Understanding the Reactivity of Trimethylsilyldiazoalkanes Participating in [3+2] Cycloaddition Reactions towards Diethylfumarate with a Molecular Electron Density Theory Perspective




Abstract
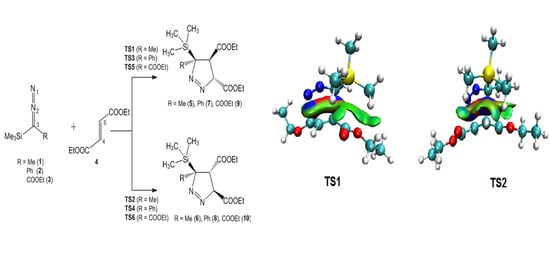
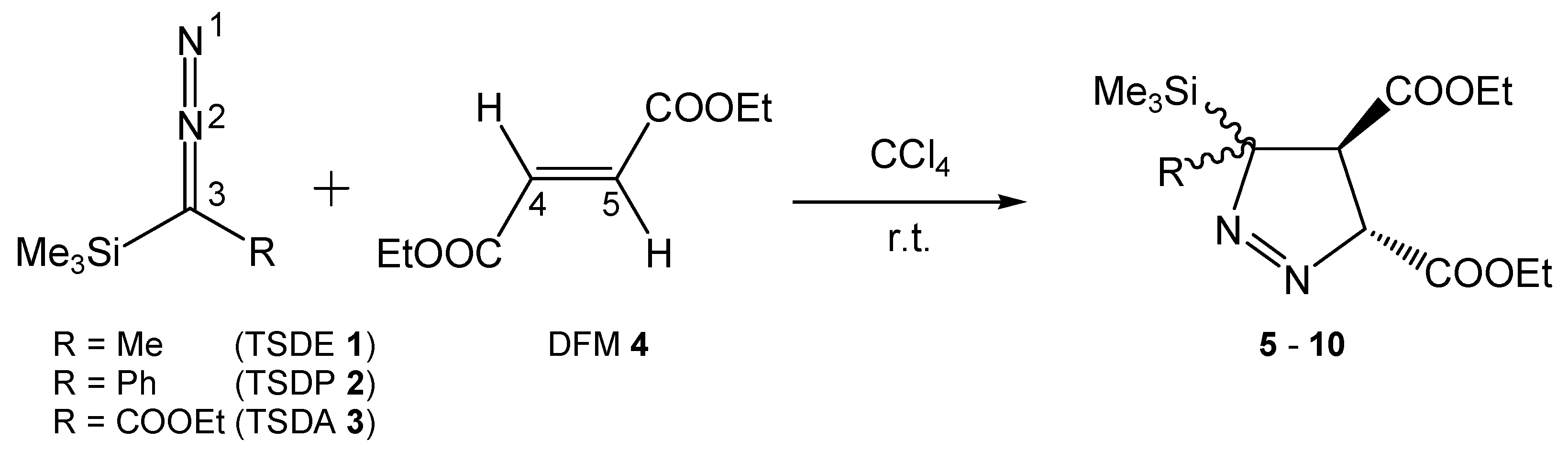
:
1. Introduction
2. Computational Methods
3. Results and Discussion
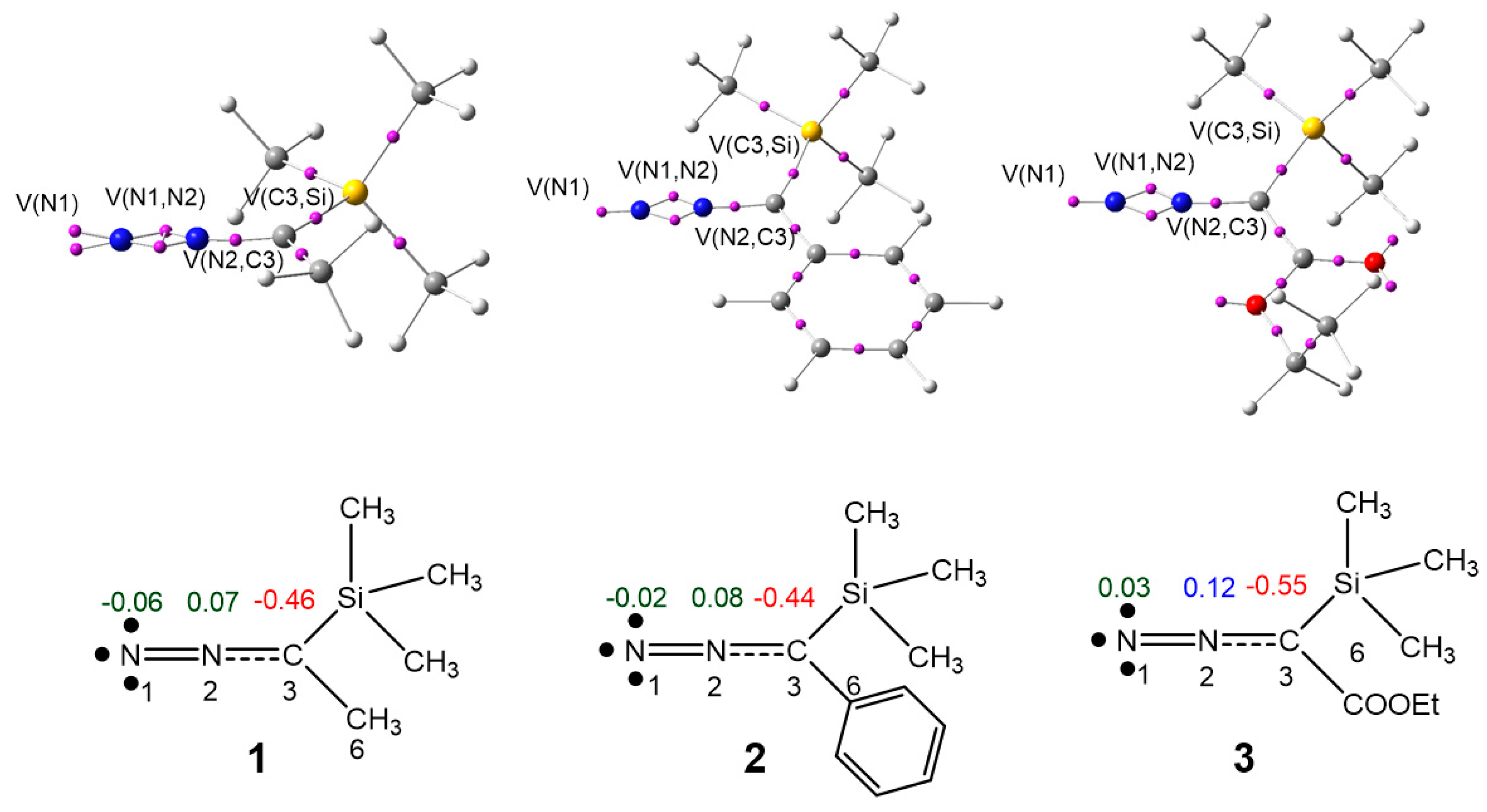
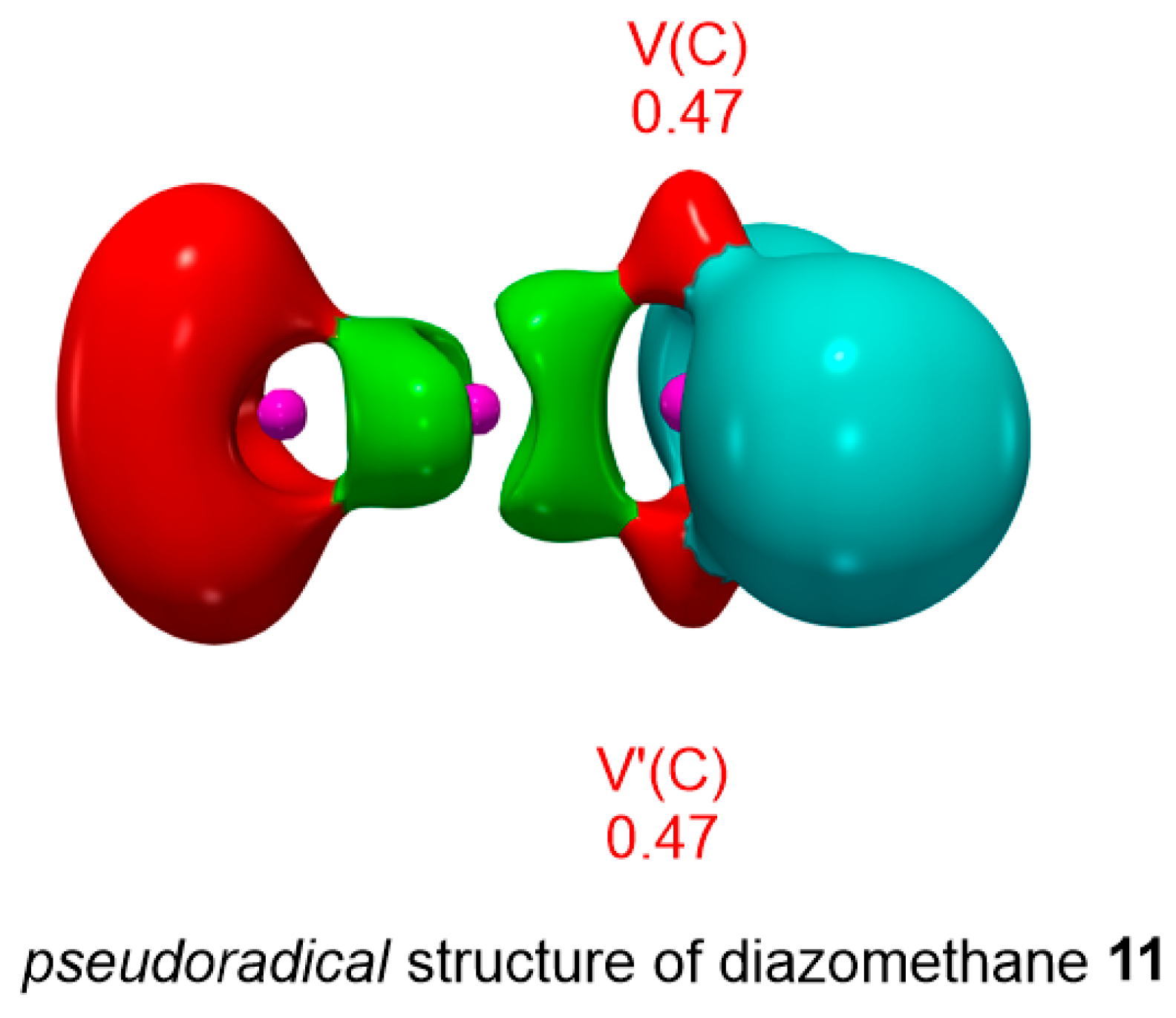
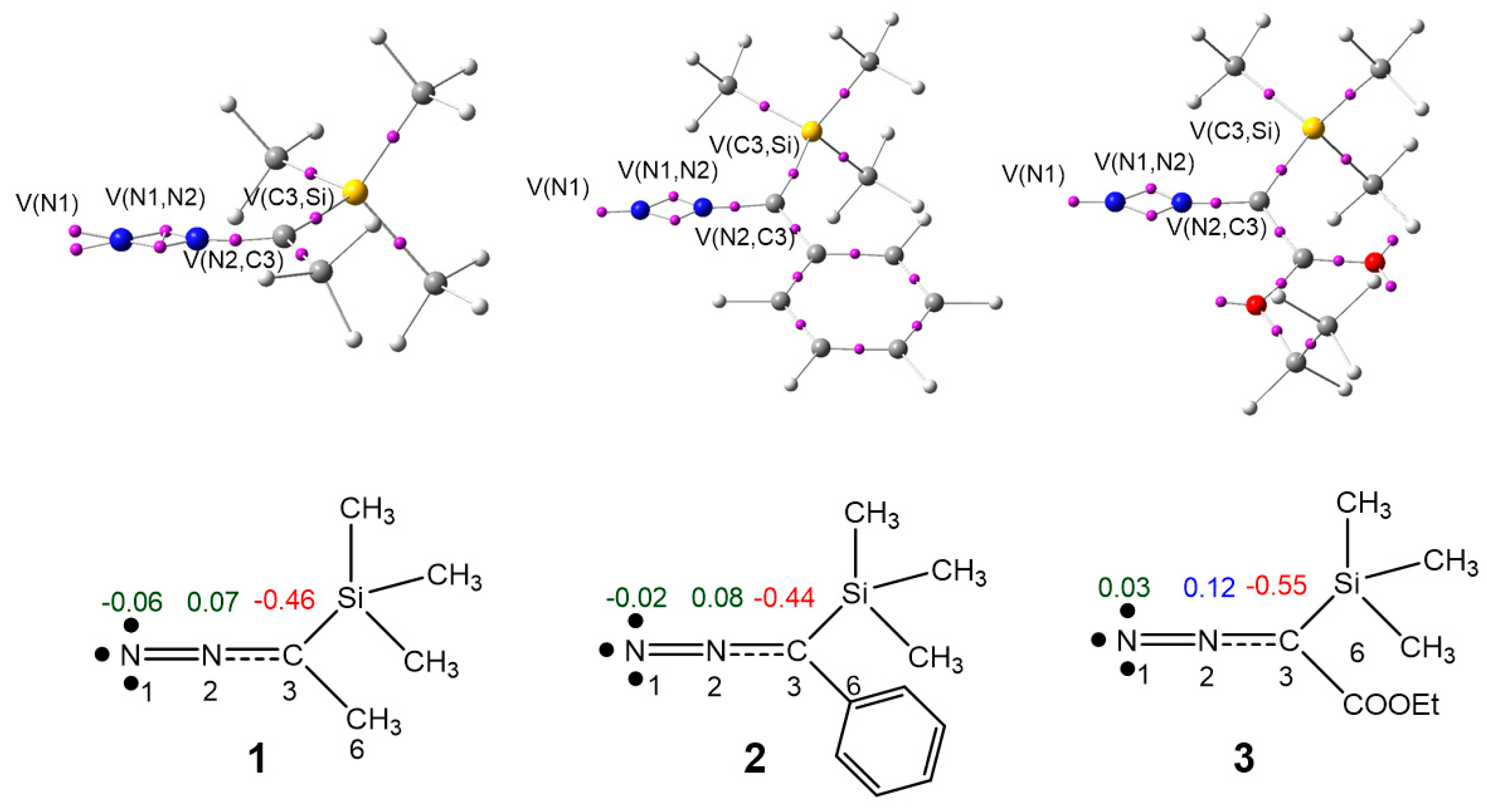
3.1. ELF Topological Analysis of Trimethylsilyldiazoalkanes TSDE 1, TSDP 2, and TSDA 3, and Diethyl Fumarate DFM 4
3.2. Analysis of the CDFT Indices of the Reactants
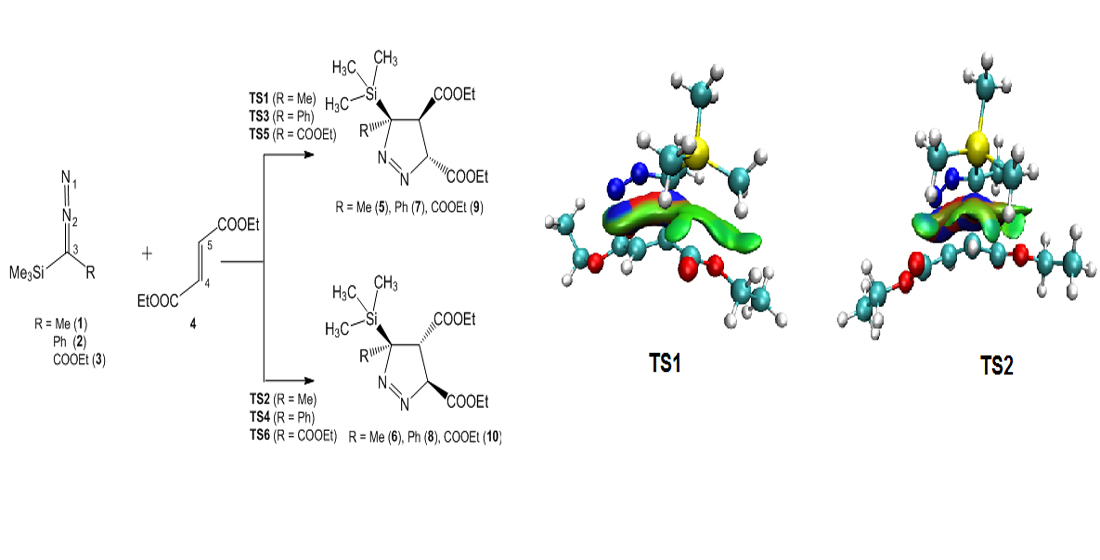
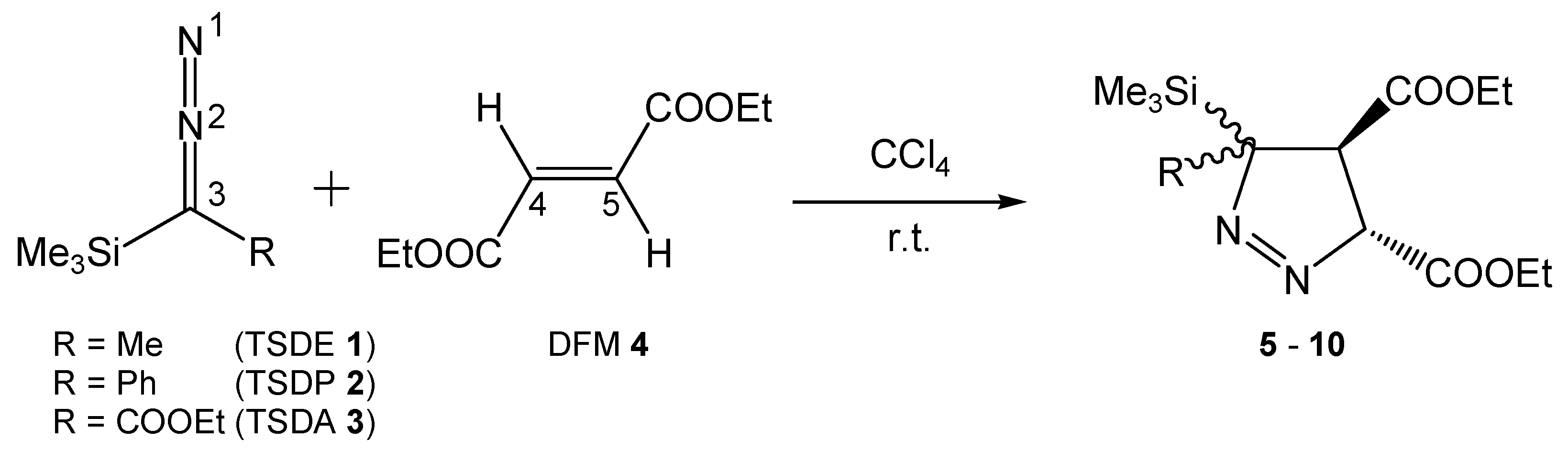
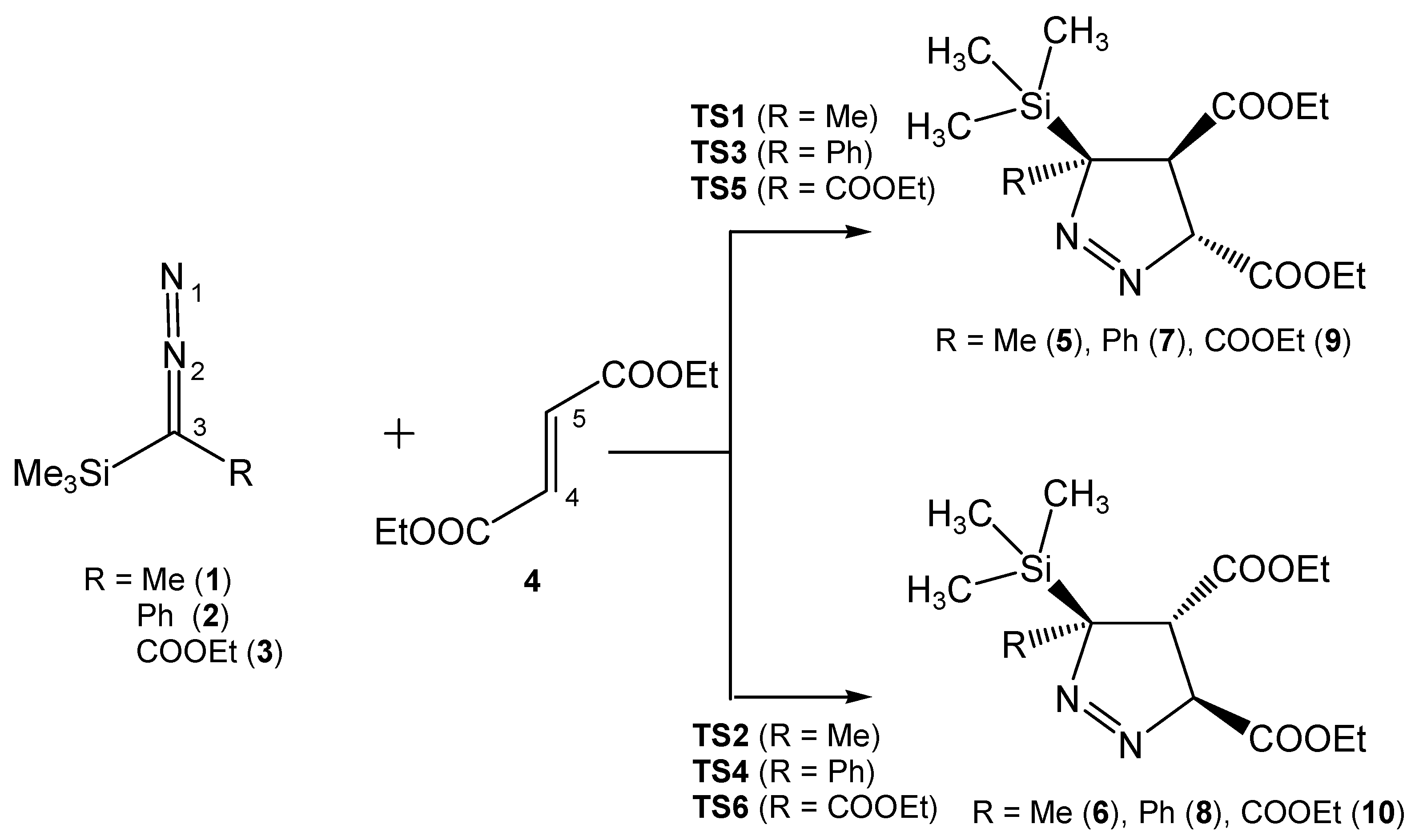
3.3. Analysis of the Energy Profile of the Stationary Points along the Feasible Reaction Paths for 32CA Reactions of TSDE 1, TSDP 2, and TSDA 3, with DFM 4
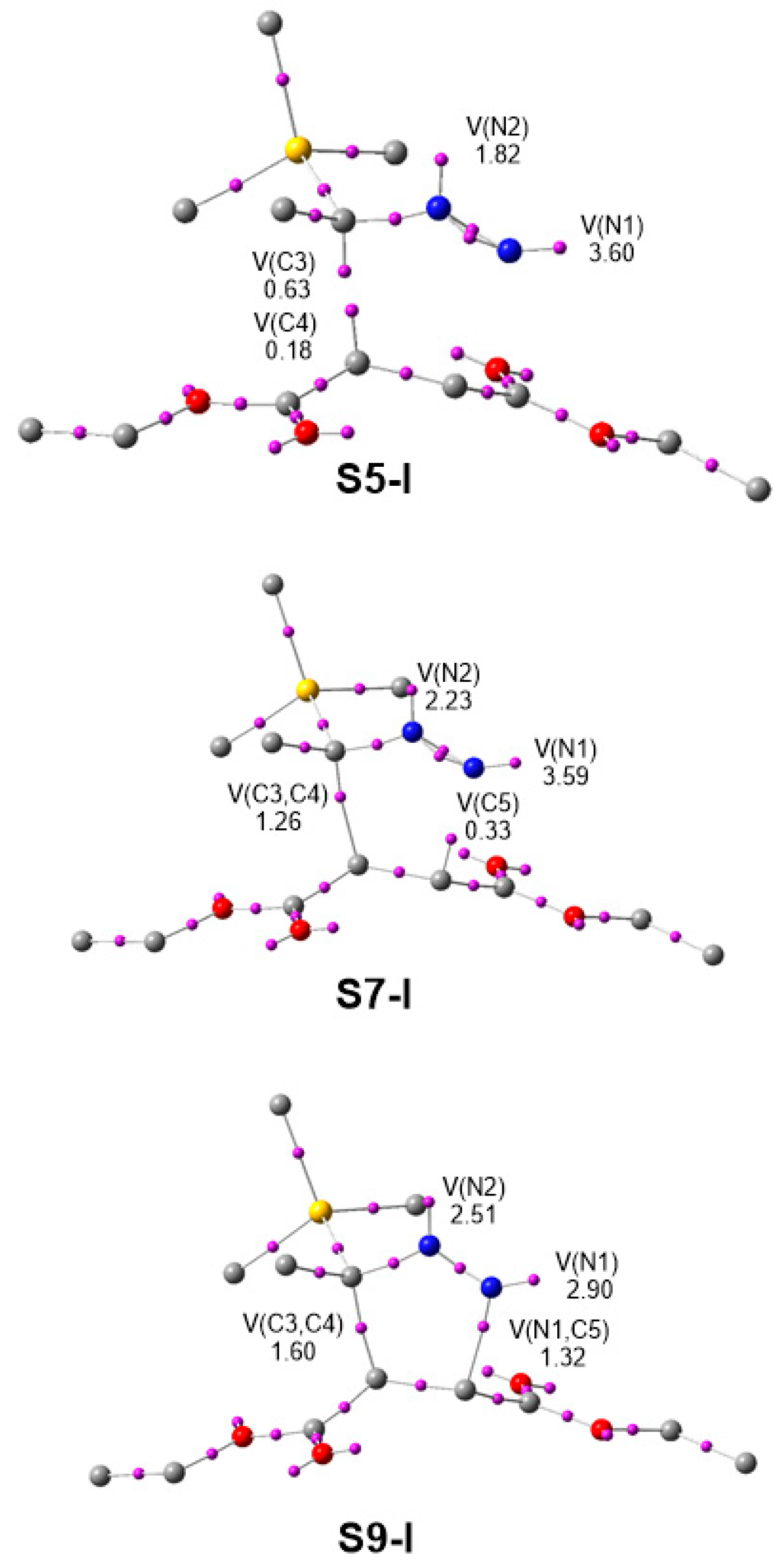
3.4. BET Study of the 32CA Reaction of TSDE 1 and TSDA 3 with DFM 4
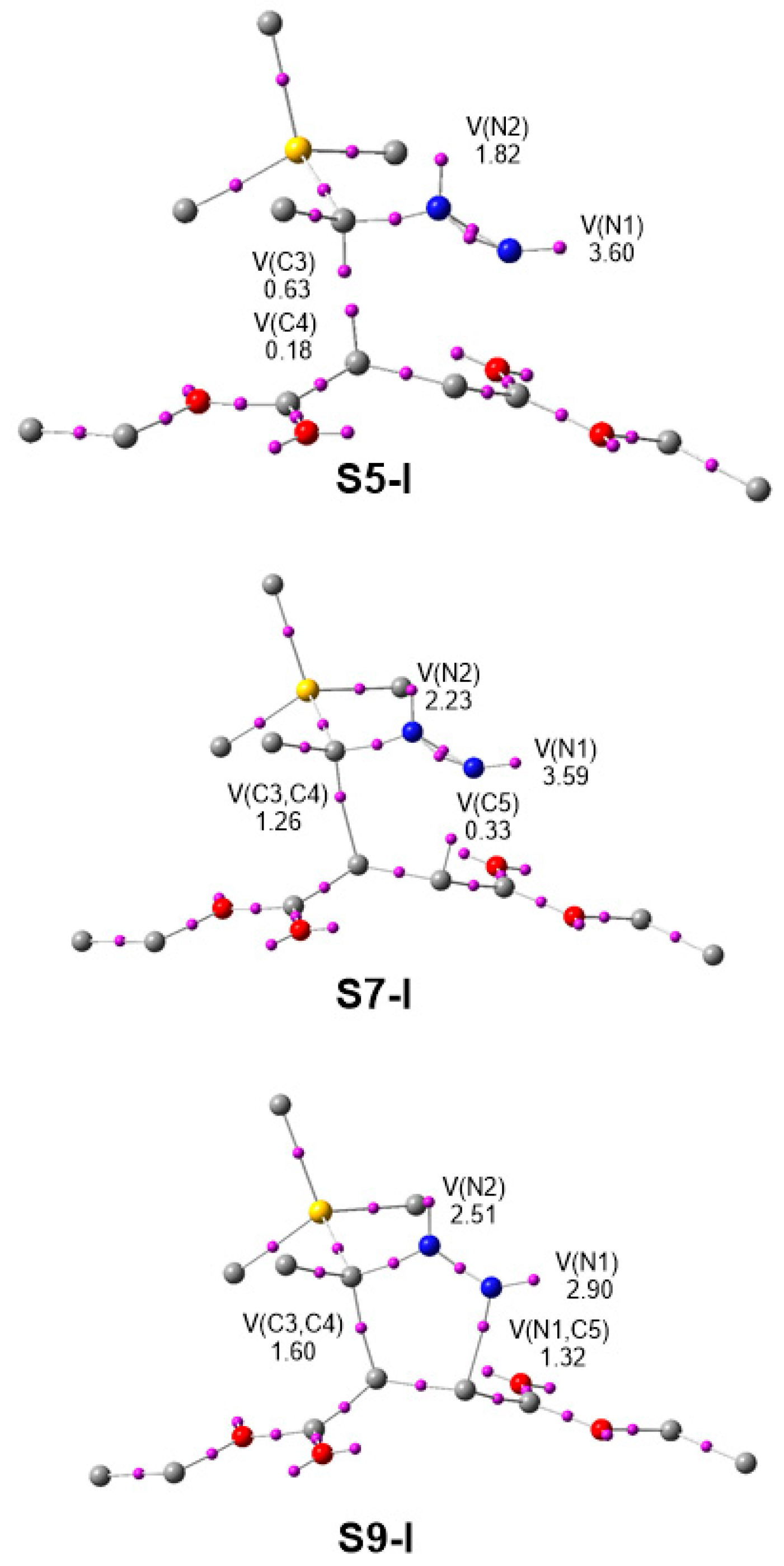
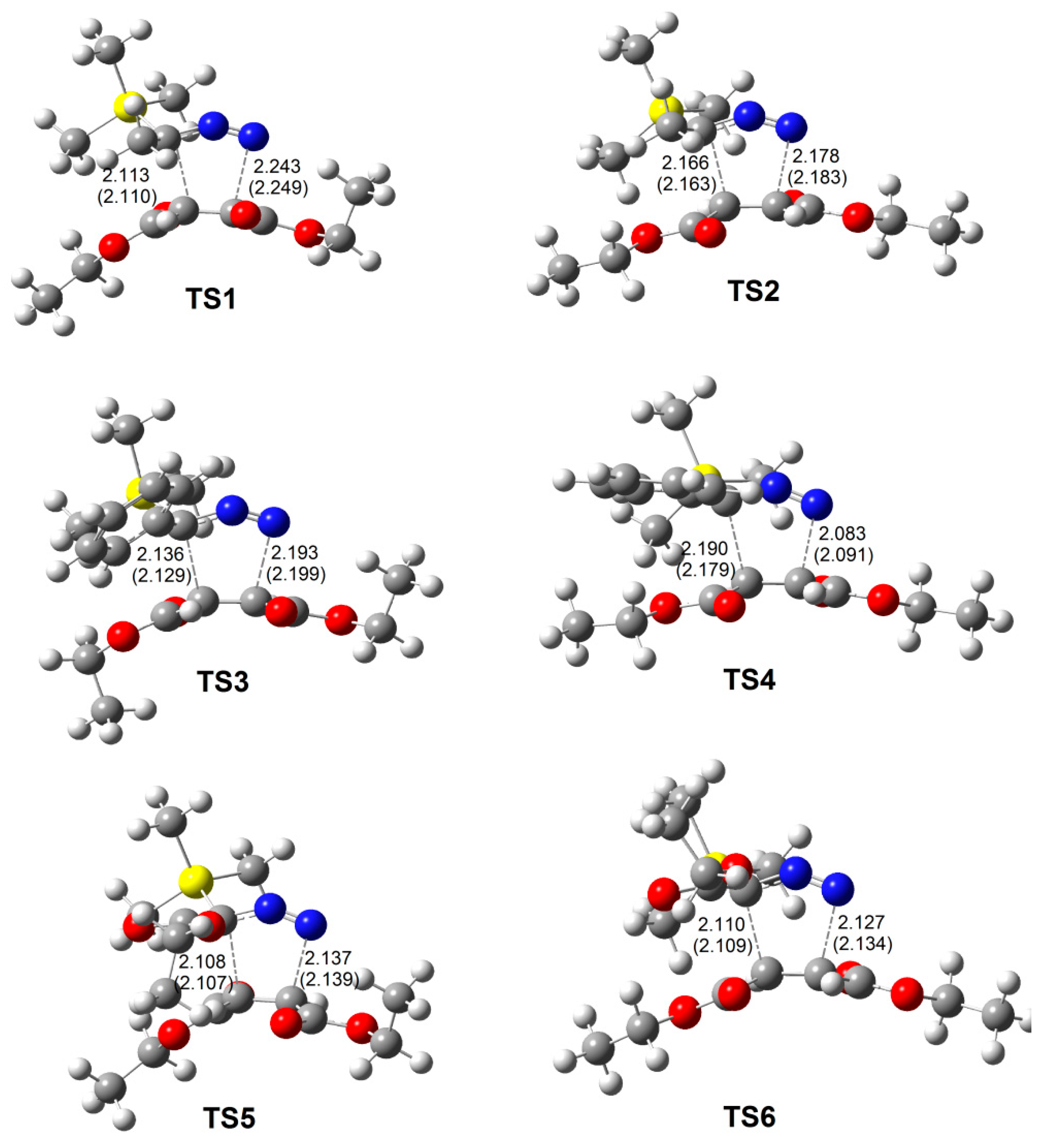
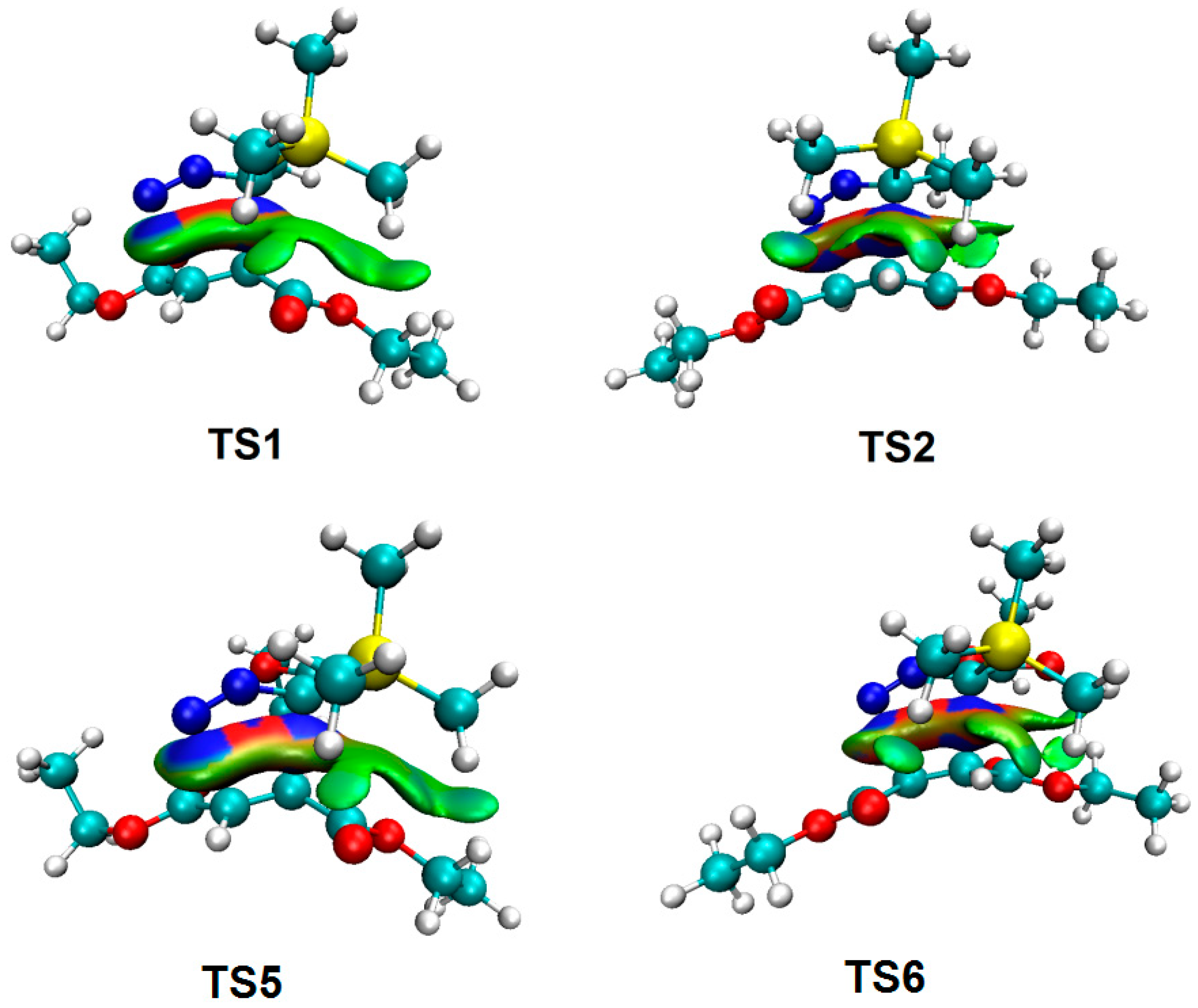
3.5. ELF Topological Analysis at the TSs
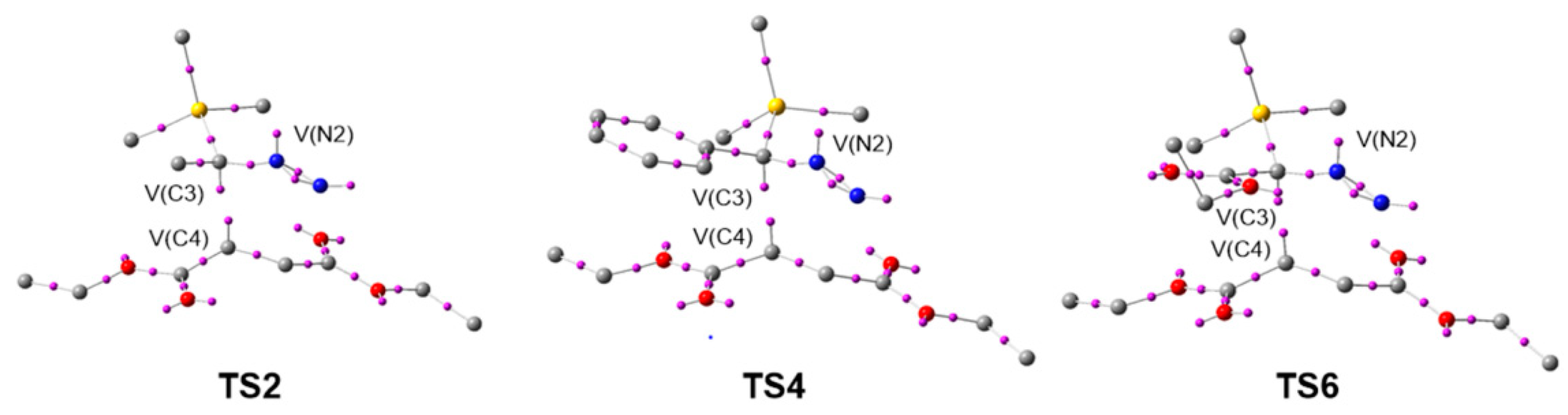
3.6. QTAIM and IGM Topological Analysis at TSs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Presser, A.; Hüfner, A. Trimethylsilyldiazomethane? A Mild and Efficient Reagent for the Methylation of Carboxylic Acids and Alcohols in Natural Products. Monatshefte Chem. Chem. Mon. 2004, 135, 1015–1022. [Google Scholar] [CrossRef]
- Kühnel, E.; Laffan, D.D.P.; Lloyd-Jones, G.C.; del Campo, T.M.; Shepperson, I.R.; Slaughter, J.L. Mechanism of Methyl Esterification of Carboxylic Acids by Trimethylsilyldiazomethane. Angew. Chem. Int. Ed. 2007, 46, 7075–7078. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.K.; Sheldon, C.G.; Macdonald, G.J.; Martin, W.P. A New Method for the Preparation of SilylEnol Ethers from Carbonyl Compounds and (Trimethylsilyl)diazomethane in a Regiospecific and Highly Stereoselective Manner. J. Am. Chem. Soc. 2002, 124, 10300–10301. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Bao, Z.; Xu, X.; Wang, J. Metal-free synthesis of gem-silylboronate esters and their Pd(0)-catalyzed cross-coupling with aryliodides. Org. Biomol. Chem. 2019, 17, 5714–5724. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Ito, S.; Furuta, H.; Kawahara, Y.; Mori, Y. Mechanism of the Regio- and Diastereoselective Ring Expansion Reaction Using Trimethylsilyldiazomethane. Org. Lett. 2012, 14, 4564–4567. [Google Scholar] [CrossRef]
- Simovic, D.; Di, M.; Marks, V.; Chatfield, A.D.C.; Rein, K.S. 1,3-Dipolar Cycloadditions of Trimethylsilyldiazomethane Revisited: Steric Demand of the Dipolarophile and the Influence on Product Distribution. J. Org. Chem. 2007, 72, 650–653. [Google Scholar] [CrossRef] [Green Version]
- Brook, A.G.; Jones, P.F. Cycloaddition Reactions of Silyldiazoalkanes. Can. J. Chem. 1971, 49, 1841–1847. [Google Scholar] [CrossRef]
- Hwu, J.R.; Wang, N. Steric influence of the trimethylsilyl group in organic reactions. Chem. Rev. 1989, 89, 1599–1615. [Google Scholar] [CrossRef]
- Padwa, A.; Wannamaker, M.W. Dipolar cycloaddition reaction of diazoalkanes with trimethylsilyl substituted alkynes. Steric control of regiochemistry by the trimethylsilyl group. Tetrahedron 1990, 46, 1145–1162. [Google Scholar] [CrossRef]
- Whitlock, G.A.; Carreira, E. Enantioselective Synthesis ofent-Stellettamide A via a Novel Dipolar Cycloaddition Reaction of (Trimethylsilyl)diazomethane. J. Org. Chem. 1997, 62, 7916–7917. [Google Scholar] [CrossRef]
- Mlostoń, G.; Pipiak, P.; Heimgartner, H. Diradical reaction mechanisms in [3+2]-cycloadditions of hetarylthioketones with alkyl- or trimethylsilyl-substituted diazomethanes. Beilstein J. Org. Chem. 2016, 12, 716–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mlostoń, G.; Celeda, M.; Jasiński, R.; Heimgartner, H. Experimental and Computational Studies on Stepwise [3+2]-Cycloadditions of Diaryldiazomethanes with Electron-Deficient Dimethyl (E)- and (Z)-2,3-Dicyanobutenedioates. Eur. J. Org. Chem. 2018, 2019, 422–431. [Google Scholar] [CrossRef] [Green Version]
- Bassindale, A.R.; Brook, A.G. Cycloaddition Reactions of Silyldiazoalkanes Involving Rearrangements from Carbon to Nitrogen. Can. J. Chem. 1974, 52, 3474–3483. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Domingo, L.R. Molecular Electron Density Theory: A Modern View of Reactivity in Organic Chemistry. Molecules 2016, 21, 1319. [Google Scholar] [CrossRef]
- Ríos-Gutiérrez, M.; Domingo, L.R.; Ríos-Gutiérez, M. Unravelling the Mysteries of the [3+2] Cycloaddition Reactions. Eur. J. Org. Chem. 2018, 2019, 267–282. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Perez, P. A Molecular Electron Density Theory Study of the Reactivity and Selectivities in [3+2] Cycloaddition Reactions of C,N-DialkylNitrones with Ethylene Derivatives. J. Org. Chem. 2018, 83, 2182–2197. [Google Scholar] [CrossRef]
- Domingo, L.R.; Acharjee, N. [3+2] Cycloaddition Reaction of C-Phenyl-N-methyl Nitrone to Acyclic-Olefin-Bearing Electron-Donating Substituent: A Molecular Electron Density Theory Study. Chemistry 2018, 3, 8373–8380. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Emamian, S. Understanding the domino reaction between 1-diazopropan-2-one and 1,1-dinitroethylene. A molecular electron density theory study of the [3+2] cycloaddition reactions of diazoalkanes with electron-deficient ethylenes. RSC Adv. 2017, 7, 15586–15595. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Geerlings, P.; de Proft, F.; Langenaeker, W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Perez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [Green Version]
- Domingo, L.R. A new C–C bond formation model based on the quantum chemical topology of electron density. RSC Adv. 2014, 4, 32415–32428. [Google Scholar] [CrossRef] [Green Version]
- Krokidis, X.; Noury, S.; Silvi, B. Characterization of Elementary Chemical Processes by Catastrophe Theory. J. Phys. Chem. A 1997, 101, 7277–7282. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon Press: Oxford, UK, 1994. [Google Scholar]
- Bader, R.F.W.; Essén, H. The characterization of atomic interactions. J. Chem. Phys. 1984, 80, 1943. [Google Scholar] [CrossRef]
- Lefebvre, C.; Khartabil, H.; Boisson, J.; Contreras-García, J.; Piquemal, J.-P.; Hénon, E. The Independent Gradient Model: A New Approach for Probing Strong and Weak Interactions in Molecules from Wave Function Calculations. ChemPhysChem 2018, 19, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. Hybrid Meta Density Functional Theory Methods for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions: The MPW1B95 and MPWB1K Models and Comparative Assessments for Hydrogen Bonding and van der Waals Interactions. J. Phys. Chem. A 2004, 108, 6908–6918. [Google Scholar] [CrossRef]
- Hehre, W.J.; Radom, L.; Schleyer, P.V.R.; Pople, J. Ab Initio Molecular Orbital Theory; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Schlegel, H.B. Optimization of equilibrium geometries and transition structures. J. Comput. Chem. 1982, 3, 214–218. [Google Scholar] [CrossRef]
- Fukui, K. Formulation of the reaction coordinate. J. Phys. Chem. 1970, 74, 4161–4163. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- González, C.; Schlegel, H.B. Improved algorithms for reaction path following: Higher-order implicit algorithms. J. Chem. Phys. 1991, 95, 5853–5860. [Google Scholar] [CrossRef] [Green Version]
- Tomasi, J.; Persico, M. Molecular Interactions in Solution: An Overview of Methods Based on Continuous Distributions of the Solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Simkin, B.I.A.; Sheikhet, I.I. Quantum Chemical and Statistical Theory of Solutions: A Computational Approach; Ellis Horwood: London, UK, 1995. [Google Scholar]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab initio study of solvated molecules: A new implementation of the polarizable continuum model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
- Cancès, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M.; Tomasi, J. Geometry optimization of molecular structures in solution by the polarizable continuum model. J. Comput. Chem. 1998, 19, 404–417. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysisa. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wave function analyzer. J. Comput. Chem. 2011, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Ahrens, J.; Geveci, B.; Law, C. ParaView: An End-User Tool for Large Data Visualization. In Visualization Handbook; Elsevier: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Ayachit, U. The ParaView Guide: A Parallel Visualization Application; Kitware: New York, NY, USA, 2015. [Google Scholar]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Domingo, L.R.; Aurell, M.J.; Perez, P.; Contreras, R. Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels–Alder reactions. Tetrahedron 2002, 58, 4417–4423. [Google Scholar] [CrossRef]
- Jaramillo, P.; Domingo, L.R.; Chamorro, E.; Perez, P. A further exploration of a nucleophilicity index based on the gas-phase ionization potentials. J. Mol. Struct. THEOCHEM 2008, 865, 68–72. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Parr, R.G.; Szentpály, L.V.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Domingo, L.R.; Chamorro, E.; Perez, P. Understanding the Reactivity of Captodative Ethylenes in Polar Cycloaddition Reactions. A Theoretical Study. J. Org. Chem. 2008, 73, 4615–4624. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Perez, P. How does the global electron density transfer diminish activation energies in polar cycloaddition reactions? A Molecular Electron Density Theory study. Tetrahedron 2017, 73, 1718–1724. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | 3 | 4 | |
|---|---|---|---|---|
| V(N1) | 3.82 | 3.74 | 3.63 | |
| V(N1,N2) | 1.79 | 1.81 | 1.89 | |
| V’(N1,N2) | 1.90 | 1.91 | 1.97 | |
| V(C3,N2) | 3.07 | 3.06 | 2.93 | |
| V(C3,C6) | 1.87 | 2.17 | 2.36 | |
| V(C3,Si) | 3.19 | 3.07 | 3.11 | |
| V(C4,C5) | 1.67 | |||
| V’(C4,C5) | 1.65 |
| μ | η | ω | N | |
|---|---|---|---|---|
| 1 | −3.12 | 4.60 | 1.06 | 3.70 |
| 2 | −3.22 | 4.11 | 1.27 | 3.84 |
| 3 | −3.89 | 4.84 | 1.56 | 2.80 |
| 4 | −4.84 | 5.39 | 2.18 | 1.58 |
| Gas Phase | CCl4 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ∆E | ∆H | ∆S | ∆G | GEDT | ∆E | ∆H | ∆S | ∆G | GEDT | |
| TS1 | 9.4 | 10.4 | −58.6 | 27.9 | 0.27 | 9.7 | 10.3 | −55.5 | 26.8 | 0.28 |
| 5 | −32.4 | −29.4 | −61.3 | −11.2 | −32.5 | −29.4 | −61.8 | −11.0 | ||
| TS2 | 7.6 | 8.1 | −56.1 | 24.9 | 0.26 | 7.6 | 8.0 | −55.7 | 24.6 | 0.27 |
| 6 | −32.3 | −29.5 | −54.7 | −13.3 | −32.4 | −29.7 | −55.1 | −13.2 | ||
| TS3 | 14.0 | 14.4 | −52.1 | 29.9 | 0.23 | 14.5 | 14.7 | −52.5 | 30.4 | 0.24 |
| 7 | −28.0 | −25.9 | −59.2 | −8.2 | −27.4 | −25.4 | −58.6 | −8.0 | ||
| TS4 | 11.3 | 10.8 | −53.4 | 26.7 | 0.21 | 11.6 | 11.8 | −53.6 | 27.8 | 0.22 |
| 8 | −25.8 | −24.6 | −62.8 | −5.9 | −25.7 | −24.5 | −63.1 | −5.7 | ||
| TS5 | 18.6 | 19.0 | −51.8 | 34.4 | 0.19 | 19.3 | 19.7 | −50.8 | 34.9 | 0.20 |
| 9 | −20.3 | −17.7 | −49.6 | −2.9 | −20.5 | −17.9 | −49.9 | −3.1 | ||
| TS6 | 15.6 | 15.2 | −54.5 | 31.4 | 0.19 | 16.4 | 16.5 | −51.0 | 31.7 | 0.20 |
| 10 | −21.4 | −19.6 | −54.0 | −3.5 | −21.6 | −19.8 | −55.1 | −3.3 | ||
| TS1 | TS2 | TS3 | TS4 | TS5 | TS6 | |
|---|---|---|---|---|---|---|
| V(N1) | 3.57 | 3.60 | 3.57 | 3.59 | 3.52 | 3.52 |
| V(N1,N2) | 1.53 | 1.44 | 1.44 | 1.31 | 1.55 | 1.55 |
| V’(N1,N2) | 1.63 | 1.67 | 1.69 | 1.72 | 1.56 | 1.55 |
| V(N2) | 1.76 | 1.82 | 1.82 | 1.94 | 1.85 | 1.88 |
| V(C3,N2) | 2.07 | 2.07 | 2.05 | 2.05 | 2.05 | 2.03 |
| V(C3,C6) | 1.91 | 1.92 | 2.13 | 2.19 | 2.41 | 2.30 |
| V(C3,Si) | 2.27 | 2.28 | 2.25 | 2.28 | 2.31 | 2.29 |
| V(C4,C5) | 2.94 | 2.96 | 2.95 | 2.93 | 2.91 | 2.91 |
| V(C3) | 0.67 | 0.63 | 0.62 | 0.55 | 0.62 | 0.62 |
| V(C4) | 0.23 | 0.18 | 0.19 | 0.20 | 0.23 | 0.20 |
| CP1 (C3-C4) | CP2 (N1-C5) | |||||
|---|---|---|---|---|---|---|
| ρ | ρ | |||||
| TS1 | 0.072 | 0.031 | −0.020 | 0.051 | 0.091 | −0.005 |
| TS2 | 0.066 | 0.036 | −0.017 | 0.058 | 0.091 | −0.008 |
| TS3 | 0.069 | 0.036 | −0.018 | 0.056 | 0.092 | −0.007 |
| TS4 | 0.063 | 0.037 | −0.016 | 0.070 | 0.091 | −0.014 |
| TS5 | 0.073 | 0.030 | −0.021 | 0.062 | 0.092 | −0.010 |
| TS6 | 0.073 | 0.031 | −0.020 | 0.064 | 0.091 | −0.011 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domingo, L.R.; Acharjee, N.; Mohammad-Salim, H.A. Understanding the Reactivity of Trimethylsilyldiazoalkanes Participating in [3+2] Cycloaddition Reactions towards Diethylfumarate with a Molecular Electron Density Theory Perspective. Organics 2020, 1, 3-18. https://0-doi-org.brum.beds.ac.uk/10.3390/org1010002
Domingo LR, Acharjee N, Mohammad-Salim HA. Understanding the Reactivity of Trimethylsilyldiazoalkanes Participating in [3+2] Cycloaddition Reactions towards Diethylfumarate with a Molecular Electron Density Theory Perspective. Organics. 2020; 1(1):3-18. https://0-doi-org.brum.beds.ac.uk/10.3390/org1010002
Chicago/Turabian StyleDomingo, Luis R., Nivedita Acharjee, and Haydar A. Mohammad-Salim. 2020. "Understanding the Reactivity of Trimethylsilyldiazoalkanes Participating in [3+2] Cycloaddition Reactions towards Diethylfumarate with a Molecular Electron Density Theory Perspective" Organics 1, no. 1: 3-18. https://0-doi-org.brum.beds.ac.uk/10.3390/org1010002