Optical Mapping in hiPSC-CM and Zebrafish to Resolve Cardiac Arrhythmias

 , , and
, , and
Abstract
:1. Introduction
2. Human-Induced Pluripotent Stem-Cell-Derived Cardiomyocytes (hiPSC-CMs) as a Model for ICA
2.1. Differentiation of hiPSC into CM
2.2. Functional Assessment of hiPSC-CM: From Patch Clamp to Multi-Electrode Arrays
3. Zebrafish as a Model for ICA
3.1. The Zebrafish Heart Electrophysiology
3.2. Functional Assessment of Zebrafish Hearts: Patch Clamp and ECG
4. Optical Mapping
4.1. Imaging Techniques for Electrophysiological Phenotyping
4.2. Fluorescent Dyes
4.3. Genetically Encoded Voltage/Calcium Indicators
4.4. Optical Mapping in hiPSC-CM and Zebrafish to Model ICAs
5. Limitations, Challenges and Future of Optical Mapping in hiPSC-CM and Zebrafish
5.1. hiPSC-CMs: From Immaturity to Organoids
5.2. Modelling ICAs in Zebrafish Models
5.3. Technical Challenges and Future Directions of Optical Mapping
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Schwartz, P.J.; Ackerman, M.J.; Antzelevitch, C.; Bezzina, C.R.; Borggrefe, M.; Cuneo, B.F.; Wilde, A.A.M. Inherited cardiac arrhythmias. Nat. Rev. Dis. Prim. 2020, 6, 1–22. [Google Scholar] [CrossRef]
- Offerhaus, J.A.; Bezzina, C.R.; Wilde, A.A.M. Epidemiology of inherited arrhythmias. Nat. Rev. Cardiol. 2020, 17, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.X.; Brown, A.; Lau, D.H.; Chugh, S.S.; Albert, C.M.; Kalman, J.M.; Sanders, P. Epidemiology of Sudden Cardiac Death: Global and Regional Perspectives. Hear. Lung Circ. 2019, 28, 6–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilde, A.A.M.; Nannenberg, E.; Van Der Werf, C. Cardiogenetics, 25 years a growing subspecialism. Neth. Hear. J. 2020, 28, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Monasky, M.M.; Micaglio, E.; Ciconte, G.; Pappone, C. Brugada Syndrome: Oligogenic or Mendelian Disease? Int. J. Mol. Sci. 2020, 21, 1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betzenhauser, M.J.; Pitt, G.S.; Antzelevitch, C. Calcium Channel Mutations in Cardiac Arrhythmia Syndromes. Curr. Mol. Pharmacol. 2015, 8, 133–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monasky, M.M.; Pappone, C.; Piccoli, M.; Ghiroldi, A.; Micaglio, E.; Anastasia, L. Calcium in Brugada Syndrome: Questions for Future Research. Front. Physiol. 2018, 9, 1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosseini, S.M.; Kim, R.; Udupa, S.; Costain, G.; Jobling, R.; Liston, E.; Jamal, S.M.; Szybowska, M.; Morel, C.F.; Bowdin, S.; et al. Reappraisal of Reported Genes for Sudden Arrhythmic Death. Circ. 2018, 138, 1195–1205. [Google Scholar] [CrossRef] [PubMed]
- Sendfeld, F.; Selga, E.; Scornik, F.S.; Pérez, G.J.; Mills, N.L.; Brugada, R. Experimental Models of Brugada syndrome. Int. J. Mol. Sci. 2019, 20, 2123. [Google Scholar] [CrossRef] [Green Version]
- Wallace, E.; Howard, L.; Liu, M.; O’Brien, T.; Ward, D.; Shen, S.; Prendiville, T. Long QT Syndrome: Genetics and Future Perspective. Pediatr. Cardiol. 2019, 40, 1419–1430. [Google Scholar] [CrossRef] [Green Version]
- Pourrier, M.; Fedida, D. The Emergence of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes (hiPSC-CMs) as a Platform to Model Arrhythmogenic Diseases. Int. J. Mol. Sci. 2020, 21, 657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, Y.; Yamanaka, S. Induced Pluripotent Stem Cells 10 Years Later. Circ. Res. 2017, 120, 1958–1968. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karagiannis, P.; Takahashi, K.; Saito, M.; Yoshida, Y.; Okita, K.; Watanabe, A.; Inoue, H.; Yamashita, J.K.; Todani, M.; Nakagawa, M.; et al. Induced Pluripotent Stem Cells and Their Use in Human Models of Disease and Development. Physiol. Rev. 2019, 99, 79–114. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wilson, G.F.; Soerens, A.G.; Koonce, C.H.; Yu, J.; Palecek, S.P.; Thomson, J.A.; Kamp, T.J. Functional Cardiomyocytes Derived From Human Induced Pluripotent Stem Cells. Circ. Res. 2009, 104, e30–e41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Y.; Lee, M.Y.; Schliffke, S.; Paavola, J.; Amos, P.J.; Ge, X.; Ye, M.; Zhu, S.; Senyei, G.; Lum, L.; et al. Small molecule Wnt inhibitors enhance the efficiency of BMP-4-directed cardiac differentiation of human pluripotent stem cells. J. Mol. Cell. Cardiol. 2011, 51, 280–287. [Google Scholar] [CrossRef] [Green Version]
- Lian, X.; Hsiao, C.; Wilson, G.; Zhu, K.; Hazeltine, L.B.; Azarin, S.M.; Raval, K.K.; Zhang, J.; Kamp, T.J.; Palecek, S.P. Cozzarelli Prize Winner: Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E1848–E1857. [Google Scholar] [CrossRef] [Green Version]
- Lian, X.; Zhang, J.; Azarin, S.M.; Zhu, K.; Hazeltine, L.B.; Bao, X.; Hsiao, C.; Kamp, T.J.; Palecek, S.P. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β-catenin signaling under fully defined conditions. Nat. Protoc. 2013, 8, 162–175. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Tang, Y.; Zhou, Y.; Zhang, J. Deciphering Role of Wnt Signalling in Cardiac Mesoderm and Cardiomyocyte Differentiation from Human iPSCs: Four-dimensional control of Wnt pathway for hiPSC-CMs differentiation. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef]
- Kumar, N.; Dougherty, J.A.; Manring, H.R.; Elmadbouh, I.; Mergaye, M.; Czirok, A.; Isai, D.G.; Belevych, A.E.; Yu, L.; Janssen, P.M.; et al. Assessment of temporal functional changes and miRNA profiling of human iPSC-derived cardiomyocytes. Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Sakmann, B.; Neher, E. Patch Clamp Techniques for Studying Ionic Channels in Excitable Membranes. Annu. Rev. Physiol. 1984, 46, 455–472. [Google Scholar] [CrossRef] [PubMed]
- Neher, E.; Sakmann, B. The Patch Clamp Technique. Sci. Am. 1992, 266, 44–51. [Google Scholar] [CrossRef]
- Laurila, E.; Ahola, A.; Hyttinen, J.; Aalto-Setälä, K. Methods for in vitro functional analysis of iPSC derived cardiomyocytes—Special focus on analyzing the mechanical beating behavior. Biochim. Biophys. Acta (BBA) Bioenerg. 2016, 1863, 1864–1872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, P.; Garg, V.; Shrestha, R.; Sanguinetti, M.C.; Kamp, T.J.; Wu, J.C. Human Induced Pluripotent Stem Cell–Derived Cardiomyocytes as Models for Cardiac Channelopathies. Circ. Res. 2018, 123, 224–243. [Google Scholar] [CrossRef] [PubMed]
- Obergrussberger, A.; Friis, S.; Brüggemann, A.; Fertig, N. Automated patch clamp in drug discovery: Major breakthroughs and innovation in the last decade. Expert Opin. Drug Discov. 2020, 1–5. [Google Scholar] [CrossRef]
- Dunlop, J.; Bowlby, M.R.; Peri, R.; Vasilyev, D.; Arias, R. High-throughput electrophysiology: An emerging paradigm for ion-channel screening and physiology. Nat. Rev. Drug Discov. 2008, 7, 358–368. [Google Scholar] [CrossRef]
- Obergrussberger, A.; Stölzle-Feix, S.; Becker, N.; Brüggemann, A.; Fertig, N.; Möller, C. Novel screening techniques for ion channel targeting drugs. Channels 2015, 9, 367–375. [Google Scholar] [CrossRef] [Green Version]
- Annecchino, L.A.; Schultz, S.R. Progress in automating patch clamp cellular physiology. Brain Neurosci. Adv. 2018, 2, 2398212818776561. [Google Scholar] [CrossRef] [Green Version]
- Mann, S.A.; Heide, J.; Knott, T.; Airini, R.; Epureanu, F.B.; Deftu, A.-F.; Deftu, A.-T.; Radu, B.M.; Amuzescu, B. Recording of multiple ion current components and action potentials in human induced pluripotent stem cell-derived cardiomyocytes via automated patch-clamp. J. Pharmacol. Toxicol. Methods 2019, 100, 106599. [Google Scholar] [CrossRef]
- Brinkwirth, N.; Takasuna, K.; Doi, M.; Becker, N.; Obergrussberger, A.; Friis, S.; Furukawa, H.; Hasegawa, Y.; Oka, T.; Ohtsuki, A.; et al. Reliable identification of cardiac liability in drug discovery using automated patch clamp: Benchmarking best practices and calibration standards for improved proarrhythmic assessment. J. Pharmacol. Toxicol. Methods 2020, 105, 106884. [Google Scholar] [CrossRef]
- Obergrussberger, A.; Haarmann, C.; Stölzle-Feix, S.; Becker, N.; Ohtsuki, A.; Brüggemann, A.; George, M.; Fertig, N. Automated Patch Clamp Recordings of Human Stem Cell-Derived Cardiomyocytes. In Bioinformatics in MicroRNA Research; Springer Science and Business Media LLC: Berlin, Germany, 2016; pp. 57–82. [Google Scholar]
- Glazer, A.; Wada, Y.; Li, B.; Muhammad, A.; Kalash, O.R.; O’Neill, M.J.; Shields, T.; Hall, L.; Short, L.; Blair, M.A.; et al. High-Throughput Reclassification of SCN5A Variants. Am. J. Hum. Genet. 2020, 107, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.-A.; Perry, M.D.; Liang, W.; Smith, N.J.; Foo, B.; Shrier, A.; Lukacs, G.L.; Hill, A.P.; Vandenberg, J.I. High-throughput phenotyping of heteromeric human ether-à-go-go-related gene potassium channel variants can discriminate pathogenic from rare benign variants. Hear. Rhythm. 2020, 17, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Bell, D.C.; Dallas, M.L. Using automated patch clamp electrophysiology platforms in pain-related ion channel research: Insights from industry and academia. Br. J. Pharmacol. 2017, 175, 2312–2321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Opbergen, C.J.M.; van der Voorn, S.M.; Vos, M.A.; de Boer, T.P.; van Veen, T.A.B. Cardiac Ca(2+) signalling in zebrafish: Translation of findings to man. Prog. Biophys. Mol. Biol. 2018, 138, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Kussauer, S.; Robert, D.; Lemcke, H. hiPSCs Derived Cardiac Cells for Drug and Toxicity Screening and Disease Modeling: What Micro- Electrode-Array Analyses Can Tell Us. Cells 2019, 8, 1331. [Google Scholar] [CrossRef] [Green Version]
- Millard, D.; Dang, Q.; Shi, H.; Zhang, X.; Strock, C.; Kraushaar, U.; Zeng, H.; Levesque, P.; Lu, H.-R.; Guillon, J.-M.; et al. Cross-Site Reliability of Human Induced Pluripotent stem cell-derived Cardiomyocyte Based Safety Assays Using Microelectrode Arrays: Results from a Blinded CiPA Pilot Study. Toxicol. Sci. 2018, 164, 550–562. [Google Scholar] [CrossRef] [Green Version]
- Ruan, J.-L.; Tulloch, N.L.; Razumova, M.V.; Saiget, M.; Muskheli, V.; Pabon, L.; Reinecke, H.; Regnier, M.; Murry, C.E. Mechanical Stress Conditioning and Electrical Stimulation Promote Contractility and Force Maturation of Induced Pluripotent Stem Cell-Derived Human Cardiac Tissue. Circulation 2016, 134, 1557–1567. [Google Scholar] [CrossRef]
- Wei, F.; Pourrier, M.; Strauss, D.G.; Stockbridge, N.; Pang, L. Effects of Electrical Stimulation on hiPSC-CM Responses to Classic Ion Channel Blockers. Toxicol. Sci. 2020, 174, 254–265. [Google Scholar] [CrossRef]
- Lieschke, G.J.; Currie, P.D. Animal models of human disease: Zebrafish swim into view. Nat. Rev. Genet. 2007, 8, 353–367. [Google Scholar] [CrossRef]
- Bradford, Y.M.; Toro, S.; Ramachandran, S.; Ruzicka, L.; Howe, D.G.; Eagle, A.; Kalita, P.; Martin, R.; Moxon, S.A.T.; Schaper, K.; et al. Zebrafish Models of Human Disease: Gaining Insight into Human Disease at ZFIN. ILAR J. 2017, 58, 4–16. [Google Scholar] [CrossRef] [Green Version]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, R.; Moyse, B.R.; Richardson, R.J. Zebrafish cardiac regeneration—looking beyond cardiomyocytes to a complex microenvironment. Histochem. Cell Biol. 2020, 154, 533–548. [Google Scholar] [CrossRef] [PubMed]
- Menke, A.L.; Spitsbergen, J.M.; Wolterbeek, A.P.M.; Woutersen, R.A. Normal Anatomy and Histology of the Adult Zebrafish. Toxicol. Pathol. 2011, 39, 759–775. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, R.N.; Jopling, C.; Van Eeden, F.J.M. Zebrafish as a Model of Cardiac Disease. Prog. Mol. Biol. Transl. Sci. 2014, 124, 65–91. [Google Scholar] [CrossRef]
- Sedmera, D.; Reckova, M.; DeAlmeida, A.; Sedmerova, M.; Biermann, M.; Volejnik, J.; Sarre, A.; Raddatz, E.; McCarthy, R.A.; Gourdie, R.G.; et al. Functional and morphological evidence for a ventricular conduction system in zebrafish and Xenopus hearts. Am. J. Physiol. Circ. Physiol. 2003, 284, H1152–H1160. [Google Scholar] [CrossRef] [Green Version]
- Sarmah, S.; Marrs, J.A. Zebrafish as a Vertebrate Model System to Evaluate Effects of Environmental Toxicants on Cardiac Development and Function. Int. J. Mol. Sci. 2016, 17, 2123. [Google Scholar] [CrossRef] [Green Version]
- Santoso, F.; Farhan, S.M.A.; Castillo, A.L.; Malhotra, N.; Saputra, F.; Kurnia, K.A.; Chen, K.H.-C.; Huang, J.-C.; Chen, J.-R.; Hsiao, C.-D. An Overview of Methods for Cardiac Rhythm Detection in Zebrafish. Biomedicines 2020, 8, 329. [Google Scholar] [CrossRef]
- Giardoglou, P.; Beis, D. On Zebrafish Disease Models and Matters of the Heart. Biomedicines 2019, 7, 15. [Google Scholar] [CrossRef] [Green Version]
- Genge, C.E.; Lin, E.; Lee, L.; Sheng, X.; Rayani, K.; Gunawan, M.; Stevens, C.M.; Li, A.Y.; Talab, S.S.; Claydon, T.; et al. The Zebrafish Heart as a Model of Mammalian Cardiac Function. Rev. Physiol. Biochem. Pharmacol. 2016, 171, 99–136. [Google Scholar] [CrossRef]
- Rayani, K.; Lin, E.; Craig, C.; Lamothe, M.; Shafaattalab, S.; Gunawan, M.; Li, A.Y.; Hove-Madsen, L.; Tibbits, G.F. Zebrafish as a model of mammalian cardiac function: Optically mapping the interplay of temperature and rate on voltage and calcium dynamics. Prog. Biophys. Mol. Biol. 2018, 138, 69–90. [Google Scholar] [CrossRef]
- Haverinen, J.; Hassinen, M.; Dash, S.N.; Vornanen, M. Expression of calcium channel transcripts in the zebrafish heart: Dominance of T-type channels. J. Exp. Biol. 2018, 221, jeb179226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnaout, R.; Ferrer, T.; Huisken, J.; Spitzer, K.; Stainier, D.Y.R.; Tristani-Firouzi, M.; Chi, N.C. Zebrafish model for human long QT syndrome. Proc. Natl. Acad. Sci. USA 2007, 104, 11316–11321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Hayashi, K.; Fujino, N.; Konno, T.; Tada, H.; Nakanishi, C.; Hodatsu, A.; Tsuda, T.; Nagata, Y.; Teramoto, R.; et al. Functional analysis of KCNH2 gene mutations of type 2 long QT syndrome in larval zebrafish using microscopy and electrocardiography. Hear. Vessel. 2019, 34, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Leong, I.U.S.; Skinner, J.R.; Shelling, A.; Love, D.R. Identification and expression analysis of kcnh2 genes in the zebrafish. Biochem. Biophys. Res. Commun. 2010, 396, 817–824. [Google Scholar] [CrossRef]
- Abramochkin, D.V.; Hassinen, M.; Vornanen, M. Transcripts of Kv7.1 and MinK channels and slow delayed rectifier K+ current (IKs) are expressed in zebrafish (Danio rerio) heart. Pflügers Arch. Eur. J. Physiol. 2018, 470, 1753–1764. [Google Scholar] [CrossRef]
- Hassinen, M.; Haverinen, J.; Hardy, M.E.; Shiels, H.A.; Vornanen, M. Inward rectifier potassium current (I K1) and Kir2 composition of the zebrafish (Danio rerio) heart. Pflügers Arch. Eur. J. Physiol. 2015, 467, 2437–2446. [Google Scholar] [CrossRef] [Green Version]
- Ravens, U. Ionic basis of cardiac electrophysiology in zebrafish compared to human hearts. Prog. Biophys. Mol. Biol. 2018, 138, 38–44. [Google Scholar] [CrossRef]
- Vornanen, M.; Hassinen, M. Zebrafish heart as a model for human cardiac electrophysiology. Channels 2016, 10, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Lin, E.; Shafaattalab, S.; Gill, J.; Al-Zeer, B.; Craig, C.; Lamothe, M.; Rayani, K.; Gunawan, M.; Li, A.Y.; Hove-Madsen, L.; et al. Physiological phenotyping of the adult zebrafish heart. Mar. Genom. 2020, 49, 100701. [Google Scholar] [CrossRef]
- Brette, F.; Luxan, G.; Cros, C.; Dixey, H.; Wilson, C.; Shiels, H.A. Characterization of isolated ventricular myocytes from adult zebrafish (Danio rerio). Biochem. Biophys. Res. Commun. 2008, 374, 143–146. [Google Scholar] [CrossRef]
- Jou, C.J.; Spitzer, K.W.; Tristani-Firouzi, M. Blebbistatin Effectively Uncouples the Excitation-Contraction Process in Zebrafish Embryonic Heart. Cell. Physiol. Biochem. 2010, 25, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Nemtsas, P.; Wettwer, E.; Christ, T.; Weidinger, G.; Ravens, U. Adult zebrafish heart as a model for human heart? An electrophysiological study. J. Mol. Cell. Cardiol. 2010, 48, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.O.; Remme, C.A. Zebrafish: A novel research tool for cardiac (patho)electrophysiology and ion channel disorders. Front. Physiol. 2012, 3, 255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda, M.; Egaña, J.T.; Allende, M.L.; Eblen-Zajjur, A. Myocardial Monophasic Action Potential Recorded by Suction Electrode for Ionic Current Studies in Zebrafish. Zebrafish 2019, 16, 427–433. [Google Scholar] [CrossRef]
- Liu, C.C.; Li, L.; Lam, Y.W.; Siu, C.W.; Cheng, S.H. Improvement of surface ECG recording in adult zebrafish reveals that the value of this model exceeds our expectation. Sci. Rep. 2016, 6, 25073. [Google Scholar] [CrossRef]
- Lin, M.-H.; Chou, H.-C.; Chen, Y.-F.; Liu, W.; Lee, C.-C.; Liu, L.Y.-M.; Chuang, Y.-J. Development of a rapid and economic in vivo electrocardiogram platform for cardiovascular drug assay and electrophysiology research in adult zebrafish. Sci. Rep. 2018, 8, 15986. [Google Scholar] [CrossRef]
- Zhao, Y.; Yun, M.; Nguyen, S.A.; Tran, M.; Nguyen, T.P. In Vivo Surface Electrocardiography for Adult Zebrafish. J. Vis. Exp. 2019, e60011. [Google Scholar] [CrossRef]
- Thorsen, K.; Dam, V.S.; Kjaer-Sorensen, K.; Pedersen, L.N.; Skeberdis, V.A.; Jurevicius, J.; Treinys, R.; Petersen, I.M.B.S.; Nielsen, M.S.; Oxvig, C.; et al. Loss-of-activity-mutation in the cardiac chloride-bicarbonate exchanger AE3 causes short QT syndrome. Nat. Commun. 2017, 8, 1696. [Google Scholar] [CrossRef]
- Bruton, J.D.; Cheng, A.J.; Westerblad, H. Measuring Ca2+ in Living Cells. Adv. Exp. Med. Biol. 2019, 1131, 7–26. [Google Scholar] [CrossRef]
- Herron, T.J.; Lee, P.; Jalife, P.M.J. Optical Imaging of Voltage and Calcium in Cardiac Cells & Tissues. Circ. Res. 2012, 110, 609–623. [Google Scholar] [CrossRef] [Green Version]
- Sanderson, M.J.; Smith, I.; Parker, I.; Bootman, M.D. Fluorescence microscopy. Cold Spring Harb. Protoc. 2014, 2014, pdb top071795. [Google Scholar] [CrossRef] [Green Version]
- Power, R.M.; Huisken, J. A guide to light-sheet fluorescence microscopy for multiscale imaging. Nat. Methods 2017, 14, 360–373. [Google Scholar] [CrossRef]
- Elisa, Z.; Toon, B.; De Smedt, S.C.; Katrien, R.; Kristiaan, N.; Braeckmans, K. Technical implementations of light sheet microscopy. Microsc. Res. Tech. 2018, 81, 941–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.-C.; Legant, W.R.; Wang, K.; Shao, L.; Milkie, D.E.; Davidson, M.W.; Janetopoulos, C.; Wu, X.S.; Hammer, J.A.; Liu, Z.; et al. Lattice light-sheet microscopy: Imaging molecules to embryos at high spatiotemporal resolution. Science 2014, 346, 1257998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toepfer, C.N.; Sharma, A.; Cicconet, M.; Garfinkel, A.C.; Mücke, M.; Neyazi, M.; Willcox, J.A.; Agarwal, R.; Schmid, M.; Rao, J.; et al. SarcTrack. Circ. Res. 2019, 124, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Sala, L.; Van Meer, B.J.; Tertoolen, L.G.; Bakkers, J.; Bellin, M.; Davis, R.P.; Denning, C.N.; Dieben, M.A.; Eschenhagen, T.; Giacomelli, E.; et al. MUSCLEMOTION. Circ. Res. 2018, 122, e5–e16. [Google Scholar] [CrossRef] [PubMed]
- Da Rocha, A.M.; Campbell, K.; Mironov, S.; Jiang, J.; Mundada, L.; Guerrero-Serna, G.; Jalife, J.; Herron, T.J. hiPSC-CM Monolayer Maturation State Determines Drug Responsiveness in High Throughput Pro-Arrhythmia Screen. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Rocha, A.M.; Creech, J.; Thonn, E.; Mironov, S.; Herron, T.J. Detection of Drug-Induced Torsades de Pointes Arrhythmia Mechanisms Using hiPSC-CM Syncytial Monolayers in a High-Throughput Screening Voltage Sensitive Dye Assay. Toxicol. Sci. 2019, 173, 402–415. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.; Upadhyay, H.; Murphy, M.; Borja, G.; Rozsahegyi, E.J.; Barnett, A.; Brookings, T.; McManus, O.B.; Werley, C.A. Simultaneous voltage and calcium imaging and optogenetic stimulation with high sensitivity and a wide field of view. Biomed. Opt. Express 2019, 10, 789–806. [Google Scholar] [CrossRef]
- Shah, D.; Prajapati, C.; Penttinen, K.; Cherian, R.M.; Koivumäki, J.T.; Alexanova, A.; Hyttinen, J.; Aalto-Setälä, K. hiPSC-Derived Cardiomyocyte Model of LQT2 Syndrome Derived from Asymptomatic and Symptomatic Mutation Carriers Reproduces Clinical Differences in Aggregates but Not in Single Cells. Cells 2020, 9, 1153. [Google Scholar] [CrossRef] [PubMed]
- Pott, A.; Bock, S.; Berger, I.M.; Frese, K.; Dahme, T.; Keßler, M.; Rinné, S.; Decher, N.; Just, S.; Rottbauer, W.; et al. Mutation of the Na+/K+-ATPase Atp1a1a.1 causes QT interval prolongation and bradycardia in zebrafish. J. Mol. Cell. Cardiol. 2018, 120, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Sleiman, Y.; Souidi, M.; Kumar, R.; Yang, E.; Jaffré, F.; Zhou, T.; Bernardin, A.; Reiken, S.; Cazorla, O.; Kajava, A.V.; et al. Modeling polymorphic ventricular tachycardia at rest using patient-specific induced pluripotent stem cell-derived cardiomyocytes. EBioMedicine 2020, 60, 103024. [Google Scholar] [CrossRef] [PubMed]
- Crestani, T.; Steichen, C.; Neri, E.; Rodrigues, M.; Fonseca-Alaniz, M.H.; Ormrod, B.; Holt, M.R.; Pandey, P.; Harding, S.; Ehler, E.; et al. Electrical stimulation applied during differentiation drives the hiPSC-CMs towards a mature cardiac conduction-like cells. Biochem. Biophys. Res. Commun. 2020, 533, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.H.; Kralj, J.M.; Douglass, A.D.; Engert, F.; Cohen, A.E. Simultaneous mapping of membrane voltage and calcium in zebrafish heart in vivo reveals chamber-specific developmental transitions in ionic currents. Front. Physiol. 2014, 5, 344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turaga, D.; Matthys, O.B.; Hookway, T.A.; Joy, D.A.; Calvert, M.; McDevitt, T.C. Single-Cell Determination of Cardiac Microtissue Structure and Function Using Light Sheet Microscopy. Tissue Eng. Part C Methods 2020, 26, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Maioli, V.; Boniface, A.; Mahou, P.; Ortas, J.F.; Abdeladim, L.; Beaurepaire, E.; Supatto, W. Fast in vivo multiphoton light-sheet microscopy with optimal pulse frequency. Biomed. Opt. Express 2020, 11, 6012–6026. [Google Scholar] [CrossRef] [PubMed]
- Logan, S.L.; Dudley, C.; Baker, R.P.; Taormina, M.J.; Hay, E.A.; Parthasarathy, R. Automated high-throughput light-sheet fluorescence microscopy of larval zebrafish. PLoS ONE 2018, 13, e0198705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lock, J.T.; Parker, I.; Smith, I. A comparison of fluorescent Ca2+ indicators for imaging local Ca2+ signals in cultured cells. Cell Calcium 2015, 58, 638–648. [Google Scholar] [CrossRef] [Green Version]
- Broyles, C.N.; Robinson, P.; Daniels, M.J. Fluorescent, Bioluminescent, and Optogenetic Approaches to Study Excitable Physiology in the Single Cardiomyocyte. Cells 2018, 7, 51. [Google Scholar] [CrossRef] [Green Version]
- Miller, E.W.; Lin, J.Y.; Frady, E.P.; Steinbach, P.A.; Kristan, W.B.; Tsien, R.Y. Optically monitoring voltage in neurons by photo-induced electron transfer through molecular wires. Proc. Natl. Acad. Sci. USA 2012, 109, 2114–2119. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, R.U.; Miller, E.W. Voltage Imaging: Pitfalls and Potential. Biochemistry 2017, 56, 5171–5177. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, C.; Kabir, S.N.; Holmes, A.P.; Lei, M.; Fabritz, L.; Rajpoot, K.; Pavlovic, D. Cardiac optical mapping —State-of-the-art and future challenges. Int. J. Biochem. Cell Biol. 2020, 126, 105804. [Google Scholar] [CrossRef] [PubMed]
- Bando, Y.; Grimm, C.; Cornejo, V.H.; Yuste, R. Genetic voltage indicators. BMC Biol. 2019, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Panzera, L.C.; Hoppa, M.B. Genetically Encoded Voltage Indicators Are Illuminating Subcellular Physiology of the Axon. Front. Cell. Neurosci. 2019, 13, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shemetov, A.A.; Monakhov, M.V.; Zhang, Q.; Canton-Josh, J.E.; Kumar, M.; Chen, M.; Matlashov, M.E.; Li, X.; Yang, W.; Nie, L.; et al. A near-infrared genetically encoded calcium indicator for in vivo imaging. Nat. Biotechnol. 2020, 2020, 1–10. [Google Scholar] [CrossRef]
- Monakhov, M.V.; Matlashov, M.E.; Colavita, M.; Song, C.; Shcherbakova, D.M.; Antic, S.D.; Verkhusha, V.V.; Knöpfel, T. Screening and Cellular Characterization of Genetically Encoded Voltage Indicators Based on Near-Infrared Fluorescent Proteins. ACS Chem. Neurosci. 2020, 11, 3523–3531. [Google Scholar] [CrossRef]
- Shinnawi, R.; Huber, I.; Maizels, L.; Shaheen, N.; Gepstein, A.; Arbel, G.; Tijsen, A.J.; Gepstein, L. Monitoring Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes with Genetically Encoded Calcium and Voltage Fluorescent Reporters. Stem Cell Rep. 2015, 5, 582–596. [Google Scholar] [CrossRef] [Green Version]
- Herron, T.J. Calcium and voltage mapping in hiPSC-CM monolayers. Cell Calcium 2016, 59, 84–90. [Google Scholar] [CrossRef]
- Goldfracht, I.; Efraim, Y.; Shinnawi, R.; Kovalev, E.; Huber, I.; Gepstein, A.; Arbel, G.; Shaheen, N.; Tiburcy, M.; Zimmermann, W.-H.; et al. Engineered heart tissue models from hiPSC-derived cardiomyocytes and cardiac ECM for disease modeling and drug testing applications. Acta Biomater. 2019, 92, 145–159. [Google Scholar] [CrossRef]
- Van Opbergen, C.J.; Koopman, C.D.; Kok, B.J.; Knöpfel, T.; Renninger, S.L.; Orger, M.B.; Vos, M.A.; Van Veen, T.A.; Bakkers, J.; De Boer, T.P. Optogenetic sensors in the zebrafish heart: A novel in vivo electrophysiological tool to study cardiac arrhythmogenesis. Theranostics 2018, 8, 4750–4764. [Google Scholar] [CrossRef]
- Lin, E.; Craig, C.; Lamothe, M.; Sarunic, M.V.; Beg, M.F.; Tibbits, G.F. Construction and use of a zebrafish heart voltage and calcium optical mapping system, with integrated electrocardiogram and programmable electrical stimulation. Am. J. Physiol. Integr. Comp. Physiol. 2015, 308, R755–R768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salgado-Almario, J.; Vicente, M.; Vincent, P.; Domingo, B.; Llopis, J. Mapping Calcium Dynamics in the Heart of Zebrafish Embryos with Ratiometric Genetically Encoded Calcium Indicators. Int. J. Mol. Sci. 2020, 21, 6610. [Google Scholar] [CrossRef] [PubMed]
- Meder, B.; Scholz, E.P.; Hassel, D.; Wolff, C.; Just, S.; Berger, I.M.; Patzel, E.; Karle, C.; Katus, H.A.; Rottbauer, W. Reconstitution of defective protein trafficking rescues Long-QT syndrome in zebrafish. Biochem. Biophys. Res. Commun. 2011, 408, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Peal, D.S.; Mills, R.W.; Lynch, S.N.; Mosley, J.M.; Lim, E.; Ellinor, P.T.; January, C.T.; Peterson, R.T.; Milan, D.J.; Elllinor, P.T. Novel Chemical Suppressors of Long QT Syndrome Identified by an In Vivo Functional Screen. Circulation 2010, 123, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Hassel, D.; Scholz, E.P.; Trano, N.; Friedrich, O.; Just, S.; Meder, B.; Weiss, D.L.; Zitron, E.; Marquart, S.; Vogel, B.; et al. Deficient ZebrafishEther-à-Go-Go–Related Gene Channel Gating Causes Short-QT Syndrome in ZebrafishReggaeMutants. Circulation 2008, 117, 866–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, K.; Teramoto, R.; Nomura, A.; Asano, Y.; Beerens, M.; Kurata, Y.; Kobayashi, I.; Fujino, N.; Furusho, H.; Sakata, K.; et al. Impact of functional studies on exome sequence variant interpretation in early-onset cardiac conduction system diseases. Cardiovasc. Res. 2020, 116, 2116–2130. [Google Scholar] [CrossRef]
- Da’As, S.I.; Thanassoulas, A.; Calver, B.L.; Beck, K.; Salem, R.; Saleh, A.; Kontogianni, I.; Al-Maraghi, A.; Nasrallah, G.K.; Safieh-Garabedian, B.; et al. Arrhythmogenic calmodulin E105A mutation alters cardiac RyR2 regulation leading to cardiac dysfunction in zebrafish. Ann. N. Y. Acad. Sci. 2019, 1448, 19–29. [Google Scholar] [CrossRef]
- Berchtold, M.W.; Zacharias, T.; Kulej, K.; Wang, K.; Torggler, R.; Jespersen, T.; Chen, J.-N.; Larsen, M.R.; La Cour, J.M. The Arrhythmogenic Calmodulin Mutation D129G Dysregulates Cell Growth, Calmodulin-dependent Kinase II Activity, and Cardiac Function in Zebrafish. J. Biol. Chem. 2016, 291, 26636–26646. [Google Scholar] [CrossRef] [Green Version]
- Juang, J.-M.J.; Binda, A.; Lee, S.-J.; Hwang, J.-J.; Chen, W.-J.; Liu, Y.-B.; Lin, L.-Y.; Yu, C.-C.; Ho, L.-T.; Huang, H.-C.; et al. GSTM3 variant is a novel genetic modifier in Brugada syndrome, a disease with risk of sudden cardiac death. EBioMedicine 2020, 57, 102843. [Google Scholar] [CrossRef]
- Estes, S.I.; Ye, D.; Zhou, W.; Dotzler, S.M.; Tester, D.J.; Bos, J.M.; Kim, C.J.; Ackerman, M.J. Characterization of the CACNA1C-R518C Missense Mutation in the Pathobiology of Long-QT Syndrome Using Human Induced Pluripotent Stem Cell Cardiomyocytes Shows Action Potential Prolongation and L-Type Calcium Channel Perturbation. Circ. Genom. Precis. Med. 2019, 12, e002534. [Google Scholar] [CrossRef] [Green Version]
- Rocchetti, M.; Sala, L.; Dreizehnter, L.; Crotti, L.; Sinnecker, D.; Mura, M.; Pane, L.S.; Altomare, C.; Torre, E.; Mostacciuolo, G.; et al. Elucidating arrhythmogenic mechanisms of long-QT syndrome CALM1-F142L mutation in patient-specific induced pluripotent stem cell-derived cardiomyocytes. Cardiovasc. Res. 2017, 113, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Spencer, C.I.; Baba, S.; Nakamura, K.; Hua, E.A.; Sears, M.A.; Fu, C.-C.; Zhang, J.; Balijepalli, S.; Tomoda, K.; Hayashi, Y.; et al. Calcium Transients Closely Reflect Prolonged Action Potentials in iPSC Models of Inherited Cardiac Arrhythmia. Stem Cell Rep. 2014, 3, 269–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yazawa, M.; Hsueh, B.; Jia, X.; Pasca, A.M.; Bernstein, J.A.; Hallmayer, J.; Dolmetsch, R.E. Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nat. Cell Biol. 2011, 471, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Kiviaho, A.L.; Ahola, A.; Larsson, K.; Penttinen, K.; Swan, H.; Pekkanen-Mattila, M.; Venäläinen, H.; Paavola, K.; Hyttinen, J.; Aalto-Setälä, K. Distinct electrophysiological and mechanical beating phenotypes of long QT syndrome type 1-specific cardiomyocytes carrying different mutations. IJC Hear. Vasc. 2015, 8, 19–31. [Google Scholar] [CrossRef] [Green Version]
- Bezzerides, V.J.; Zhang, A.; Xiao, L.; Simonson, B.; Khedkar, S.A.; Baba, S.; Ottaviano, F.; Lynch, S.; Hessler, K.; Rigby, A.C.; et al. Inhibition of serum and glucocorticoid regulated kinase-1 as novel therapy for cardiac arrhythmia disorders. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Barajas-Martinez, H.; Smith, M.; Hu, D.; Goodrow, R.J.; Puleo, C.; Hasdemir, C.; Antzelevitch, C.; Pfeiffer, R.; Treat, J.A.; Cordeiro, J.M. Susceptibility to Ventricular Arrhythmias Resulting from Mutations in FKBP1B, PXDNL, and SCN9A Evaluated in hiPSC Cardiomyocytes. Stem Cells Int. 2020, 2020, 1–16. [Google Scholar] [CrossRef]
- Liang, P.; Sallam, K.; Wu, H.; Li, Y.; Itzhaki, I.; Garg, P.; Zhang, Y.; Vermglinchan, V.; Lan, F.; Gu, M.; et al. Patient-Specific and Genome-Edited Induced Pluripotent Stem Cell–Derived Cardiomyocytes Elucidate Single-Cell Phenotype of Brugada Syndrome. J. Am. Coll. Cardiol. 2016, 68, 2086–2096. [Google Scholar] [CrossRef]
- El-Battrawy, I.; Müller, J.; Zhao, Z.; Cyganek, L.; Zhong, R.; Zhang, F.; Kleinsorge, M.; Lan, H.; Li, X.; Xu, Q.; et al. Studying Brugada Syndrome With an SCN1B Variants in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Front. Cell Dev. Biol. 2019, 7, 261. [Google Scholar] [CrossRef]
- Zhang, X.-H.; Morad, M. Calcium signaling in human stem cell-derived cardiomyocytes: Evidence from normal subjects and CPVT afflicted patients. Cell Calcium 2016, 59, 98–107. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.-H.; Wei, H.; Xia, Y.; Morad, M. Calcium signaling consequences of RyR2 mutations associated with CPVT1 introduced via CRISPR/Cas9 gene editing in human-induced pluripotent stem cell–derived cardiomyocytes: Comparison of RyR2-R420Q, F2483I, and Q4201R. Hear. Rhythm. 2020. [Google Scholar] [CrossRef]
- Pölönen, R.P.; Penttinen, K.; Swan, H.; Aalto-Setälä, K. Antiarrhythmic Effects of Carvedilol and Flecainide in Cardiomyocytes Derived from Catecholaminergic Polymorphic Ventricular Tachycardia Patients. Stem Cells Int. 2018, 2018, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ahola, A.; Pölönen, R.P.; Aalto-Setälä, K.; Hyttinen, J. Simultaneous Measurement of Contraction and Calcium Transients in Stem Cell Derived Cardiomyocytes. Ann. Biomed. Eng. 2017, 46, 148–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itzhaki, I.; Maizels, L.; Huber, I.; Gepstein, A.; Arbel, G.; Caspi, O.; Miller, L.; Belhassen, B.; Nof, E.; Glikson, M.; et al. Modeling of Catecholaminergic Polymorphic Ventricular Tachycardia With Patient-Specific Human-Induced Pluripotent Stem Cells. J. Am. Coll. Cardiol. 2012, 60, 990–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penttinen, K.; Swan, H.; Vanninen, S.; Paavola, J.; Lahtinen, A.M.; Kontula, K.; Aalto-Setälä, K. Antiarrhythmic Effects of Dantrolene in Patients with Catecholaminergic Polymorphic Ventricular Tachycardia and Replication of the Responses Using iPSC Models. PLoS ONE 2015, 10, e0125366. [Google Scholar] [CrossRef]
- Bezzerides, V.J.; Caballero, A.; Wang, S.; Ai, Y.; Hylind, R.J.; Lu, F.; Heims-Waldron, D.A.; Chambers, K.D.; Zhang, D.; Abrams, D.J.; et al. Gene Therapy for Catecholaminergic Polymorphic Ventricular Tachycardia by Inhibition of Ca 2+ /Calmodulin-Dependent Kinase II. Circulation 2019, 140, 405–419. [Google Scholar] [CrossRef] [PubMed]
- Lan, H.; Xu, Q.; El-Battrawy, I.; Zhong, R.; Li, X.; Lang, S.; Cyganek, L.; Borggrefe, M.; Zhou, X.; Akin, I. Ionic Mechanisms of Disopyramide Prolonging Action Potential Duration in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes From a Patient With Short QT Syndrome Type 1. Front. Pharmacol. 2020, 11, 554422. [Google Scholar] [CrossRef]
- El-Battrawy, I.; Lan, H.; Cyganek, L.; Zhao, Z.; Li, X.; Buljubasic, F.; Lang, S.; Yücel, G.; Sattler, K.; Zimmermann, W.-H.; et al. Modeling Short QT Syndrome Using Human-Induced Pluripotent Stem Cell–Derived Cardiomyocytes. J. Am. Hear. Assoc. 2018, 7, 7. [Google Scholar] [CrossRef] [Green Version]
- Shinnawi, R.; Shaheen, N.; Huber, I.; Shiti, A.; Arbel, G.; Gepstein, A.; Ballan, N.; Setter, N.; Tijsen, A.J.; Borggrefe, M.; et al. Modeling Reentry in the Short QT Syndrome With Human-Induced Pluripotent Stem Cell–Derived Cardiac Cell Sheets. J. Am. Coll. Cardiol. 2019, 73, 2310–2324. [Google Scholar] [CrossRef]
- Wu, P.; Deng, G.; Sai, X.; Guo, H.; Huang, H.; Zhu, P. Maturation strategies and limitations of induced pluripotent stem cell-derived cardiomyocytes. Biosci. Rep. 2020. [Google Scholar] [CrossRef]
- Magdy, T.; Schuldt, A.J.; Wu, J.C.; Bernstein, D.; Burridge, P.W. Human Induced Pluripotent Stem Cell (hiPSC)-Derived Cells to Assess Drug Cardiotoxicity: Opportunities and Problems. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 83–103. [Google Scholar] [CrossRef]
- Marchianò, S.; Bertero, A.; Murry, C.E. Learn from Your Elders: Developmental Biology Lessons to Guide Maturation of Stem Cell-Derived Cardiomyocytes. Pediatr. Cardiol. 2019, 40, 1367–1387. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.E.; Anzai, T.; Chanthra, N.; Uosaki, H. A Brief Review of Current Maturation Methods for Human Induced Pluripotent Stem Cells-Derived Cardiomyocytes. Front. Cell Dev. Biol. 2020, 8, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gintant, G.; Traebert, M. The roles of human induced pluripotent stem cell-derived cardiomyocytes in drug discovery: Managing in vitro safety study expectations. Expert Opin. Drug Discov. 2020, 15, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Richards, D.J.; Li, Y.; Kerr, C.M.; Yao, J.; Beeson, G.C.; Coyle, R.C.; Chen, X.; Jia, J.; Damon, B.; Wilson, R.; et al. Human cardiac organoids for the modelling of myocardial infarction and drug cardiotoxicity. Nat. Biomed. Eng. 2020, 4, 446–462. [Google Scholar] [CrossRef]
- Jou, C.J.; Barnett, S.M.; Bian, J.-T.; Weng, H.C.; Sheng, X.; Tristani-Firouzi, M. An In Vivo Cardiac Assay to Determine the Functional Consequences of Putative Long QT Syndrome Mutations. Circ. Res. 2013, 112, 826–830. [Google Scholar] [CrossRef] [Green Version]
- Nesmith, H.W.; Zhang, H.; Rogers, J.M. Optical mapping of electromechanics in intact organs. Exp. Biol. Med. 2019, 245, 368–373. [Google Scholar] [CrossRef]
- Várkuti, B.H.; Képiró, M.; Horváth, I.Á.; Végner, L.; Ráti, S.; Zsigmond, Á.; Hegyi, G.; Lenkei, Z.; Varga, M.; Málnási-Csizmadia, A. A highly soluble, non-phototoxic, non-fluorescent blebbistatin derivative. Sci. Rep. 2016, 6, 26141. [Google Scholar] [CrossRef] [Green Version]
- Weber, M.; Scherf, N.; Meyer, A.M.; Panáková, D.; Kohl, P.; Huisken, J. Cell-accurate optical mapping across the entire developing heart. eLife 2017, 6, e28307. [Google Scholar] [CrossRef]
- Kappadan, V.; Telele, S.; Uzelac, I.; Fenton, F.; Parlitz, U.; Luther, S.; Christoph, J. High-Resolution Optical Measurement of Cardiac Restitution, Contraction, and Fibrillation Dynamics in Beating vs. Blebbistatin-Uncoupled Isolated Rabbit Hearts. Front. Physiol. 2020, 11, 464. [Google Scholar] [CrossRef]
- Antinucci, P.; Hindges, R. A crystal-clear zebrafish for in vivo imaging. Sci. Rep. 2016, 6, 29490. [Google Scholar] [CrossRef] [Green Version]
- Lazzari-Dean, J.; Gest, A.M.M.; Miller, E.W. Optical estimation of absolute membrane potential using fluorescence lifetime imaging. eLife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
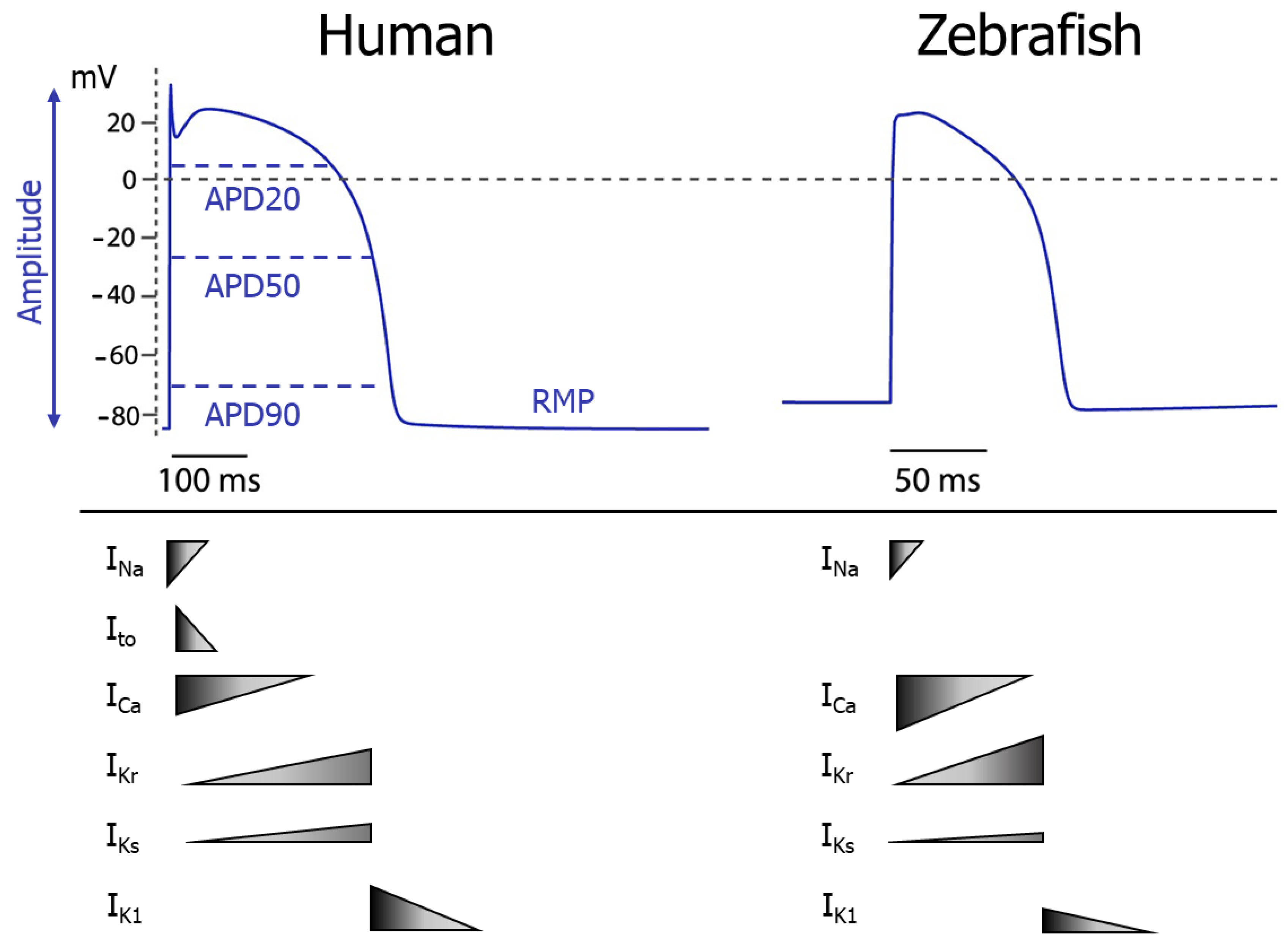

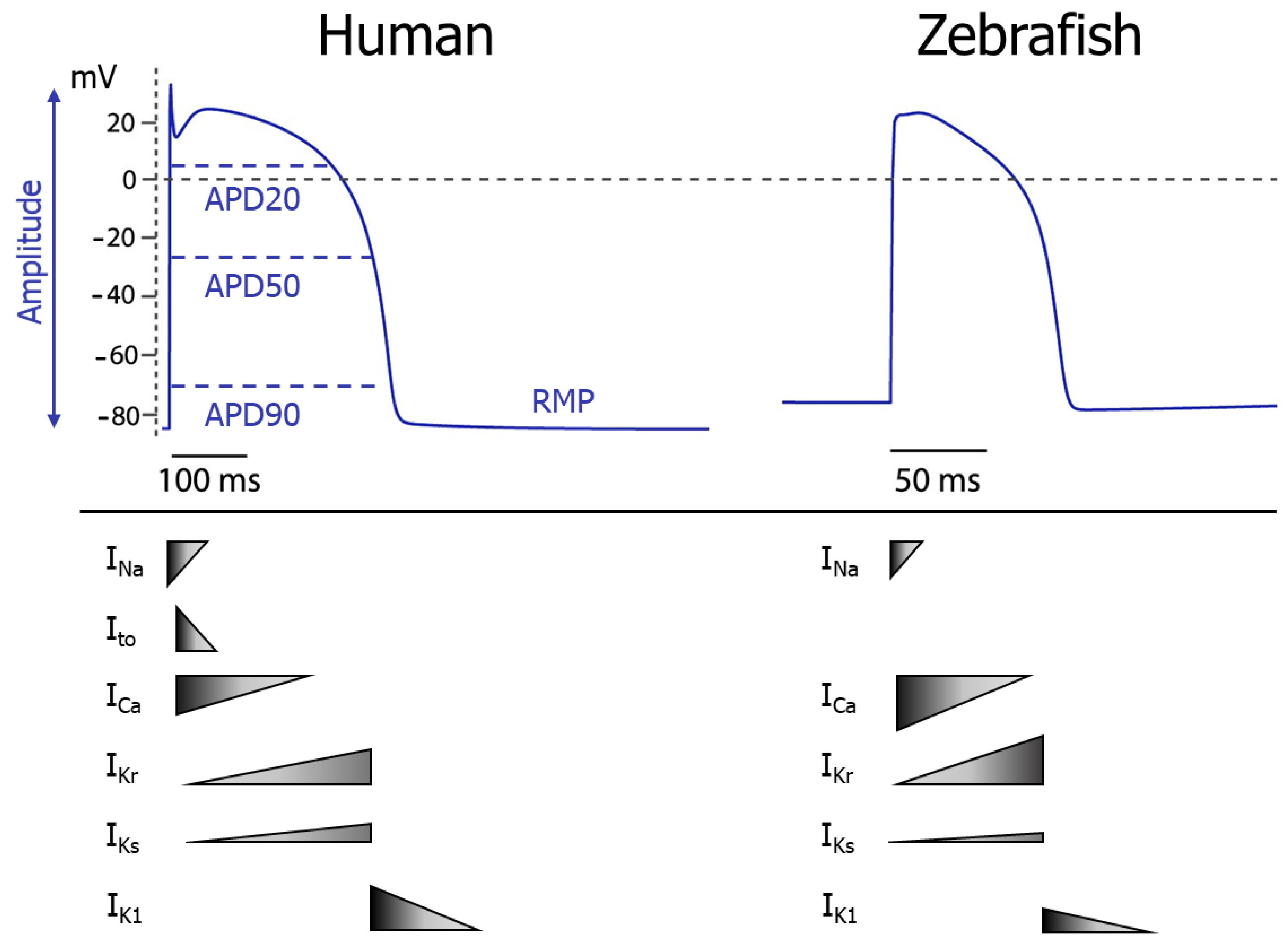{kind=link}
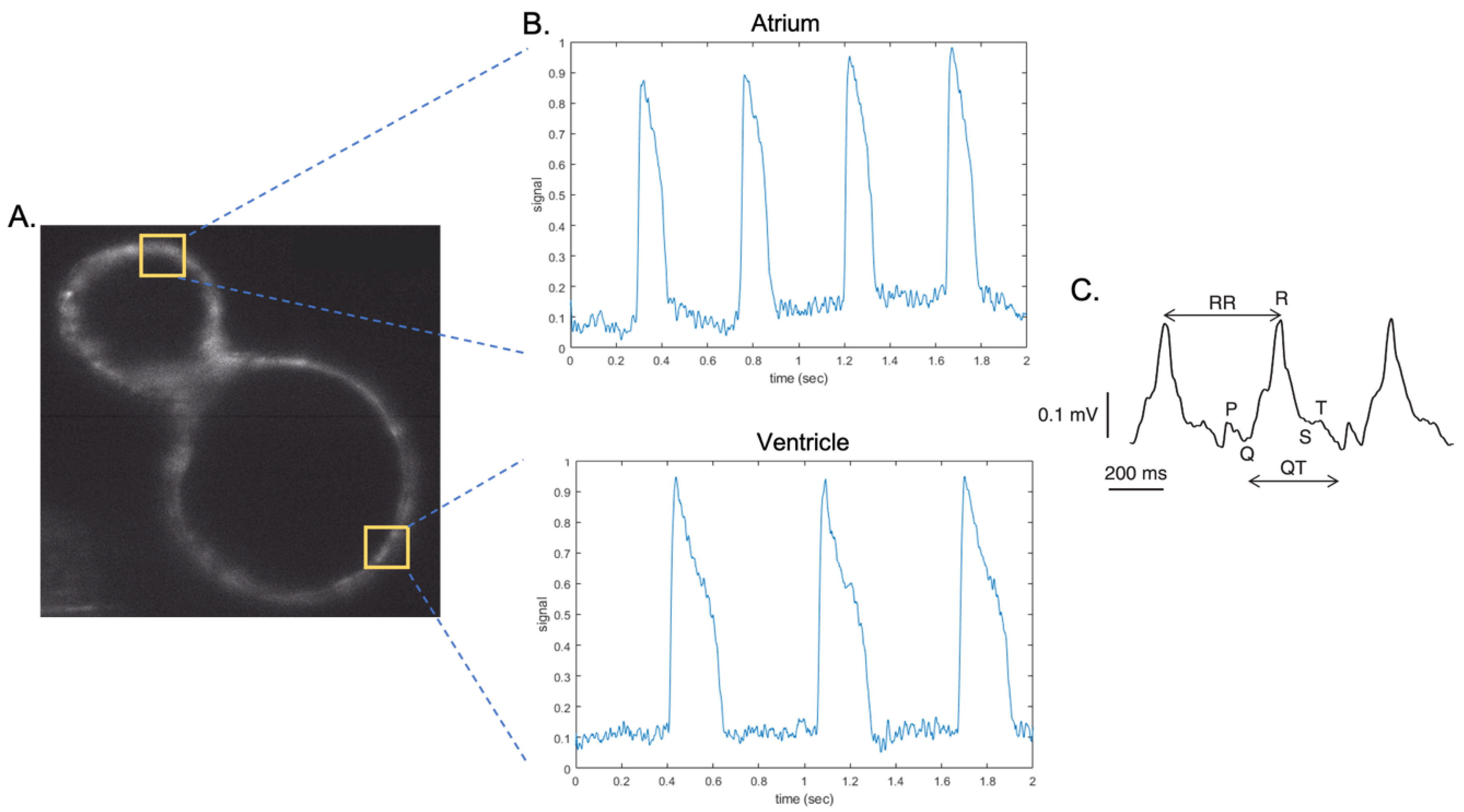
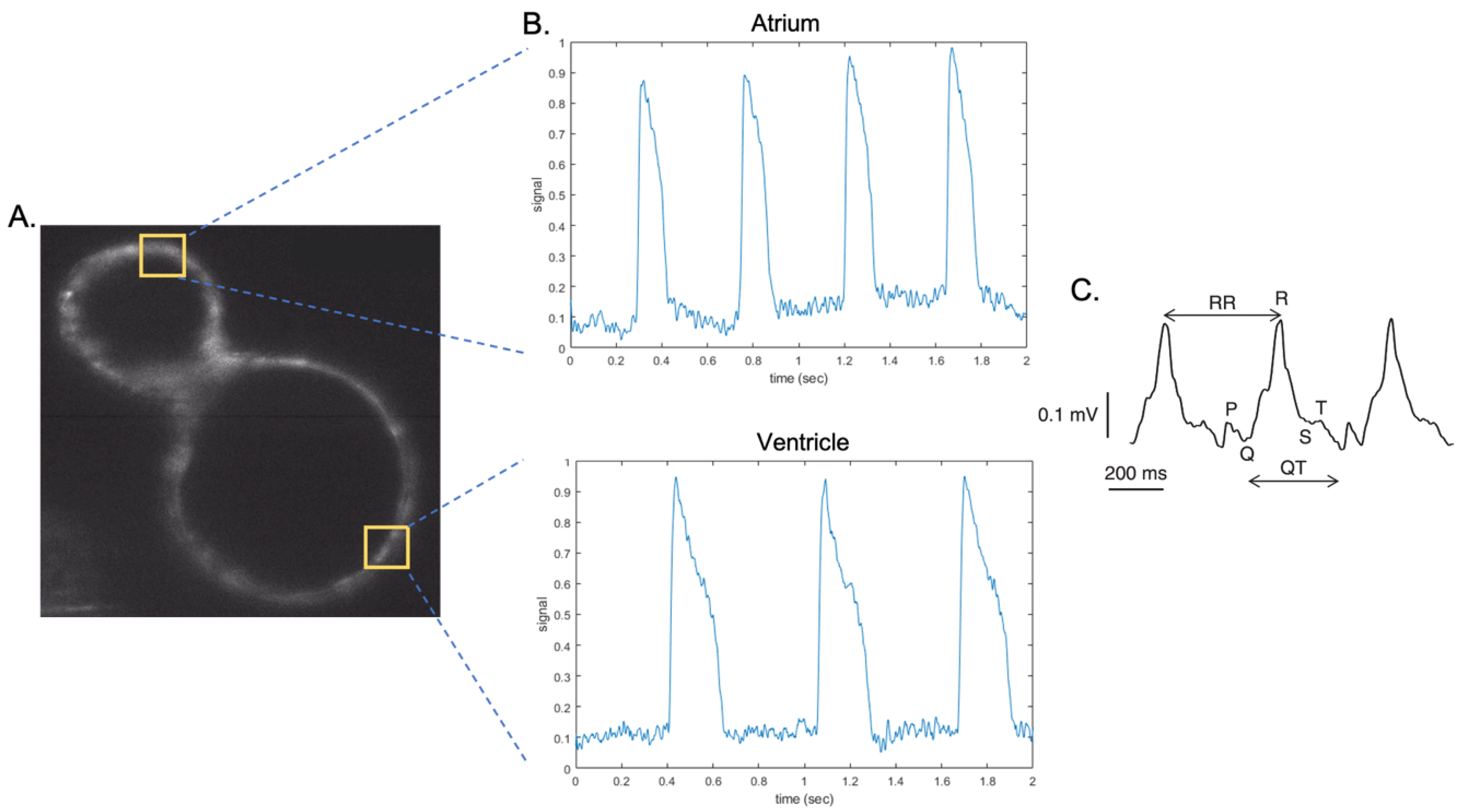{kind=link}
| Method | Advantages | Disadvantages | References |
|---|---|---|---|
| Video recording | Easy to perform | Limited info (heart rate) | hiPSC-CM [76] Zebrafish [77] |
| EFM | Easy to perform Ideal for 2D cell monolayers Add-on on conventional microscopes Available in every lab/university | Background fluorescence Out-of-focus light detection Difficult with 3D structures | hiPSC-CM [78,79,80,81] Zebrafish [82] |
| CLSM | Optical sectioning (3D samples) Little out-of-focus light detection Line scan method Often available in every university | Dedicated set-up High intensity light source Cytotoxic/bleaching (less with line-scan method) | hiPSC-CM [83,84] Zebrafish [62,85] |
| LSFM | Optical sectioning (3D samples) Minimal out-of-focus light detection Illuminating only the field of focus Live specimen imaging | Very dedicated set-up Not available in every institution Lower throughput | hiPSC-CM (organoid) [86] Zebrafish [73,87,88] |
| ICA | Model | Calcium | Voltage | ||
|---|---|---|---|---|---|
| Dye | GECI | Dye | GEVI | ||
| LQTS | hiPSC-CM | Fluo-4 AM [81,111,112,113,114] Fura-2 AM [115] | / | / | Arclight [111,116] |
| Zebrafish | Calcium Green Dextran [82,104] | gCaMP [53] | di-4 ANEPPS [105] | / | |
| BrS | hiPSC-CM | Fluo-4 AM [117,118] Fluo-3 AM [119] | / | / | / |
| CPVT | hiPSC-CM | Fluo-4 AM [83,120,121,122,123,124] Fura-2 AM [125] | GCaMP6f-Junctin [126] | / | / |
| SQTS | hiPSC-CM | Fluo-3 AM [127,128] | FluoVolt [129] | / | / |
| Zebrafish | Calcium Green Dextran [106] | / | / | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vandendriessche, B.; Sieliwonczyk, E.; Alaerts, M.; Loeys, B.L.; Snyders, D.; Schepers, D. Optical Mapping in hiPSC-CM and Zebrafish to Resolve Cardiac Arrhythmias. Hearts 2020, 1, 181-199. https://0-doi-org.brum.beds.ac.uk/10.3390/hearts1030018
Vandendriessche B, Sieliwonczyk E, Alaerts M, Loeys BL, Snyders D, Schepers D. Optical Mapping in hiPSC-CM and Zebrafish to Resolve Cardiac Arrhythmias. Hearts. 2020; 1(3):181-199. https://0-doi-org.brum.beds.ac.uk/10.3390/hearts1030018
Chicago/Turabian StyleVandendriessche, Bert, Ewa Sieliwonczyk, Maaike Alaerts, Bart L. Loeys, Dirk Snyders, and Dorien Schepers. 2020. "Optical Mapping in hiPSC-CM and Zebrafish to Resolve Cardiac Arrhythmias" Hearts 1, no. 3: 181-199. https://0-doi-org.brum.beds.ac.uk/10.3390/hearts1030018