Deficiency in gp91Phox (NOX2) Protects against Oxidative Stress and Cardiac Dysfunction in Iron Overloaded Mice

Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal and Iron-Treatment Models, and gp91phox Knockout Strain
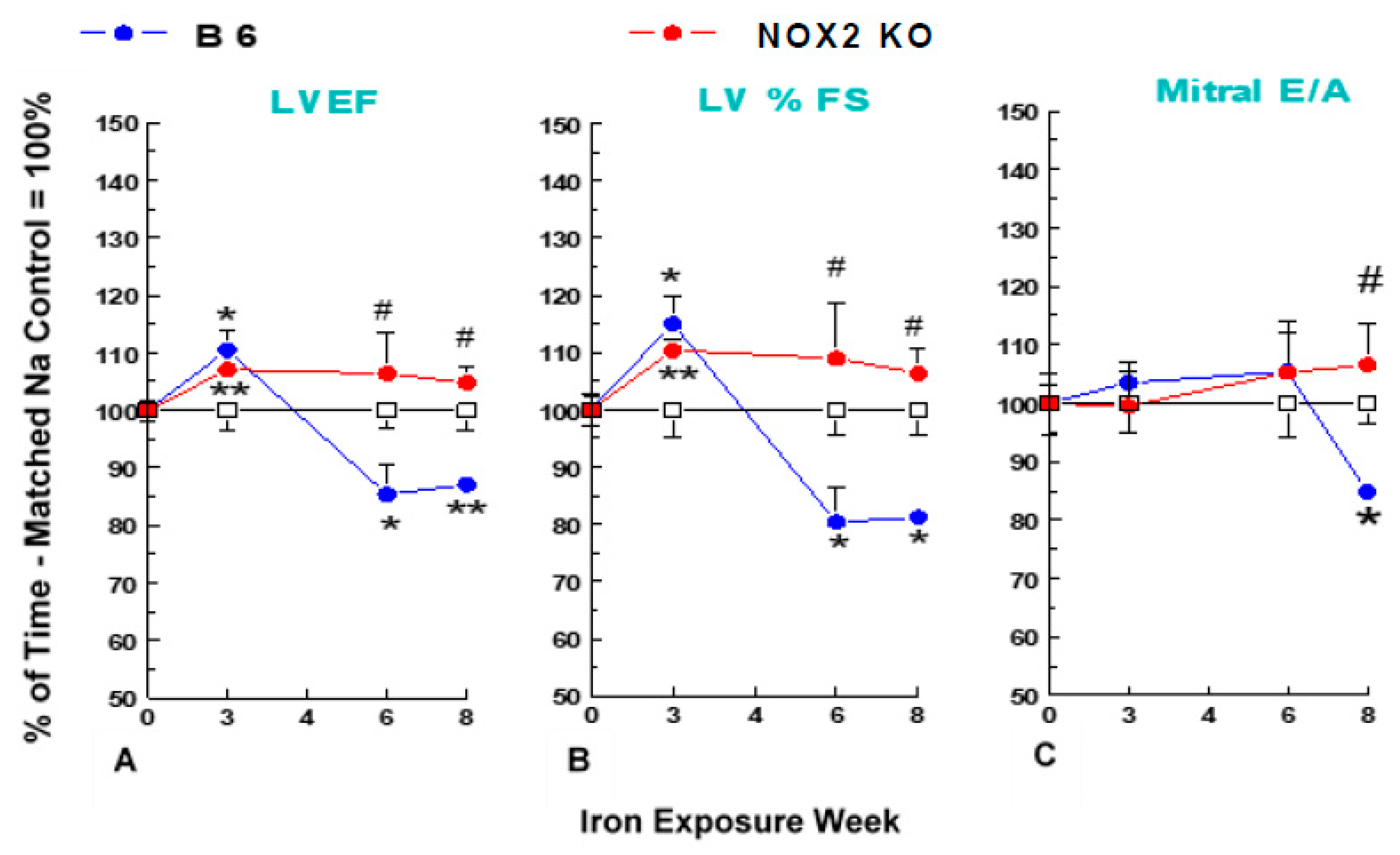
2.2. Non-Invasive Echocardiography
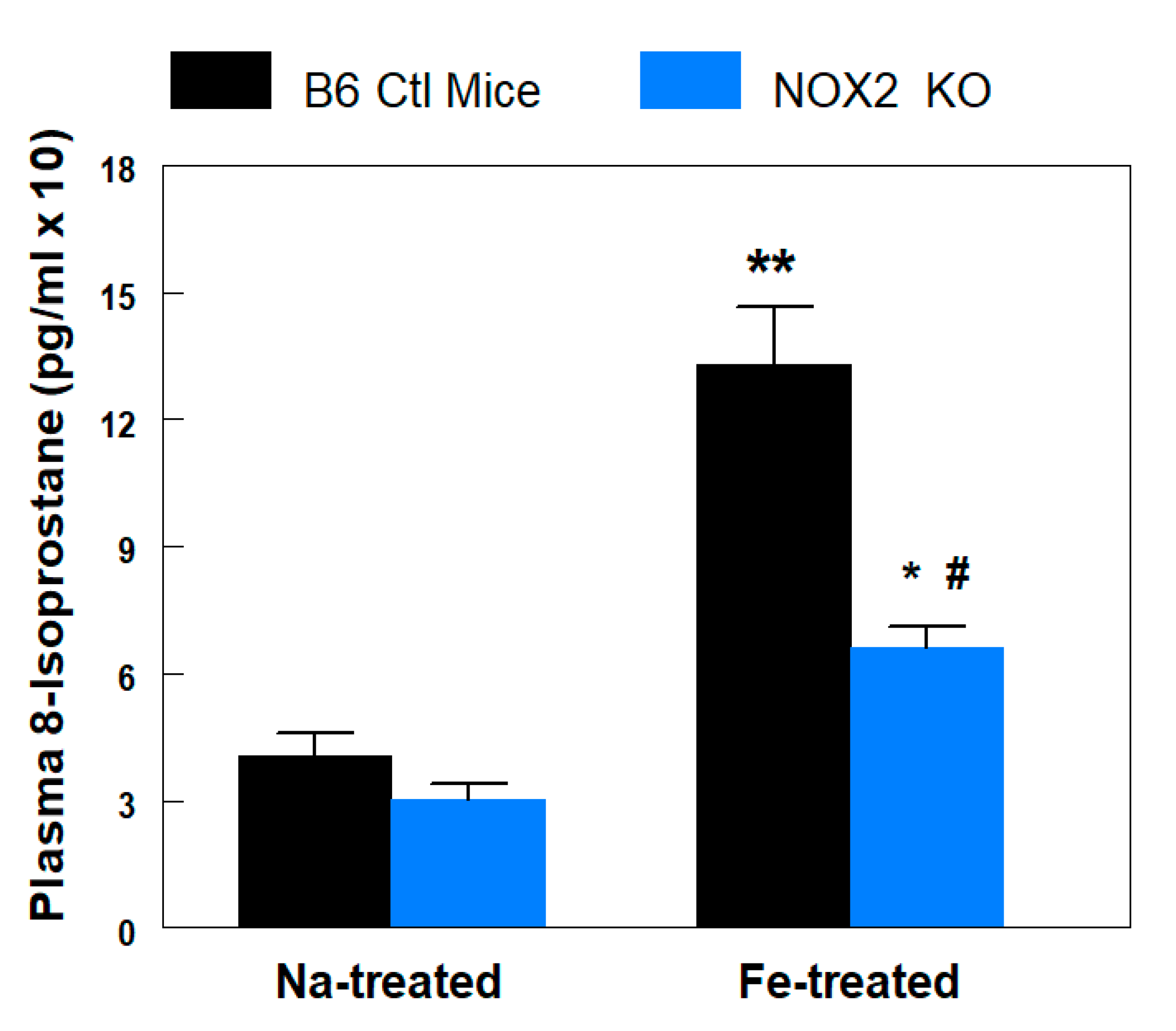
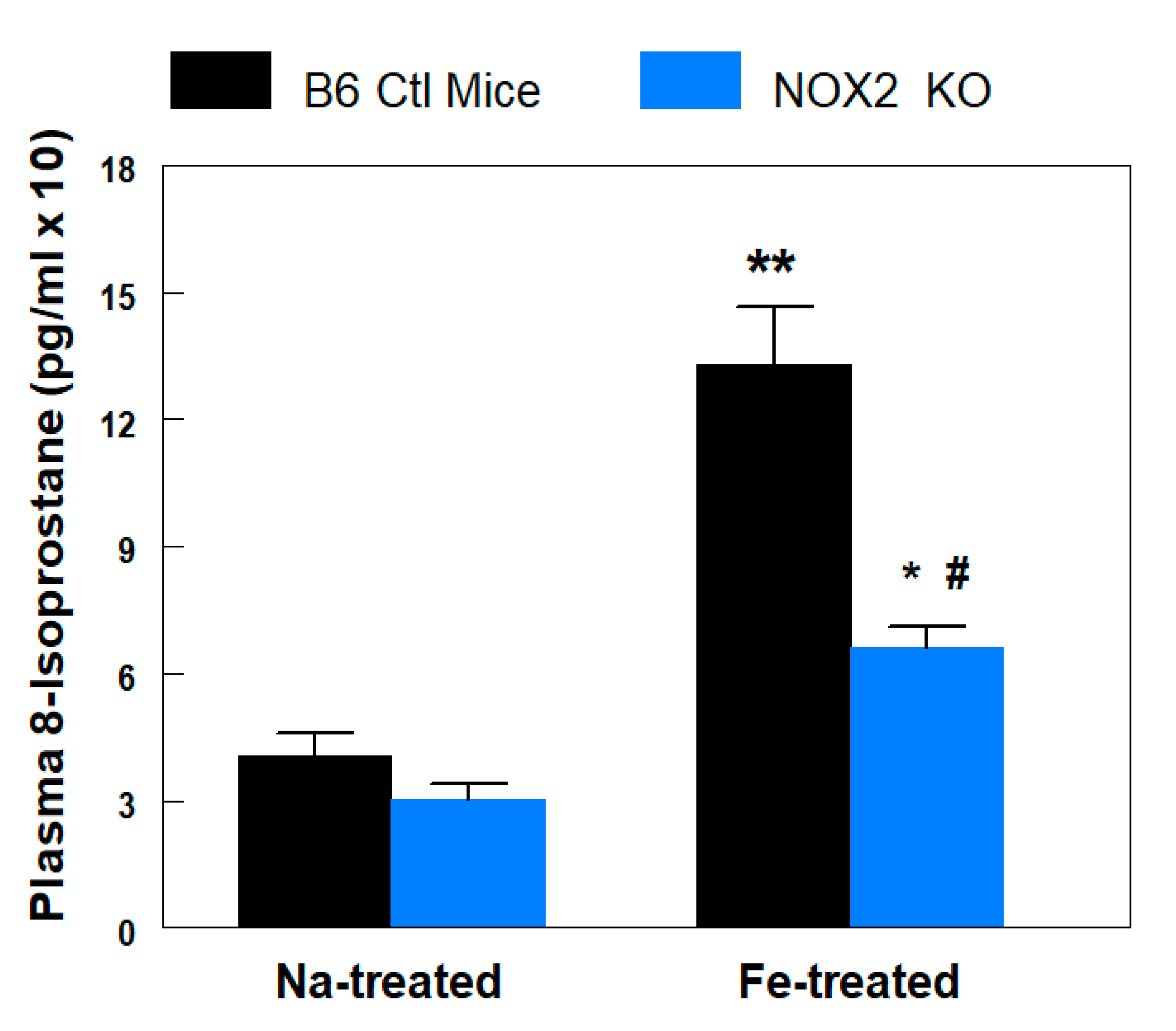
2.3. Plasma 8-Isoprostane Assay
2.4. Iron Determinations
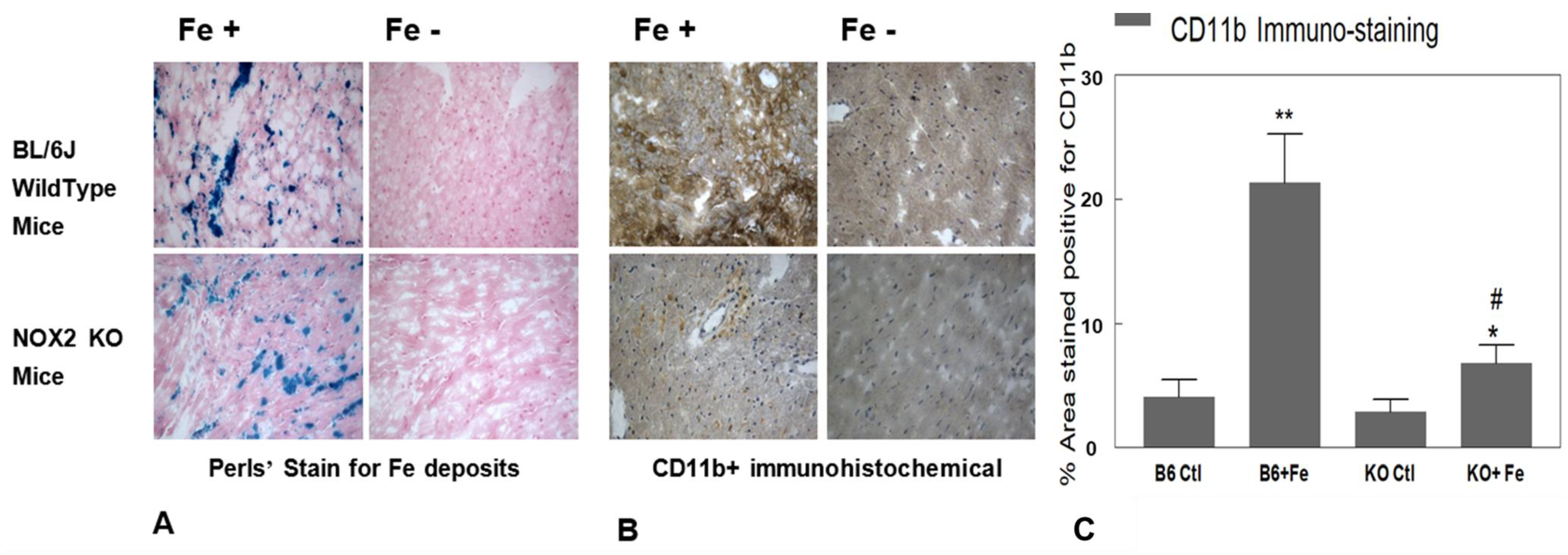
2.5. Cardiac Tissue CD11b+ Staining and Analysis
2.6. Statistical Assessments
3. Results
3.1. Weight Gain
3.2. Plasma 8-Isoprostane Levels
3.3. Cardiac Contractile Dysfunction
3.4. Fe Accumulation and White Blood Cell (WBC) Infiltration
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Zhang, H.; Zhabyeyev, P.; Wang, S.; Oudit, G.Y. Role of iron metabolism in heart failure: From iron deficiency to iron overload. Biochim. Biophys. Acta Mol. Basis. Dis. 2019, 1865, 1925–1937. [Google Scholar] [CrossRef] [PubMed]
- Coates, T.D. Iron overload in transfusion-dependent patients. Hematol. Am. Soc. Hematol. Educ. Program 2019, 2019, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Saliba, A.N. Iron overload in thalassemia: Different organs at different rates. Hematology Am. Soc. Hematol. Educ. Program 2017, 2017, 265–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imam, M.U.; Zhang, S.; Ma, J.; Wang, H.; Wang, F. Antioxidants mediate both iron homeostasis and oxidative stress. Nutrients 2017, 9, 671. [Google Scholar] [CrossRef]
- Gunther, T.; Hollriegl, V.; Vormann, J.; Disch, G.; Classen, H.G. Effects of Fe loading on vitamin E and malondialdehyde of liver, heart and kidney from rats fed diets containing various amounts of magnesium and vitamin E. Magnes. Bull. 1992, 14, 88–93. [Google Scholar]
- Halliwell, B.; Gutteridge, J.M.C. Oxygen free radicals and iron in relation to biology and medicine: Some problems and concepts. Arch. Biochem. Biophys. 1986, 246, 501–514. [Google Scholar] [CrossRef]
- Berdoukas, V.; Coates, T.D.; Cabantchik, Z.I. Iron and oxidative stress in cardiomyopathy in thalassemia. Free Radic. Biol. Med. 2015, 88, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Kramer, J.H.; Misik, V.; Weglicki, W.B. Lipid peroxidation-derived free radical production and post-ischemic myocardial reperfusion injury. Ann. N. Y. Acad. Sci. 1994, 723, 180–196. [Google Scholar] [CrossRef]
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. NADPH oxidase: Role in cardiovascular biology and disease. Circ. Res. 2000, 86, 494–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rupin, A.; Paysant, J.; Sansilverstri-Morel, P.; Lembrez, N.; Lacoste, J.M.; Cordi, A.; Verbeureu, T.J. Role of NADPH oxidase-mediated superoxide production in the regulation of E-selectin expression by Endothelial cells subjected to anoxia/reoxygenation. Cardiovasc. Res. 2004, 63, 323–330. [Google Scholar] [CrossRef]
- Zhang, Y.; Murugesan, P.; Huang, K.; Cai, H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: Novel therapeutic targets. Nat. Rev. Cardiol. 2020, 17, 170–194. [Google Scholar] [CrossRef] [PubMed]
- Kramer, J.H.; Spurney, C.F.; Iantorno, M.; Tziros, C.; Mak, I.T.; Weglicki, W.B. D-propranolol protects against oxidative stress and progressive cardiac dysfunction in Fe-overloaded rats. Can. J. Physiol. Pharmacol. 2012, 90, 1257–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollock, J.D.; Williams, D.A.; Gifford, M.A.; Li, L.L.; Du, X.; Fisherman, J.; Orkin, S.H.; Doerschuk, C.M.; Dinauer, M.C. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat. Genet. 1995, 9, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Spurney, C.F.; Sali, A.; Guerron, A.D.; Iantorno, M.; Yu, Q.; Gordish-Dressman, H.; Rayavarapu, S.; van der Meulen, J.; Hoffman, E.P.; Nagaraju, K. Losartan decreases cardiac muscle fibrosis and improves cardiac function in dystrophin-deficient mdx mice. J. Cardiovasc. Pharmacol. Ther. 2011, 16, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Zghoul, N.; Alam-Eldin, N.; Mak, I.T.; Silver, B.; Weglicki, W.B. Hypomagnesemia in diabetes patients: Comparison of serum and intracellular measurement of responses to magnesium supplementation and its role in inflammation. Diabetes Metab. Syndr. Obes. 2018, 11, 389–400. [Google Scholar] [CrossRef] [Green Version]
- Mak, I.T.; Chmielinska, J.J.; Spurney, C.F.; Weglicki, W.B.; Kramer, J.H. Combination ART-induced oxidative/nitrosative stress, neurogenic inflammation and cardiac dysfunction in HIV-1 transgenic (Tg) rats: Protection by Mg. Int. J. Mol. Sci. 2018, 19, 2409. [Google Scholar] [CrossRef] [Green Version]
- Luna, L.G. (Ed.) Manual of Histologic Staining Methods of the Armed Forces Institute of Pathology, 3rd ed.; Blakiston Division, McGraw-Hill: New York, NY, USA, 1968; pp. 184–185. [Google Scholar]
- Mak, I.T.; Kramer, J.H.; Chen, X.; Chmielinska, J.J.; Spurney, C.F.; Weglicki, W.B. Mg-supplementation attenuates ritonavir-induced hyperlipidemia, oxidative stress and cardiac dysfunction in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R1102–R1111. [Google Scholar] [CrossRef]
- Mak, I.T.; Chmielinska, J.J.; Kramer, J.H.; Weglicki, W.B. AZT-induced oxidative cardiovascular toxicity: Attenuation by Mg-supplementation. Cardiovasc. Toxicol. 2009, 9, 78–85. [Google Scholar] [CrossRef]
- Chmielinska, J.J.; Kramer, J.H.; Mak, I.T.; Spurney, C.F.; Weglicki, W.B. Substance P receptor blocker, aprepitant, inhibited cutaneous and other neurogenic inflammation side effects of the EGFR1-TKI, erlotinib. Mol. Cell Biochem. 2020, 465, 175–185. [Google Scholar] [CrossRef]
- Pepping, J.K.; Freeman, L.R.; Gupta, S.; Keller, J.N.; Bruce-Keller, A.J. NOX2 deficiency attenuates markers of adiposopathy and brain injury induced by high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E392–E404. [Google Scholar] [CrossRef] [Green Version]
- El-Benna, J.; Dang, P.M.; Gougerot-Pocidalo, M.A.; Marie, J.C.; Braut-Boucher, F. P47phox, the phagocyte NADPH oxidase/NOX2 organizer: Structure, phosphorylation and implication in diseases. Exp. Mol. Med. 2009, 41, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.M. Parsing the role of NADPH oxidase enzymes and reactive oxygen species in heart failure. Circulation 2015, 131, 602–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Günthel, M.; Barnett, P.; Christoffels, V.M. Development, proliferation, and growth of the mammalian heart. Mol. Ther. 2018, 26, 1599–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brissot, P.; Ropert, M.; Le Lan, C.; Loréal, O. Non-transferrin bound iron: A key role in iron overload and iron toxicity. Biochim. Biophys. Acta 2012, 1820, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Knutson, M.D. Non-transferrin-bound iron transporters. Free Radic. Biol. Med. 2019, 133, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Dimitrios, T.; Kremastinos, D.T.; Farmakis, D. Iron overload cardiomyopathy in clinical practice. Circulation 2011, 124, 2253–2263. [Google Scholar]
- Di Meo, S.; Venditti, P.; De Leo, T. Tissue protection against oxidative stress. Experientia 1996, 52, 786–794. [Google Scholar] [CrossRef]
- Aronow, W.S. Management of cardiac hemochromatosis. Arch. Med. Sci. 2018, 14, 560–568. [Google Scholar]
- Kolnagou, A.; Michaelides, Y.; Kontos, C.; Kyriacou, K.; Kontoghiorghes, G.J. Myocyte damage and loss of myofibers is the potential mechanism of iron overload toxicity in congestive cardiac failure in thalassemia. Complete reversal of the cardiomyopathy and normalization of iron load by deferiprone. Hemoglobin 2008, 32, 17–28. [Google Scholar] [CrossRef]
- Cavdar, Z.; Oktan, M.A.; Ural, C.; Calisir, M.; Kocak, A.; Heybeli, C.; Yildiz, S.; Arici, A.; Ellidokuz, H.; Celik, A.; et al. Renoprotective effects of alpha lipoic acid on iron overload-induced kidney injury in rats by suppressing NADPH oxidase 4 and p38 MAPK signaling. Biol. Trace Elem. Res. 2020, 193, 483–493. [Google Scholar] [CrossRef]
- Ribeiro Júnior, R.F.; Marques, V.B.; Nunes, D.O.; Stefanon, I.; Dos Santos, L. Chronic iron overload induces functional and structural vascular changes in small resistance arteries via NADPH oxidase-dependent O2− production. Toxicol. Lett. 2017, 279, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Looi, Y.H.; Grieve, D.J.; Siva, A.; Walker, S.J.; Anilkumar, N.; Cave, A.C. Involvement of Nox2 NADPH oxidase in adverse cardiac remodeling after myocardial infarction. Hypertension 2008, 51, 319–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirker, A.; Murdoch, C.E.; Protti, A.; Sawyer, G.J.; Santos, C.X.; Martin, D.; Zhang, X.; Brewer, A.C.; Zhang, M.; Shah, A.M. Cell-specific effects of Nox2 on the acute and chronic response to myocardial infarction. J. Mol. Cell. Cardiol. 2016, 98, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Krijnen, P.A.J.; Meischl, C.; Hack, C.E.; Meijer, C.J.L.M.; Visser, C.A.; Roos, D. Increased Nox2 expression in human cardiomyocytes after acute myocardial infarction. J. Clin. Pathol. 2003, 56, 194–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, N.R. Advances in iron chelation therapy: Transitioning to a new oral formulation. Drugs Context 2017, 6, 212502. [Google Scholar] [CrossRef] [Green Version]
- Mobarra, N.; Shanaki, M.; Ehteram, H.; Nasiri, H.; Sahmani, M.; Saeidi, M.; Goudarzi, M.; Pourkarim, H.; Azad, M. A review on iron chelators in treatment of iron overload syndromes. Int. J. Hematol. Oncol. Stem Cell Res. 2016, 10, 239–247. [Google Scholar]
- Altenhöfer, S.; Radermacher, K.A.; Kleikers, P.W.; Wingler, K.; Schmidt, H.H. Evolution of NADPH oxidase inhibitors: Selectivity and mechanisms for target engagement. Antioxid. Redox Signal. 2015, 23, 406–427. [Google Scholar]
- Hirano, K.; Chen, W.S.; Chueng, A.L.; Dunne, A.A.; Seredinina, T.; Filippova, A.; Ramachandran, S.; Brideges, A.; Chaudry, L.; Pettman, G.; et al. Discovery of GSK2795039, a novel small molecule NADPH oxidase 2 inhibitor. Antioxid. Redox Signal. 2015, 23, 358–374. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Echo Parameter | B6 Ctl Mice | NOX2 KO |
|---|---|---|
| Ao Peak Grad (mmHg) | 3.40 ± 0.17 | 3.48 ± 0.34 |
| Endocard. CO (mL/min) | 8.14 ± 0.73 | 8.76 ± 0.75 |
| LVEF (%) | 70.49 ± 1.17 | 68.25 ± 1.21 |
| LV FS (%) | 38.63 ± 0.87 | 37.12 ± 0.97 |
| LV Mass (uncorrected) (g) | 70.80 ± 5.50 | 76.77 ± 4.80 |
| PA Peak Grad (mmHg) | 1.52 ± 0.11 | 1.68 ± 0.11 |
| Heart Rate (bpm) | 446 ± 24.9 | 421 ± 6.30 |
| MV A (cm/s) | 367.7 ± 26.0 | 379.6 ± 22.6 |
| MV E at A (cm/s) | 589.1 ± 24.0 | 623.8 ± 27.9 |
| MV E/A | 1.60 ± 0.09 | 1.64 ± 0.08 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mak, I.T.; Kramer, J.H.; Iantorno, M.; Chmielinska, J.J.; Weglicki, W.B.; Spurney, C.F. Deficiency in gp91Phox (NOX2) Protects against Oxidative Stress and Cardiac Dysfunction in Iron Overloaded Mice. Hearts 2020, 1, 117-125. https://0-doi-org.brum.beds.ac.uk/10.3390/hearts1020012
Mak IT, Kramer JH, Iantorno M, Chmielinska JJ, Weglicki WB, Spurney CF. Deficiency in gp91Phox (NOX2) Protects against Oxidative Stress and Cardiac Dysfunction in Iron Overloaded Mice. Hearts. 2020; 1(2):117-125. https://0-doi-org.brum.beds.ac.uk/10.3390/hearts1020012
Chicago/Turabian StyleMak, I. Tong, Jay H. Kramer, Micaela Iantorno, Joanna J. Chmielinska, William B. Weglicki, and Christopher F. Spurney. 2020. "Deficiency in gp91Phox (NOX2) Protects against Oxidative Stress and Cardiac Dysfunction in Iron Overloaded Mice" Hearts 1, no. 2: 117-125. https://0-doi-org.brum.beds.ac.uk/10.3390/hearts1020012