
Porous Polymer Gel Electrolytes Influence Lithium Transference Number and Cycling in Lithium-Ion Batteries



Abstract
:1. Introduction
2. Materials and Methods
2.1. Vinyl Sulfonate Salt Preparation
2.2. Synthesis of Condensed, Transparent Membrane PEGDMA-VS-0
2.3. Synthesis of Porous, Opaque Membrane PEGDMA-VS-0
2.4. Synthesis of Opaque and Condensed Membrane PEGDMA
2.5. Ion Exchange Process
2.6. Solvent and Salt Drying
2.7. Electrolyte Preparation
2.8. Electrolyte Uptake Test
2.9. Elemental Analysis
2.10. Cathode Preparation—LiFePO4
2.11. Conductivity Measurement
2.12. Fourier-Transform Infrared Spectroscopy (FTIR)
2.13. Scanning Electron Microscope (SEM)
2.14. Lithium Symmetric Cells—Electrochemical Impedance Spectroscopy
2.15. Li/LiFePO4 Cells—Galvanostatic Cycling
3. Results and Discussion
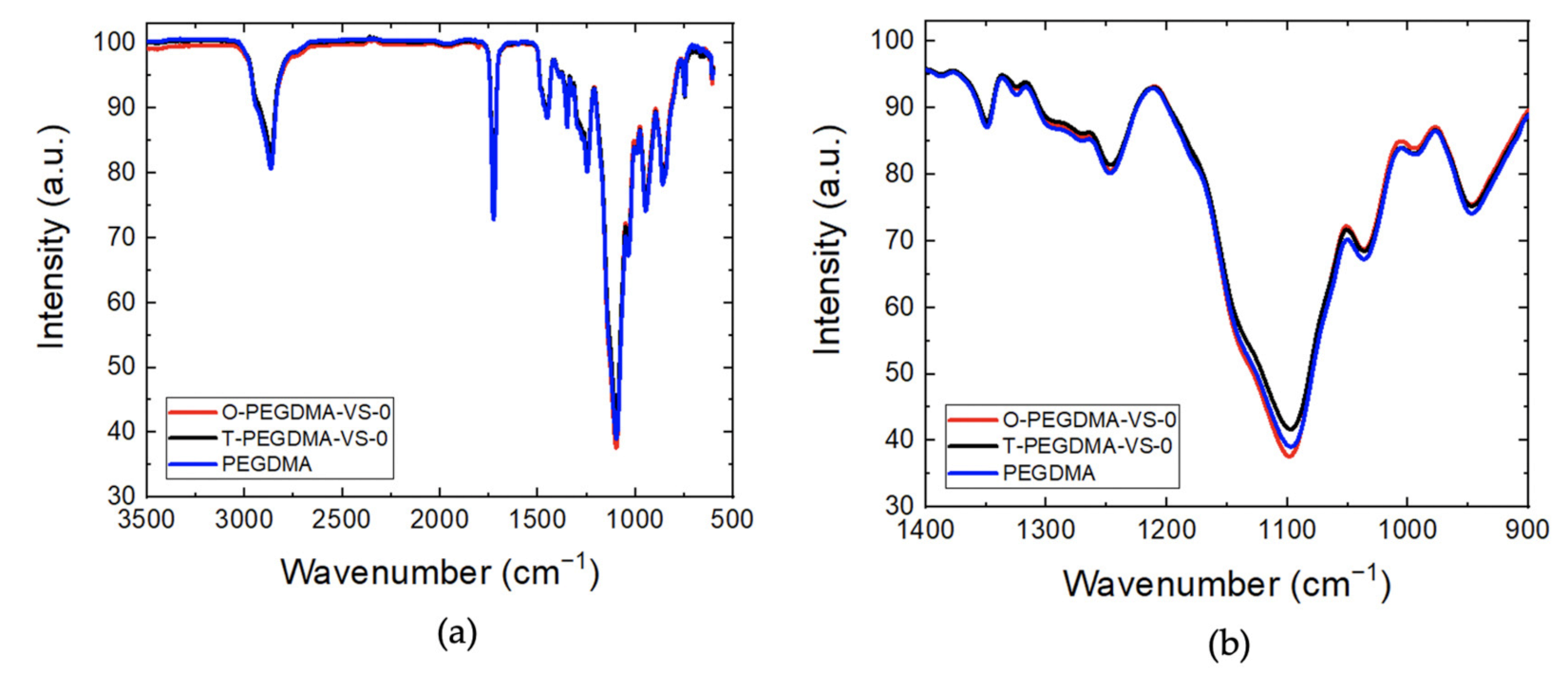
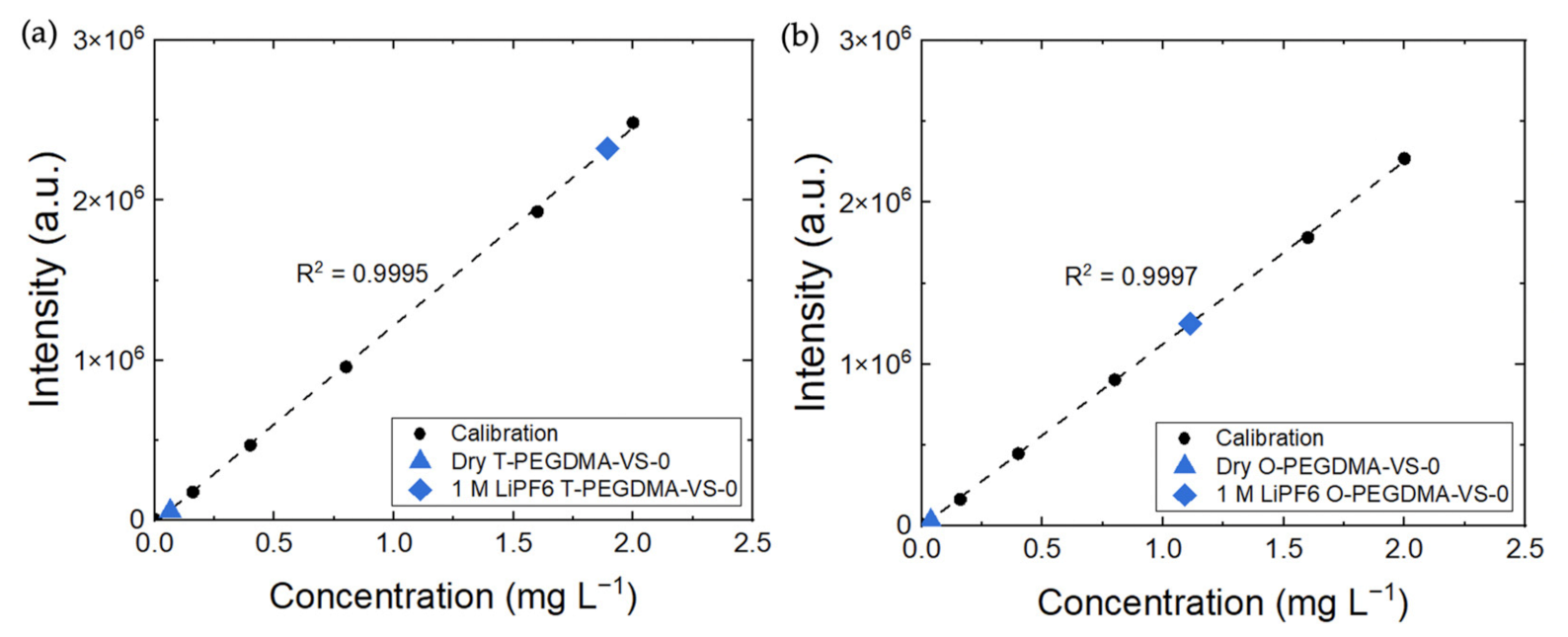
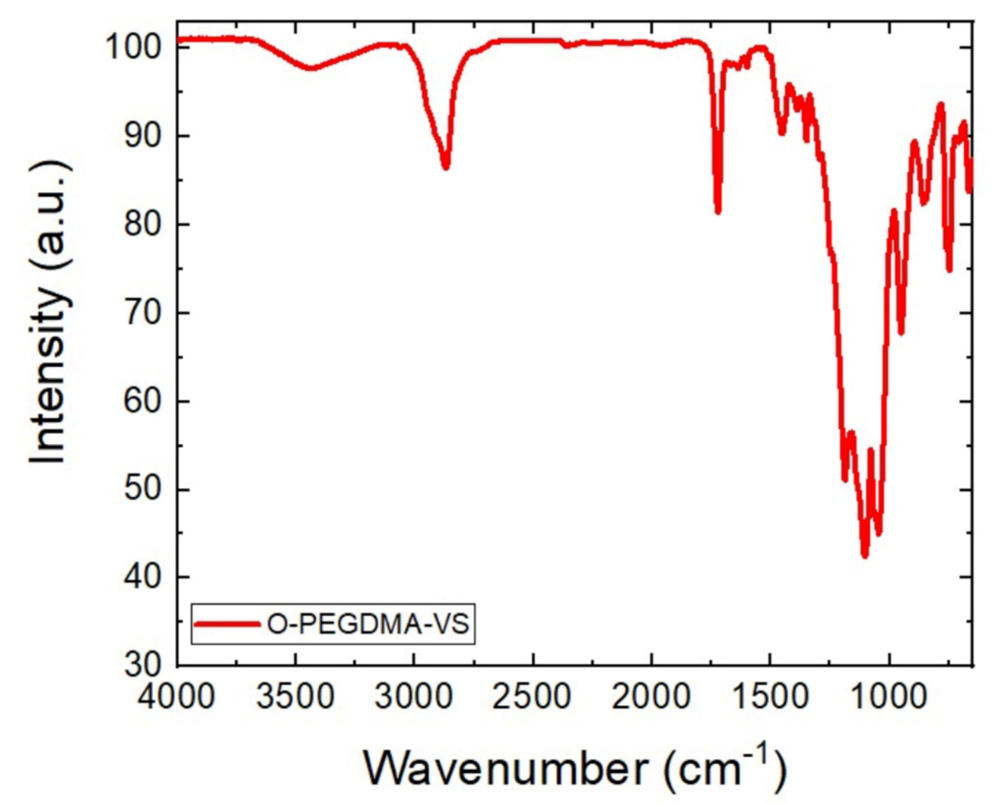
3.1. FTIR (Fourier Transform Infrared Spectroscopy) and ICP-OES (Inductively Coupled Plasma-Optical Emission Spectroscopy) Analysis
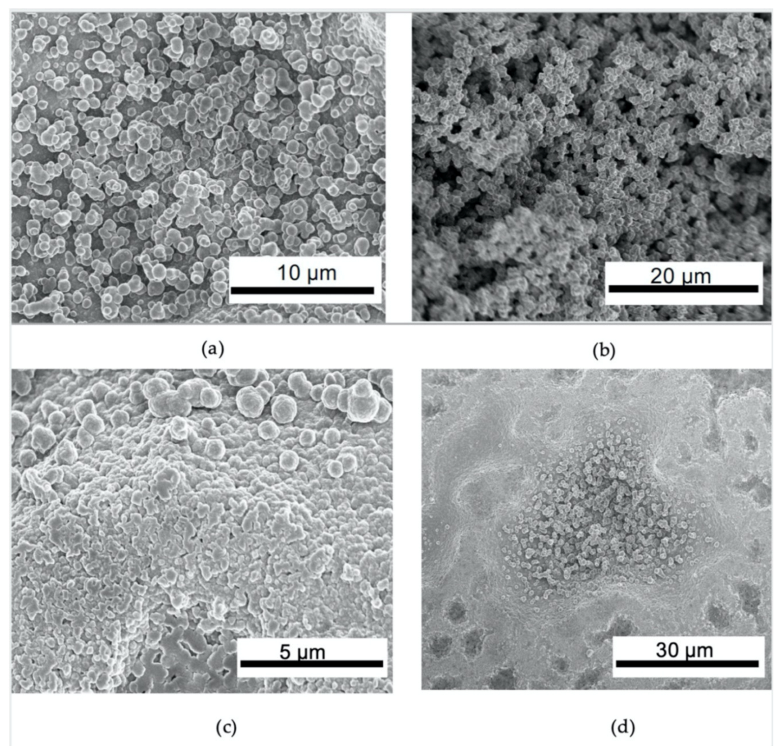
3.2. Structure Analysis of O-PEGDMA-VS-0 and T-PEGDMA-VS-0
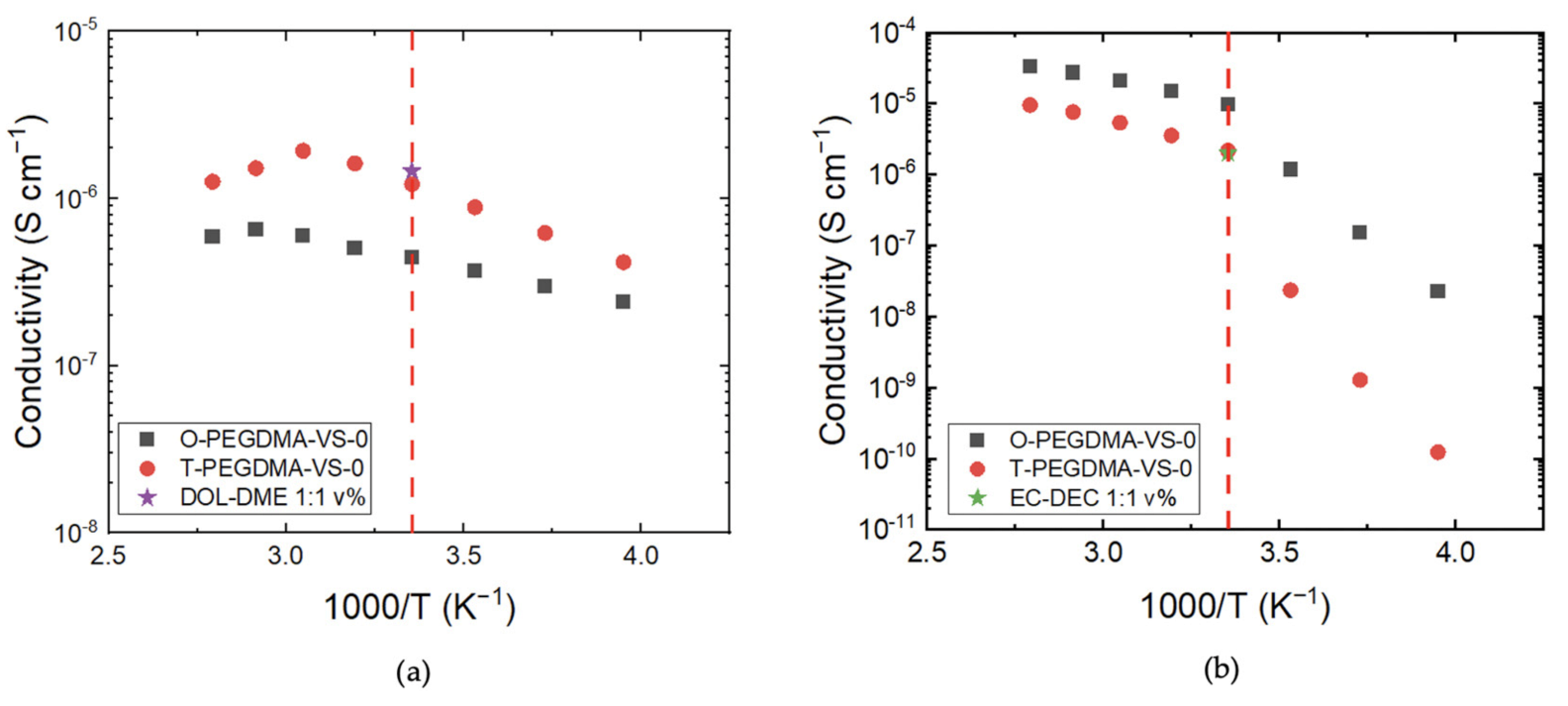
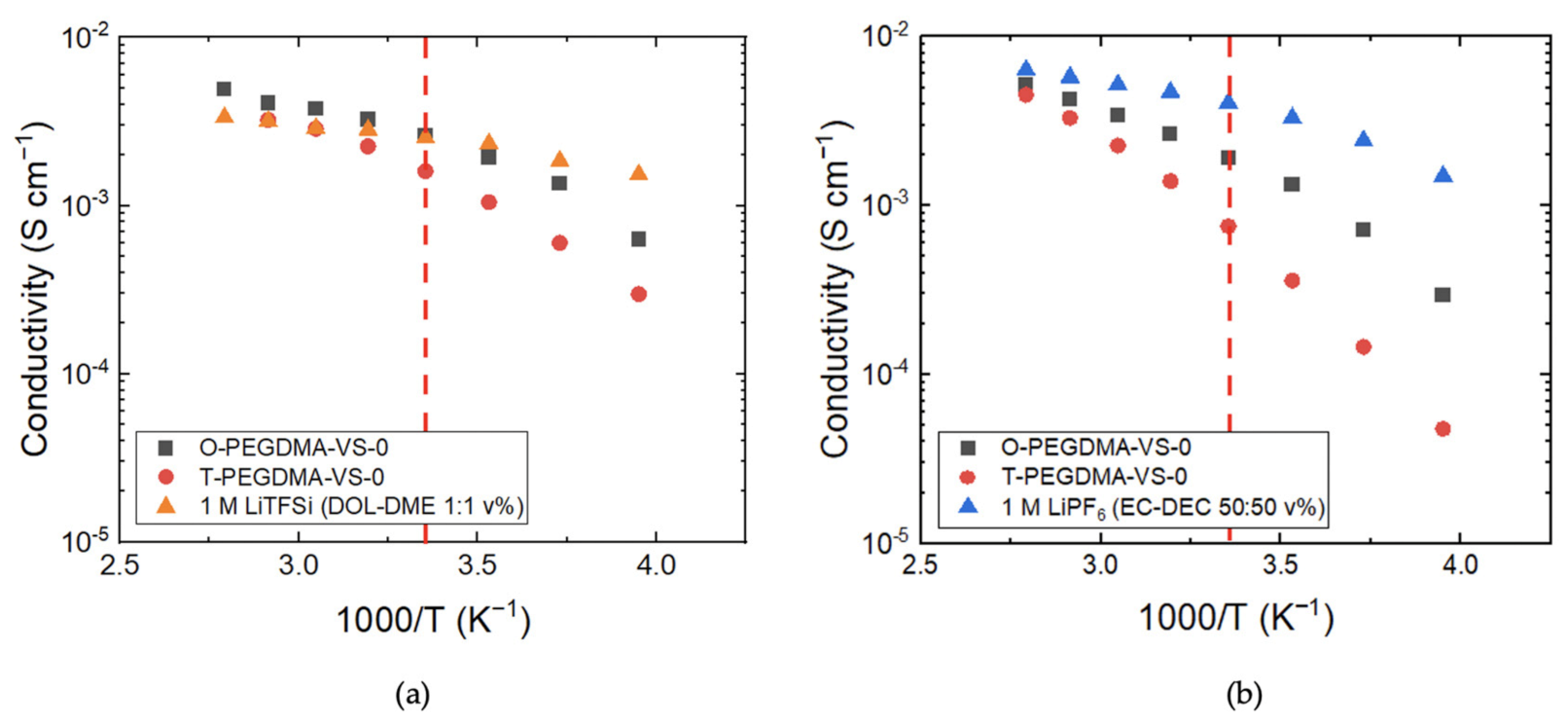
3.3. Conductivity Comparison of O-PEGDMA-VS-0 and T-PEGDMA-VS-0
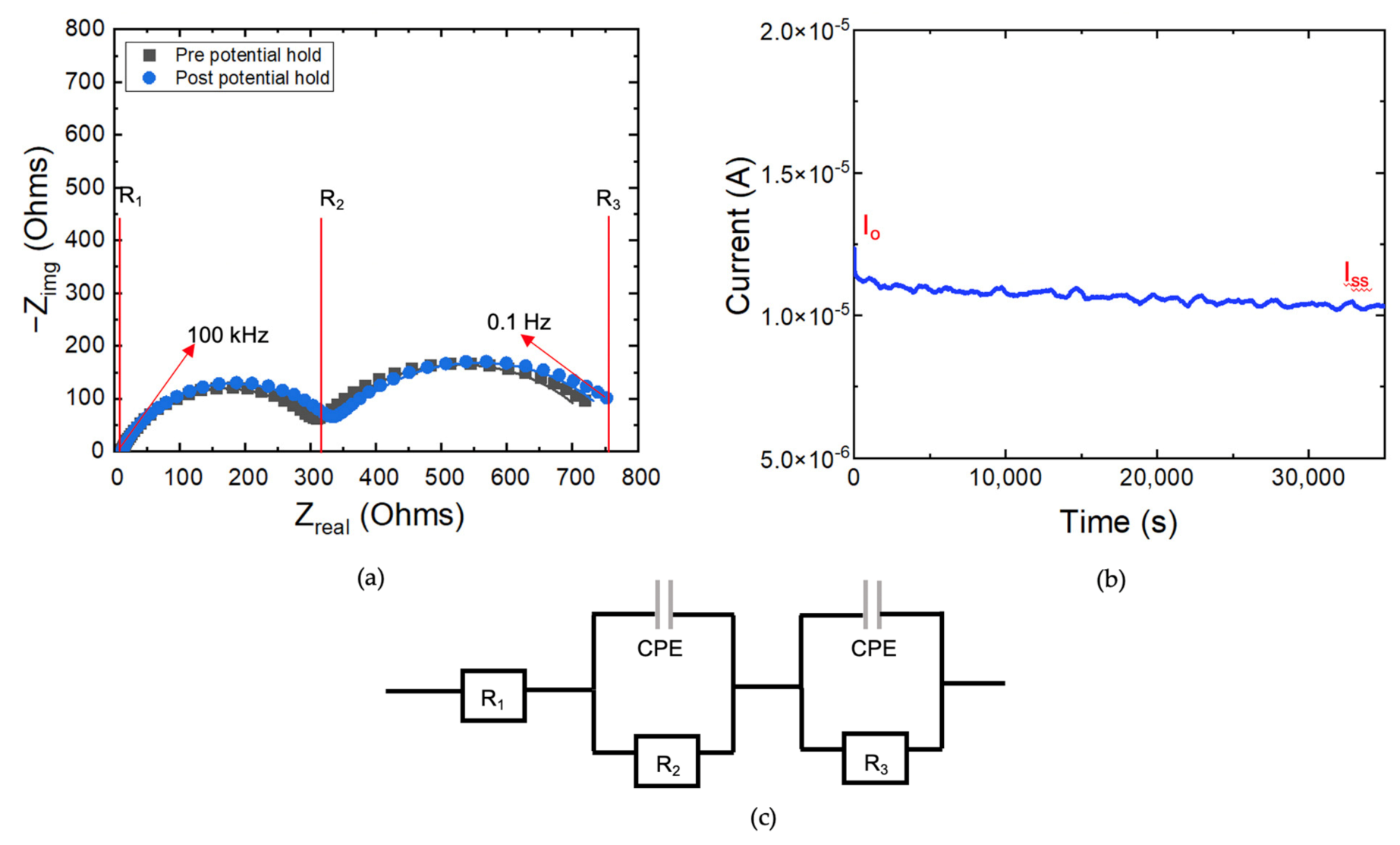
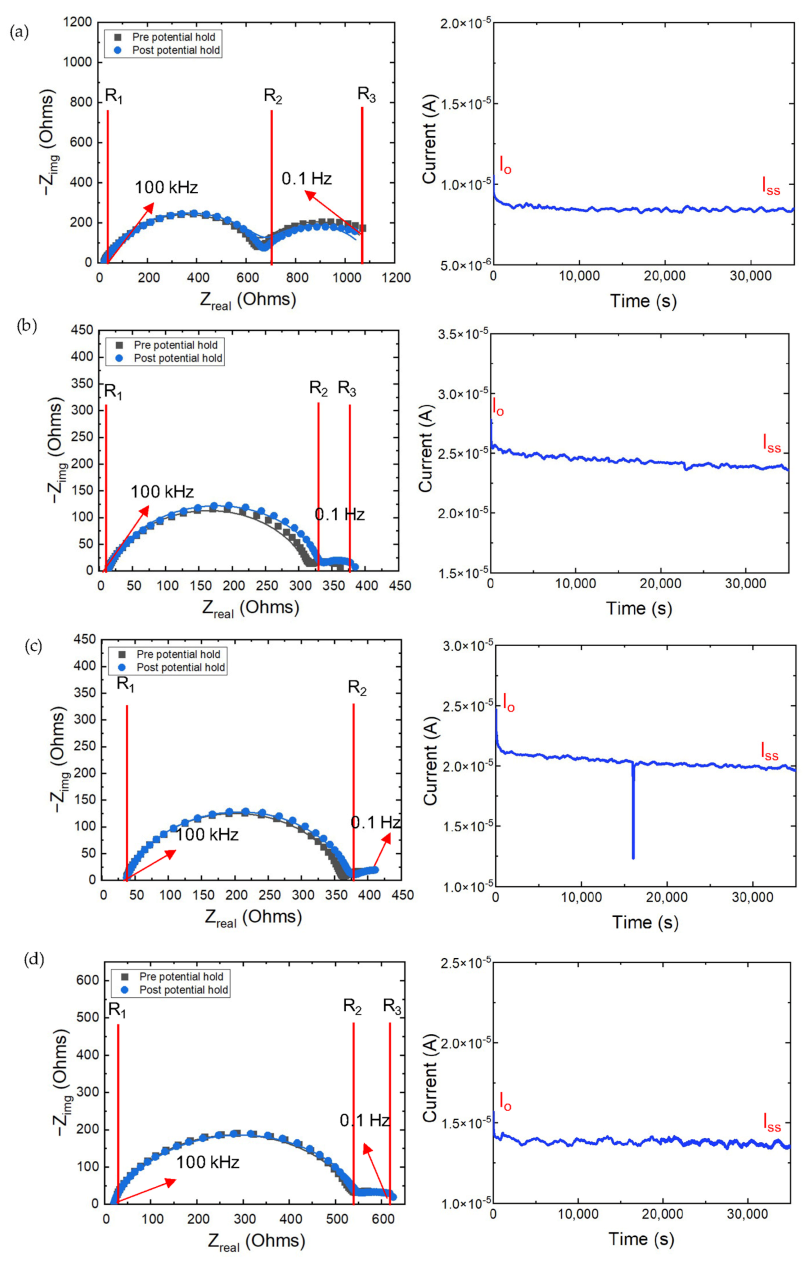
3.4. Electrochemical Impedance Spectroscopy—Transference Number Measurements
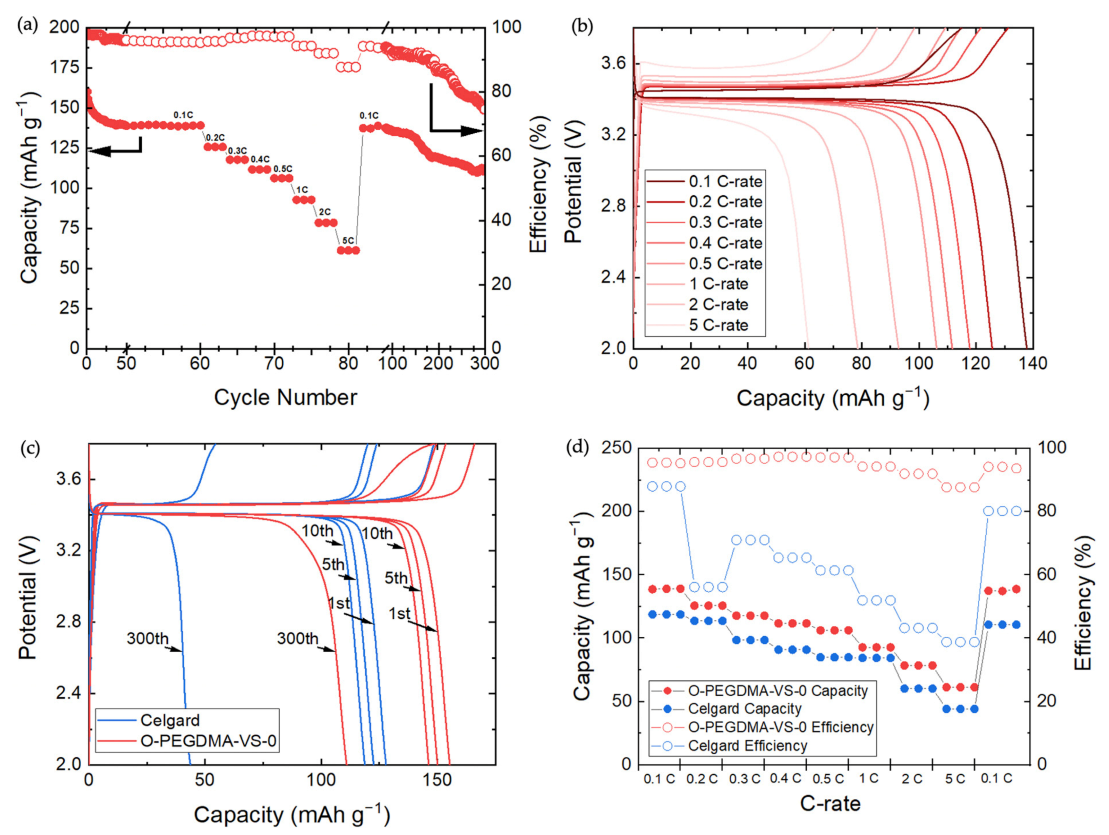
3.5. Li/LiFePO4 Cycling Performance
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Expected Li+ Concentration (mol mg−1) | Measured Li+ Concentration (mol/mg) | |
|---|---|---|---|
| T-PEGDMA-VS-0 | 4% | ||
| O-PEGDMA-VS-0 | 3% |
| Sample Name | Expected Li+ Concentration (mol mg−1) | Measured Li+ Concentration (mol mg−1) | |
|---|---|---|---|
| O-PEGDMA-VS-0 in 1 M LiTFSI (DOL-DME) | 8% | ||
| O-PEGDMA-VS-0 in 2 M LiOH | 3% |
| Sample Name | Measured Li+ Concentration (mol mg−1) | |
|---|---|---|
| T-PEGDMA-VS-0 in 1 M LiPF6 (EC-DEC) | 265% | |
| O-PEGDMA-VS-0 in 1 M LiPF6 (EC-DEC) | 531% |
| Sample Name | Mass Increase % | Volume Increase % |
|---|---|---|
| O-PEGDMA-VS-0 | 329 | 166 |
| T-PEGDMA-VS-0 | 245 | 155 |
| Sample Name | R1,EIS1 (Ohms) | R2,EIS1 (Ohms) | R1,EIS2 (Ohms) | R2,EIS2 (Ohms) | Ri,EIS2 = R2,EIS1 − R1,EIS1 (Ohms) | Rss,EIS2 = R2,EIS2 − R1, EIS2 (Ohms) | Iss (A) | Io (A) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| O-PEGDMA-VS-0 in 1 M LiPF6 (EC-DEC) (1st trial) | 14 | 312 | 14 | 336 | 298 | 322 | 0.0098 | 0.79 | ||
| O-PEGDMA-VS-0 in 1 M LiPF6 (EC-DEC) (2nd trial) | 18 | 382 | 18 | 405 | 364 | 387 | 0.011 | 0.78 | ||
| O-PEGDMA-VS-0 in 1 M LiPF6 (EC-DEC) (3rd trial) | 13 | 551 | 13 | 555 | 538 | 542 | 0.0098 | 0.79 | ||
| T-PEGDMA-VS-0 in 1 M LiPF6 (EC-DEC) | 29 | 671 | 29 | 691 | 642 | 662 | 0.011 | 0.65 | ||
| O-PEGDMA-VS-0 in 1 M LiTFSI (DOL-DME) | 13 | 317 | 13 | 332 | 304 | 318 | 0.0098 | 0.51 | ||
| T-PEGDMA-VS-0 in 1 M LiTFSI (DOL-DME) | 37 | 363 | 37 | 377 | 326 | 340 | 0.0098 | 0.47 | ||
| Celgard in 1 M LiPF6 (EC-DEC) | 26 | 552 | 4 | 590 | 526 | 586 | 0.010 | 0.47 |


Appendix B. The Discussion on the Impacts of the Crosslinking Procedure
| Scheme | Measured Li+ Concentration (mol mg−1) | |
|---|---|---|
| * O-PEGDMA-VS-0 | 3% | |
| ** O-PEGDMA-VS (wet) | 4% |

References
- Cano, Z.P.; Banham, D.; Ye, S.; Hintennach, A.; Lu, J.; Fowler, M.; Chen, Z. Batteries and fuel cells for emerging electric vehicle markets. Nat. Energy 2018, 3, 279–289. [Google Scholar] [CrossRef]
- Schmuch, R.; Wagner, R.; Hörpel, G.; Placke, T.; Winter, M. Performance and cost of materials for lithium-based rechargeable automotive batteries. Nat. Energy 2018, 3, 267–278. [Google Scholar] [CrossRef]
- Zhang, Z.; Shao, Y.; Lotsch, B.; Hu, Y.-S.; Li, H.; Janek, J.; Nazar, L.F.; Nan, C.; Maier, J.; Armand, M.; et al. New horizons for inorganic solid state ion conductors. Energy Envrion. Sci. 2018, 11, 1945–1976. [Google Scholar] [CrossRef] [Green Version]
- Xu, K. Electrolytes and interphases in li-Ion batteries and beyond. Chem. Rev. 2014, 114, 11503–11618. [Google Scholar] [CrossRef] [PubMed]
- Baik, J.-H.; Kim, S.; Hong, D.G.; Lee, J.-C. Gel Polymer electrolytes based on polymerizable lithium salt and poly(ethylene glycol) for lithium battery applications. ACS Appl. Mater. Interfaces 2019, 11, 29718–29724. [Google Scholar] [CrossRef]
- Ford, H.O.; Park, B.; Jiang, J.; Seidler, M.E.; Schaefer, J.L. Enhanced Li+ conduction within single-son conducting polymer gel electrolytes via reduced cation-polymer interaction. ACS Mater. Lett. 2020, 2, 272–279. [Google Scholar] [CrossRef]
- Deng, K.; Wang, S.; Ren, S.; Han, D.; Xiao, M.; Meng, Y. Network type sp3 boron-based single-ion conducting polymer electrolytes for lithium ion batteries. J. Power Sources 2017, 360, 98–105. [Google Scholar] [CrossRef]
- Zhang, H.; Li, C.; Piszcz, M.; Coya, E.; Rojo, T.; Rodriguez-Martinez, L.M.; Armand, M.; Zhou, Z. Single lithium-ion conducting solid polymer electrolytes: Advances and perspectives. Chem. Soc. Rev. 2017, 46, 797–815. [Google Scholar] [CrossRef]
- Bouchet, R.; Maria, S.; Meziane, R.; Aboulaich, A.; Lienafa, L.; Bonnet, J.-P.; Phan, T.T.N.; Bertin, D.; Gigmes, D.; Devaux, D.; et al. Single-ion BAB triblock copolymers as highly efficient electrolytes for lithium-metal batteries. Nat. Mater. 2013, 12, 452–457. [Google Scholar] [CrossRef]
- Lin, M.; Fu, C.; Li, L.; Mayilvahanan, K.S.; Watkins, T.; Perdue, B.R.; Zavadil, K.R.; Helms, B.A. Nanoporous polymer films with a high cation transference number stabilize lithium metal anodes in light-weight batteries for electrified transportation. Nano Lett. 2019, 19, 1387–1394. [Google Scholar] [CrossRef]
- Ma, L.; Nath, P.; Tu, Z.; Tikekar, M.; Archer, L.A. Highly conductive, sulfonated, UV-Cross-Linked separators for Li–S batteries. Chem. Mater. 2016, 28, 5147–5154. [Google Scholar] [CrossRef]
- Li, L.; Wang, M.; Wang, J.; Ye, F.; Wang, S.; Xu, Y.; Liu, J.; Xu, G.; Zhang, Y.; Zhang, Y.; et al. Asymmetric gel polymer electrolyte with high lithium ion conductivity for dendrite-free lithium metal batteries. J. Mater. Chem. A 2020, 8, 8033–8040. [Google Scholar] [CrossRef]
- Wang, Y.; Fu, L.; Shi, L.; Wang, Z.; Zhu, J.; Zhao, Y.; Yuan, S. Gel polymer electrolyte with high li transference number enhancing the cycling stability of lithium anodes. ACS Appl. Mater. Interfaces 2019, 11, 5168–5175. [Google Scholar] [CrossRef]
- Baran, M.J.; Carrington, M.E.; Sahu, S.; Baskin, A.; Song, J.; Baird, M.A.; Teat, S.J.; Meckler, S.M.; Fu, C.; Prendergast, D.; et al. Diversity-oriented synthesis of polymers of intrinsic microporosity with explicit solid solvation cages for lithium ions. Nature 2021, 592, 225–233. [Google Scholar] [CrossRef]
- Du, H.; Shi, S.; Liu, W.; Che, G.; Piao, M. Hydrophobic-force-driven adsorption of bisphenol A from aqueous solution by polyethylene glycol diacrylate hydrogel microsphere. Environ. Sci. Pollut. Res. 2019, 26, 22362–22371. [Google Scholar] [CrossRef]
- Aydın, D.; Kızılel, S. Water-in-water emulsion based synthesis of hydrogel nanospheres with tunable release kinetics. Jom 2017, 69, 1185–1194. [Google Scholar] [CrossRef]
- Nam, C.; Yoon, J.; Ryu, S.A.; Choi, C.-H.; Lee, H. Water and oil insoluble PEGDA-based microcapsule: Biocompatible and multicomponent encapsulation. ACS Appl. Mater. Interfaces 2018, 10, 40366–40371. [Google Scholar] [CrossRef]
- Merrill, L.C.; Ford, H.O.; Schaefer, J.L. Application of single-ion conducting gel polymer electrolytes in magnesium batteries. ACS Appl. Energy Mater. 2019, 2, 6355–6363. [Google Scholar] [CrossRef]
- Bruce, P.G.; Evans, J.; Vincent, C.A. Conductivity and transference number measurements on polymer electrolytes. Solid State Ion. 1988, 28, 918–922. [Google Scholar] [CrossRef]
- Diederichsen, K.M.; McShane, E.J.; McCloskey, B.D. Promising routes to a high Li + transference number electrolyte for lithium ion batteries. ACS Energy Lett. 2017, 2, 2563–2575. [Google Scholar] [CrossRef]
- Strauss, E.; Menkin, S.; Golodnitsky, D. On the way to high-conductivity single lithium-ion conductors. J. Solid State Electrochem. 2017, 21, 1879–1905. [Google Scholar] [CrossRef]
- Zaleski, R.; Krasucka, P.; Skrzypiec, K.; Goworek, J. Macro- and nanoscopic studies of porous polymer swelling. Macromolecules 2017, 50, 5080–5089. [Google Scholar] [CrossRef]
- Sienkiewicz, A.; Krasucka, P.; Charmas, B.; Stefaniak, W.; Goworek, J. Swelling effects in cross-linked polymers by thermogravimetry. J. Therm. Anal. Calorim. 2017, 130, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Markevich, E.; Salitra, G.; Chesneau, F.; Schmidt, M.; Aurbach, D. Very stable lithium metal stripping–plating at a high rate and high areal capacity in fluoroethylene carbonate-based organic electrolyte solution. ACS Energy Lett. 2017, 2, 1321–1326. [Google Scholar] [CrossRef]
- Liu, Y.; Lin, D.; Yuen, P.Y.; Liu, K.; Xie, J.; Dauskardt, R.H.; Cui, Y. An artificial solid electrolyte interphase with high Li-ion conductivity, mechanical strength, and flexibility for stable lithium metal anodes. Adv. Mater. 2017, 29, 1605531. [Google Scholar] [CrossRef]






| Sample Name | Crosslinker | Crosslinker Mass (g) | * VS (g) | Anhydrous DMSO (g) | Photoinitiator (g) | Water (µL) |
|---|---|---|---|---|---|---|
| T-PEGDMA-VS-0 | PEGDMA 750 g/mol | 0.536 | 0.214 | 0.850 | 0.0075 | - |
| O-PEGDMA-VS-0 | PEGDMA 750 g/mol | 0.536 | 0.214 | 0.799 | 0.0075 | 51.4 |
| Sample Name/Electrolyte | |
|---|---|
| O-PEGDMA-VS-0 in 1 M LiPF6 (EC-DEC) | 0.78 |
| T-PEGDMA-VS-0 in 1 M LiPF6 (EC-DEC) | 0.65 |
| O-PEGDMA-VS-0 in 1 M LiTFSI (DOL-DME) | 0.51 |
| T-PEGDMA-VS-0 in 1 M LiTFSI (DOL-DME) | 0.47 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boz, B.; Ford, H.O.; Salvadori, A.; Schaefer, J.L. Porous Polymer Gel Electrolytes Influence Lithium Transference Number and Cycling in Lithium-Ion Batteries. Electron. Mater. 2021, 2, 154-173. https://0-doi-org.brum.beds.ac.uk/10.3390/electronicmat2020013
Boz B, Ford HO, Salvadori A, Schaefer JL. Porous Polymer Gel Electrolytes Influence Lithium Transference Number and Cycling in Lithium-Ion Batteries. Electronic Materials. 2021; 2(2):154-173. https://0-doi-org.brum.beds.ac.uk/10.3390/electronicmat2020013
Chicago/Turabian StyleBoz, Buket, Hunter O. Ford, Alberto Salvadori, and Jennifer L. Schaefer. 2021. "Porous Polymer Gel Electrolytes Influence Lithium Transference Number and Cycling in Lithium-Ion Batteries" Electronic Materials 2, no. 2: 154-173. https://0-doi-org.brum.beds.ac.uk/10.3390/electronicmat2020013