
ERβ in Triple-Negative Breast Cancer: Emerging Concepts and Therapeutic Possibilities

 , , , , and
, , , , and 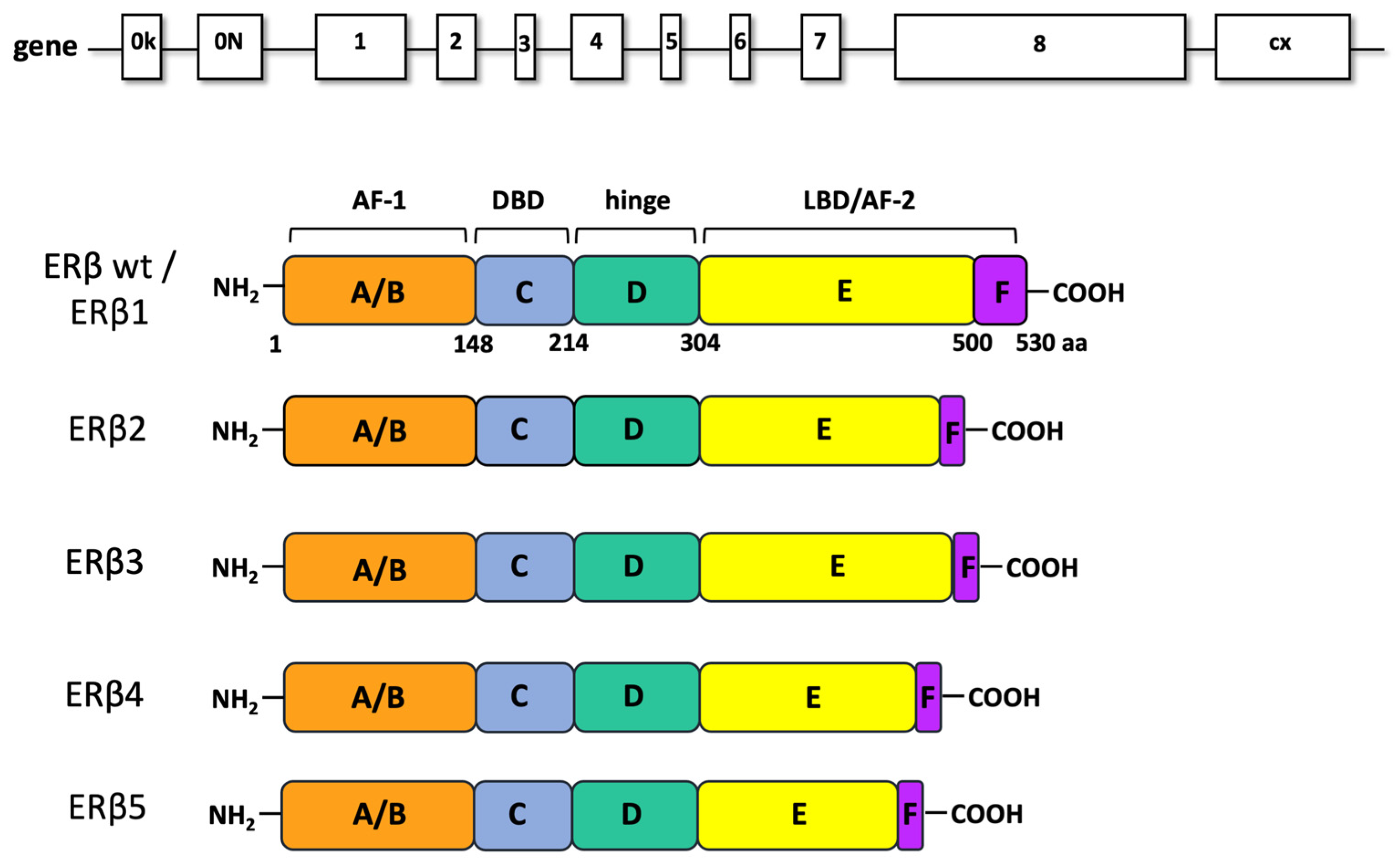{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Estrogen Action in Target Cells
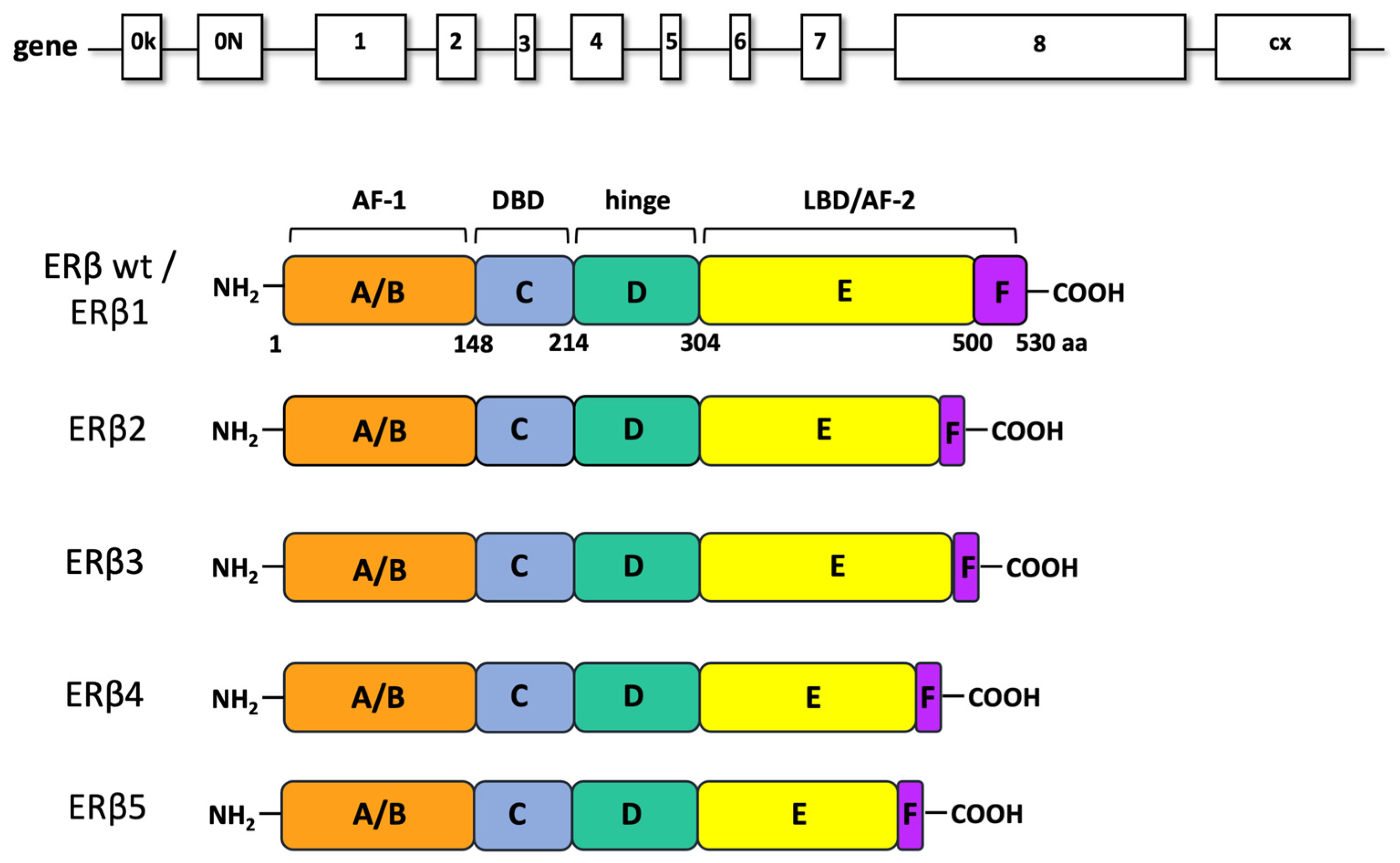
3. ERβ: Structure and Function
4. ERβ in TNBC
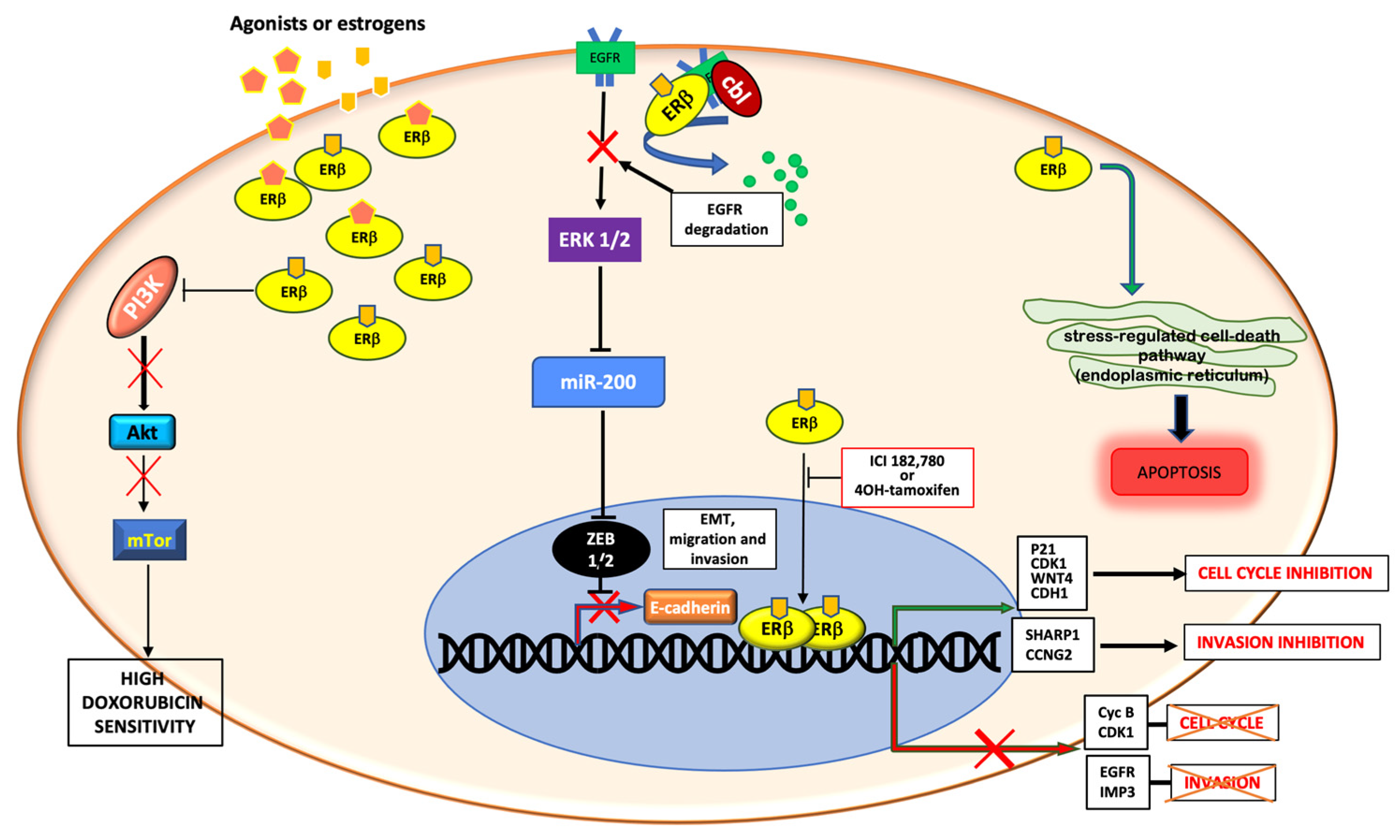
4.1. Genomic Action of ERβ in TNBC
4.2. Non-Genomic Actions of ERβ in TNBC
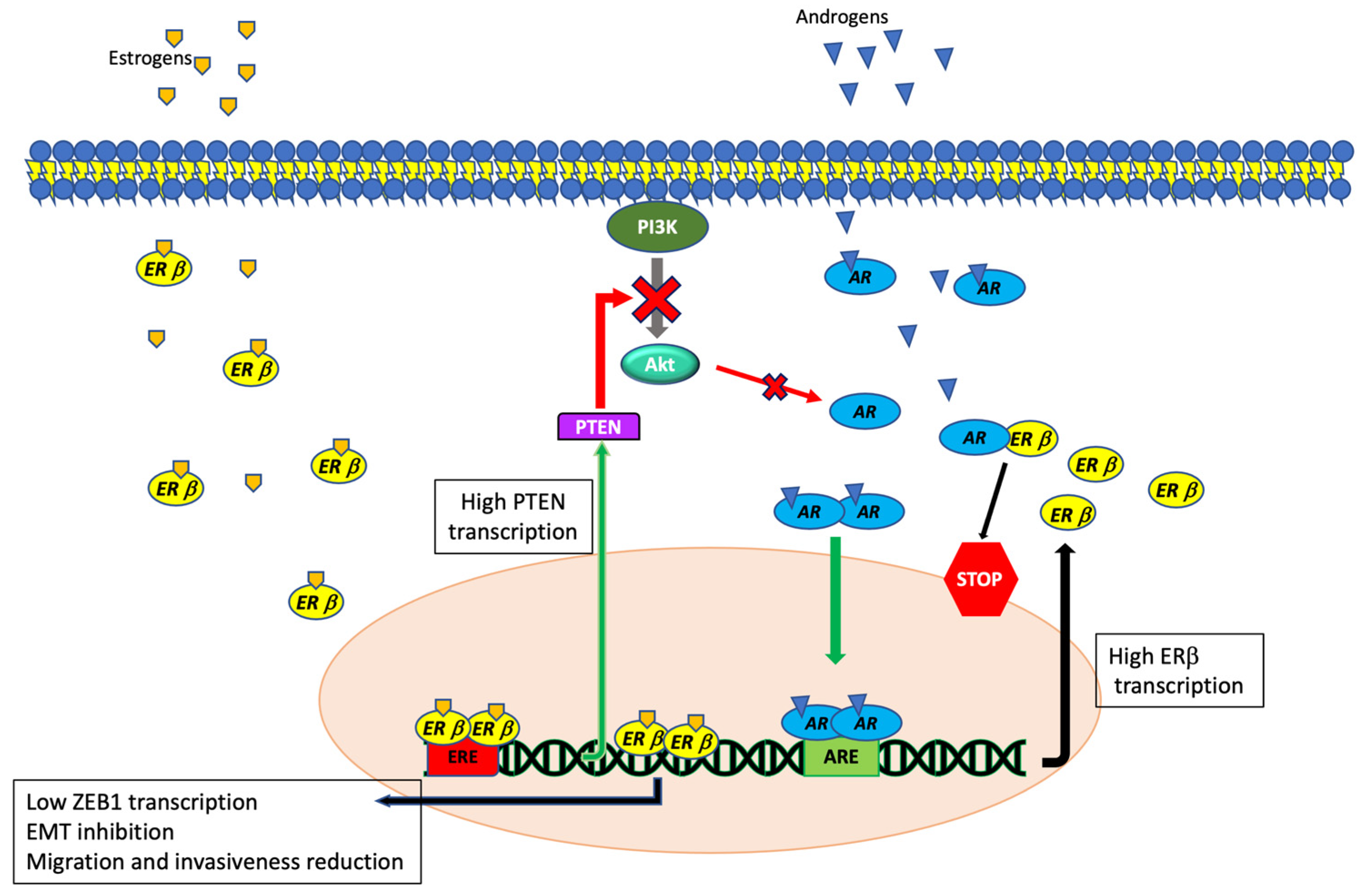
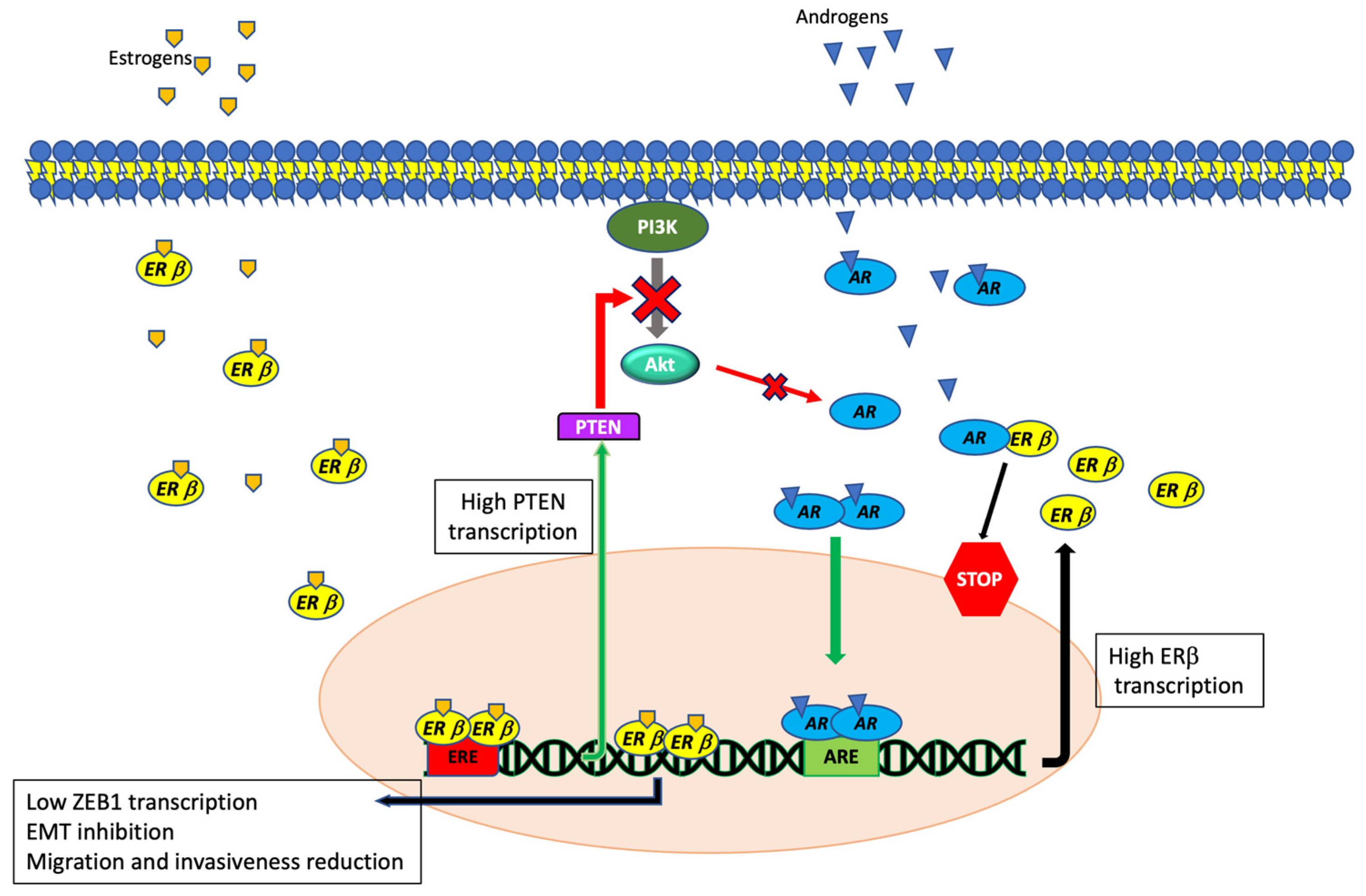
4.3. ERβ and Androgen Receptor Interaction in TNBCs
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AR | androgen receptor |
| BC | breast cancer |
| EnR | endoplasmic reticulum |
| ER | estradiol receptor |
| TNBC | triple negative breast cancer |
References
- Sharma, R. Global, regional, national burden of breast cancer in 185 countries: Evidence from GLOBOCAN 2018. Breast Cancer Res. Treat. 2021, 187, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Salvi, S.; Bonafè, M.; Bravaccini, S. Androgen receptor in breast cancer: A wolf in sheep’s clothing? A lesson from prostate cancer. Semin. Cancer Biol. 2020, 60, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Chan, P.S.; Lok, V.; Chen, X.; Ding, H.; Jin, Y.; Yuan, J.; Lao, X.-Q.; Zheng, Z.-J.; Wong, M.C. Global incidence and mortality of breast cancer: A trend analysis. Aging 2021, 13, 5748–5803. [Google Scholar] [CrossRef] [PubMed]
- Rivenbark, A.G.; O’Connor, S.M.; Coleman, W.B. Molecular and cellular heterogeneity in breast cancer: Challenges for personalized medicine. Am. J. Pathol. 2013, 183, 1113–1124. [Google Scholar] [CrossRef] [Green Version]
- Giovannelli, P.; Di Donato, M.; Galasso, G.; di Zazzo, E.; Bilancio, A.; Migliaccio, A. The Androgen Receptor in Breast Cancer. Front. Endocrinol. 2018, 9, 492. [Google Scholar] [CrossRef] [Green Version]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-Negative Breast Cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [Green Version]
- Migliaccio, A.; Di Domenico, M.; Castoria, G.; Nanayakkara, M.; Lombardi, M.; De Falco, A.; Bilancio, A.; Varricchio, L.; Ciociola, A.; Auricchio, F. Steroid Receptor Regulation of Epidermal Growth Factor Signaling through Src in Breast and Prostate Cancer Cells: Steroid Antagonist Action. Cancer Res. 2005, 65, 10585–10593. [Google Scholar] [CrossRef] [Green Version]
- di Zazzo, E.; Galasso, G.; Giovannelli, P.; Di Donato, M.; Castoria, G. Estrogens and Their Receptors in Prostate Cancer: Therapeutic Implications. Front. Oncol. 2018, 8, 2. [Google Scholar] [CrossRef] [Green Version]
- Hartman, J.; Edvardsson, K.; Lindberg, K.; Zhao, C.; Williams, C.; Ström, A.; Gustafsson, J.-Å. Tumor Repressive Functions of Estrogen Receptor β in SW480 Colon Cancer Cells. Cancer Res. 2009, 69, 6100–6106. [Google Scholar] [CrossRef] [Green Version]
- Paruthiyil, S.; Parmar, H.; Kerekatte, V.; Cunha, G.R.; Firestone, G.L.; Leitman, D.C. Estrogen Receptor β Inhibits Human Breast Cancer Cell Proliferation and Tumor Formation by Causing a G2 Cell Cycle Arrest. Cancer Res. 2004, 64, 423–428. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhang, C.; Chen, K.; Tang, H.; Tang, J.; Song, C.; Xie, X. ERβ1 inversely correlates with PTEN/PI3K/AKT pathway and predicts a favorable prognosis in triple-negative breast cancer. Breast Cancer Res. Treat. 2015, 152, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, J.-A.; Strom, A.; Warner, M. Update on ERbeta. J. Steroid Biochem. Mol. Biol. 2019, 191, 105312. [Google Scholar] [CrossRef]
- Yan, S.; Dey, P.; Ziegler, Y.; Jiao, X.; Kim, S.H.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Contrasting activities of estrogen receptor beta isoforms in triple negative breast cancer. Breast Cancer Res. Treat. 2021, 185, 281–292. [Google Scholar] [CrossRef]
- Hamilton, K.J.; Hewitt, S.C.; Arao, Y.; Korach, K.S. Estrogen Hormone Biology. Curr. Top. Dev. Biol. 2017, 125, 109–146. [Google Scholar] [CrossRef]
- Di Croce, L.; Okret, S.; Kersten, S.; Gustafsson, J.-Å.; Parker, M.; Wahli, W.; Beato, M. Steroid and nuclear receptors. Villefranche-sur-Mer, France, May 25–27, 1999. EMBO J. 1999, 18, 6201–6210. [Google Scholar] [CrossRef] [Green Version]
- Szego, C.M.; Davis, J.S. Adenosine 3′,5′-monophosphate in rat uterus: Acute elevation by estrogen. Proc. Natl. Acad. Sci. USA 1967, 58, 1711–1718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Romancer, M.; Poulard, C.; Cohen, P.; Sentis, S.; Renoir, J.-M.; Corbo, L. Cracking the Estrogen Receptor’s Posttranslational Code in Breast Tumors. Endocr. Rev. 2011, 32, 597–622. [Google Scholar] [CrossRef] [Green Version]
- Giovannelli, P. Targeting rapid action of sex-steroid receptors in breast and prostate cancers. Front. Biosci. 2012, E4, 453–461. [Google Scholar] [CrossRef]
- Migliaccio, A.; Castoria, G.; Auricchio, F. Src-dependent signalling pathway regulation by sex-steroid hormones: Therapeutic implications. Int. J. Biochem. Cell Biol. 2007, 39, 1343–1348. [Google Scholar] [CrossRef]
- Levin, E.R.; Hammes, S.R. Nuclear receptors outside the nucleus: Extranuclear signalling by steroid receptors. Nat. Rev. Mol. Cell Biol. 2016, 17, 783–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicent, G.P.; Zaurin, R.; Nacht, A.S.; Font-Mateu, J.; Le Dily, F.; Beato, M. Nuclear Factor 1 Synergizes with Progesterone Receptor on the Mouse Mammary Tumor Virus Promoter Wrapped around a Histone H3/H4 Tetramer by Facilitating Access to the Central Hormone-responsive Elements. J. Biol. Chem. 2010, 285, 2622–2631. [Google Scholar] [CrossRef] [Green Version]
- Castoria, G.; Auricchio, F.; Migliaccio, A. Extranuclear partners of androgen receptor: At the crossroads of proliferation, migration, and neuritogenesis. FASEB J. 2017, 31, 1289–1300. [Google Scholar] [CrossRef] [Green Version]
- Elappano, R.; Episano, A.; Maggiolini, M. GPER Function in Breast Cancer: An Overview. Front. Endocrinol. 2014, 5, 66. [Google Scholar] [CrossRef] [Green Version]
- Kuiper, G.G.; Shughrue, P.J.; Merchenthaler, I.; Gustafsson, J.A. The estrogen receptor beta subtype: A novel mediator of estrogen action in neuroendocrine systems. Front. Neuroendocrinol. 1998, 19, 253–286. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.T.; McKee, D.D.; Slentz-Kesler, K.; Moore, L.B.; Jones, S.A.; Horne, E.L.; Su, J.-L.; Kliewer, S.A.; Lehmann, J.M.; Willson, T.M. Cloning and Characterization of Human Estrogen Receptor β Isoforms. Biochem. Biophys. Res. Commun. 1998, 247, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Leygue, E.; Murphy, L.C. A bi-faceted role of estrogen receptor β in breast cancer. Endocrine-Relat. Cancer 2013, 20, R127–R139. [Google Scholar] [CrossRef] [PubMed]
- Leung, Y.-K.; Mak, P.; Hassan, S.; Ho, S.-M. Estrogen receptor (ER)-beta isoforms: A key to understanding ER-beta signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 13162–13167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuiper, G.; Enmark, E.; PeltoHuikko, M.; Nilsson, S.; Gustafsson, J.A. Cloning of a novel estrogen receptor expressed in rat prostate and ovary. Proc. Natl. Acad. Sci. USA 1996, 93, 5925–5930. [Google Scholar] [CrossRef] [Green Version]
- Božović, A.; Mandušić, V.; Todorović, L.; Krajnović, M. Estrogen Receptor Beta: The Promising Biomarker and Potential Target in Metastases. Int. J. Mol. Sci. 2021, 22, 1656. [Google Scholar] [CrossRef] [PubMed]
- Warner, M.; Gustafsson, J.A. The role of estrogen receptor beta (ERbeta) in malignant diseases—A new potential target for antipro-liferative drugs in prevention and treatment of cancer. Biochem. Biophys. Res. Commun. 2010, 396, 63–66. [Google Scholar] [CrossRef]
- Wisinski, K.B.; Xu, W.; Tevaarwerk, A.J.; Saha, S.; Kim, K.; Traynor, A.; Dietrich, L.; Hegeman, R.; Patel, D.; Blank, J.; et al. Targeting Estrogen Receptor Beta in a Phase 2 Study of High-Dose Estradiol in Metastatic Triple-Negative Breast Cancer: A Wisconsin Oncology Network Study. Clin. Breast Cancer 2016, 16, 256–261. [Google Scholar] [CrossRef] [Green Version]
- Chantzi, Ν.Ι.; Tiniakos, D.G.; Palaiologou, M.; Goutas, N.; Filippidis, T.; Vassilaros, S.D.; Dhimolea, E.; Mitsiou, D.J.; Alexis, Μ.N. Estrogen receptor beta 2 is associated with poor prognosis in estrogen receptor alpha-negative breast carcinoma. J. Cancer Res. Clin. Oncol. 2013, 139, 1489–1498. [Google Scholar] [CrossRef]
- Tong, D.; Schuster, E.; Seifert, M.; Czerwenka, K.; Leodolter, S.; Zeillinger, R. Expression of estrogen receptor beta isoforms in human breast cancer tissues and cell lines. Breast Cancer Res. Treat. 2002, 71, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Faria, M.; Karami, S.; Granados-Principal, S.; Dey, P.; Verma, A.; Choi, D.S.; Elemento, O.; Bawa-Khalfe, T.; Chang, J.C.; Strom, A.M.; et al. The ERβ4 variant induces transformation of the normal breast mammary epithelial cell line MCF-10A; the ERβ variants ERβ2 and ERβ5 increase aggressiveness of TNBC by regulation of hypoxic signaling. Oncotarget 2018, 9, 12201–12211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaşar, P.; Ayaz, G.; User, S.D.; Güpür, G.; Muyan, M. Molecular mechanism of estrogen-estrogen receptor signaling. Reprod. Med. Biol. 2017, 16, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Shanle, E.K.; Zhao, Z.; Hawse, J.; Wisinski, K.; Keles, S.; Yuan, M.; Xu, W. Research Resource: Global Identification of Estrogen Receptor β Target Genes in Triple Negative Breast Cancer Cells. Mol. Endocrinol. 2013, 27, 1762–1775. [Google Scholar] [CrossRef] [Green Version]
- Reese, J.M.; Bruinsma, E.S.; Monroe, D.G.; Negron, V.; Suman, V.J.; Ingle, J.N.; Goetz, M.P.; Hawse, J.R. ERβ inhibits cyclin dependent kinases 1 and 7 in triple negative breast cancer. Oncotarget 2017, 8, 96506–96521. [Google Scholar] [CrossRef] [Green Version]
- Lazennec, G.; Bresson, D.; Lucas, A.; Chauveau, C.; Vignon, F. ERβ Inhibits Proliferation and Invasion of Breast Cancer Cells. Endocrinology 2001, 142, 4120–4130. [Google Scholar] [CrossRef]
- Reese, J.M.; Bruinsma, E.S.; Nelson, A.W.; Chernukhin, I.; Carroll, J.S.; Li, Y.; Subramaniam, M.; Suman, V.J.; Negron, V.; Monroe, D.G.; et al. ERβ-mediated induction of cystatins results in suppression of TGFβ signaling and inhibition of triple-negative breast cancer metastasis. Proc. Natl. Acad. Sci. USA 2018, 115, E9580–E9589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schüler-Toprak, S.; Häring, J.; Inwald, E.C.; Moehle, C.; Ortmann, O.; Treeck, O. Agonists and knockdown of estrogen receptor β differentially affect invasion of triple-negative breast cancer cells in vitro. BMC Cancer 2016, 16, 951. [Google Scholar] [CrossRef] [Green Version]
- Samanta, S.; Sharma, V.M.; Khan, A.; Mercurio, A.M. Regulation of IMP3 by EGFR signaling and repression by ERβ: Implications for triple-negative breast cancer. Oncogene 2012, 31, 4689–4697. [Google Scholar] [CrossRef] [PubMed]
- Bado, I.; Nikolos, F.; Rajapaksa, G.; Gustafsson, J.-Å.; Thomas, C. ERβ decreases the invasiveness of triple-negative breast cancer cells by regulating mutant p53 oncogenic function. Oncotarget 2016, 7, 13599–13611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, W.; Tang, L.; Xu, Y.; Sun, Q.; Yang, F.; Guan, X. ERβ1 inhibits metastasis of androgen receptor-positive triple-negative breast cancer by suppressing ZEB1. J. Exp. Clin. Cancer Res. 2017, 36, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, V.; Di Zazzo, E.; Galasso, G.; De Rosa, C.; Abbondanza, C.; Sinisi, A.A.; Altucci, L.; Migliaccio, A.; Castoria, G. Estrogens Modulate Somatostatin Receptors Expression and Synergize with the Somatostatin Analog Pasireotide in Prostate Cells. Front. Pharmacol. 2019, 10, 28. [Google Scholar] [CrossRef] [Green Version]
- Improta-Brears, T.; Whorton, A.R.; Codazzi, F.; York, J.D.; Meyer, T.; McDonnell, D.P. Estrogen-induced activation of mitogen-activated protein kinase requires mobilization of intracellular calcium. Proc. Natl. Acad. Sci. USA 1999, 96, 4686–4691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aronica, S.M.; Kraus, W.L.; Katzenellenbogen, B.S. Estrogen action via the cAMP signaling pathway: Stimulation of adenylate cyclase and cAMP-regulated gene transcription. Proc. Natl. Acad. Sci. USA 1994, 91, 8517–8521. [Google Scholar] [CrossRef] [Green Version]
- Migliaccio, A.; Castoria, G.; Di Domenico, M.; De Falco, A.; Bilancio, A.; Lombardi, M.; Barone, M.V.; Ametrano, D.; Zannini, M.S.; Abbondanza, C.; et al. Steroid-induced androgen receptor-oestradiol receptor beta-Src complex triggers prostate cancer cell proliferation. EMBO J. 2000, 19, 5406–5417. [Google Scholar] [CrossRef] [Green Version]
- Stellato, C.; Nassa, G.; Tarallo, R.; Giurato, G.; Ravo, M.; Rizzo, F.; Marchese, G.; Alexandrova, E.; Cordella, A.; Baumann, M.; et al. Identification of cytoplasmic proteins interacting with unliganded estrogen receptor α and β in human breast cancer cells. Proteomics 2015, 15, 1801–1807. [Google Scholar] [CrossRef]
- Castoria, G.; Migliaccio, A.; Bilancio, A.; Di Domenico, M.; De Falco, A.; Lombardi, M.; Fiorentino, R.; Varricchio, L.; Barone, M.V.; Auricchio, F. PI3-kinase in concert with Src promotes the S-phase entry of oestradiol-stimulated MCF-7 cells. EMBO J. 2001, 20, 6050–6059. [Google Scholar] [CrossRef] [Green Version]
- Le Romancer, M.; Treilleux, I.; Leconte, N.; Robin-Lespinasse, Y.; Sentis, S.; Bouchekioua-Bouzaghou, K.; Goddard, S.; Gobert-Gosse, S.; Corbo, L. Regulation of Estrogen Rapid Signaling through Arginine Methylation by PRMT1. Mol. Cell 2008, 31, 212–221. [Google Scholar] [CrossRef]
- Dillon, R.L.; White, D.E.; Muller, W.J. The phosphatidyl inositol 3-kinase signaling network: Implications for human breast cancer. Oncogene 2007, 26, 1338–1345. [Google Scholar] [CrossRef]
- Lei, S.; Fan, P.; Wang, M.; Zhang, C.; Jiang, Y.; Huang, S.; Fang, M.; He, Z.; Wu, A. Elevated estrogen receptor β expression in triple negative breast cancer cells is associated with sensitivity to doxorubicin by inhibiting the PI3K/AKT/mTOR signaling pathway. Exp. Ther. Med. 2020, 20, 1630–1636. [Google Scholar] [CrossRef]
- Greish, K.; Nehoff, H.; Bahman, F.; Pritchard, T.; Taurin, S. Raloxifene nano-micelles effect on triple-negative breast cancer is mediated through estrogen receptor-β and epidermal growth factor receptor. J. Drug Target. 2019, 27, 903–916. [Google Scholar] [CrossRef] [PubMed]
- Rajapaksa, G.; Nikolos, F.; Bado, I.; Clarke, R.; Gustafsson, J.-Å.; Thomas, C. ERβ decreases breast cancer cell survival by regulating the IRE1/XBP-1 pathway. Oncogene 2015, 34, 4130–4141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, C.; Rajapaksa, G.; Nikolos, F.; Hao, R.; Katchy, A.; Mccollum, C.W.; Bondesson, M.; Quinlan, P.; Thompson, A.; Krishnamurthy, S.; et al. ERβ1 represses basal-like breast cancer epithelial to mesenchymal transition by destabilizing EGFR. Breast Cancer Res. 2012, 14, R148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesei, A.; Castoria, G. Editorial: The Androgen Receptor in Breast Cancer. Front. Endocrinol. 2021, 11, 636480. [Google Scholar] [CrossRef] [PubMed]
- Traina, T.A.; Miller, K.; Yardley, D.A.; Eakle, J.; Schwartzberg, L.S.; O’Shaughnessy, J.; Gradishar, W.; Schmid, P.; Winer, E.; Kelly, C.; et al. Enzalutamide for the Treatment of Androgen Receptor–Expressing Triple-Negative Breast Cancer. J. Clin. Oncol. 2018, 36, 884–890. [Google Scholar] [CrossRef]
- Giovannelli, P.; Di Donato, M.; Auricchio, F.; Castoria, G.; Migliaccio, A. Androgens Induce Invasiveness of Triple Negative Breast Cancer Cells Through AR/Src/PI3-K Complex Assembly. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, M.; Wu, J.; Ling, R.; Li, N. Quadruple negative breast cancer. Breast Cancer 2020, 27, 527–533. [Google Scholar] [CrossRef]
- Anestis, A.; Sarantis, P.; Theocharis, S.; Zoi, I.; Tryfonopoulos, D.; Korogiannos, A.; Koumarianou, A.; Xingi, E.; Thomaidou, D.; Kontos, M.; et al. Estrogen receptor beta increases sensitivity to enzalutamide in androgen receptor-positive triple-negative breast cancer. J. Cancer Res. Clin. Oncol. 2019, 145, 1221–1233. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monaco, A.; Licitra, F.; Di Gisi, M.; Galasso, G.; Di Donato, M.; Giovannelli, P.; Migliaccio, A.; Castoria, G. ERβ in Triple-Negative Breast Cancer: Emerging Concepts and Therapeutic Possibilities. Endocrines 2021, 2, 356-365. https://0-doi-org.brum.beds.ac.uk/10.3390/endocrines2030033
Monaco A, Licitra F, Di Gisi M, Galasso G, Di Donato M, Giovannelli P, Migliaccio A, Castoria G. ERβ in Triple-Negative Breast Cancer: Emerging Concepts and Therapeutic Possibilities. Endocrines. 2021; 2(3):356-365. https://0-doi-org.brum.beds.ac.uk/10.3390/endocrines2030033
Chicago/Turabian StyleMonaco, Alessandra, Fabrizio Licitra, Martina Di Gisi, Giovanni Galasso, Marzia Di Donato, Pia Giovannelli, Antimo Migliaccio, and Gabriella Castoria. 2021. "ERβ in Triple-Negative Breast Cancer: Emerging Concepts and Therapeutic Possibilities" Endocrines 2, no. 3: 356-365. https://0-doi-org.brum.beds.ac.uk/10.3390/endocrines2030033