The Role of Genetics in Risk Stratification of Thoracic Aortic Aneurysm Dissection
by
 , , ,
, , ,
Jotte Rodrigues Bento
1 ,
,
Josephina A.N. Meester
1,
Ilse Luyckx
1,
Aline Verstraeten
1 and
Bart L. Loeys
1,2,* 1
Center for Medical Genetics, Faculty of Medicine and Health Sciences, Antwerp University Hospital and University of Antwerp, 2610 Antwerp, Belgium
2
Department of Human Genetics, Radboud University Medical Centre, 6500 Nijmegen, The Netherlands
*
Author to whom correspondence should be addressed.
Hearts 2020, 1(2), 50-61; https://0-doi-org.brum.beds.ac.uk/10.3390/hearts1020007
Submission received: 22 June 2020
/
Revised: 27 July 2020
/
Accepted: 29 July 2020
/
Published: 3 August 2020
(This article belongs to the Special Issue Thoracic Aorta)
Abstract
:Thoracic aortic aneurysms are prevalent in the Western population and are often caused by genetic defects. If undetected, aneurysms can dissect or rupture, which are events associated with a high mortality rate. Hitherto no cure exists other than elective surgery if aneurysm dimensions reach a certain threshold. In the past decades, genotype-phenotype associations have emerged that enable clinicians to start stratifying patients according to risk for dissection. Nonetheless, risk assessment is—to this day—confounded by the lack of full comprehension of underlying genetics and modifying genetic risk factors that complicate the yet established genotype-phenotype correlations. Further research that focuses on identifying these additional risk markers is crucial.
1. Introduction—Thoracic Aortic Aneurysms
An aortic aneurysm is an abnormal enlargement of the aorta, caused by progressive dilatation of a weakened vascular wall [1]. Aortic aneurysms can be categorised into two subtypes according to anatomical location: abdominal aortic aneurysms (AAAs, below the diaphragm) and thoracic aortic aneurysms (TAAs, above the diaphragm). The prevalence, aetiology and pathology of AAAs and TAAs differ substantially: whereas AAA occurrence is typically attributed to advanced age and risk factors, such as smoking and atherosclerotic disease, TAAs appear in all age categories and have a prominent genetic predisposition [2]. Consequently, in the past decades, gene discovery efforts uncovered a vast number of monogenic disease causes and risk loci involved in TAA pathology of both syndromic and non-syndromic forms. Functional characterization of disease genes revealed aberrant extracellular matrix (ECM) homeostasis, TGF-β signalling and vascular smooth muscle cell contractility as fundamental processes underlying aortic wall weakening (Figure 1B) [1,3].
If TAAs remain unnoticed, they entail a significant risk for dissections, which associate with catastrophic consequences such as acute aortic valve insufficiency, heart failure, stroke, aortic rupture and a 24 h mortality rate of 25%. Dynamic factors, such as smoking status, dyslipidemia and high blood pressure, increase the risk for these events even more (Figure 1A) [4]. Although medical blood pressure control with β-blockers or angiotensin II receptor blockers can slow down aneurysm progression to some extent, the only preventive measure with regard to aortic dissection is to perform elective surgery in patients with an aortic diameter reaching a certain threshold (5.5 cm being the general recommendation) [5]. It is, however, important that identified TAAs are serially monitored by means of cardiovascular echocardiography, CT, or MRI, since TAA growth rate, severity and dissection hazard can differ greatly [1].
By studying known molecular defects underlying varying TAA phenotypes, several genotype-phenotype associations were discovered. Therefore, not only the rate of TAA progression, but also family history for dissection, gender and genotype must be taken into consideration to improve clinical risk stratification and decision-making [6]. For instance, the clinically overlapping Marfan syndrome (MFS), Loeys–Dietz syndrome (LDS) and Meester–Loeys syndrome (MRLS) with co-occurring TAA, are caused by pathogenic variants in different key players of the same pathway. Nevertheless, it is important to distinguish these different syndromes, since TAA location and severity differ significantly according to which pathway component is affected. Furthermore, the location and type of variant in the same gene can influence the risk of aortic dissection: haplo-insufficient (e.g., out-of-frame splice site mutations, premature stopcodons) or dominant negative mutations (e.g., inframe indels and missense mutations) can affect survival rates in a different manner in TAA diseases as demonstrated in the vascular Ehlers–Danlos syndrome [7]. These associations enable clinicians to stratify patients according to risk, establish well-defined clinical guidelines and determine endpoints for surgical intervention.
In the following sections, a short overview of current risk stratification strategies based on established genotype-phenotype correlations will be provided for MFS, LDS, MRLS, vEDS and familial TAA caused by ACTA2 mutations (Table 1). Lastly, genetic modifiers and emerging non-genetic risk factors (Figure 1A) will be discussed.
2. Current Genetic Risk Stratification Strategies
2.1. Stratification by Affected Genes: Marfan Syndrome and Loeys–Dietz Syndrome
Multiple syndromes presenting with TAAs exist, which are often clinically highly overlapping. However, TAA severity and the location of aneurysm formation can differ significantly between phenotypes, urging discrimination by identifying the causal mutation. This is illustrated by MFS and LDS, two connective tissue disorders involving TAA that are assumedly caused by dysregulation of the TGF-β pathway—a pathway that is involved in a multitude of cellular functions (Figure 2) [8,9]. MFS is an autosomal dominant disorder caused by mutations in the fibrillin-1 gene (FBN1), which encodes a protein that is assembled into microfibrils. Patients with this multisystem disorder primarily present with skeletal overgrowth, ectopia lentis and aortic root aneurysms at the level of the Sinuses of Valsalva. The risk of acute dissections is presumed to be directly proportional to the maximum diameter of the aorta and prophylactic surgery is recommended at a diameter of 5 cm, or if the aneurysmal growth rate exceeds 1 cm per annum [9]. Surgery should be considered in patients who have aortic root aneurysm, with maximal ascending aortic diameters 4.5 cm for patients with Marfan syndrome with risk factors [9]. Although it was initially assumed that aberrant fibrillin-1 solely caused a defect in structural organization of the medial matrix, imbalanced TGF-β signaling is now also suggested to be a disease culprit. Fibrillin-1 microfibrils normally sequester TGF-β in the extracellular matrix and, thus, regulate its bioavailability. The observed increase of TGF-β signaling in MFS causes an overall degeneration of the media due to elastic fiber fragmentation, deposition of mucoid ECM components and an excess of disorganized collagen; ultimately leading to weakening, aneurysm formation and dissection of the aortic wall [8,10].
The histological abnormalities mentioned above also appear in LDS, first described in 2005 as a consequence of dysfunctional TGF-β receptors 1 and 2 (encoded by TGFBR1 and TGFBR2). The presence of a bifid uvula or cleft palate, hypertelorism and more prominent arterial tortuosity with widespread aneurysms distinguish LDS patients from MFS patients. Of great importance is that, in patients suffering from LDS, aneurysms occur not only at the sinuses of Valsalva, but also appear beyond the aortic root with pronounced involvement of aortic side branches [6,11]. Moreover, aneurysms tend to be more aggressive and dissections occur at a younger age and smaller diameter. The latter observations instigate imaging of the entire arterial tree and surgical intervention at an aneurysmal diameter of 4cm—or if growth rate exceeds 0.5 cm/year [6].
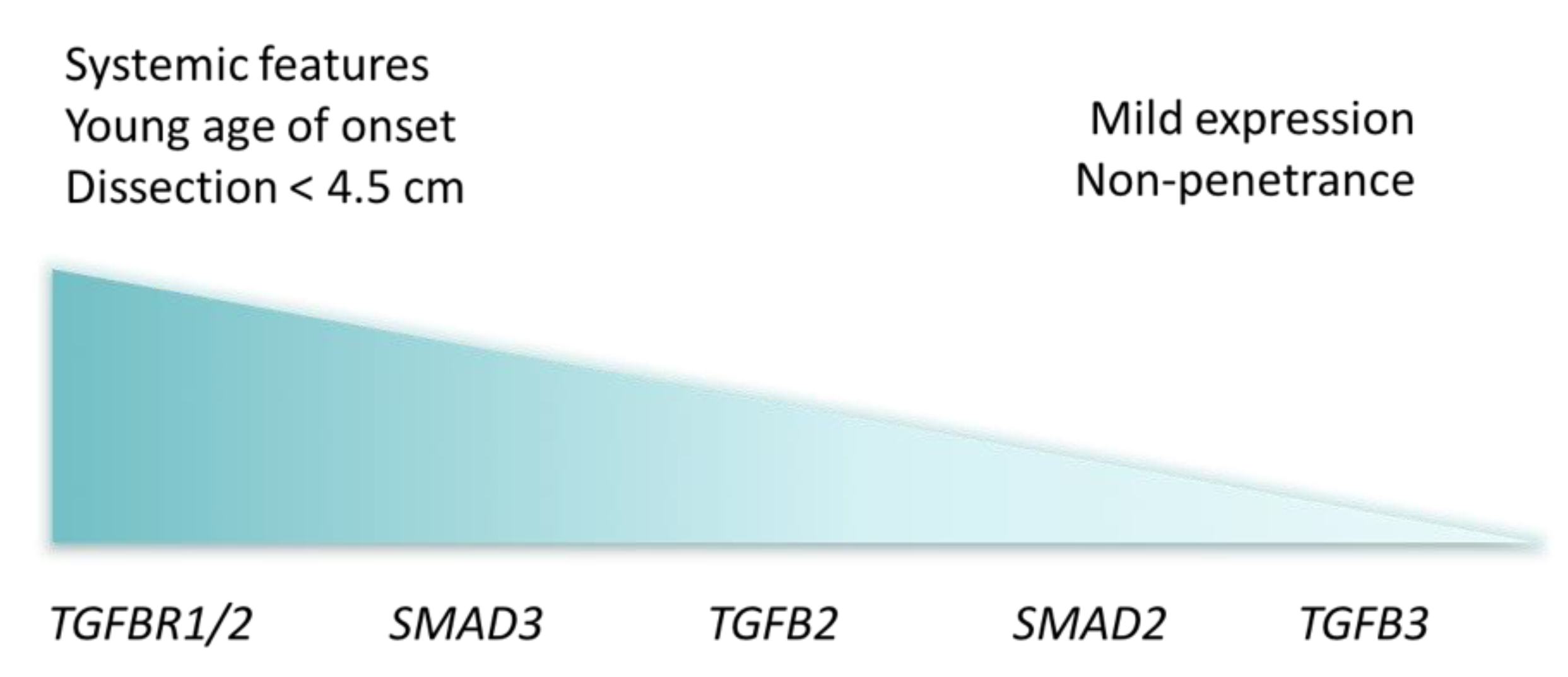
Since the initial discovery of TGFBR1 and 2 mutations, variants in four more genes (i.e., TGFB2, TGFB3, SMAD2 and SMAD3) have been linked to LDS. Although severely affected LDS patients have been described to carry pathogenic variants in any of these genes, certain genes typically cause milder phenotypes (Figure 3) [12,13]. Defects in TGFBR1 and 2 cause LDS1 and LDS2, respectively, with an equally more severe clinical presentation. Dissections occur at a young age—even as young as three months old—and at small aortic dimensions [6]. Bicuspid aortic valve, patent ductus arteriosus and atrial septal defect occur more frequently in these patients and during the course of disease, numerous vascular surgeries are necessary. LDS3 is the result of affected SMAD3 function, which is essential for signal propagation in the TGF-β pathway. Here, phenotypes are somewhat comparable to LDS1 and LDS2 and patients have a stronger predisposition for osteoarthritis [14]. Pathogenic variants in SMAD2, TGFB2 (LDS4) and TGFB3 (LDS5) give rise to milder LDS phenotypes, with dissections at a larger diameter compared to other subtypes, less systemic features and higher incidence of non-penetrance [15,16,17,18].
As such, LDS types exhibit a phenotypic spectrum of severity on their own, complicating clinical delineation even more. A recent publication pointed out that TGFB3 carriers show profound intrafamilial variable expressivity and that homozygous mutations in the TGFB3 gene cause a worse disease course [19]. Furthermore, the ever expanding clinical spectrum of LDS-genes challenges the well-established understanding that LDS patients display more aggressive TAA dissections, as demonstrated by Muhlstadt et al., who performed a case-matched comparison between LDS and MFS [20]. These two studies encourage further reseach and large-scale comparative studies to fine tune clinical considerations, apart from the current surgical endpoints being the aortic dimensions and growth rate.
2.2. Stratification by Gender: Meester-Loeys Syndrome and Bicuspid Valve Associated TAA
Albeit TAA is mostly inherited in an autosomal dominant matter, an X-linked syndromic form was recently identified in a patient cohort of male probands with molecularly unexplained LDS-like features. The condition, now termed Meester–Loeys syndrome (MRLS), is characterized by early onset aneurysm formation (from one year old) and dissection (as early as 15 years old) and strongly resembles MFS and LDS with typical features such as pectus deformities, joint hypermobility, aortic root dilatation and mitral valve insufficiency [21]. Distinctive LDS features, such as bifid uvula, hypertelorism and cervical spine instability, are also found, and non-typical features, such as brain ventricular dilatation, gingival hypertrophy and platyspondyly, differentiate MRLS from LDS and MFS. The strong resemblance to these two other connective tissue disorders hinted towards involvement of another, X-linked ECM gene with possible involvement in TGF-β signaling. Looking more closely at structural and functional changes in the aortic wall, a low to normal collagen content and normal elastic fibers are noted in contrast to the increased collagen and fragmented elastin in MFS and LDS; but enhanced TGF-β signaling was indeed demonstrated by increased SMAD2 phosphorylation. Targeted resequencing of candidate ECM and TGF-β pathway genes identified loss-of-function mutations of the BGN gene as the cause of disease. BGN encodes biglycan, a small leucine-rich protein sequestered in and responsible for ECM assembly and maintenance and, like fibrillin, regulation of growth factors and signaling proteins. The phenotype of female carriers of a BGN loss-of-function mutation can range from unaffected to dilatation of the aortic root and death due to dissection, but is usually of milder cardiovascular severity [21]. As indicated before, due to the different levels of severity between MFS, LDS, MRLS but also MRLS males versus MRLS females, differentiation with a molecular diagnosis is crucial—not only for clinical guidelines regarding the aortic phenotype, but also to monitor other MRLS findings such as cervical spine instability.
Another striking gender difference is observed in patients with a bicuspid aortic valve (BAV), a congenital anomaly with a prevalence of 1–2%. Patients with this malformation have an aortic valve comprising of two cusps instead of three, which can result in an elevation of shear stress on the aortic wall due to abnormal blood flow patterns. Regions with increased shear stress display significant remodeling and deterioration of the ECM with elastic fiber degeneration, rendering the aortic wall more fragile and often resulting in dilatation of the aorta [22,23,24]. Consequently, TAA is 86 times more prevalent in patients with BAV compared to normal individuals having a tricuspid aortic valve and aortic surgery is performed at a smaller diameter [25]. Of note, BAV occurs three times more and associated TAA occurrence is significantly higher among men compared to women, which is attributable to a gender-dependent aortic remodeling: the matrix metalloproteases and their inhibitors that regulate remodeling of the ECM are differentially expressed, resulting in decreased integrity of collagen fibres compared to females. Lower survival of VSMC also contributes to a larger dilatation in males. Hence, specific focus should be drawn to aortic dimensions in males with BAV, with regular follow up to monitor growth rate and to plan timely interventions [26].
Overall, it is well established that gender plays a significant role in aneurysm development: the male-to-female ratio in AAA is 6:1 and for TAA a lesser pronounced ratio of 1.7:1 [27,28]. There have been animal studies where involvement of sex hormones in aortic aneurysm formation has been indicated, suggesting either an aneurysm promoting role of testosterone, or a preventive role of estrogen through reduction of ECM degrading enzymes [26]. The observation of a higher occurrence of BAV, aortic dilatation, aneurysm and dissection in Turner syndrome patients (only one X chromosome, no Y chromosome) compared to females with two X-chromosomes hints towards a protective role of the X chromosome in aortopathy [29]. However, most of the genetic factors underpinning the observed gender differences have yet to be uncovered.
2.3. Stratification by Variant Type: Vascular Ehlers–Danlos Syndrome, Haploinsufficiency and Dominant Negative Mutations
2.3.1. Effect of Variant Types on the Presentation of Vascular Ehlers–Danlos Syndrome
With deficient ECM regulation being at the heart of TAA formation, pathogenic variants in genes encoding important structural molecules of the connective tissue such as collagens, can cause frailty of the aortic and arterial wall. One such disorder, caused by autosomal dominant mutations in the COL3A1 gene, is termed vascular Ehlers–Danlos syndrome (vEDS) and presents with life-threatening complications such as arterial dissections, bowel perforations and uterine ruptures. Aortic dissections and ruptures are responsible for almost 70% of deaths with a mean survival of 50 years [7,30]. COL3A1 encodes the alpha chain of type 3 collagen which trimerizes into a triple helical configuration to eventually form a type III collagen molecule, an essential ECM component in hollow organs, such as blood vessels and the intestine [31]. Interestingly, a role for collagen III in sequestering BMP (bone morphogenic protein), analogously to fibrillin-1 with TGF-β has been hypothesized.
In two large-scale studies with 572 [7] and 215 [30] vEDS subjects, genotype-phenotype association analyses were carried out. Splice variants resulting in in frame exon-skipping or a shortened but stably expressed product give rise to the most severe phenotype with a short lifespan and high risk for dissection. Specifically, patients with a donor splice site mutation have mean survival age of 37 years, whereas acceptor splice site mutation carriers have a mean survival age of 52 years. The latter is comparable to the mean survival of patients with glycine substitutions in the canonical Gly-X-Y repeat of the triple helical domain [7]. Seemingly, the amino acid that replaces a helical glycine can affect disease severity as well: when the survival per replacement amino acid was analyzed, it was discovered that a replacement serine had a more detrimental effect on survival compared to arginine or aspartic acid, and that valine had a less severe consequence than all three previously mentioned amino acids [7].
Of note, vEDS patients with a null mutation as a result of frameshift, nonsense substitutions or large deletions have the best survival rate of all vEDS patients: 70% had an event-free survival at the age of 40 [7] and overall survival could not be estimated due to the low number of deaths [30]. That is because splice site and glycine mutations hamper the stable assembly of type III collagen molecules into procollagen homotrimers and thus act in a dominant-negative (DN) manner on the overall formation of connective tissue. Contrariwise, null mutations—i.e., haploinsufficiency (HI)—just result in a reduced amount of correctly assembled mature collagen [7,30].
2.3.2. Haploinsufficiency and Dominant-Negative FBN1 Mutations
Similar to vEDS, mutations in the FBN1 gene can be categorized into two subtypes. Whole gene deletions, unstable mutant protein and nonsense mediated decay of abnormal mRNA all result in HI, whereas variants that produce an aberrant protein that is stably expressed are classified as DN mutations [32]. Similar to type III collagen, fibrillin-1 molecules interact to form fibrils. Consequently, structurally disturbed molecules alter the entire fibril structure resulting in disorganized ECM and a weakened aortic wall [33]. Although for many years it was suspected that, for MFS too, patients with a DN genotype present with a more severe phenotype, recent studies with analyses of large patient cohorts suggested that HI patients have an increased risk for aortic complication and cardiovascular death [34,35]. Most recently, Meester et al. conducted a comparative study between patients with HI and DN mutations, where no significant differences in cardiovascular phenotypes were discovered (unpublished data).
Recently, a clinical trial on a Dutch cohort of 357 adult MFS patients (age > 18 years) was performed to study the effect of genotype on responsiveness to Losartan—an angiotensin II receptor 1 blocker that lowers blood pressure and TGF-β signaling. In this study, patients with HI FBN1 mutations demonstrated a marked decrease in aortic root dilatation rate compared to DN patients [32,36], which indicates that the mutation type can determine whether a specific therapy could exert a beneficial effect. However, in a younger cohort (age 6–25 years) of similar size we were unable to confirm this difference (unpublished data).
2.4. Stratification by Variant Location: From Low-Penetrant Familial Thoracic Aortic Aneurysms to Severe Early Onset Syndrome
As mentioned in the introduction, the location of the variant within a certain gene can affect the phenotype significantly, ranging from incomplete penetrance or isolated aortic disease to an early-onset multisystem disorder. A striking example is the missense mutation p.R179H in the ACTA2 gene, which encodes smooth muscle specific actin (α-SMA). α-SMA is critical for the function of smooth muscle cells within the aortic wall. Hence, apart from the TGF-β pathway and ECM matrix, impairment in smooth muscle cell function also contributes to TAA development, as illustrated in Figure 1 and the section below.
Smooth muscle cells in the aortic wall and other visceral organs perform their function by contracting in response to mechanical stretch. Smooth muscle-specific actin plays a pivotal role in this process, as interactions between actin and myosin filaments result in contraction [37]. α-SMA consists of free monomers organized into filaments by means of inter-strand bonds. When the concentration free monomers is high in comparison with assembled filaments, a proliferative cell state is promoted due to the increased expression of growth-responsive genes. When monomers are predominantly polymerized into filaments, transcription and translation of contractile proteins is induced—essential for functional performance of a mature, contractile smooth muscle cell.
Heterozygous mutations in ACTA2 account for up to 20% of familial non syndromic TAA cases. Phenotypes in mutation carriers are age-related and often show incomplete penetrance (up to 50%) [37]. Here, surgery is recommended when the maximal diameter of the aneurysm reaches 4.5–5.0 cm [38]. However, one particular amino acid substitution (p.R179H) elicits a severe early onset and highly penetrant phenotype with multisystemic smooth muscle dysfunction. In addition to TAA, patients suffer from pulmonary hypertension, malrotation and hypoperistalsis of the gastrointestinal system, hypotonic bladder, congenital mydriasis and cerebrovascular disease [38,39,40]. The amino acid change in question, p.R179H, alters a critical interaction site for establishment of the inter-strand binding of free actin monomers into filaments [39,41]. Accordingly, actin is unable to form filaments and smooth muscle cells are inhibited to mature, proliferate continuously and secrete abnormal ECM components [41].
3. Genetic Modifiers
Patients and families with Mendelian segregation of the same TAA-causing mutation often show profound inter- and intra-familial phenotypic variability, going from non-penetrance to sudden death at young age due to dissection. This hints towards the presence of additional genetic variation which contributes to the final clinical presentation, complicating risk assessment and patient management. As proof of principle for the existence of genetic modifiers in rare monogenic conditions, Drumm et al. found that variants in the TGFB1 gene modify pulmonary disease and clinical outcome in cystic fibrosis patients with the same CTFR mutation, thus identifying TGFB1 as a so-called genetic modifier [42]. As availability of patient samples and sufficient sample size for statistical significance are necessary, genetic modifier studies are often challenging for rare diseases such as vEDS, MFS and LDS. Nonetheless, a recent paper studying genetic factors explaining clinical variability in MFS identified several genetic modifier loci of which risk alleles exert a more severe phenotype. For example, a second pathogenic event in the COL4A1 gene was found to worsen disease [43]. In another publication, Landis et al. describe their efforts to identify genetic modifiers controlling syndromic and familial TAA penetrance and expressivity. By means of exome sequencing, candidate genetic modifiers were discovered, among which a rare variant in ADCK4 that segregated with mild TAA in several families. The association between ADCK4 and TAA can be explained by the observed expression of this gene in aortic smooth muscle cells and the suspected involvement in functioning and survival of these cells through regulation of mitochondrial activity [44]. These two studies already represent an advancement towards a complete understanding of the complex molecular fundament underlying TAA.
4. Emerging Non-Genetic Risk Determinants
As mentioned above, the aortic diameter is currently the mainstay for timing preventative operations, since a fairly linear correlation was found between dilation and risk of dissection or rupture. However, a recent study examined aortic dilatation at the time of dissection and noted that the average aortic diameter at dissection was smaller than 5.5 cm, with 40% of patients even below 5 cm [5]. Aortic size alone, whether or not depending on an underlying molecular defect is, thus, not sufficient for clinical decision-making and better risk predictors are necessary.
Where the currently uncovered genotype-phenotype associations and aortic diameters fail to fully grasp risk and prognosis, several other non-genetic disease properties emerge as complementary means to assess risk as precisely as possible. For instance, the stiffness of the ascending aorta—which is calculated using blood pressure values and dimensions of the aorta in systole and diastole—has been suggested to correlate with the growth rate and clinical outcomes [45,46]. At least one year before the event of dissection, Emerel et al. identified increased stiffness and wall stress in the region where tearing occurred in patients with TAA dissections by creating biomechanical property maps derived from echocardiographical and computed tomography images [45]. Likewise, in the clinical study from Tierney et al., MFS patients were shown to exhibit increased wall stiffness compared to controls—even with normal aortic root dimensions. Higher stiffness associated with higher frequency of surgery, dissection and death [46].
Franken et al. recently proposed another non-genetic indicator of cardiovascular severity in MFS. In their study, a method to determine aortic tortuosity by 3D visualization of the aorta with magnetic resonance imaging (MRI) was developed and validated. The aortic tortuosity index in MFS patients was significantly higher compared to control and correlated with a higher risk for aortic complications [47]. Moreover, Heuts et al. suggest aortic lengthening as a predictive marker of TAA dissection as they observed an average increase of 2 cm of the ascending aorta in patients with calculations performed on computed tomography images [48].
As previously described in Section 2.2, abnormal blood flow patterns can have a deleterious effect on aortic wall integrity due to regional deviation of wall shear stress distribution and flow eccentricity, inducing ECM remodeling and elastic fiber degeneration. 4D cardiovascular MRI enables to depict these abberant outflow patterns and to calculate resulting shear stress, hemodynamic markers that can be highly relevant for patients with a bicuspid aortic valve [22,49]. Elastic fiber degeneration itself poses opportunities for biomarkers and imaging markers. One study investigated the relation between aortic microcalcification (which co-localizes with aortic elastin degradation) and the risk for dissection in MFS and hypothesizes that microcalcification may serve as a marker for aortic disease severity [50]. Additionally, an association was found between fibrillin-1 fragment concentrations in the blood circulation of TAA patients and the frequency and dissection of aneurysms [51].
The above results of the recent clinical studies demonstrate that measurement of predisposing biomechanical markers with non-invasive methods offers opportunities to predict aortic aneurysm dissection.
5. Towards Gene-Tailored Management
The difference in disease severity, survival and therapeutic response underlines yet again the importance of genetics in prognosis, risk estimation and tailored therapeutic strategies for TAA and other conditions. However, despite few but promising uncovered genotype-phenotype associations, accurate assessment of aneurysm and dissection risk still remains difficult. Clarification of conflicting results, identification of modifier genes and optimization of non-invasive bio- and imaging markers will pave the way to accurate risk assessment and tailored clinical guidelines.
Author Contributions
J.R.B.: writing—original draft preparation; writing—review and editing: J.A.N.M., I.L., A.V., B.L.L.; funding acquisition: A.V., B.L.L. All authors have read and agreed to the published version of the manuscript.
Funding
This review effort is supported by funding from the University of Antwerp (GOA, Methusalem-OEC grant “Genomed” FFB190208), the Research Foundation Flanders (FWO, Belgium, G.0356.17), the Dutch Heart Foundation (2013T093), the Marfan Foundation. B.L. is a senior clinical investigator of FWO and holds a consolidator grant from the European Research Council (Genomia—ERC-COG-2017-771945).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Verstraeten, A.; Luyckx, I.; Loeys, B. Aetiology and management of hereditary aortopathy. Nat. Rev. Cardiol. 2017, 14, 197–208. [Google Scholar] [CrossRef]
- Lindsay, M.E.; Dietz, H.C. The genetic basis of aortic aneurysm. Cold Spring Harb. Perspect. Med. 2014, 4, a015909. [Google Scholar] [CrossRef] [Green Version]
- Milewicz, D.M.; Regalado, E. Heritable Thoracic Aortic Disease Overview. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Hiratzka, L.F.; Bakris, G.L.; Beckman, J.A.; Bersin, R.M.; Carr, V.F.; Casey, D.E., Jr.; Eagle, K.A.; Hermann, L.K.; Isselbacher, E.M.; Kazerooni, E.A.; et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with Thoracic Aortic Disease: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation 2010, 121, e266–e369. [Google Scholar]
- Pape, L.A.; Tsai, T.T.; Isselbacher, E.M.; Oh, J.K.; O’Gara, P.T.; Evangelista, A.; Fattori, R.; Meinhardt, G.; Trimarchi, S.; Bossone, E.; et al. Aortic diameter >= 5.5 cm is not a good predictor of type A aortic dissection - Observations from the international registry of acute aortic dissection (IRAD). Circulation 2007, 116, 1120–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacCarrick, G.; Black, J.H.; Bowdin, S.; El-Hamamsy, I.; Frischmeyer-Guerrerio, P.A.; Guerrerio, A.L.; Sponseller, P.D.; Loeys, B.; Dietz, H.C. Loeys-Dietz syndrome: A primer for diagnosis and management. Genet. Med. 2014, 16, 576–587. [Google Scholar] [CrossRef] [PubMed]
- Pepin, M.G.; Schwarze, U.; Rice, K.M.; Liu, M.; Leistritz, D.; Byers, P.H. Survival is affected by mutation type and molecular mechanism in vascular Ehlers-Danlos syndrome (EDS type IV). Genet. Med. 2014, 16, 881–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Judge, D.P.; Dietz, H.C. Marfan’s syndrome. Lancet 2005, 366, 1965–1976. [Google Scholar]
- Van Laer, L.; Dietz, H.; Loeys, B. Loeys-Dietz syndrome. Adv. Exp. Med. Biol. 2014, 802, 95–105. [Google Scholar]
- Waters, K.M.; Rooper, L.M.; Guajardo, A.; Halushka, M.K. Histopathologic differences partially distinguish syndromic aortic diseases. Cardiovasc. Pathol. 2017, 30, 6–11. [Google Scholar]
- Loeys, B.L.; Schwarze, U.; Holm, T.; Callewaert, B.L.; Thomas, G.H.; Pannu, H.; De Backer, J.F.; Oswald, G.L.; Symoens, S.; Manouvrier, S.; et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N. Engl. J. Med. 2006, 355, 788–798. [Google Scholar] [CrossRef]
- Loeys, B.L.; Dietz, H.C. Loeys-Dietz Syndrome. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Hostetler, E.M.; Regalado, E.S.; Guo, D.C.; Hanna, N.; Arnaud, P.; Muino-Mosquera, L.; Callewaert, B.L.; Lee, K.; Leal, S.M.; Wallace, S.E.; et al. SMAD3 pathogenic variants: Risk for thoracic aortic disease and associated complications from the Montalcino Aortic Consortium. J. Med. Genet. 2019, 56, 252–260. [Google Scholar] [CrossRef] [PubMed]
- van de Laar, I.M.; Oldenburg, R.A.; Pals, G.; Roos-Hesselink, J.W.; de Graaf, B.M.; Verhagen, J.M.; Hoedemaekers, Y.M.; Willemsen, R.; Severijnen, L.A.; Venselaar, H.; et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat. Genet. 2011, 43, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, M.E.; Schepers, D.; Bolar, N.A.; Doyle, J.J.; Gallo, E.; Fert-Bober, J.; Kempers, M.J.; Fishman, E.K.; Chen, Y.; Myers, L.; et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat. Genet. 2012, 44, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Bertoli-Avella, A.M.; Gillis, E.; Morisaki, H.; Verhagen, J.M.A.; de Graaf, B.M.; van de Beek, G.; Gallo, E.; Kruithof, B.P.T.; Venselaar, H.; Myers, L.A.; et al. Mutations in a TGF-beta ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J. Am. Coll. Cardiol. 2015, 65, 1324–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schepers, D.; Tortora, G.; Morisaki, H.; MacCarrick, G.; Lindsay, M.; Liang, D.; Mehta, S.G.; Hague, J.; Verhagen, J.; van de Laar, I.; et al. A mutation update on the LDS-associated genes TGFB2/3 and SMAD2/3. Hum. Mutat. 2018, 39, 621–634. [Google Scholar] [CrossRef] [Green Version]
- Micha, D.; Guo, D.C.; Hilhorst-Hofstee, Y.; van Kooten, F.; Atmaja, D.; . Overwater, E.; Cayami, F.K.; Regalado, E.S.; van Uffelen, R.; Venselaar, H.; et al. SMAD2 Mutations Are Associated with Arterial Aneurysms and Dissections. Hum. Mutat. 2015, 36, 1145–1149. [Google Scholar] [CrossRef]
- Marsili, L.; Overwater, E.; Hanna, N.; Baujat, G.; Baars, M.J.H.; Boileau, C.; Bonneau, D.; Brehin, A.C.; Capri, Y.; Cheung, H.Y.; et al. Phenotypic spectrum of TGFB3 disease-causing variants in a Dutch-French cohort and first report of a homozygous patient. Clin. Genet. 2020, 97, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Muhlstadt, K.; De Backer, J.; von Kodolitsch, Y.; Kutsche, K.; Muino Mosquera, L.; Brickwedel, J.; Girdauskas, E.; Mir, T.S.; Mahlmann, A.; Tsilimparis, N.; et al. Case-matched Comparison of Cardiovascular Outcome in Loeys-Dietz Syndrome versus Marfan Syndrome. J. Clin. Med. 2019, 8, 2079. [Google Scholar]
- Meester, J.A.; Vandeweyer, G.; Pintelon, I.; Lammens, M.; Van Hoorick, L.; De Belder, S.; Waitzman, K.; Young, L.; Markham, L.W.; Vogt, J.; et al. Loss-of-function mutations in the X-linked biglycan gene cause a severe syndromic form of thoracic aortic aneurysms and dissections. Genet. Med. 2017, 19, 386–395. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Palomares, J.F.; Dux-Santoy, L.; Guala, A.; Kale, R.; Maldonado, G.; Teixido-Tura, G.; Galian, L.; Huguet, M.; Valente, F.; Gutierrez, L.; et al. Aortic flow patterns and wall shear stress maps by 4D-flow cardiovascular magnetic resonance in the assessment of aortic dilatation in bicuspid aortic valve disease. J. Cardiovasc. Magn. Reson. 2018, 20, 28. [Google Scholar] [CrossRef]
- Hope, M.D.; Sigovan, M.; Wrenn, S.J.; Saloner, D.; Dyverfeldt, P. MRI hemodynamic markers of progressive bicuspid aortic valve-related aortic disease. J. Magn. Reson. Imaging 2014, 40, 140–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzzardi, D.G.; Barker, A.J.; van Ooij, P.; Malaisrie, S.C.; Puthumana, J.J.; Belke, D.D.; Mewhort, H.E.; Svystonyuk, D.A.; Kang, S.; Verma, S.; et al. Valve-Related Hemodynamics Mediate Human Bicuspid Aortopathy: Insights From Wall Shear Stress Mapping. J. Am. Coll. Cardiol. 2015, 66, 892–900. [Google Scholar]
- Lee, J.; Shen, M.; Parajuli, N.; Oudit, G.Y.; McMurtry, M.S.; Kassiri, Z. Gender-dependent aortic remodelling in patients with bicuspid aortic valve-associated thoracic aortic aneurysm. J. Mol. Med. 2014, 92, 939–949. [Google Scholar] [PubMed]
- Kong, W.K.; Regeer, M.V.; Ng, A.C.; McCormack, L.; Poh, K.K.; Yeo, T.C.; Shanks, M.; Parent, S.; Enache, R.; Popescu, B.A.; et al. Sex Differences in Phenotypes of Bicuspid Aortic Valve and Aortopathy: Insights From a Large Multicenter, International Registry. Circ. Cardiovasc. Imaging 2017, 10, 2079. [Google Scholar]
- Kuivaniemi, H.; Ryer, E.J.; Elmore, J.R.; Hinterseher, I.; Smelser, D.T.; Tromp, G. Update on abdominal aortic aneurysm research: From clinical to genetic studies. Scientifica 2014, 2014, 564734. [Google Scholar]
- Steckmeier, B. Epidemiology of aortic disease: Aneurysm, dissection, occlusion. Radiologe 2001, 41, 624–632. [Google Scholar] [CrossRef]
- Carlson, M.; Silberbach, M. Dissection of the aorta in Turner syndrome: Two cases and review of 85 cases in the literature. BMJ Case Rep. 2009, 2009, bcr0620091998. [Google Scholar] [CrossRef] [Green Version]
- Frank, M.; Albuisson, J.; Ranque, B.; Golmard, L.; Mazzella, J.M.; Bal-Theoleyre, L.; Fauret, A.L.; Mirault, T.; Denarie, N.; Mousseaux, E.; et al. The type of variants at the COL3A1 gene associates with the phenotype and severity of vascular Ehlers-Danlos syndrome. Eur. J. Hum. Genet. 2015, 23, 1657–1664. [Google Scholar] [CrossRef]
- Kuivaniemi, H.; Tromp, G. Type III collagen (COL3A1): Gene and protein structure, tissue distribution, and associated diseases. Gene 2019, 707, 151–171. [Google Scholar] [CrossRef]
- Franken, R.; den Hartog, A.W.; Radonic, T.; Micha, D.; Maugeri, A.; van Dijk, F.S.; Meijers-Heijboer, H.E.; Timmermans, J.; Scholte, A.J.; van den Berg, M.P.; et al. Beneficial Outcome of Losartan Therapy Depends on Type of FBN1 Mutation in Marfan Syndrome. Circ. Cardiovasc. Genet. 2015, 8, 383–388. [Google Scholar] [CrossRef] [Green Version]
- Byers, P.H. Determination of the molecular basis of Marfan syndrome: A growth industry. J. Clin. Investig. 2004, 114, 161–163. [Google Scholar] [CrossRef] [PubMed]
- Franken, R.; Groenink, M.; de Waard, V.; Feenstra, H.M.; Scholte, A.J.; van den Berg, M.P.; Pals, G.; Zwinderman, A.H.; Timmermans, J.; Mulder, B.J. Genotype impacts survival in Marfan syndrome. Eur. Heart J. 2016, 37, 3285–3290. [Google Scholar] [CrossRef] [PubMed]
- Franken, R.; Teixido-Tura, G.; Brion, M.; Forteza, A.; Rodriguez-Palomares, J.; Gutierrez, L.; Garcia Dorado, D.; Pals, G.; Mulder, B.J.; Evangelista, A. Relationship between fibrillin-1 genotype and severity of cardiovascular involvement in Marfan syndrome. Heart 2017, 103, 1795–1799. [Google Scholar] [CrossRef] [PubMed]
- den Hartog, A.W.; Franken, R.; van den Berg, M.P.; Zwinderman, A.H.; Timmermans, J.; Scholte, A.J.; de Waard, V.; Spijkerboer, A.M.; Pals, G.; Mulder, B.J.; et al. The effect of losartan therapy on ventricular function in Marfan patients with haploinsufficient or dominant negative FBN1 mutations. Neth. Heart J. 2016, 24, 675–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, D.C.; Papke, C.L.; Tran-Fadulu, V.; Regalado, E.S.; Avidan, N.; Johnson, R.J.; Kim, D.H.; Pannu, H.; Willing, M.C.; Sparks, E.; et al. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am. J. Hum. Genet. 2009, 84, 617–627. [Google Scholar] [CrossRef] [Green Version]
- van de Laar, I.; Arbustini, E.; Loeys, B.; Bjorck, E.; Murphy, L.; Groenink, M.; Kempers, M.; Timmermans, J.; Roos-Hesselink, J.; Benke, K.; et al. European reference network for rare vascular diseases (VASCERN) consensus statement for the screening and management of patients with pathogenic ACTA2 variants. Orphanet J. Rare Dis. 2019, 14, 264. [Google Scholar] [CrossRef]
- Regalado, E.S.; Mellor-Crummey, L.; De Backer, J.; Braverman, A.C.; Ades, L.; Benedict, S.; Bradley, T.J.; Brickner, M.E.; Chatfield, K.C.; Child, A.; et al. Clinical history and management recommendations of the smooth muscle dysfunction syndrome due to ACTA2 arginine 179 alterations. Genet. Med. 2018, 20, 1206–1215. [Google Scholar] [CrossRef] [Green Version]
- Milewicz, D.M.; Ostergaard, J.R.; Ala-Kokko, L.M.; Khan, N.; Grange, D.K.; Mendoza-Londono, R.; Bradley, T.J.; Olney, A.H.; Ades, L.; Maher, J.F.; et al. De novo ACTA2 mutation causes a novel syndrome of multisystemic smooth muscle dysfunction. Am. J. Med. Genet. A 2010, 152, 2437–2443. [Google Scholar] [CrossRef] [Green Version]
- Georgescu, M.M.; Pinho Mda, C.; Richardson, T.E.; Torrealba, J.; Buja, L.M.; Milewicz, D.M.; Raisanen, J.M.; Burns, D.K. The defining pathology of the new clinical and histopathologic entity ACTA2-related cerebrovascular disease. Acta Neuropathol. Commun. 2015, 3, 81. [Google Scholar] [CrossRef] [Green Version]
- Drumm, M.L.; Konstan, M.W.; Schluchter, M.D.; Handler, A.; Pace, R.; Zou, F.; Zariwala, M.; Fargo, D.; Xu, A.; Dunn, J.M.; et al. Genetic modifiers of lung disease in cystic fibrosis. N. Engl. J. Med. 2005, 353, 1443–1453. [Google Scholar] [CrossRef]
- Aubart, M.; Gazal, S.; Arnaud, P.; Benarroch, L.; Gross, M.S.; Buratti, J.; Boland, A.; Meyer, V.; Zouali, H.; Hanna, N.; et al. Association of modifiers and other genetic factors explain Marfan syndrome clinical variability. Eur. J. Hum. Genet. 2018, 26, 1759–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landis, B.J.; Schubert, J.A.; Lai, D.; Jegga, A.G.; Shikany, A.R.; Foroud, T.; Ware, S.M.; Hinton, R.B. Exome Sequencing Identifies Candidate Genetic Modifiers of Syndromic and Familial Thoracic Aortic Aneurysm Severity. J. Cardiovasc. Transl. Res. 2017, 10, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Emerel, L.; Thunes, J.; Kickliter, T.; Billaud, M.; Phillippi, J.A.; Vorp, D.A.; Maiti, S.; Gleason, T.G. Predissection-derived geometric and distensibility indices reveal increased peak longitudinal stress and stiffness in patients sustaining acute type A aortic dissection: Implications for predicting dissection. J. Thorac. Cardiovasc. Surg. 2019, 158, 355–363. [Google Scholar] [CrossRef]
- Selamet Tierney, E.S.; Levine, J.C.; Sleeper, L.A.; Roman, M.J.; Bradley, T.J.; Colan, S.D.; Chen, S.; Campbell, M.J.; Cohen, M.S.; De Backer, J.; et al. Influence of Aortic Stiffness on Aortic-Root Growth Rate and Outcome in Patients With the Marfan Syndrome. Am. J. Cardiol. 2018, 121, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Franken, R.; El Morabit, A.; de Waard, V.; Timmermans, J.; Scholte, A.J.; van den Berg, M.P.; Marquering, H.; Planken, N.R.; Zwinderman, A.H.; Mulder, B.J.; et al. Increased aortic tortuosity indicates a more severe aortic phenotype in adults with Marfan syndrome. Int. J. Cardiol. 2015, 194, 7–12. [Google Scholar] [CrossRef]
- Heuts, S.; Adriaans, B.P.; Gerretsen, S.; Natour, E.; Vos, R.; Cheriex, E.C.; Crijns, H.; Wildberger, J.E.; Maessen, J.G.; Schalla, S.; et al. Aortic elongation part II: The risk of acute type A aortic dissection. Heart 2018, 104, 1778–1782. [Google Scholar] [CrossRef]
- Mahadevia, R.; Barker, A.J.; Schnell, S.; Entezari, P.; Kansal, P.; Fedak, P.W.; Malaisrie, S.C.; McCarthy, P.; Collins, J.; Carr, J.; et al. Bicuspid aortic cusp fusion morphology alters aortic three-dimensional outflow patterns, wall shear stress, and expression of aortopathy. Circulation 2014, 129, 673–682. [Google Scholar] [CrossRef] [Green Version]
- Wanga, S.; Hibender, S.; Ridwan, Y.; van Roomen, C.; Vos, M.; van der Made, I.; van Vliet, N.; Franken, R.; van Riel, L.A.; Groenink, M.; et al. Aortic microcalcification is associated with elastin fragmentation in Marfan syndrome. J. Pathol. 2017, 243, 294–306. [Google Scholar] [CrossRef] [Green Version]
- Marshall, L.M.; Carlson, E.J.; O’Malley, J.; Snyder, C.K.; Charbonneau, N.L.; Hayflick, S.J.; Coselli, J.S.; Lemaire, S.A.; Sakai, L.Y. Thoracic aortic aneurysm frequency and dissection are associated with fibrillin-1 fragment concentrations in circulation. Circ. Res. 2013, 113, 1159–1168. [Google Scholar] [CrossRef] [Green Version]
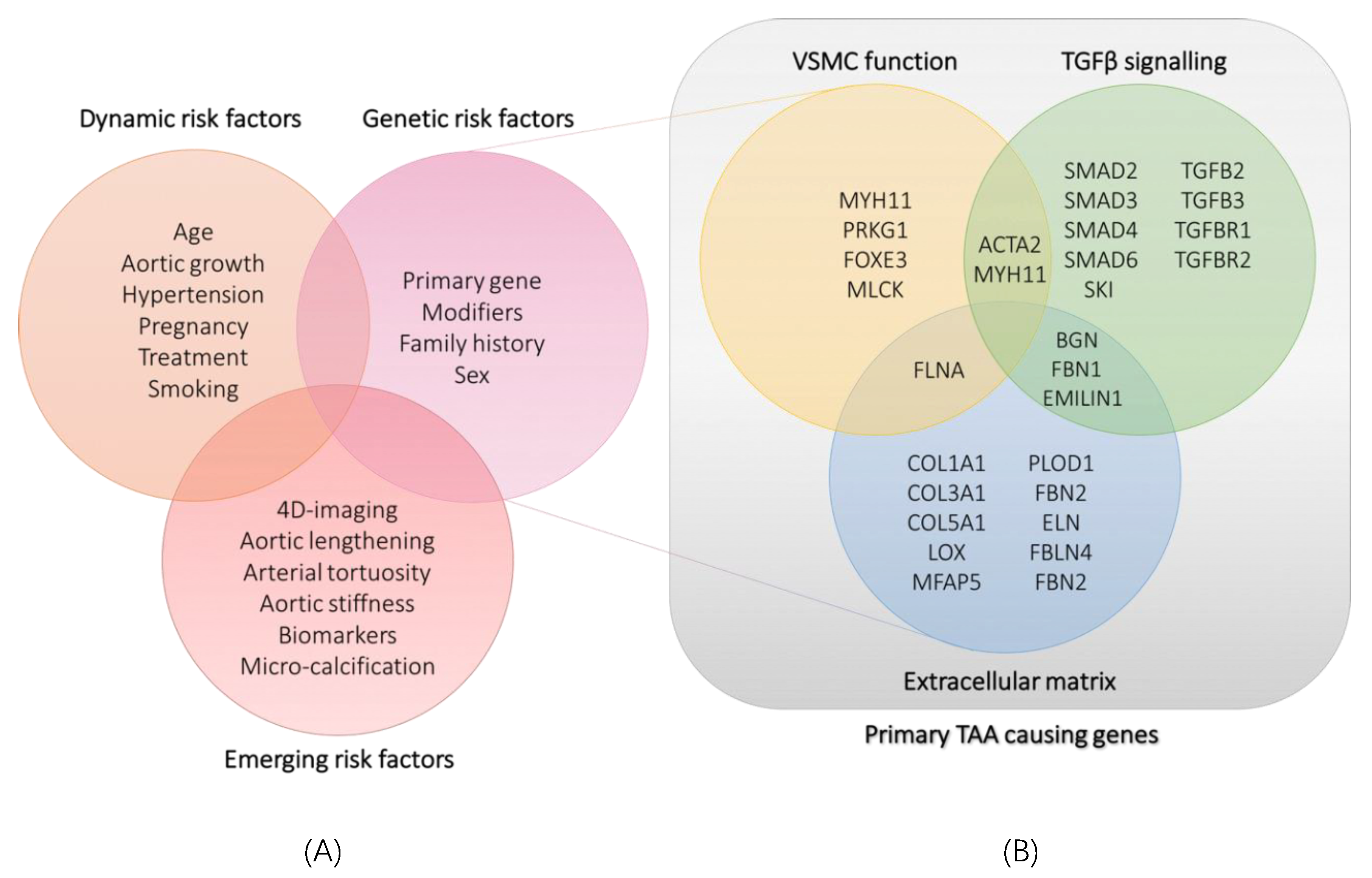
Figure 1.
(A): classification of currently known dynamic and genetic risk factors and emerging risk factors for thoracic aortic aneurysm and dissection. (B): classification of identified genes involved in syndromic and non-syndromic thoracic aortic aneurysms according to the pathway or cellular process they are involved in. TAA: thoracic aortic aneurysm; VSMC: vascular smooth muscle cell.
Figure 1.
(A): classification of currently known dynamic and genetic risk factors and emerging risk factors for thoracic aortic aneurysm and dissection. (B): classification of identified genes involved in syndromic and non-syndromic thoracic aortic aneurysms according to the pathway or cellular process they are involved in. TAA: thoracic aortic aneurysm; VSMC: vascular smooth muscle cell.

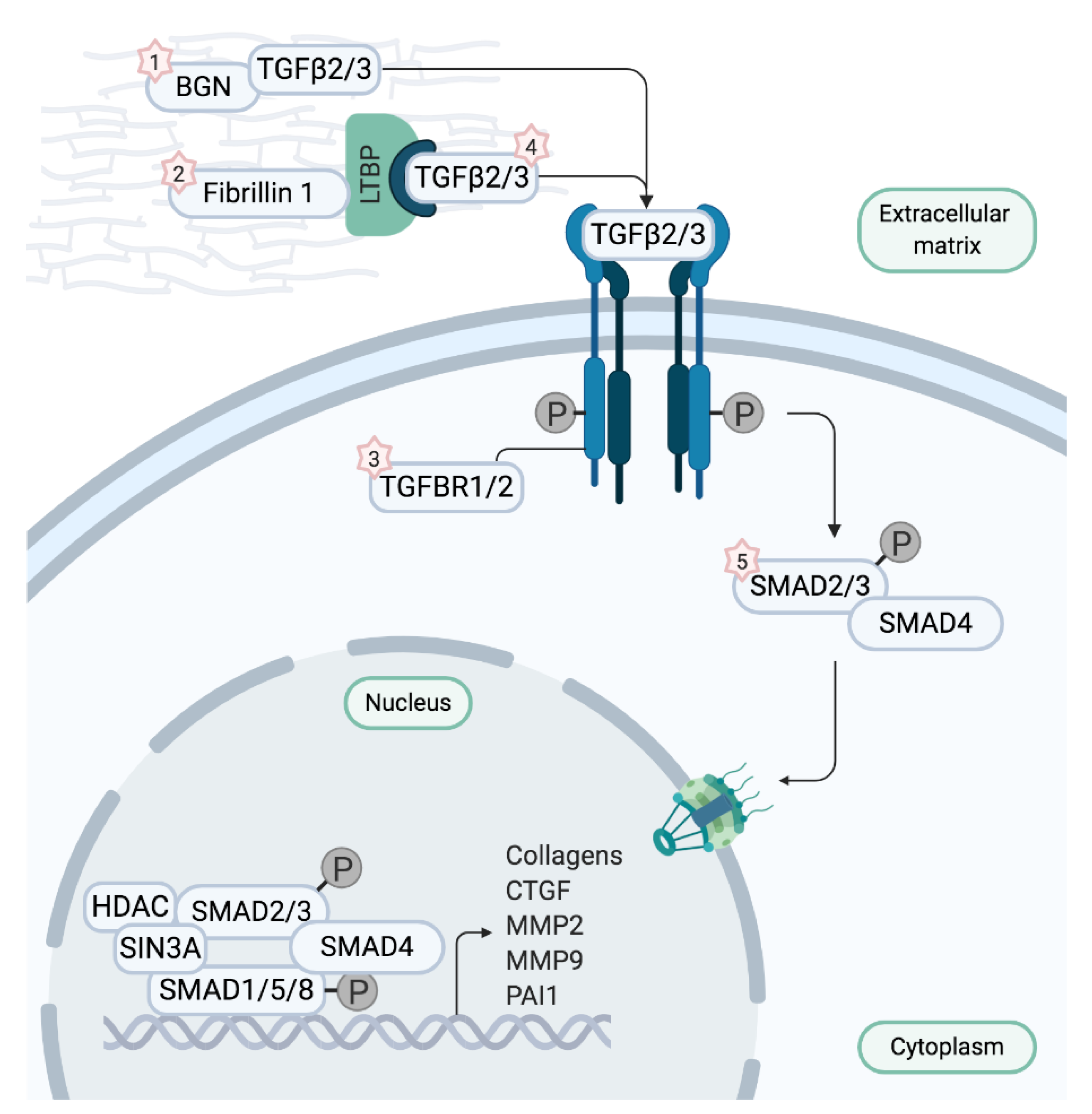
Figure 2.
Depiction of the canonical TGF-β signaling pathway. Biglycan and Fibrillin 1 sequester transforming growth factor-β ligands (TGF-β) in physiological conditions. When TGF-β is dissociated, it binds to the TGF-β receptor complex encoded by TGFBR1 and 2, thereby initiating signaling cascades and inducing transcription of TGF-β target genes. Upon binding of TGF-β, the TGF-β receptor complex phosphorylates transcription factors SMAD2 and 3 which translocate to the nucleus to start transcription after forming a complex with SMAD4. When aberrant fibrillin 1 is unable to sequester TGF-β, signaling cascades are uncontrollably activated, resulting in overexpression of the target genes. Intriguingly, defects in ligands TGF-β2 and TGF-β3, TGF-β receptors, or SMAD2 and 3 also appear to augment TGF-β signaling. Mutations in BGN cause Meester–Loeys syndrome (1), mutations in FBN1 cause Marfan syndrome (2), mutations in TGFBR1 and 2 cause Loeys–Dietz syndrome 1 and 2 (3), respectively (LDS1 and LDS2), mutations in TGFB2 and TGFB3 cause LDS4 and LDS5, respectively (4), and mutations in SMAD3 cause LDS3 (5). Figure adapted from Verstraeten et al. [1].
Figure 2.
Depiction of the canonical TGF-β signaling pathway. Biglycan and Fibrillin 1 sequester transforming growth factor-β ligands (TGF-β) in physiological conditions. When TGF-β is dissociated, it binds to the TGF-β receptor complex encoded by TGFBR1 and 2, thereby initiating signaling cascades and inducing transcription of TGF-β target genes. Upon binding of TGF-β, the TGF-β receptor complex phosphorylates transcription factors SMAD2 and 3 which translocate to the nucleus to start transcription after forming a complex with SMAD4. When aberrant fibrillin 1 is unable to sequester TGF-β, signaling cascades are uncontrollably activated, resulting in overexpression of the target genes. Intriguingly, defects in ligands TGF-β2 and TGF-β3, TGF-β receptors, or SMAD2 and 3 also appear to augment TGF-β signaling. Mutations in BGN cause Meester–Loeys syndrome (1), mutations in FBN1 cause Marfan syndrome (2), mutations in TGFBR1 and 2 cause Loeys–Dietz syndrome 1 and 2 (3), respectively (LDS1 and LDS2), mutations in TGFB2 and TGFB3 cause LDS4 and LDS5, respectively (4), and mutations in SMAD3 cause LDS3 (5). Figure adapted from Verstraeten et al. [1].

Figure 3.
Visual representation of Loeys–Dietz syndrome cardiovascular severity by affected genes, with most severe clinical presentation to the left and less severe to the right.
Figure 3.
Visual representation of Loeys–Dietz syndrome cardiovascular severity by affected genes, with most severe clinical presentation to the left and less severe to the right.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Genotype/phenotype characteristics of thoracic aortic aneurysm syndromes. LDS, Loeys–Dietz syndrome; vEDS, vascular Ehlers–Danlos syndrome; MRLS, Meester–Loeys syndrome; FTAA, familial thoracic aortic aneurysm; MSMDS multisystemic smooth muscle dysfunction syndrome; AD, autosomal dominant; XL, X-linked.
Table 1.
Genotype/phenotype characteristics of thoracic aortic aneurysm syndromes. LDS, Loeys–Dietz syndrome; vEDS, vascular Ehlers–Danlos syndrome; MRLS, Meester–Loeys syndrome; FTAA, familial thoracic aortic aneurysm; MSMDS multisystemic smooth muscle dysfunction syndrome; AD, autosomal dominant; XL, X-linked.
| Disease | MIM | Gene | Mode of Inheritance (% De Novo) | Key Features | Vascular Involvement |
|---|---|---|---|---|---|
| Marfan | #154700 | FBN1 | AD (30%) | Ectopia lentis—skeletal overgrowth | Sinus of Valsalva aneurysm/dissection |
| LDS1 | #609192 | TGFBR1 | AD (25%) | Hypertelorism, bifid uvula/cleft palate, arterial tortuosity | Widespread aortic and arterial aneurysm/dissection |
| LDS2 | #610168 | TGFBR2 | |||
| LDS3 | #613795 | SMAD3 | |||
| LDS4 | #614816 | TGFB2 | |||
| LDS5 | #615582 | TGFB3 | |||
| vEDS | #130050 | COL3A1 | AD (50%) | Thin skin—joint hypermobility | Predominant dissection of middle-sized arteries |
| MRLS | #300989 | BGN | XL | Hypertelorism, skeletal findings | Aortic aneurysm |
| FTAA6 | #611788 | ACTA2 | AD (rare) | Levido reticularis, iris flocculi | Ascending aortic aneurysm |
| MSMDS | #613834 | ACTA2-R179 | AD (100%) | Mydriasis, white matter densities, bowel dysmotility, hypotonic bladder | Persistent ductus arteriosus, aortic aneurysm |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Rodrigues Bento, J.; Meester, J.A.N.; Luyckx, I.; Verstraeten, A.; Loeys, B.L. The Role of Genetics in Risk Stratification of Thoracic Aortic Aneurysm Dissection. Hearts 2020, 1, 50-61. https://0-doi-org.brum.beds.ac.uk/10.3390/hearts1020007
AMA Style
Rodrigues Bento J, Meester JAN, Luyckx I, Verstraeten A, Loeys BL. The Role of Genetics in Risk Stratification of Thoracic Aortic Aneurysm Dissection. Hearts. 2020; 1(2):50-61. https://0-doi-org.brum.beds.ac.uk/10.3390/hearts1020007
Chicago/Turabian StyleRodrigues Bento, Jotte, Josephina A.N. Meester, Ilse Luyckx, Aline Verstraeten, and Bart L. Loeys. 2020. "The Role of Genetics in Risk Stratification of Thoracic Aortic Aneurysm Dissection" Hearts 1, no. 2: 50-61. https://0-doi-org.brum.beds.ac.uk/10.3390/hearts1020007