
A Partial Phenotype of adFNDI Related to the Signal Peptide c.55G>A Variant of the AVP Gene

 ,
,  ,
,  , ,
, , 
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Patients and Methods
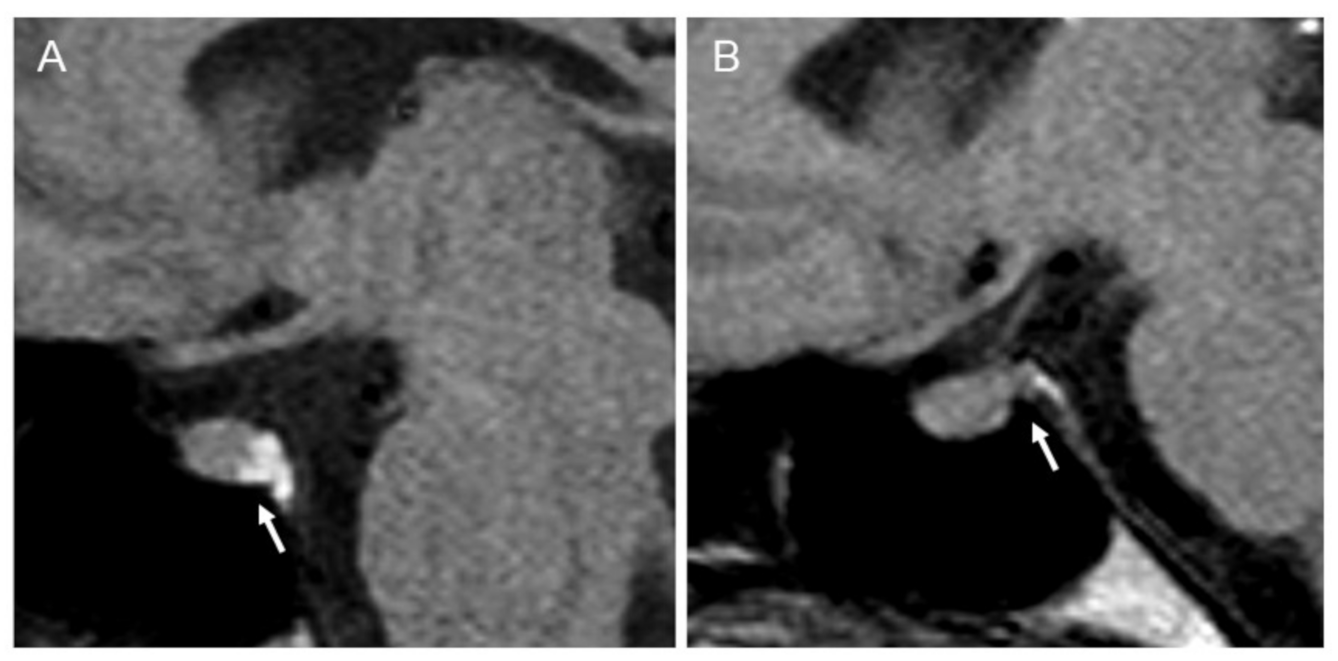
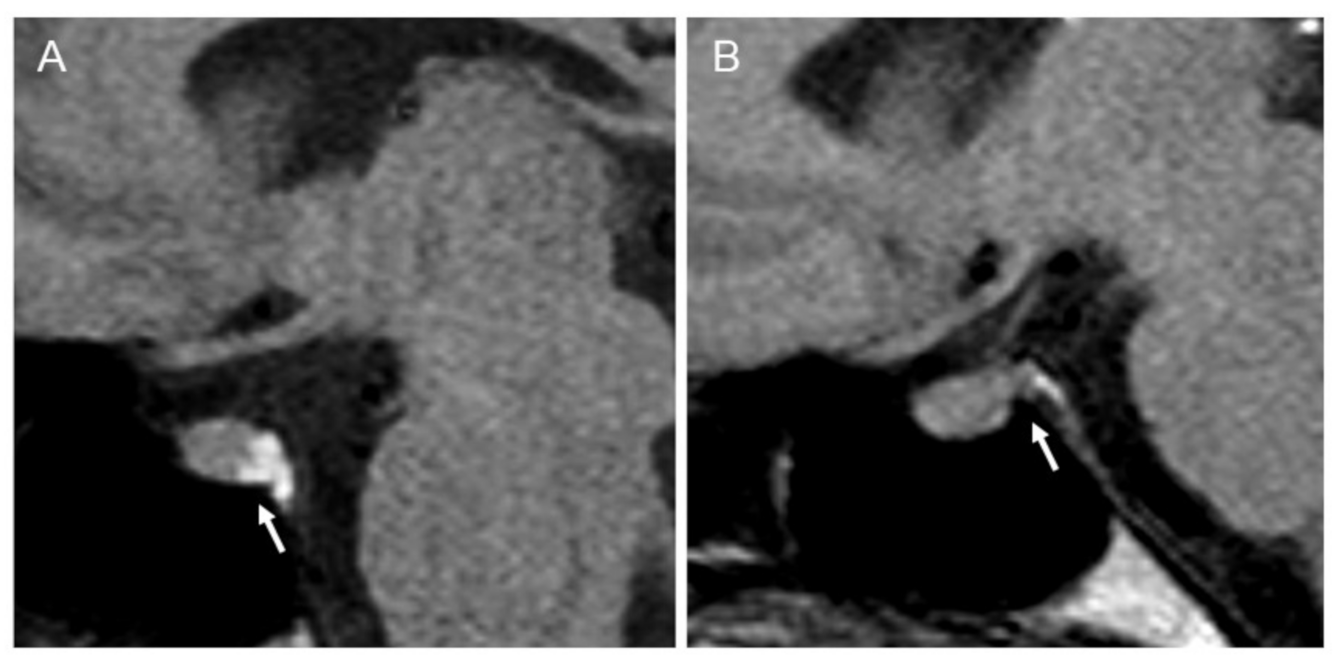
2.1. Clinical and Radiological Assessments
2.2. Genetic Testing
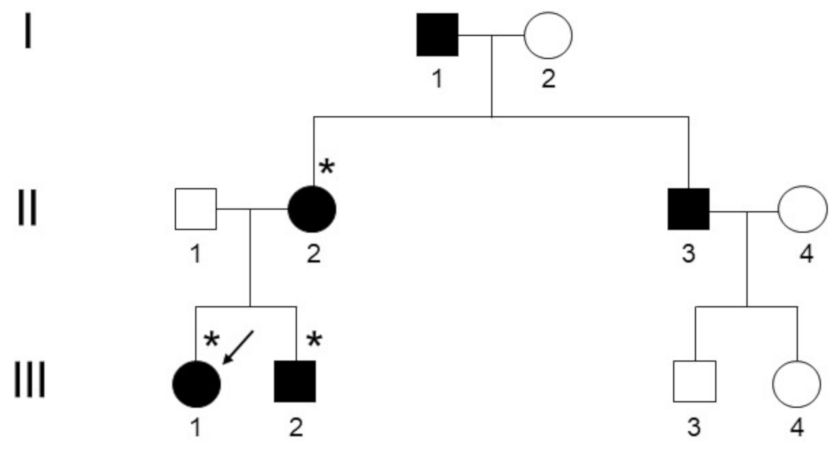
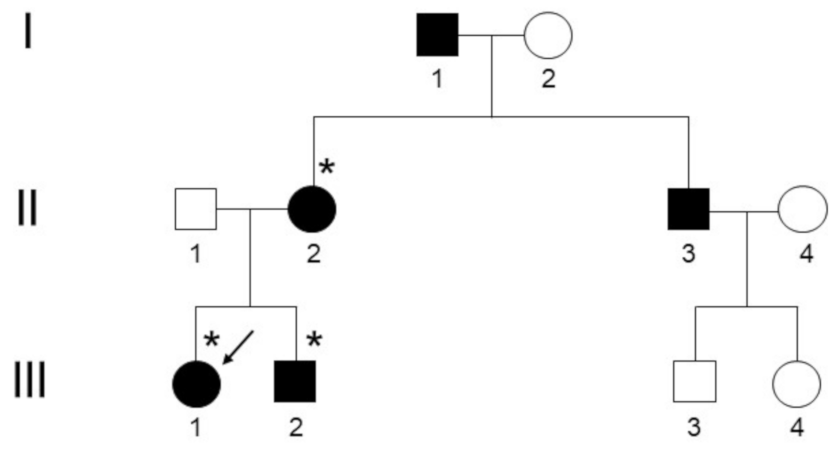
3. Results
3.1. Clinical and Radiological Assessments
3.2. Genetic Testing
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Babey, M.; Kopp, P.; Robertson, G.L. Familial forms of diabetes insipidus: Clinical and molecular characteristics. Nat. Rev. Endocrinol. 2011, 7, 701–714. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, S.; Galiveeti, S.; Bichet, D.G.; Roth, J. Diabetes insipidus: Celebrating a century of vasopressin therapy. Endocrinology 2014, 155, 4605–4621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christ-Crain, M.; Bichet, D.G.; Fenske, W.K.; Goldman, M.B.; Rittig, S.; Verbalis, J.G.; Verkman, A.S. Diabetes insipidus. Nat. Rev. Dis. Prim. 2019, 5, 54. [Google Scholar] [CrossRef]
- Burbach, J.P.; Luckman, S.M.; Murphy, D.; Gainer, H. Gene regulation in the magnocellular hypothalamo-neurohypophysial system. Physiol. Rev. 2001, 81, 1197–1267. [Google Scholar] [CrossRef]
- Knepper, M.A.; Kwon, T.H.; Nielsen, S. Molecular physiology of water balance. N. Engl. J. Med. 2015, 372, 1349–1358. [Google Scholar] [CrossRef]
- Miyata, T.; Hagiwara, D.; Hodai, Y.; Miwata, T.; Kawaguchi, Y.; Kurimoto, J.; Ozaki, H.; Mitsumoto, K.; Takagi, H.; Suga, H.; et al. Degradation of Mutant Protein Aggregates within the Endoplasmic Reticulum of Vasopressin Neurons. iScience. 2020, 23, 101648. [Google Scholar] [CrossRef] [PubMed]
- Russell, T.A.; Ito, M.; Ito, M.; Yu, R.N.; Martinson, F.A.; Weiss, J.; Jameson, J.L. A murine model of autosomal dominant neurohypophyseal diabetes insipidus reveals progressive loss of vasopressin-producing neurons. J. Clin. Investig. 2003, 112, 1697–1706. [Google Scholar] [CrossRef]
- García-Castaño, A.; Madariaga, L.; de Nanclares, G.P.; Vela, A.; Rica, I.; Gaztambide, S.; Martínez, R.; De Lapiscina, I.M.; Urrutia, I.; Aguayo, A.; et al. Forty-One Individuals with Mutations in the AVP-NPII Gene Associated with Familial Neurohypophyseal Diabetes Insipidus. J. Clin. Endocrinol. Metab. 2020, 105, dgaa069. [Google Scholar] [CrossRef]
- Perrotta, S.; Di Iorgi, N.; Ragione, F.D.; Scianguetta, S.; Borriello, A.; Allegri, A.E.M.; Ferraro, M.; Santoro, C.; Napoli, F.; Calcagno, A.; et al. Early-onset central diabetes insipidus is associated with de novo arginine vasopressin-neurophysin II or Wolfram syndrome 1 gene mutations. Eur. J. Endocrinol. 2015, 172, 461–472. [Google Scholar] [CrossRef] [Green Version]
- Toustrup, L.B.; Kvistgaard, H.; Palmfeldt, J.; Bjerre, C.K.; Gregersen, N.; Rittig, S.; Corydon, T.J.; Christensen, J.H. The Novel Ser18del AVP Variant Causes Inherited Neurohypophyseal Diabetes Insipidus by Mechanisms Shared with Other Signal Peptide Variants. Neuroendocrinology 2018, 106, 167–186. [Google Scholar] [CrossRef]
- Siggaard, C.; Rittig, S.; Corydon, T.J.; Andreasen, P.H.; Jensen, T.G.; Andresen, B.S.; Robertson, G.L.; Gregersen, N.; Bolund, L.; Pedersen, E.B. Clinical and molecular evidence of abnormal processing and trafficking of the vasopressin preprohormone in a large kindred with familial neurohypophyseal diabetes insipidus due to a signal peptide mutation. J. Clin. Endocrinol. Metab. 1999, 84, 2933–2941. [Google Scholar] [CrossRef] [Green Version]
- Tian, D.; Cen, J.; Nie, M.; Gu, F. Identification of five novel arginine vasopressin gene mutations in patients with familial neurohypophyseal diabetes insipidus. Int. J. Mol. Med. 2016, 38, 1243–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AME Guidelines. Available online: http://www.associazionemediciendocrinologi.it/materiali/manuali/2006-b/download_file_144.pdf (accessed on 21 December 2020).
- Jendle, J.; Christensen, J.H.; Kvistgaard, H.; Gregersen, N.; Rittig, S. Late-onset familial neurohypophyseal diabetes insipidus due to a novel mutation in the AVP gene. Clin. Endocrinol. 2012, 77, 586–592. [Google Scholar] [CrossRef]
- Maghnie, M.; Villa, A.; Arico, M.; Larizza, D.; Pezzotta, S.; Beluffi, G.; Genovese, E.; Severi, F. Correlation between magnetic resonance imaging of posterior pituitary and neurohypophyseal function in children with diabetes insipidus. J. Clin. Endocrinol. Metab. 1992, 74, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Colombo, N.; Berry, I.; Kucharczyk, J.; De Groot, J.; Larson, T.; Norman, D.; Newton, T.H.; Kucharczyk, W. Posterior pituitary gland: Appearance on MR images in normal and pathologic states. Radiology 1987, 165, 481–485. [Google Scholar] [CrossRef]
- Cizmarova, M.; Nagyova, G.; Janko, V.; Pribilincova, Z.; Virgova, D.; Ilencikova, D.; Kovacs, L. Late onset of familial neurogenic diabetes insipidus in monozygotic twins. Endocr. Regul. 2013, 47, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J.H.; Siggaard, C.; Corydon, T.J.; DeSanctis, L.; Kovacs, L.; Robertson, G.L.; Gregersen, N. Six novel mutations in the arginine vasopressin gene in 15 kindreds with autosomal dominant familial neurohypophyseal diabetes insipidus give further insight into the pathogenesis. Eur. J. Hum. Genet. 2004, 12, 44–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patti, G.; Scianguetta, S.; Roberti, D.; Di Mascio, A.; Balsamo, A.; Brugnara, M.; Cappa, M.; Casale, M.; Cavarzere, P.; Cipriani, S.; et al. Familial neurohypophyseal diabetes insipidus in 13 kindreds and 2 novel mutations in the vasopressin gene. Eur. J. Endocrinol. 2019, 181, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Juul, K.V.; Klein, B.M.; Sandström, R.; Erichsen, L.; Nørgaard, J.P. Gender difference in antidiuretic response to desmopressin. Am. J. Physiol. Renal. Physiol. 2011, 300, F1116–F1122. [Google Scholar] [CrossRef] [PubMed]
- Chiefari, E.; Ventura, V.; Capula, C.; Randazzo, G.; Scorcia, V.; Fedele, M.; Arcidiacono, B.; Nevolo, M.T.; Bilotta, F.L.; Vitiello, M.; et al. A polymorphism of HMGA1 protects against proliferative diabetic retinopathy by impairing HMGA1-induced VEGFA expression. Sci. Rep. 2016, 6, 39429. [Google Scholar] [CrossRef] [Green Version]
- Mirabelli, M.; Chiefari, E.; Caroleo, P.; Arcidiacono, B.; Corigliano, D.M.; Giuliano, S.; Brunetti, F.S.; Tanyolaç, S.; Foti, D.P.; Puccio, L.; et al. Long-Term Effectiveness of Liraglutide for Weight Management and Glycemic Control in Type 2 Diabetes. Int. J. Environ. Res. Public Health 2019, 17, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruni, A.C.; Momeni, P.; Bernardi, L.; Tomaino, C.; Frangipane, F.; Elder, J.; Kawarai, T.; Sato, C.; Pradella, S.; Wakutani, Y.; et al. Heterogeneity within a large kindred with frontotemporal dementia: A novel progranulin mutation. Neurology 2007, 69, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Chiefari, E.; Tanyolaç, S.; Paonessa, F.; Pullinger, C.R.; Capula, C.; Iiritano, S.; Mazza, T.; Forlin, M.; Fusco, A.; Durlach, V.; et al. Functional variants of the HMGA1 gene and type 2 diabetes mellitus. JAMA 2011, 305, 903–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutishauser, J.; Kopp, P.; Gaskill, M.B.; Kotlar, T.J.; Robertson, G.L. Clinical and molecular analysis of three families with autosomal dominant neurohypophyseal diabetes insipidus associated with a novel and recurrent mutations in the vasopressin-neurophysin II gene. Eur. J. Endocrinol. 2002, 146, 649–656. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tocci, V.; Mirabelli, M.; Giuliano, S.; Chiefari, E.; Hagelskjær Knudsen, J.; Kvistgaard, H.; La Torre, D.; Aversa, A.; Foti, D.P.; Hvarregaard Christensen, J.; et al. A Partial Phenotype of adFNDI Related to the Signal Peptide c.55G>A Variant of the AVP Gene. Endocrines 2021, 2, 37-43. https://0-doi-org.brum.beds.ac.uk/10.3390/endocrines2010004
Tocci V, Mirabelli M, Giuliano S, Chiefari E, Hagelskjær Knudsen J, Kvistgaard H, La Torre D, Aversa A, Foti DP, Hvarregaard Christensen J, et al. A Partial Phenotype of adFNDI Related to the Signal Peptide c.55G>A Variant of the AVP Gene. Endocrines. 2021; 2(1):37-43. https://0-doi-org.brum.beds.ac.uk/10.3390/endocrines2010004
Chicago/Turabian StyleTocci, Vera, Maria Mirabelli, Stefania Giuliano, Eusebio Chiefari, Jane Hagelskjær Knudsen, Helene Kvistgaard, Domenico La Torre, Antonio Aversa, Daniela Patrizia Foti, Jane Hvarregaard Christensen, and et al. 2021. "A Partial Phenotype of adFNDI Related to the Signal Peptide c.55G>A Variant of the AVP Gene" Endocrines 2, no. 1: 37-43. https://0-doi-org.brum.beds.ac.uk/10.3390/endocrines2010004