Vitamin A as a Transcriptional Regulator of Cardiovascular Disease


Drug Research Program, Division of Pharmacology and Pharmacotherapy, Faculty of Pharmacy, University of Helsinki, 00790 Helsinki, Finland
*
Author to whom correspondence should be addressed.
Hearts 2020, 1(2), 126-145; https://0-doi-org.brum.beds.ac.uk/10.3390/hearts1020013
Submission received: 28 August 2020
/
Revised: 17 September 2020
/
Accepted: 17 September 2020
/
Published: 21 September 2020
(This article belongs to the Special Issue Nutrient Deficiency and Drug Induced Cardiac Injury and Dysfunction)
Abstract
:Vitamin A is a micronutrient and signaling molecule that regulates transcription, cellular differentiation, and organ homeostasis. Additionally, metabolites of Vitamin A are utilized as differentiation agents in the treatment of hematological cancers and skin disorders, necessitating further study into the effects of both nutrient deficiency and the exogenous delivery of Vitamin A and its metabolites on cardiovascular phenotypes. Though vitamin A/retinoids are well-known regulators of cardiac formation, recent evidence has emerged that supports their role as regulators of cardiac regeneration, postnatal cardiac function, and cardiovascular disease progression. We here review findings from genetic and pharmacological studies describing the regulation of both myocyte- and vascular-driven cardiac phenotypes by vitamin A signaling. We identify the relationship between retinoids and maladaptive processes during the pathological hypertrophy of the heart, with a focus on the activation of neurohormonal signaling and fetal transcription factors (Gata4, Tbx5). Finally, we assess how this information might be leveraged to develop novel therapeutic avenues.
1. Introduction
Vitamin A and its retinoid metabolites are known differentiation inducers and antioxidants used in the treatment of blood cancers and skin disorders [1,2,3]. Additionally, Vitamin A/retinoids are essential for mammalian embryogenesis [4,5], and excessive vitamin A/retinoids are known to cause congenital heart diseases (CHDs, full list of abbreviations in Table 1) in rodents and humans [6,7,8,9]. Intriguingly, population studies have identified a relationship between serum levels of vitamin A/retinoids and adult cardiovascular diseases (CVDs) [10], the global leading cause of death and one for which regenerative therapies are lacking. Thus, an improved understanding of teratogenic and postnatal mechanisms-of-action of vitamin A/retinoid signaling is of importance to both fetal and adult health.
We here review the molecular mechanisms of vitamin A/retinoid signaling in mammals and describe how these contribute to both cardiogenesis and postnatal cardiovascular function. Furthermore, we review evidence from population, genetic, and pharmacological studies on the role of vitamin A/retinoids in cardiac regeneration, myocardial/arterial homeostasis, and CVDs. Finally, we highlight the interaction of vitamin A and prototypical disease signaling pathways and examine how this evidence contributes to an understanding of the pathophysiology of CVDs, micronutrient deficiency, and therapeutic cardiac regeneration.
2. Molecular Mechanisms of Vitamin A/Retinoid Signaling and Homeostasis
Cell- and tissue-level effects of vitamin A/retinoid signaling are dependent on a complex series of steps, encompassing dietary intake, storage, mobilization, transport, metabolism to active forms, and activation of retinoic acid receptors (Figure 1). Dietary vitamin A/retinoids are ingested in the form of β-carotene, retinol, or retinyl esters [1], and a lack of dietary vitamin A is one of the most common micronutrient deficiencies [11]. After ingestion, β-carotene and retinol are converted into retinyl esters, and these are stored in the liver until mobilized, at which point they are usually re-converted into retinol [1].
After mobilization, retinol is transported from the liver to tissues bound to retinol binding proteins (RBPs) [12], though retinyl esters may also be transported to distant tissues within chylomicrons [13]. Intracellularly, retinol is bound to cellular retinol binding proteins (CRBPs) [12], and retinol and retinal dehyrodgenases oxidize retinol into active retinoid isoforms, the most predominant of which are all-trans retinoic acid (ATRA) and its stereoisomers, 9-cis retinoic acid (9CRA) and 13-cis retinoic acid [1]. In order to exert its effects on gene expression, ATRA/9CRA bind to homo- or heterodimers of retinoid x receptors (RXRs) and retinoic acid receptors (RARs), which themselves include various subtypes (α, β, γ) [1]. Interestingly, both ATRA and 9CRA activate RARs, whereas only 9CRA is a natural activator of RXRs [14,15,16]. Moreover, a diverse set of RAR/RXR receptor homo- and heterodimers can be formed in response to ligand binding, and these bind to DNA to regulate transcriptional activity [14]. RARs/RXRs are expressed in numerous tissues/organ systems, and gene expression and genetic loss-of-function analyses have revealed tissue-specific roles of specific combinations of RARs/RXRs, expertly reviewed elsewhere [17,18]. Importantly, synthetic retinoids have been developed which are selective for RARα, RARβ-γ, RARγ and RXRs [19,20,21,22,23], and these serve as tool compounds for probing specific receptor activity.
The biological activity of retinoids is also regulated by their degradation. Cell and tissue levels of active retinoid forms are controlled by Cyp26a1, Cyp26b1, and Cyp26c1, enzymes that degrade both ATRA and 9CRA [24]. Interestingly, Cyp26 enzymes are themselves under the transcriptional control of RAR/RXR, thus forming a negative feedback loop that regulates levels of active retinoids at the cellular and tissue level [24]. Though in vitro studies allow precise perturbation of retinoic acid (RA) signaling, in vivo effects are regulated by the mobilization of vitamin A in the liver [25]. Interestingly, vitamin A levels were reported to increase in the hearts of rats following myocardial infarction (MI), in conjunction with increased hydrolysis of vitamin A stores in the liver and the transcription of RA-dependent reporter genes [26,27]. Furthermore, there were increased levels of retinol in the hearts of mice lacking β-carotene-15,15′-dioxygenase, the enzyme responsible for converting β-carotene into active retinoids [28]. Thus, RA activity is a function of dietary intake, the synthesis of active ATRA/9CRA forms, degradation, binding of retinoids to nuclear RAR/RXR receptors, and modulation of transcriptional outcomes.
3. Effects of Vitamin A/Retinoids on the Formation of the Heart and Differentiation of Multipotent Cardiovascular Progenitor Cells
Maternal vitamin A deficiency was long ago identified as a cause of congenital heart abnormalities in rodents [6,7], and the exposure of the fetus to exogenous ATRA is highly teratogenic to the embryonic human heart [8]. In addition to providing insights into the onset of CHDs, a more thorough understanding of the molecular mechanisms by which retinoids exert effects on the formation of the heart could provide novel strategies for regeneration of the adult heart following MI. Furthermore, knowledge of these mechanisms could guide efforts to impede the re-activation of developmental gene regulatory networks in the failing heart [29].
The effects of retinoids on the formation of the heart have been well-defined by embryological studies. During early cardiogenesis, RA signaling is integral to the following developmental processes: (1) negative regulation of multipotent cardiac progenitor cells (CPCs)/promotion of cardiomyocyte differentiation [30,31,32,33]; (2) patterning of multipotent progenitors into anterior (atrial) and posterior (ventricular) cell fates [34,35,36]; (3) rightward looping of the heart [32,37]; and (4) Formation of mature structures in the heart, such as septa, atrioventricular cushions, epicardium, ductus arteriosus, and mature ventricular muscle [38,39,40,41,42].
Interestingly, RA receptors appear to have specific functions during cardiogenesis. For instance, RXRα is necessary for embryonic viability, and the deletion of RXRα led to ventricular dysfunction, persistent atrial gene expression in the ventricles, thin muscular walls more resembling atria, and defects in the formation of the ventricular septum [43,44]. However, both single and co-deletion of RARα1 and RARβ do not result in cardiac phenotypes in embryos and adults [45], suggesting that RXRα is a predominant mediator of RA signaling in the developing heart. Interestingly, embryonic RA signaling is subject to complex feedback mechanisms defined by the presence or absence of specific RA receptors, evidenced by zebrafish experiments in which the deletion of RARα led to subsequent overall increases in RA signaling [46].
Developmental effects of RA signaling are also determined by the degradation of RA by Cyp26 enzymes. Deletion of both Cyp26a1 and Cyp26c1 in zebrafish led to an increase in atrial cells and a decrease in vascular cells, in addition to a loss of ventricular cardiomyocytes derived from first heart field progenitor cells [47,48]. Interestingly, overexpression studies of Cyp26a1 revealed that this enzyme regulates RA levels at a tissue, rather than a cellular level, and this further helped to shape developmental gradients [49]. Thus, Cyp26 enzymes regulate the availability of retinoids during development, thereby affecting tissue formation.
The effects of RA during development appear to occur partially through the activation of HOX genes, key drivers of developmental gene activation in the formation of diverse tissues, including the heart [50,51,52]. RA target genes within second-heart field progenitor cells include Hoxa1, Hoxa3, and Hoxb1 (mouse), as well as Hoxb5b (zebrafish) [52,53]. Interestingly, the overexpression of Hoxb5b exerted similar phenotypes to that of excessive RA in zebrafish embryos, suggesting that HOX genes are major target genes of RA signaling during early embryogenesis [54]. In addition to HOX genes, RA signaling is upstream of several transcription factors key to cardiogenesis, such as Gata4, Tbx5, Pitx2, and Nr2f2 [55,56,57,58]. Thus, vitamin A/retinoids exhibit effects on key developmental processes during the formation of the heart, and this occurs via the activation of cardiac transcription factors.
4. Vitamin A Signaling and Regeneration of the Myocardium
The regeneration of lost myocardium has recently emerged as a potential therapeutic strategy for the treatment of MI, and vitamin A/retinoids have been implicated in cardiac regeneration, driven by both endogenous mechanisms and cell transplantation (Figure 2a). Expression of the retinoic acid synthesis gene Raldh2 was shown to be induced in the epicardium of the ventricle, atrium, and outflow tract of the regenerating zebrafish heart following myocardial injury [59]. A follow-up study showed that Raldh2 is activated in proliferating cells of the endocardium of fish following cardiac injury, and the inhibition of RARα led to decreased cardiomyocyte proliferation and impeded regeneration of the myocardium [60]. Indeed, the secretion of RA by the epicardium was demonstrated to be required for the proliferation of embryonic cardiomyocytes in the chick model [61]. Furthermore, a population of Raldh2-expressing epicardial cells were shown to migrate into the ventricular myocardium in the chick model, and the authors suggested that these cells may serve as a source of retinoic acid that promotes the thickening of the embryonic myocardium. Moreover, the authors observed that Raldh2+ epicardial cells differentiate into smooth muscle and endothelial cells of the coronary vessels [62]. Additionally, RA signaling regulated secretion of erythropoietin by the liver during embryogenesis, and this in turn stimulated expression of Igf2 in the epicardium, resulting in cardiomyocyte proliferation [63]. Thus, RA signaling has been implicated in endogenous cardiomyocyte proliferation and regenerative processes native to the hearts of lower vertebrates.
In addition to the induction of endogenous proliferation, the injection of stem cell-derived cardiomyocytes into the infarcted heart has recently emerged as a therapeutic strategy to replace the cardiomyocytes lost following MI [64]. Importantly, RA was identified as a general inducer of differentiation of pluripotent stem cells (PSCs) [65,66,67]. Moreover, RA has been shown in numerous studies to promote the directed differentiation of PSC- and CPC-derived cardiomyocytes. For instance, ATRA promoted the differentiation of H9c2 cardiomyoblast cells, leading to the expression of cardiomyocyte gene expression programs and providing evidence that ATRA is a differentiation inducer of CPCs [68,69]. Additionally, several in vitro studies in PSCs have reported retinoids as specific inducers of the atrial cardiomyocyte cell fate [58,70,71] (Figure 2b). In our own study, dose- and stage-specific effects of retinoids were observed on the differentiation of atrial and ventricular PSC-derived cardiomyocytes using markers of early cardiogenesis to detect differentiation status, mirroring embryonic phenotypes [70]. Studies by a separate group showed that the titration of BMP and Activin A were shown to regulate the levels of Cyp26 RA degrading enzymes, thereby potentiating RA response in CPCs and determining whether cells assumed an atrial or ventricular fate [72]. A phenotypic screen of 10,000 compounds for the induction of proliferation of human multipotent CPCs led to the identification of RAR, but not RXR agonists as potent inducers of CPC proliferation. Importantly, the authors found that treating CPCs with retinoids on day 4 of differentiation promoted the atrial cardiomyocyte fate, whereas day 5 treatment led to the induction of cardiac progenitor proliferation [73]. Therefore, the effect of manipulation of RA signaling in PSCs and CPCs is determined by the stage of differentiation, and this pathway can be manipulated chemically to generate specific cardiac cell subtypes. Moreover, in vitro platforms provide tractable systems for the examination of temporal effects on vitamin A/retinoid signaling and serve as a potential source of cell subtypes to regenerate the heart following myocardial injury.
5. Population-Based Studies Implicating Vitamin A Deficiency in the Onset of CVDs
In addition to the evidence for the role of RA signaling in cardiogenesis, population-based studies have implicated RA signaling in the development of adult CVDs. A Finnish study of men aged 46–65 reported that low serum β-carotene levels were associated with an increased risk of acute MI [74], and low serum β-carotene levels were associated with an increased risk of cardiovascular mortality, especially among smokers [75]. In a Chinese study, low serum RA levels (median 21 nM vs. 39 nM) were associated with an increased risk of mortality in patients with coronary artery disease, and high RA levels were associated with a decreased risk of overall mortality [10]. A separate study in a US population of over-50-year-olds showed that both low (<30 µg/dL) and high (>80 µg/dL) serum RA levels were associated with increased mortality due to CVDs [76]. Indeed, patients with elevated serum vitamin A (>3.10 µM) were also observed to have elevated apolipoprotein B and to be at a greater risk for CVDs [77]. However, not all population studies display a relationship between vitamin A and coronary artery disease, as evidenced by a Singaporean study in which no significant differences in vitamin A levels were observed in the livers of patients who died of coronary artery disease [78]. Collectively, these studies indicate the potential relevance of RA signaling to CVDs in human patients.
6. Effects of Vitamin A/Retinoids in the Healthy and Diseased Postnatal Myocardium
In addition to the relationship between low serum RA and CVDs identified in population studies, there is ample evidence from animal and in vitro studies demonstrating that RA signaling affects myocardial function and CVDs. A recent study showed that RARα decreases in wild type mice during aging and when subjected to metabolic stress, and the genetic deletion of RARα in adult mouse myocardium resulted in diastolic dysfunction, impaired Ca2+ handling, and increased oxidative stress [79]. Similarly, Cellular retinoic acid binding protein 1 (Crabp1) knockout mice were more susceptible to heart failure and remodeling induced by isoproterenol, an agonist of adrenergic signaling, and this was attenuated by treatment with ATRA (5 mg/kg/day delivered intraperitoneally) [80].
Animal models have also implicated RA signaling in the pathogenesis of MI. A retinoic acid response element (RARE)-luciferase reporter was activated in the heart following MI in mice, in addition to the RA synthesis gene Aldh1a2 and the RA degradation gene Cyp26b1 [27]. RA receptors RARα and RARγ were more prominently upregulated in cardiac fibroblasts, whereas RARβ was specifically upregulated in cardiomyocytes [27]. Importantly, separate studies showed that exogenously administered ATRA (1 µM; 0.1 µM−10 µM) decreased the proliferation of primary cardiac fibroblasts in vitro [27,81], suggesting that ATRA might have anti-fibrotic properties in the infarcted heart. A separate study demonstrated that dietary vitamin A supplementation (0.3 mg/kg given daily) in rats subjected to MI led to decreased myocyte hypertrophy and an increased maximum rate of rise of left ventricular pressure six months after injury [82].
Evidence exists that RA-signaling is an important component of cardiomyopathies. Dilated cardiopathy was observed in a transgenic mouse line containing a cardiac-specific overexpression cassette of RARα [83], suggesting deleterious effects of prolonged activation of RA signaling. In mouse models of obesity, daily delivery of ATRA by stomach intubation (2 μg/g/day) restored natriuretic peptide levels and prevented adverse cardiac remodeling [84]. Similarly, dietary supplementation with RA decreased cardiac injury in mouse models of gestational diabetes mellitus (3 mg/kg/day), tobacco smoke, and methylglyoxal (1 mg/kg/day) [85,86,87]. Beneficial effects of RA in the MI model might be attributed to its anti-hypertrophic effects in cardiomyocytes (Figure 3a). Exogenous ATRA (0.1 µM−1 µM) attenuated phenylephrine-induced hypertrophy in cultured neonatal cardiomyocytes, leading to reductions in cell size and activation of atrial natriuretic peptide, suggesting that ATRA prevents adverse effects due to excessive activation of adrenergic receptors [88]. Similarly, ATRA (10 µM) attenuated endothelin-induced hypertrophy and the activation of natriuretic peptide transcription in neonatal cardiomyocytes in vitro, and this effect was more pronounced in combination with 1,25(OH)2 vitamin D3 (VD3) [89]. Thus, RA signaling interferes with pro-hypertrophic effects of neurohormonal signaling characteristically activated in the context of heart failure.
Beneficial effects of modulation of RA signaling have also been observed in other CVD models in vivo. In rats subjected to aortic banding, ATRA (30 mg/kg/day by gavage) prevented deterioration in systolic and diastolic dysfunction, in addition to inhibiting hypertrophic gene expression [90]. ATRA (5 mg or 10 mg/kg/day by intraperitoneal injection) impeded cardiac damage caused by isoproterenol treatment in mice and rats [91,92] and regulated calcium signaling by inhibiting phosphorylation of Ca2+/calmodulin-dependent protein kinase II (CaMKII) and phospholamban [91]. Furthermore, ATRA (10 µM–20 µM) attenuated pro-arrhythmic effects of isoproterenol and lysophosphatidylcholine in cultured neonatal cardiomyocytes, and reperfusion-induced ventricular tachycardia and fibrillation were attenuated by the intravenous infusion of ATRA (1 mM delivered by perfusion at 8 mL/hour) in vivo [93]. Thus, ATRA has been reported in various studies to provide beneficial effects in diverse CVD models in vivo.
In addition to the study of exogenous delivery of ATRA in disease models, several studies have investigated the effects of Vitamin A deficiency and/or dietary supplementation. Separate studies found adverse remodeling was induced, rather than impeded by depleted vitamin A stores, and Vitamin-A depleted hearts underwent more severe adverse remodeling following MI [94,95], though another study reported beneficial effects of vitamin A deficiency [96]. Additionally, in contrast to other studies, ATRA dietary supplementation (24 µg/kg/day for 90 days or 0.3 mg–10 mg/kg/day for two months) in normal rats induced adverse remodeling of the ventricle [97,98]. These studies indicate context-dependent effects of manipulation of retinoids/Vitamin A signalling in the healthy vs diseased heart. Additionally, these findings broadly support the complex relationship between RA levels and CV function, mirrored by human studies in which both low (<30µg/dL) and high (>80µg/dL) serum RA levels led to adverse effects [76]. Further experimentation might be necessary to fully optimize therapeutic concentrations of serum RA upon exogenous treatment. Regardless, animal studies broadly indicate a role for retinoids in maintaining myocardial homeostasis, and potentially indicate RA signaling as a therapeutic target for CVDs.
7. Effects of Vitamin A on Arterial Homeostasis
Thickening of the intima via the proliferation of vascular smooth muscle cells is an important component of the pathophysiology of atherosclerosis, the underlying cause of a majority of CVDs [99]. Importantly, ATRA has been shown to inhibit proliferative and adverse phenotypical changes of vascular smooth muscle cells (SMCs), in vitro and in vivo (Figure 3b). For instance, rat aortic SMCs were shown to express RARα, RARγ, RXRα, and RXRβ, and treatment with ATRA (2 µM–20 µM) inhibited the proliferation of rat aortic SMCs [100]. Though RARα was suggested as the major mediator of antiproliferative effects of ATRA (0.125 µM–2 µM) on SMCs [101], a separate study demonstrated that RARγ agonists also inhibited the proliferation of SMCs [102]. Importantly, several studies confirmed anti-proliferative as well as anti-apoptotic effects of ATRA (1 µM; 5 µM–10 µM; 2 µM) on cultured rat aortic SMCs [103,104,105]. Anti-proliferative effects were also observed in human cells, as ATRA (0.022 µM, 0.01 µM–1 µM, 0.12 µM) inhibited the proliferation and migration of human arterial smooth muscle cells in vitro [106,107,108]. Interestingly, the proliferation of human arterial endothelial cells was not affected by ATRA, suggesting differential responses of stromal cells to retinoids [106]. This was supported by the observation that retinol (1.25 µM–20 µM) inhibited the proliferation of intimal SMCs but increased the proliferation of medial SMCs, reportedly due to different abilities of these cell types to metabolize retinol to ATRA [109]. In a separate study, ATRA (1 µM) inhibited the proliferation of human aortic SMCs and adult murine cardiofibroblasts, while increasing the expression of RA target genes (Rarβ, Cyp26b1) in mouse adult cardiomyocytes [110]. However, in human umbilical vein endothelial cells, exogenous ATRA (1 µM) led to increased vascular endothelial growth factor (VEGF) signaling, thereby potentiating angiogenesis [111]. Thus, in vitro studies are largely supportive of an anti-proliferative effect of retinoids in aortic SMCs, in addition to showing cell-type specific phenotypic changes.
Exogenous retinoids have also shown beneficial effects on diseases of the arteries in vivo. Dietary ATRA (1.5 mg/day, 30 mg/kg/day) reduced neointima area in both the carotid artery and aorta of rats subjected to balloon injury [112,113]. Similarly, following balloon angioplasty or jugular vein bypass grafts, the oral administration of ATRA (0.6 mg/kg/day, 10 mg/kg/day by gavage) and the local delivery of ATRA (10 mL of 10 µM infusion) decreased intimal thickening in rabbits by decreasing proliferative activity [114,115,116]. Oral administration of Am80, a RARα agonist, impeded formation of the neointima following vascular injury and modulated SMC phenotypes in a rabbit model of stent placement [117]. In a separate study, daily ATRA (5 mg–10 mg/kg/day by gavage for three months) attenuated the thickening of intramyocardial and intrarenal arteries, as well as ventricular fibrosis in spontaneously hypertensive rats, though cardiac function was not improved [118].
Interestingly, some studies support the beneficial effects of retinoids in arterial pathologies which may not be accounted for by their anti-proliferative activities. For instance, ATRA (6 mg/kg/day) was reported to achieve favorable vascular remodeling without inhibiting proliferation [119]. Additionally, a separate study reported the upregulation of vascular integrins in SMCs by ATRA (1 mg/kg every other day for 3 weeks) in adult rats, suggesting an alternative mechanism by which ATRA can induce vascular remodeling [120]. ATRA (1 µM–10 µM) was also reported to modulate contractility of the aorta in organ culture, suggesting another mechanism by which ATRA could regulate blood pressure [121]. Indeed, vitamin A-deficient rats appeared to have SMCs with reduced differentiation capacity and decreased contractility [122]. ATRA stimulation (0.1 µM–1 µM) was also reported to impede osteogenic differentiation of cultured human coronary artery SMCs, leading to the suppression of calcification [123]. However, this may be a delicate balance, as excess dietary vitamin A caused aortic valve calcification in mice fed retinyl palmitate (200 IU/g for 12 months) [124].
Despite the abundance of preclinical studies in rodent and animal models, only a limited number of human studies have examined the involvement of RA signaling in atherosclerosis. Measurement of vitamin A metabolites in human samples revealed that ATRA was increased in the left ventricles of patients with end-stage heart failure caused by coronary artery disease, in addition to describing the upregulation of RA genes (ALDH1A2, CRABP2, etc.) in atherosclerotic plaques [110]. Furthermore, a polymorphism in Cyp26b1, which leads to increased RA degradation, was implicated in the size of atherosclerotic lesions [125]. Further studies with human biopsies or human induced pluripotent stem cell models might help to uncover the specific roles of retinoids in regulating the proliferative capacity and phenotype of human smooth muscle cells.
8. Effects of Vitamin A/Retinoids on Prototypical Disease Signaling Pathways
In line with the beneficial effects observed in CVD cell and animal models, RA signaling interacts with signaling pathways known to regulate CVD progression. For instance, neurohormonal signaling is activated during maladaptive pathological hypertrophy [126], and several studies indicate that these pathways can be modulated by retinoids. In separate studies, ATRA (0.1 µM–1 µM; 10 µM) inhibited the deleterious effects of adrenergic and endothelin signaling on cardiomyocytes [88,89]. Similarly, ATRA (0.01 µM–2 µM) attenuated the endothelin-induced proliferation of rat ASMCs [127]. Additionally, an analysis of the hearts of LRAT (−/−) mice lacking hepatic retinol stores and fed a vitamin A-depleted diet identified vitamin A as a potential regulator of natriuretic peptides Nppa and Nppb [96]. In a separate study, exogenous ATRA (0.5 µM) was shown to regulate the transcription of natriuretic peptide receptor A by direct binding to the promoter and the modulation of histone acetylation [128]. Thus, RA signaling modulates the neurohormonal signaling characteristic of maladaptive remodeling in the post-MI heart.
Antagonists of the renin-angiotensin-aldosterone system are commonly used as heart failure treatments in order to support vasodilation and a favorable hemodynamic state [126]. Furthermore, angiotensin II is a key mediator of pathological ventricular remodeling during heart failure [126]. Exogenous ATRA (0.1 µM–10 µM) was reported to inhibit effects of angiotensin II on both cardiac fibroblasts and neonatal cardiomyocytes [129], and exogenous ATRA (1 µM) also inhibited the proliferation of vascular SMCs induced by angiotensin II [130]. In primary neonatal cardiomyocytes subjected to stretch, ATRA (0.1 µM–5 µM) inhibited renin-angiotensin signaling by the transcriptional repression of renin, angiotensinogen, angiotensin-converting enzyme, and the angiotensin type 1 receptor [90]. Thus, RA signaling is also an inhibitor of the angiotensin pathway, an important mediator of ventricular remodeling during heart failure.
A hallmark of maladaptive cardiac remodeling is the re-activation of fetal transcription factors [29,131], and RA signaling has been proven to modulate the activity of fetal transcription factors. For instance, ATRA (0.2 µM, 0.01 µM–0.1 µM) induced the expression and DNA binding of Gata4 [132,133], a key regulator of cardiomyocyte hypertrophy [134]. Additionally, vitamin A-deficient embryos exhibited the downregulation of Gata4 [55], suggesting that Gata4 is downstream of RA in both the embryonic and adult contexts. Importantly, the overexpression of Gata4 following MI induced angiogenesis and prevented adverse cardiac remodeling [135], suggesting that the upregulation of Gata4 by RA signaling could engage therapeutic transcriptional pathways. Similarly, Tbx5, a core cardiac transcription factor and the causative factor in Holt–Oram syndrome, cardiomyocyte differentiation, and formation of the interventricular septum was also downstream of RA signaling [56,136,137,138]. RA signaling was also reported to activate the transcription of Pitx2 in the embryo, another important developmental cardiac transcription factor [57]. Importantly, Pitx2 and Tbx5 were recently shown to regulate atrial rhythm [139], providing another potential mechanism by which activated RA signaling could modulate cardiovascular function.
The effects of RA signaling might also be via the modulation of downstream kinases. The anti-hypertrophic effects of ATRA (5 µM) in neonatal cardiomyocytes were reported to occur in conjunction with upregulation of expression of MAP kinase phosphatase-1 and 2, thereby inhibiting MAP kinase signaling [140]. ATRA (1 µM–4 µM) was also reported to activate AMP-activated protein kinases during the impediment of proliferation in mouse vascular SMCs [141]. Finally, ATRA (0.1 µM–1 µM) was reported to modulate plasminogen activator inhibitor 1 gene expression, and this was impeded by tyrosine kinase inhibitors [142]. Thus, beneficial effects of the activation of vitamin A/retinoid signaling may be accounted for by the regulation of neurohormonal signaling, developmental transcription factors, and downstream kinases.
9. Antioxidant and Metabolic Properties of Vitamin A/Retinoids
Some beneficial effects of vitamin A/retinoids may be due to their antioxidant properties or other changes to metabolism rather than interference with disease signaling pathways. Vitamin A/retinoids are well known antioxidants [143,144,145,146], and antioxidant properties of vitamin A given to rats (25 IU/kg/day) were reported to underlie its ability to protect the heart from doxorubicin-induced toxicity [147]. Similarly, antioxidant activity of endogenous vitamin A was increased in response to oxidative stress induced by doxorubicin, a common cancer treatment with dose-limiting cardiotoxicity [148]. Importantly, ATRA (10 mg/kg/day) attenuated the oxidative stress-induced apoptosis associated with myocardial ischemia/reperfusion injury. However, anti-apoptotic effects were more clearly linked to RA-driven upregulation of pro-survival signals at the transcriptional level, rather than direct antioxidant activity of vitamin A [149]. In another study, vitamin A deficiency led to an increased proportion of saturated fatty acids and phospholipids in the heart, in conjunction with an upregulation of peroxisome proliferator-activated receptor [150]. Furthermore, in conjunction with a cardiac-specific binding partner CERIP, RXR regulated transcription of the human MCAD gene, an important component of fatty acid oxidation in the mature heart [151]. Thus, vitamin A/retinoids might also exert beneficial effects, due to their antioxidant effects and the modulation of metabolic pathways.
10. Exogenous Retinoids or Retinoid Inhibition as Therapeutic Agents in Cardiovascular Diseases
Despite ample evidence for the role of RA signaling in diverse CVD pathologies, clinical trials examining the effects of β-carotene supplementation on CVDs have not shown any benefits [152,153,154,155]. However, such results are not necessarily in contrast to preclinical findings (especially in vitro), as β-carotene supplementation may not result in elevated RA signaling at a cellular or organ level, due to the multiple levels of control of active retinoid concentrations. Indeed, differences might exist between the systemic and local effects of retinoids in CVDs, and RA-containing formulations that are directly injected into the heart might represent a feasible therapeutic avenue. For instance, hyaluronan conjugated to RA and butyric acid enhanced the differentiation of PSCs and mesenchymal stem cells to spontaneously differentiating cardiomyocytes, and this led to myocardial repair in vivo [156,157,158]. Additionally, intramyocardial delivery of a hyaluronan mixed ester of butyric acid and RA was beneficial following MI, and led to the transcriptional upregulation of VEGF and the stem-cell marker KDR, potentially by increasing histone acetylation [159]. However, in a separate study intramyocardial injection of ATRA to the infarction zone (10 injections of 30 µL 3.3 µM) worsened cardiac function following MI, whereas intramyocardial injection of a novel inhibitor of ATRA uptake (5′-methoxyleoligin) was cardioprotective [160]. Interestingly, 5′-methoxyleoligin was also reported to promote arteriogenesis, and in vitro angiogenic effects were dependent on the transcriptional upregulation of the ATRA-degrading enzyme Cyp26b1 [161]. It is unclear what underlies the discrepancy between these studies, though developmental findings indicating complex feedback networks governing RA signaling could provide clues. More careful optimization of dosing might be required to ensure adequate tissue levels and mirror anti-hypertrophic activity seen in vitro. Regardless, local administration of RA formulations, with or without the co-injection of stem cells, might serve to enhance therapeutic cardiac repair without inducing systemic effects characteristic of retinoid differentiation therapy. This would also avoid the need for mobilization, transport, and metabolism needed with β-carotene supplementation.
11. Conclusions
Though long known to affect the formation of the heart, studies in past decades have implicated vitamin A/retinoid signaling as a modulator of adult cardiac function, including a major role in the pathophysiology of responses to injury of the myocardium and arterial walls. This is supported by population studies implicating that both excesses and deficiencies of serum RA levels are associated with CVD dysfunction. The ability of modulators of RA signaling to control endogenous processes such as proliferation and differentiation hold promise for their use in the development of therapies for CVDs, though challenges regarding negative systemic effects will likely impede advancement in this area unless targeted formulations that can be delivered intramyocardially are further refined. Additionally, a better understanding of how Cyp26 and retinol dehydrogenases maintain concentrations of ATRA/9CRA in the diseased heart could allow for the targeted modulation of retinoid levels. Further basic research in these areas and into mechanisms by which RA signaling modulates classical disease pathways could shed light on the pathophysiology of CVDs and provide further opportunities for therapeutic modulation.
Author Contributions
Conceptualization, R.S.L. and B.L.K.; writing—original draft preparation, R.S.L. and B.L.K.; writing—review and editing, R.S.L. and B.L.K.; visualization, R.S.L. and B.L.K.; funding acquisition, R.S.L. and B.L.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by R.S.L. grants from the Finnish Foundation for Cardiovascular Research and the Finnish Cultural Foundation; B.L.K., grants from Biocentrum Helsinki and Biocenter Finland. Open access funding provided by University of Helsinki.
Acknowledgments
We thank Heikki Ruskoaho for valuable comments on the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bushue, N.; Wan, Y.J. Retinoid pathway and cancer therapeutics. Adv. Drug Deliv. Rev. 2010, 62, 1285–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, L.A.; Sigman, C.C.; Andreola, F.; Ross, S.A.; Kelloff, G.J.; De Luca, L.M. Retinoids in chemoprevention and differentiation therapy. Carcinogenesis 2000, 21, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- Kolli, S.S.; Pecone, D.; Pona, A.; Cline, A.; Feldman, S.R. Topical Retinoids in Acne Vulgaris: A Systematic Review. Am. J. Clin. Dermatol. 2019, 20, 345–365. [Google Scholar] [CrossRef] [PubMed]
- Niederreither, K.; Subbarayan, V.; Dollé, P.; Chambon, P. Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nat. Genet. 1999, 21, 444–448. [Google Scholar] [CrossRef]
- Niederreither, K.; Vermot, J.; Schuhbaur, B.; Chambon, P.; Dollé, P. Embryonic retinoic acid synthesis is required for forelimb growth and anteroposterior patterning in the mouse. Development 2002, 129, 3563–3574. [Google Scholar]
- Wilson, J.G.; Warkany, J. Aortic-arch and cardiac anomalies in the offspring of vitamin A deficient rats. Am. J. Anat. 1949, 85, 113–155. [Google Scholar] [CrossRef]
- Wilson, J.G.; Roth, C.B.; Warkany, J. An analysis of the syndrome of malformations induced by maternal vitamin A deficiency. Effects of restoration of vitamin A at various times during gestation. Am. J. Anat. 1953, 92, 189–217. [Google Scholar] [CrossRef]
- Lammer, E.J.; Chen, D.T.; Hoar, R.M.; Agnish, N.D.; Benke, P.J.; Braun, J.T.; Curry, C.J.; Fernhoff, P.M.; Grix, A.W., Jr.; Lott, I.T.; et al. Retinoic acid embryopathy. N. Engl. J. Med. 1985, 313, 837–841. [Google Scholar] [CrossRef]
- Rothman, K.J.; Moore, L.L.; Singer, M.R.; Nguyen, U.S.; Mannino, S.; Milunsky, A. Teratogenicity of high vitamin A intake. N. Engl. J. Med. 1995, 333, 1369–1373. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, H.; Mu, D.; Li, D.; Zhong, Y.; Jiang, N.; Zhang, Y.; Xia, M. Association of Serum Retinoic Acid With Risk of Mortality in Patients With Coronary Artery Disease. Circ. Res. 2016, 119, 557–563. [Google Scholar] [CrossRef] [Green Version]
- Wirth, J.P.; Petry, N.; Tanumihardjo, S.A.; Rogers, L.M.; McLean, E.; Greig, A.; Garrett, G.S.; Klemm, R.D.; Rohner, F. Vitamin A Supplementation Programs and Country-Level Evidence of Vitamin A Deficiency. Nutrients 2017, 9, 190. [Google Scholar] [CrossRef]
- Noy, N. Retinoid-binding proteins: Mediators of retinoid action. Biochem. J. 2000, 348, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wongsiriroj, N.; Blaner, W.S. The multifaceted nature of retinoid transport and metabolism. Hepatobiliary Surg. Nutr. 2014, 3, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Das, B.C.; Thapa, P.; Karki, R.; Das, S.; Mahapatra, S.; Liu, T.C.; Torregroza, I.; Wallace, D.P.; Kambhampati, S.; Van Veldhuizen, P.; et al. Retinoic acid signaling pathways in development and diseases. Bioorg. Med. Chem. 2014, 22, 673–683. [Google Scholar] [CrossRef] [Green Version]
- Heyman, R.A.; Mangelsdorf, D.J.; Dyck, J.A.; Stein, R.B.; Eichele, G.; Evans, R.M.; Thaller, C. 9-cis retinoic acid is a high affinity ligand for the retinoid X receptor. Cell 1992, 68, 397–406. [Google Scholar] [CrossRef]
- Levin, A.A.; Sturzenbecker, L.J.; Kazmer, S.; Bosakowski, T.; Huselton, C.; Allenby, G.; Speck, J.; Kratzeisen, C.; Rosenberger, M.; Lovey, A.; et al. 9-cis retinoic acid stereoisomer binds and activates the nuclear receptor RXR alpha. Nature 1992, 355, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Dollé, P. Developmental expression of retinoic acid receptors (RARs). Nucl. Recept. Signal. 2009, 7, e006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mark, M.; Ghyselinck, N.B.; Chambon, P. Function of retinoic acid receptors during embryonic development. Nucl. Recept. Signal. 2009, 7, e002. [Google Scholar] [CrossRef] [Green Version]
- Boehm, M.F.; Zhang, L.; Zhi, L.; McClurg, M.R.; Berger, E.; Wagoner, M.; Mais, D.E.; Suto, C.M.; Davies, J.A.; Heyman, R.A.; et al. Design and synthesis of potent retinoid X receptor selective ligands that induce apoptosis in leukemia cells. J. Med. Chem. 1995, 38, 3146–3155. [Google Scholar] [CrossRef] [PubMed]
- Delescluse, C.; Cavey, M.T.; Martin, B.; Bernard, B.A.; Reichert, U.; Maignan, J.; Darmon, M.; Shroot, B. Selective high affinity retinoic acid receptor alpha or beta-gamma ligands. Mol. Pharmacol. 1991, 40, 556–562. [Google Scholar]
- Lehmann, J.M.; Jong, L.; Fanjul, A.; Cameron, J.F.; Lu, X.P.; Haefner, P.; Dawson, M.I.; Pfahl, M. Retinoids selective for retinoid X receptor response pathways. Science 1992, 258, 1944–1946. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.; Duong, T.T.; Johnson, A.T.; Klein, E.S.; Wang, L.; Khalifa, B.; Chandraratna, R.A. Identification of highly potent retinoic acid receptor alpha-selective antagonists. J. Med. Chem. 1997, 40, 2445–2451. [Google Scholar] [CrossRef] [PubMed]
- Thoreau, E.; Arlabosse, J.M.; Bouix-Peter, C.; Chambon, S.; Chantalat, L.; Daver, S.; Dumais, L.; Duvert, G.; Feret, A.; Ouvry, G.; et al. Structure-based design of Trifarotene (CD5789), a potent and selective RARγ agonist for the treatment of acne. Bioorg. Med. Chem. Lett. 2018, 28, 1736–1741. [Google Scholar] [CrossRef]
- Ross, A.C.; Zolfaghari, R. Cytochrome P450s in the regulation of cellular retinoic acid metabolism. Annu. Rev. Nutr. 2011, 31, 65–87. [Google Scholar] [CrossRef] [Green Version]
- Widjaja-Adhi, M.A.K.; Golczak, M. The molecular aspects of absorption and metabolism of carotenoids and retinoids in vertebrates. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 23, 158571. [Google Scholar] [CrossRef]
- Palace, V.P.; Hill, M.F.; Khaper, N.; Singal, P.K. Metabolism of vitamin A in the heart increases after a myocardial infarction. Free Radic. Biol. Med. 1999, 26, 1501–1507. [Google Scholar] [CrossRef]
- Bilbija, D.; Haugen, F.; Sagave, J.; Baysa, A.; Bastani, N.; Levy, F.O.; Sirsjö, A.; Blomhoff, R.; Valen, G. Retinoic acid signalling is activated in the postischemic heart and may influence remodelling. PLoS ONE 2012, 7, e44740. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.A.; Jiang, H.; Trent, C.M.; Yuen, J.J.; Narayanasamy, S.; Curley, R.W., Jr.; Harrison, E.H.; Goldberg, I.J.; Maurer, M.S.; Blaner, W.S. Cardiac dysfunction in β-carotene-15,15′-dioxygenase-deficient mice is associated with altered retinoid and lipid metabolism. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1675–H1684. [Google Scholar] [CrossRef] [Green Version]
- Oka, T.; Xu, J.; Molkentin, J.D. Re-employment of developmental transcription factors in adult heart disease. Semin. Cell Dev. Biol. 2007, 18, 117–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keegan, B.R.; Feldman, J.L.; Begemann, G.; Ingham, P.W.; Yelon, D. Retinoic acid signaling restricts the cardiac progenitor pool. Science 2005, 307, 247–249. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.C.; Dollé, P.; Ryckebüsch, L.; Noseda, M.; Zaffran, S.; Schneider, M.D.; Niederreither, K. Endogenous retinoic acid regulates cardiac progenitor differentiation. Proc. Natl. Acad. Sci. USA 2010, 107, 9234–9239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryckebusch, L.; Wang, Z.; Bertrand, N.; Lin, S.C.; Chi, X.; Schwartz, R.; Zaffran, S.; Niederreither, K. Retinoic acid deficiency alters second heart field formation. Proc. Natl. Acad. Sci. USA 2008, 105, 2913–2918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wobus, A.M.; Kaomei, G.; Shan, J.; Wellner, M.C.; Rohwedel, J.; Ji, G.; Fleischmann, B.; Katus, H.A.; Hescheler, J.; Franz, W.M. Retinoic acid accelerates embryonic stem cell-derived cardiac differentiation and enhances development of ventricular cardiomyocytes. J. Mol. Cell. Cardiol. 1997, 29, 1525–1539. [Google Scholar] [CrossRef] [PubMed]
- Gassanov, N.; Er, F.; Zagidullin, N.; Jankowski, M.; Gutkowska, J.; Hoppe, U.C. Retinoid acid-induced effects on atrial and pacemaker cell differentiation and expression of cardiac ion channels. Differentiation 2008, 76, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Hochgreb, T.; Linhares, V.L.; Menezes, D.C.; Sampaio, A.C.; Yan, C.Y.; Cardoso, W.V.; Rosenthal, N.; Xavier-Neto, J. A caudorostral wave of RALDH2 conveys anteroposterior information to the cardiac field. Development 2003, 130, 5363–5374. [Google Scholar] [CrossRef] [Green Version]
- Xavier-Neto, J.; Neville, C.M.; Shapiro, M.D.; Houghton, L.; Wang, G.F.; Nikovits, W., Jr.; Stockdale, F.E.; Rosenthal, N. A retinoic acid-inducible transgenic marker of sino-atrial development in the mouse heart. Development 1999, 126, 2677–2687. [Google Scholar]
- Niederreither, K.; Vermot, J.; Messaddeq, N.; Schuhbaur, B.; Chambon, P.; Dollé, P. Embryonic retinoic acid synthesis is essential for heart morphogenesis in the mouse. Development 2001, 128, 1019–1031. [Google Scholar]
- Colbert, M.C.; Kirby, M.L.; Robbins, J. Endogenous retinoic acid signaling colocalizes with advanced expression of the adult smooth muscle myosin heavy chain isoform during development of the ductus arteriosus. Circ. Res. 1996, 78, 790–798. [Google Scholar] [CrossRef]
- Guadix, J.A.; Ruiz-Villalba, A.; Lettice, L.; Velecela, V.; Muñoz-Chápuli, R.; Hastie, N.D.; Pérez-Pomares, J.M.; Martínez-Estrada, O.M. Wt1 controls retinoic acid signalling in embryonic epicardium through transcriptional activation of Raldh2. Development 2011, 138, 1093–1097. [Google Scholar] [CrossRef] [Green Version]
- Gruber, P.J.; Kubalak, S.W.; Pexieder, T.; Sucov, H.M.; Evans, R.M.; Chien, K.R. RXR alpha deficiency confers genetic susceptibility for aortic sac, conotruncal, atrioventricular cushion, and ventricular muscle defects in mice. J. Clin. Investig. 1996, 98, 1332–1343. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Pashmforoush, M.; Sucov, H.M. Retinoic acid regulates differentiation of the secondary heart field and TGFbeta-mediated outflow tract septation. Dev. Cell 2010, 18, 480–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Gise, A.; Zhou, B.; Honor, L.B.; Ma, Q.; Petryk, A.; Pu, W.T. WT1 regulates epicardial epithelial to mesenchymal transition through β-catenin and retinoic acid signaling pathways. Dev. Biol. 2011, 356, 421–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyson, E.; Sucov, H.M.; Kubalak, S.W.; Schmid-Schönbein, G.W.; DeLano, F.A.; Evans, R.M.; Ross, J., Jr.; Chien, K.R. Atrial-like phenotype is associated with embryonic ventricular failure in retinoid X receptor alpha -/- mice. Proc. Natl. Acad. Sci. USA 1995, 92, 7386–7390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sucov, H.M.; Dyson, E.; Gumeringer, C.L.; Price, J.; Chien, K.R.; Evans, R.M. RXR alpha mutant mice establish a genetic basis for vitamin A signaling in heart morphogenesis. Genes Dev. 1994, 8, 1007–1018. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.Y.; Luo, J.; Evans, R.M.; Giguere, V.; Sucov, H.M. Compartment-selective sensitivity of cardiovascular morphogenesis to combinations of retinoic acid receptor gene mutations. Circ. Res. 1997, 80, 757–764. [Google Scholar] [CrossRef]
- D’Aniello, E.; Rydeen, A.B.; Anderson, J.L.; Mandal, A.; Waxman, J.S. Depletion of retinoic acid receptors initiates a novel positive feedback mechanism that promotes teratogenic increases in retinoic acid. PLoS Genet. 2013, 9, e1003689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rydeen, A.B.; Waxman, J.S. Cyp26 enzymes are required to balance the cardiac and vascular lineages within the anterior lateral plate mesoderm. Development 2014, 141, 1638–1648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rydeen, A.B.; Waxman, J.S. Cyp26 Enzymes Facilitate Second Heart Field Progenitor Addition and Maintenance of Ventricular Integrity. PLoS Biol. 2016, 14, e2000504. [Google Scholar] [CrossRef]
- Rydeen, A.; Voisin, N.; D’Aniello, E.; Ravisankar, P.; Devignes, C.S.; Waxman, J.S. Excessive feedback of Cyp26a1 promotes cell non-autonomous loss of retinoic acid signaling. Dev. Biol. 2015, 405, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Marshall, H.; Nonchev, S.; Sham, M.H.; Muchamore, I.; Lumsden, A.; Krumlauf, R. Retinoic acid alters hindbrain Hox code and induces transformation of rhombomeres 2/3 into a 4/5 identity. Nature 1992, 360, 737–741. [Google Scholar] [CrossRef]
- Marshall, H.; Studer, M.; Pöpperl, H.; Aparicio, S.; Kuroiwa, A.; Brenner, S.; Krumlauf, R. A conserved retinoic acid response element required for early expression of the homeobox gene Hoxb-1. Nature 1994, 370, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, N.; Roux, M.; Ryckebüsch, L.; Niederreither, K.; Dollé, P.; Moon, A.; Capecchi, M.; Zaffran, S. Hox genes define distinct progenitor sub-domains within the second heart field. Dev. Biol. 2011, 353, 266–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waxman, J.S.; Keegan, B.R.; Roberts, R.W.; Poss, K.D.; Yelon, D. Hoxb5b acts downstream of retinoic acid signaling in the forelimb field to restrict heart field potential in zebrafish. Dev. Cell 2008, 15, 923–934. [Google Scholar] [CrossRef] [Green Version]
- Waxman, J.S.; Yelon, D. Increased Hox activity mimics the teratogenic effects of excess retinoic acid signaling. Dev. Dyn. 2009, 238, 1207–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostetskii, I.; Jiang, Y.; Kostetskaia, E.; Yuan, S.; Evans, T.; Zile, M. Retinoid signaling required for normal heart development regulates GATA-4 in a pathway distinct from cardiomyocyte differentiation. Dev. Biol. 1999, 206, 206–218. [Google Scholar] [CrossRef] [PubMed]
- De Bono, C.; Thellier, C.; Bertrand, N.; Sturny, R.; Jullian, E.; Cortes, C.; Stefanovic, S.; Zaffran, S.; Théveniau-Ruissy, M.; Kelly, R.G. T-box genes and retinoic acid signaling regulate the segregation of arterial and venous pole progenitor cells in the murine second heart field. Hum. Mol. Genet. 2018, 27, 3747–3760. [Google Scholar] [CrossRef] [PubMed]
- Matt, N.; Dupé, V.; Garnier, J.M.; Dennefeld, C.; Chambon, P.; Mark, M.; Ghyselinck, N.B. Retinoic acid-dependent eye morphogenesis is orchestrated by neural crest cells. Development 2005, 132, 4789–4800. [Google Scholar] [CrossRef] [Green Version]
- Devalla, H.D.; Schwach, V.; Ford, J.W.; Milnes, J.T.; El-Haou, S.; Jackson, C.; Gkatzis, K.; Elliott, D.A.; Chuva de Sousa Lopes, S.M.; Mummery, C.L.; et al. Atrial-like cardiomyocytes from human pluripotent stem cells are a robust preclinical model for assessing atrial-selective pharmacology. EMBO Mol. Med. 2015, 7, 394–410. [Google Scholar] [CrossRef]
- Lepilina, A.; Coon, A.N.; Kikuchi, K.; Holdway, J.E.; Roberts, R.W.; Burns, C.G.; Poss, K.D. A dynamic epicardial injury response supports progenitor cell activity during zebrafish heart regeneration. Cell 2006, 127, 607–619. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, K.; Holdway, J.E.; Major, R.J.; Blum, N.; Dahn, R.D.; Begemann, G.; Poss, K.D. Retinoic acid production by endocardium and epicardium is an injury response essential for zebrafish heart regeneration. Dev. Cell 2011, 20, 397–404. [Google Scholar] [CrossRef] [Green Version]
- Stuckmann, I.; Evans, S.; Lassar, A.B. Erythropoietin and retinoic acid, secreted from the epicardium, are required for cardiac myocyte proliferation. Dev. Biol. 2003, 255, 334–349. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Pomares, J.M.; Phelps, A.; Sedmerova, M.; Carmona, R.; González-Iriarte, M.; Muñoz-Chápuli, R.; Wessels, A. Experimental studies on the spatiotemporal expression of WT1 and RALDH2 in the embryonic avian heart: A model for the regulation of myocardial and valvuloseptal development by epicardially derived cells (EPDCs). Dev. Biol. 2002, 247, 307–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brade, T.; Kumar, S.; Cunningham, T.J.; Chatzi, C.; Zhao, X.; Cavallero, S.; Li, P.; Sucov, H.M.; Ruiz-Lozano, P.; Duester, G. Retinoic acid stimulates myocardial expansion by induction of hepatic erythropoietin which activates epicardial Igf2. Development 2011, 138, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.W.; Chen, B.; Yang, X.; Fugate, J.A.; Kalucki, F.A.; Futakuchi-Tsuchida, A.; Couture, L.; Vogel, K.W.; Astley, C.A.; Baldessari, A.; et al. Human embryonic stem cell-derived cardiomyocytes restore function in infarcted hearts of non-human primates. Nat. Biotechnol. 2018, 36, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Jones-Villeneuve, E.M.; McBurney, M.W.; Rogers, K.A.; Kalnins, V.I. Retinoic acid induces embryonal carcinoma cells to differentiate into neurons and glial cells. J. Cell Biol. 1982, 94, 253–262. [Google Scholar] [CrossRef]
- McBurney, M.W.; Jones-Villeneuve, E.M.; Edwards, M.K.; Anderson, P.J. Control of muscle and neuronal differentiation in a cultured embryonal carcinoma cell line. Nature 1982, 299, 165–167. [Google Scholar] [CrossRef]
- Strickland, S.; Mahdavi, V. The induction of differentiation in teratocarcinoma stem cells by retinoic acid. Cell 1978, 15, 393–403. [Google Scholar] [CrossRef]
- Branco, A.F.; Pereira, S.P.; Gonzalez, S.; Gusev, O.; Rizvanov, A.A.; Oliveira, P.J. Gene Expression Profiling of H9c2 Myoblast Differentiation towards a Cardiac-Like Phenotype. PLoS ONE 2015, 10, e0129303. [Google Scholar] [CrossRef] [Green Version]
- Ménard, C.; Pupier, S.; Mornet, D.; Kitzmann, M.; Nargeot, J.; Lory, P. Modulation of L-type calcium channel expression during retinoic acid-induced differentiation of H9C2 cardiac cells. J. Biol. Chem. 1999, 274, 29063–29070. [Google Scholar] [CrossRef] [Green Version]
- Leigh, R.S.; Ruskoaho, H.J.; Kaynak, B.L. A novel dual reporter embryonic stem cell line for toxicological assessment of teratogen-induced perturbation of anterior-posterior patterning of the heart. Arch. Toxicol. 2020, 94, 631–645. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Jiang, J.; Han, P.; Yuan, Q.; Zhang, J.; Zhang, X.; Xu, Y.; Cao, H.; Meng, Q.; Chen, L.; et al. Direct differentiation of atrial and ventricular myocytes from human embryonic stem cells by alternating retinoid signals. Cell Res. 2011, 21, 579–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Protze, S.I.; Laksman, Z.; Backx, P.H.; Keller, G.M. Human Pluripotent Stem Cell-Derived Atrial and Ventricular Cardiomyocytes Develop from Distinct Mesoderm Populations. Cell Stem Cell 2017, 21, 179–194. [Google Scholar] [CrossRef]
- Drowley, L.; McPheat, J.; Nordqvist, A.; Peel, S.; Karlsson, U.; Martinsson, S.; Müllers, E.; Dellsén, A.; Knight, S.; Barrett, I.; et al. Discovery of retinoic acid receptor agonists as proliferators of cardiac progenitor cells through a phenotypic screening approach. Stem Cells Transl. Med. 2020, 9, 47–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karppi, J.; Laukkanen, J.A.; Mäkikallio, T.H.; Kurl, S. Low serum lycopene and β-carotene increase risk of acute myocardial infarction in men. Eur. J. Public Health 2012, 22, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Karppi, J.; Laukkanen, J.A.; Mäkikallio, T.H.; Ronkainen, K.; Kurl, S. Low β-carotene concentrations increase the risk of cardiovascular disease mortality among Finnish men with risk factors. Nutr. Metab. Cardiovasc. Dis. 2012, 22, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Min, K.B.; Min, J.Y. Relation of serum vitamin A levels to all-cause and cause-specific mortality among older adults in the NHANES III population. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 1197–1203. [Google Scholar] [CrossRef]
- Olsen, T.; Vinknes, K.J.; Svingen, G.F.T.; Pedersen, E.R.; Tell, G.S.; Blomhoff, R.; Drevon, C.A.; Ueland, P.M.; Midttun, Ø.; Refsum, H.; et al. Cardiovascular disease risk associated with serum apolipoprotein B is modified by serum vitamin A. Atherosclerosis 2017, 265, 325–330. [Google Scholar] [CrossRef] [Green Version]
- Saha, N.; Ng, T.B.; Tan, P.Y.; Wee, K.P. Vitamin A reserve of liver in health and coronary heart disease among ethnic groups in Singapore. Br. J. Nutr. 1988, 60, 407–412. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Guleria, R.S.; Thomas, C.M.; Roth, A.; Gerilechaogetu, F.; Kumar, R.; Dostal, D.E.; Baker, K.M.; Pan, J. Loss of myocardial retinoic acid receptor α induces diastolic dysfunction by promoting intracellular oxidative stress and calcium mishandling in adult mice. J. Mol. Cell. Cardiol. 2016, 99, 100–112. [Google Scholar] [CrossRef] [Green Version]
- Park, S.W.; Persaud, S.D.; Ogokeh, S.; Meyers, T.A.; Townsend, D.; Wei, L.N. CRABP1 protects the heart from isoproterenol-induced acute and chronic remodeling. J. Endocrinol. 2018, 236, 151–165. [Google Scholar] [CrossRef]
- He, Y.; Huang, Y.; Zhou, L.; Lu, L.M.; Zhu, Y.C.; Yao, T. All-trans retinoic acid inhibited angiotensin II-induced increase in cell growth and collagen secretion of neonatal cardiac fibroblasts. Acta Pharmacol. Sin. 2006, 27, 423–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paiva, S.A.; Matsubara, L.S.; Matsubara, B.B.; Minicucci, M.F.; Azevedo, P.S.; Campana, A.O.; Zornoff, L.A. Retinoic acid supplementation attenuates ventricular remodeling after myocardial infarction in rats. J. Nutr. 2005, 135, 2326–2328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colbert, M.C.; Hall, D.G.; Kimball, T.R.; Witt, S.A.; Lorenz, J.N.; Kirby, M.L.; Hewett, T.E.; Klevitsky, R.; Robbins, J. Cardiac compartment-specific overexpression of a modified retinoic acid receptor produces dilated cardiomyopathy and congestive heart failure in transgenic mice. J. Clin. Investig. 1997, 100, 1958–1968. [Google Scholar] [CrossRef] [Green Version]
- Manolescu, D.C.; Jankowski, M.; Danalache, B.A.; Wang, D.; Broderick, T.L.; Chiasson, J.L.; Gutkowska, J. All-trans retinoic acid stimulates gene expression of the cardioprotective natriuretic peptide system and prevents fibrosis and apoptosis in cardiomyocytes of obese ob/ob mice. Appl. Physiol. Nutr. Metab. 2014, 39, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, J.; Lu, M.; Wang, H.; Tang, F. Retinoic acid attenuates cardiac injury induced by hyperglycemia in pre- and post-delivery mice. Can. J. Physiol. Pharmacol. 2020, 98, 6–14. [Google Scholar] [CrossRef]
- Oliveira, L.C.; Azevedo, P.S.; Minicucci, M.E.; Rafacho, B.P.; Duarte, D.R.; Matsubara, L.S.; Matsubara, B.B.; Paiva, S.A.; Zornoff, L.A. Retinoic acid prevents ventricular remodelling induced by tobacco smoke exposure in rats. Acta Cardiol. 2011, 66, 3–7. [Google Scholar] [CrossRef]
- Subramanian, U.; Nagarajan, D. All-Trans Retinoic Acid supplementation prevents cardiac fibrosis and cytokines induced by Methylglyoxal. Glycoconj. J. 2017, 34, 255–265. [Google Scholar] [CrossRef]
- Zhou, M.D.; Sucov, H.M.; Evans, R.M.; Chien, K.R. Retinoid-dependent pathways suppress myocardial cell hypertrophy. Proc. Natl. Acad. Sci. USA 1995, 92, 7391–7395. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Garami, M.; Cheng, T.; Gardner, D.G. 1,25(OH)2 vitamin D3, and retinoic acid antagonize endothelin-stimulated hypertrophy of neonatal rat cardiac myocytes. J. Clin. Investig. 1996, 97, 1577–1588. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, R.; Palm-Leis, A.; Scott, R.C., 3rd; Guleria, R.S.; Rachut, E.; Baker, K.M.; Pan, J. All-trans retinoic acid prevents development of cardiac remodeling in aortic banded rats by inhibiting the renin-angiotensin system. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H633–H644. [Google Scholar] [CrossRef] [Green Version]
- Park, S.W.; Nhieu, J.; Lin, Y.W.; Wei, L.N. All-trans retinoic acid attenuates isoproterenol-induced cardiac dysfunction through Crabp1 to dampen CaMKII activation. Eur. J. Pharmacol. 2019, 858, 172485. [Google Scholar] [CrossRef]
- Sultan, F.; Kaur, R.; Mir, A.H.; Maqbool, I.; Lonare, M.; Singh, D.; Rampal, S.; Dar, J.A. Rosuvastatin and retinoic acid may act as ‘pleiotropic agents’ against β-adrenergic agonist-induced acute myocardial injury through modulation of multiple signalling pathways. Chem. Biol. Interact. 2020, 318, 108970. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.X.; Leaf, A. Protective effects of All-trans-retinoic acid against cardiac arrhythmias induced by isoproterenol, lysophosphatidylcholine or ischemia and reperfusion. J. Cardiovasc. Pharmacol. 1995, 26, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, P.S.; Minicucci, M.F.; Chiuso-Minicucci, F.; Justulin, L.A., Jr.; Matsubara, L.S.; Matsubara, B.B.; Novelli, E.; Seiva, F.; Ebaid, G.; Campana, A.O.; et al. Ventricular remodeling induced by tissue vitamin A deficiency in rats. Cell. Physiol. Biochem. 2010, 26, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Minicucci, M.F.; Azevedo, P.S.; Oliveira, S.A., Jr.; Martinez, P.F.; Chiuso-Minicucci, F.; Polegato, B.F.; Justulin, L.A., Jr.; Matsubara, L.S.; Matsubara, B.B.; Paiva, S.A.; et al. Tissue vitamin A insufficiency results in adverse ventricular remodeling after experimental myocardial infarction. Cell. Physiol. Biochem. 2010, 26, 523–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asson-Batres, M.A.; Ryzhov, S.; Tikhomirov, O.; Duarte, C.W.; Congdon, C.B.; Lessard, C.R.; McFarland, S.; Rochette-Egly, C.; Tran, T.L.; Galindo, C.L.; et al. Effects of vitamin A deficiency in the postnatal mouse heart: Role of hepatic retinoid stores. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H1773–H1789. [Google Scholar] [CrossRef] [Green Version]
- de Paiva, S.A.; Zornoff, L.A.; Okoshi, M.P.; Okoshi, K.; Matsubara, L.S.; Matsubara, B.B.; Cicogna, A.C.; Campana, A.O. Ventricular remodeling induced by retinoic acid supplementation in adult rats. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H2242–H2246. [Google Scholar] [CrossRef] [Green Version]
- Silva, R.A.C.; Gonçalves, A.F.; Dos Santos, P.P.; Rafacho, B.; Claro, R.F.T.; Minicucci, M.F.; Azevedo, P.S.; Polegato, B.F.; Zanati, S.G.; Fernandes, A.A.; et al. Cardiac Remodeling Induced by All-Trans Retinoic Acid is Detrimental in Normal Rats. Cell. Physiol. Biochem. 2017, 43, 1449–1459. [Google Scholar] [CrossRef]
- Doran, A.C.; Meller, N.; McNamara, C.A. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 812–819. [Google Scholar] [CrossRef] [Green Version]
- Miano, J.M.; Topouzis, S.; Majesky, M.W.; Olson, E.N. Retinoid receptor expression and all-trans retinoic acid-mediated growth inhibition in vascular smooth muscle cells. Circulation 1996, 93, 1886–1895. [Google Scholar] [CrossRef]
- Neuville, P.; Yan, Z.; Gidlöf, A.; Pepper, M.S.; Hansson, G.K.; Gabbiani, G.; Sirsjö, A. Retinoic acid regulates arterial smooth muscle cell proliferation and phenotypic features in vivo and in vitro through an RARalpha-dependent signaling pathway. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1430–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pakala, R.; Benedict, C.R. RAR gamma agonists inhibit proliferation of vascular smooth muscle cells. J. Cardiovasc. Pharmacol. 2000, 35, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, C.; Sasaguri, T.; Komiyama, Y.; Takahashi, H. All-trans retinoic acid inhibits vascular smooth muscle cell proliferation targeting multiple genes for cyclins and cyclin-dependent kinases. Hypertens. Res. 2001, 24, 579–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlandi, A.; Francesconi, A.; Cocchia, D.; Corsini, A.; Spagnoli, L.G. Phenotypic heterogeneity influences apoptotic susceptibility to retinoic acid and cis-platinum of rat arterial smooth muscle cells in vitro: Implications for the evolution of experimental intimal thickening. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1118–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, H.; Haendeler, J.; Aebly, M.R.; Kelly, L.A.; Cholewa, B.C.; Koike, G.; Kwitek-Black, A.; Jacob, H.J.; Berk, B.C.; Miano, J.M. Retinoic acid-induced tissue transglutaminase and apoptosis in vascular smooth muscle cells. Circ. Res. 2000, 87, 881–887. [Google Scholar] [CrossRef] [Green Version]
- Axel, D.I.; Frigge, A.; Dittmann, J.; Runge, H.; Spyridopoulos, I.; Riessen, R.; Viebahn, R.; Karsch, K.R. All-trans retinoic acid regulates proliferation, migration, differentiation, and extracellular matrix turnover of human arterial smooth muscle cells. Cardiovasc. Res. 2001, 49, 851–862. [Google Scholar] [CrossRef]
- Johst, U.; Betsch, A.; Wiskirchen, J.; Schöber, W.; Vonthein, R.; Rinkert, N.; Kehlbach, R.; Claussen, C.D.; Duda, S.H. All-trans and 9-cis retinoid acids inhibit proliferation, migration, and synthesis of extracellular matrix of human vascular smooth muscle cells by inducing differentiation in vitro. J. Cardiovasc. Pharmacol. 2003, 41, 526–535. [Google Scholar] [CrossRef]
- Wakino, S.; Kintscher, U.; Kim, S.; Jackson, S.; Yin, F.; Nagpal, S.; Chandraratna, R.A.; Hsueh, W.A.; Law, R.E. Retinoids inhibit proliferation of human coronary smooth muscle cells by modulating cell cycle regulators. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 746–751. [Google Scholar] [CrossRef] [Green Version]
- Gidlof, A.C.; Ocaya, P.; Olofsson, P.S.; Torma, H.; Sirsjo, A. Differences in retinol metabolism and proliferative response between neointimal and medial smooth muscle cells. J. Vasc. Res. 2006, 43, 392–398. [Google Scholar] [CrossRef]
- Bilbija, D.; Elmabsout, A.A.; Sagave, J.; Haugen, F.; Bastani, N.; Dahl, C.P.; Gullestad, L.; Sirsjö, A.; Blomhoff, R.; Valen, G. Expression of retinoic acid target genes in coronary artery disease. Int. J. Mol. Med. 2014, 33, 677–686. [Google Scholar] [CrossRef]
- Saito, A.; Sugawara, A.; Uruno, A.; Kudo, M.; Kagechika, H.; Sato, Y.; Owada, Y.; Kondo, H.; Sato, M.; Kurabayashi, M.; et al. All-trans retinoic acid induces in vitro angiogenesis via retinoic acid receptor: Possible involvement of paracrine effects of endogenous vascular endothelial growth factor signaling. Endocrinology 2007, 148, 1412–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.W.; Park, S.J.; Park, S.W.; Kim, J.J.; Hong, M.K.; Song, J.K. All-trans-retinoic acid attenuates neointima formation with acceleration of reendothelialization in balloon-injured rat aorta. J. Korean Med. Sci. 2000, 15, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Miano, J.M.; Kelly, L.A.; Artacho, C.A.; Nuckolls, T.A.; Piantedosi, R.; Blaner, W.S. All-Trans-retinoic acid reduces neointimal formation and promotes favorable geometric remodeling of the rat carotid artery after balloon withdrawal injury. Circulation 1998, 98, 1219–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; He, B.; Zheng, D.; Zhang, S.; Liu, J.; Zhu, S. All-trans retinoic acid reduces intimal thickening after balloon angioplasty in atherosclerotic rabbits. Chin. Med. J. 1999, 112, 121–123. [Google Scholar] [PubMed]
- Herdeg, C.; Oberhoff, M.; Baumbach, A.; Schroeder, S.; Leitritz, M.; Blattner, A.; Siegel-Axel, D.I.; Meisner, C.; Karsch, K.R. Effects of local all-trans-retinoic acid delivery on experimental atherosclerosis in the rabbit carotid artery. Cardiovasc. Res. 2003, 57, 544–553. [Google Scholar] [CrossRef] [Green Version]
- Leville, C.D.; Dassow, M.S.; Seabrook, G.R.; Jean-Claude, J.M.; Towne, J.B.; Cambria, R.A. All-trans-retinoic acid decreases vein graft intimal hyperplasia and matrix metalloproteinase activity in vivo. J. Surg. Res. 2000, 90, 183–190. [Google Scholar] [CrossRef]
- Fujiu, K.; Manabe, I.; Ishihara, A.; Oishi, Y.; Iwata, H.; Nishimura, G.; Shindo, T.; Maemura, K.; Kagechika, H.; Shudo, K.; et al. Synthetic retinoid Am80 suppresses smooth muscle phenotypic modulation and in-stent neointima formation by inhibiting KLF5. Circ. Res. 2005, 97, 1132–1141. [Google Scholar] [CrossRef] [Green Version]
- Lü, L.; Yao, T.; Zhu, Y.Z.; Huang, G.Y.; Cao, Y.X.; Zhu, Y.C. Chronic all-trans retinoic acid treatment prevents medial thickening of intramyocardial and intrarenal arteries in spontaneously hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1370–H1377. [Google Scholar] [CrossRef] [Green Version]
- Wiegman, P.J.; Barry, W.L.; McPherson, J.A.; McNamara, C.A.; Gimple, L.W.; Sanders, J.M.; Bishop, G.G.; Powers, E.R.; Ragosta, M.; Owens, G.K.; et al. All-trans-retinoic acid limits restenosis after balloon angioplasty in the focally atherosclerotic rabbit: A favorable effect on vessel remodeling. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 89–95. [Google Scholar] [CrossRef] [Green Version]
- Medhora, M.M. Retinoic acid upregulates beta(1)-integrin in vascular smooth muscle cells and alters adhesion to fibronectin. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H382–H387. [Google Scholar] [CrossRef] [Green Version]
- Wright, G.; Wang, S.; Bailey, G.; Reichenbecher, V.; Wright, G.L. Effect of retinoic acid on contractile competence of vascular smooth muscle. Am. J. Physiol. 1996, 270, H1363–H1370. [Google Scholar] [CrossRef]
- Wright, G.L.; Wang, S.; Fultz, M.E.; Arif, I.; Matthews, K.; Chertow, B.S. Effect of vitamin A deficiency on cardiovascular function in the rat. Can. J. Physiol. Pharmacol. 2002, 80, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.A.; Chen, J.; Nallamshetty, S.; Pham, T.; Goto, S.; Muehlschlegel, J.D.; Libby, P.; Aikawa, M.; Aikawa, E.; Plutzky, J. Retinoids Repress Human Cardiovascular Cell Calcification With Evidence for Distinct Selective Retinoid Modulator Effects. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 656–669. [Google Scholar] [CrossRef] [PubMed]
- Huk, D.J.; Hammond, H.L.; Kegechika, H.; Lincoln, J. Increased dietary intake of vitamin A promotes aortic valve calcification in vivo. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Krivospitskaya, O.; Elmabsout, A.A.; Sundman, E.; Söderström, L.A.; Ovchinnikova, O.; Gidlöf, A.C.; Scherbak, N.; Norata, G.D.; Samnegård, A.; Törmä, H.; et al. A CYP26B1 polymorphism enhances retinoic acid catabolism and may aggravate atherosclerosis. Mol. Med. 2012, 18, 712–718. [Google Scholar] [CrossRef] [PubMed]
- Kaye, D.M.; Krum, H. Drug discovery for heart failure: A new era or the end of the pipeline? Nat. Rev. Drug Discov. 2007, 6, 127–139. [Google Scholar] [CrossRef]
- Chen, S.; Gardner, D.G. Retinoic acid uses divergent mechanisms to activate or suppress mitogenesis in rat aortic smooth muscle cells. J. Clin. Investig. 1998, 102, 653–662. [Google Scholar] [CrossRef]
- Kumar, P.; Garg, R.; Bolden, G.; Pandey, K.N. Interactive roles of Ets-1, Sp1, and acetylated histones in the retinoic acid-dependent activation of guanylyl cyclase/atrial natriuretic peptide receptor-A gene transcription. J. Biol. Chem. 2010, 285, 37521–37530. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.J.; Zhu, Y.C.; Yao, T. Effects of all-trans retinoic acid on angiotensin II-induced myocyte hypertrophy. J. Appl. Physiol. 2002, 92, 2162–2168. [Google Scholar] [CrossRef] [Green Version]
- Haxsen, V.; Adam-Stitah, S.; Ritz, E.; Wagner, J. Retinoids inhibit the actions of angiotensin II on vascular smooth muscle cells. Circ. Res. 2001, 88, 637–644. [Google Scholar] [CrossRef] [Green Version]
- Oka, T.; Maillet, M.; Watt, A.J.; Schwartz, R.J.; Aronow, B.J.; Duncan, S.A.; Molkentin, J.D. Cardiac-specific deletion of Gata4 reveals its requirement for hypertrophy, compensation, and myocyte viability. Circ. Res. 2006, 98, 837–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arceci, R.J.; King, A.A.; Simon, M.C.; Orkin, S.H.; Wilson, D.B. Mouse GATA-4: A retinoic acid-inducible GATA-binding transcription factor expressed in endodermally derived tissues and heart. Mol. Cell. Biol. 1993, 13, 2235–2246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molkentin, J.D.; Kalvakolanu, D.V.; Markham, B.E. Transcription factor GATA-4 regulates cardiac muscle-specific expression of the alpha-myosin heavy-chain gene. Mol. Cell. Biol. 1994, 14, 4947–4957. [Google Scholar] [CrossRef] [Green Version]
- Pikkarainen, S.; Tokola, H.; Majalahti-Palviainen, T.; Kerkela, R.; Hautala, N.; Bhalla, S.S.; Charron, F.; Nemer, M.; Vuolteenaho, O.; Ruskoaho, H. GATA-4 is a nuclear mediator of mechanical stretch-activated hypertrophic program. J. Biol. Chem. 2003, 278, 23807–23816. [Google Scholar] [CrossRef] [Green Version]
- Rysä, J.; Tenhunen, O.; Serpi, R.; Soini, Y.; Nemer, M.; Leskinen, H.; Ruskoaho, H. GATA-4 is an angiogenic survival factor of the infarcted heart. Circ. Heart Fail. 2010, 3, 440–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruneau, B.G.; Nemer, G.; Schmitt, J.P.; Charron, F.; Robitaille, L.; Caron, S.; Conner, D.A.; Gessler, M.; Nemer, M.; Seidman, C.E.; et al. A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell 2001, 106, 709–721. [Google Scholar] [CrossRef] [Green Version]
- Koshiba-Takeuchi, K.; Mori, A.D.; Kaynak, B.L.; Cebra-Thomas, J.; Sukonnik, T.; Georges, R.O.; Latham, S.; Beck, L.; Henkelman, R.M.; Black, B.L.; et al. Reptilian heart development and the molecular basis of cardiac chamber evolution. Nature 2009, 461, 95–98. [Google Scholar] [CrossRef]
- Luna-Zurita, L.; Stirnimann, C.U.; Glatt, S.; Kaynak, B.L.; Thomas, S.; Baudin, F.; Samee, M.A.; He, D.; Small, E.M.; Mileikovsky, M.; et al. Complex Interdependence Regulates Heterotypic Transcription Factor Distribution and Coordinates Cardiogenesis. Cell 2016, 164, 999–1014. [Google Scholar] [CrossRef] [Green Version]
- Nadadur, R.D.; Broman, M.T.; Boukens, B.; Mazurek, S.R.; Yang, X.; van den Boogaard, M.; Bekeny, J.; Gadek, M.; Ward, T.; Zhang, M.; et al. Pitx2 modulates a Tbx5-dependent gene regulatory network to maintain atrial rhythm. Sci. Transl. Med. 2016, 8, 354ra115. [Google Scholar] [CrossRef] [Green Version]
- Palm-Leis, A.; Singh, U.S.; Herbelin, B.S.; Olsovsky, G.D.; Baker, K.M.; Pan, J. Mitogen-activated protein kinases and mitogen-activated protein kinase phosphatases mediate the inhibitory effects of all-trans retinoic acid on the hypertrophic growth of cardiomyocytes. J. Biol. Chem. 2004, 279, 54905–54917. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Deng, B.; Jiang, X.; Cai, M.; Liu, N.; Zhang, S.; Tan, Y.; Huang, G.; Jin, W.; Liu, B.; et al. All-Trans-Retinoic Acid Suppresses Neointimal Hyperplasia and Inhibits Vascular Smooth Muscle Cell Proliferation and Migration via Activation of AMPK Signaling Pathway. Front. Pharmacol. 2019, 10, 485. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, A.; Kanai, H.; Arai, M.; Sekiguchi, K.; Uchiyama, T.; Nagai, R.; Kurabayashi, M. Retinoids induce the PAI-1 gene expression through tyrosine kinase-dependent pathways in vascular smooth muscle cells. J. Cardiovasc. Pharmacol. 2002, 39, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Das, N.P. Effects of vitamin A and its analogs on nonenzymatic lipid peroxidation in rat brain mitochondria. J. Neurochem. 1989, 52, 585–588. [Google Scholar] [CrossRef]
- Halevy, O.; Sklan, D. Inhibition of arachidonic acid oxidation by beta-carotene, retinol and alpha-tocopherol. Biochim. Biophys. Acta 1987, 918, 304–307. [Google Scholar] [CrossRef]
- Samokyszyn, V.M.; Marnett, L.J. Inhibition of liver microsomal lipid peroxidation by 13-cis-retinoic acid. Free Radic. Biol. Med. 1990, 8, 491–496. [Google Scholar] [CrossRef]
- Vile, G.F.; Winterbourn, C.C. Inhibition of adriamycin-promoted microsomal lipid peroxidation by beta-carotene, alpha-tocopherol and retinol at high and low oxygen partial pressures. FEBS Lett. 1988, 238, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Tesoriere, L.; Ciaccio, M.; Valenza, M.; Bongiorno, A.; Maresi, E.; Albiero, R.; Livrea, M.A. Effect of vitamin A administration on resistance of rat heart against doxorubicin-induced cardiotoxicity and lethality. J. Pharmacol. Exp. Ther. 1994, 269, 430–436. [Google Scholar]
- Danelisen, I.; Palace, V.; Lou, H.; Singal, P.K. Maintenance of myocardial levels of vitamin A in heart failure due to adriamycin. J. Mol. Cell. Cardiol. 2002, 34, 789–795. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhu, J.; Zhao, X.; Yang, K.; Lu, L.; Zhang, F.; Shen, W.; Zhang, R. All-Trans Retinoic Acid Ameliorates Myocardial Ischemia/Reperfusion Injury by Reducing Cardiomyocyte Apoptosis. PLoS ONE 2015, 10, e0133414. [Google Scholar] [CrossRef]
- Vega, V.A.; Anzulovich, A.C.; Varas, S.M.; Bonomi, M.R.; Giménez, M.S.; Oliveros, L.B. Effect of nutritional vitamin A deficiency on lipid metabolism in the rat heart: Its relation to PPAR gene expression. Nutrition 2009, 25, 828–838. [Google Scholar] [CrossRef]
- Cresci, S.; Clabby, M.L.; Kelly, D.P. Evidence for a novel cardiac-enriched retinoid X receptor partner. J. Biol. Chem. 1999, 274, 25668–25674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alpha-Tocopherol Beta Carotene Cancer Prevention Study Group. The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. N. Engl. J. Med. 1994, 330, 1029–1035. [Google Scholar] [CrossRef]
- Greenberg, E.R.; Baron, J.A.; Karagas, M.R.; Stukel, T.A.; Nierenberg, D.W.; Stevens, M.M.; Mandel, J.S.; Haile, R.W. Mortality associated with low plasma concentration of beta carotene and the effect of oral supplementation. JAMA 1996, 275, 699–703. [Google Scholar] [CrossRef]
- Hennekens, C.H.; Buring, J.E.; Manson, J.E.; Stampfer, M.; Rosner, B.; Cook, N.R.; Belanger, C.; LaMotte, F.; Gaziano, J.M.; Ridker, P.M.; et al. Lack of effect of long-term supplementation with beta carotene on the incidence of malignant neoplasms and cardiovascular disease. N. Engl. J. Med. 1996, 334, 1145–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virtamo, J.; Rapola, J.M.; Ripatti, S.; Heinonen, O.P.; Taylor, P.R.; Albanes, D.; Huttunen, J.K. Effect of vitamin E and beta carotene on the incidence of primary nonfatal myocardial infarction and fatal coronary heart disease. Arch. Intern. Med. 1998, 158, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Simioniuc, A.; Campan, M.; Lionetti, V.; Marinelli, M.; Aquaro, G.D.; Cavallini, C.; Valente, S.; Di Silvestre, D.; Cantoni, S.; Bernini, F.; et al. Placental stem cells pre-treated with a hyaluronan mixed ester of butyric and retinoic acid to cure infarcted pig hearts: A multimodal study. Cardiovasc. Res. 2011, 90, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Ventura, C.; Maioli, M.; Asara, Y.; Santoni, D.; Scarlata, I.; Cantoni, S.; Perbellini, A. Butyric and retinoic mixed ester of hyaluronan. A novel differentiating glycoconjugate affording a high throughput of cardiogenesis in embryonic stem cells. J. Biol. Chem. 2004, 279, 23574–23579. [Google Scholar] [CrossRef] [Green Version]
- Ventura, C.; Cantoni, S.; Bianchi, F.; Lionetti, V.; Cavallini, C.; Scarlata, I.; Foroni, L.; Maioli, M.; Bonsi, L.; Alviano, F.; et al. Hyaluronan mixed esters of butyric and retinoic Acid drive cardiac and endothelial fate in term placenta human mesenchymal stem cells and enhance cardiac repair in infarcted rat hearts. J. Biol. Chem. 2007, 282, 14243–14252. [Google Scholar] [CrossRef] [Green Version]
- Lionetti, V.; Cantoni, S.; Cavallini, C.; Bianchi, F.; Valente, S.; Frascari, I.; Olivi, E.; Aquaro, G.D.; Bonavita, F.; Scarlata, I.; et al. Hyaluronan mixed esters of butyric and retinoic acid affording myocardial survival and repair without stem cell transplantation. J. Biol. Chem. 2010, 285, 9949–9961. [Google Scholar] [CrossRef] [Green Version]
- Danzl, K.; Messner, B.; Doppler, C.; Nebert, C.; Abfalterer, A.; Sakic, A.; Temml, V.; Heinz, K.; Streitwieser, R.; Edelmann, T.; et al. Early inhibition of endothelial retinoid uptake upon myocardial infarction restores cardiac function and prevents cell, tissue, and animal death. J. Mol. Cell. Cardiol. 2019, 126, 105–117. [Google Scholar] [CrossRef] [Green Version]
- Messner, B.; Kern, J.; Wiedemann, D.; Schwaiger, S.; Türkcan, A.; Ploner, C.; Trockenbacher, A.; Aumayr, K.; Bonaros, N.; Laufer, G.; et al. 5-Methoxyleoligin, a lignan from Edelweiss, stimulates CYP26B1-dependent angiogenesis in vitro and induces arteriogenesis in infarcted rat hearts in vivo. PLoS ONE 2013, 8, e58342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Overview of Vitamin A/retinoid signaling. Dietary intake of retinoids is commonly in the form of β-carotene, Retinyl esters, or retinol. These are stored in the liver and transported to target tissues via retinol binding proteins. Active retinoid forms of all-trans retinoic acid (ATRA) and 9-cis retinoic acid (9CRA) bind to homo and heterodimers of retinoic acid receptors (RARs) and retinoid X receptors (RXRs). Levels of ATRA and 9CRA are controlled via Cyp26-mediated degradation of active metabolites. Following myocardial infarction, vitamin A levels have been shown to increase in the heart, as evidenced by direct measurement of vitamin A levels and RA-induced transcription. All-trans retinoic acid (ATRA), 9-cis retinoic acid (9CRA), Retinoic acid receptor (RAR), Retinoid X receptor (RXR).
Figure 1.
Overview of Vitamin A/retinoid signaling. Dietary intake of retinoids is commonly in the form of β-carotene, Retinyl esters, or retinol. These are stored in the liver and transported to target tissues via retinol binding proteins. Active retinoid forms of all-trans retinoic acid (ATRA) and 9-cis retinoic acid (9CRA) bind to homo and heterodimers of retinoic acid receptors (RARs) and retinoid X receptors (RXRs). Levels of ATRA and 9CRA are controlled via Cyp26-mediated degradation of active metabolites. Following myocardial infarction, vitamin A levels have been shown to increase in the heart, as evidenced by direct measurement of vitamin A levels and RA-induced transcription. All-trans retinoic acid (ATRA), 9-cis retinoic acid (9CRA), Retinoic acid receptor (RAR), Retinoid X receptor (RXR).

Figure 2.
Vitamin A signaling and cardiac regeneration. (a) During zebrafish heart regeneration, Raldh2 increases in the epicardium of injured hearts, promoting cardiomyocyte proliferation and regeneration of lost myocardial tissue. Secreted retinoic acid has also been shown to promote proliferation of the embryonic chick heart, indicating the possibility to manipulate this pathway to induce therapeutic cardiac regeneration via cardiomyocyte proliferation. (b) Effects of Vitamin A/retinoids on the differentiation of pluripotent-stem cell-derived cardiomyocytes observed in Leigh et al. 2020. Vitamin A/retinoids promote the differentiation of pluripotent stem cells to atrial cardiomyocytes in a process involving the expression of retinoic acid-degrading Cyp26 enzymes in multipotent cardiac progenitor cells. Reporter cell lines in Leigh et al. allow assessment of differentiation to atrial (atrRFP) and ventricular (venGFP) cell fates and demonstrate the stage-specific effects of retinoids. Atria (A), Ventricle (V), Sinus Venosus (SV), Bulbus arteriosus (BA), Retinoic Acid (RA), Cardiomyocyte (CM), Pluripotent Stem Cells (PSCs), Cardiac Progenitor Cells (CPCs), Retinoic acid (RA), All-trans retinoic acid (ATRA), Ventricular GFP reporter (venGFP), atrial RFP reporter (atrRFP).
Figure 2.
Vitamin A signaling and cardiac regeneration. (a) During zebrafish heart regeneration, Raldh2 increases in the epicardium of injured hearts, promoting cardiomyocyte proliferation and regeneration of lost myocardial tissue. Secreted retinoic acid has also been shown to promote proliferation of the embryonic chick heart, indicating the possibility to manipulate this pathway to induce therapeutic cardiac regeneration via cardiomyocyte proliferation. (b) Effects of Vitamin A/retinoids on the differentiation of pluripotent-stem cell-derived cardiomyocytes observed in Leigh et al. 2020. Vitamin A/retinoids promote the differentiation of pluripotent stem cells to atrial cardiomyocytes in a process involving the expression of retinoic acid-degrading Cyp26 enzymes in multipotent cardiac progenitor cells. Reporter cell lines in Leigh et al. allow assessment of differentiation to atrial (atrRFP) and ventricular (venGFP) cell fates and demonstrate the stage-specific effects of retinoids. Atria (A), Ventricle (V), Sinus Venosus (SV), Bulbus arteriosus (BA), Retinoic Acid (RA), Cardiomyocyte (CM), Pluripotent Stem Cells (PSCs), Cardiac Progenitor Cells (CPCs), Retinoic acid (RA), All-trans retinoic acid (ATRA), Ventricular GFP reporter (venGFP), atrial RFP reporter (atrRFP).
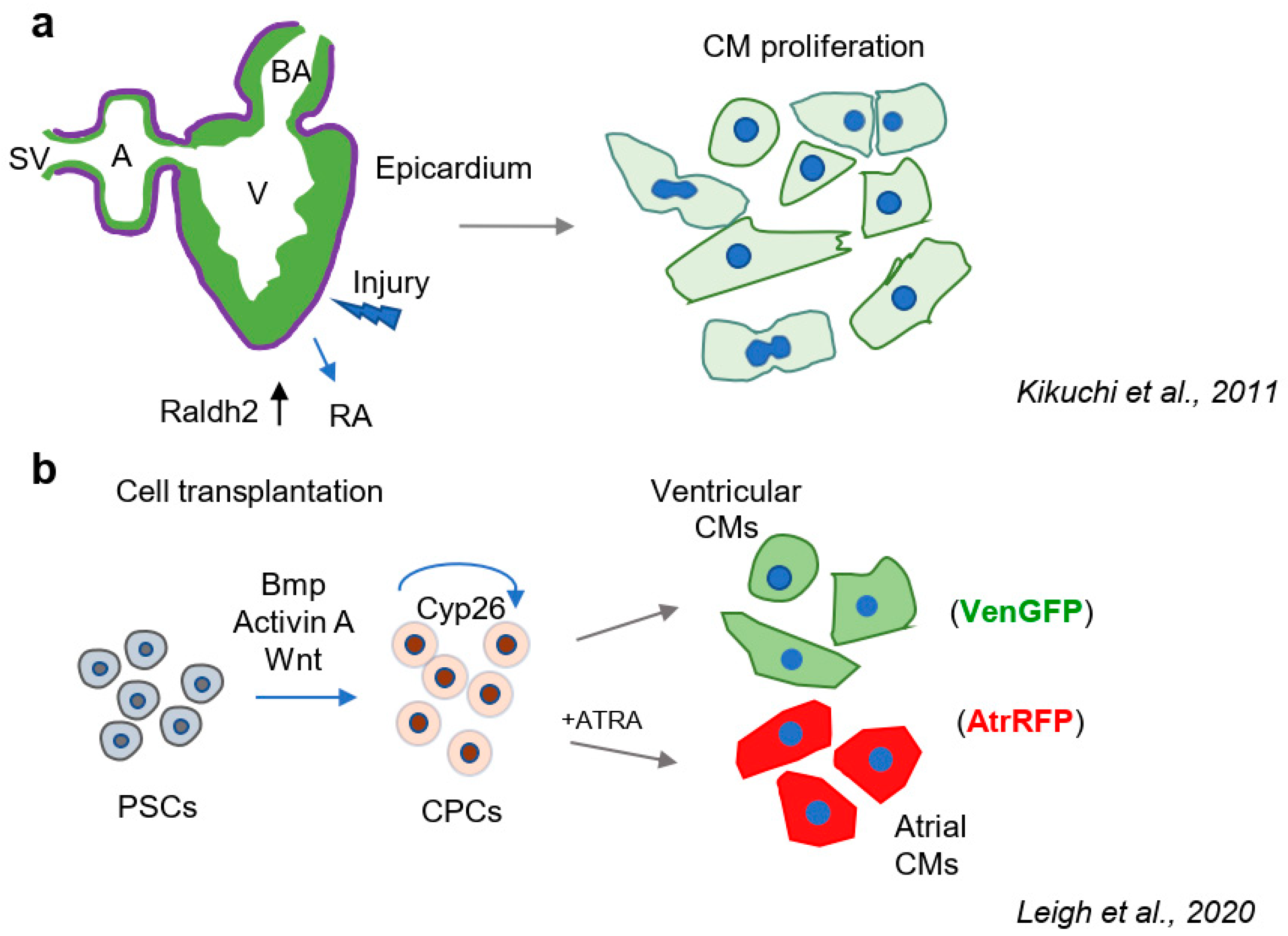
Figure 3.
Anti-hypertrophic and anti-proliferative effects of Vitamin A/retinoid signaling in the postnatal mammalian heart. (a) Anti-hypertrophic effects of retinoids on neonatal cardiomyocytes. Retinoids interfere with the induction of cellular hypertrophy induced by physical stretch and neurohormonal signals, such as Angiotensin II, Phenylephrine, and Endothelin. This suggests a mechanism through which retinoids could promote favorable changes in the diseased heart; (b) Anti-proliferative effects of retinoids on vascular smooth muscle cells. ATRA inhibits the proliferation of smooth muscle cells in vitro and in vivo. Inhibition of smooth muscle cell proliferation impedes thickening of the arteries and pathological vascular remodeling. Cardiomyocytes (CMs), All-trans retinoic acid (ATRA), Retinoic acid receptor (RAR), Retinoid x receptor (RXR).
Figure 3.
Anti-hypertrophic and anti-proliferative effects of Vitamin A/retinoid signaling in the postnatal mammalian heart. (a) Anti-hypertrophic effects of retinoids on neonatal cardiomyocytes. Retinoids interfere with the induction of cellular hypertrophy induced by physical stretch and neurohormonal signals, such as Angiotensin II, Phenylephrine, and Endothelin. This suggests a mechanism through which retinoids could promote favorable changes in the diseased heart; (b) Anti-proliferative effects of retinoids on vascular smooth muscle cells. ATRA inhibits the proliferation of smooth muscle cells in vitro and in vivo. Inhibition of smooth muscle cell proliferation impedes thickening of the arteries and pathological vascular remodeling. Cardiomyocytes (CMs), All-trans retinoic acid (ATRA), Retinoic acid receptor (RAR), Retinoid x receptor (RXR).
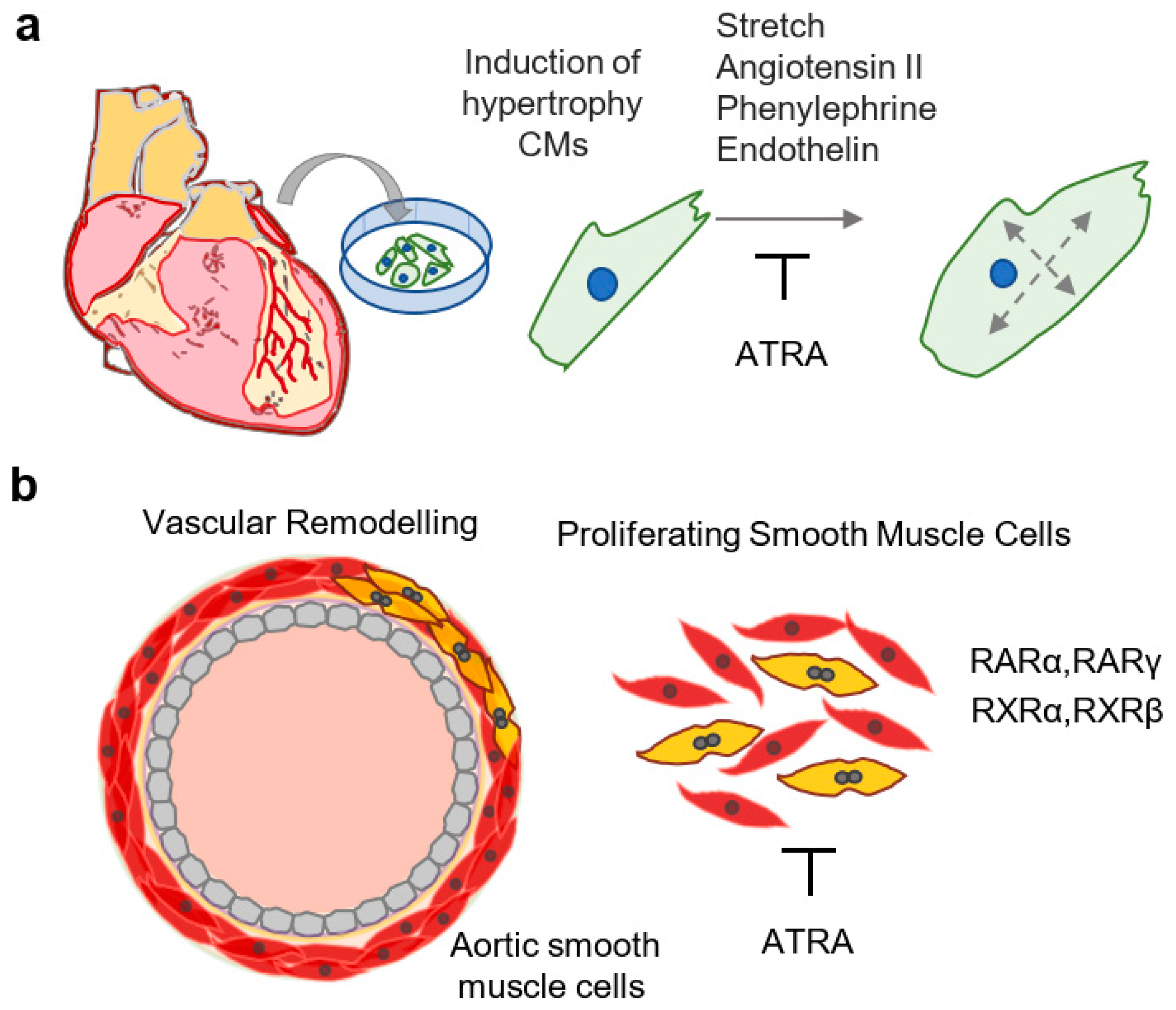

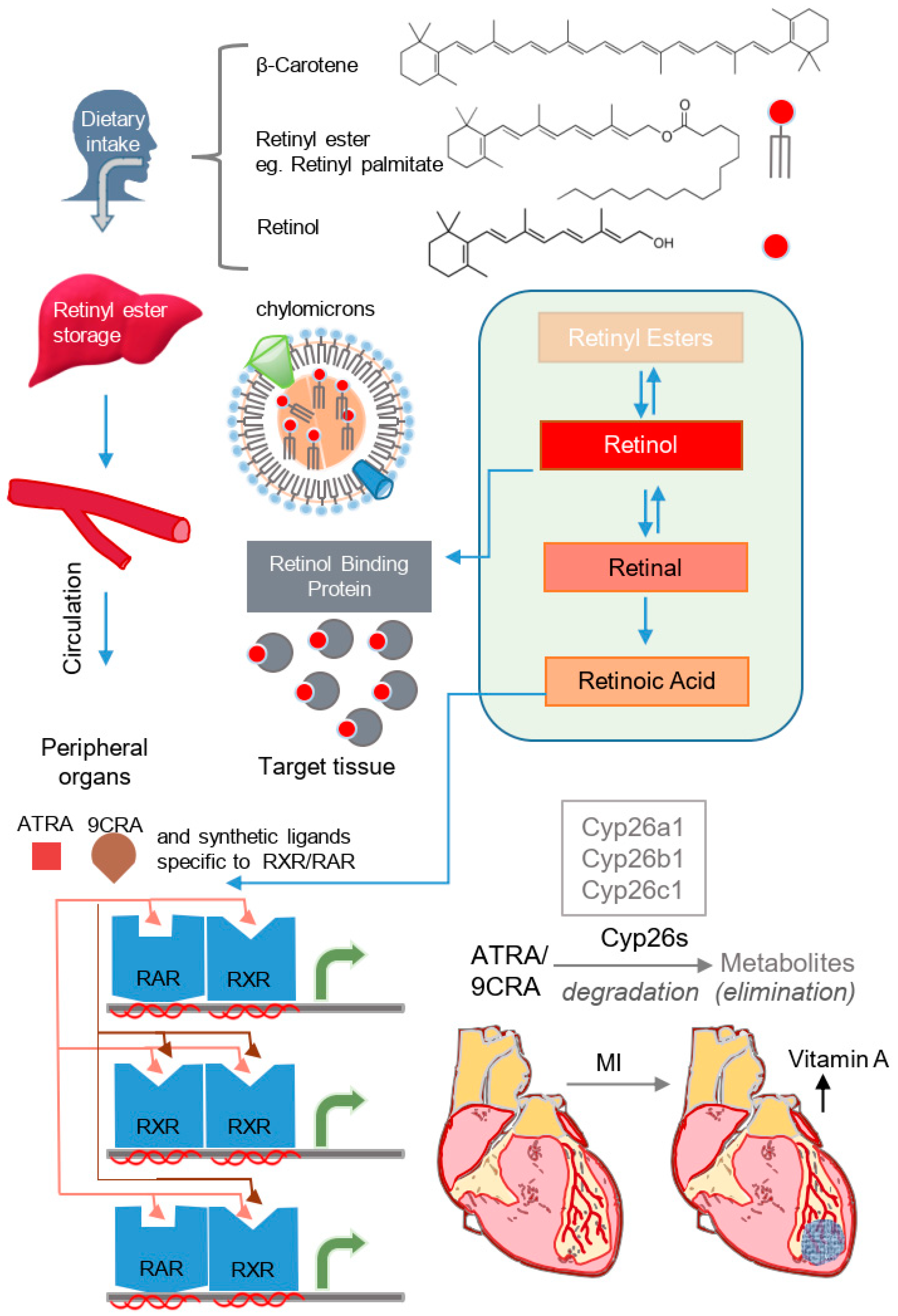{kind=link}
{kind=link}
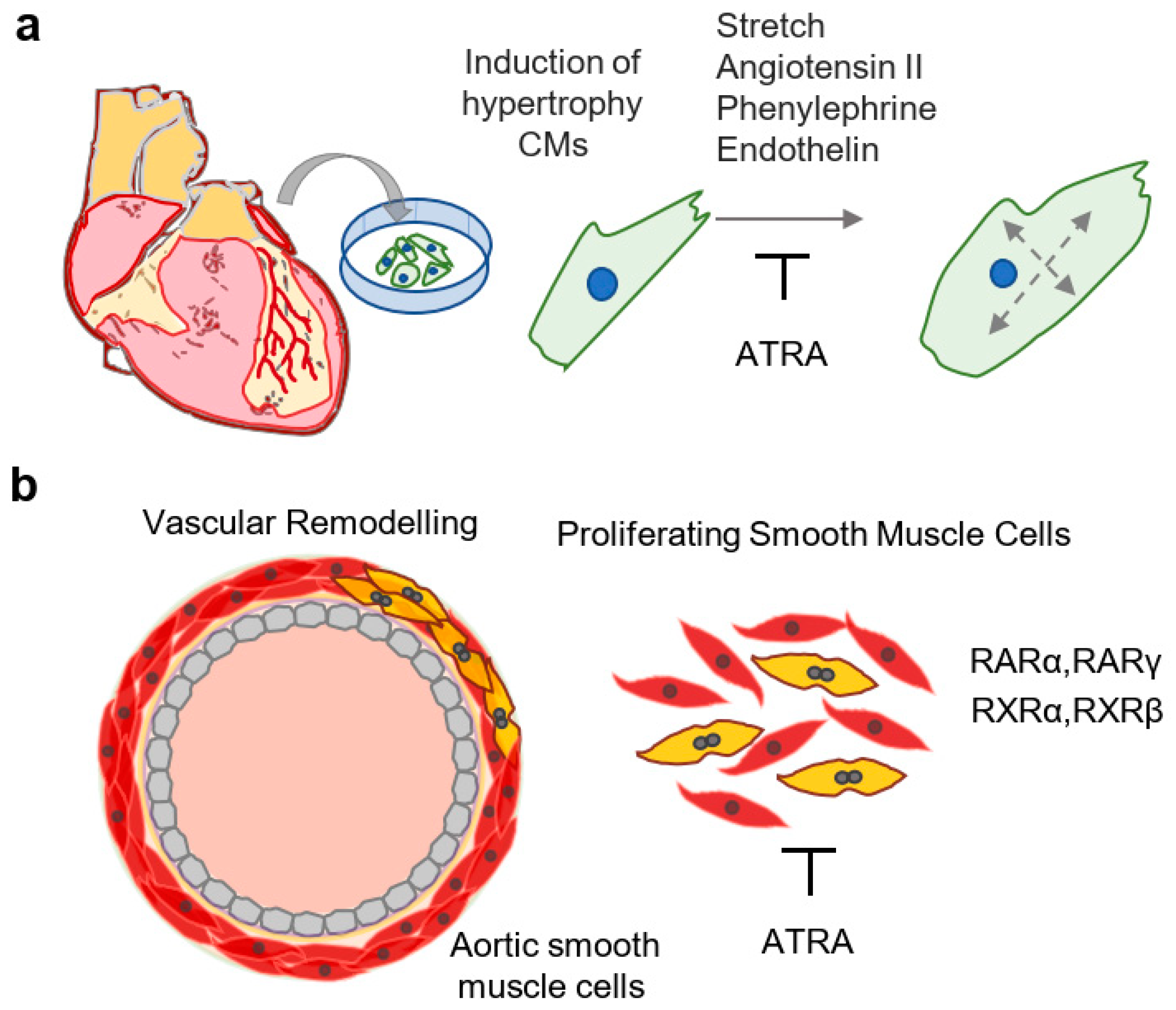{kind=link}
Table 1.
Abbreviations used in the current study.
| Abbreviation | Full Name | Abbreviation | Full Name |
|---|---|---|---|
| 9CRA | 9-cis retinoic acid | PSC | pluripotent stem cell |
| ATRA | all-trans retinoic acid | RA | retinoic acid |
| atrRFP | atrial RFP reporter | RAR | retinoic acid receptor |
| CaMKII | Ca2+/calmodulin-dependent protein kinase II | RARE | retinoic acid response element |
| CHD | congenital heart disease | RBP | retinol binding protein |
| CPC | cardiac progenitor cell | RXR | retinoid x receptor |
| CRBP | cellular retinol binding protein | SMC | smooth muscle cell |
| CVD | cardiovascular disease | venGFP | ventricular GFP reporter |
| MI | myocardial infarction | VD3 | vitamin D3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Leigh, R.S.; Kaynak, B.L. Vitamin A as a Transcriptional Regulator of Cardiovascular Disease. Hearts 2020, 1, 126-145. https://0-doi-org.brum.beds.ac.uk/10.3390/hearts1020013
AMA Style
Leigh RS, Kaynak BL. Vitamin A as a Transcriptional Regulator of Cardiovascular Disease. Hearts. 2020; 1(2):126-145. https://0-doi-org.brum.beds.ac.uk/10.3390/hearts1020013
Chicago/Turabian StyleLeigh, Robert S., and Bogac L. Kaynak. 2020. "Vitamin A as a Transcriptional Regulator of Cardiovascular Disease" Hearts 1, no. 2: 126-145. https://0-doi-org.brum.beds.ac.uk/10.3390/hearts1020013