experiences significant decrease in ventricular wall thickness, there is no evidence that the decrease in wall thickness is preceded by a hypertrophic phase that leads to myocardial enlargement. Rather, the taurine-deficient mouse heart undergoes significant atrophy, with cardiomyocyte atrophy being a prominent characteristic of the cardiomyopathic heart [6].
2.1. Impaired Calcium Handling Contributes to Cardiomyopathy
A major cause of contractile dysfunction of the failing heart is impaired delivery of Ca2+ to the muscle protein, troponin [9]. Excitation contraction coupling accounts for most of the Ca2+ that triggers contraction. However, there are other sources of Ca2+ that can influence contractile function, one related to taurine [10]. The taurine transporter of the cell membrane catalyzes the co-transport of taurine and Na+, which, combined with the actions of the Na+/Ca2+ exchanger, leads to a net increase in the intracellular content of both taurine and Ca2+. Another possible scenario would be a slowing of taurine and Na+ efflux, which would preserve [Na+]i and enhance [Ca2+]i. Both mechanisms are functional in the normal heart but are inoperative in the TauTKO mouse, which lacks an active taurine transporter. According to Chapman et al. [10], taurine regulates [Na+]I, which in turn is linked to contractile function through the actions of the Na+/Ca2+ and Na+/H+ exchangers. These effects provide a logical explanation for the development of taurine-deficient cardiomyopathy.
One of the central Ca2+ transporters involved in the regulation of [Ca2+]i. during excitation-contraction coupling is the sarcoplasmic reticular Ca2+ ATPase, which pumps Ca2+ from the cytosol into the sarcoplasmic reticular (SR) vesicles, promoting myocardial relaxation. The loading of the SR vesicles serves another function: to increase the size of the SR Ca2+ pool available for release upon stimulation, which enhances contractile function. A prominent mechanism leading to stimulation of SR Ca2+ ATPase activity is the activation of phospholamban, a SR protein whose dephosphorylation form is capable of inactivating SR Ca2+ ATPase. However, the activity of the SR Ca2+ ATPase in the taurine-deficient heart is only modestly depressed at pCa = 6.5 and not at all at pCa = 6.0 or at the lower concentrations of Ca2+ (Figure 1). These data suggest that diminished SR Ca2+ ATPase activity unlikely plays a central role in the development of the taurine-deficient cardiomyopathy of the TauTKO mouse.
Figure 1 shows that taurine deficiency causes a significant reduction in systolic function of the TauTKO heart. Similarly, systolic function, as well as diastolic function, are adversely affected by dietary taurine depletion of the cat heart [7]. While both the TauTKO mouse and the dietary cat model are taurine-deficient, nutritionally deficient cats harbor a normal taurine transporter but a reduction in plasma taurine content. Thus, in the nutritionally deficient cat the reduction in intracellular taurine content is associated with a decline in taurine efflux, which according to Chapman et al. [10] results in diminished [Na+]i and [Ca2+]i. Although the TauTKO heart lacks the taurine transporter, there is reason to believe that the heart contains reduced [Na+]i, as exposure of neonatal cardiomyocytes to medium containing 5 mM β-alanine, an inhibitor of the taurine transporter, leads to a significant decrease in both intracellular taurine levels and [Na+]i, [11]. Nonetheless, [Ca2+]i, is not decreased in the β-alanine treated cardiomyocyte, suggesting that knocking out or inhibiting the taurine transporter may alter the central mechanism underlying the development of the taurine-deficient cardiomyopathy.
2.2. Taurine-Deficient Heart Is Energy-Deficient
Mitochondrial taurine content is high and largely exists as a free amino acid [12]. However, a small amount of taurine conjugates with a uridine residue in the wobble position of tRNALeu(UUR) [13,14]. In the absence of the taurine conjugate, the interaction between the anticodon AAU of the tRNA and the UUG codon is weak. By strengthening the interaction between the anticodon and codon, taurine promotes the biosynthesis of proteins whose mRNAs contain multiple UUG codons [14]. The most taurine-sensitive mitochondria encoded protein is ND6, whose mRNA contains 8 UUG codons, representing 42% of all leucine residues of the protein. The primary function of the 13 mitochondria encoded proteins is to serve as subunits of respiratory chain complexes, with ND6 existing as 1 of 44 protein subunits that make up complex I of the electron transport chain [14]. Mitochondria obtained from taurine-deficient hearts contain reduced levels of ND6 and diminished activity of complex I (NADH: ubiquinone oxidoreductase) [14,15]. Although taurine deficiency has no direct effect on the activities of complexes II-V, the flux of electrons through downstream complexes of taurine-deficient mitochondria is reduced. This occurs because there are fewer electrons generated by complex I and the flow of complex I-generated electrons into the rest of the electron transport chain is diminished [16,17,18]. Nonetheless, one can bypass the defect in complex I by using permeabilized cardiomyocytes exposed to the complex II substrate, succinate. As seen in Table 1, the oxidation of complex I substrates, glutamate/malate, are decreased in permeabilized, taurine-deficient cardiomyocytes (β-alanine group) while the oxidation of succinate is unaffected by taurine deficiency, which was generated by exposure of cardiomyocytes to medium containing the taurine transport inhibitor, β-alanine [17].
Energy metabolism of the taurine-deficient heart resembles that of the hypoxic myocardium, largely because both conditions are associated with reduced oxygen consumption and respiratory chain flux. In hypoxia, the lack of oxygen decreases flux through complex IV while backing up the flow of electrons through complexes I and III, leading to an increase in the NADH/NAD+ ratio. The heart responds to the accumulation of reducing equivalents (NADH) by inhibiting further generation of NADH. Consequently, oxidative metabolism is severely suppressed in the hypoxic heart. Similarly, oxidative metabolism of virtually every substrate of the heart, including the primary substrates, glucose and fatty acids, is downregulated in the taurine-deficient heart (Figure 2). While the metabolism of glucose to pyruvate is enhanced by mild hypoxia and taurine deficiency, pyruvate is largely converted to lactate rather than being oxidized by the citric acid cycle. This series of events, in combination with reduced fatty acid metabolism, diminished citric acid cycle activity and slowing of electron flux, contributes to the depletion of ATP stores (Figure 3). Although the delivery of electrons from NADH to the electron transport chain results in the normal generation of 3 moles of ATP/NADH oxidized in both normal and taurine-deficient cardiomyocytes, less ATP is produced in the taurine-deficient cardiomyocyte because of diminished generation of reducing equivalents, inhibition of complex I and impaired delivery of electrons to the electron transport chain [18].
Experiments utilizing isolated LV working hearts perfused with buffer containing 5 mM 3H-glucose and 5 mM 14C-acetate (the oxidation of acetate is a measure citric acid cycle flux) confirm the ability of taurine-deficient hearts to readily convert glucose to pyruvate without adequately catalyzing the oxidation of pyruvate by pyruvate dehydrogenase. The bottleneck in glucose oxidation is caused by NADH-mediated inhibition of pyruvate dehydrogenase activity, a form of feedback inhibition related to diminished complex I activity and elevated NADH levels [19]. The high NADH/NAD+ ratio also diminishes the rates of endogenous fatty acid oxidation and citric acid cycle flux, which are other sources of reducing equivalents for ATP generation. Because feedback regulation of cardiac metabolism by NADH/NAD ratio and ATP/ADP ratio in the heart is prominent, contractile function plays a minor role in regulation of energy metabolism in the taurine-deficient heart. Hence, the taurine-deficient heart is energy-deficient, with the primary defect in complex I of the respiratory chain and secondary defects in NADH generation by fatty acid and glucose metabolism. The net effect is reduced rates of electron transport and oxidative phosphorylation, leading to ATP deficiency (Figure 3). Based on recent studies examining the metabolic states of unique models of heart failure, it has been concluded that impaired glucose and fatty acid metabolism contribute to the development of cardiomyopathies [20,21,22,23]. Therefore, it is not surprising that the taurine-deficient heart also develops a cardiomyopathy [6,7].
2.3. Role of Mitochondria, Oxidative Stress and Apoptosis in Development of Taurine-Deficient Cardiomyopathy
Dysfunctional mitochondria play a central role in the development of heart failure [24]. Among the properties that contribute to this response are reduced ATP generation, enhanced superoxide generation, impaired cell signaling and cell death (both apoptosis and necrosis). Superoxide is generated when electrons, which leak from complexes I and III of the electron transport chain, are picked up by the acceptor oxygen [25,26]. Although electron transport is normally quite efficient, the mitochondria are a major source of superoxide even in the normal heart. Oxidative stress arises when considerably greater levels of superoxide are generated, as the rate of electron flux through the respiratory chain is significantly reduced. Indeed, the standard inhibitor of complex I, rotenone, increases the generation of superoxide despite reductions in the oxidation of NADH and the delivery of electrons to coenzyme Q and complex III, indicating that it diverts electrons away from the respiratory chain in favor of superoxide generation [18]. The same pattern develops in taurine-deficient hearts and fibroblasts, as delivery of electrons to the electron transport chain are diminished while mitochondrial superoxide production increases [17,27].
Superoxide belongs to a group of oxidants known as reactive oxygen species (ROS). The electron of superoxide is readily transferred to a cellular acceptor, but particularly noteworthy is its spontaneous reaction with the intracellular messenger, nitric oxide, to produce the potent oxidant, peroxynitrite (ONOO-). The affinity of nitric oxide for superoxide is so strong that it competes with the enzyme, superoxide dismutase, for superoxide. In the presence of superoxide dismutase, superoxide is converted to the moderate oxidant, hydrogen peroxide. While hydrogen peroxide can damage mitochondria, its exposure to iron leads to the formation of the highly reactive oxidant, hydroxy radical, which can cause extensive damage to the cardiomyocyte. Hydrogen peroxide is neutralized by the enzyme, glutathione peroxide, which utilizes reduced glutathione and NADPH to catalyze the conversion of hydrogen peroxide to water. Catalase also catalyzes the breakdown of hydrogen peroxide, converting it to water and oxygen.
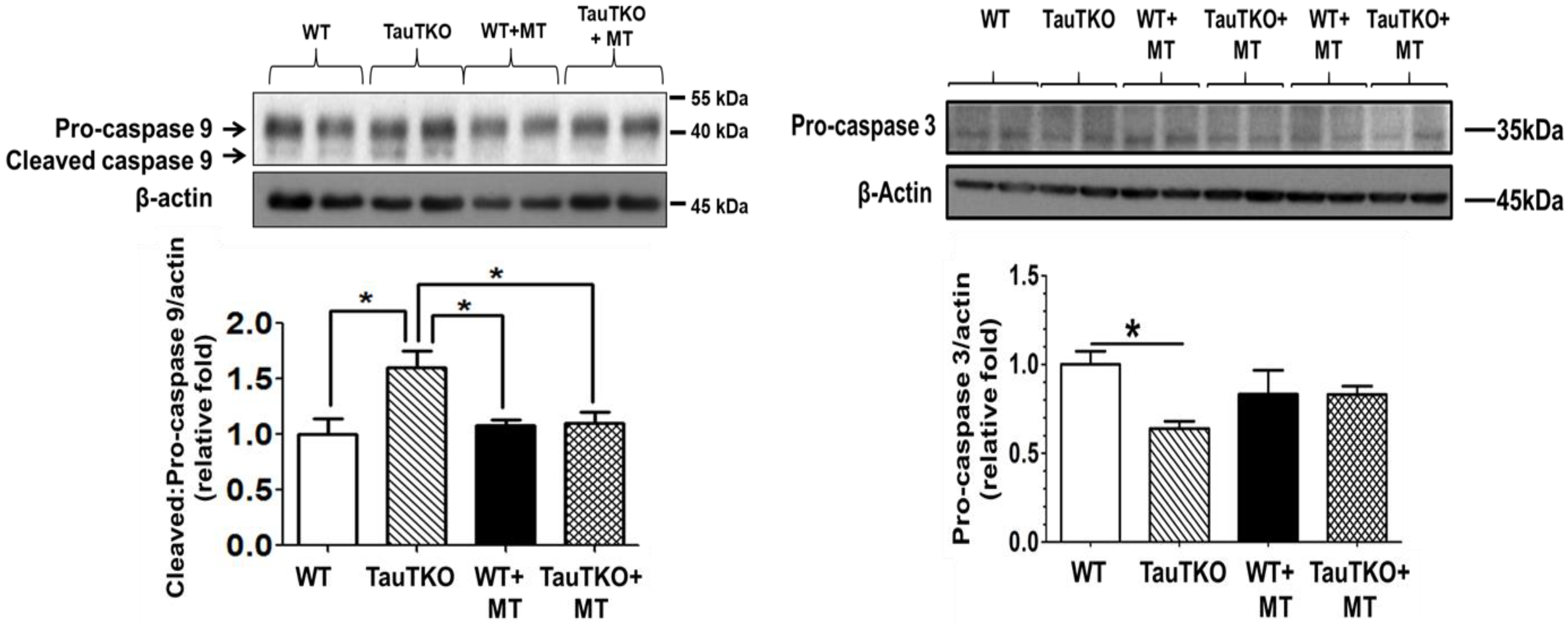
Mitochondrial oxidative stress and fragmentation are symptoms of unhealthy mitochondria, which are capable of initiating apoptosis [24,28]. Although mitochondrial fragmentation can occur in the absence of apoptosis, it is usually associated with an early step in apoptosis, which is detected shortly around the time of cytochrome c release from the mitochondria [29]. The release of cytochrome c is preceded by the permeabilization of the mitochondria by one of two mechanisms, either the mitochondrial permeability transition or creation of a Bax/Bak pore [30]. The mitochondrial permeability transition pore (mPTP), which is located in the inner mitochondrial membrane, normally remains in its closed state. Opening of the pore is mediated by excessive calcium accumulation in the mitochondrial matrix, ATP depletion, increased phosphate content and oxidative stress. Although the protein composition of the mPTP remains controversial, it is recognized that pore opening allows the movement of cytoplasmic water and solutes (≤1500 Da) into the mitochondrial matrix. Chronic opening of the pore leads to catastrophic changes to the cell, including the loss of mitochondrial membrane potential, increased production of ROS, severe ATP depletion, mitochondrial swelling, outer mitochondrial membrane rupture and the release of pro-apoptotic factors from the cell [30]. According to Jong et al. [31], initiation of apoptosis by taurine depletion is blocked by cyclosporin A, indicating that the mPTP is activated by taurine depletion. One likely candidate for this action is oxidative stress, as oxidative stress is a common feature of taurine deficiency [15]. Moreover, treatment of 3-month old taurine-deficient mice (TauTKO mice) with the mitochondrial-specific antioxidant, MitoTempo, prevents the stimulation of myocardial caspases 9 and 3 (Figure 4) [15]. Furthermore, restoration of taurine content of taurine-deficient fibroblasts is associated with reversal of mitochondrial fragmentation and improvement in viability [17].
The Bcl-2 family consists of three groups of small molecular weight proteins that regulate the balance between cell survival and death. The anti-apoptotic family members inhibit the BH3 pro-apoptotic family members and the pro-apoptotic Bax and Bak proteins. During activation by BH3 pro-apoptotic family members, Bax and Bak are converted from harmless monomers to harmful polymers, which become imbedded in the mitochondrial outer membrane where they form a pore that permeabilizes the membrane. The pore is large enough to allow the escape of pro-apoptotic proteins, such as cytochrome c, from the inter-mitochondrial space to the cytosol. In the cytosol, cytochrome c interacts with APAF 1 (apoptotic protease-activating factor 1) and ATP to form an apoptosome, which activates caspase 9. The effector caspase 3 is then activated by caspase 9 to initiate apoptosis. However, a role of Bax-mediated mitochondrial permeabilization in the onset of apoptosis during taurine deficiency seems unlikely, as taurine deficiency is not accompanied by large changes in the Bcl-2/Bax ratio [17].
The facilitation of mitochondrial fragmentation in taurine-deficient fibroblasts is mediated in part by a 50% increase in the levels of Dynamin related protein 1 (Drp1), a large GTPase that drives mitochondrial fragmentation in mammalian cells [32]. Upon stimulation of apoptosis, Drp1 is recruited to the outer membrane of the mitochondria, where it initiates mitochondrial fragmentation and the release of cytochrome c [29]. Therefore, a close relationship exists between Drp1-mediated mitochondrial fragmentation and both mitochondrial permeabilization and mitochondrial apoptosis. In support of this idea, Shetewy et al. [17] found that Drp1 levels were elevated 50% in taurine-deficient fibroblasts, an effect associated with mitochondrial fragmentation, oxidative stress and apoptosis.
2.4. Clinical Importance of Taurine as Therapy in Treatment of Congestive Heart Failure
Taurine has been approved for the treatment of congestive heart failure in Japan [33]. This decision was based on compelling evidence showing a beneficial effect of taurine in both animal models of heart failure [34,35,36] and humans suffering from congestive heart failure [33,37]. In one human study, a double-blind, randomized crossover, placebo-controlled format was employed to evaluate the beneficial effect of taurine therapy (6 g/day) on several symptoms of congestive heart failure, including dyspnea, crackles and edema [37]. Taurine treatment not only reduced the severity of common symptoms, but was also associated with an improvement in the NY Heart Association functional classification and the need for other therapeutic agents, such as digitalis and diuretics [37,38].
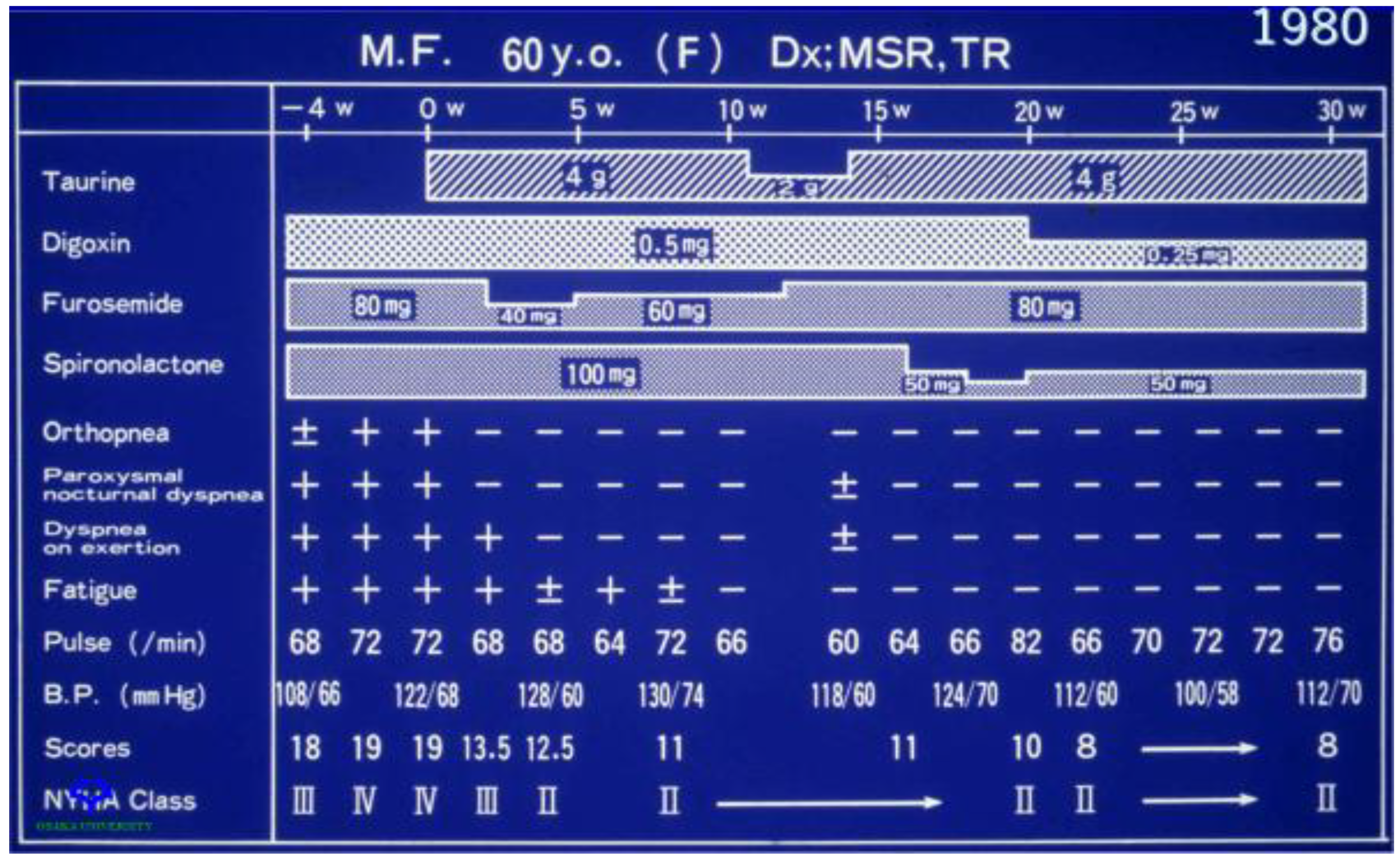
Representative data from a 60-year-old female patient with New York Heart Association class IV congestive heart failure who was treated with digoxin (0.5 mg/day), furosemide (80 mg/day) and spironolactone (100 mg/day) prior to the onset of taurine treatment are shown in Figure 5. Three symptoms, orthopnea, dyspnea on exertion (breathlessness after climbing one flight of stairs) and fatigue, were examined. Within two weeks of taurine therapy, the patient had reverted to a class II NY Heart Association classification, as there were improvements in all three symptoms. A reduction in the dose of taurine after 11 weeks of therapy from 4 g/day to 2 g/day worsened dyspnea on exertion, but the effect was reversible, as restoration of taurine to 4 g/day also led to an improvement in dyspnea on exertion. Subsequent reductions in the dose of digoxin and spironolactone did not alter the condition of the patient.
Congestive heart failure describes a pathophysiological state arising from a diverse group of cardiac conditions, including ischemic heart disease, hypertension, diabetes mellitus, valvular heart disease, atrial and ventricular fibrillation and thyroid disease. These conditions cause a reduction in cardiac output, which in turn activates neurohumoral systems (sympathetic nervous system and renin-angiotensin II-aldosterone system) that initially increase myocardial contractility, enhance myocardial relaxation and promote vasoconstriction. However, excessive neurohumoral stimulation leads to a series of adverse events, such as calcium mishandling, myocardial remodeling and generation of reactive oxygen species, which cause deterioration of contractile function. Because angiotensin II and catecholamine blockade significantly improve the long-term morbidity and mortality of patients with congestive heart failure, inhibition of the neurohumoral agents has become the mainstay for treatment of congestive heart failure. Ito et al. [38] reviewed the evidence that taurine benefits patients with congestive heart failure by inhibiting the actions of the same neurohumoral agents. In addition, taurine improves mitochondrial function and ATP biosynthesis, which are also adversely affected in the failing heart [15,27]. Yet, many subjects with congestive heart failure may be taurine-deficient because they are often anorexic and eat sparingly [39]. This led to the suggestion that taurine therapy merely functions to restore taurine levels in subjects with congestive heart failure. However, poor intake of taurine does not appear to cause overt taurine deficiency in humans. Rather, poor nutrition can lead to other maladies, such as folate deficiency [40]. According to Schaffer et al. [14], folic acid plays an essential role in the formation of the 5-taurinomethyuridine conjugate of tRNALeu(UUR). Therefore, an improvement in the diet of patients with congestive heart failure, including an increase in dietary taurine and folate, should enhance mitochondrial function of the failing myocardium, as well as reduce the toxic actions of the neurohumoral agents.
2.5. Taurine Involvement in the Disease, MELAS
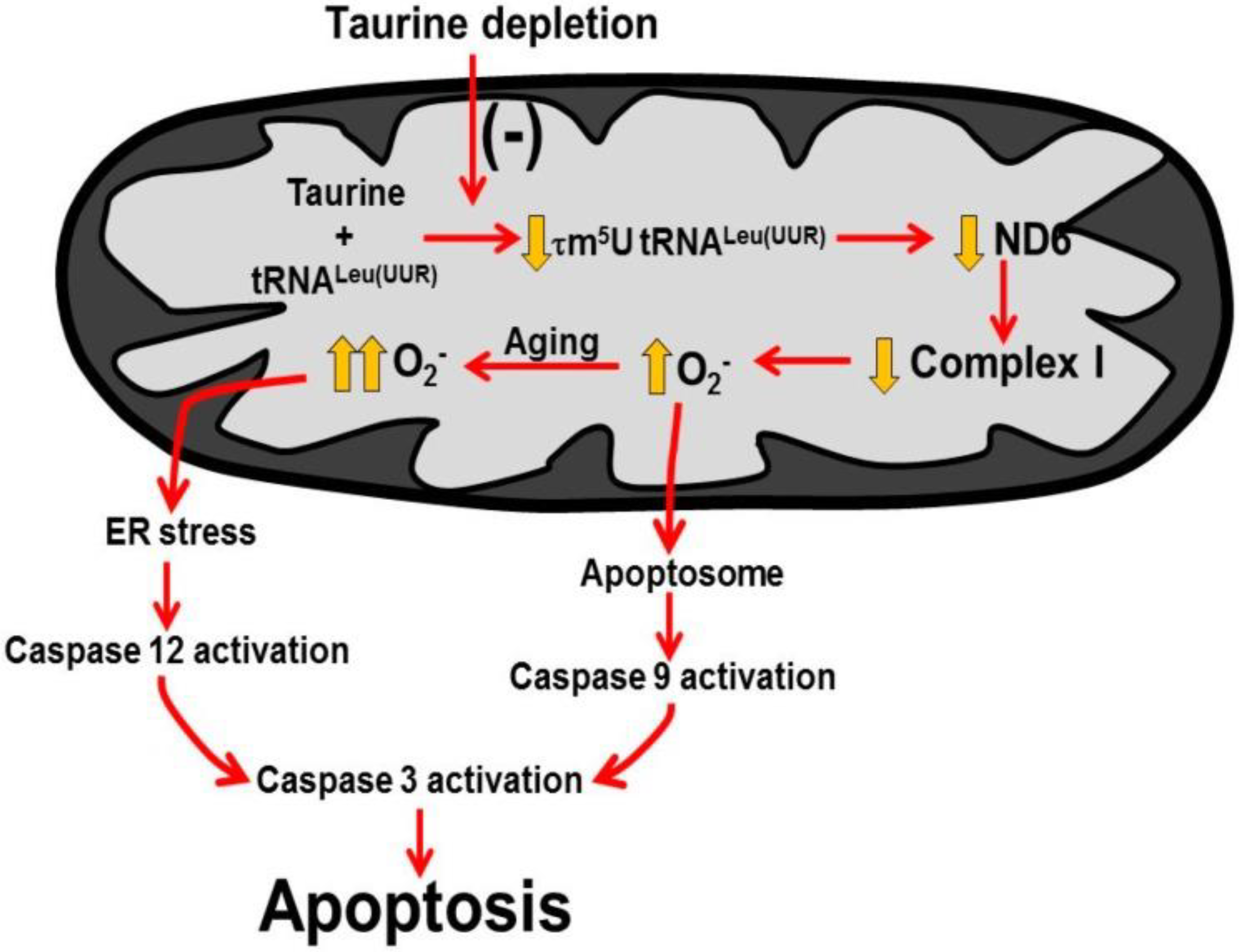
The phenotype that develops in taurine deficiency mimics that of the mitochondrial disease, MELAS [14]. This is not surprising because both conditions share a common pathology (Figure 6). The primary defect in 80% of MELAS cases (myopathy, encephalopathy, lactic acidosis and stroke-like episodes) is a point mutation in the gene for tRNALeu(UUR), which interferes with the conjugation of a uridine residue of tRNALeu(UUR) with taurine, forming 5-taurinomethyluridine-tRNALeu(UUR) [41]. The modified uridine residue is located at the wobble position of the anticodon UAA, which is capable of forming a base pair with two codons, UUU and UUG. The 5-taurinomethyluridine conjugate has no effect on the interaction of UAA with UUU, however, it significantly strengthens the interaction between UAA and UUG, thereby improving UUG decoding [13]. The biosynthesis of one mitochondria encoded protein, ND6, is heavily dependent on proper UUG decoding. Hence, impaired formation of 5-taurinomethyluridine-tRNALeu(UUR) in MELAS, likely reduces the content of ND6, resulting in a decrease in complex I activity [14]. Taurine deficiency also reduces ND6 levels and complex I activity by lowering the content of the 5-taurinomethyluridine-tRNALeu(UUR) conjugate in the mitochondria [15].
Although the use of taurine as a therapeutic agent in the treatment of MELAS has been limited to date, the few clinical studies performed have been extremely promising. The logic behind the studies is the presumption that administration of high amounts of taurine overcomes the block in 5-taurinomethyluridine-tRNALeu(UUR) formation caused by the MELAS point mutation. One of the patients studied, who had a A3243G point mutation, presented with stroke-like episodes that did not respond to anticonvulsant therapy. Shortly after the onset of taurine therapy, the epileptic and stroke-like episodes cease and the lactic acidosis symptom disappears [42]. In a related study, a 21-year-old male harboring a A3243G mutation was treated successfully with taurine despite years of unsuccessful treatment with anticonvulsant therapy. Cybrids (cytoplasmic hybrids) prepared from the second patient exhibited impaired oxygen consumption and mitochondrial membrane potential. Addition of 40 mM taurine to the incubation medium containing the abnormal cybrids, partially corrected electron transport chain function, resulting in a reduction in oxidative stress. The few clinical studies performed using taurine as a therapeutic agent provide evidence that the defect in 5-taurinomethyluridine-tRNALeu(UUR) formation can be at least partially reversed by administration of high doses of taurine. It is possible that taurine therapy could develop into a first-line treatment for MELAS. However, advances in the field require additional studies.
Author Contributions
S.W.S. and J.H.K., were involved in conception and/or design of research; S.W.S., C.J.J., J.A. and T.I. performed experiments; S.W.S. and C.J.J. analyzed data; J.A. and S.W.S. interpreted results of experiments; J.A., S.W.S. and C.J.J. prepared figures; S.W.S. drafted manuscript as first author; S.W.S. and J.H.K. edited and revised manuscript; S.W.S. and J.H.K., approved the final version of manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
Funding for these studies was provided by The American Heart Association.
Acknowledgments
We dedicate this manuscript to the memory of our esteemed colleague, Junichi Azuma. He carried out the clinical trials that established taurine as a therapeutic agent in the treatment of congestive heart failure, and his contributions to taurine research will be missed.
Conflicts of Interest
The authors declare no conflict of interest. The sponsors had no role in study design, in the collection, analyses, or interpretation of data, in the writing of the manuscript and in the decision to publish results.
References
- Kramer, G.A.; Kittleson, M.D.; Fox, P.R.; Lewis, J.; Pion, P.D. Plasma taurine concentrations in normal dog and in dogs with heart disease. J. Vet. Intern. Med. 1995, 9, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Lubec, L.; Ya-hue, Z.; Pertti, T.; Kitzmueller, E.; Lubec, G. Distribution and disappearance of the radiolabeled carbon derived from -arginine and taurine in the mouse. Life Sci. 1997, 60, 2273–2381. [Google Scholar] [CrossRef]
- Kocsis, J.J.; Kostos, V.J.; Baskin, S.I. Taurine levels in the heart tissues of various species. In Taurine; Huxtable, R., Barbeau, A., Eds.; Raven Press: New York, NY, USA, 1976; pp. 145–153. [Google Scholar]
- Schaffer, S.; Solodushko, V.; Azuma, J. Taurine-deficient cardiomyopathy: Role of phospholipids, calcium and osmotic stress. Adv. Expt. Med. Biol. 2000, 483, 57–69. [Google Scholar]
- Pion, P.D.; Kittleson, M.D.; Rogers, Q.R.; Morris, J.G. Myocardial failure in cats associated with low plasma taurine: A reversible cardiomyopathy. Science 1987, 237, 764–768. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Kimura, Y.; Uozumi, I.Y.; Takai, M.; Muraoka, S.; Matsuda, T.; Ueki, K.; Yoshiyama, M.; Ikawa, M.; Okabe, M.; et al. Taurine depletion caused by knocking out the taurine transporter gene leads to a cardiomyopathy with cardiac atrophy. J. Mol. Cell Cardiol. 2008, 44, 927–937. [Google Scholar] [CrossRef]
- Novotny, M.J.; Hogan, P.A.; Paley, D.M.; Adams, H.R. Systolic and diastolic dysfunction of the left ventricle induced by taurine deficiency in cats. Am. J. Physiol. 1991, 261, H121–H127. [Google Scholar] [CrossRef]
- Mihl, C.; Dassen, W.R.H.; Kuipers, H. Cardiac remodelling: Concentric versus eccentric hypertrophy in strength and endurance athletes. Neth. Heart J. 2008, 16, 129–133. [Google Scholar] [CrossRef] [Green Version]
- Gwathmey, J.K.; Copelas, L.; MacKinnon, R.; Schoen, F.J.; Feldman, M.D.; Grossman, W.; Morgan, J.P. Abnormal intracellular calcium handling in myocardium from patients with end-stage heart failure. Circ. Res. 1987, 61, 70–76. [Google Scholar] [CrossRef] [Green Version]
- Chapman, R.A.; Suleiman, M.-S.; Earm, Y.E. Taurine and the heart. Cardiovasc. Res. 1993, 27, 358–363. [Google Scholar] [CrossRef]
- Schaffer, S.W.; Solodushko, V.; Kakhniashvili, D. Beneficial effect of taurine depletion on osmotic sodium and calcium loading during chemical hypoxia. Am. J. Physiol. 2002, 282, C1113–C1120. [Google Scholar] [CrossRef] [Green Version]
- Jong, C.J.; Ito, T.; Mozaffari, M.; Azuma, J.; Schaffer, S. Effect of β-alanine treatment on mitochondrial taurine level and 5-taurinomethyluridine content. J. Biomed. Sci. 2011, 17 (Suppl. S1), S25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurata, S.; Ohtsuki, T.; Wada, T.; Kirino, Y.; Takai, K.; Saigo, K.; Watanabe, K.; Suzuki, T. Decoding property of C5 uridine modification at the wobble position of tRNA anticodon. Nucleic Acids Res. 2003, 3, 245–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaffer, S.W.; Jong, C.J.; Ito, T.; Azuma, J. Role of taurine in the pathogenesis of MELAS and MERRF. Amino Acids 2014, 46, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Jong, C.J.; Ito, T.; Prentice, H.; Wu, J.-Y.; Schaffer, S.W. Role of Mitochondria and Endoplasmic Reticulum in Taurine-Deficiency-Mediated Apoptosis. Nutrients 2017, 9, 795. [Google Scholar] [CrossRef] [Green Version]
- Schaffer, S.W.; Shimada-Takaura, K.; Jong, C.J.; Ito, T.; Takahashi, K. Impaired energy metabolism of the taurine-deficient heart. Amino. Acids 2016, 48, 549–558. [Google Scholar] [CrossRef]
- Shetewy, A.; Shimada-Takaura, K.; Warner, D.; Jong, C.J.; Mehdi, A.B.; Alexeyev, M.; Takahashi, K.; Schaffer, S.W. Mitochondrial defects associated with B-alanine toxicity: Relevance to hyper-beta-alaninemia. Mol. Cell Biochem. 2016, 416, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Radad, K.; Rausch, W.-D.; Gille, G. Rotenone induces cell death in primary dopaminergic culture by increasing ROS production and inhibiting mitochondrial respiration. Neurochem. Int. 2006, 49, 379–386. [Google Scholar] [CrossRef]
- Patel, M.S.; Korotchkina, L.G. Regulation of the pyruvate dehydrogenase complex. Biochem. Soc. Trans. 2006, 34, 217–222. [Google Scholar] [CrossRef]
- Hansson, A.; Hance, N.; Dufour, E.; Rantanen, A.; Hultenby, K.; Clayton, D.A.; Wibom, R.; Larsson, N.G. A switch in metabolism precedes increased mitochondrial biogenesis in respiratory chain-deficient mouse hearts. Proc. Natl. Acad. Sci. USA 2004, 101, 3136–3141. [Google Scholar] [CrossRef] [Green Version]
- Ingwall, J.S.; Weiss, R.G. Is the failing heart energy starved? Circ. Res. 2004, 95, 135–145. [Google Scholar] [CrossRef]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 2005, 85, 1093–1199. [Google Scholar] [CrossRef] [PubMed]
- Doenst, T.; Pytel, G.; Schrepper, A.; Amorium, P.; Farber, G.; Shingu, Y.; Mohr, F.W.; Schwarzer, M. Decreased Rates of Substrate Oxidation Ex Vivo Predict the Onset of Heart Failure and Contractile in Rats With Pressure Overload Dysfunction in rats with pressure overload. Cardiovasc. Res. 2010, 86, 461–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosca, M.G.; Hoppel, C.L. Mitochondrial dysfunction in heart failure. Heart Fail Rev. 2013, 18, 1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grivernnikova, V.G.; Vinogradov, A.D. Generation of superoxide by the mitochondrial complex I. Biochim. Biophys. Acta 2006, 1757, 553–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleier, L.; Drose, B. Superoxide generation by complex III. From mechanistic rationales to functional consequences. Biochem. Biophys. Acta 2013, 1827, 1320–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jong, C.J.; Azuma, J.; Schaffer, S. Mechanism underlying the antioxidant activity of taurine: Prevention of mitochondrial oxidant production. Amino Acids 2012, 42, 2223–2232. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Sheu, S.S.; Robotham, J.L.; Yoon, Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc. Res. 2008, 79, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Suen, D.-F.; Norris, K.L.; Youle, R.J. Mitochondria dynamics and apoptosis. Genes Dev. 2008, 22, 1577–1590. [Google Scholar] [CrossRef] [Green Version]
- Henry-Mowatt, J.; Dive, C.; Martinou, J.-C.; James, D. Role of mitochondrial membrane permeabilization in apoptosis and cancer. Oncogene 2004, 23, 2850–2860. [Google Scholar] [CrossRef] [Green Version]
- Jong, C.J.; Azuma, J.; Schaffer, S.W. Role of Mitochondrial Permeability Transition in Taurine Deficiency-Induced Apoptosis. Exp. Clin. Cardiol. 2011, 16, 125–128. [Google Scholar]
- Hoppins, S.; Lackner, L.; Nunnari, J. The machines that dividend fuse mitochondria. Annu. Rev. Biochem. 2007, 7, 751–780. [Google Scholar] [CrossRef] [PubMed]
- Azuma, J.; Hasegawa, H.; Sawamura, A.; Awata, N.; Harada, H.; Ogura, K.; Kishimoto, S. Taurine for treatment of congestive heart failure. Int. J. Cardiol. 1982, 2, 303–304. [Google Scholar] [CrossRef]
- Takihara, K.; Azuma, J.; Awata, N.; Ohta, H.; Hamaguchi, T.; Sawamura, A.; Tanaka, Y.; Kishimoto, S.; Sperelakis, N. Beneficial effect of taurine in rabbits with chronic congestive heart failure. Am. Heart J. 1986, 112, 11278–11284. [Google Scholar] [CrossRef]
- Ohta, H.; Azuma, J.; Onishi, S.; Awata, N.; Takihara, K.; Kishimoto, S. Protective effect of taurine against isoprenaline-induced myocardial damage. Basic Res. Cardiol. 1986, 81, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Ohta, H.; Azuma, J.; Awata, N.; Hamaguchi, T.; Tanaka, Y.; Sawamura, A.; Kishimoto, S.; Sperelakis, N. Mechanism of the protective action of taurine against isoprenaline induced myocardial damage. Cardiovasc. Res. 1988, 22, 407–413. [Google Scholar] [CrossRef]
- Azuma, J.; Sawamura, A.; Awata, N.; Ohta, H.; Hamaguchi, T.; Harada, H.; Takihara, K.; Hasegawa, H.; Yamagami, T.; Ishiyama, T.; et al. Therapeutic effect of taurine in congestive heart failure: A double blind crossover trial. Clin. Cardiol. 1985, 8, 276–282. [Google Scholar] [CrossRef]
- Ito, T.; Schaffer, S.; Azuma, J. The effect of taurine on chronic heart failure: Actions of taurine against catecholamine and angiotensin II. Amino Acids 2013, 46, 111–119. [Google Scholar] [CrossRef]
- Jeejeebhoy, F.; Keith, M.; Freeman, M.; Barr, A.; McCall, M.; Kurian, R.; Mazer, D.; Errett, L. Nutritional supplementation with MyoVive repletes essential cardiac myocyte nutrients and reduces left ventricular size in patients with left ventricular dysfunction. Am. Heart. J. 2002, 143, 1092–1100. [Google Scholar] [CrossRef]
- Brody, J.I.; Soltys, H.D.; Zinsser, H.F. Folic acid deficiency in congestive heart failure. Brit. Heart J. 1969, 31, 741–745. [Google Scholar] [CrossRef] [Green Version]
- Kirino, Y.; Goto, Y.-I.; Campos, Y.; Aremos, J.; Suzuki, T. Specific correlation between the wobble modification deficiency in mutant tRNAs and the clinical features of a human mitochondrial disease. Proc. Natl. Acad. Sci. USA 2005, 1029, 7127–7132. [Google Scholar] [CrossRef] [Green Version]
- Rikimaru, M.; Ohsawa, Y.; Wolf, A.M.; Ichimiya, H.; Kamimura, N.; Nishimatsu, S.; Ohta, S.; Sunada, Y. Taurine ameliorates impaired mitochondrial function and prevents stroke-like episodes in patients with MELAS. Intern. Med. 2012, 51, 3351–3357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
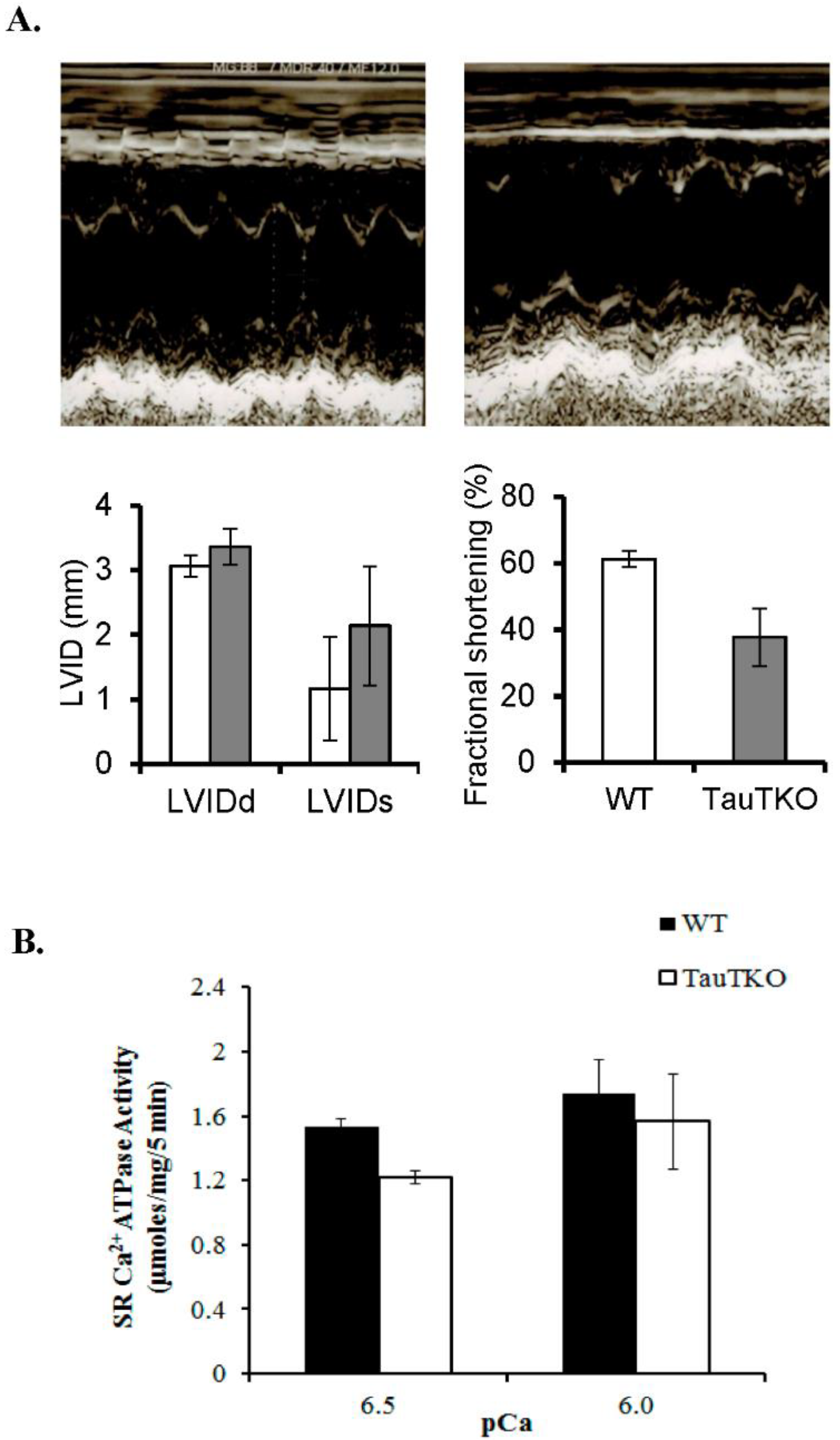
Figure 1.
Reduction of cardiac and sarcoplasmic function in taurine transporter knockout (TauTKO) mice. (A) Left ventricular function was assessed by echocardiography. Shown are representative M-mode echocardiograms of wildtype (WT) (left panel) and TauTKO mice (right panel). LVDd, LV (LV end diastolic dimensions); LVDs (LV end systolic dimension) and LVFS (LV fractional shortening). (B) Sarcoplasmic reticular Ca2+ ATPase activity of WT and TauTKO hearts. The recovery of SR protein was similar between WT and TauTKO hearts. SR Ca2+ ATPase activity of isolated SR from WT and TauTKO hearts was assayed at pCa 6.5 and 6.0. Significant differences exist between SR Ca2+ ATPase activity of WT and TauTKO hearts (p < 0.05). Reprinted from “Role of protein phosphorylation in excitation-contraction coupling in taurine deficient hearts” by K.C. Ramila, C. J. Jong, V. Pastukh, T. Ito, J. Azuma, S.W. Schaffer, 2015, American Journal of Physiology-Heart and Circulatory Physiology, 308(3), p. H232.
Figure 1.
Reduction of cardiac and sarcoplasmic function in taurine transporter knockout (TauTKO) mice. (A) Left ventricular function was assessed by echocardiography. Shown are representative M-mode echocardiograms of wildtype (WT) (left panel) and TauTKO mice (right panel). LVDd, LV (LV end diastolic dimensions); LVDs (LV end systolic dimension) and LVFS (LV fractional shortening). (B) Sarcoplasmic reticular Ca2+ ATPase activity of WT and TauTKO hearts. The recovery of SR protein was similar between WT and TauTKO hearts. SR Ca2+ ATPase activity of isolated SR from WT and TauTKO hearts was assayed at pCa 6.5 and 6.0. Significant differences exist between SR Ca2+ ATPase activity of WT and TauTKO hearts (p < 0.05). Reprinted from “Role of protein phosphorylation in excitation-contraction coupling in taurine deficient hearts” by K.C. Ramila, C. J. Jong, V. Pastukh, T. Ito, J. Azuma, S.W. Schaffer, 2015, American Journal of Physiology-Heart and Circulatory Physiology, 308(3), p. H232.
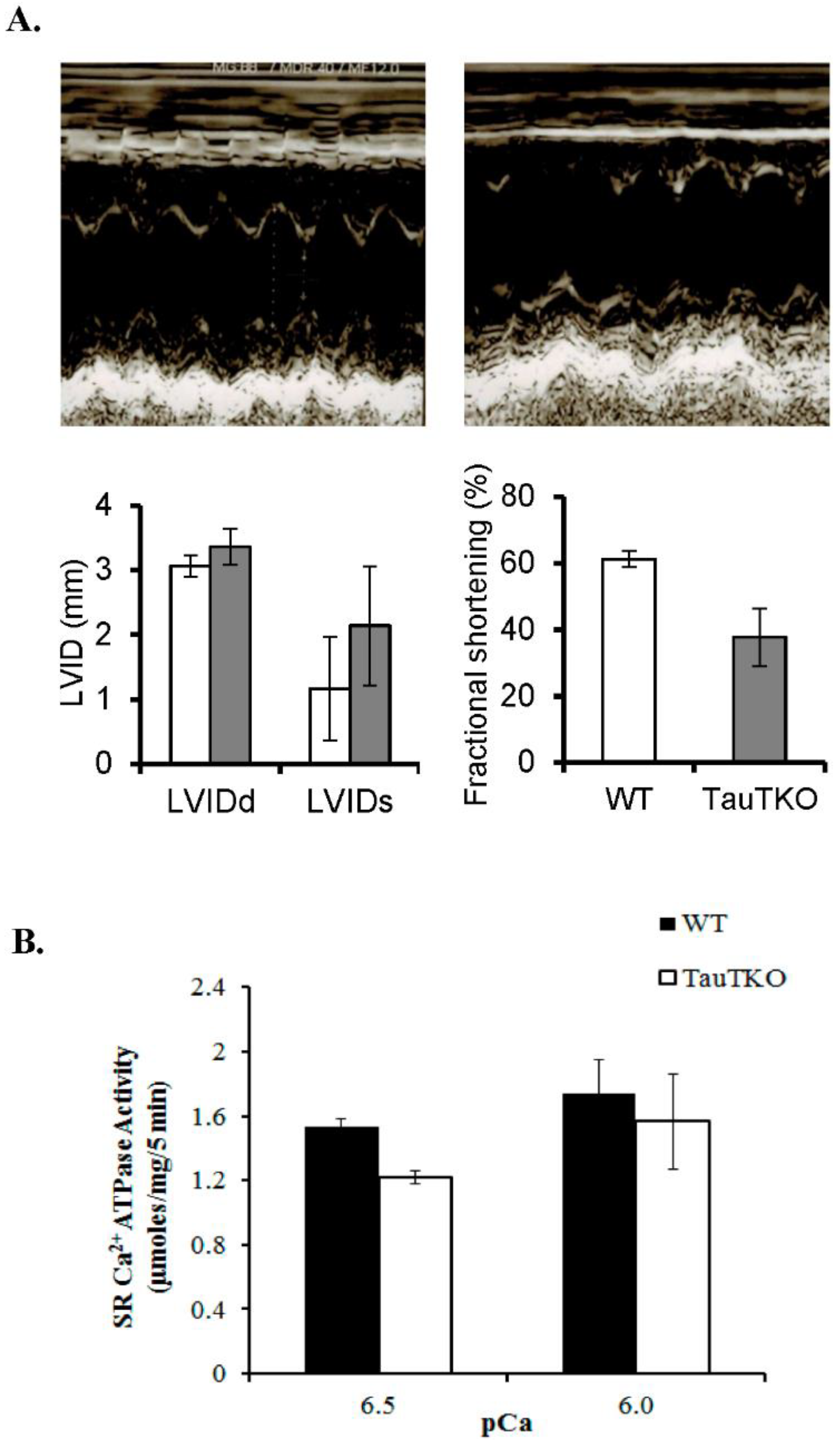
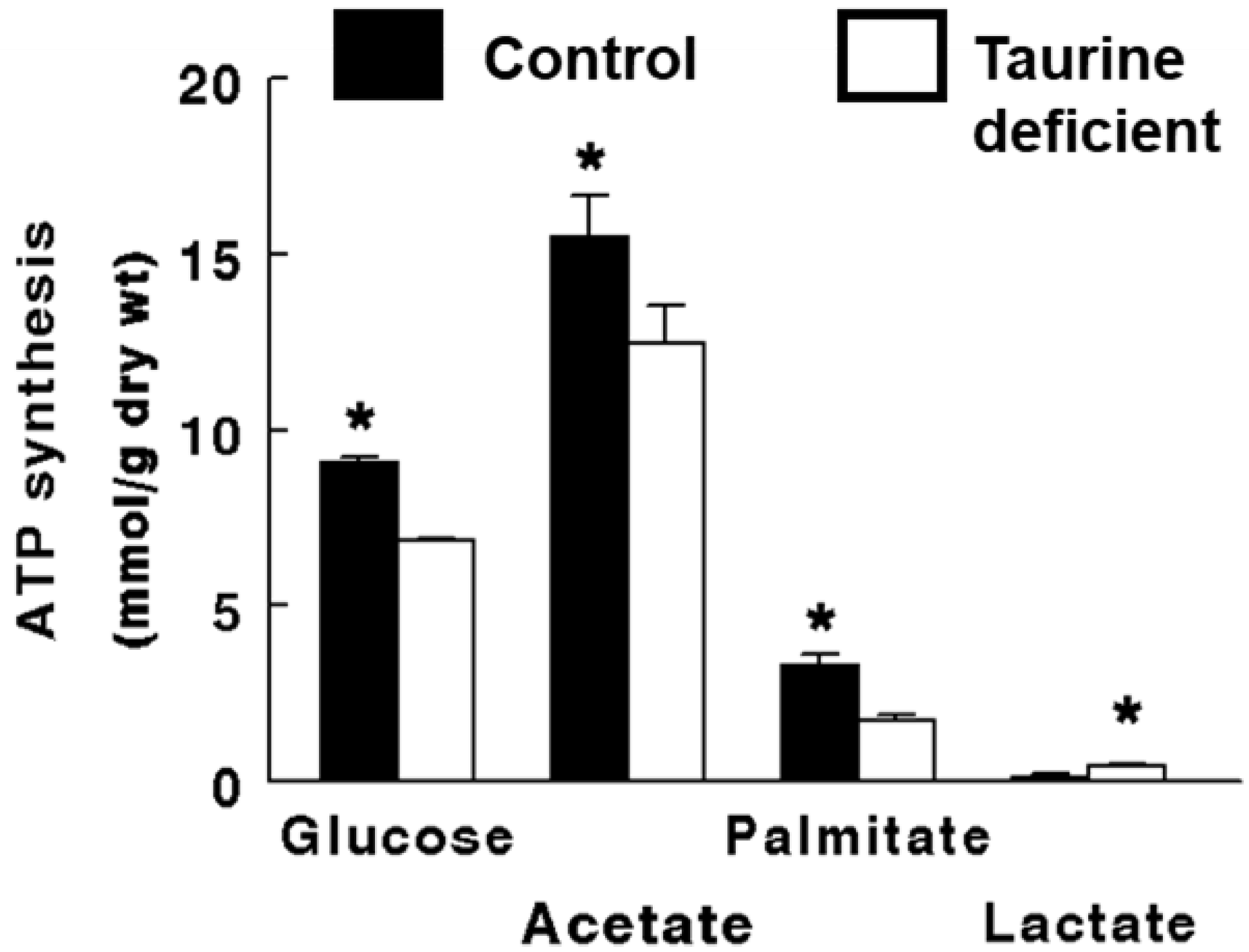
Figure 2.
Taurine deficiency reduces ATP synthesis in working hearts perfused with Krebs–Henseleit buffer containing 5 mM 3H-glucose and 5 mM 14C-acetate. Preload and afterload were fixed at 20 cm and 90 cm H2O, respectively, and the hearts were paced at 300 beats/min. The rates of glucose utilization and citric acid cycle flux were calculated from the amount of 3H-H2O and 14C-CO2 found in the perfusate, respectively The rate of ATP synthesis from glucose and acetate is based on the oxidation of the two exogenous substrates. ATP synthesis from lactate refers to the amount of ATP generated from the metabolism of glucose to lactate. Based on the model described by Schaffer et al. (2016), palmitate oxidation was determined from the rate of oxygen consumption. The rate of ATP synthesis from palmitate represents the oxidation of endogenous stores of fatty acid. The P/O ratio used in calculating ATP synthesis from palmitate was 2.8. A P/O ratio used for glucose was 3.17 and for acetate was 2.5. Lactate and pyruvate levels of the perfusate were determined and the production of ATP from lactate was calculated assuming 2 μmol ATP/μmol of glucose. * Asterisks denote a significant difference between the taurine-deficient and control hearts. Reprinted from “Impaired energy metabolism of the taurine-deficient heart” by S. Schaffer, K. Shimada-Takaura, C.J. Jong, T. Ito, K. Takahashi, 2016, Amino Acids, 48(2), p. 549.
Figure 2.
Taurine deficiency reduces ATP synthesis in working hearts perfused with Krebs–Henseleit buffer containing 5 mM 3H-glucose and 5 mM 14C-acetate. Preload and afterload were fixed at 20 cm and 90 cm H2O, respectively, and the hearts were paced at 300 beats/min. The rates of glucose utilization and citric acid cycle flux were calculated from the amount of 3H-H2O and 14C-CO2 found in the perfusate, respectively The rate of ATP synthesis from glucose and acetate is based on the oxidation of the two exogenous substrates. ATP synthesis from lactate refers to the amount of ATP generated from the metabolism of glucose to lactate. Based on the model described by Schaffer et al. (2016), palmitate oxidation was determined from the rate of oxygen consumption. The rate of ATP synthesis from palmitate represents the oxidation of endogenous stores of fatty acid. The P/O ratio used in calculating ATP synthesis from palmitate was 2.8. A P/O ratio used for glucose was 3.17 and for acetate was 2.5. Lactate and pyruvate levels of the perfusate were determined and the production of ATP from lactate was calculated assuming 2 μmol ATP/μmol of glucose. * Asterisks denote a significant difference between the taurine-deficient and control hearts. Reprinted from “Impaired energy metabolism of the taurine-deficient heart” by S. Schaffer, K. Shimada-Takaura, C.J. Jong, T. Ito, K. Takahashi, 2016, Amino Acids, 48(2), p. 549.

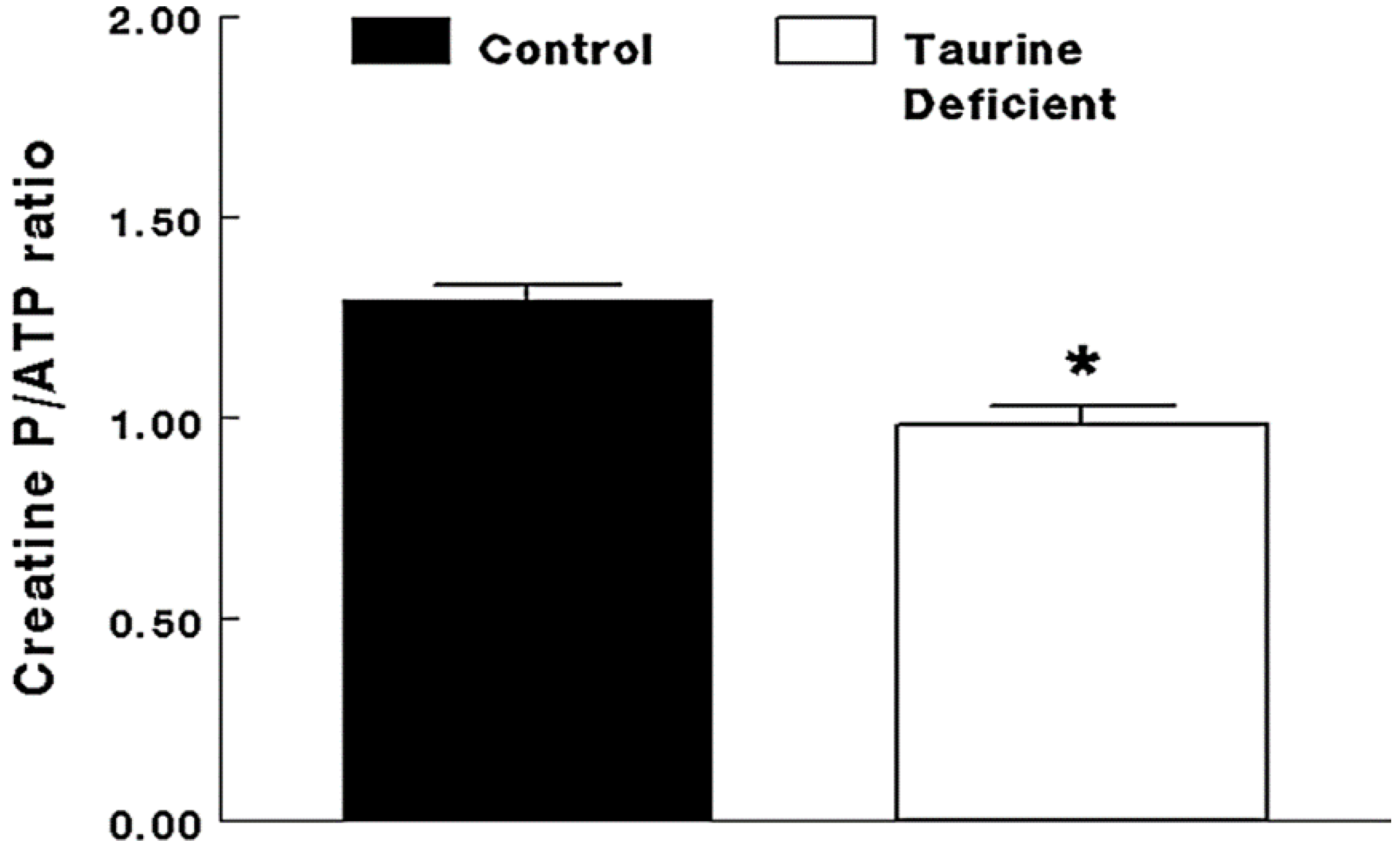
Figure 3.
Taurine-deficient hearts are energy-deficient. Taurine-deficient and normal control hearts were perfused with Krebs–Henseleit buffer containing 5 mM glucose and 5 mM acetate. Hearts were frozen with tongs placed in liquid nitrogen. The hearts were extracted with perchloric acid and the extract neutralized before assaying for creatine phosphate and ATP levels. The data are expressed as a common measure of high energy phosphate, the creatine phosphate/ATP ratio. * The asterisk denotes a significant difference between taurine-deficient and normal hearts. Reprinted from “Impaired energy metabolism of the taurine-deficient heart” by S. Schaffer, K. Shimada-Takaura, C.J. Jong, T. Ito, K. Takahashi, 2016, Amino Acids, 48(2), p. 549.
Figure 3.
Taurine-deficient hearts are energy-deficient. Taurine-deficient and normal control hearts were perfused with Krebs–Henseleit buffer containing 5 mM glucose and 5 mM acetate. Hearts were frozen with tongs placed in liquid nitrogen. The hearts were extracted with perchloric acid and the extract neutralized before assaying for creatine phosphate and ATP levels. The data are expressed as a common measure of high energy phosphate, the creatine phosphate/ATP ratio. * The asterisk denotes a significant difference between taurine-deficient and normal hearts. Reprinted from “Impaired energy metabolism of the taurine-deficient heart” by S. Schaffer, K. Shimada-Takaura, C.J. Jong, T. Ito, K. Takahashi, 2016, Amino Acids, 48(2), p. 549.
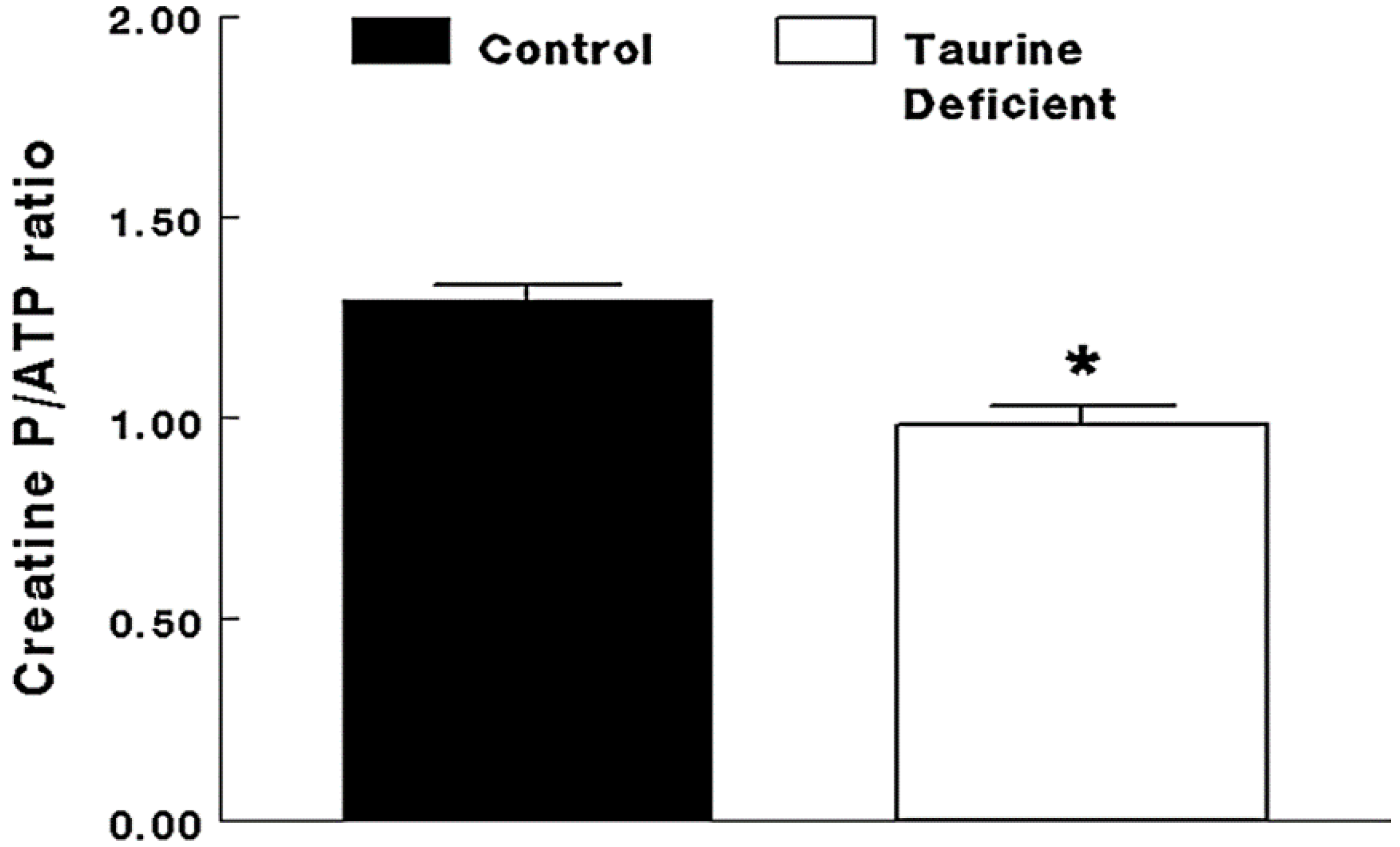
Figure 4.
MitoTempo suppresses the activation of caspases 9 and 3 in taurine-deficient hearts. Three-month-old TauTKO and wildtype mice were administered i.p. the mitochondrial-specific antioxidant, MitoTempo (1.4 mg/kg/day), for 7 days. Lysates of treated and untreated hearts were subjected to western blot analyses for the active, cleaved form of caspase 9, as well as the inactive pro-caspase 9 and 3 forms. * Asterisks denote significant differences between the taurine-deficient and wildtype control hearts. Reprinted from “Role of mitochondria and endoplasmic reticulum in taurine-deficiency-mediated apoptosis” by C.J. Jong, T. Ito, H. Prentice, J.Y. Wu, S. W. Schaffer, 2017, Nutrients, 9(8), p. 795.
Figure 4.
MitoTempo suppresses the activation of caspases 9 and 3 in taurine-deficient hearts. Three-month-old TauTKO and wildtype mice were administered i.p. the mitochondrial-specific antioxidant, MitoTempo (1.4 mg/kg/day), for 7 days. Lysates of treated and untreated hearts were subjected to western blot analyses for the active, cleaved form of caspase 9, as well as the inactive pro-caspase 9 and 3 forms. * Asterisks denote significant differences between the taurine-deficient and wildtype control hearts. Reprinted from “Role of mitochondria and endoplasmic reticulum in taurine-deficiency-mediated apoptosis” by C.J. Jong, T. Ito, H. Prentice, J.Y. Wu, S. W. Schaffer, 2017, Nutrients, 9(8), p. 795.
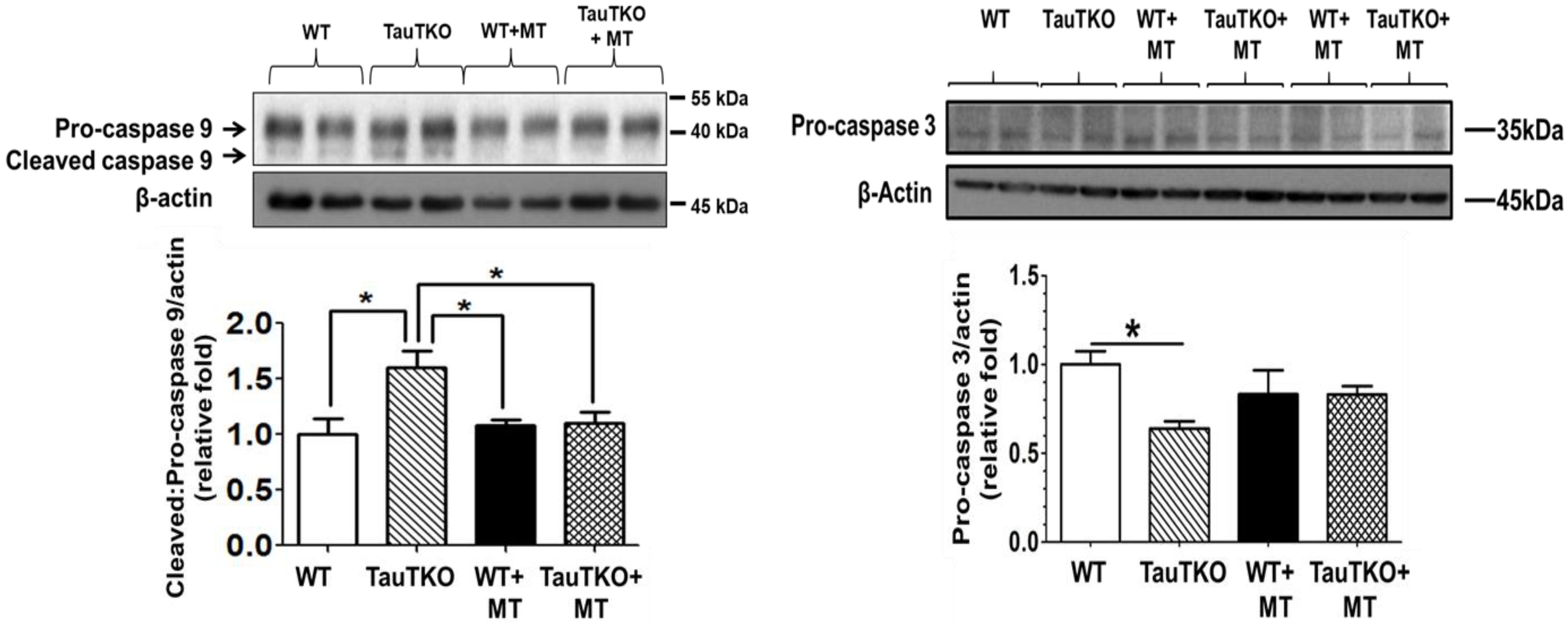
Figure 5.
Effect of taurine therapy on symptoms of congestive heart failure. A 60-year-old female with New York Heart Association class IV congestive heart failure entered on week 0 into a clinical trial evaluating the effect of taurine therapy (either 2 g/day or 4 g/day) on symptoms of congestive heart failure. The baseline condition of the patient is represented by values at weeks −4 and 0. Prior to the onset of taurine therapy, the patient was administered digoxin (80 mg/day), furosemide (80 mg/day) and spironolactone (100 mg/day). Throughout the course of the 30-week study, changes were made in the treatment protocol. The patient responded favorably to taurine therapy, as the NY Heart Association classification was reduced from class IV to class II and there was a corresponding improvement in orthopnea, dyspnea on exertion and fatigue.
Figure 5.
Effect of taurine therapy on symptoms of congestive heart failure. A 60-year-old female with New York Heart Association class IV congestive heart failure entered on week 0 into a clinical trial evaluating the effect of taurine therapy (either 2 g/day or 4 g/day) on symptoms of congestive heart failure. The baseline condition of the patient is represented by values at weeks −4 and 0. Prior to the onset of taurine therapy, the patient was administered digoxin (80 mg/day), furosemide (80 mg/day) and spironolactone (100 mg/day). Throughout the course of the 30-week study, changes were made in the treatment protocol. The patient responded favorably to taurine therapy, as the NY Heart Association classification was reduced from class IV to class II and there was a corresponding improvement in orthopnea, dyspnea on exertion and fatigue.
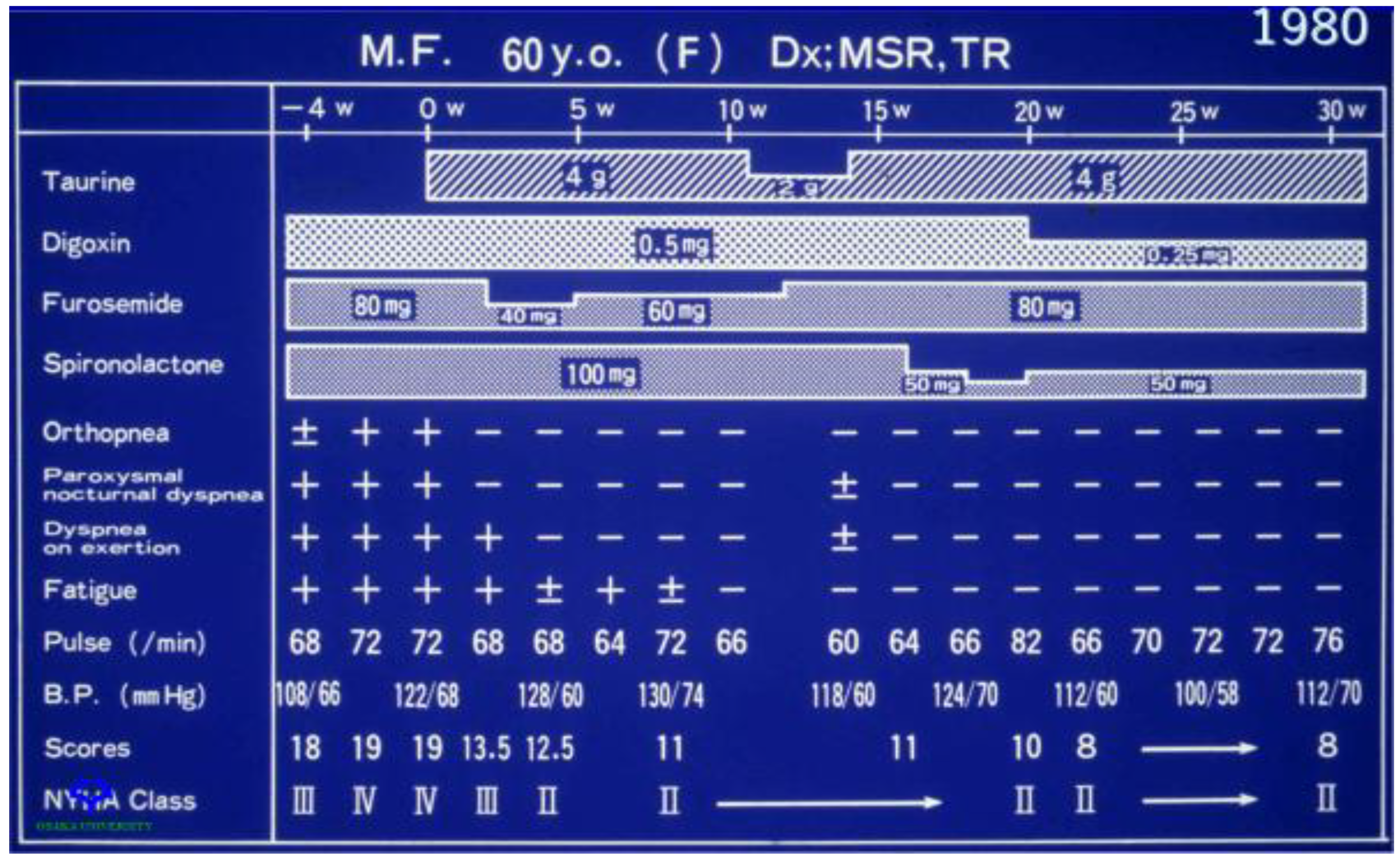
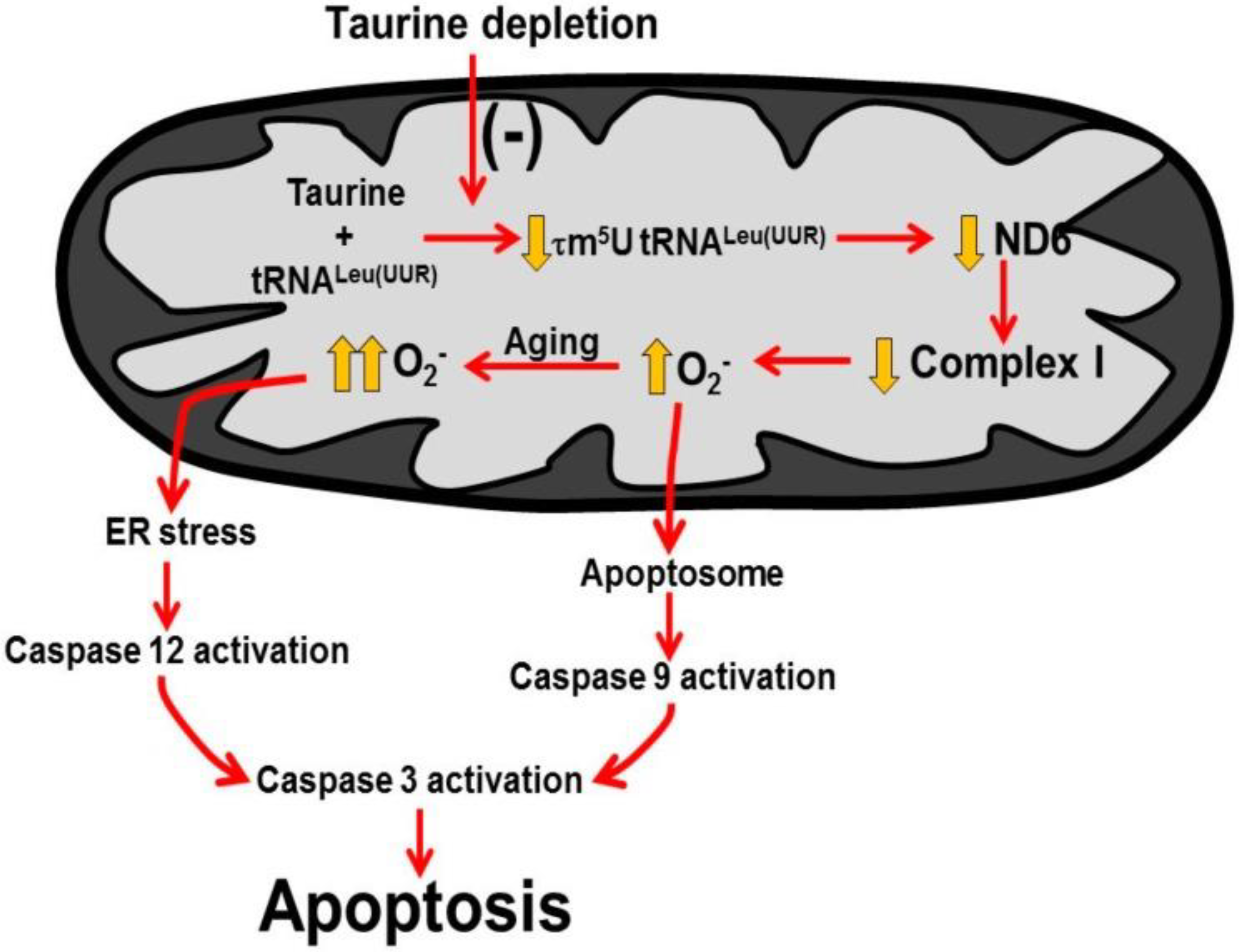
Figure 6.
Taurine deficiency induces myocardial cell death. A decline in mitochondrial taurine content decreases the formation of the taurine conjugate, 5-taurinomethyluridine-RNALeu(UUR) that alters the binding of the anticodon of the tRNA to the UUG codon of the mitochondrial mRNAs. This reduces the biosynthesis of ND6, a subunit of complex I of the electron transport chain. As a result, complex I activity decreases and disrupts the electron flow through the respiratory chain, causing the diversion of electrons to oxygen to form excessive superoxide. Mitochondrial damage leads to increased mitochondrial membrane permeability that promotes cytochrome c release and formation of the apoptosome to activate caspase 9 and 3 to stimulate apoptosis. In aging, oxidative stress initiates ER (endoplasmic reticular) stress that activates caspase 12, which is an initiating caspase that further stimulates caspase 3 and apoptosis. Reprinted from “Role of mitochondria and endoplasmic reticulum in taurine-deficiency-mediated apoptosis” by C.J. Jong, T. Ito, H. Prentice, J.Y. Wu, S. W. Schaffer, 2017, Nutrients, 9(8), p. 795.
Figure 6.
Taurine deficiency induces myocardial cell death. A decline in mitochondrial taurine content decreases the formation of the taurine conjugate, 5-taurinomethyluridine-RNALeu(UUR) that alters the binding of the anticodon of the tRNA to the UUG codon of the mitochondrial mRNAs. This reduces the biosynthesis of ND6, a subunit of complex I of the electron transport chain. As a result, complex I activity decreases and disrupts the electron flow through the respiratory chain, causing the diversion of electrons to oxygen to form excessive superoxide. Mitochondrial damage leads to increased mitochondrial membrane permeability that promotes cytochrome c release and formation of the apoptosome to activate caspase 9 and 3 to stimulate apoptosis. In aging, oxidative stress initiates ER (endoplasmic reticular) stress that activates caspase 12, which is an initiating caspase that further stimulates caspase 3 and apoptosis. Reprinted from “Role of mitochondria and endoplasmic reticulum in taurine-deficiency-mediated apoptosis” by C.J. Jong, T. Ito, H. Prentice, J.Y. Wu, S. W. Schaffer, 2017, Nutrients, 9(8), p. 795.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Differential effects of β-alanine-mediated taurine depletion on mitochondrial respiration in permeabilized cardiomyocytes using either a complex I substrate combination, glutamate/malate, or a complex II substrate, succinate. Normal and taurine-deficient neonatal rat cardiomyocytes were prepared by incubating cells in buffer supplemented with 0 or 5 mM of beta-alanine for 48 h. The cells were then suspended in buffer containing either 4 mM glutamate and 2 mM malate or 5 mM succinate before being placed in a chamber fitted with a Clark oxygen electrode. The cell membrane was permeabilized with saponin, allowing the substrates to enter the mitochondria. After obtaining a stable rate of state 2 respiration, 1 µmol ADP (Adenosine diphosphate) was added to initiate state 3 respiration. A new slower rate of respiration (state 4) develops when ADP is completely converted to ATP. Actual state 3 and 4 respiration rates expressed as μatom/min/mg protein are shown in parentheses. RCR is the respiratory control ratio (state 3/state 4 ratio). * Asterisks denote a significant difference between the taurine-deficient and control groups (p < 0.05). Reprinted from “Mitochondrial defects associated with β-alanine toxicity: relevance to hyper-beta-alaninemia” by A. Shetewy, K. Shimada-Takaura, D. Warner, C.J. Jong, A. Al-Mehdi, M. Alexeyev, K. Takahashi, S. Schaffer, 2016, Molecular and Cellular Biochemistry, 416(1–2), p. 11.
Table 1.
Differential effects of β-alanine-mediated taurine depletion on mitochondrial respiration in permeabilized cardiomyocytes using either a complex I substrate combination, glutamate/malate, or a complex II substrate, succinate. Normal and taurine-deficient neonatal rat cardiomyocytes were prepared by incubating cells in buffer supplemented with 0 or 5 mM of beta-alanine for 48 h. The cells were then suspended in buffer containing either 4 mM glutamate and 2 mM malate or 5 mM succinate before being placed in a chamber fitted with a Clark oxygen electrode. The cell membrane was permeabilized with saponin, allowing the substrates to enter the mitochondria. After obtaining a stable rate of state 2 respiration, 1 µmol ADP (Adenosine diphosphate) was added to initiate state 3 respiration. A new slower rate of respiration (state 4) develops when ADP is completely converted to ATP. Actual state 3 and 4 respiration rates expressed as μatom/min/mg protein are shown in parentheses. RCR is the respiratory control ratio (state 3/state 4 ratio). * Asterisks denote a significant difference between the taurine-deficient and control groups (p < 0.05). Reprinted from “Mitochondrial defects associated with β-alanine toxicity: relevance to hyper-beta-alaninemia” by A. Shetewy, K. Shimada-Takaura, D. Warner, C.J. Jong, A. Al-Mehdi, M. Alexeyev, K. Takahashi, S. Schaffer, 2016, Molecular and Cellular Biochemistry, 416(1–2), p. 11.
| Mitochondrial Respiration of Beta-Alanine Treated and Untreated Permeabilized Cardiomyocytes | ||||
|---|---|---|---|---|
| Condition | Substrate | State 3 rate | State 4 rate | RCR |
| Control | Glu-Mal | 1.0 (43.61) | 1.0 (11.03) | 1.0 |
| B-alanine | Glu-Mal | 0.78 ± 0.08 * | 0.9 ± 0.09 | 0.8 |
| Control | Succinate | 1.0 (65.4) | 1.0 (15.8) | 1.0 |
| B-alanine | Succinate | 0.98 ± 0.09 | 1.2 ± 0.1 | 0.95 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Schaffer, S.W.; Ito, T.; Azuma, J.; Jong, C.J.; Kramer, J.H. Mechanisms Underlying Development of Taurine-Deficient Cardiomyopathy. Hearts 2020, 1, 86-98. https://0-doi-org.brum.beds.ac.uk/10.3390/hearts1020010
AMA Style
Schaffer SW, Ito T, Azuma J, Jong CJ, Kramer JH. Mechanisms Underlying Development of Taurine-Deficient Cardiomyopathy. Hearts. 2020; 1(2):86-98. https://0-doi-org.brum.beds.ac.uk/10.3390/hearts1020010
Chicago/Turabian StyleSchaffer, Stephen W., Takashi Ito, Junichi Azuma, Chian Ju Jong, and Jay H. Kramer. 2020. "Mechanisms Underlying Development of Taurine-Deficient Cardiomyopathy" Hearts 1, no. 2: 86-98. https://0-doi-org.brum.beds.ac.uk/10.3390/hearts1020010