
The Stress Axis in Obesity and Diabetes Mellitus: An Update


1
Division of Endocrinology, Diabetes & Metabolism, Department of Medicine, A.O. S. Croce & Carle, 12100 Cuneo, Italy
2
Department of Medical Sciences, University of Turin, 10100 Turin, Italy
3
Division of Internal Medicine, Department of Medicine, A.O. S. Croce & Carle, 12100 Cuneo, Italy
*
Author to whom correspondence should be addressed.
Endocrines 2021, 2(3), 334-347; https://0-doi-org.brum.beds.ac.uk/10.3390/endocrines2030031
Submission received: 25 June 2021
/
Revised: 2 August 2021
/
Accepted: 30 August 2021
/
Published: 6 September 2021
(This article belongs to the Special Issue Neuroregulation of the Hypothalamus-Pituitary-Adrenal)
{kind=link}
Abstract
:The hypothalamic–pituitary–adrenal axis is a tightly regulated system that represents one of the body’s mechanisms for responding to acute and chronic stress. Prolonged stress and/or inadequate regulation of the stress system can lead to a condition of chronic hypercortisolism or, in some cases, a blunted cortisol response to stress, contributing to insulin resistance, increased adiposity and type 2 diabetes mellitus. Moreover, acute and chronic stress can exacerbate or worsen metabolic conditions by supporting an inflammatory state and a tight relationship between stress, inflammation and adipose tissue has been reported and has been a growing subject of interest in recent years. We reviewed and summarized the evidence supporting hypothalamic–pituitary–adrenal axis dysregulation as an important biological link between stress, obesity, inflammation and type 2 diabetes mellitus. Furthermore, we emphasized the possible role of infectious-related stress such as SarsCov2 infection in adrenal axis dysregulation, insulin resistance and diabetes in a bidirectional link. Understanding and better defining the links between stress and obesity or diabetes could contribute to further definition of the pathogenesis and the management of stress-related complications, in which the HPA axis dysregulation has a primary role.
1. Introduction
The hypothalamic-pituitary-adrenal (HPA) axis is one of the main systems which, in concert with the sympathetic nervous system (SNS), is activated in acute stress situations and is thus defined as the stress axis [1].
When chronically activated or dysregulated at central or peripheral level, it can affect the individual’s health. In fact, through its action on the neuroendocrine, metabolic and immune systems, chronic stress can contribute, especially in vulnerable individuals, to the development of several diseases, including obesity (OB) and diabetes (DM) [1,2].
Chronic exposure to stressors and the subsequent prolonged secretion of glucocorticoids (GCs) can result in changes in certain gene expressions involved in cellular pathways which are crucial to the body’s intermediate metabolism. These alterations are associated with the development of adiposity, insulin resistance, depletion of lean mass and coagulation disorders resulting in hypercoagulability, dyslipidaemia, hypertension and increased release of inflammatory cytokines [1,2]. The Cushingoid phenotype, which summarizes these alterations well, commonly characterizes subjects with abdominal OB, metabolic syndrome (MS) or type 2 DM (T2DM), as well as subjects suffering from depression and alcoholism, i.e., the so-called “pseudo-Cushing’s” conditions [2,3,4]. It should be noted that the chronic stress-induced Cushingoid phenotype may be influenced not only by chronic hypercortisolism, but also by changes in corticosteroid-binding globulin levels, GC receptor sensitivity in peripheral tissues and by the activity of 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1), a key enzyme in the peripheral GC metabolism [2,3,4]. These alterations are also associated with a kind of ‘genetic programming’ of the HPA axis that occurs in the fetal and perinatal period [5], influencing the activity of this axis in adulthood and promoting the onset of OB and MS [6,7].
The traditional factors that link OB to DM are also related to a hypercaloric diet, a sedentary lifestyle and socioeconomic status (SES). Individuals of lower SES have restricted autonomy and more limited opportunities, which could lead to more stress and consequently bring about an increase in stress hormones, in addition to cortisol and catecholamines, such as glucagon and growth hormone. This condition might ultimately change fat deposition, increasing visceral fat and raising the risk of developing DM [8,9]. A decreased quantity or quality of sleep, such as in shift work or in conditions such as sleep apnea, can also be related to weight gain and OB. This may lead to a desire for highly caloric food, owing to an imbalance of appetite hormones. Other effects include increased HPA axis reactivity and growth hormone axis impairment, which again may enhance visceral fat mass and lead to insulin resistance [10]
In this article we highlighted and reviewed the most recent data on the relationship between HPA axis activity during chronic stress and its role in the development of MS, OB and T2DM. We also emphasized the possible role of infectious-related stress, such as from SarsCov2 infection, in HPA axis dysregulation, insulin resistance and T2DM, in a dual inverse link.
2. Methods
We reviewed articles from the last three decades up to July 2021 published in PubMed/Medline. These included original articles, review articles, systematic review articles, meta-analysis and book chapters. We reported the most interesting and recent findings. This literature search was performed using the following search string syntax: (“stress” OR “stress axis” OR “cortisol” OR “glucocorticoid receptors”) AND (“diabetes” OR “metabolic syndrome” OR “metabolism” OR “obesity” OR “adiposity”) AND (“COVID-19” OR “SarsCov2 infection”) AND (“inflammation” OR “cytokines”).
2.1. Stress and Adiposity: The Role of Cortisol in Increasing Visceral Fat Accumulation
The adaptive response to stress in humans is significantly determined by the action of cortisol. This hormone strongly influences the whole organism through a wide spectrum of effects, most of which are catabolic and directed at assuring the maximum energy available for the adaptive response to the stressful events [1,2]. GCs stimulate hepatic gluconeogenesis by increasing plasma glucose concentrations. They also induce lipolysis in some areas of the body while promoting adipose tissue accumulation in others (namely in the abdominal, perivisceral and dorso-cervical areas). Furthermore, they lead to protein catabolism in muscle, bone and skin by promoting the use of amino acids as substrates for oxidative pathways. In addition to their known catabolic functions, GCs also antagonize the anabolic effects of growth hormone, insulin, thyroid and sex hormones during stress [1,2].
In general, the consequences of excessive exposure to cortisol or excessive sensitivity to the action of GCs can be summarized as an activation of lipoprotein-lipase with an accumulation of triglycerides and lipids in adipocytes and an inhibition of fatty acid mobilization, in concert with insulin [11]. For this reason, the prolonged action of cortisol is associated with an increase in abdominal adiposity, which is further determined by the concomitant endocrine alterations associated with chronic hypercortisolism, in particular the inhibition of the gonadal and somatotropic axis. In addition, the latter facilitates the appearance of metabolic alterations and increases cardiovascular risk of MS and T2DM [2,3,4,11,12].
2.2. Hpa Axis in Obesity and Diabetes
OB has long been considered a condition connected with functional hypercortisolim or pseudo-Cushing’s condition, although no definitive and confirming data on HPA axis hyperactivity are available and contrasting data are present [11,12]. Besides OB, other endocrine diseases including poorly controlled DM and polycystic ovary syndrome, as well as other conditions such as chronic alcoholism and psychiatric disorders, may show functional hypercortisolism with some clinical and metabolic related signs, giving rise to what is called pseudo-Cushing’s syndrome [13]. Subjects with visceral OB may show increased 24 h urinary cortisol levels and an increased urinary cortisone to cortisol ratio [8]. Accordingly, some studies show a positive association between postprandial salivar cortisol and body max index (BMI), waist to hip ratio (WHR), blood glucose, serum insulin and lipids in obese adult male patients with visceral adiposity [3,11,12,14]. Cortisol metabolic clearance has been reported as being negatively correlated with insulin sensitivity. Furthermore, an increased response of the HPA axis to different stimuli, including food, low dose ACTH and CRH, have been reported in obese subjects [3]. Thus, a general pattern of findings emerged where greater abdominal fat is associated with higher cortisol levels and greater responsivity of the HPA axis, as reflected in morning awakening and acute stress reactivity, though some studies did conversely show under-responsiveness [3].
In particular, the cortisol awakening response (CAR) has been shown to be related to life stress and has been studied in OB. CAR represents a spike in salivary cortisol concentrations just after waking [15]. Irregularities in CAR occur in the form of either a blunted or an exaggerated response that can be related to HPA axis dysregulation [15]. These alterations have been described in various mental and physical illnesses, such as depression [11,16]. In OB, CAR has been reported as being negatively related to abdominal fat mass [17]. Conversely, other studies have found evidence of the opposite, i.e., an exaggerated response correlated with BMI and WHR. Other studies found OB and CAR to be unrelated [17]. However, all these results come from studies with methodological limits. One of the most serious studies in terms of methodology [18], which conducted multi-point diurnal saliva sampling over three days in a large sample of men and women, actually showed a negative relationship between OB and CAR. As a result, a blunted CAR seems to be more likely in OB [17].
Another important marker of HPA axis activity is the diurnal slope that measures the daily pattern of cortisol concentrations. In normal conditions the pattern of daily cortisol concentrations follows a negative slope [19], i.e., cortisol concentrations increase sharply immediately upon waking (CAR) and then show an attenuated decline throughout the day [20]. Flat slopes—as observed in those lacking a robust CAR or those failing to reach sufficiently low levels by evening—are indicative of HPA dysregulation and are associated with negative outcomes in cases of abdominal OB, namely a higher risk of cardiovascular disease, T2DM and stroke [21].
As concerns daily cortisol production evaluated through 24 h urinary free cortisol (UFC) or by multiple days of diurnal blood or salivary cortisol sampling, inconsistent findings are obtained in OB, some findings showing hypercortisolism and others hypocortisolism [17].
With reference to the dexamethasone suppression test that explores GC feedback sensitivity, studies in patients with OB have again yielded mixed results regarding both general and abdominal OB when using dexamethasone at different dosages (1 mg standard dose p.o. or doses per kilogram of body weight). A lower inhibition after dexamethasone was shown in female but not in male obese subjects [22].
Perhaps the perturbation in cortisol suppression occurs only at certain dosages of dexamethasone and dysregulation may be detectable only at certain thresholds. In general, null results tend to emerge when examining the relationship between feedback sensitivity and generalized OB, while abdominal OB seems to be associated with poorer feedback sensitivity [17].
2.3. Tendency of GC Receptors and Their Polymorphisms to Modulate GC Sensitivity, Increasing Susceptibility to Likelihood of Adiposity and Insulin Resistance
Polymorphisms in the GC receptor (GR) gene have been associated with altered GC sensitivity and changes in body composition and in metabolic variables, through either up- or downregulation of the effect of GCs on target tissues. An example of a potentially up-regulating GR polymorphism (thus favouring metabolic complications) is the BclI (rs41423247) polymorphism [23,24,25]. Although several studies have analyzed the effects of the BclI polymorphism on body composition and metabolic variables (namely a worse metabolic profile and increased cardiovascular risk), somewhat inconsistent results have been reported [24,25]. In 2013 a Dutch cohort study showed that homozygous carriers of the BclI polymorphism of the GR gene have significantly greater total body fat, contributing to higher HOMA2-IR, compared to heterozygous carriers and noncarriers [26]. Conversely, other polymorphisms such as A3669G and ER22/23EK have been reported to be associated with reduced GR sensitivity and a better metabolic profile [23,24,25].
2.4. Role of 11β-Hsd1 Enzyme Dysregulation in the Pathogenesis of Obesity and Insulin Resistance
11β-hydroxysteroid dehydrogenases enzymes (11β-HSD) catalyze the interconversion of hormonally active cortisol and inactive cortisone at pre-receptor levels. Two isoforms of 11β-HSD have been identified. 11β-HSD1 is a low-affinity NADP(H)-dependent dehydrogenase/oxoreductase that interconverts inactive cortisone and active cortisol with a bidirectional function predominantly expressed in hepatic, gonadal, and central-nervous-system tissues [27]. It has a mainly reductase function in vivo, generating active glucocorticoid at a prereceptor level. Conversely 11β-HSD2 is a high-affinity NAD-dependent dehydrogenase which inactivates cortisol, protecting the non-selective mineralocorticoid receptor in the kidney and colon from cortisol excess [27]. Since 1997, Stewart et al. have hypothesized that visceral adipose stromal cells (from omental fat), but not subcutaneous fat, can generate active cortisol from inactive cortisone through the expression of 11β-HSD1, suggesting that central OB may reflect “Cushing’s disease of the omentum” [28].
In animal models, the impact of the 11β-HSD1 enzyme on the development of abdominal OB is well defined. In 11β-HSD1 knockout mice a certain resistance to the development of OB-induced hyperglycemia is observed. In particular 11β-HSD1 knockout mice are protected from increased adiposity at both the omental and subcutaneous levels, unlikely to have elevated serum free fatty acids [29].
On the contrary, transgenic mice selectively overexpressing 11β-HSD-1 in adipose tissue (AT) developed visceral OB, pronounced insulin-resistant diabetes and hyperlipidemia [30]. Other studies in animals showed that N-(6-Substituted-1,3-benzothiazol-2-yl)benzenesulfonamide derivatives-1-8 significantly reduce plasma glucose level in a non-insulin-dependent DM rat model, by inhibiting 11beta-HSD1 [31]. Other selective inhibitors of 11beta-HSD1 such as 4-(phenyl-sulfonamido-methyl) benzamides also showed improved glycemic control, decreased serum lipids, and enhanced insulin sensitivity in diabetic OB/OB mice [32]. Lastly, N-(Pyridin-2-yl) as well as arylsulfonamides play an 11betaHSD1 inhibiting action [33].
4′-cyano-biphenyl-4-sulfonic acid (6-amino-pyridin-2-yl)-amide (PF-915275), the potent 11betaHSD1 inhibitor that is selective for primate and human enzymes, reduced insulin levels in a dose-related manner in cynomolgus monkeys, reinforcing the hypothesis that 11betaHSD1 inhibitors may be useful in the treatment of DM and other related metabolic diseases [34].
In humans, a higher expression of the enzyme is described in omental fat rather than in the subcutaneous one [35]. However, in simple OB, circulating levels of cortisol are actually normal or low in some studies, with increased cortisol secretion rates, thus suggesting an impairment of cortisol metabolism in peripheral tissues via 11b-HSDs [36]. It has been hypothesized that, in AT, there is an increase in 11β-HSD1 synthesis in OB in accordance with studies performed on subcutaneous abdominal biopsies from obese men and women showing higher levels of enzyme activity [37,38]. However, other authors were unable to correlate whole-tissue or adipocyte 11β-HSD1 synthesis with OB [39]. A recent study, which took into consideration single-nucleotide polymorphisms (SNPs) in the 11β-HSD1-coding gene (HSD11B1), shows that genetic variation in the 11β-HSD1 gene determines NAFLD and visceral OB [40].
Peroxisome proliferator-activated receptor (PPAR) agonists could exert a suppressive effect on 11β-HSD1. Long-term treatment with PPARγ agonists could consequently contribute to the decrease of 11β-HSD1 adipose tissue in humans [41,42].
Novel compounds, amongst which are adamantyl-triazoles, are currently under investigation and preliminary findings from both experimental and human studies show a favourable effect on glucose and lipid metabolism, weight reduction and adipokine levels [43]. Some clinical trials have been conducted on humans with selective 11β-HSD1 inhibitors. For instance, two selective 11β-HSD1 inhibitors (RO5093151/RO-151 and RO5027383/RO-838) were investigated in a randomized, controlled study in metformin-treated patients with T2DM. Slight metabolic improvements were seen in HbA1c, body weight and insulin sensitivity parameters [44].
Therefore, on the whole, studies in animals and humans provide evidence of a pathogenetic role of 11β-HSD1 in the development of abdominal OB and related metabolic alterations. For this reason, scientific-pharmaceutical research is focusing on 11β-HSD1 as a significant target for therapeutic strategy development in patients with OB and T2DM.
2.5. Stress, Inflammation and Metabolic Syndrome
The relationship between stress, inflammation and AT has been a growing subject of interest in recent years. Considerable data indicate a potential role of inflammation and increased levels of cytokines, produced by both immune cells and adipocytes, in the pathogenesis of MS and T2DM [45,46,47,48]. Furthermore, acute and chronic stress can exacerbate or worsen metabolic conditions by supporting an inflammatory state [45,46].
AT has traditionally been divided into two types, white AT (WAT) and brown AT (BAT), based on tissue color, though cytological, biochemical and functional differences also exist. A third type of adipocyte, beige or brite adipocyte, has similar characteristics to BAT, although it differs in developmental origins and body distribution [49].
In humans and mammals, WAT accounts for the majority of AT and is responsible for energy storage, endocrine signalling and insulin sensitivity [47]. Depending on its distribution in the body, WAT itself can be broadly classified into two subtypes: visceral WAT (vWAT) and subcutaneous WAT (scWAT). vWAT consists of perirenal, perigonadal, mesenteric and retroperitoneal WAT and is more represented in men (android fat deposition). scWAT deposits are found in the cranial, nasal, gastrointestinal, gluteal and femoral areas and are predominant in females (gynoid fat deposition) [50].
In contrast, BAT is widely distributed in mammals in the postnatal period and during hibernation. However, imaging studies have revealed that metabolically active BAT is also found in adults, between anterior neck muscles and in the supraclavicular and thoracic regions [51,52]. As opposed to BAT which develops embryonically, beige fat develops postnatally and arises from different progenitor cells [53]. Beige adipocytes are distributed in the cervical–supraclavicular, shoulder-blades, axillary, mediastinal, paravertebral, perirenal, and peri-aortic regions [54].
In healthy humans, BAT and beige fat activity contributes to whole-body fat oxidation and diet-induced thermogenesis [55]. In physiological conditions, while insulin-sensitive WAT is the main site for energy storage and is involved in fatty acid (FA) biosynthesis by storing triglyceride, BAT and beige fat play a critical role in energy homeostasis, glucose uptake and FA breakdown, causing energy dissipation and heat production [56]. This is due to the thermogenic-promoting properties in BAT and beige fat, including dense mitochondria, thermogenic uncoupling protein 1 (UCP1) and lipid droplets that provide chemical energy [57].
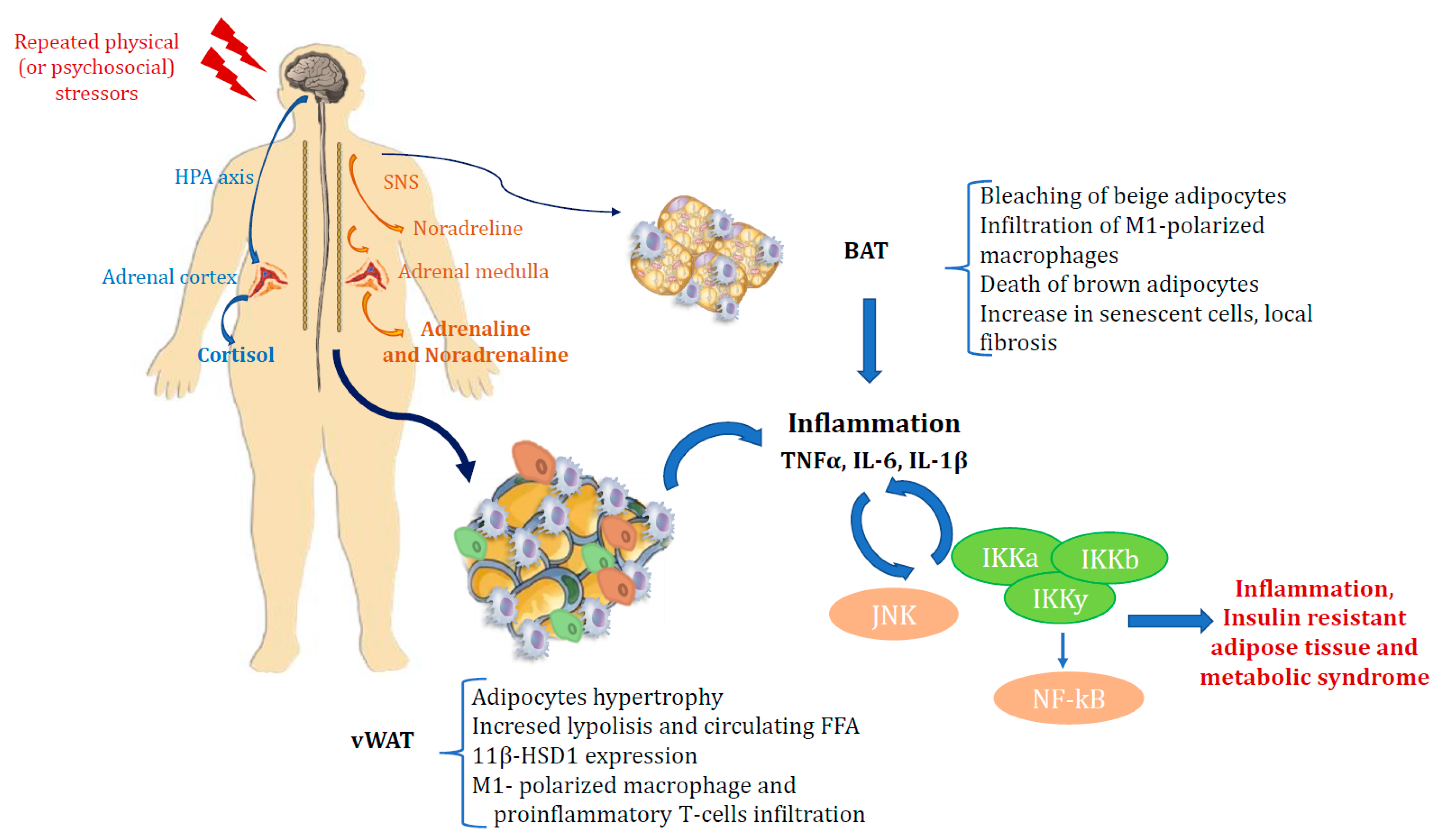
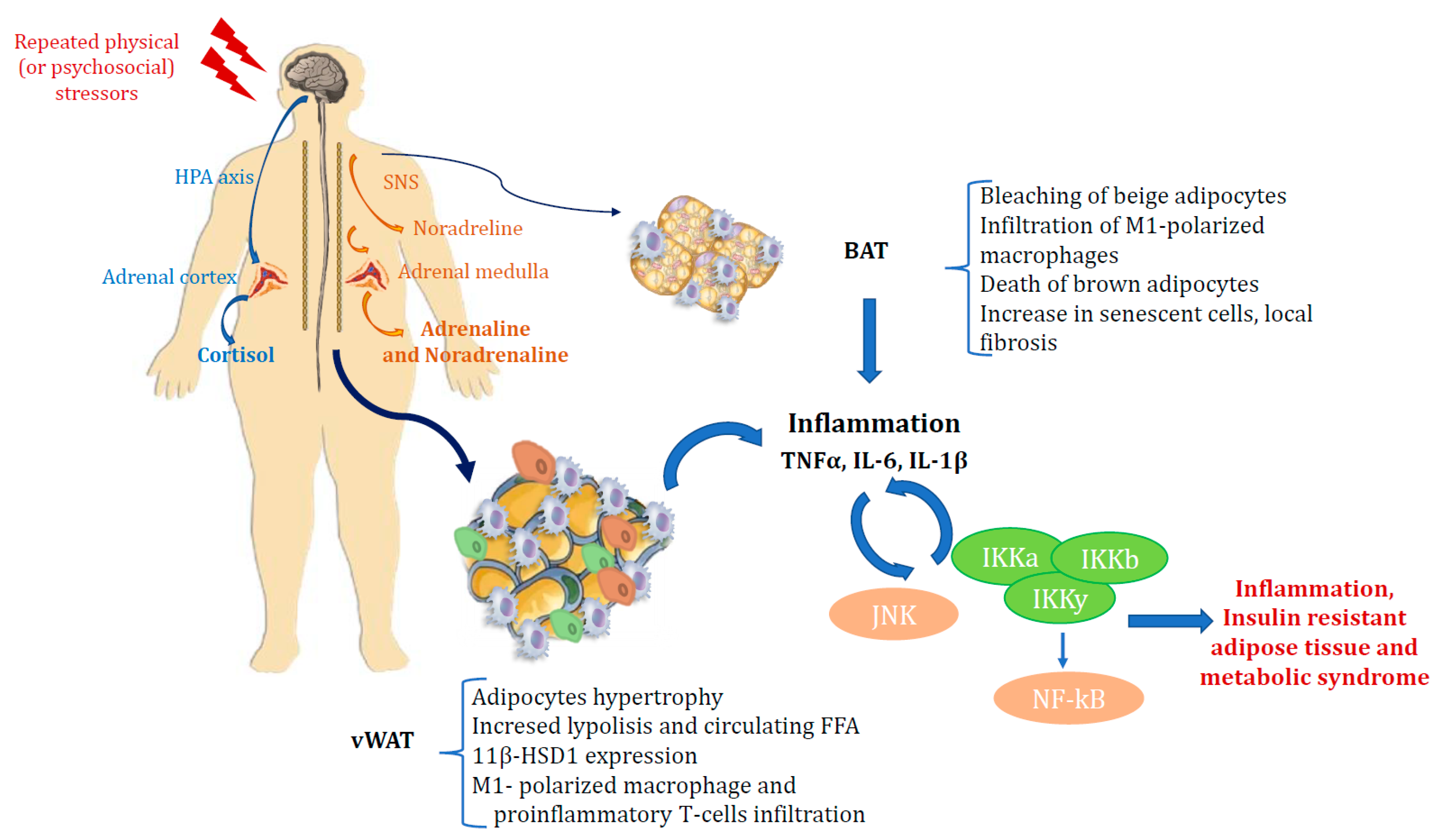
If AT becomes insulin resistant, the capacity of insulin to suppress adipocyte lipolysis and reduce serum levels of FA and glycerol is impaired. This results in a sustained exposure of liver and muscle to high levels of FA and a further ectopic uptake and storage of lipids in these tissues. Ectopic lipids impair insulin signaling, and thus insulin resistance in adipocytes through increased lipolysis may play a major role in whole-body insulin resistance [58,59,60]. In WAT, M1-polarized macrophages are believed to contribute to insulin resistance while M2-polarised macrophages contrast with the action of M1 macrophages and enhance insulin sensitivity [61,62,63]. It has also been observed that M1-polarized macrophages can infiltrate WAT when mice are switched to an obesogenic high-fat diet. Hence the idea emerged that AT and inflammation are closely interconnected and that chronic inflammation in AT is responsible for insulin resistance in OB [48,59,60]. Catecholamines also carry out a well known role in the regulation of lipolysis. Depending on which β1/2-adrenergic (β-AR) or α2-adrenergic receptor they bind to, they can respectively activate or inhibit lipolysis. This depends on both the receptor affinity and the number of the two different receptors on the adipocyte plasmalemma. In patients with MS and insulin resistance, the lipolytic effect of catecholamines occurs particularly in vAT [58]. Activation of the SNS also leads to cortisol increase, thus reflecting a slight hypercortisolism, with effects on AT which depend on the duration of hypercortisolemia as well as on circulating insulin levels. High insulin levels correspond to a lipogenic effect of cortisol in WAT, but chronic hypercortisolism induces lipolysis in adipocytes, with increased levels of circulating FA. In addition, high cortisol levels induce expression of the 11beta-HSD1 enzyme in AT, which converts cortisone to cortisol, thus leading to visceral OB, insulin resistance and DM, as previously reported in transgenic mice overexpressing 11beta-HSD1 [29,30,35]. SNS also plays a role in BAT. It promotes thermogenesis in brown and beige adipocytes by stimulating β3 adrenergic receptors, inducing the expression of UCP1 and mitochondrial respiration via the cAMP/PKA pathway [64,65]. The β-adrenergic sensitivity of brown and beige adipocytes has been shown to decrease in OB [66], which may explain, at least in part, the reduction in energy consumption that manifests itself in these conditions. In contrast, activation of α2 adrenergic receptors, unlike β adrenergic receptors, suppresses thermogenesis in BAT [67]. Furthermore, hyperactivation of the SNS pathway in AT, as occurs during acute stress, also induces bleaching of beige adipocytes with subsequent infiltration of M1-polarized macrophages, death of brown adipocytes and an increase in senescent cells, local fibrosis and inflammation [68]. BAT and beige adipocytes also express receptors for glucocorticoids, and cold stress can activate the HPA axis, increasing plasma corticosterone levels [69]. Glucocorticoids increase the ability of isoprenaline to stimulate thermogenesis in primary human brown adipocyte cultures, and acute treatment with glucocorticoids stimulates BAT function in healthy humans [57,70,71]. Increased lipolysis can modulate adipocyte secretion of molecules including adipokines or lipokines, as well as cytokine production by macrophages [57,58,59,71]. Indeed, it has long been known that the immune system and metabolism are closely linked and that OB is associated with the accumulation of M1-polarized macrophages in AT, which have been found to be the main source of cytokines, including TNF-a, interleukin 6, interleukin 1b, monocyte chemotactic, protein 1 (MCP-1, CCL2), and the macrophage inhibitory factor (MIF) [57].
T lymphocytes and macrophages are closely interconnected in AT, and T-cell infiltration is involved at this level by regulating macrophage number and activity. In one study, it was observed that the macrophage content in visceral fat of obese subjects was 12% compared to 4% in lean subjects [40]. In obese subjects, CD8+ cytotoxic T lymphocytes (known as cytotoxic T cells) and CD4+ helper 1 (Th1) T lymphocytes stimulate adipose macrophage activation and produce pro-inflammatory cytokines, inducing insulin resistance. It has also been shown that in OB, the number of anti-inflammatory and insulin-sensitizing lymphocytes, such as CD4+ T helper 2 (Th2) lymphocytes and Foxp3+ regulatory T cells (Tregs) in vAT, is reduced [57]. The synthesis of inflammatory cytokines by macrophages in AT is mediated by the activation of two major intracellular inflammatory pathways, c-Jun N-terminal kinase (JNK)-activating protein 1 (AP1) and IKappa B kinase beta (IKK)-nuclear factor kappa-activated B-cell light chain-enhancer (NF-kB). The latter is in turn triggered by the same molecules implicated in the development of insulin resistance, including reactive oxygen species (ROS), saturated FA, and inflammatory cytokines, emphasizing their relevance in the onset of the disease [72]. Among the inflammatory cytokines, IL-6, IL-1b, and TNF-α are those most involved in modulating metabolic activities. IL-6 is considered the critical regulatory cytokine that mediates the stimulatory relationship between the immune system and the HPA axis [73]. IL-6 stimulates the HPA axis both acutely and chronically. As a result, a marked increase in circulating ACTH has been observed even after just a single subcutaneous administration of IL-6. In contrast, anti-IL-6 antibodies almost completely block the stimulatory effect on the axis induced by lipopolysaccharide bacteria [74].
A relationship between IL-6 and insulin resistance has been described in OB and MS, although data are not consistent. Circulating concentrations of IL-6 and PCR are increased in subjects with OB and are predictive of T2DM in predisposed subjects. IL-6 production from liver and tissue correlates with and promotes insulin resistance, while IL-6 produced by skeletal muscle has a protective effect [73]. Hyperglycaemia is known to trigger an inflammatory process in pancreatic insulae and to facilitate β-cell apoptosis. This pro-apoptotic mechanism is up-regulated by the glucose-induced increase in IL-1β through its stimulating effect on the 11β-HSD1 enzyme [74,75].
Repeated stress induces increased levels of IL-1b and other inflammatory cytokines in subcutaneous WAT. This impaires its regular lipogenesis and adipogenesis functions, thus resulting in impaired insulin signalling and a shift of circulating lipids toward visceral adipose tissue with subsequent development of visceral obesity [74,76].
TNFa is a proinflammatory cytokine released by macrophages in response to physiological and pathological processes such as infections, apoptosis, tumor-induced cachexia, autoimmune and chronic inflammatory diseases [77]. TNFa is also involved in hepatic and peripheral insulin resistance. In particular TNFa has been shown to impair insulin signalling in murine adipocytes, hepatoma and muscle cells by serine phosphorylation of the insulin receptor substrate 1(IRS-1) and of protein phosphatase 1 (PP-1), and through the activation of protein tyrosine phosphates (SH-PTPase) [78,79,80]. In humans, several authors have described increased TNF- mRNA concentrations in the AT of obese or insulin-resistant patients [80,81]. Treatment with TNF-a neutralizing antibodies has been shown to improve insulin sensitivity in obese rats [82], highlighting that inflammation may be involved in OB-induced glucose intolerance (Figure 1).
In conclusion, AT represents an extremely active metabolic and endocrine organ capable of synthesizing hormones, cytokines and adipokines that interact in an integrated network with the nervous system through the HPA axis and the immune system, influencing metabolic homeostasis. Several studies support the idea that systemic and metabolic inflammation are among the major causes of OB-induced insulin resistance and beta-cell dysfunction. Accordingly, specific anti-inflammatory treatment strategies with different molecular targets in obese patients with T2DM may improve insulin sensitivity as well as beta-cell function [82,83,84,85,86].
Acute and chronic stress through the HPA axis and SNS can exacerbate or worsen metabolic conditions by supporting an inflammatory state. AT and inflammation are closely interconnected and chronic inflammation in AT is responsible for insulin resistance in OB, where excessive expansion of vWAT induces a cascade of biological processes including mitochondrial dysfunction, hypoxia and adipocyte death, all of which are thought to contribute to inflammation.
2.6. COVID-19, Stress and Diabetes: A Bidirectional Relationship
Since the beginning of the SARSCoV-2 pandemic, DM emerged as one of the most common comorbidities and a potential cause of poor outcomes. A dangerous relationship between COVID-19 and DM has definitely been defined [87,88,89,90,91].
The infection per se may lead to a full cascade of the stress responses, including the activation of the neuroendocrine HPA stress axis and other systems that maintain homeostasis [92] with the previously described metabolic consequences. In this context, the SARS-CoV-2 viral infection features peculiar metabolic and vascular consequences in obese and diabetic subjects.
In actual fact, the prevalence of DM was higher among patients hospitalized for COVID-19 who were admitted to the intensive care unit (ICU) or died [88,89] while, on the whole, DM conferred a two–threefold increased rate of poor disease outcome [90,91].
Individuals with DM who became infected with COVID-19 were reported to have a higher non-survival prevalence (ranging between 22% and 31%) compared to non-diabetic subjects [93]. In particular, mortality rates from COVID-19 were highest in elderly people with DM [94]. Besides DM and elderly age, other risk factors for COVID-19 adverse outcomes were OB, hypertension and cardiovascular disease, all very common conditions in T2DM [95]. Age is the strongest risk factor for developing T2DM and the effect of ageing on immune function might be equally important for COVID-19 susceptibility and severity [96].
OB and increased visceral fat are independent risk factors for COVID-19 complications in young patients because abdominal adiposity may promote and amplify the inflammatory response [97]. During the first phase of infection, the marked IL-6 release, which regulates the hepatic production of acute-phase reactants, provides a link between vAT and inflammation [98]. Furthermore, ACE2 receptor expression is upregulated on alveolar epithelial cells and in visceral fat, particularly in obese subjects. OB has also been linked to a condition of increased risk of thrombosis and coagulopathy. Individuals with diabetes and infected with COVID-19 had higher D-dimer levels than those without diabetes. Other critical findings observed in obese subjects with Covid19 include myocardial inflammation, lymphocyte infiltration in the liver, macrophage clustering in the brain, axonal injuries, microthrombi in glomeruli and focal pancreatitis [99]. Therefore, the presence of both DM and OB represented a poor prognostic outcome in the presence of SARS-CoV-2 infection, with potential progression of thrombotic events [100].
Other factors have been shown to contribute to poor prognosis in patients with COVID-19 and DM. These include impairment of innate immunity, pro-inflammatory cytokine milieu, reduced expression of ACE2 and use of renin–angiotensin–aldosterone system antagonists [87,88,90,91]. In a population of patients with COVID 19, serum levels of interleukin-6 (IL-6), C-reactive protein and ferritin were significantly higher in patients with DM than in those without [100]. Moreover, direct SarsCov2-beta cell damage detectable through immunohistochemistry and in-situ hybridization, from the pancreas of patients who had died of SARS, was shown [99]. This last factor, together with others such as cytokine-induced insulin resistance, hypokalemia and the very drugs used in the treatment of COVID-19 (e.g., corticosteroids, lopinavir/ritonavir) have been hypothesized to worsen glucose control in people with DM [87,88,89,90,91].
Another important aspect in the relationship between DM and COVID-19 is COVID-induced hyperglycemia, which includes stress- and drug-induced-hyperglycaemia. The presence of hyperglycaemia and DM were found to be independent predictors of morbidity and mortality in patients with SARS-CoV-2 infection [101]. It has also been reported that high blood glucose levels directly increased the concentration of glucose within airway secretions, thus further impairing respiratory drive [101,102,103].
The presence of typical complications of DM (cardiovascular disease, heart failure and chronic kidney disease) further increases COVID-19 mortality, because the individuals affected may be subject to worse COVID-19 complications including vasculopathy, coagulopathy, and increased psychological stress [104,105,106].
It is worthy of mention that the COVID-19 related lockdown induced a further worsening of the pathology in patients with OB and DM. This stressful period decreased quality of life, with alterations in behavioral and eating patterns, changes in daily life, reduced physical exercise, as well as increased feelings of stress and anxiety, all influencing diabetes self-management, body weight and glycemic control [87,88,89,90,91,105,106].
On the whole, these findings suggest a bidirectional relationship between COVID-19 and metabolic disease: on the one hand, pre-existing metabolic diseases potentiate the severity of COVID-19, and, on the other, owing to different mechanisms, including lockdown-induced behavioral patterns, this viral infection exacerbates preexisting metabolic frailty.
3. Conclusions
Prolonged stress and/or inadequate regulation of stress can lead to a condition of chronic hypercortisolism or, in some cases, to an inadequate cortisol response to stress, contributing to insulin resistance, increased adiposity and T2DM. In fact, through its action on the neuroendocrine, metabolic and immune systems, chronic stress can contribute, especially in vulnerable individuals, to an altered biological and behavioral condition, characterized by epigenetic changes in cellular pathways crucial to the body’s glucose and lipid metabolism and a release of inflammatory cytokines. Other factors including changes in GC receptor sensitivity and 11β-HSD1 activity may lead to the onset of OB and metabolic alterations in vulnerable subjects.
The emerging role for inflammation and cytokines, produced by both immune cells and adipocytes, has been defined as one of the major causes of OB-induced insulin resistance and beta-cell dysfunction. Therefore, new antidiabetic therapy has addressed specific anti-inflammatory treatment strategies with the aim of reducing blood glucose levels as well as improving insulin sensitivity and beta-cell function.
Finally, the role of infections in inflammation, hypercortisolism and DM has been defined. Since the beginning of the SARSCoV-2 pandemic, OB and DM have emerged as the most common comorbidities and potential drivers of poor outcomes in COVID-19, thus underlying the existence of a dangerous interrelationship between COVID-19 and OB or DM.
Understanding and better defining the links between stress and OB or DM could contribute to further defining the pathogenesis and the management of stress-related complications, in which HPA axis dysregulation plays a primary role.
Author Contributions
Conceptualization, L.G.; methodology, L.G.; writing—original draft preparation, L.G., S.D., S.B. and F.T.; writing—review and editing, L.G., S.D., S.B. and F.T.; visualization, L.G. and S.D.; supervision, L.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Ethical review and approval by Ethics Committee were waived for this study, due to the characteristics of the manuscript as narrative review.
Informed Consent Statement
Not applicable.
Data Availability Statement
We exclude this statement.
Acknowledgments
The authors wish to thank Anna Racca for the linguistic revision of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Nicolaides, N.C.; Kyratzi, E.; Lamprokostopoulou, A.; Chrousos, G.P.; Charmandari, E. Stress, the stress system and the role of glucocorticoids. Neuroimmunomodulation 2015, 22, 6–19. [Google Scholar] [CrossRef] [PubMed]
- Kyrou, I.; Chrousos, G.P.; Tsigos, C. Stress, visceral obesity and metabolic complications. Ann. N. Y. Acad. Sci. USA 2006, 1083, 77–110. [Google Scholar] [CrossRef] [PubMed]
- Bose, M.; Olivan, B.; Laferre, B. Stress and obesity: The role of hypothalamic-pituitary adrenal axis in metabolic disease. Curr. Opin. Endocrinol. Diabetes Obes. 2009, 16, 340–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anagnostis, P.; Athyros, V.G.; Tziomalos, K.; Karagiannis, A.; Mikhailidis, D.P. The pathogenetic role of cortisol in the metabolic syndrome: A hypothesis. J. Clin. Endocrinol. Metab. 2009, 94, 2692–2701. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, R.M. Glucocorticoid excess and the developmental origins of disease: Two decades of testing the hypothesis—2012 Curt Richter Award Winner. Psychoneuroendocrinology 2013, 38, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fogelman, N.; Canli, T. Early life stress and cortisol: A meta-analysis. Horm. Behav. 2018, 98, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Gentile, S. Untreated depression during pregnancy: Short- and long-term effects in offspring. A systematic review. Neuroscience 2017, 342, 154–166. [Google Scholar] [CrossRef]
- Anekwe, C.H.; Jarrell, A.R.; Townsend, M.J.; Gaudier, G.I.; Hiserodt, J.M.; Stanford, C.S. Socioeconomics of obesity. Curr. Obes. Rep. 2020, 9, 272–279. [Google Scholar] [CrossRef]
- Hjelholt, A.; Høgild, M.; Bak, M.; Christiansen, M.; Bæk, A.A.-S.; Jessen, N.; Richelsen, B.; Bønløkke, S.; Møller, P.N.; Jørgensen, J.O.L. Growth hormone and obesity. Endocrinol. Metab. Clin. N. Am. 2020, 49, 239–250. [Google Scholar] [CrossRef]
- Van Der Valk, E.S.; Akker, E.L.V.D.; Savas, M.; Kleinendorst, L.; Visser, J.A.; Van Haelst, M.M.; Sharma, A.M.; Van Rossum, E.F. A comprehensive diagnostic approach to detect underlying causes of obesity in adults. Obes. Rev. 2019, 20, 795–804. [Google Scholar] [CrossRef]
- Björntorp, P. Do stress reactions cause abdominal obesity and comorbidities? Obes. Rev. 2001, 2, 73–86. [Google Scholar] [CrossRef]
- Rosmond, R.; Dallman, M.F.; Björntorp, P. Stress-related cortisol secretion in men: Relationships with abdominal obesity and en-docrine metabolic and hemodynamic abnormalities. J. Clin. Endocrinol. Metab. 1998, 83, 1853–1859. [Google Scholar] [PubMed] [Green Version]
- Scaroni, C.; Albiger, N.M.; Palmieri, S.; Iacuaniello, D.; Graziadio, C.; Damiani, L.; Zilo, M.; Stigliano, A.; Colao, A.M.; Pivonello, E.; et al. Approach to patients with pseudo-Cushing’s states. Endocr. Connect. 2020, 9, R1–R13. [Google Scholar] [CrossRef] [Green Version]
- Rosmond, R.; Radulovic, V.; Holm, G. A brief update of glucocorticoid receptor variants and obesity risk. Ann. N. Y. Acad. Sci. USA 2006, 1083, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Stalder, T.; Kirschbaum, C.; Kudielka, B.M.; Adam, E.K.; Pruessner, J.; Wüst, S.; Dockray, S.; Smyth, N.; Evans, P.; Hellhammer, D.H.; et al. Assessment of the cortisol awakening response: Expert consensus guidelines. Psychoneuroendocrinology 2015, 63, 414–432. [Google Scholar] [CrossRef] [PubMed]
- Fries, E.; Dettenborn, L.; Kirschbaum, C. The cortisol awakening response (CAR): Facts and future directions. Int. J. Psychophysiol. 2009, 72, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Rodrigueza, A.C.I.; Epelb, E.S.; Whitea, M.L.; Standena, E.C.; Secklc, J.R.; Tomiyamaa, A.J. Hypothalamic-pituitary-adrenal axis dysregulation and cortisol activity in obesity: A systematic review. Psychoneuroendocrinology 2015, 62, 301–308. [Google Scholar] [CrossRef] [Green Version]
- Champaneri, S.; Xu, X.; Carnethon, M.; Bertoni, A.; Seeman, T.; DeSantis, A.S.; Roux, A.D.; Shrager, S.; Golden, S.H. Diurnal salivary cortisol is associated with body mass index and waist circumference: The multiethnic study of atherosclerosis. Obesity 2012, 21, E56–E63. [Google Scholar] [CrossRef]
- Stone, A.A.; Schwartz, J.E.; Smyth, J.; Kirschbaum, C.; Cohen, S.; Hellhammer, D.; Grossman, S. Individual differences in the diurnal cycle of salivary freecortisol: A replication of flattened cycles for some individuals. Psychoneuroendocrinology 2001, 26, 295–306. [Google Scholar] [CrossRef]
- Pruessner, J.C.; Kirschbaum, C.; Meinlschmid, G.; Hellhammer, D.H. Two formulas for computation of the area under the curve represent measures of total hormone concentration versus time-dependent change. Psychoneuroendocrinology 2003, 28, 916–931. [Google Scholar] [CrossRef]
- Rosmond, R.; Bjorntorp, P. The hypothalamic-pituitary-adrenal axis activity as a predictor of cardiovascular disease, type 2 diabetes and stroke. J. Intern. Med. 2000, 247, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Pasquali, R.; Ambrosi, B.; Armanini, D.; Cavagnini, F.; Uberti, E.D.; Rio, G.D.; de Pergola, G.; Maccario, M.; Mantero, F.; Marugo, M.; et al. Cortisol and ACTH response to oral dexamethasone in obesity and effectsof sex, body fat distribution, and dexamethasone concentrations: Adose-response study. J. Clin. Endocrinol. Metab. 2002, 87, 166–175. [Google Scholar] [CrossRef]
- Manenschijn, L.; Akker, E.L.T.V.D.; Lamberts, S.W.J.; Van Rossum, E.F.C. Clinical features associated with glucocorticoid receptor polymorphisms. Ann. N. Y. Acad. Sci. USA 2009, 1179, 179–198. [Google Scholar] [CrossRef] [PubMed]
- Van Rossum, E.F.; Koper, J.W.; Van Den Beld, A.W.; Uitterlinden, A.G.; Arp, P.; Ester, W.; Janssen, J.A.M.J.L.; Brinkmann, A.O.; De Jong, F.H.; Grobbee, D.E.; et al. Identification of the BclI polymorphism in the glucocorticoid receptor gene: Association with sensitivity to glucocorticoids in vivo and body mass index. Clin. Endocrinol. 2003, 59, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Weaver, J.U.; Hitman, G.A.; Kopelman, P.G. An association between a BclI restriction fragment length polymorphism of the gluco-corticoid receptor locus and hyperinsulinaemia in obese women. J. Mol. Endocrinol. 1992, 9, 295–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geelen, C.C.; van Greevenbroek, M.M.; van Rossum, E.F.; Schaper, N.C.; Nijpels, G.; Hart, L.M.; Schalkwijk, C.G.; Ferreira, I.; van der Kallen, C.J.; Sauerwein, H.P.; et al. BclI glucocorticoid receptor polymorphism is associated with greater body fatness: The hoorn and CODAM studies. J. Clin. Endocrinol. Metab. 2013, 98, E595–E599. [Google Scholar] [CrossRef] [Green Version]
- Tomlinson, J.; Walker, E.A.; Bujalska, I.J.; Draper, N.; Lavery, G.G.; Cooper, M.S.; Hewison, M.; Stewart, P.M. 11β-Hydroxysteroid Dehydrogenase Type 1: A tissue-specific regulator of glucocorticoid response. Endocr. Rev. 2004, 25, 831–866. [Google Scholar] [CrossRef] [PubMed]
- Bujalska, I.J.; Kumar, S.; Stewart, P.M. Does central obesity reflect “Cushing’s disease of the omentum”? Lancet 1997, 349, 1210–1213. [Google Scholar] [CrossRef]
- Morgan, S.A.; McCabe, E.L.; Gathercole, L.L.; Hassan-Smith, Z.K.; Larner, D.P.; Bujalska, I.J.; Stewart, P.M.; Tomlinson, J.W.; Lavery, G.G. 11beta-HSD1 is the major regulator of the tissue-specific effects of circulating glucocorticoid excess. Proc. Natl. Acad. Sci. USA 2014, 324, E2482–E2491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuzaki, H.; Paterson, J.; Shinyama, H.; Morton, N.M.; Mullins, J.J.; Seckl, J.R.; Flier, J.S. A Transgenic model of visceral obesity and the metabolic syndrome. Science 2001, 294, 2166–2170. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Díaz, H.; Villalobos-Molina, R.; Ortiz-Andrade, R.; Díaz-Coutiño, D.; Medina-Franco, J.L.; Webster, S.; Binnie, M.; Estrada-Soto, S.; Ibarra-Barajas, M.; León-Rivera, I.; et al. Antidiabetic activity of N-(6-substituted-1,3-benzothiazol-2-yl)benzenesulfonamides. Bioorganic Med. Chem. Lett. 2008, 18, 2871–2877. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhou, Z.; Yang, H.; Chen, J.; Feng, Y.; Du, L.; Leng, Y.; Shen, J. 4-(Phenylsulfonamidomethyl)benzamides as potent and selective inhibitors of the 11β-hydroxysteroid dehydrogenase type 1 with efficacy in diabetic ob/ob mice. Bioorganic Med. Chem. Lett. 2009, 19, 4455–4458. [Google Scholar] [CrossRef]
- Siu, M.; Johnson, T.O.; Wang, Y.; Nair, S.K.; Taylor, W.D.; Cripps, S.J.; Matthews, J.J.; Edwards, M.P.; Pauly, T.A.; Ermolieff, J.; et al. N-(Pyridin-2-yl) arylsulfonamide inhibitors of 11β-hydroxysteroid dehydrogenase type 1: Discovery of PF-915275. Bioorganic Med. Chem. Lett. 2009, 19, 3493–3497. [Google Scholar] [CrossRef]
- Bhat, B.G.; Hosea, N.; Fanjul, A.; Herrera, J.; Chapman, J.; Thalacker, F.; Stewart, P.M.; Rejto, P.A. Demonstration of proof of mechanism and pharmacokinetics and pharmacodynamic relationship with 4′-Cyano-biphenyl-4-sulfonic Acid (6-Amino-pyridin-2-yl)-amide (PF-915275), an Inhibitor of 11β-Hydroxysteroid Dehydrogenase Type 1, in Cynomolgus Monkeys. J. Pharmacol. Exp. Ther. 2007, 324, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.D.; Azevedo, I.; Monteiro, R.; Martins, M.J. 11β-Hydroxysteroid dehydrogenase type 1: Relevance of its modulation in the pathophysiology of obesity, the metabolic syndrome and type 2 diabetes mellitus. Diabetes Obes. Metab. 2012, 14, 869–881. [Google Scholar] [CrossRef]
- Fraser, R.; Ingram, M.C.; Anderson, N.H.; Morrison, C.; Davies, E.; Connell, J.M. Connell cortisol effects on body mass, blood pressure, and cholesterol in the general population. Hypertension 1999, 33, 1364–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rask, E.; Olsson, T.; Soderberg, S.; Andrew, R.; Livingstone, D.E.; Johnson, O.; Walker, B.R. Tissue-specific dysregulation of cortisol metabolism in human obesity. J. Clin. Endocrinol. Metab. 2001, 86, 1418–1421. [Google Scholar] [CrossRef]
- Rask, E.; Walker, B.R.; Söderberg, S.; Livingstone, D.E.W.; Eliasson, M.; Johnson, O.; Andrew, R.; Olsson, T. Tissue-specific changes in peripheral cortisol metabolism in obese women: Increased adipose 11b-hydroxysteroid dehydrogenase type 1 activity. J. Clin. Endocrinol. Metab. 2002, 87, 3330–3336. [Google Scholar]
- Tomlinson, J.W.; Sinha, B.; Bujalska, I.; Hewison, M.; Stewart, P.M. Expression of 11b-hydroxysteroiddehydrogenase type 1 in adipose tissue is not increased in human obesity. J. Clin. Endocrinol. Metab. 2002, 87, 5630–5635. [Google Scholar] [CrossRef] [Green Version]
- Lutz, S.Z.; Peter, A.; Machicao, F.; Lamprinou, A.; Machann, J.; Schick, F.; Königsrainer, I.; Königsrainer, A.; Fritsche, A.; Staiger, H.; et al. Genetic Variation in the 11β-hydroxysteroid-dehydrogenase 1 Gene Determines NAFLD and Visceral Obesity. J. Clin. Endocrinol. Metab. 2016, 101, 4743–4751. [Google Scholar] [CrossRef] [Green Version]
- Mai, K.; Andres, J.; Bobbert, T.; Maser-Gluth, C.; Möhlig, M.; Bähr, V.; Pfeiffer, A.F.H.; Spranger, J.; Diederich, S. Rosiglitazone decreases 11beta-hydroxysteroid dehydrogenase type 1 in subcutaneous adipose tissue. Clin. Endocrinol. 2007, 67, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Wake, D.J.; Stimson, R.H.; Tan, G.D. Effects of peroxisome proliferator-activated receptor-alpha and -gamma agonists on 11beta-hydroxysteroid dehydrogenase type 1 in subcutaneous adipose tissue in men. J. Clin. Endocrinol. Metab. 2007, 92, 1848–1856. [Google Scholar] [CrossRef] [Green Version]
- Anagnostis, P.; Katsiki, N.; Adamidou, F.; Athyros, V.G.; Karagiannis, A.; Kita, M.; Mikhailidis, D.P. 11beta-Hydroxysteroid dehydrogenase type 1 inhibitors: Novel agents for the treatment of metabolic syndrome and obesity-related disorders? Metabolism 2012, 62, 21–33. [Google Scholar] [CrossRef]
- Heise, T.; Morrow, L.; Hompesch, M.; Häring, H.-U.; Kapitza, C.; Abt, M.; Ramsauer, M.; Magnone, M.-C.; Fuerst-Recktenwald, S. Safety, efficacy and weight effect of two 11β-HSD1 inhibitors in metformin-treated patients with type 2 diabetes. Diabetes Obes. Metab. 2014, 16, 1070–1077. [Google Scholar] [CrossRef]
- Donath, M.Y.; Shoelson, S.E. Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 2011, 11, 98–107. [Google Scholar] [CrossRef]
- Ferrante, A.W. Macrophages, fat, and the emergence of immunometabolism. J. Clin. Investig. 2013, 123, 4992–4993. [Google Scholar] [CrossRef] [PubMed]
- Richard, A.J.; White, U.; Elks, C.M.; Stephens, J.M. Adipose tissue: Physiology to metabolic dysfunction. In The Insulin Receptor and Its Signal Transduction Network; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Eds.; Endotext [Internet] MD Text.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar] [PubMed]
- Kumari, M.; Heeren, J.; Scheja, L. Regulation of immunometabolism in adipose tissue. Semin. Immunopathol. 2017, 40, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Cohen, P.; Spiegelman, B.M. Adaptive thermogenesis in adipocytes: Is beige the new brown? Genes Dev. 2013, 27, 234–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandão, B.; Poojari, A.; Rabiee, A. Thermogenic fat: Development, physiological function, and therapeutic potential. Int. J. Mol. Sci. 2021, 22, 5906. [Google Scholar] [CrossRef]
- Nedergaard, J.; Bengtsson, T.; Cannon, B. Unexpected evidence for active brown adipose tissue in adult humans. Am. J. Physiol. Metab. 2007, 293, E444–E452. [Google Scholar] [CrossRef] [PubMed]
- Cypess, A.M.; Lehman, S.; Williams, G.; Tal, I.; Rodman, D.; Goldfine, A.B.; Kuo, F.C.; Palmer, E.L.; Tseng, Y.-H.; Doria, A.; et al. Identification and importance of brown ad-ipose tissue in adult humans. N. Engl. J. Med. 2009, 360, 1509–1517. [Google Scholar] [CrossRef] [Green Version]
- Rothwell, N.J.; Stock, M.J. Surgical removal of brown fat results in rapid and complete compensation by other depots. Am. J. Physiol. Integr. Comp. Physiol. 1989, 257, R253–R258. [Google Scholar] [CrossRef]
- Rogers, N.H. Brown adipose tissue during puberty and with aging. Ann. Med. 2014, 47, 142–149. [Google Scholar] [CrossRef]
- Hibi, M.; Oishi, S.; Matsushita, M.; Yoneshiro, T.; Yamaguchi, T.; Usui, C.; Yasunaga, K.; Katsuragi, Y.; Kubota, K.; Tanaka, S.; et al. Brown adipose tissue is involved in diet-induced thermogenesis and whole-body fat utilization in healthy humans. Int. J. Obes. 2016, 40, 1655–1661. [Google Scholar] [CrossRef] [Green Version]
- Rosen, E.D.; Spiegelman, B.M. What we talk about when we talk about fat. Cell 2014, 156, 20–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rui, L. Brown and beige adipose tissues in health and disease. Compr. Physiol. 2017, 7, 1281–1306. [Google Scholar] [CrossRef] [PubMed]
- Morigny, P.; Houssier, M.; Mouisel, E.; Langin, D. Adipocyte lipolysis and insulin resistance. Biochimie 2015, 125, 259–266. [Google Scholar] [CrossRef] [PubMed]
- McNelis, J.C.; Olefsky, J.M. Macrophages, immunity, and metabolic disease. Immunity 2014, 41, 36–48. [Google Scholar] [CrossRef] [Green Version]
- Boutens, L.; Stienstra, R. Adipose tissue macrophages: Going off track during obesity. Diabetologia 2016, 59, 879–894. [Google Scholar] [CrossRef] [Green Version]
- Glass, C.K.; Olefsky, J.M. Inflammation and lipid signaling in the etiology of insulin resistance. Cell Metab. 2012, 15, 635–645. [Google Scholar] [CrossRef] [Green Version]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Saltiel, A. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [CrossRef] [Green Version]
- Cao, W.; Daniel, K.W.; Robidoux, J.; Puigserver, P.; Medvedev, A.V.; Bai, X.; Floering, L.M.; Spiegelman, B.M.; Collins, S. p38 mitogen-activated protein kinase is the cen-tral regulator of cyclic AMP-dependent transcription of the brown fat uncoupling protein 1 gene. Mol. Cell. Biol. 2004, 24, 3057–3067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puigserver, P.; Rhee, J.; Lin, J.; Wu, Z.; Yoon, J.C.; Zhang, C.Y.; Krauss, S.; Mootha, V.K.; Lowell, B.B.; Spiegelman, B.M. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARgamma coactivator-1. Mol. Cell. 2001, 8, 971–982. [Google Scholar] [CrossRef]
- Carey, A.L.; Formosa, M.F.; Van Every, B.; Bertovic, D.; Eikelis, N.; Lambert, G.W.; Kalff, V.; Duffy, S.J.; Cherk, M.H.; Kingwell, B.A. Ephedrine activates brown adipose tissue in lean but not obese humans. Diabetologia 2013, 56, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Zylan, K.D.; Carlisle, H.J. Effect of ambient temperature on the paradoxical metabolic responses to norepinephrine. Pharmacol. Biochem. Behav. 1992, 43, 577–582. [Google Scholar] [CrossRef]
- Kotzbeck, P.; Giordano, A.; Mondini, E.; Murano, I.; Severi, I.; Venema, W.; Cecchini, M.P.; Kershaw, E.E.; Barbatelli, G.; Haemmerle, G.; et al. Brown adipose tissue whitening leads to brown adi-pocyte death and adipose tissue inflammation. J. Lipid Res. 2018, 59, 784–794. [Google Scholar] [CrossRef] [Green Version]
- van den Beukel, J.C.; Grefhorst, A.; Quarta, C.; Steenbergen, J.; Mastroberardino, P.G.; Lombès, M.; Delhanty, P.J.; Mazza, R.; Pagotto, U.; van der Lely, A.J.; et al. Direct activating effects of adrenocorticotropic hormone (ACTH) on brown adipose tissue are attenuated by corticosterone. FASEB J. Off. Public Fed. Am. Soc. Exp. Biol. 2014, 28, 4857–4867. [Google Scholar]
- Ramage, L.E.; Akyol, M.; Fletcher, A.M.; Forsythe, J.; Nixon, M.; Carter, R.N.; van Beek, E.J.; Morton, N.M.; Walker, B.R.; Stimson, R.H. Glucocorticoids acutely increase brown adipose tissue activity in humans, revealing species-specific differences in UCP-1 regulation. Cell Metab. 2016, 24, 130–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Yu, X.; Chen, Y. Recruitment of thermogenic fat: Trigger of fat burning. Front. Endocrinol. 2021, 12, 696505. [Google Scholar] [CrossRef]
- Solinas, G.; Karin, M. JNK1 and IKKbeta: Molecular links between obesity and metabolic dysfunction. FASEB J. Off. Public. Fed. Am. Soc. Exp. Biol. 2010, 24, 2596–2611. [Google Scholar]
- Osborn, O.; Olefsky, J.M. The cellular and signaling networks linking the immune system and metabolism in disease. Nat. Med. 2012, 18, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Speaker, K.J.; Fleshner, M. Interleukin-1 beta: A potential link between stress and the development of visceral obesity. BMC Physiol. 2012, 12, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomlinson, J.W.; Moore, J.; Cooper, M.S.; Bujalska, I.; Shahmanesh, M.; Burt, C.; Strain, A.; Hewison, M.; Stewart, P.M. Regulation of expression of 11beta-hydroxysteroid dehydrogenase type 1 in adipose tissue: Tissue-specific induction by cytokines. Endocrinology 2001, 142, 1982–1989. [Google Scholar] [CrossRef]
- Jager, J.; Grémeaux, T.; Cormont, M.; Le Marchand-Brustel, Y.; Tanti, J.-F. Interleukin-1beta-induced insulin resistance in adipocytes through down-regulation of insulin receptor substrate-1 expression. Endocrinology 2007, 148, 241–251. [Google Scholar] [CrossRef]
- Old, L.J. Tumor necrosis factor. Sci. Am. 1988, 258, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.T.; Ree, D.; Kolls, J.K.; Fuselier, J.; Coy, D.H.; Bryer-Ash, M. An in vivo model for elucidation of the mechanism of tumor necrosis factor-α (TNF-α)-induced insulin resistance: Evidence for differential regulation of insulin signaling by TNF-α. Endocrinology 1998, 139, 4928–4935. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.H.; Dobrescu, C.; Bagby, G.J. Tumor necrosis factor impairs insulin action on peripheral glucose disposal and hepatic glucose output. Endocrinology 1992, 130, 43–52. [Google Scholar] [CrossRef]
- Borst, S.E. The role of TNF-α in insulin resistance. Endocrine 2004, 23, 177–182. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Arner, P.; Caro, J.F.; Atkinson, R.L.; Spiegelman, B.M. Increased adipose tissue expression of tumor necrosis fac-tor-alpha in human obesity and insulin resistance. J. Clin. Investig. 1995, 95, 2409–2415. [Google Scholar] [CrossRef]
- Hotamisligil, G.; Shargill, N.; Spiegelman, B. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef]
- Fleischman, A.; Shoelson, S.E.; Bernier, R.; Goldfine, A.B. Salsalate improves glycemia and inflammatory parameters in obese young adults. Diabetes Care 2007, 31, 289–294. [Google Scholar] [CrossRef] [Green Version]
- Larsen, C.M.; Faulenbach, M.; Vaag, A.A.; Vølund, A.; Ehses, J.; Seifert, B.; Mandrup-Poulsen, T.; Donath, M.Y. Interleukin-1–Receptor Antagonist in Type 2 Diabetes Mellitus. N. Engl. J. Med. 2007, 356, 1517–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneto, H.; Nakatani, Y.; Miyatsuka, T.; Kawamori, D.; Matsuoka, T.-A.; Matsuhisa, M.; Kajimoto, Y.; Ichijo, H.; Yamasaki, Y.; Hori, M. Possible novel therapy for diabetes with cell-permeable JNK-inhibitory peptide. Nat. Med. 2004, 10, 1128–1132. [Google Scholar] [CrossRef]
- Yin, M.J.; Yamamoto, Y.; Gaynor, R.B. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature 1998, 396, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Pal, R.; Bhadada, S.K. COVID-19 and diabetes mellitus: An unholy interaction of two pandemics. Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 513–517. [Google Scholar] [CrossRef]
- Fadini, G.P.; Morieri, M.L.; Longato, E.; Avogaro, A. Prevalence and impact of diabetes among people infected with SARS-CoV-2. J. Endocrinol. Investig. 2020, 43, 867–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fadini, G.P.; Morieri, M.L.; Boscari, F.; Fiorettoa, P.; Marana, A.; Busettoa, L.; Bonoraa, B.M.; Selmina, E.; Arcidiacono, G.; Pinelli, S.; et al. Newly diagnosed diabetes and admission hyperglicemia predict COVID severity by ag-gravating repiratory dysfunction. Diabetes Res. Clin. Prat. 2020, 168, 108374. [Google Scholar] [CrossRef]
- Mantovani, A.; Byrne, C.D.; Zheng, M.-H.; Targher, G. Diabetes as a risk factor for greater COVID-19 severity and in-hospital death: A meta-analysis of observational studies. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 1236–1248. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; She, Z.-G.; Cheng, X.; Qin, J.-J.; Zhang, X.-J.; Cai, J.; Lei, F.; Wang, H.; Xie, J.; Wang, W.; et al. Association of Blood Glucose Control and Outcomes in Patients with COVID-19 and Pre-existing Type 2 Diabetes. Cell Metab. 2020, 31, 1068–1077.e3. [Google Scholar] [CrossRef]
- Steenblock, C.; Todorov, V.; Kanczkowski, W.; Eisenhofer, G.; Schedl, A.; Wong, M.-L.; Licinio, J.; Bauer, M.; Young, A.; Gainetdinov, R.R.; et al. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and the neuroendocrine stress axis. Mol. Psychiatry 2020, 25, 1611–1617. [Google Scholar] [CrossRef]
- Cuschieri, S.; Grech, S. COVID-19 and diabetes: The why, the what and the how. J. Diabetes Complicat. 2020, 34, 107637. [Google Scholar] [CrossRef]
- Yang, J.; Zheng, Y.; Gou, X.; Puab, K.; Chen, Z.; Guo, Q.; Jia, R.; Wang, H.; Wang, Y.; Zhou, Y.; et al. Prevalence of comorbidities and its effects in patients infected with SARS-CoV-2: A systematic review and meta-analysis. Int. J. Infect. Dis. 2020, 94, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Gerich, J.E. Type 2 diabetes mellitus is associated with multiple cardiometabolic risk factors. Clin. Cornerstone 2007, 8, 53–68. [Google Scholar] [CrossRef]
- Drucker, D.J. Coronavirus infections and type 2 diabetes—Shared pathways with therapeutic implications. Endocr. Rev. 2020, 41, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Iacobellis, G.; Malavazos, A.E.; Ferreira, T. COVID-19 rise in younger adults with obesity: Visceral adiposity can predict the risk. Obesity 2020, 28, 1795. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Eagon, J.C.; Trujillo, M.E.; Scherer, P.E.; Klein, S. Visceral fat adipokine secretion is associated with systemic inflammation in obese humans. Diabetes 2007, 56, 1010–1013. [Google Scholar] [CrossRef] [Green Version]
- Eketunde, A.O.; Mellacheruvu, S.P.; Oreoluwa, P. A review of postmortem findings in patients with COVID-19. Cureus 2020, 12, 9438. [Google Scholar] [CrossRef]
- Guo, W.; Li, M.; Dong, Y.; Zhou, H.; Zhang, Z.; Tian, C.; Qin, R.; Wang, H.; Shen, Y.; Du, K.; et al. Diabetes is a risk factor for the progression and prognosis of COVID-19. Diabetes Metab. Res. Rev. 2020, 36, e3319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.-K.; Feng, Y.; Yuan, M.Y.; Fu, H.J.; Wu, B.Y.; Sun, G.Z.; Yang, G.R.; Zhang, X.L.; Wang, L.; Xu, X.P.; et al. Plasma glucose levels and diabetes are independent predictors for mortality and morbidity in patients with SARS. Diabet. Med. 2006, 23, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Morra, M.E.; Thanh, L.; Kamel, M.G.; Ghazy, A.A.; Altibi, A.M.; Dat, L.M.; Thy, T.N.X.; Vuong, N.L.; Mostafa, M.R.; Ahmed, S.I.; et al. Clinical outcomes of current medical approaches for Middle East respiratory syndrome: A systematic review and meta-analysis. Rev. Med. Virol. 2018, 28, e1977. [Google Scholar] [CrossRef] [PubMed]
- Philips, B.J.; Meguer, J.-X.; Redman, J.; Baker, E. Factors determining the appearance of glucose in upper and lower respiratory tract secretions. Intensiv. Care Med. 2003, 29, 2204–2210. [Google Scholar] [CrossRef] [PubMed]
- Holman, N.; Knighton, P.; Kar, P.; O’Keefe, J.; Curley, M.; Weaver, A.; Barron, E.; Bakhai, C.; Khunti, K.; Wareham, N.J.; et al. Risk factors for COVID-19-related mortality in people with type 1 and type 2 diabetes in England: A population-based cohort study. Lancet Diabetes Endocrinol. 2020, 8, 823–833. [Google Scholar] [CrossRef]
- le Roux, C.W. COVID-19 alters thinking and management in metabolic diseases. Nat. Rev. Endocrinol. 2020, 17, 71–72. [Google Scholar] [CrossRef]
- Landstra, C.P.; de Koning, J.P. COVID-19 and diabetes: Understanding the interrelationship and risks for a severe course. Front. Endocrinol. 2021, 12, 599. [Google Scholar] [CrossRef]
Figure 1.
Relationship between the stress axis, inflammation, obesity and diabetes.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Gianotti, L.; Belcastro, S.; D’Agnano, S.; Tassone, F. The Stress Axis in Obesity and Diabetes Mellitus: An Update. Endocrines 2021, 2, 334-347. https://0-doi-org.brum.beds.ac.uk/10.3390/endocrines2030031
AMA Style
Gianotti L, Belcastro S, D’Agnano S, Tassone F. The Stress Axis in Obesity and Diabetes Mellitus: An Update. Endocrines. 2021; 2(3):334-347. https://0-doi-org.brum.beds.ac.uk/10.3390/endocrines2030031
Chicago/Turabian StyleGianotti, Laura, Sara Belcastro, Salvatore D’Agnano, and Francesco Tassone. 2021. "The Stress Axis in Obesity and Diabetes Mellitus: An Update" Endocrines 2, no. 3: 334-347. https://0-doi-org.brum.beds.ac.uk/10.3390/endocrines2030031