Pathological Maintenance and Evolution of Breast Cancer: The Convergence of Irreversible Biological Actions of ER Alpha


Institute Jules Bordet, Cancer Center of the Université Libre de Bruxelles, 1000 Brussels, Belgium
Endocrines 2021, 2(1), 1-14; https://0-doi-org.brum.beds.ac.uk/10.3390/endocrines2010001
Submission received: 18 September 2020
/
Revised: 30 November 2020
/
Accepted: 14 December 2020
/
Published: 24 December 2020
(This article belongs to the Special Issue Molecular Mechanisms of Estrogen Signaling Pathways)
{kind=link}
Abstract
:Estrogen receptor alpha (ERα) is a modulator of breast cancer maintenance and evolution. Hence, analysis of underlying mechanisms by which ERα operates is of importance for the improvement of the hormonal therapy of the disease. This review focuses on the irreversible character of the mechanism of action of ERα, which also concerns other members of the steroid hormones receptors family. ERα moves in permanence between targets localized especially at the chromatin level to accomplish gene transcriptions imposed by the estrogenic ligands and specific antagonists. Receptor association as at the plasma membrane, where it interacts with other recruitment sites, extends its regulatory potency to growth factors and related peptides through activation of signal transductions pathways. If the latter procedure is suitable for the transcriptions in which the receptor operates as a coregulator of another transcription factor, it is of marginal influence with regard to the direct estrogenic regulation procedure, especially in the context of the present review. Irreversibility of the successive steps of the underlying transcription cycle guarantees maintenance of homeostasis and evolution according to vital necessities. To justify this statement, reported data are essentially described in a holistic view rather than in the context of exhaustive analysis of a molecular event contributing to a specific function as well as in a complementary perspective to elaborate new therapeutic approaches with antagonistic potencies against those tumors promoting ERα properties.
1. Introduction
From birth to death, anabolic and catabolic processes generate or eliminate a variety of cellular elements, regulating age-dependent functions that confer to life its irreversible character. They contribute, indeed, to the elimination of partially altered (“old”) as well as inadequate elements to favor their replacement by newly synthetized (operative) substituents. To satisfy this requirement, life modulators often operate within cyclic procedures with high thermodynamic efficiency. Steroid hormones are prototypes of such molecules because their production is cyclically regulated, giving rise to repeated transient associations with a family of specific receptors, the mechanism by which they operate requiring a complementary assistance of various coregulators to establish an irreversible character to the whole regulatory procedure (recent reviews: [1,2,3,4]). At the time of discovery, these receptors were classified as “nuclear receptors”. Their capacity to regulate the expression of a panel of genes ([5], especially Chap. V) justified this terminology, which may appear today as quite obsolete. It is indeed known that these receptors act not solely as transcription factors but also as inducers of signal transduction pathways issued from the activation of a peculiar pool of receptors anchored within plasma membrane lipid rafts [4]. Close contact between this receptor pool and membrane recruitment sites for a multitude of regulatory agents extends the sensitivity of the whole receptor population to non-steroidal molecules, especially calcium [6] and growth factors [7], as described with the two ER forms (α and β) extensively investigated in the context of the breast cancer [8,9] topic of this review. An evident cooperative action of the plasma membrane and nuclear receptor pools to generate some responses (membrane receptors operate largely before transcriptions) [10,11,12] conjugated with a complementary identification of other pools in microsomes [13] and mitochondria [14] lead us to wonder about a suitable terminology adaptation: “intracellular receptors” would obviously be a better fit than “nuclear receptors”. The fact that gene transcriptions results from space-time traffics of receptors between various docking sites localized onto the membrane as well as within the nucleus [9] supports this view.
Successive irreversible conformational changes imposed by extracellular messages to the receptors favor their ability to recruit coregulators, of which catalytic activities (kinases, phosphatases, methylases, acetylases, ubiquitin ligases) are required for the achievement of transcriptions [8]. In this regard, two mechanisms have been identified: one in which the receptor directs the whole procedure, the other where it assists the action of another transcription factor [15,16]. In the first case, the receptor usually regulates the expression of genes under the control of a palindromic sequence of nucleotides of the DNA (response element) localized within their promoter region. In the second case, it acts as a coregulator of a transcription factor bound to its own response element, especially when the activating message implicates a signal transduction pathway issued from the plasma membrane. These two transcriptional procedures, which may cooperate [15], act cyclically until the initial regulatory request has been satisfied [8]. While this property is established for a substantial set of genes (Section 4), a recent report [17] proposed that it may not concern a genome-wide activation because, in this case, recorded transcription displayed an amplified sustained aspect without any temporal decrease. This report, based on an unusual approach to monitor ERα binding to chromatin, has not been confirmed nor subjected to any debate, which has largely limited its conceptual impact. In this regard, one may consider that it may explain the spectacular chromatin dispersion observed by electronic microscopy under ERα activation [18,19] as well as the strong increase of nuclear staining recorded in parallel by autoradiography with a tritiated preparation of the intercalating agent actinomycin D [19,20], two properties extremely difficult to justify for a limited expression of genes. Whatever the issue of this potent controversy concerning the mechanism regulating an extended expression of the genome, one may consider that it will not invalidate the biological relevance of data related to conformational changes, dimerization, intracellular movements, and coregulators recruitments, which all concur to the irreversibility of action of the receptors. Hence, description of these properties in the context of the present review would not suffer criticism.
ERα was selected as a representative prototype for such a purpose; long-term interest in this receptor in the context of hormone dependence of breast cancer justifies this option. Even if some mechanisms described in this review may solely concern this topic, most reported data may be extrapolated to other poles of interest providing valuable information for many investigators in endocrinology and fundamentalists, as well as clinicians.
2. Biological Irreversibility: A Concept Applied to Regulatory Functions
At first, to avoid any ambiguity, it should be stressed that the concept to which this review is devoted is totally distinct from the philosophical/theological notion of determinism, which considers that major life events are programmed at birth, during infancy, or at a peculiar moment; moreover, determinism implies the banishment of all efforts to escape to imposed unsuitable events. Biological irreversibility evoked here is in total opposition with this liberticide (antiregulatory) view because it refers to a metabolic procedure of which a physiological flexibility may satisfy biological requirements. Impeding all steps of the metabolic pathway to return to its starting point until a requested function has been accomplished, irreversibility protects the dynamic character of the whole regulatory procedure against contradictory messages. If such an interference is nevertheless absolutely needed, this protection vanishes at the level of the starting point of the pathway to facilitate the emergence a substitutive way that will progressively limit the impact of the previous one, thereby abrogating a chaotic behavior. This regulatory advantage is enhanced by the cyclisation of the procedure because its starting and ending points become identical; at this point, lack of sensitivity to a new message maintains homeostasis while its acceptance facilitates evolution. Hence, in the context of estrogens and related antagonists, irreversibility facilitates the transition from growth to differentiation that these hormones may impose.
Unfortunately, under pathological promoting conditions, such a physiological protective irreversibility may generate major deleterious effects, as largely commented here in the context of breast cancer. In conventional monolayer cell culture of ERα-positive MCF-7 cells, this factor largely limits the chance for a major change of receptor-mediated response under successive expositions to ligands conferring distinct configurations adequate for such a change [21], thereby reducing the potential impact of a hormonal therapy. When such a property occurs (i.e., rapid receptor accumulation recorded with hydroxytamoxifen in MCF-7 cells previously maintained in a condition of receptor down-regulation through exposure to estradiol or a pure antiestrogen [22]), one may consider that it is relevant to an absence of cellular synchronization that may not palliate such a curative deficiency.
Such an absence of synchronization indeed allows the simultaneous existence of cellular populations in other phases of the cell cycle, of which the statistical distribution does not vary along the logarithmic growth phase, each of these populations being sensitive to distinct growth/differentiation promoting agents. Such a status generates a proliferative pattern without visible waives (cf. multi-cylindric motorization), which are also detectable at the molecular level; electrophoretic separation of proteins from whole cell extracts by SDS-PAGE indeed gives identical pictures that are moreover reflected at the level of ERα expression [23]. The complementary observation of a same growth promoting effect in response to an estrogenic pulse along the logarithmic cell growth phase implies a regulation of the whole receptor machinery as the majority of proteins. Hence, a relationship between cell cycle progression and expression of ERα and coregulators would logically exist, contributing to the cyclic character of their action. In fact, this conclusion has been confirmed without any ambiguity in experiments conducted with synchronized cells, in which successive recruitment waives for coregulators are easily detectable [24].
3. Relationship between Cell Cycle Progression and ERα Expression in Breast Cancer Cells
Interactions between ERα, cyclins, and cyclin kinases (CDK), as well as CDK inhibitors, has been established [reviews: 25–28]. If functions of a lot of these interactions in the context of cell growth is still undecrypted, accumulated data has left no doubt that they contribute to a triangular relationship between the receptor turnover rate, its influence on transcription, and the progression of the cell cycle. To briefly illustrate this statement in the context of breast cancer cells, I selected an interaction of the receptor with the cyclins A and D1 that enhances the phosphorylation of the retinoblastoma protein (pRb; G1/S transition checkpoint), a step promoting cell division; for CDK inhibition, I pointed ERα binding to the C-terminal region of the p27 CDK inhibitor to prevent accumulation of the p27 inhibitor. For exhaustive information on such interactions, as well as the perspectives they open for therapeutic programs, readers should consult the reviews cited above.
The relationship between tumor evolution and ERα expression has been extensively investigated in large cohorts of patients to evaluate its prognostic value (recent reports: ref. [25,26]). Data recorded here focus on Ki-67 as a specific marker of proliferation, the receptor as an index of estrogen sensitivity, and the histological grading because the cellular differentiation to which it refers is of prime importance (Grade 1 relates to well differentiated tumors looking like normal tissue, Grade 2 displays a moderate differentiated aspect strongly different from the aspect recorded with Grade 3, which drastically differs from the normal tissue). Ki-67 level increases with the G1-G3 transition in contrast to the ERα expression, which shows an inverse tendency giving rise essentially to a high Ki-67/ERα-negative status at G3, which is in agreement with its known bad prognosis. On the other hand, a lack of any information relative to a potential intranuclear interaction between Ki-67 and ERα suggests an implication of the mechanisms governing cell differentiation in the coordination of their actions. Such a view stresses the importance of an identification of commune regulatory partners as well as the phases of the cell cycle at which such regulatory actors would operate. While ERα data are relatively abundant (see below), reports on Ki-67 are largely insufficient [27] for such a purpose. Studies conducted with ERα-positive and -negative breast cancer cell lines would be accurate to palliate this gap [28,29,30,31,32] (see next paragraph). For in vivo experimentations, the transplantable MXT mouse mammary tumor model might be selected because it mimics human behavior [33,34]. Cellular aspect of stored samples of the original MXT tumor (available in tumors banks) display a well differentiated status that progressively vanishes during successive transplantations, giving rise to an undifferentiated, basal-like, pattern. Growth increases in parallel with a progressive decrease of ERα expression, giving rise finally to the ERα-negative status associated with the emergence of a low molecular weight receptor variant, whose main characteristics are very similar to those of the human ERα36 (Section 8).
Phases of the cell cycle at which ERα is expressed, as well as the influence of ligands on this expression, has been investigated [28,29,30,31,32]. In the absence of ligand, ERα level increases during the G0/G1-phase before declining until the late S-phase, allowing the onset of a second increase at the time of the S/G2 transition, which is less sensitive to the promoting effect of estradiol than the former [32]. Interestingly, ERα ligands provoke the emergence of a nuclear receptor pool devoid of the estradiol binding ability, which seems to be implicated in the regulation of the receptor turnover rate [35]. Accumulation of this pool at G0/G1 provides an explanation for the weak sensitivity of the hormone at S/G2, a hypothesis supported by the fact that this loss of estrogen binding ability is a first step of the mechanism of action of Erα, contributing to its irreversible character (Section 5).
Latest reported data relative to this topic concerns the effect of estradiol, hydroxytamoxifen, and fulvestrant on the cell cycle progression [36]. They confirm an expected relationship between this kinetic parameter and the influence of these ligands on ERα expression: estradiol: Strong cell cycle acceleration/strong ERα downregulation resulting from a decrease of its mRNA stability [37] and enhanced proteosomal degradation [38,39] by a process requiring a nucleo-cytoplasmic shuttling [40]; tamoxifen: No major kinetic effect/no proteasomal ERα elimination provoking its accumulation in the cell nucleus in an estradiol non-binding form [41]; fulvestrant: Cell cycle acceleration/proteasomal ERα degradation [38] by a procedure differing from the one induced by estradiol [39]. These operational differences are probably relevant to the distinct ubiquitine-dependent shipments of the receptor to proteasomes required for the accomplishment of its ligand-induced missions [42,43]. In fact, multiple facets of this suspected regulation are insufficient to elaborate a regulatory model, as proven by a study with bortezomid [44]. This proteasome inhibitor provokes indeed an ERα elimination rather than the stabilization observed with usual inhibitors (MG 132, lactacystin, LLnL). This property results from a loss of the ERα gene (ESR1) expression by an inactivation of chromatin environment at a −150 Kb distal enhancer of the transcription start site.
4. Factors Implicated in the Irreversible Character of ERα-Mediated Transcriptions
Molecular aspects of ERα-mediated transcriptions include allosteric mechanisms related to the intracellular traffics of the receptor and cofactor recruitments [8,9,45,46,47,48,49,50,51]. These events analyzed in details in Section 5, Section 6 and Section 7 are briefly described here under to stress their importance to satisfy homeostasis or evolution. Figure 1 illustrates schematically these events which, as already evocated, must be abrogated in a therapeutic perspective.
The starting point of the transcription procedure is the abrogation of a repressive function exerted by a few chaperones on the native cytoplasmic ERα. This liberation provokes a loss of its ligand binding ability to protect the pursuit of the procedure against potential antagonistic messages. ERα homo-dimerization occurs in parallel to satisfy receptor association with estrogen response elements (EREs) of genes to transcribe, apparently without a detrimental effect on complementary ERE-independent transcriptions in which the receptor plays a role of coregulator [15]. A pool of such liberated ERα is subjected to palmitoylation [11] for its transient anchorage within caveolae of the plasma membrane [4] to contribute to signal transductions pathways without any loss of capacity to migrate thereafter to the nucleus to coordinate the integration of messages issued from this transient localization in genes expressions [10,11,12]. To this end, ERα is subjected to successive conformtional changes, mainly through phosphorylations, methylations, acetylations, and ubiquitilations [46] and related coactivators recruitments, some of them containing a LxxLL motif operating at the AF-2 site of the receptor [8,9]. These changes occur at least in part on a “hub/platform” [47] localized in front of the ligand binding domain [46,48,49] to facilitate cooperative actions or exchanges between the receptor in its partners according to a well-defined program, a function that would also protect the transcriptional cycle against inappropriate interferences (Section 6). Rapidity of the regulatory “dialog” at this level most likely limits such potential deleterious effects. Subsequent monoubiquitilation through non-covalent interactions with Leu 429 and Ala 330 implicated in signal transductions and Lys 302 and 303 of the exchange hub (“couple monoubiquitilation”) favors a cooperation between the AF-1 and AF-2 sites of ERα for the final accomplishment of its transcriptional mission [48]. In this regard, monoubiquitilation distinguishes from polyubiquitilation, which arrests transcription by a proteosomal receptor degradation.
When the requested gene expression appears satisfied, polyubiquitilation [49] displaces ERα from its DNA anchorage site for a shuttling to a cytoplasmic proteasome where it is degraded, thereby stopping its action. The underlying mechanism implicates now a covalent interaction with Lys 302 and 303, confirming the major role of ubiquitilationsat at this level in the mechanism of action of ERα. Note in this context that all facets of ubiquitilations have been largely viewed in publications with pertinent schematic illustrations [43,50].
An identical transcription cycle implying the participation of a newly synthetized receptor pool may then occur if the original request maintains (homeostasis) while a new program devoted to the expression of other genes may start if required (evolution). Interactions of proteolytic ERα fragments issued from the receptor degradation, most likely through an assistance of procedures regulating its synthesis, maturation, and turnover rate [8], would contribute to these events [51] (interaction of such a fragment with the chaperone Hsp 70 has been identified, Section 6). An extra cellular release of some of these peptides, perhaps in cooperation with a recently reported lysosomal autophagy focusing on the receptor pool implicated in signal transductions [52,53,54], may play a complementary role via an autocrine regulatory loop (Section 7).
Of note, a large panel of natural and synthetic ERα ligands initiate these procedures. In fact, this paradoxical lack of selectivity satisfied functions identified in ancestral receptor operating when the metabolic production of steroidal estrogens did not exist [55]. Endocrine disruptors (phytoestrogens, pesticides, heavy metallic ions) pertain to actual xenoestrogens for which a great interest is evident [56].
5. ERα “Activation”: Loss of Ligand Binding Capacity and Related Receptor Dimerization
ERα “activation” refers to its irreversible liberation from repressive constrains exercised especially by the chaperone Hsp90 with which it associates in an oligomeric structure, regulating its ligand binding ability and transactivation potency [9,57,58], as already evoked in Section 4. Abrogation of ERα~Hsp90 complexion by the Hsp90 antagonist radicicol indeed decreases estradiol binding as well as gene transcription by a procedure, implying in fine a proteosomal elimination of the receptor, the mechanism of which has been subjected to some controversy [58,59] (complementary or alternative possibility: lysosomal degradation, Section 7). Selective Estrogen Receptor Modulators (SERMs), known to stabilize ERα in the cell nucleus in an estradiol non-binding form, were found to antagonize this radicicol-induced elimination [59], suggesting that the abrogation of ERa~Hsp90 complexion may affects ligand binding without detrimental action on the nuclear receptor anchorage. Lack of strong estradiol binding ability, an essential characteristic of the nuclear receptor form [35], supports such a view. Hence, loss of ligand binding ability and dimerization acquired for nuclear anchorage appear to be the early steps of the mechanism of action of ERα, the cooperation of which would promote its irreversible character.
This concept derives from a pilot investigation designed by Elwood V. Jensen (“the father of ERα”), which invites me to assess the exactitude of its opinion that the estrogen-induced ERα dimerization required for its transactivation potency maintains when the estrogen is removed from its binding pocket (i.e., maintenance of the 5S dimeric ERα form in sucrose gradient sedimentation). This was, in fact, a study devoted to validating the irreversibility of this early step in the activation of the receptor. Our data confirmed this concept, justifying the inclusion of our experimental details in a Supplementary Materials of the present review because they were solely reported in the proceedings of a meeting [60]. Interestingly, total restoration of the original estrogen labeling of the receptor was unsuccessful for its dimeric “stripped” form, provoking a temporal arrest of our investigation.
Personal experiments aimed to identify the origin of this property focused on the assessment of mutual exchanges between bound estrogens and antiestrogens [21]. This approach revealed that a strong anchorage of a ligand within its binding pocket largely limits its displacement by a potential competitor, a concept in agreement with a report showing that the association of ERα to tandem ERE copies slows ligand dissociation and synergistically activates gene expression [61]. Such a property could obviously not be ascribed to the 5S stripped ERα of our study because it was devoid of any residual bound estrogen, a status more closely related to a recent report revealing that ligand dissociation from an activated receptor is related to its dimerization [62], a property that is in agreement with our finding that the absence of restoration of original labeling of the dimeric “stripped” receptor results essentially from a strong loss of estrogen binding affinity.
This loss of binding affinity would favor signal transductions and related ERE-independent transcriptions in contrast to ERE-dependent transcription, of which anchorage of ligands within the binding pocket of the receptor seems be primordial. Such a view indeed finds some support in our recent studies conducted with estrothiazine [63], a peculiar estrogen without any detectable competitive potency for estradiol binding to ERα but which promotes a loss of its estradiol binding ability as well as its dimerization consequently inducing an ERK1/2 activation that precedes AP1- and ERE-dependent transcriptions [11,63], two activities related respectively to ERα dimers localized onto the plasma membrane [64] (ERK1/2) and within nucleus (AP1; ERE). Interestingly, the adjunction of a sulfone to this molecule at a position that slightly enhanced its complexion with the ligand binding pocket negated growth promotion without any similar effect on ERE-dependent transcription [65]. This property recorded with another compound [66] pertaining to a large class ERα mediators devoid of apparent competitive capacity for estradiol binding [67] established a prominent role of ERα dimerization in the mediation of its diverse functions, which is apparently relate to the selective binding properties of the ligands.
6. Allosteric ERα Activation Implying Successive Coregulator Recruitments
The hub, evoked in Section 4, localizes at the extreme left side of the ligand binding domain of ERα (Pro 295-Threo 311; PLMIKRSKKNSLALSLT) in close contact with another recruitment site of this domain (β turn/H4; “BF3”) to regulate coregulators recruitment at the activation function AF-2, which plays a major role in the onset of estrogen-induced responses [68,69,70,71]. BF3 nomination [70,71], was selected for its distinction from the activation functions AF-1 and AF-2 implicated respectively in estrogen-independent and-dependent mechanisms. The association of agents of various chemical natures with the hub and/or the BF3 provokes a “relaxation” of the molecular conformation of the ligand binding domain, allowing enhanced AF-2 expression with concomitant pursue of the initiated transcription cycle [71]. This structural change may perhaps concern the global structure of the receptor because AF-1 and AF-2 often cooperate. This allosteric activation results from the abrogation of a repressive ERα conformation imposed by the docking of a set of amino acids of the hub (S301-T311) within the BF3 β turn/H4 [69,70]; a repressive docking procedure that seems to concern the whole family of steroid hormone receptors [70,71].
Calmodulin (CaM), an ERα partner that associates with the hub [72,73], was thought to largely contribute to this relaxation through its ability to stabilize the receptor in its dimeric form without strong estrogen binding affinity in a conformation accurate for association with EREs [74,75], thereby contributing to subsequent transcriptions and enhanced cell proliferation [72,73] (Section 5). Studies conducted with a synthetic P295-T311 peptide (ERα17p) aimed to displace CaM from its binding motif by a competitive procedure produced an unexpected ERα activation as observed with estrogens, suggesting the implication of a CaM-independent relaxation procedure [73]. The finding that alanine or glycine substitutions of the couple of lysines 302 and 303 present in ERα17p, which are of major importance for CaM binding [73,76,77], maintain this apparent estrogenic activity, suggest the implication of other ERα partners in a suspected transient CaM-assisted relaxation procedure [73]. Hsp70 and PNRCs have been identified as potential candidates because they associate with ERα17p [77,78]. The subsequent observation that the upstream PLMI module of ERα17p favors the interaction of the receptor with the plasma membrane GPR30 estrogen binding protein [79], which is also subjected to CaM binding, provided complementary information relative to this intriguing ERα activation mechanism; ERα~GPR30 interaction activates the ERK1/2 signal transduction pathways, giving rise to cell proliferation enhancement.
The molecular aspect of this apparent ERα~GPR30-induced abrogation of the repressive action of the P295-T311~BF3 axis on cell proliferation is unknown. A model proposed to explain signal transduction pathways issued from the membrane as well as transcriptions generated by the steroid hormone receptors might be a key to orient research. According to this model [80], the ligand binding sites of these receptors are composed of two cavities corresponding respectively to a channel entrance and a docking chamber separated by a barrier. The ligand entrance within the channel activates signal transductions while a ligand whose chemical structure [81,82,83] may open the barrier engulfs easily within the chamber to favor transcription. One may speculate that GPR30 linked to ERα could adopt a conformation containing a kind of channel to orient the ligand’s entrance within an adjacent receptor chamber for transcriptions, probably under the supervision of CaM. Of course, other hypotheses could be proposed, such as an intracellular mobility of CaM and GPR30 [84], which would facilitate the traffic of ERα from the membrane to the nucleus, ultimately provoking AF1/AF2 cooperative actions.
On the other hand, the finding that ERα17p generates estradiol-like responses [73,77] largely contributes to the concept that ERα proteolysis may contribute to the cyclic character of its mechanism of action through intracellular procedures such as an interference of such a degradation peptide at the Hsp70/90 level that manages the maturation of the newly synthetized receptor [8,77]. Extracellular secretion of such peptides, especially those acting at the ERa~GPR30 level as ERα17p [79], would favor the initiation of a new transcription cycle through an autocrine procedure [51]. Of course, experimental studies are needed to validate this view, even though a few data reported in the next section support it.
Whatever the issues of future investigations aimed at validating the concepts evoked here, one may already consider that recorded procedures would contribute to the irreversibility of the mechanism of action of the receptor.
7. Implication of Proteolytic ERα Fragments in Autocrine and Paracrine Regulations
Estrogens induce an extracellular protein secretion; some of these proteins exist at the time of the hormonal stimulation, others are induced by this stimulation [85,86]. The presence of detectable ERα/β in the blood of women [87] indicates that this secretion concerns these receptors as well as peptides issued from their proteasomal or lysosomal degradation [51,52,53,54], as already suggested. The finding that the ERα17p peptide (Section 6) generates a variety of responses in ERα+ and ERα− breast cancer cells [88] suggests that such a proteolytic elimination may modulate the evolution of human tumors through autocrine and paracrine procedures, according to a growth factor-like effect [7].
Moreover, the detection of natural anti-ERα natural antibodies with estrogenic activities in the blood of women [89,90] introduces the concept of an auto-immunological reaction against secreted ERα proteolysis products, thereby contributing to breast cancer development. The complementary finding that these antibodies, which associate with the estrogen binding motif of the receptor pool localized on the plasma membrane, generate a tamoxifen resistance [91] is of importance, especially in the context of patients suffering from this resistance. Research on this thematic may perhaps offer a chance for new treatments.
These remarks, related to products issued from the final step of the transcriptional cycle of ERα, support the view outlined in the previous section that this step does not arrest the action of the receptor but, on the contrary, opens new ways for its pursuit and amplification.
8. Concluding Remarks and Conclusions
The purpose of this work was to provide an extended overview of the irreversible character of the mechanism of action of ERα, including a few speculative proposals to promote investigations to enhance knowledge of the underlying procedures that could contribute to new therapeutic developments, even reported data do not concern all facets of the topic. Influence of receptor variants extensively described in several reviews [92,93,94,95] were not evoked because the multitude of accumulated data could logically not be addressed in the present context (about 1000 references relative to splicing variants affecting the primary structure of the receptor as well as point mutations at the level of amino acids subjected to post-transcriptional modifications). Such ERα alterations, detected in various tissues, concur to evolutive pathologies including breast cancer, suggesting that a critical analysis of these data in the present context might be extremely fruitful. As proof of such a view, I mention the properties of a few variants as representative examples: (1) a splicing of ERα36 present in most aggressive breast cancers (negative for ERα, PR, and HER-2) localized essentially at the plasma membrane to induce signal transductions, giving rise to a constitutive growth and tamoxifen resistance, confirming the importance of the trafficking of the receptor in the hormonal regulation of growth [96,97,98,99], (2) R538G and Y537S punctual mutations localized in the ligand binding domain that provoke its maintenance in an adequate conformation for antiestrogen resistance [100,101,102], (3) K303R punctual mutation of the P295-T311 regulatory hub (Section 6) confers estrogen hypersensitivity to breast cancers with concomitant poor outcomes; phosphorylation of its adjacent S305 induces low curative responses to tamoxifen and anti-aromatase inhibitors [103,104].
Potential interference of ERα/ERβ hetero-dimerization in the action of the alpha dimer was also not addressed because a large number of readers would have difficulty to understand underlying molecular events. Hence, the present work may be assimilated to an initiation phase for complementary reports. Nevertheless, I consider conceptual data provided here would be largely sufficient to satisfy the scientific and clinical communities, especially in the hope of increasing our therapeutic armamentarium [105,106] against breast cancer emergence and development.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2673-396X/2/1/1/s1, S1: Dimeric Structure of Unoccupied Activated Estrogen Receptor.
Funding
This research received no external funding.
Acknowledgments
Elwood Jensen passed away some years ago. This review may be considered as a tribute to him because he invited me to verify his opinion that dimerization irreversibility is a capital step in the mechanism of the action of steroid hormone receptors. I wish also to warmly thank Elias Castanas and Yves Jacquot, who contributed to decrypting the procedures by which ERα17p induces its peculiar functions. Acknowledgements also go to the colleagues and PhD students whose experimental investigations helped me to elaborate on the concepts recorded in this review.
Conflicts of Interest
As a retired Honorary Professor, I declare a total lack of contact with any pharmaceutical company.
Abbreviations
| AF | Activation function |
| AP 1 | Activation Protein 1 |
| BF3 | Binding Function 3 |
| CDK | Cyclin-Dependent Kinase |
| ER | Estrogen Receptor |
| ERE | Estrogen Response Element |
| ERK | Extra Celluar Regulated Kinase |
| ERα 17p | Peptide corresponding to the P295-T311 amino acid sequence of ERα |
| GPR | G-Protein Coupled Receptor |
| HER 2 | Human Epidermal Growth Factor 2 |
| Hsp | Heat Shock Protein |
| Ki-67 | Protein identified with an antibody raised against a Hodgkin’ lymphoma protein |
| LxxLL | binding motif of coregulators L refers to Leucine, x to any other amino acid |
| PRNC | Proline-rich Nuclear Receptor Regulatory Protein |
| PR | Progesterone Receptor |
| pRb | Retinoblastoma Protein |
| SERM | Selective Estrogen Receptor Modulator |
References
- Arnal, J.-F.; Lenfant, F.; Metivier, R.; Flouriot, G.; Henrion, D.; Adlanmerini, M.; Gourdy, P.; Chambon, P.; Katzenellenbogen, B.; Katzenellenbogen, J. Membrane and nuclear estrogen receptor alpha actions: From tissue specificity to medical implications. Physiol. Rev. 2017, 17, 1045–1087. [Google Scholar] [CrossRef] [PubMed]
- Weikum, E.R.; Liu, X.; Ortlund, E.A. The nuclear receptor superfamily: A structural perspective. Protein Sci. 2018, 27, 1876–1892. [Google Scholar] [CrossRef] [PubMed]
- Hager, G.L.; Lim, C.M.; Bauman, C.T. Trafficking of nuclear receptors in living cells. J. Steroid Biochem. Mol. Biol. 2000, 74, 249–254. [Google Scholar] [CrossRef]
- Levin, E.R. Extranuclear steroid receptors are essential for steroid hormone actions. Annu. Rev. Med. 2015, 66, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.H.; Peck, E.J., Jr. Female Sex Steroids. Receptors and Functions. In Monographs in Endocrinology; Springer: Berlin/Heidelberg, Germany, 1979; Volume 14. [Google Scholar]
- Leclercq, G. Calcium-induced activation of estrogen captor alpha—New insight. Steroids 2012, 77, 924–927. [Google Scholar] [CrossRef]
- Lippman, M.E.; Dickson, R.B.; Kassid, A.; Gelmann, R.E.; Davidson, N.; McManaway, M.; Huff, K.; Bonzert, D.; Bates, S.; Swain, S.; et al. Autocrine and paracrine growth regulation of human breast cancer. J. Steroid Biochem. 1986, 24, 147–154. [Google Scholar] [CrossRef]
- Leclercq, G.; Lacroix, M.; Laïos, I.; Laurent, G. Estrogen receptor alpha: Impact of Ligands on intracellular shuttling and turnover rate in breast cancer cells. Curr. Cancer Drug Targets 2006, 6, 39–64. [Google Scholar] [CrossRef] [Green Version]
- Kampa, M.; Pelekanou, V.; Notas, G.; Stathopoulos, E.N.; Castanas, E. The estrogen receptor: Two or more molecules, multiple variants, diverse localizations, signaling and functions. Are we undergoing a paradigm-shift as regards their significance in breast cancer? Hormones 2013, 12, 69–85. [Google Scholar] [CrossRef]
- Seo, H.-S.; Leclercq, G. Evaluation of potential implication of membrane estrogen binding sites on ERE-dependent transcriptional activity and intracellular estrogen receptor-alpha regulation in MCF-7 breast cancer cells. J. Steroid Biochem. Mol. Biol. 2002, 80, 109–123. [Google Scholar] [CrossRef]
- La Rosa, P.; Perisi, V.; Leclercq, G.; Marino, M.; Acconcia, F. Palmitoylation regulates 17β-estradiol-induced estrogen receptor-a degradation and transcriptional activity. Mol. Endocrinol. 2012, 26, 762–764. [Google Scholar] [CrossRef] [Green Version]
- Winkeldfeld, S.R.; Lin, F.D.E. Communication between genomic and non-genomic signaling events coordinate steroid hormone actions. Steroids 2018, 133, 2–7. [Google Scholar]
- Watson, G.H.; Muldoon, T.G. Specific binding of estrogen and estrogen receptor complex by microsomes from estrogen responsive tissues of the rat. Endocrinology 1985, 117, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Klinge, C.M. Estrogens regulate life and death in mitochondria. J. Bioenerg. 2017, 49, 307–324. [Google Scholar] [CrossRef] [PubMed]
- Kushner, P.J.; Agard, D.A.; Greene, G.L.; Scanlan, T.S.; Shiau, A.K.; Uth, R.M.; Webb, P. Estrogen receptor pathways to AP-1. J. Steroid Biochem. Mol. Biol. 2000, 74, 311–317. [Google Scholar] [CrossRef]
- Safe, S.; Kim, K. Non classical ER/Sp and AR/AP-1 signaling pathways. J. Mol. Endocrinol. 2008, 41, 263–275. [Google Scholar] [CrossRef]
- Holding, A.N.; Cullen, A.E.; Markowitz, M. Genome-Wide estrogen receptor alpha activation is sustained not cyclical. Elife. 2018, 7, e40854. [Google Scholar] [CrossRef]
- Vic, P.; Garcia, M.; Humeau, C.; Rochefort, H. Early effect of estrogen on chromatin ultrastructure in endometrial nuclei. Mol. Cell. Endocrinol. 1980, 19, 79–92. [Google Scholar] [CrossRef]
- Verrijdt, A.; Leclercq, G.; Devleeschouwer, N.; Danguy, A. Tritiated actinomycin-D staining method: A valuable tool to study estrogen receptor-induced modifications of transcriptional activity in normal and neoplastic cells. Arch. Int. Physiol. Biochim. 1985, 93, 65–73. [Google Scholar] [CrossRef]
- Leclercq, G.; Hulin, N.; Heuson, J.-C. Interaction of activated of estradiol receptor complex and chromatin in isolated uterine nuclei. Eur. J. Cancer. 1973, 9, 681–685. [Google Scholar] [CrossRef]
- El Khissin, A.; Leclercq, G. Exchange of bound estrogens and antiestrogens in MCF-7 cells: Evidence for ligand–induced stable configurations of the estrogen receptor. Steroids 1998, 63, 65–574. [Google Scholar] [CrossRef]
- Laïos, I.; Journé, F.; Laurent, G.; Nonclercq, D.; Toillon, R.-A.; Seo, H.-S.; Leclercq, G. Mechanism governing the accumulation of estrogen receptor alpha in MCF-7 breast cancer cells treated with hydroxytamoxifen and related antiestrogens. J. Steroid Biochem. Mol. Biol. 2003, 87, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Madeddu, L.; Legros, N.; De Vleeschouwer, N.; Bosman, C.; Piccart, M.J.; Leclercq, G. Estrogen receptor status and estradiol sensitivity of MCF-7 cells in exponential growth phase. Eur. J. Cancer Clin. Oncol. 1988, 23, 185–190. [Google Scholar] [CrossRef]
- Metivier, R.; Penot, G.; Hubner, M.R.; Reid, G.; Rand, H.; Kos, M.; Gannon, F. Estrogen receptor-alpha directs ordered, cyclic and combinatorial recruitment of cofactors on a natural target promotor. Cell 2003, 115, 751–763. [Google Scholar] [CrossRef] [Green Version]
- Inwald, E.C.; Klinkhammer-Schalke, M.; Hofstädter, F.; Zeman, F.; Koller, M.; Gersternhauer, M.; Ortmann, O. Ki-67 is a prognostic parameter in breast cancer patients: Results of a large population-based cohort of cancer registry. Brest Cancer Res Treat. 2013, 139, 539–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Wu, S.; Zhou, J.; Sun, J.; Lin, Q.; Lin, H.; Guan, X.; He, Z. Prognostic value of Ki-67 in breast cancer patients with positive lymph nodes: A retrospective cohort study. PLoS ONE 2014, 9, e87264. [Google Scholar] [CrossRef]
- Sun, X.; Kaufman, P.D. Ki-67: More than a proliferation marker. Chromosoma 2018, 127, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Doisneau-Sixou, S.F.; Sergio, S.M.; Caroll, J.S.; Hui, R.; Mushgrove, E.A.; Sutherland, R.L. Estrogens and antiestrogens regulation of cell cycle progression in breast cancer cells. Endocr. Relat. Cancer 2003, 10, 179–186. [Google Scholar] [CrossRef]
- Zheng, Y.; Murphy, L.C. Regulation of steroid hormones receptors and co regulators during cycle highlights potential novel function in addition to roles as transcription factors. Nuclear Recept. Signal. 2016, 14, e001. [Google Scholar] [CrossRef] [Green Version]
- Maynadier, M.; Ramirez, J.-M.; Cathiard, A.-M.; Platet, N.; Gras, D.; Geizes, M.M.; Saeed, S.; Nirde, P.; Garcia, M. Unliganded estrogen receptor alpha inhibits breast cancer cell growth through interaction with a cyclin-dependent kinase inhibitor (p21(WAF1)). FASEB J. 2007, 22, 671–681. [Google Scholar] [CrossRef]
- Wasierska-Gadek, J.; Mauritz, M. Why (multi) targeting of cyclin-dependent kinases is a promising therapeutic option for hormone-positive breast cancer and beyond. Future Med. Chem. 2016, 8, 55–72. [Google Scholar] [CrossRef]
- Rostagno, P.; Moll, J.I.; Birtwisle-Peyrottes, I.; Ettore, F.; Caldani, C. Cell cycle expression of estrogen receptors determined by image analysis on human breast cancer cells in vitro and in vivo. Breast Cancer Res. Treat. 1996, 39, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Danguy, A.; Kiss, R.; Leclercq, G.; Heuson, J.C.; Pasteels, J.L. Morphology of MXT mammary tumors. Correlation with growth characteristics and hormone sensitivity. Eur. J. Cancer Clin. Oncol. 1986, 22, 69–75. [Google Scholar] [PubMed]
- Piccart, M.J.; Tivedi, S.; Maaroufi, Y.; Debbaudt, A.; Leclercq, G. Evolution towards hormone independence of the MXT mouse mammary tumor is associated with a gradual change in its estrogen receptor molecular polymorphism. Cancer Biochem. Biophys. 1998, 16, 169–182. [Google Scholar] [PubMed]
- El Khissiin, A.; Journé, F.; Laïos, I.; Seo, H.-S.; Leclercq, G. Evidence of an estrogen receptor form devoid of estrogen binding ability in MCF-7 cell. Steroids 2000, 65, 903–913. [Google Scholar] [CrossRef]
- JavanModoghadam, S.; Weihua, Z.; Hunt, K.K.; Keyomarsi, K. Estrogen receptor alpha is cell cycle–regulated and regulates cell cycle in a ligand-dependent fashion. Cell Cycle 2016, 15, 1579–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saceda, M.; Lindsey, R.K.; Solomon, H.; Angeloni, S.; Martin, M.B. Estradiol regulates estrogen receptor mRNA stability. J. Steroid Biochem. Mol. Biol. 1998, 66, 113–120. [Google Scholar] [CrossRef]
- Laïos, I.; Journe, F.; Nonclercq, D.; Salazar Vidal, D.; Toillon, R.-A.; Leclercq, G. Role of the proteasome in the regulation of estrogen receptor α turnover and function in MCF-7 breast cancer cells. J. Steroids Biochem. Molec. Biol. 2005, 94, 347–359. [Google Scholar] [CrossRef] [PubMed]
- El Khissiin, A.; Leclercq, G. Implication of proteasome in estrogen receptor degradation. FEBS Lett. 1999, 448, 160–166. [Google Scholar] [CrossRef] [Green Version]
- Nonclercq, D.; Journé, F.; Laïos, I.; Chaboteaux, C.; ToillonR, A.; Leclercq, G.; Laurent, G. Effect of nuclear receptor inhibition on estrogen rececptor regulation in breast cancer cells. J. Mol. Endocrocrinol. 2007, 39, 105–118. [Google Scholar] [CrossRef] [Green Version]
- Leclercq, G.; Legros, N.; Piccart, M.J. Accumulation of a non-binding form of estrogen receptor in MCF-7 cells under hydroxytamoxifen treatment. J. Steroid Biochem. Mol. Biol. 1992, 41, 545–552. [Google Scholar] [CrossRef]
- Tecalco-Cruz, A.C.; Pérez-Alvarado, I.A.; Ramirez-Jarquin, J.O. Nucleo-cytoplasmic transport of estrogen receptor alpha in breast cancer cells. Cell. Signal. 2017, 34, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Kondavoka, I.V.; Shashova, E.E.; Sidenko, E.A.; Astahova, T.M.; Zakarova, L.A.; Sharova, N.P. Estrogen receptors and ubiquitin proteasome system. Mutual regulation. Biomolecules 2020, 10, 500. [Google Scholar]
- Powers, G.L.; Rajbhandari, P.; Solodin, N.M.; Brickford, B.; Alarid, E.T. The proteasome inhibitor Bortesomib induces an inhibitory chromatin environment at a distal enhancer of the estrogen receptor-α gene. PLoS ONE 2013, 8, e81110. [Google Scholar] [CrossRef] [PubMed]
- Ayaz, G.; Vasar, P.; Karakya, B.; Kars, G.; Razizaden, N.; Yavuz, K.; Turan, G.; Muyan, M. Dynamic transcriptional events mediated by estrogen receptor alpha. Front. Biosci. 2019, 24, 245–276. [Google Scholar]
- Le Romancer, M.; Poulard, C.; Cohen, P.; Sentis, S.; Renoir, J.-M.; Corbo, L. Cracking the estrogen receptors posttranslational code in breast tumors. Endocr. Rev. 2011, 32, 597–622. [Google Scholar] [CrossRef] [Green Version]
- Le Dilly, F.; Beato, M. Signaling by steroid hormones in the 3D nuclear space. Int. J. Mol. Sci. 2018, 19, 306. [Google Scholar] [CrossRef]
- Pesiri, V.; Di Muzio, E.; Polticelli, F.; Acconcia, F. Selective binding of estrogen receptor α. IUBMB Life 2016, 68, 497–501. [Google Scholar] [CrossRef] [Green Version]
- Tecalco-Cruz, A.C.; Ramirez-Jarquin, J.O. Polyubiquitination inhibition of esytogen receptor alpha and its implication in breast cancer. World J. Clin. Oncol. 2018, 9, 60–70. [Google Scholar] [CrossRef]
- Tecalco-Cruz, A.C.; Ramirez-Jarquin, J.O.; Cruz-Ramos, E. Estrogen receptor alpha and its ubiquitination in breast cancer. Curr. Drug Targets 2019, 20, 690–704. [Google Scholar] [CrossRef]
- Gallo, D.; Haddad, I.; Laurent, G.; Vinh, J.; Jacquemotte, F.; Jacquot, Y.; Leclercq, G. Regulatory function of the P295-T311 motif of the estrogen receptor α—Does proteosomal degradation of the receptor induce peptides implicated in estrogenic responses? Nuclear Recept. Signal. 2018, e007. [Google Scholar] [CrossRef] [Green Version]
- Totta, P.; Perisi, V.; Marino, M.; Acconcia, F. Lysosomial function is involved in 17 bêta estradiol-induced estrogen receptor alpha degradation and cell proliferation. PLoS ONE 2014, 9, e94880. [Google Scholar] [CrossRef] [PubMed]
- Totta, P.; Busonero, C.; Leone, S.; Marino, M.; Acconcia, F. Dynamin II is required for 17β-estradiol signaling and autophagy-based ERα degradation. Sci. Rep. 2018, 6, 23727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampayo, R.G.; Toscani, A.M.; Rubashkin, M.G.; Thi, K.; Masullo, L.A.; Violi, I.L.; Lakins, J.N.; Caceres, A.A.; Hines, W.C.; Leskow, F.C.; et al. Fibronectine rescues estrogen receptor α from lysosomal degradation in breast cancer cells. J. Cell. Biol. 2018, 217, 2777–2798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eick, G.; Thornton, J.W. Evolution of steroid receptors from an estrogen-sensitive ancestral receptor. Mol. Cell. Endocrinol. 2011, 334, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Albini, A.; Rosano, C.; Angelini, A.; Amaro, A.; Esposito, A.I.; Maramotti, S.; Noonan, D.M.; Pfeffer, U. Exogenous hormonal regulation in breast cancer cells by phytoestrogens and endocrine disruptors. Curr. Med. Chem. 2014, 21, 1129–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiss, A.F.; Benzeno, S.; Rao, J.; Caplan, A.J. Control of estrogen ligand binding by Hsp90. J. Steroid Biochem. Mol. Biol. 2000, 72, 223–230. [Google Scholar]
- Lee, M.O.; Kim, E.O.; Kwon, H.J.; Kim, Y.M.; Kang, H.J.; Kang, H.; Lee, J.L. Radicicol represses transcriptional activity of estrogen receptor the by suppressing the stabilization of the receptor by heat shock protein 90. Mol. Cell. Endocrinol. 2002, 188, 47–54. [Google Scholar] [CrossRef]
- Nonclercq, D.; Journé, F.; Body, J.-C.; Leclercq, G.; Laurent, G. Ligand-independent and agonist-mediated degradation of estrogen receptor -α in breast carcinoma cells; evidence for distinct degradative pathways. Mol. Cell. Endocrinol. 2004, 227, 53–65. [Google Scholar] [CrossRef]
- Leclercq, G.; Jensen, E.V. Dimeric structure of unoccupied; activated estrogen receptor. In Proceedings of the Fifth International CBT Symposium on Intracellular Hormone Receptors and Ligand-Dependent Transcriptional Regulation, Huddinnge, Sweden, 6–8 September 1993; p. 91. [Google Scholar]
- Klinge, C.M. Estrogen binding to estrogen response elements slows ligand dissociation and synergically activates reporter gene expression. Mol. Cell Endocrinol. 1999, 150, 99–111. [Google Scholar] [CrossRef]
- Sonoda, M.T.; Martinez, L.; Webb, P.; Skaf, M.S.; Polikarpov, I. Ligand dissociation from estrogen receptor is mediated by receptor dimerization: Evidence from molecular dynamics simulations. Molec. Endorinol. 2008, 22, 1565–1578. [Google Scholar] [CrossRef] [Green Version]
- Leclercq, G.; Laïos, I.; Elie-Caille, C.; Leiber, D.; Laurent, G.; Lesnivska, E.; Tanfin, Z.; Jacquot, Y. ERα dimerisation: A key factor for the weak estrogenic activity of an ERα modulator unable to compete with estradiol in binding assays. J. Recept. Signal Transdcuct. Res. 2017, 37, 149–166. [Google Scholar] [CrossRef] [PubMed]
- Razandi, M.; Pedram, A.; Merchenthaler, I.; Greene, G.L.; Levin, E.R. Plasma membrane estrogens receptors exist and function as dimers. Mol. Endocrinol. 2004, 18, 2854–2865. [Google Scholar] [CrossRef] [PubMed]
- Jacquot, Y.; Spaggiari, D.; Schappler, J.; Lesniewska, E.; Rudaz, S.; Leclercq, G. ERE-dependent transcription and cell proliferation: Independence of these two processes mediated by the introduction of a sulfone function into the weak estrogen estrothazine. Eur. J. Pharm. Sci. 2017, 109, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Jacquot, Y.; Laïos, I.; Cleeren, A.; Nonclercq, D.; Bermont, L.; Refoulevet, B.; Boubekeur, K.; Xicluna, A.; Leclercq, G.; Laurent, G. Synthesis, structure, and estrogenic activity of 4-amino-3-(2 methylbenzyl) coumarins on human breast cancer cells. Bioorg. Med. Chem. 2007, 15, 2269–2282. [Google Scholar] [CrossRef] [PubMed]
- Laïos, I.; Cleeren, A.; Leclercq, G.; Nonclercq, D.; Laurent, G.; Schlenk, M.; Wellner, A.; Gust, R. Effects of (R/S)/(S,R)-4,5-bis(2-chloro_4-Hydroxyphenyl)piperazines on estrogen receptor alpha level and transcriptional activity in MCF-7 cells. Biochem. Pharmacol. 2000, 74, 1029–1038. [Google Scholar]
- Jacquot, Y.; Gallo, D.; Leclercq, G. Estrogen receptor alpha—Identification by a modeling approach of a potential polyproline II recognizing domain within the AF-2 region of the receptor that would play a role of prime importance in its mechanism of action. J. Steroid Biochem. Mol. Biol. 2007, 104, 1–10. [Google Scholar] [CrossRef]
- Gallo, D.; Leclercq, G.; Jacquot, Y. The N-Terminal part of the ligand binding domain of the the human estrogen receptor α: A new target for estrogen disruptors. In Medical Chemistry Research Progress; Colombo, G.P., Ricci, S., Eds.; Nova Science Publishers: Hauppauge, NY, USA, 2008. [Google Scholar]
- Buzon, V.; Carbo, L.R.; Estruch, S.B.; FLetterick, R.J.; Estebanez-Perpina, E. A conserved surface on the ligand binding domain of nuclear receptors for allosteric controls. Mol. Cell. Endocrinol. 2012, 348, 394–402. [Google Scholar] [CrossRef]
- Mackinmon, J.A.; Gallastegui, N.; Osguthorpe, D.J.; Hagler, A.T.; Estebanez-Perpina, E. Allosteric mechanisms of nuclear receptors: Insights from computional simulations. Mol. Cell. Endocrinol. 2014, 393, 75–78. [Google Scholar] [CrossRef]
- Li, I.; Sacks, D.B. Functional interactions between calmodulin and estrogen receptor-alpha. Cell Signal. 2007, 19, 439–443. [Google Scholar] [CrossRef]
- Gallo, D.; Jacquot, Y.; Laurent, G.; Leclercq, G. Calmodulin, a regulatory partner of the estrogen receptor alpha in breast cancer cells. Mol. Cell. Endocrinol. 2008, 268, 20–26. [Google Scholar] [CrossRef] [Green Version]
- Carlier, L.; Byrne, C.; Miclet, E.; Bourgouin-Voillard, S.; Nicaise, M.; Tabet, J.C.; Desmadrol, M.; Leclercq, G.; Lequin, O.; Jacquot, Y. Studies of the interaction between calmodulin and the R287-T311 region of human estrogen receptor α reveal an atypical binding process. Biochem. Biophys. Res. Commun. 2012, 419, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Y.; Hedman, A.C.; Ames, J.B.; Sacks, D.B. Calmodulin lobes facilitate dimerization and activation of estrogen receptor-α. J. Biol. Chem. 2017, 292, 4614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourgouin-Voillard, S.; Fournier, F.; Alfonso, C.; Jacquot, Y.; Leclercq, G.; Tabet, J.C. Calmodulin association with the synthetic ERα 17p peptide investigated by Mass Spectrometry. Int. J. Mass Spectrom. 2011, 305, 87–94. [Google Scholar] [CrossRef]
- Gallo, D.; Haddad, I.; Duvillier, H.; Jacquemotte, F.; Laïos, I.; Laurent, G.; Jacquot, Y.; Vinh, H.; Leclercq, G. Trophic effect in MCF-7 cells of ERalpha17p, a peptide corresponding to a platform regulatory motif of the estrogen receptor alpha–Underlying mechanism. J. Steroid Mol. Biol. 2008, 109, 138–149. [Google Scholar] [CrossRef]
- Byrne, C.; Miclet, E.; Broutin, I.; Gallo, D.; Pelekanou, V.; Kampa, M.; Leclercq, G.; Jacquot, Y. Identification of polyproline II regions derived from the proline-rich nuclear coactivators PNRC and PNRC2: New insights for ERα coactivators interactions. Cherality 2013, 25, 628–642. [Google Scholar] [CrossRef]
- Leiber, D.; Burlina, F.; Byrne, C.; Robin, P.; Piesse, C.; Gonzalez, L.; Leclercq, G.; Tafin, Z.; Jacquot, Y. The sequence Pro298-Thr311 of the hinge of oestrogen receptor a involved in ERK1/2 activation via GPR30 in leiomyoma cells. Biochem. J. 2015, 472, 97–104. [Google Scholar] [CrossRef]
- Norman, A.W.; Mizicki, M.T.; Norman, D.P. Steroid-hormone rapid actions. Membrane receptors and conformational ensemble model. Nat. Rev. Drug Discov. 2004, 3, 27–41. [Google Scholar] [CrossRef]
- Jordan, V.C.; Schafer, J.M.; Levenson, A.S.; Liu, H.; Pease, K.M.; Simons, L.A.; Zapf, J.W. Molecular classification of estrogens. Cancer Res. 2001, 61, 6619–6623. [Google Scholar]
- Bai, Z.; Gust, R. Breast cancer, estrogen receptor and ligands. Arch. Pharm. 2009, 342, 133–149. [Google Scholar] [CrossRef]
- Bourgoin-Voillard, S.; Gallo, D.; Laïos, I.; Cleeren, A.; El Bali, L.; Jacquot, Y.; Nonclercq, D.; Laurent, G.; Tabet, J.-C.; Leclercq, G. Capacity of type I and II to confer to estrogen receptor alpha an appropriate conformation for the recruitment of coactivators containing a LxxLL motif—Relationship with the regulation of receptor level and ERE-dependent transcription in MCF-7 cells. Bichem. Pharmacol. 2010, 79, 746–757. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.-B.; Graber, C.T.; Quinn, J.A.; Filardo, E.J. Retrograde transport of the membrane estrogen receptor, G-protein-coupled-receptor-30 (GPR30/GPR) from the plasma membrane towards the nucleus. Steroids 2011, 76, 892–896. [Google Scholar] [PubMed]
- Mairesse, N.; Devleeschouwer, N.; Leclercq, G.; Galand, P. Estogen-induced synthesis and secretion of proteins in the human breast cancer cell line MCF-7. J. Steroid Biochem. 1981, 15, 375–381. [Google Scholar] [CrossRef]
- Olea-Serrano, N.; Leclercq, G.; Mairesse, N.; Heuson, J.-C. Bulk protein secretion induced by estradiol in MCF-7 cells. Eur. J. Cancer Clin. Oncol. 1985, 21, 1267–1271. [Google Scholar] [CrossRef]
- Linares, P.M.; Algaba, A.; Urzainqui, A.; Guijarro-Rojas, M.; Fernandez-Contreras, M.E.; Garrido, J.; Chaparro, M.; Gisbert, J.P.; Bermejo, F.; Guerra, I.; et al. Ratio of circulating estrogen receptors bêta and alpha (ERβ/ERα) indicates endoscopic activity in patients with Crohn’s disease. Dig. Dis. Sci. 2017, 62, 2744–2754. [Google Scholar] [CrossRef] [PubMed]
- Notas, G.; Kampa, M.; Pelekanou, V.; Toullinaki, M.; Jacquot, Y.; Leclercq, G.; Castanas, E. Whole transcriptome analysis of the ERα synthetic fragment P295-T311 (ERa17p) identifies specific ERα-isoform (ERα, ERα36)-dependent and independent actions in breast cancer cells. Mol. Oncol. 2013, 7, 595–610. [Google Scholar] [CrossRef]
- Maselli, A.; Cappoccia, S.; Pugliese, P.; Raggi, C.; Cirulli, F.; Fabi, A.; Maloni, W.; Perdomicini, M.; Ortona, E. Antibodies specific to estrogen receptor alpha act as estrogen agonists and their levels correlate with breast cancer cell proliferation. Oncoimmunology 2015, 12, e107375. [Google Scholar]
- Leclercq, G. Natural anti-estrogen receptor alpha antibodies able to induce estrogenic responses in breast cancer cells: Hypotheses concerning their mechanisms of action and emergence. Int. J. Mol Sci. 2018, 30, 411. [Google Scholar] [CrossRef] [Green Version]
- Maselli, A.; Perlato, S.; Pugglisi, R.; Riggi, C.; Spada, M.; Macchia, D.; Pontecorvi, G.; Iessi, E.; Pagano, M.T.; Cirulli, F.; et al. Antibodies specific to ERα are involved in tamoxifen resistance in hormone positive breast cancer. Cells 2019, 8, 750. [Google Scholar] [CrossRef] [Green Version]
- Herynck, M.H.; Fuqua, S.A.W. Estrogen receptor mutations in human disease. Endocr. Rev. 2004, 25, 869–898. [Google Scholar] [CrossRef] [Green Version]
- Barone, I.; Brusco, L.; Fuqua, S.A.W. Estrogen receptor mutations and changes in downstream gene expression signaling. Clin. Cancer Res. 2010, 16, 2702–2708. [Google Scholar] [CrossRef] [Green Version]
- Reinert, T.; Saad, E.A.; Barrios, C.H.; Bines, J. Cinical implications of ESR1 mutations in hormone receptor-positive advanced breast cancers. Front. Oncol. 2017, 7, e00026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, V.C.; Corpan, R.; Maximov, P.Y. Estrogen receptor mutations found in breast cancer metastases integrated with molecular pharmacology of selective ER modulators. J. Natl. Cancer Inst. 2015, 107, 075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.-Y.; Yin, L. Estrogen receptor alpha-36 (ER-α36): Anew player in human breast cancer. Mol. Cell. Endocrinol. 2015, 418, 193–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelekanou, V.; Notas, G.; Kampa, M.; Tsentelierou, E.; Radojiric, J.; Leclercq, G.; Castanas, E.; Stathopoulos, E. ERα, a new variant of the ERα is expressed in triple negative breast carcinomas and has a specific transcriptomic signature in breast cancer cell lines. Steroids 2012, 77, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Chaudhri, R.A.; Haddadi, A.; Lobachev, K.S.; Schwartz, Z.; Boyan, B.D. Estrogen receptor-alpha 36 mediates the ant-apoptotic effect of estradiol in triple negative cancer cells via a membrane-Aassociated mechanism. Biochim. Biophys. Acta Mol. Res. 2014, 1843, 2796–2806. [Google Scholar] [CrossRef] [Green Version]
- Pagano, M.T.; Ortona, E.; Dupuis, M.L. A role for estrogen receptor alpha36 in cancer progression. Font. Endocrinol. 2020, 11, e00506. [Google Scholar] [CrossRef]
- Merenbakh-Lamin, K.; Ben-Baruch, N.; Yesheskel, A.; Dvir, A.; Soussan-Gutman, L.; Jeselsohn, R.; Yelensky, R.; Brown, M.; Miller, V.A.; Sarid, D.; et al. D538G mutation in estrogen receptor-α: A novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res. 2013, 73, 6856–6864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fanning, S.W.; Mayne, C.G.; Dharmarajan, V.; Carlson, K.E.; Martin, T.A.; Novick, S.J.; Toy, W.; Green, B.; Panchamukhi, S.; Katzenellenbogen, B.S.; et al. Estrogen receptor alpha somatic Mutations Y537S and D538G confer breast cancer endocrine resistance by stabilising the activating function2 binding conformation. Elife 2016, 5, e122792. [Google Scholar] [CrossRef] [PubMed]
- Pavlin, M.; Gelsomino, L.; Barone, I.; Spinello, A.; Catalano, S.; Ando, S.; Magisrato, A. Structural Thermodynamic and kinetic traits of antiestogen-componds selectivity targeting the Y537S mutant estrogen receptor α transcritional activity in breast cancer cell lines. Front. Chem. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordano, G.; Cui, Y.; Barone, I.; Ando, S.; Mancini, A.M.; Berno, V.; Fuqua, A.W. Growth factor-indued to tamoxifen is associated with a mutation of estrogen receptor alpha and its phosphorylation at serine 305. Breast Cancer Res. Treat. 2010, 119, 71–85. [Google Scholar] [CrossRef] [Green Version]
- Barone, I.; Iacopetta, D.; Covington, K.R.; Cui, Y.; Tsimelzon, A.; Beyer, A.; Ando, S.; Fuqua, A.W. Phosphorylation of the mutant K303R estrogen receptor alpha at serune 305 affects aromatase inhibitor activity. Oncogene 2010, 29, 404–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leclercq, G.; Gallo, D.; Cossy, J.; Laïos, I.; Larsimont, D.; Laurent, G.; Jacquot, Y. Peptides targeting estrogen receptor alpha–potential applications for breast cancer treatment. Curr. Pharm. Des. 2011, 17, 2632–2653. [Google Scholar] [CrossRef] [PubMed]
- Busoner, C.; Leone, S.; Bartolini, S.; Acconcia, F. Strategies to degrade estrogen receptor α in primary and ESR1 mutant-expressing metastatic breast cancer. Mol. Cell. Endocrinol. 2019, 480, 107–121. [Google Scholar] [CrossRef] [PubMed]
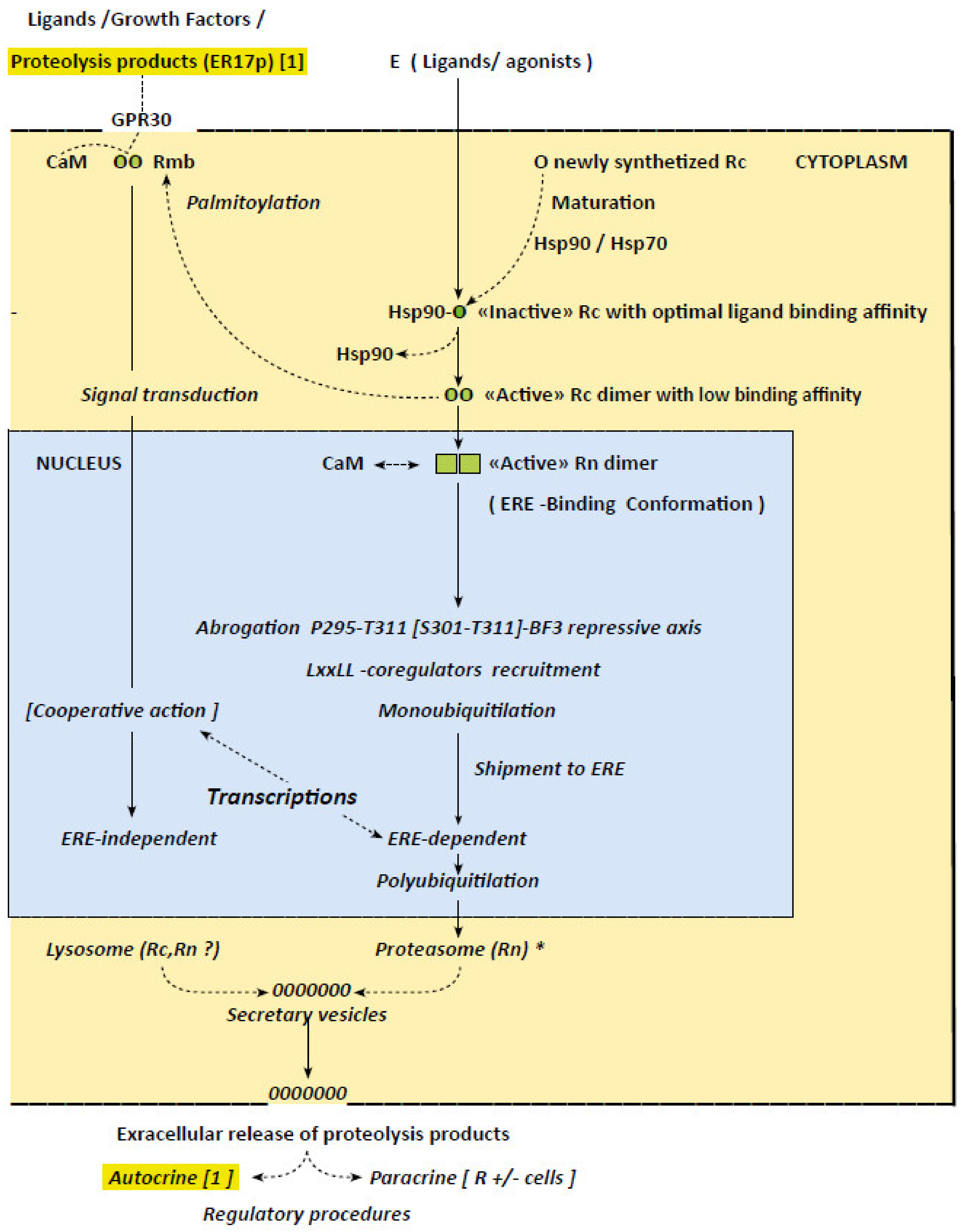
Figure 1.
Schematic overview of the concepts relative to the irreversible character of the mechanisms by which ERα operate. Figure depicts steps of the procedures giving rise in fine to ERE- dependent and -independent transcription induced, respectively, by agonist ligands and growth factors, implying a signal transduction pathway issued from the plasma membrane. Proteosomal degradation of a receptor having accomplished its mission is also recorded, as is the the suspected extracellular secretion of resulting peptides in the cyclic character of the transcription procedures via an autocrine regulation (eventually in cooperation with a lysosomal lysis). Sustained intracellular traffics are identified (full lines) to distinguish them from those solely evoked or suspected (dotted lines). Circles and squares symbolize the receptor, respectively, in its cytoplasmic and nuclear form. Open circles refers to the native/“inactive” receptor; green circles refer to estrogen bound receptors of which the green intensity is related to their complexion stability. Vertical full lines refer to the successive events giving rise in fine to transcriptions, while dotted lines focus on related interactions and conformational changes. The asterisk on (E-Rn)* indicates that the chemical structures of the activating ligands may orient the receptor to distinct proteasomes. Concepts recorded in the figure: Primary cytoplasmic events: Maturation of native, newly synthetized, receptor in an “inactive” form with optimal ligand binding affinity (Hsp90~R complex; Section 4 and Section 7, ref. [7,8]). Estrogen-induced abrogation of a repressive action on this inactive receptor exerted by Hsp90 (Section 4 and Section 5) giving rise to an “active” dimer with low binding affinity (Section 5; Supplementary Materials: 5S form). Palmitoylation of a receptor pool (Section 4) for shipment to the plasma membrane and signal transductions activations (Section 1 and Section 5: ref. [63,64]).) Nuclear events: CaM-induced stabilization of active dimer in a form accurate for complexion with EREs and most probably anchorage on the plasma membrane for interactions with GPR30 (Section 6, ref. [74,75] and [79], respectively). Related successive transient conformational changes on hubs required for ERE-dependent transcriptions (i.e., abrogation of repressive P295-T311~BF3 axis; LxxLL-coregulators recruitment; monoubiquitilation-induced shipment for ERE-dependent transcription; ERE-independent transcription in which the receptor operates as a coregulator (Section 1 and Section 4, Section 5 and Section 6). Arrest of transcription through polyubiquitilation-induced translocation to a cytoplasmic proteasome, ensuring its degradation (Section 4). Cytoplasmic proteosomal and lysosomal receptor degradations: Extracellular release of resulting products. Proteosomal degradation operates essentially on the nuclear receptor pool while lysomal procedure focus on the pool implicated in signal transductions (Section 4). Extra cellular secretion of resulting peptides implicated in autocrine and paracrine regulations are suspected, even an autocrine procedure [1] contributing to the repetitive character of the receptor action seems likely (Section 7).
Figure 1.
Schematic overview of the concepts relative to the irreversible character of the mechanisms by which ERα operate. Figure depicts steps of the procedures giving rise in fine to ERE- dependent and -independent transcription induced, respectively, by agonist ligands and growth factors, implying a signal transduction pathway issued from the plasma membrane. Proteosomal degradation of a receptor having accomplished its mission is also recorded, as is the the suspected extracellular secretion of resulting peptides in the cyclic character of the transcription procedures via an autocrine regulation (eventually in cooperation with a lysosomal lysis). Sustained intracellular traffics are identified (full lines) to distinguish them from those solely evoked or suspected (dotted lines). Circles and squares symbolize the receptor, respectively, in its cytoplasmic and nuclear form. Open circles refers to the native/“inactive” receptor; green circles refer to estrogen bound receptors of which the green intensity is related to their complexion stability. Vertical full lines refer to the successive events giving rise in fine to transcriptions, while dotted lines focus on related interactions and conformational changes. The asterisk on (E-Rn)* indicates that the chemical structures of the activating ligands may orient the receptor to distinct proteasomes. Concepts recorded in the figure: Primary cytoplasmic events: Maturation of native, newly synthetized, receptor in an “inactive” form with optimal ligand binding affinity (Hsp90~R complex; Section 4 and Section 7, ref. [7,8]). Estrogen-induced abrogation of a repressive action on this inactive receptor exerted by Hsp90 (Section 4 and Section 5) giving rise to an “active” dimer with low binding affinity (Section 5; Supplementary Materials: 5S form). Palmitoylation of a receptor pool (Section 4) for shipment to the plasma membrane and signal transductions activations (Section 1 and Section 5: ref. [63,64]).) Nuclear events: CaM-induced stabilization of active dimer in a form accurate for complexion with EREs and most probably anchorage on the plasma membrane for interactions with GPR30 (Section 6, ref. [74,75] and [79], respectively). Related successive transient conformational changes on hubs required for ERE-dependent transcriptions (i.e., abrogation of repressive P295-T311~BF3 axis; LxxLL-coregulators recruitment; monoubiquitilation-induced shipment for ERE-dependent transcription; ERE-independent transcription in which the receptor operates as a coregulator (Section 1 and Section 4, Section 5 and Section 6). Arrest of transcription through polyubiquitilation-induced translocation to a cytoplasmic proteasome, ensuring its degradation (Section 4). Cytoplasmic proteosomal and lysosomal receptor degradations: Extracellular release of resulting products. Proteosomal degradation operates essentially on the nuclear receptor pool while lysomal procedure focus on the pool implicated in signal transductions (Section 4). Extra cellular secretion of resulting peptides implicated in autocrine and paracrine regulations are suspected, even an autocrine procedure [1] contributing to the repetitive character of the receptor action seems likely (Section 7).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Leclercq, G. Pathological Maintenance and Evolution of Breast Cancer: The Convergence of Irreversible Biological Actions of ER Alpha. Endocrines 2021, 2, 1-14. https://0-doi-org.brum.beds.ac.uk/10.3390/endocrines2010001
AMA Style
Leclercq G. Pathological Maintenance and Evolution of Breast Cancer: The Convergence of Irreversible Biological Actions of ER Alpha. Endocrines. 2021; 2(1):1-14. https://0-doi-org.brum.beds.ac.uk/10.3390/endocrines2010001
Chicago/Turabian StyleLeclercq, Guy. 2021. "Pathological Maintenance and Evolution of Breast Cancer: The Convergence of Irreversible Biological Actions of ER Alpha" Endocrines 2, no. 1: 1-14. https://0-doi-org.brum.beds.ac.uk/10.3390/endocrines2010001