
Novel Insights into the Molecular Mechanisms of Ischemia/Reperfusion Injury in Kidney Transplantation

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Ischemia and Reperfusion
2.1. Ischemia
2.2. Reperfusion
2.3. Endothelial Dysfunction
3. Immune Response
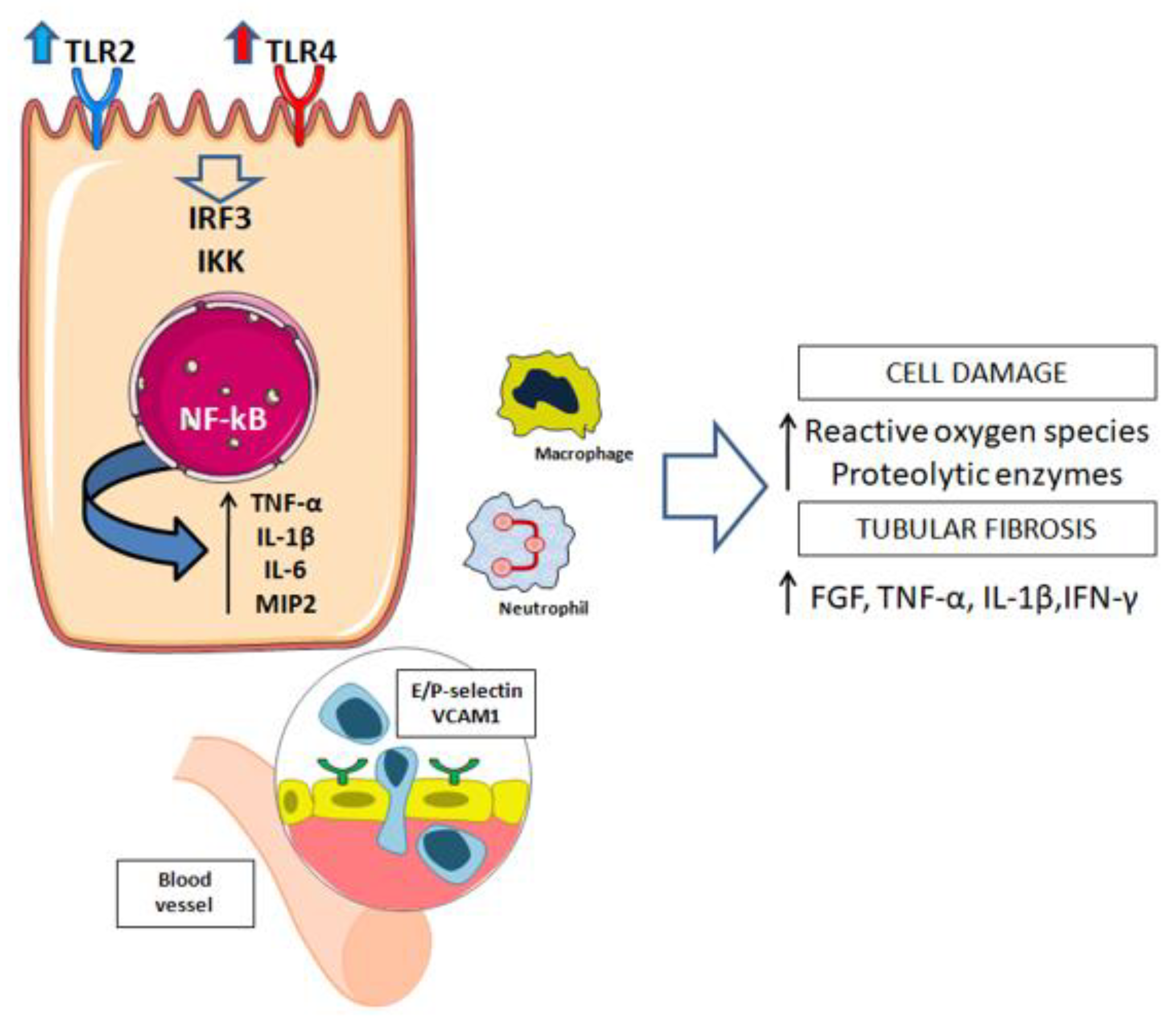
3.1. Innate Immune Response
3.2. Innate to Adaptive Immune Response Translation
3.3. Adaptive Immune Response
4. Complement System
5. Cellular Metabolism Alterations in Ischemia-Reperfusion Injury
6. Potential Prevention and Therapeutic Strategies
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- GBD Chronic Kidney Disease Collaboration. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef] [Green Version]
- Global Observatory of Donation and Transplantation. Available online: http://www.transplant-observatory.org/ (accessed on 15 March 2021).
- Losappio, V.; Stallone, G.; Infante, B.; Schena, A.; Rossini, M.; Maiorano, A.; Fiorentino, M.; Ditonno, P.; Lucarelli, G.; Battaglia, M.; et al. A single-center cohort study to define the role of pretransplant biopsy score in the long-term outcome of kidney transplantation. Transplantation 2014, 97, 934–939. [Google Scholar] [CrossRef]
- Vavallo, A.; Lucarelli, G.; Spilotros, M.; Bettocchi, C.; Palazzo, S.; Selvaggi, F.P.; Battaglia, M.; Ditonno, P. Impact of donor-recipient gender on kidney graft and patient survival: Short- and long-term outcomes. World J. Urol. 2014, 32, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.J.; Courtney, A.E.; Cardwell, C.R.; Maxwell, A.P.; Lucarelli, G.; Veroux, M.; Furriel, F.; Cannon, R.M.; Hoogeveen, E.K.; Doshi, M.; et al. Recipient obesity and outcomes after kidney transplantation: A systematic review and meta-analysis. Nephrol. Dial. Transpl. 2015, 30, 1403–1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Impedovo, S.V.; De Lorenzis, E.; Volpe, A.; Gesualdo, L.; Grandaliano, G.; Palazzo, S.; Lucarelli, G.; Bettocchi, C.; Terrone, C.; Stratta, P.; et al. Middle and long-term outcomes of dual kidney transplant: A multicenter experience. Transpl. Proc. 2013, 45, 1237–1241. [Google Scholar] [CrossRef] [PubMed]
- Lucarelli, G.; Bettocchi, C.; Battaglia, M.; Impedovo, S.V.; Vavallo, A.; Grandaliano, G.; Castellano, G.; Schena, F.P.; Selvaggi, F.P.; Ditonno, P. Extended criteria donor kidney transplantation: Comparative outcome analysis between single versus double kidney transplantation at 5 years. Transpl. Proc. 2010, 42, 1104–1107. [Google Scholar] [CrossRef]
- Ditonno, P.; Lucarelli, G.; Impedovo, S.V.; Spilotros, M.; Grandaliano, G.; Selvaggi, F.P.; Bettocchi, C.; Battaglia, M. Obesity in kidney transplantation affects renal function but not graft and patient survival. Transpl. Proc. 2011, 43, 367–372. [Google Scholar] [CrossRef]
- Querard, A.H.; Foucher, Y.; Combescure, C.; Dantan, E.; Larmet, D.; Lorent, M.; Pouteau, L.; Giral, M.; Gillaizeau, F. Comparison of survival outcomes between Expanded Criteria Donor and Standard Criteria Donor kidney transplant recipients: A systematic review and meta-analysis. Transpl. Int. 2016, 29, 403–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heylen, L.; Pirenne, J.; Samuel, U.; Tieken, I.; Naesens, M.; Sprangers, B.; Jochmans, I. The Impact of Anastomosis Time During Kidney Transplantation on Graft Loss: A Eurotransplant Cohort Study. Am. J. Transpl. 2017, 17, 724–732. [Google Scholar] [CrossRef]
- Summers, D.M.; Watson, C.J.; Pettigrew, G.J.; Johnson, R.J.; Collett, D.; Neuberger, J.M.; Bradley, J.A. Kidney donation after circulatory death (DCD): State of the art. Kidney Int. 2015, 88, 241–249. [Google Scholar] [CrossRef]
- Breda, A.; Lucarelli, G.; Rodriguez-Faba, O.; Guirado, L.; Facundo, C.; Bettocchi, C.; Gesualdo, L.; Castellano, G.; Grandaliano, G.; Battaglia, M.; et al. Clinical and pathological outcomes of renal cell carcinoma (RCC) in native kidneys of patients with end-stage renal disease: A long-term comparative retrospective study with RCC diagnosed in the general population. World J. Urol. 2015, 33, 1–7, Correction in 2015, 33, 9. [Google Scholar] [CrossRef] [PubMed]
- Lucarelli, G.; Ditonno, P. Editorial comment from Dr Lucarelli and Dr Ditonno to Impact of graft nephrectomy on outcomes of second kidney transplantation. Int. J. Urol. 2014, 21, 802–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucarelli, G.; Vavallo, A.; Bettocchi, C.; Losappio, V.; Gesualdo, L.; Grandaliano, G.; Selvaggi, F.P.; Battaglia, M.; Ditonno, P. Impact of transplant nephrectomy on retransplantation: A single-center retrospective study. World J. Urol. 2013, 31, 959–963. [Google Scholar] [CrossRef]
- Vavallo, A.; Lucarelli, G.; Bettocchi, C.; Tedeschi, M.; Palazzo, S.; Losappio, V.; Gesualdo, L.; Grandaliano, G.; Selvaggi, F.P.; Battaglia, M.; et al. Allograft nephrectomy: What is the best surgical technique? Transpl. Proc. 2012, 44, 1922–1925. [Google Scholar] [CrossRef] [PubMed]
- Ponticelli, C. Ischemia-reperfusion injury: A major protagonist in kidney transplantation. Nephrol. Dial. Transpl. 2014, 29, 1134–1140. [Google Scholar] [CrossRef]
- Cooper, J.E.; Wiseman, A.C. Acute kidney injury in kidney transplantation. Curr. Opin. Nephrol. Hypertens. 2013, 22, 698–703. [Google Scholar] [CrossRef]
- Salvadori, M.; Rosso, G.; Bertoni, E. Update on ischemia-reperfusion injury in kidney transplantation: Pathogenesis and treatment. World J. Transpl. 2015, 5, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Edelstein, C.L.; Ling, H.; Schrier, R.W. The nature of cell injury. Kidney Int. 1997, 51, 1341–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, L.B. New concepts in reactive oxygen species and cardiovascular reperfusion physiology. Cardiovasc. Res. 2004, 61, 461–470. [Google Scholar] [CrossRef]
- Alkaitis, M.S.; Crabtree, M.J. Recoupling the cardiac nitric oxide synthases: Tetrahydrobiopterin synthesis and recycling. Curr. Heart Fail. Rep. 2012, 9, 200–210. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Jackson, R.M. Reactive species mechanisms of cellular hypoxia-reoxygenation injury. Am. J. Physiol. Cell Physiol. 2002, 282, C227–C241. [Google Scholar] [CrossRef] [Green Version]
- Simone, S.; Rascio, F.; Castellano, G.; Divella, C.; Chieti, A.; Ditonno, P.; Battaglia, M.; Crovace, A.; Stafferi, F.; Oortwijn, B.; et al. Complement-dependent NADPH oxidase enzyme activation in renal ischemia/reperfusion injury. Free Radic. Biol. Med. 2014, 74, 263–273. [Google Scholar] [CrossRef]
- Martin, J.L.; Gruszczyk, A.V.; Beach, T.E.; Murphy, M.P.; Saeb-Parsy, K. Mitochondrial mechanisms and therapeutics in ischaemia reperfusion injury. Pediatr. Nephrol. 2019, 34, 1167–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinlich, R.; Oberst, A.; Beere, H.M.; Green, D.R. Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Faller, D.V. Endothelial cell responses to hypoxic stress. Clin. Exp. Pharmacol. Physiol. 1999, 26, 74–84. [Google Scholar] [CrossRef]
- Kwon, O.; Hong, S.M.; Ramesh, G. Diminished NO generation by injured endothelium and loss of macula densa nNOS may contribute to sustained acute kidney injury after ischemia-reperfusion. Am. J. Physiol. Renal. Physiol. 2009, 296, 25–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonventre, J.V.; Yang, L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Investig. 2011, 121, 4210–4221. [Google Scholar] [CrossRef]
- Legrand, M.; Mik, E.G.; Johannes, T.; Payen, D.; Ince, C. Renal hypoxia and dysoxia after reperfusion of the ischemic kidney. Mol. Med. 2008, 14, 502–516. [Google Scholar] [CrossRef]
- Kwon, O.; Hong, S.M.; Sutton, T.A.; Temm, C.J. Preservation of peritubular capillary endothelial integrity and increasing pericytes may be critical to recovery from postischemic acute kidney injury. Am. J. Physiol. Renal. Physiol. 2008, 295, F351–F359. [Google Scholar] [CrossRef] [Green Version]
- Curci, C.; Castellano, G.; Stasi, A.; Divella, C.; Loverre, A.; Gigante, M.; Simone, S.; Cariello, M.; Montinaro, V.; Lucarelli, G.; et al. Endothelial-to-mesenchymal transition and renal fibrosis in ischaemia/reperfusion injury are mediated by complement anaphylatoxins and Akt pathway. Nephrol. Dial. Transpl. 2014, 29, 799–808. [Google Scholar] [CrossRef] [Green Version]
- Eltzschig, H.K.; Collard, C.D. Vascular ischemia and reperfusion injury. Br. Med. Bull. 2004, 70, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Carden, D.L.; Granger, D.N. Pathophysiology of ischemia reperfusion injury. J. Pathol. 2000, 190, 255–266. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Bowie, A.G. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar] [CrossRef]
- Delneste, Y.; Beauvillain, C.; Jeannin, P. Innate immunity: Structure and function of TLRs. Med. Sci. 2007, 23, 67–73. [Google Scholar]
- Kawasaki, T.; Kawai, T. Toll-Like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- Rusai, K.; Sollinger, D.; Baumann, M.; Wagner, B.; Strobl, M.; Schmaderer, C.; Roos, M.; Kirschning, C.; Heemann, U.; Lutz, J. Toll-like receptors 2 and 4 in renal ischemia/reperfusion injury. Pediatr. Nephrol. 2010, 25, 853–860. [Google Scholar] [CrossRef]
- Kruger, B.; Krick, S.; Dhillon, N.; Lerner, S.M.; Ames, S.; Bromberg, J.S.; Lin, M.; Walsh, L.; Vella, J.; Fischereder, M.; et al. Donor Toll-like receptor 4 contributes to ischemia and reperfusion injury following human kidney transplantation. Proc. Natl. Acad. Sci. USA 2009, 106, 3390–3395. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Chen, G.; Wyburn, K.R.; Yin, J.; Bertolino, P.; Eris, J.M.; Alexander, S.I.; Sharland, A.F.; Chadban, S.J. TLR4 activation mediates kidney ischemia/reperfusion injury. J. Clin. Investig. 2007, 117, 2847–2859. [Google Scholar] [CrossRef]
- Chen, J.; John, R.; Richardson, J.A.; Shelton, J.M.; Zhou, X.J.; Wang, Y.; Wu, Q.Q.; Hartono, J.R.; Winterberg, P.D.; Lu, C.Y. Toll-like receptor 4 regulates early endothelial activation during ischemic acute kidney injury. Kidney Int. 2011, 79, 288–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, H.R.; Rabb, H. The innate immune response in ischemic acute kidney injury. Clin. Immunol. 2009, 130, 41–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, S.K.; Sung, S.A.; Cho, W.Y.; Go, K.J.; Kim, H.K. Macrophages contribute to the initiation of ischemic acute renal failure in rats. Nephrol. Dial. Transpl. 2006, 21, 1231–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y. Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 2011, 7, 684–696. [Google Scholar] [CrossRef]
- Leemans, J.C.; Butter, L.M.; Pulskens, W.P.; Teske, G.J.D.; Claessen, N.; van der Poll, T.; Florquin, S. The role of Toll-like receptor 2 in inflammation and fibrosis during progressive renal injury. PLoS ONE 2009, 4, e5704. [Google Scholar] [CrossRef]
- Leemans, J.C.; Stokman, G.; Claessen, N.; Rouschop, K.M.; Teske, G.J.D.; Kirschning, C.J.; Akira, S.; van der Poll, T.; Weening, J.J.; Florquin, S. Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J. Clin. Investig. 2005, 115, 2894–2903. [Google Scholar] [CrossRef] [Green Version]
- Reilly, M.; Miller, R.M.; Thomson, M.H.; Patris, V.; Ryle, P.; McLoughlin, L.; Mutch, P.; Gilboy, P.; Miller, C.; Broekema, M.; et al. Randomized, double-blind, placebo-controlled, dose-escalating phase I, healthy subjects study of intravenous OPN-305, a humanized Anti-TLR2 antibody. Clin. Pharmacol. Ther. 2013, 94, 593–600. [Google Scholar] [CrossRef] [Green Version]
- Snelgrove, S.L.; Lo, C.; Hall, P.; Lo, C.Y.; Alikhan, M.A.; Coates, P.T.; Holdsworth, S.R.; Hickey, M.J.; Kitching, A.R. Activated Renal Dendritic Cells Cross Present Intrarenal Antigens After Ischemia-Reperfusion Injury. Transplantation 2017, 101, 1013–1024. [Google Scholar] [CrossRef] [PubMed]
- Damman, J.; Daha, M.R.; van Son, W.J.; Leuvenink, H.G.; Ploeg, R.J.; Seelen, M.A. Crosstalk between complement and Toll-like receptor activation in relation to donor brain death and renal ischemia-reperfusion injury. Am. J. Transpl. 2011, 11, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Batal, I.; De Serres, S.A.; Safa, K.; Bijol, V.; Ueno, T.; Onozato, M.L.; Iafrate, A.J.; Herter, J.M.; Lichtman, A.H.; Mayadas, T.N.; et al. Dendritic Cells in Kidney Transplant Biopsy Samples Are Associated with T Cell Infiltration and Poor Allograft Survival. J. Am. Soc. Nephrol. 2015, 26, 3102–3113. [Google Scholar] [CrossRef] [Green Version]
- Bajwa, A.; Huang, L.; Ye, H.; Dondeti, K.; Song, S.; Rosin, D.L.; Lynch, K.R.; Lobo, P.I.; Li, L.; Okusa, M.D. Dendritic cell sphingosine 1-phosphate receptor-3 regulates Th1-Th2 polarity in kidney ischemia-reperfusion injury. J. Immunol. 2012, 189, 2584–2596. [Google Scholar] [CrossRef] [Green Version]
- Benichou, G.; Thomson, A.W. Direct versus indirect allorecognition pathways: On the right track. Am. J. Transpl. 2009, 9, 655–656. [Google Scholar] [CrossRef] [Green Version]
- Ysebaert, D.K.; De Greef, K.E.; De Beuf, A.; Van Rompay, A.R.; Vercauteren, S.; Persy, V.P.; De Broe, M.E. T-cells as mediators in renal ischemia/reperfusion injury. Kidney Int. 2004, 66, 491–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Perrot, M.; Young, K.; Imai, Y.; Liu, M.; Waddell, T.K.; Fischer, S.; Zhang, L.; Keshavjee, S. Recipient T cells mediate reperfusion injury after lung transplantation in the rat. J. Immunol. 2003, 171, 4995–5002. [Google Scholar] [CrossRef] [Green Version]
- Fiorina, P.; Ansari, M.J.; Jurewicz, M.; Barry, M.; Ricchiuti, V.; Smith, R.N.; Shea, S.; Means, T.K.; Auchincloss, H., Jr.; Luster, A.D.; et al. Role of CXC chemokine receptor 3 pathway in renal ischemic injury. J. Am. Soc. Nephrol. 2006, 17, 716–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabb, H. The T cell as a bridge between innate and adaptive immune systems: Implications for the kidney. Kidney Int. 2002, 61, 1935–1946. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.W.; Bae, E.; Kwon, S.H.; Yu, M.Y.; Cha, R.H.; Lee, H.; Kim, D.K.; Lee, J.P.; Ye, S.K.; Yoo, J.Y.; et al. Transcriptional modulation of the T helper 17/interleukin 17 axis ameliorates renal ischemia-reperfusion injury. Nephrol. Dial. Transpl. 2019, 34, 1481–1498. [Google Scholar] [CrossRef]
- Loverre, A.; Divella, C.; Castellano, G.; Tataranni, T.; Zaza, G.; Rossini, M.; Ditonno, P.; Battaglia, M.; Palazzo, S.; Gigante, M.; et al. T helper 1, 2 and 17 cell subsets in renal transplant patients with delayed graft function. Transpl. Int. 2011, 24, 233–242. [Google Scholar] [CrossRef]
- Afzali, B.; Lombardi, G.; Lechler, R.I.; Lord, G.M. The role of T helper 17 (Th17) and regulatory T cells (Treg) in human organ transplantation and autoimmune disease. Clin. Exp. Immunol. 2007, 148, 32–46. [Google Scholar] [CrossRef] [Green Version]
- Loong, C.C.; Hsieh, H.G.; Lui, W.Y.; Chen, A.; Lin, C.Y. Evidence for the early involvement of interleukin 17 in human and experimental renal allograft rejection. J. Pathol. 2002, 197, 322–332. [Google Scholar] [CrossRef]
- Tang, J.L.; Subbotin, V.M.; Antonysamy, M.A.; Troutt, A.B.; Rao, A.S.; Thomson, A.W. Interleukin-17 antagonism inhibits acute but not chronic vascular rejection. Transplantation 2001, 72, 348–350. [Google Scholar] [CrossRef]
- Ko, G.J.; Linfert, D.; Jang, H.R.; Higbee, E.; Watkins, T.; Cheadle, C.; Liu, M.; Racusen, L.; Grigoryev, D.N.; Rabb, H. Transcriptional analysis of infiltrating T cells in kidney ischemia-reperfusion injury reveals a pathophysiological role for CCR5. Am. J. Physiol. Renal. Physiol. 2012, 302, F762–F773. [Google Scholar] [CrossRef] [Green Version]
- Waldmann, H.; Hilbrands, R.; Howie, D.; Cobbold, S. Harnessing FOXP3þ regulatory T cells for transplantation tolerance. J. Clin. Investig. 2014, 124, 1439–1445. [Google Scholar] [CrossRef] [Green Version]
- Wood, K.J.; Sakaguchi, S. Regulatory T cells in transplantation tolerance. Nat. Rev. Rheumatol. 2003, 3, 199–210. [Google Scholar] [CrossRef]
- Ferrer, I.R.; Hester, J.; Bushell, A.; Wood, K.J. Induction of transplantation tolerance through regulatory cells: From mice to men. Immunol. Rev. 2014, 258, 102–116. [Google Scholar] [CrossRef]
- Kinsey, G.; Sharma, R.; Huang, L.; Li, L.; Vergis, A.L.; Ye, H.; Ju, S.T.; Okusa, M.D. Regulatory T Cells Suppress Innate Immunity in Kidney Ischemia-Reperfusion Injury. J. Am. Soc. Nephrol. 2009, 20, 1744–1753. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Wang, Y.M.; Wang, Y.; Zhang, G.Y.; Zheng, G.; Yi, S.; O’Connell, P.J.; Harris, D.C.; Alexander, S.I. Regulatory T cells in kidney disease and transplantation. Kidney Int. 2016, 90, 502–514. [Google Scholar] [CrossRef] [PubMed]
- Braza, F.; Dugast, E.; Panov, I.; Paul, C.; Vogt, K.; Pallier, A.; Chesneau, M.; Baron, D.; Guerif, P.; Lei, H.; et al. Central role of CD45RA- Foxp3hi memory regulatory T cells in clinical kidney transplantation tolerance. J. Am. Soc. Nephrol. 2015, 26, 1795–1805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duhen, T.; Duhen, R.; Lanzavecchia, A.; Sallusto, F.; Campbell, D.J. Functionally distinct subsets of human FOXP3 Treg cells that phenotypically mirror effector Th cells. Blood 2012, 119, 4430–4440. [Google Scholar] [CrossRef] [Green Version]
- Kawai, M.; Kitade, H.; Mathieu, C.; Waer, M.; Pirenne, J. Inhibitory and stimulatory effects of cyclosporine A on the development of regulatory T cells in vivo. Transplantation 2005, 79, 1073–1077. [Google Scholar] [CrossRef] [PubMed]
- Nauser, C.L.; Farrar, C.A.; Sacks, S.H. Complement Recognition Pathways in Renal Transplantation. J. Am. Soc. Nephrol. 2017, 28, 2571–2578. [Google Scholar] [CrossRef]
- Farrar, C.A.; Tran, D.; Li, K.; Wu, W.; Peng, Q.; Schwaeble, W.; Zhou, W.; Sacks, S.H. Collectin-11 detects stress-induced l-fucose pattern to trigger renal epithelial injury. J. Clin. Investig. 2016, 126, 1911–1925. [Google Scholar] [CrossRef] [Green Version]
- Asgari, E.; Farrar, C.A.; Lynch, N.; Ali, Y.M.; Roscher, S.; Stover, C.; Zhou, W.; Schwaeble, W.J.; Sacks, S.H. Mannan-binding lectin-associated serine protease 2 is critical for the development of renal ischemia reperfusion injury and mediates tissue injury in the absence of complement C4. FASEB J. 2014, 28, 3996–4003. [Google Scholar] [CrossRef]
- Wallis, R. Interactions between mannose binding lectin and MASPs during complement activation by the lectin pathway. Immunobiology 2007, 212, 289–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janewit, D.; Surovtseva, Y.V.; Qin, L.; Li, G.; Liu, R.; Clark, P.; Manes, T.D.; Wang, C.; Kashgarian, M.; Kirkiles-Smith, N.C.; et al. Complement membrane attack complexes activate noncanonical NF-kB by forming an Akt+ NIK+ signalosome on Rab5+ endosomes. Proc. Natl. Acad. Sci. USA 2015, 112, 9686–9969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanaka, K.; Kakuta, Y.; Miyagawa, S.; Nakazawa, S.; Kato, T.; Abe, T.; Imamura, R.; Okumi, M.; Maeda, A.; Okuyama, H.; et al. Depression of complement regulatory factors in rat and human renal grafts is associated with the progress of acute T-cell mediated rejection. PLoS ONE 2016, 11, e0148881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellano, G.; Franzin, R.; Stasi, A.; Divella, C.; Sallustio, F.; Pontrelli, P.; Lucarelli, G.; Battaglia, M.; Staffieri, F.; Crovace, A.; et al. Complement Activation During Ischemia/ Reperfusion Injury Induces Pericyte-to-Myofibroblast Transdifferentiation Regulating Peritubular Capillary Lumen Reduction Through pERK Signaling. Front. Immunol. 2018, 9, 1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Divella, C.; Stasi, A.; Franzin, R.; Rossini, M.; Pontrelli, P.; Sallustio, F.; Netti, G.S.; Ranieri, E.; Lacitignola, L.; Staffieri, F.; et al. Pentraxin-3-mediated complement activation in a swine model of renal ischemia/reperfusion injury. Aging 2021, 13, 10920–10933. [Google Scholar] [CrossRef] [PubMed]
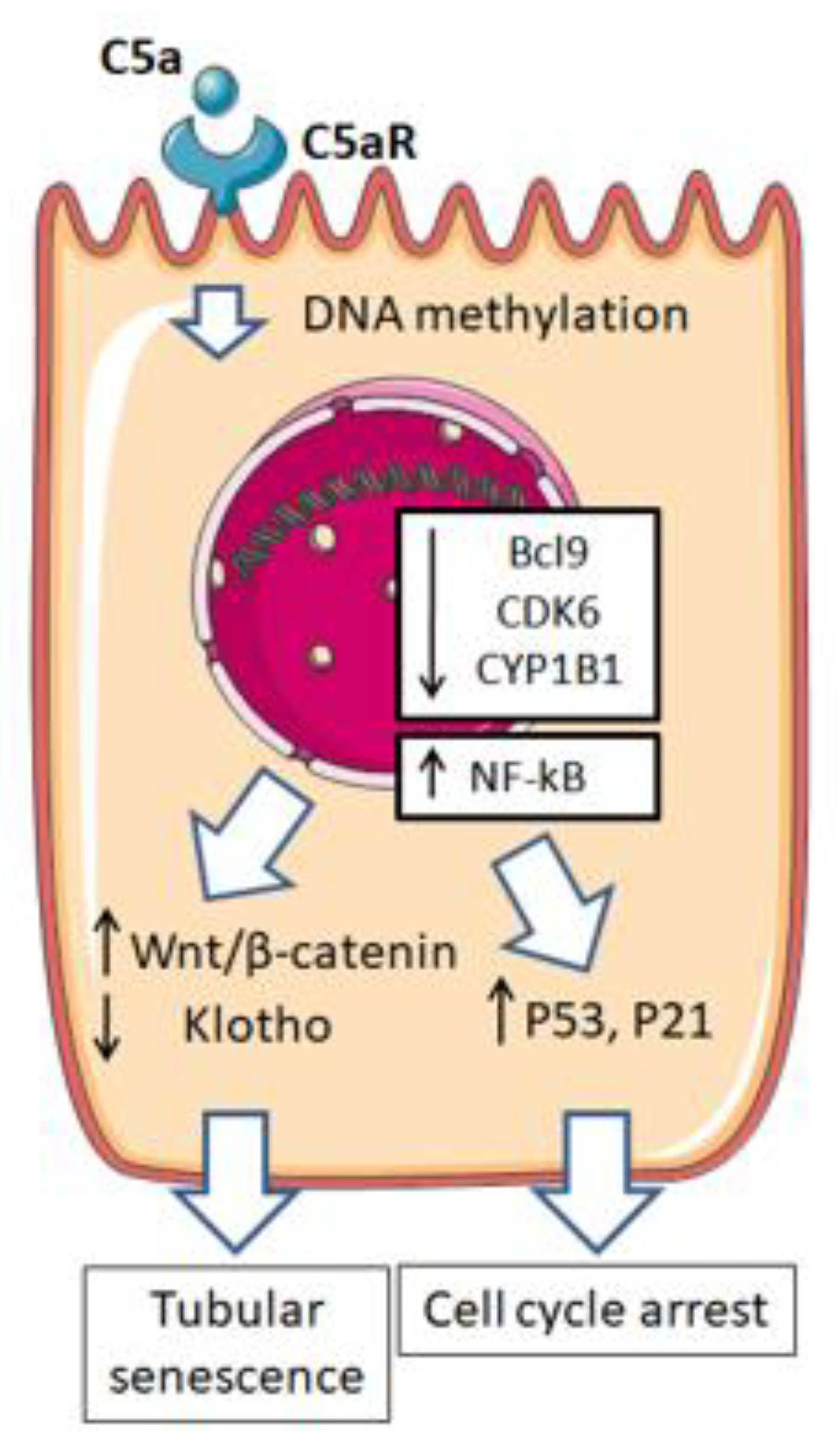
- Castellano, G.; Franzin, R.; Sallustio, F.; Stasi, A.; Banelli, B.; Romani, M.; De Palma, G.; Lucarelli, G.; Divella, C.; Battaglia, M.; et al. Complement component C5a induces aberrant epigenetic modifications in renal tubular epithelial cells accelerating senescence by Wnt4/βcatenin signaling after ischemia/reperfusion injury. Aging 2019, 11, 4382–4406. [Google Scholar] [CrossRef]
- Castellano, G.; Intini, A.; Stasi, A.; Divella, C.; Gigante, M.; Pontrelli, P.; Franzin, R.; Accetturo, M.; Zito, A.; Fiorentino, M.; et al. Complement Modulation of Anti-Aging Factor Klotho in Ischemia/Reperfusion Injury and Delayed Graft Function. Am. J. Transpl. 2016, 16, 325–333. [Google Scholar] [CrossRef]
- Lucarelli, G.; Galleggiante, V.; Rutigliano, M.; Sanguedolce, F.; Cagiano, S.; Bufo, P.; Lastilla, G.; Maiorano, E.; Ribatti, D.; Giglio, A.; et al. Metabolomic profile of glycolysis and the pentose phosphate pathway identifies the central role of glucose-6-phosphate dehydrogenase in clear cell-renal cell carcinoma. Oncotarget 2015, 6, 13371–13386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucarelli, G.; Rutigliano, M.; Galleggiante, V.; Giglio, A.; Palazzo, S.; Ferro, M.; Simone, C.; Bettocchi, C.; Battaglia, M.; Ditonno, P. Metabolomic profiling for the identification of novel diagnostic markers in prostate cancer. Expert Rev. Mol. Diagn. 2015, 15, 1211–1224. [Google Scholar] [CrossRef]
- Lucarelli, G.; Rutigliano, M.; Sanguedolce, F.; Galleggiante, V.; Giglio, A.; Cagiano, S.; Bufo, P.; Maiorano, E.; Ribatti, D.; Ranieri, E.; et al. Increased Expression of the Autocrine Motility Factor is Associated with Poor Prognosis in Patients with Clear Cell-Renal Cell Carcinoma. Medicine 2015, 94, e2117. [Google Scholar] [CrossRef]
- Ragone, R.; Sallustio, F.; Piccinonna, S.; Rutigliano, M.; Vanessa, G.; Palazzo, S.; Lucarelli, G.; Ditonno, P.; Battaglia, M.; Fanizzi, F.P.; et al. Renal Cell Carcinoma: A Study through NMR-Based Metabolomics Combined with Transcriptomics. Diseases 2016, 4, 7. [Google Scholar] [CrossRef]
- Lucarelli, G.; Rutigliano, M.; Ferro, M.; Giglio, A.; Intini, A.; Triggiano, F.; Palazzo, S.; Gigante, M.; Castellano, G.; Ranieri, E.; et al. Activation of the kynurenine pathway predicts poor outcome in patients with clear cell renal cell carcinoma. Urol. Oncol. 2017, 35, 461.e15–461.e27. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, C.; Meregalli, C.; Bombelli, S.; Di Stefano, V.; Salerno, F.; Torsello, B.; De Marco, S.; Bovo, G.; Cifola, I.; Mangano, E.; et al. The glucose and lipid metabolism reprogramming is grade-dependent in clear cell renal cell carcinoma primary cultures and is targetable to modulate cell viability and proliferation. Oncotarget 2017, 8, 113502–113515. [Google Scholar] [CrossRef] [Green Version]
- Lucarelli, G.; Loizzo, D.; Franzin, R.; Battaglia, S.; Ferro, M.; Cantiello, F.; Castellano, G.; Bettocchi, C.; Ditonno, P.; Battaglia, M. Metabolomic insights into pathophysiological mechanisms and biomarker discovery in clear cell renal cell carcinoma. Expert Rev. Mol. Diagn. 2019, 19, 397–407. [Google Scholar] [CrossRef]
- Lucarelli, G.; Loizzo, D.; Ferro, M.; Rutigliano, M.; Vartolomei, M.D.; Cantiello, F.; Buonerba, C.; Di Lorenzo, G.; Terracciano, D.; De Cobelli, O.; et al. Metabolomic profiling for the identification of novel diagnostic markers and therapeutic targets in prostate cancer: An update. Expert Rev. Mol. Diagn. 2019, 19, 377–387. [Google Scholar] [CrossRef]
- Lucarelli, G.; Rutigliano, M.; Sallustio, F.; Ribatti, D.; Giglio, A.; Lepore Signorile, M.; Grossi, V.; Sanese, P.; Napoli, A.; Maiorano, E.; et al. Integrated multi-omics characterization reveals a distinctive metabolic signature and the role of NDUFA4L2 in promoting angiogenesis, chemoresistance, and mitochondrial dysfunction in clear cell renal cell carcinoma. Aging 2018, 10, 3957–3985. [Google Scholar] [CrossRef]
- Bombelli, S.; Torsello, B.; De Marco, S.; Lucarelli, G.; Cifola, I.; Grasselli, C.; Strada, G.; Bovo, G.; Perego, R.A.; Bianchi, C. 36-kDa Annexin A3 Isoform Negatively Modulates Lipid Storage in Clear Cell Renal Cell Carcinoma Cells. Am. J. Pathol. 2020, 190, 2317–2326. [Google Scholar] [CrossRef] [PubMed]
- Lucarelli, G.; Ferro, M.; Loizzo, D.; Bianchi, C.; Terracciano, D.; Cantiello, F.; Bell, L.N.; Battaglia, S.; Porta, C.; Gernone, A.; et al. Integration of Lipidomics and Transcriptomics Reveals Reprogramming of the Lipid Metabolism and Composition in Clear Cell Renal Cell Carcinoma. Metabolites 2020, 10, 509. [Google Scholar] [CrossRef]
- Ferro, M.; Terracciano, D.; Buonerba, C.; Lucarelli, G.; Bottero, D.; Perdonà, S.; Autorino, R.; Serino, A.; Cantiello, F.; Damiano, R.; et al. The emerging role of obesity, diet and lipid metabolism in prostate cancer. Future Oncol. 2017, 13, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Lucarelli, G.; Ferro, M.; Battaglia, M. Multi-omics approach reveals the secrets of metabolism of clear cell-renal cell carcinoma. Transl. Androl. Urol. 2016, 5, 801–803. [Google Scholar] [CrossRef] [Green Version]
- Barin-Le Guellec, C.; Largeau, B.; Bon, D.; Marquet, P.; Hauet, T. Ischemia/reperfusion-associated tubular cells injury in renal transplantation: Can metabolomics inform about mechanisms and help identify new therapeutic targets? Pharmacol. Res. 2018, 129, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Guy, A.J.; Nath, J.; Cobbold, M.; Ludwig, C.; Tennant, D.A.; Inston, N.G.; Ready, A.R. Metabolomic analysis of perfusate during hypothermic machine perfusion of human cadaveric kidneys. Transplantation 2015, 99, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Malagrino, P.A.; Venturini, G.; Yogi, P.S.; Dariolli, R.; Padilha, K.; Kiers, B.; Gois, T.C.; Motta-Leal-Filho, J.M.; Takimura, C.K.; Girardi, A.C.C.; et al. Metabolomic characterization of renal ischemia and reperfusion in a swine model. Life Sci. 2016, 156, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Domański, L.; Safranow, K.; Ostrowski, M.; Pawlik, A.; Olszewska, M.; Dutkiewicz, G.; Ciechanowski, K. Oxypurine and purine nucleoside concentrations in renal vein of allograft are potential markers of energy status of renal tissue. Arch. Med. Res. 2007, 38, 240–246. [Google Scholar] [CrossRef]
- Zhang, J.; Wei, X.; Tang, Z.; Miao, B.; Luo, H.; Hu, X.; Luo, Y.; Zhou, Y.; Na, N. Elucidating the molecular pathways and immune system transcriptome during ischemia-reperfusion injury in renal transplantation. Int. Immunopharmacol. 2020, 81, 106246. [Google Scholar] [CrossRef] [PubMed]
- Glotz, D.; Russ, G.; Rostaing, L.; Legendre, C.; Tufveson, G.; Chadban, S.; Grinyó, J.; Mamode, N.; Rigotti, P.; Couzi, L.; et al. Safety and effcacy of eculizumab for the prevention of antibody-mediated rejection after deceased-donor kidney transplantation in patients with preformed donor-specific antibodies. Am. J. Transpl. 2019, 19, 2865–2875. [Google Scholar] [CrossRef] [Green Version]
- Marks, W.H.; Mamode, N.; Montgomery, R.A.; Stegall, M.D.; Ratner, L.E.; Cornell, L.D.; Rowshani, A.T.; Colvin, R.B.; Dain, B.; Boice, J.A.; et al. Safety and efficacy of eculizumab in the prevention of antibody-mediated rejection in living-donor kidney transplant recipients requiring desensitization therapy: A randomized trial. Am. J. Transpl. 2019, 19, 2876–2888. [Google Scholar] [CrossRef] [Green Version]
- Schröppel, B.; Akalin, E.; Baweja, M.; Bloom, R.D.; Florman, S.; Goldstein, M.; Haydel, B.; Hricik, D.E.; Kulkarni, S.; Levine, M.; et al. Peritransplant eculizumab does not prevent delayed graft function in deceased donor kidney transplant recipients: Results of two randomized controlled pilot trials. Am. J. Transpl. 2020, 20, 564–572. [Google Scholar] [CrossRef]
- Jordan, S.C.; Kucher, K.; Bagger, M.; Hockey, H.U.; Wagner, K.; Ammerman, N.; Vo, A. Intravenous immunoglobulin significantly reduces exposure of concomitantly administered anti-C5 monoclonal antibody tesidolumab. Am. J. Transpl. 2020, 20, 2581–2588. [Google Scholar] [CrossRef]
- Jordan, S.C.; Choi, J.; Aubert, O.; Haas, M.; Loupy, A.; Huang, E.; Peng, A.; Kim, I.; Louie, S.; Ammerman, N.; et al. A phase I/II, double-blind, placebo-controlled study assessing safety and efficacy of C1 esterase inhibitor for prevention of delayed graft function in deceased donor kidney transplant recipients. Am. J. Transpl. 2018, 18, 2955–2964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brüggenwirth, I.M.A.; Martins, P.N. RNA interference therapeutics in organ transplantation: The dawn of a new era. Am. J. Transpl. 2020, 20, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Cucchiari, D.; Rovira, J.; Diekmann, F. Regulated Cell Death at the Crossroad Between Ischemia-reperfusion Injury and Innate Immunity in Kidney Transplantation. Transplantation 2020, 104, 1772–1773. [Google Scholar] [CrossRef]
- Chen, Y.; Shi, J.; Xia, T.C.; Xu, R.; He, X.; Xia, Y. Preservation Solutions for Kidney Transplantation: History, Advances and Mechanisms. Cell Transpl. 2019, 28, 1472–1489. [Google Scholar] [CrossRef]
- Faure, A.; Bruzzese, L.; Steinberg, J.G.; Jammes, Y.; Torrents, J.; Berdah, S.V.; Garnier, E.; Legris, T.; Loundou, A.; Chalopin, M.; et al. Effectiveness of pure argon for renal transplant preservation in a preclinical pig model of heterotopic autotransplantation. J. Transl. Med. 2016, 14, 40. [Google Scholar] [CrossRef] [Green Version]
- Ozaki, K.S.; Yoshida, J.; Ueki, S.; Pettigrew, G.L.; Ghonem, N.; Sico, R.M.; Lee, L.Y.; Shapiro, R.; Lakkis, F.G.; Pacheco-Silva, A.; et al. Carbon monoxide inhibits apoptosis during cold storage and protects kidney grafts donated after cardiac death. Transpl. Int. 2012, 25, 107–117. [Google Scholar] [CrossRef]
- Yoshida, J.; Ozaki, K.S.; Nalesnik, M.A.; Ueki, S.; Castillo-Rama, M.; Faleo, G.; Ezzelarab, M.; Nakao, A.; Ekser, B.; Echeverri, G.J.; et al. Ex vivo application of carbon monoxide in UW solution prevents transplant-induced renal ischemia/reperfusion injury in pigs. Am. J. Transpl. 2010, 10, 763–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAnulty, J.F.; Reid, T.W.; Waller, K.R.; Murphy, C.J. Successful six-day kidney preservation using trophic factor supplemented media and simple cold storage. Am. J. Transpl. 2002, 2, 712–718. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, K.; Koo, D.D.; Davies, D.R.; Gray, D.W.; McLaren, A.J.; Welsh, K.I.; Morris, P.J.; Fuggle, S.V. Lecithinized superoxide dismutase reduces cold ischemia-induced chronic allograft dysfunction. Kidney Int. 2002, 61, 1160–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Hosgood, S.A.; Nicholson, M.L. Naked small interfering RNA of caspase-3 in preservation solution and autologous blood perfusate protects isolated ischemic porcine kidneys. Transplantation 2011, 91, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Moser, M.A.; Arcand, S.; Lin, H.B.; Wojnarowicz, C.; Sawicka, J.; Banerjee, T.; Luo, Y.; Beck, G.R.; Luke, P.P.; Sawicki, G. Protection of the transplant kidney from preservation injury by inhibition of matrix metalloproteinases. PLoS ONE 2016, 11, e0157508. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Phase | Metabolites (+/−) | Model | Reference |
|---|---|---|---|
| Ischemia | Lactate, Choline (+) Amino acids (valine, alanine, glutamate, glycine) (+) Glutathione (−) | Swine kidney auto-transplantation | [94] |
| Reperfusion (kidney with DGF) | Glucose (+) Glutathione (−) | Human cadaveric perfusate | [95] |
| Reperfusion (kidney with IGF pool) | 3-hydroxybutyrate (+) Lactate (+) Citrate, glutamate (+) | Human cadaveric perfusate | [95] |
| Ischemia | Glutamate (+) Serine (+) Isovalerylglycine (+) Proline (+) Aminobutyrate (+) Choline (+) | Swine kidney IRI serum | [96] |
| Reperfusion | Citrate (+) Pyruvate (+) 3-hydroxybutyrate (+) 3-aminoisobutyrate (+) Betaine (+) Carnosine (+) N-phenylacetylglycine (+) | Swine kidney IRI urine | [96] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loizzo, D.; di Meo, N.A.; Peluso, M.R.; Rutigliano, M.; Matera, M.; Miacola, C.; Palella, G.; Tedeschi, M.; Spilotros, M.; Ferro, M.; et al. Novel Insights into the Molecular Mechanisms of Ischemia/Reperfusion Injury in Kidney Transplantation. Transplantology 2021, 2, 191-207. https://0-doi-org.brum.beds.ac.uk/10.3390/transplantology2020018
Loizzo D, di Meo NA, Peluso MR, Rutigliano M, Matera M, Miacola C, Palella G, Tedeschi M, Spilotros M, Ferro M, et al. Novel Insights into the Molecular Mechanisms of Ischemia/Reperfusion Injury in Kidney Transplantation. Transplantology. 2021; 2(2):191-207. https://0-doi-org.brum.beds.ac.uk/10.3390/transplantology2020018
Chicago/Turabian StyleLoizzo, Davide, Nicola Antonio di Meo, Mattia Rocco Peluso, Monica Rutigliano, Matteo Matera, Carlos Miacola, Gaetano Palella, Michele Tedeschi, Marco Spilotros, Matteo Ferro, and et al. 2021. "Novel Insights into the Molecular Mechanisms of Ischemia/Reperfusion Injury in Kidney Transplantation" Transplantology 2, no. 2: 191-207. https://0-doi-org.brum.beds.ac.uk/10.3390/transplantology2020018