
Autoimmune Hypophysitis with Late Renal Involvement: A Case Report

 , ,
, ,  ,
,  and
and
Abstract
:1. Introduction

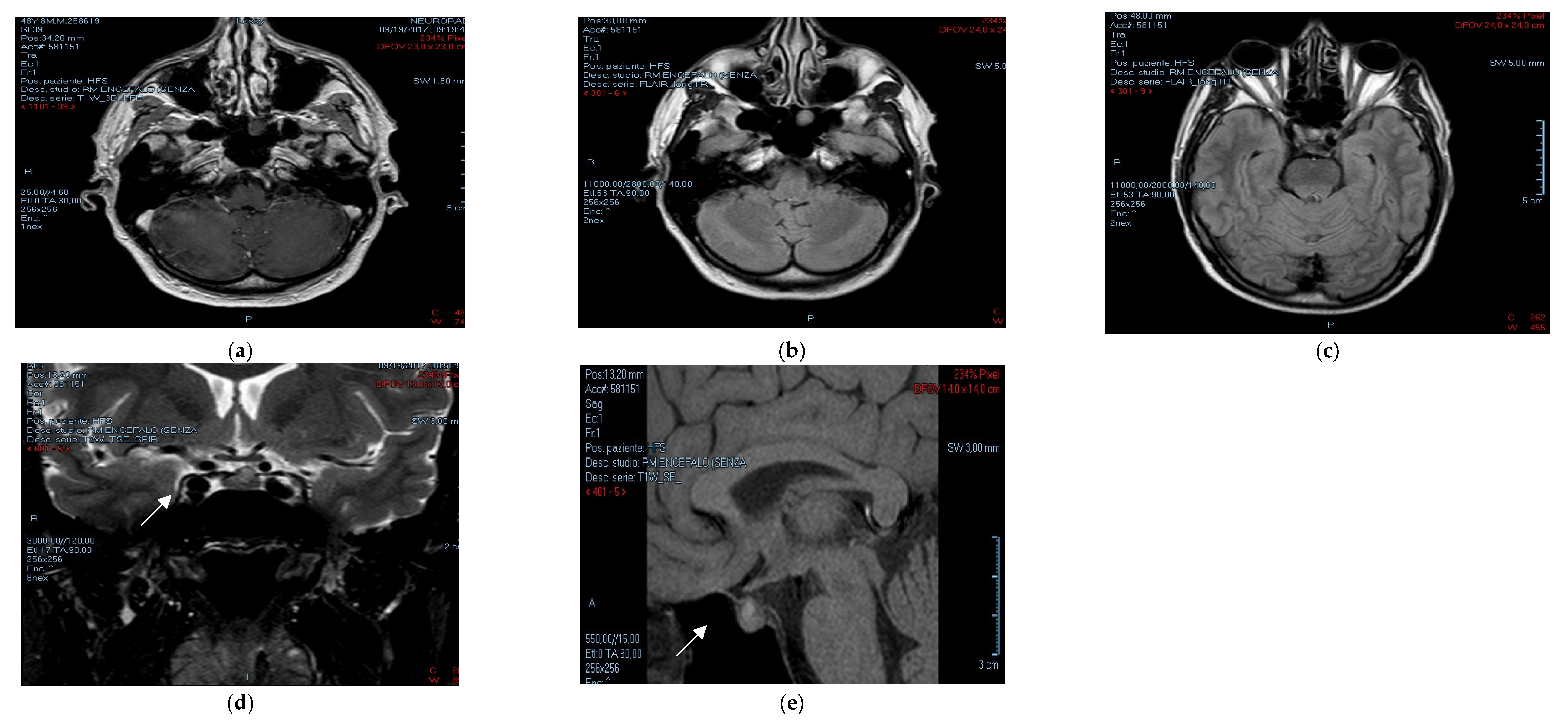{kind=link}
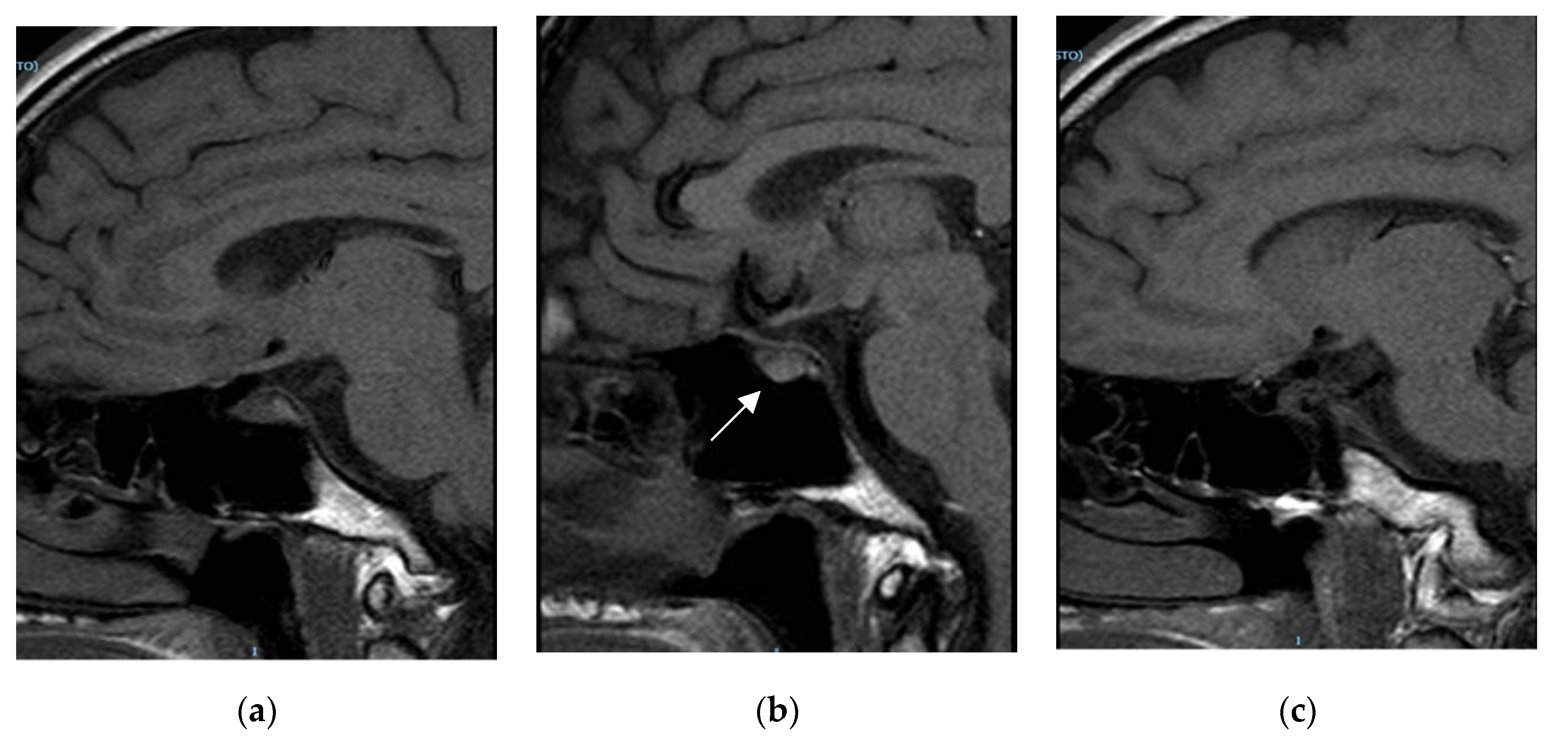{kind=link}
{kind=link}
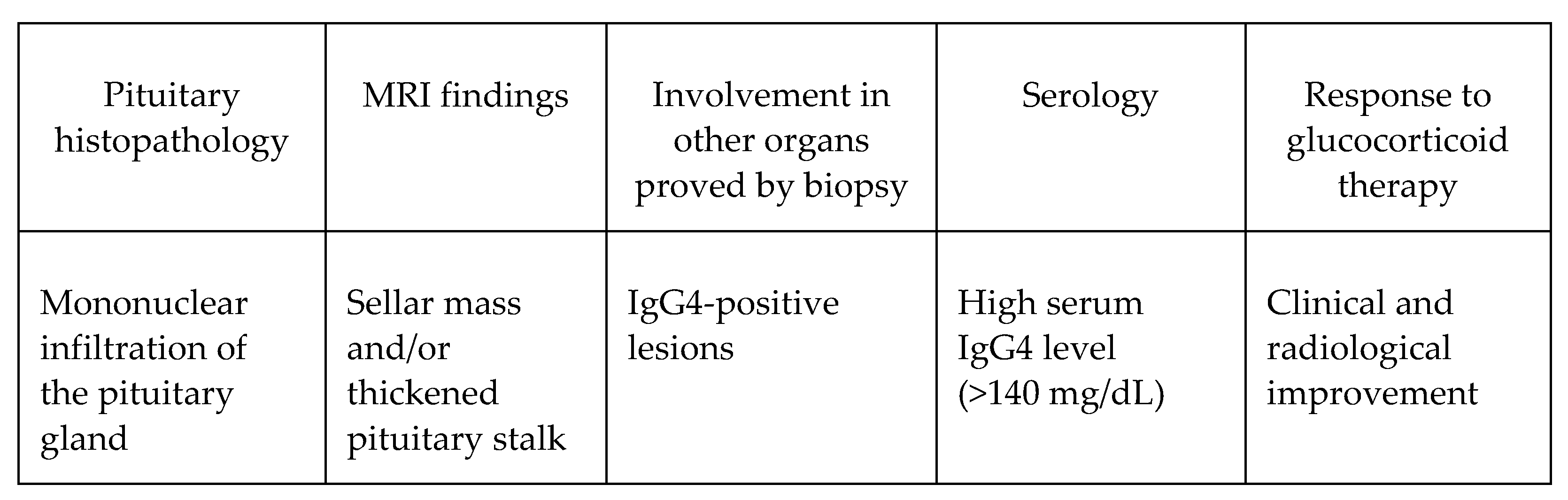{kind=link}
| Anatomic Location | Adenohypophysitis |
| Neurohypophysitis | |
| Panhypophysitis | |
| Histological Findings | Lymphocytic |
| Granulomatous | |
| Xanthomatous | |
| Necrotizing | |
| IgG4 plasmacytic | |
| Etiology | Primary |
| Secondary to systemic diseases, immunomodulatory drugs or sellar involvement |
2. Patients and Methods
2.1. Hormonal Assessment
2.2. Radiological Assessment
2.3. Nephrological Assessment
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shimatsu, A.; Oki, Y.; Fujisawa, I.; Sano, T. Pituitary and stalk lesions (infundibulo-hypophysitis) associated with immunoglobulin G4-related systemic disease: An emerging clinical entity. Endocr. J. 2009, 56, 1033–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Vliet, H.J.; Perenboom, R.M. Multiple pseudotumors in IgG4-associated multifocal systemic fibrosis. Ann. Intern. Med. 2004, 141, 896–897. [Google Scholar] [CrossRef] [PubMed]
- Leporati, P.; Landek-Salgado, M.A.; Lupi, I.; Chiovato, L.; Caturegli, P. IgG4-related hypophysitis: A new addition to the hypophysitis spectrum. J. Clin. Endocrinol. Metab. 2011, 96, 1971–1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lojou, M.; Bonneville, J.F.; Ebbo, M.; Schleinitz, N.; Castinetti, F. IgG4 hypophysitis: Diagnosis and management. Presse Med. 2020, 49, 104016. [Google Scholar] [CrossRef] [PubMed]
- Shikuma, J.; Kan, K.; Ito, R.; Hara, K.; Sakai, H.; Miwa, T.; Kanazawa, A.; Odawara, M. Critical review of IgG4-related hypophysitis. Pituitary 2017, 20, 282–291. [Google Scholar] [CrossRef]
- Joshi, M.N.; Whitelaw, B.C.; Carroll, P.V. MECHANISMS IN ENDOCRINOLOGY: Hypophysitis: Diagnosis and treatment. Eur. J. Endocrinol. 2018, 179, R151–R163. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Sze, G.; Endo, K. Hypophysitis: Endocrinologic and dynamic MR findings. Am. J. Neuroradiol. 1998, 19, 439–444. [Google Scholar] [PubMed]
- Appelman-Dijkstra, N.M.; Claessen, K.M.; Roelfsema, F.; Pereira, A.M.; Biermasz, N.R. Long-term effects of recombinant human GH replacement in adults with GH deficiency: A systematic review. Eur. J. Endocrinol. 2013, 169, R1–R14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koltowska-Häggström, M.; Mattsson, A.F.; Monson, J.P.; Kind, P.; Badia, X.; Casanueva, F.F.; Busschbach, J.; Koppeschaar, H.P.; Johannsson, G. Does long-term GH replacement therapy in hypopituitary adults with GH deficiency normalise quality of life? Eur. J. Endocrinol. 2006, 155, 109–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosman, F.; Post, K.D.; Holub, D.A.; Wardlaw, S.L. Lymphocytic Hypophysitis. Report of 3 New Cases and Review of the Literature. Medicine 1989, 68, 240. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gao, H.; Li, Z.; Zhang, X.; Ding, Y.; Li, F. Clinical Characteristics of 76 Patients with IgG4-Related Hypophysitis: A Systematic Literature Review. Int. J. Endocrinol. 2019, 2019, 5382640. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Lu, S.; Chen, H.; Wu, Q.; Fei, Y.; Li, M.; Zhang, X.; Tian, X.; Zheng, W.; Leng, X.; et al. Clinical characteristics of immunoglobulin G4–related disease: A prospective study of 118 Chinese patients. Rheumatology 2015, 54, 1982–1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Viignera, S.; Izzo, G.; Emerenziani, G.P.; Cannarella, R.; Condorelli, R.A.; Calogero, A.E.; Aversa, A. Male hypogonadism: Therapeutic choices and pharmacological mangement. Minerva Endocrinol. 2020, 45, 189–203. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iuliano, S.; Zagari, M.C.; Vergine, M.; Comi, A.; Andreucci, M.; Patella, G.; Giuliano, S.; La Vignera, S.; Brunetti, A.; Aversa, A.; et al. Autoimmune Hypophysitis with Late Renal Involvement: A Case Report. Endocrines 2021, 2, 160-166. https://0-doi-org.brum.beds.ac.uk/10.3390/endocrines2020016
Iuliano S, Zagari MC, Vergine M, Comi A, Andreucci M, Patella G, Giuliano S, La Vignera S, Brunetti A, Aversa A, et al. Autoimmune Hypophysitis with Late Renal Involvement: A Case Report. Endocrines. 2021; 2(2):160-166. https://0-doi-org.brum.beds.ac.uk/10.3390/endocrines2020016
Chicago/Turabian StyleIuliano, Stefano, Maria Carmela Zagari, Margherita Vergine, Alessandro Comi, Michele Andreucci, Gemma Patella, Stefania Giuliano, Sandro La Vignera, Antonio Brunetti, Antonio Aversa, and et al. 2021. "Autoimmune Hypophysitis with Late Renal Involvement: A Case Report" Endocrines 2, no. 2: 160-166. https://0-doi-org.brum.beds.ac.uk/10.3390/endocrines2020016