Participation of Phosphorylated Analogues of Nitroethene in Diels–Alder Reactions with Anthracene: A Molecular Electron Density Theory Study and Mechanistic Aspect


Abstract
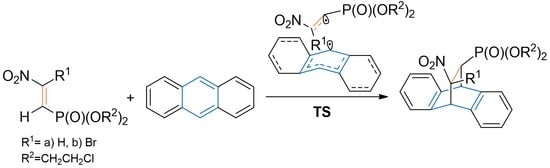
:
1. Introduction
2. Computational Details
3. Results and Discussion
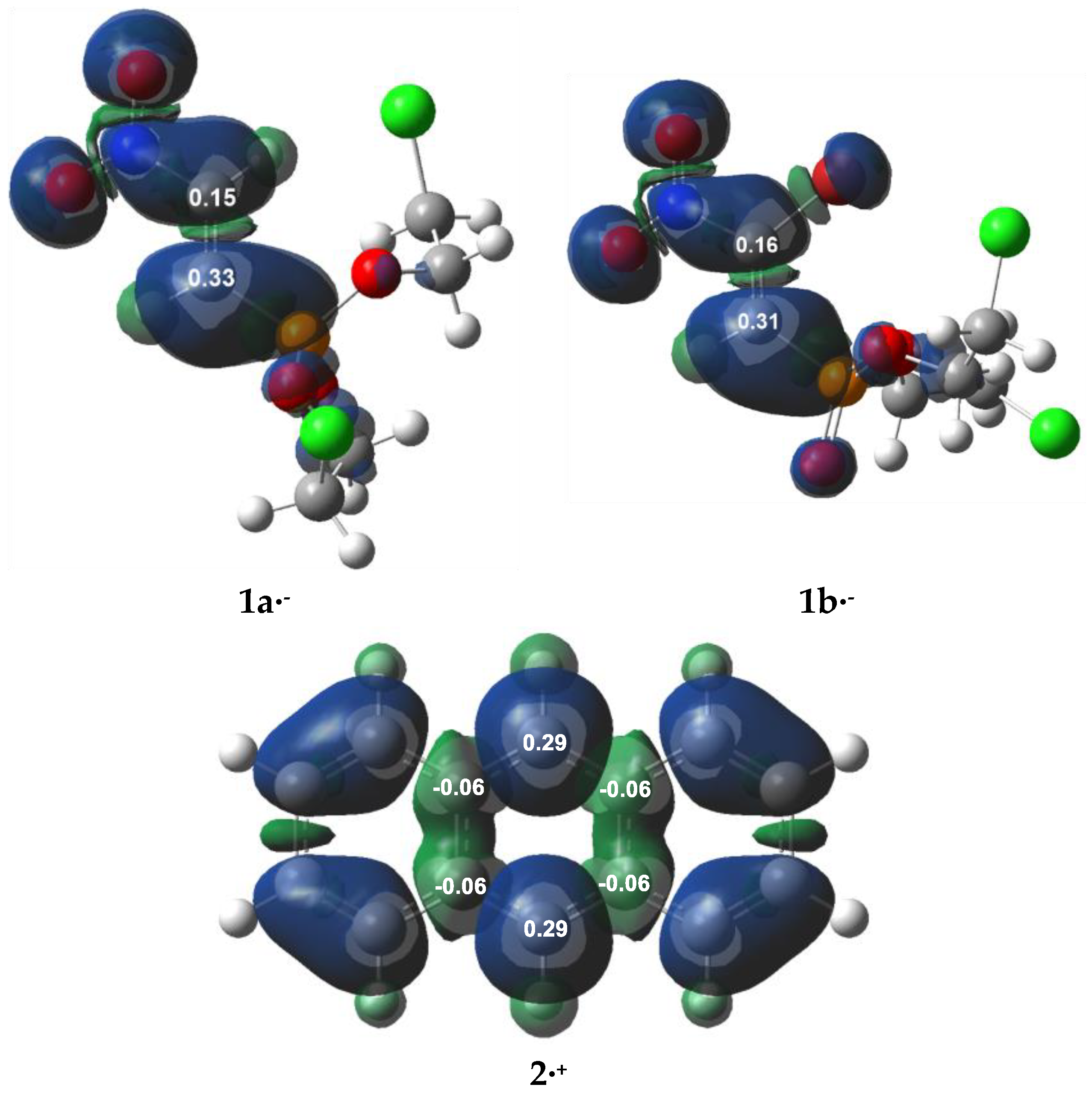
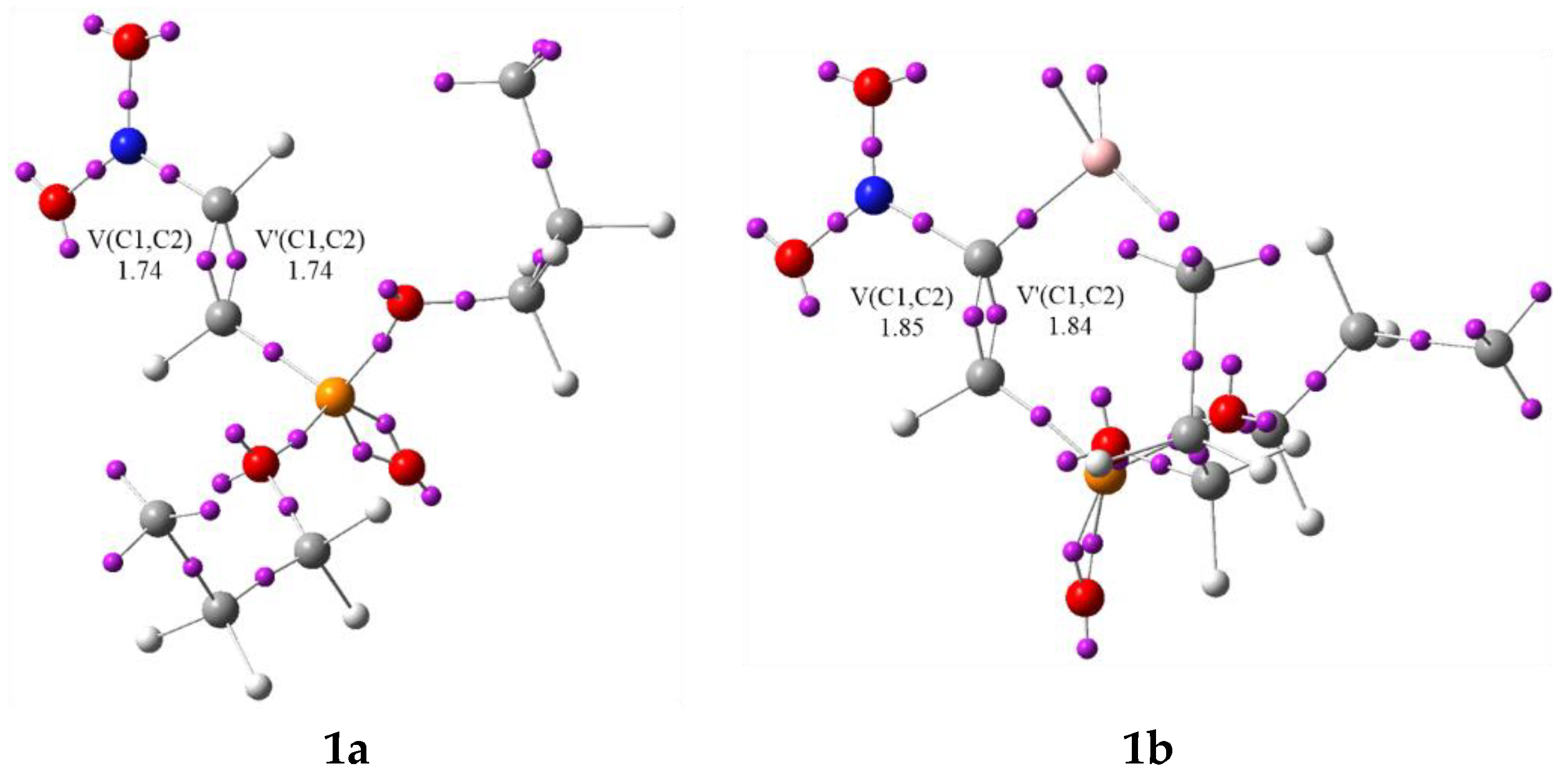
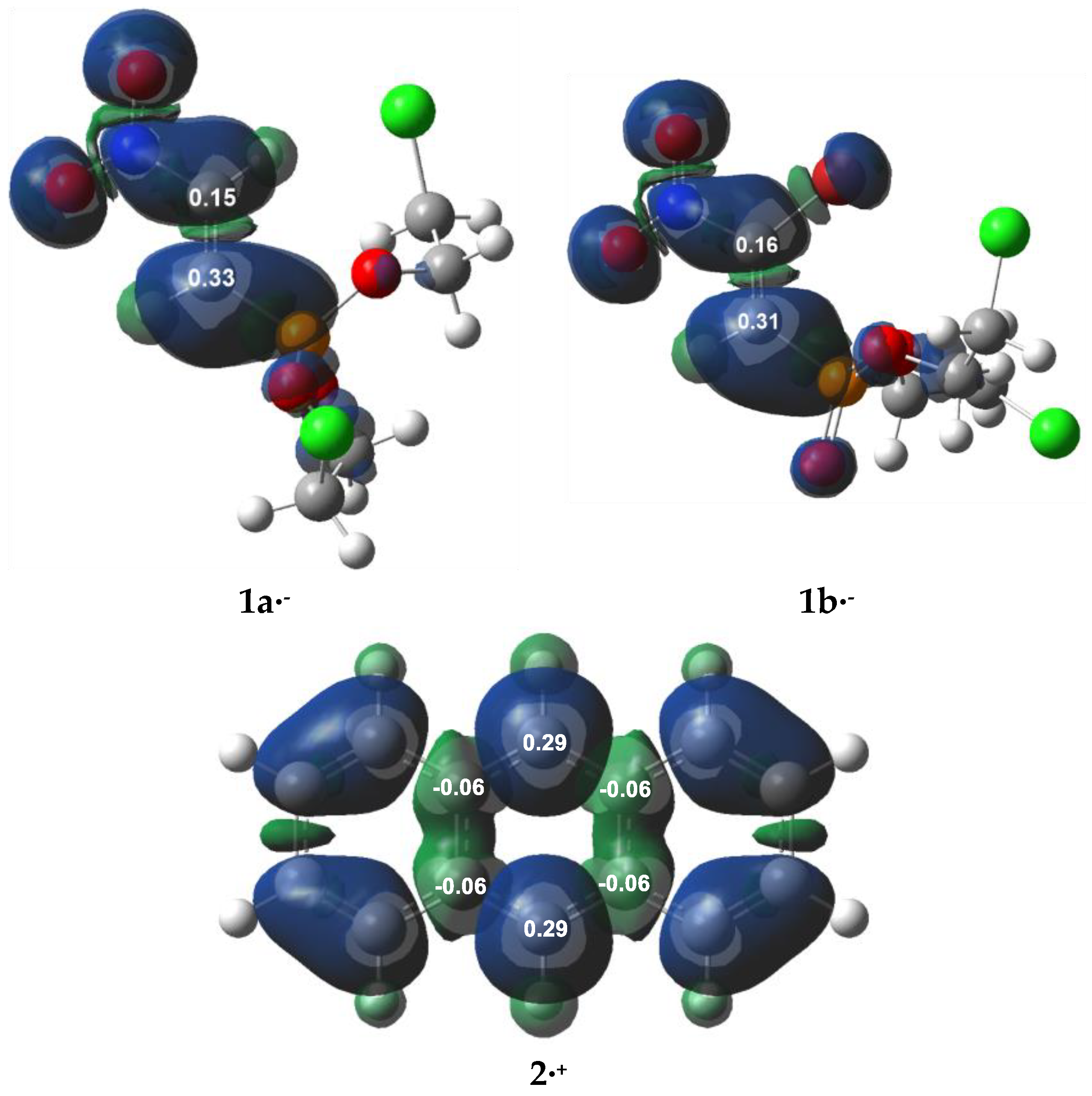
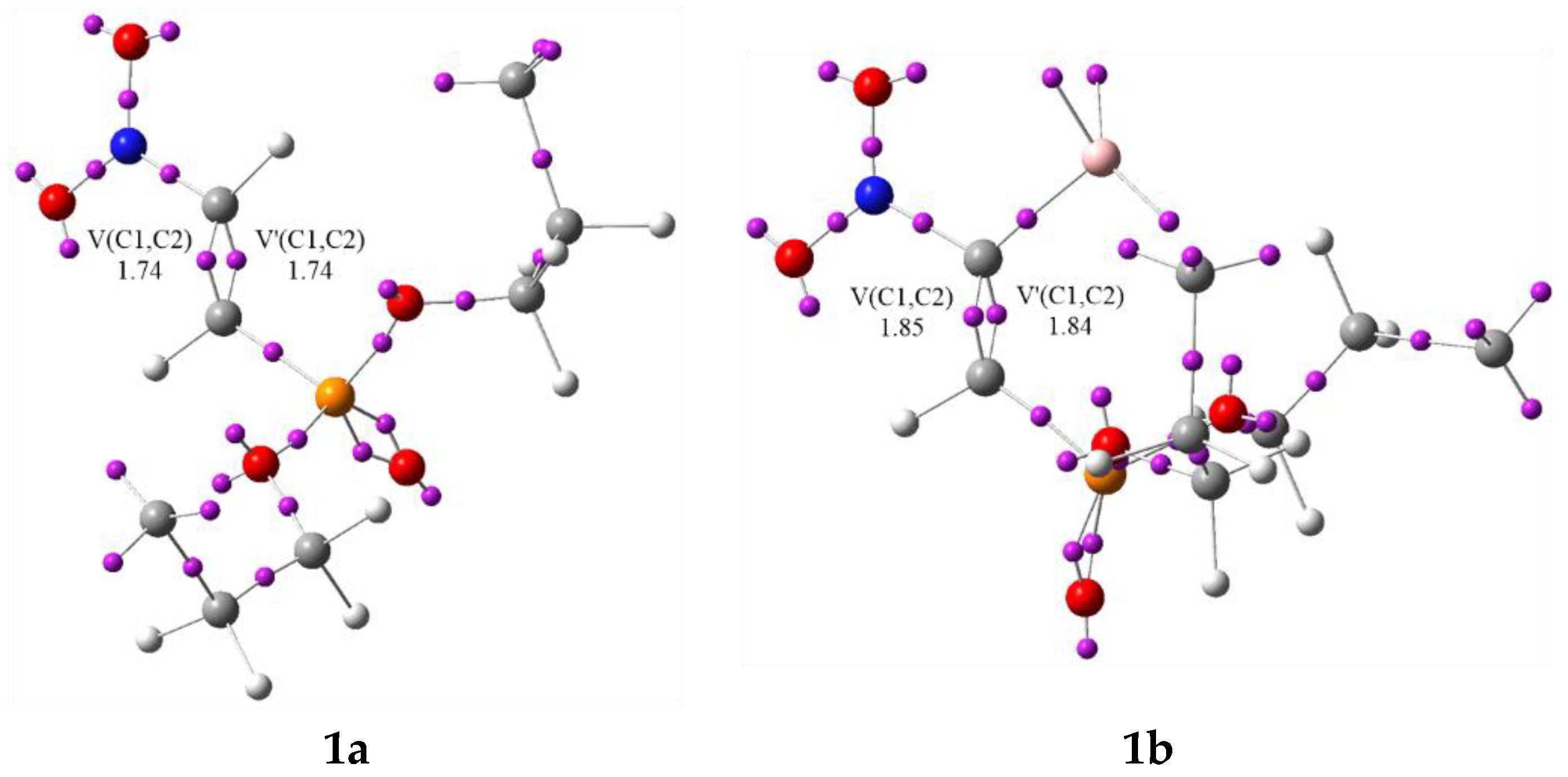
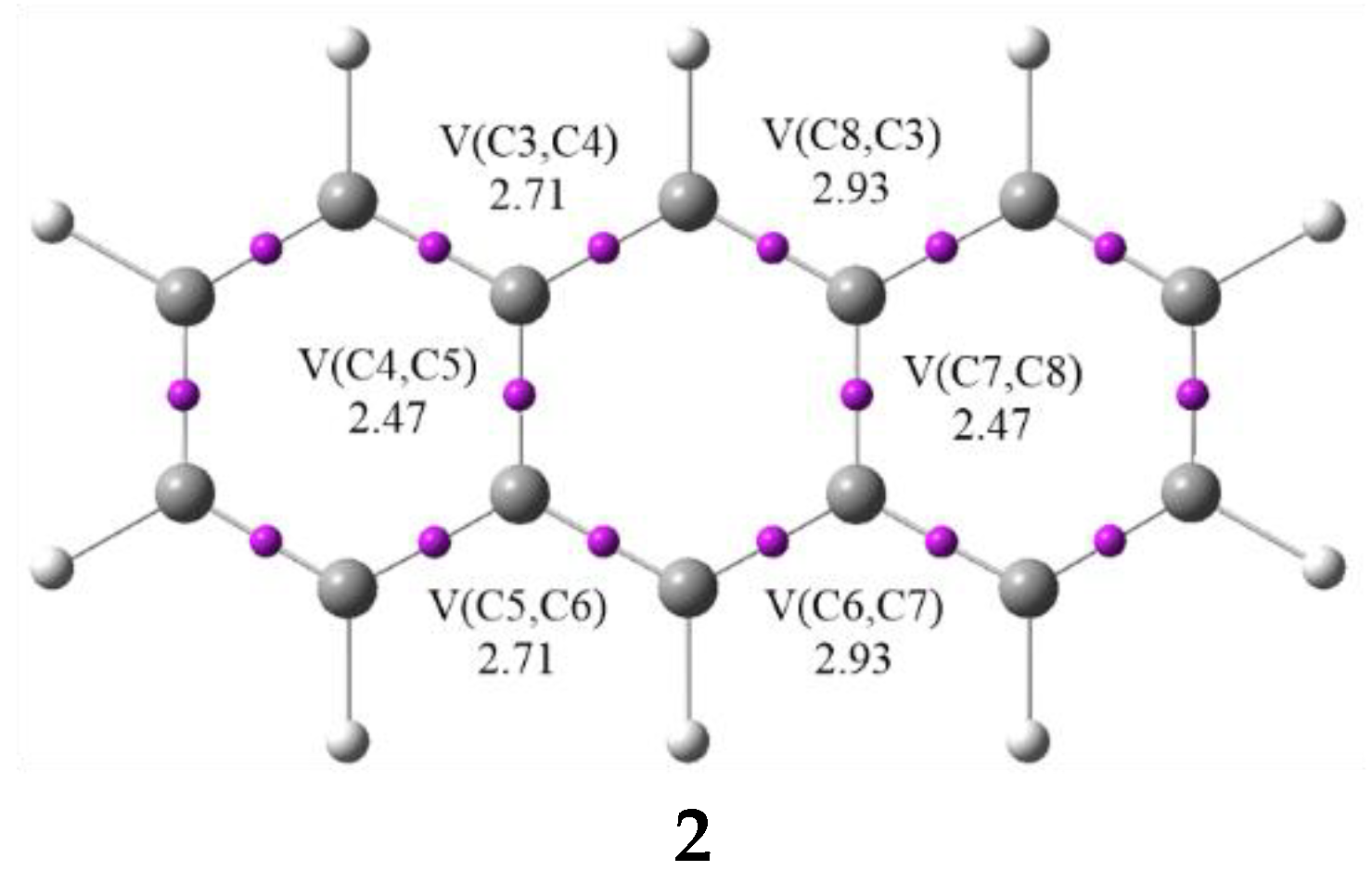
3.1. Analysis of the CDFT Reactivity Indices and ELF Characterization of the Reactants
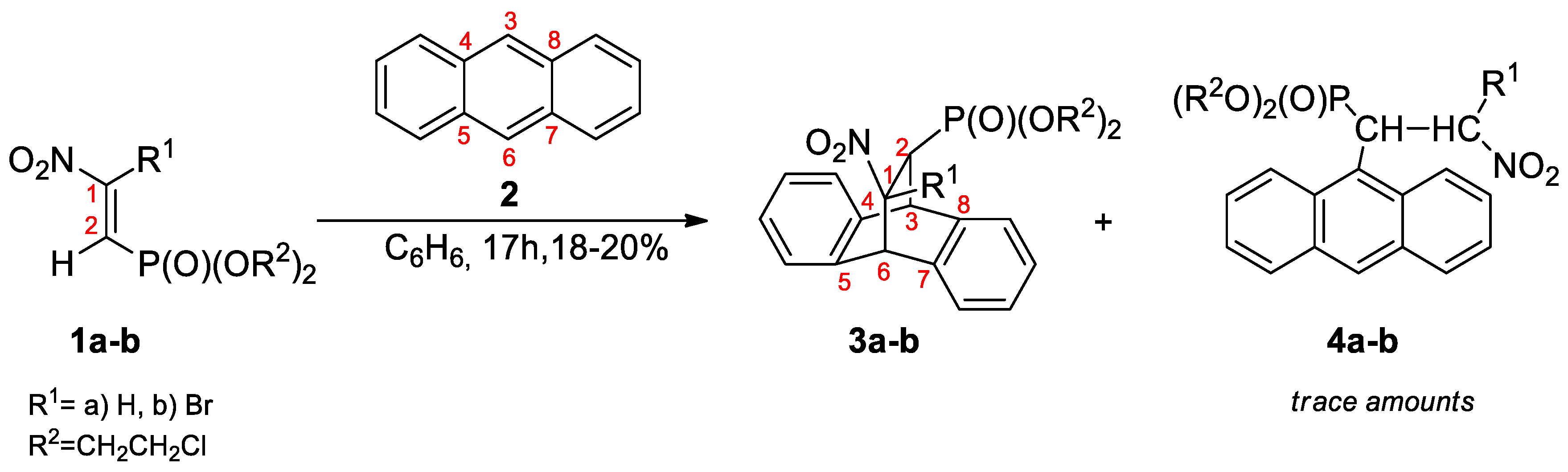
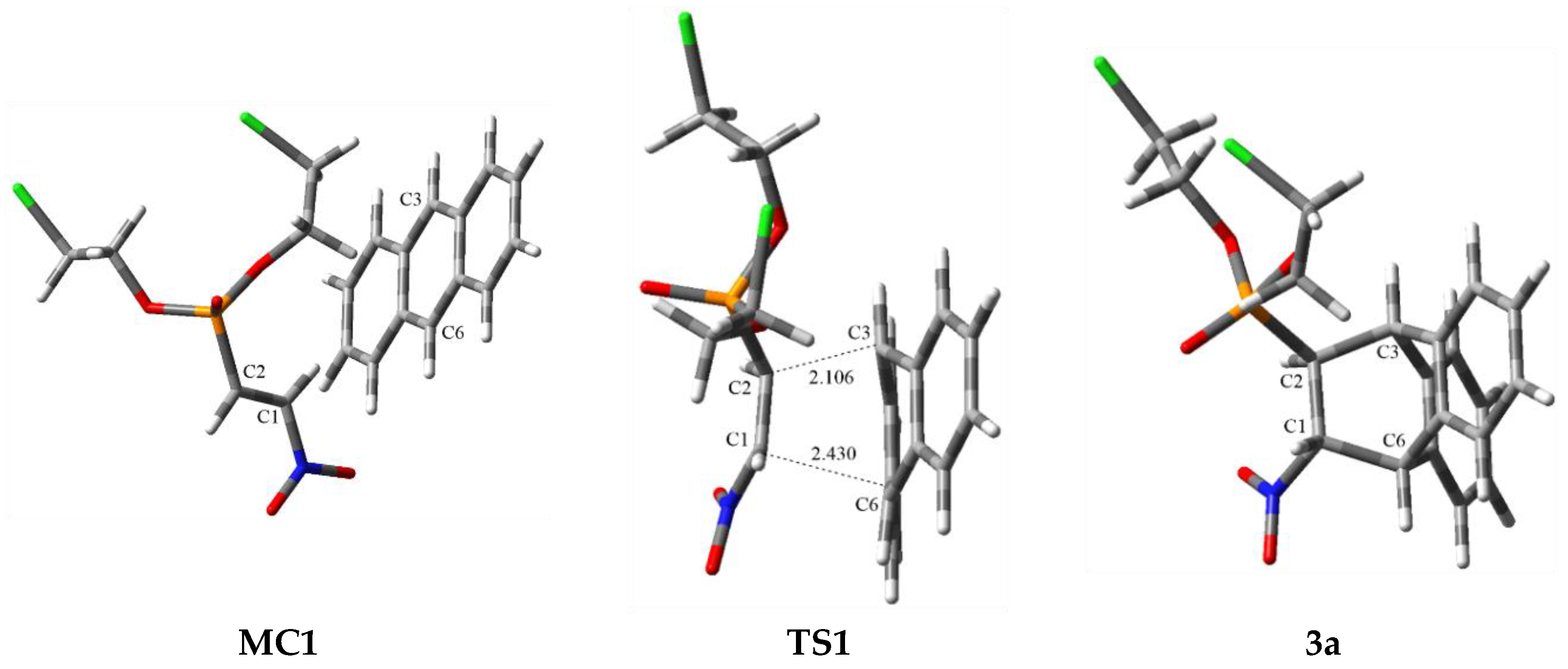
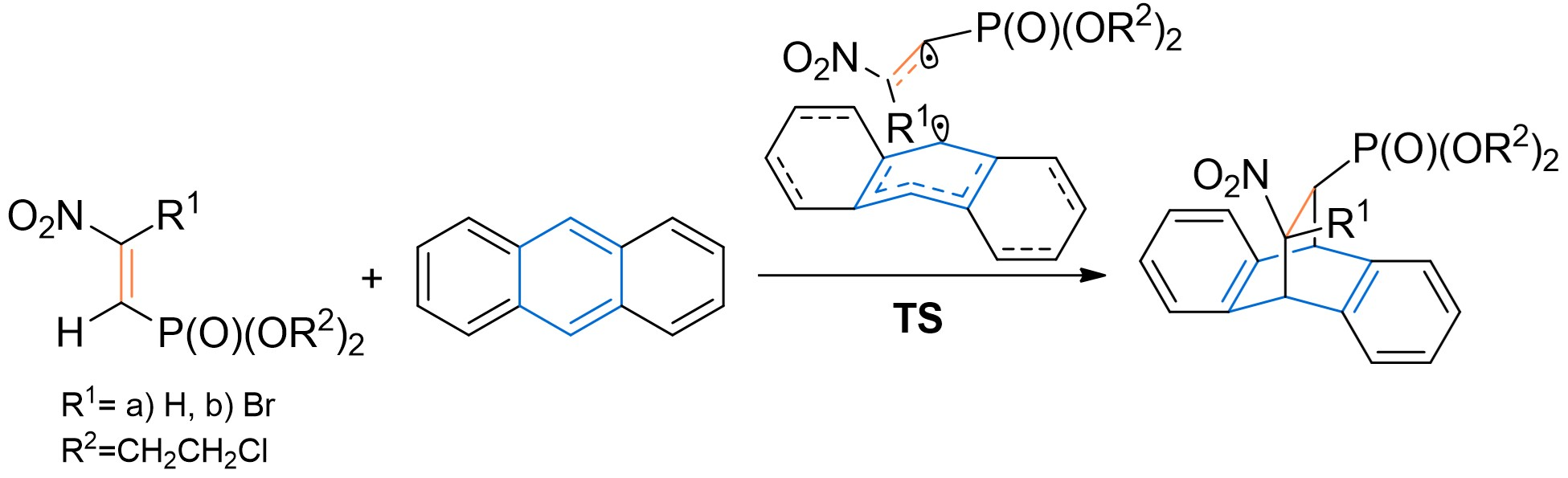
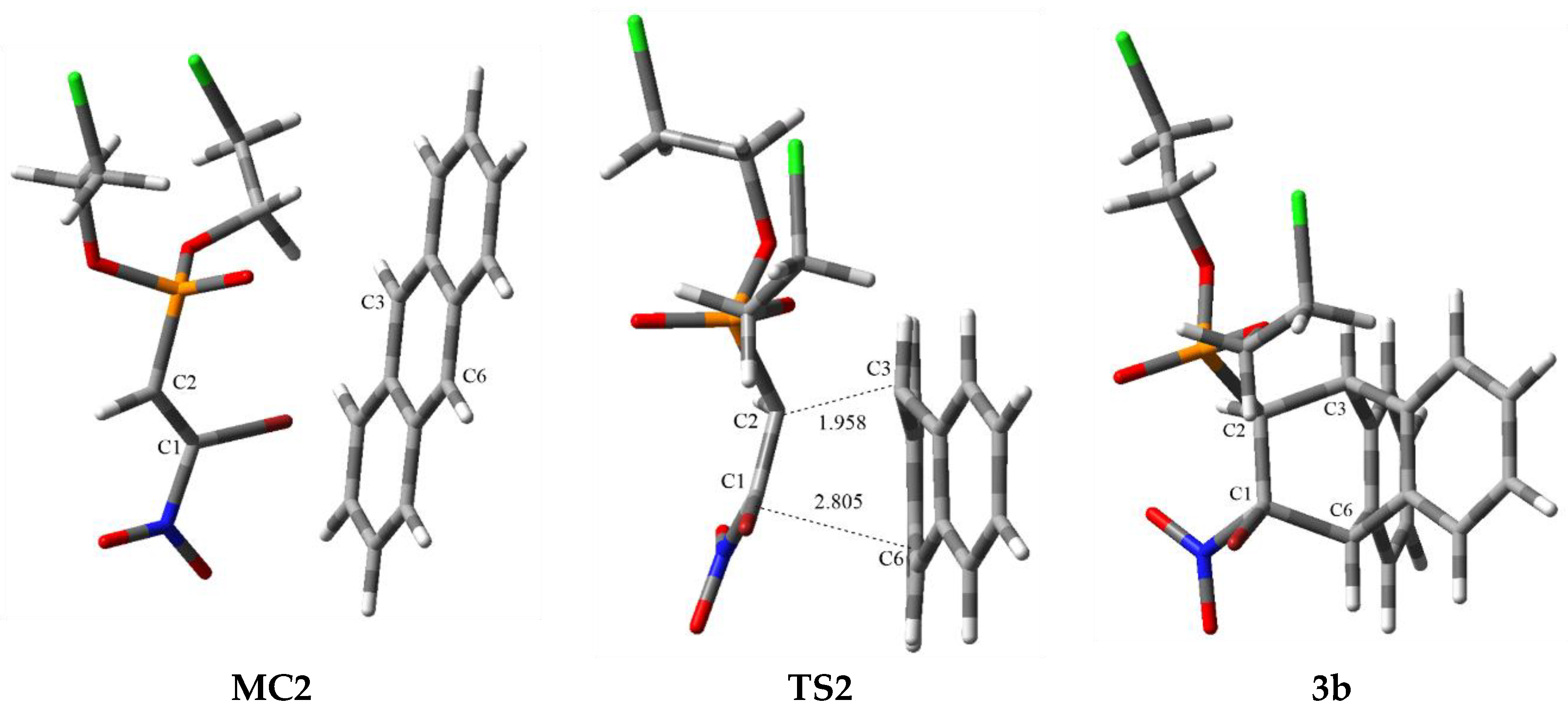
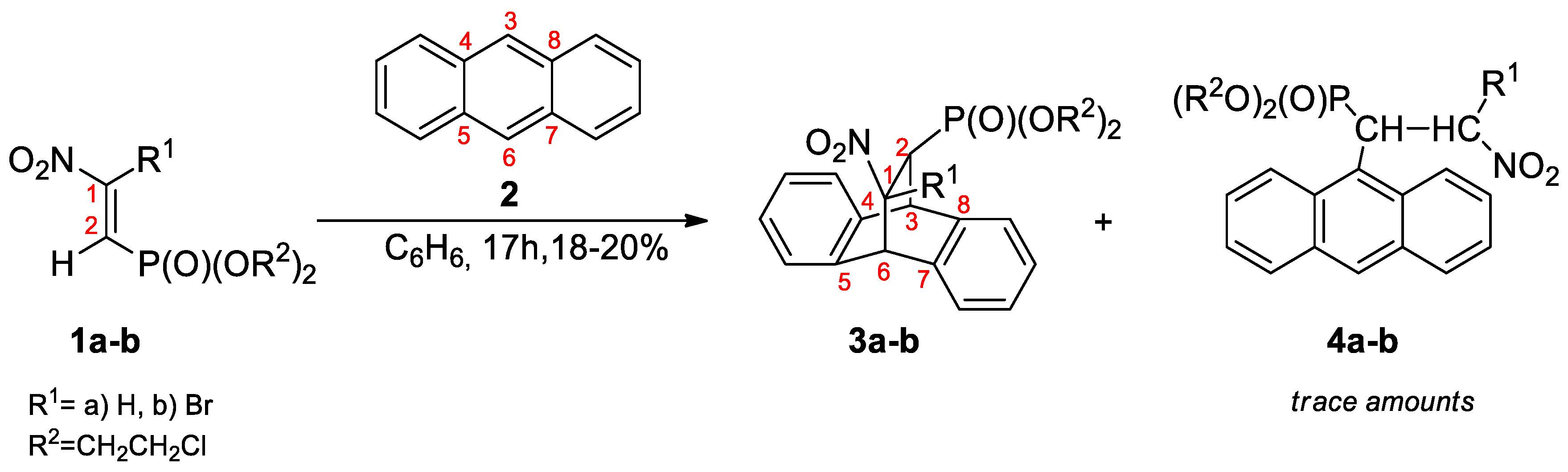
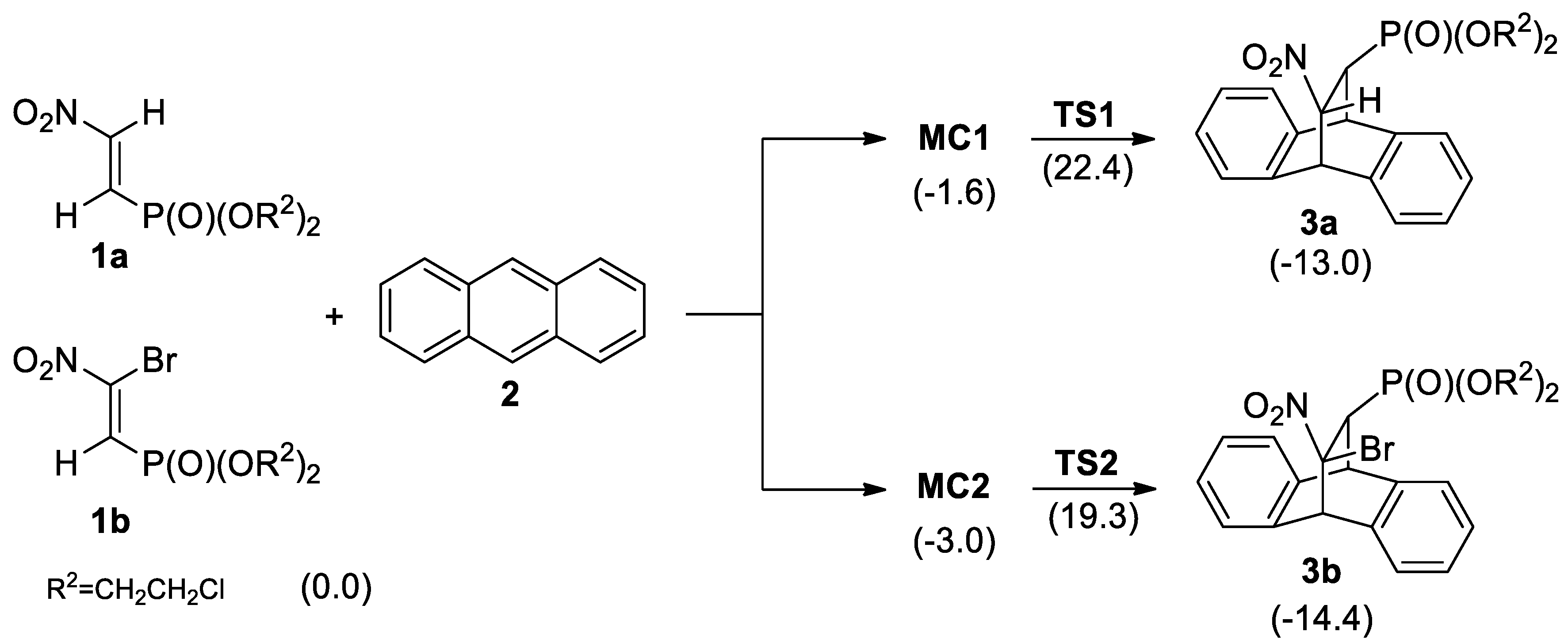
3.2. DA Reaction Profiles of the bis(2-chloroethyl) 2-nitro- 1a and bis(2-chloroethyl) 2-bromo-2-Nitroethenylphosphonates 1b with Anthracene 2
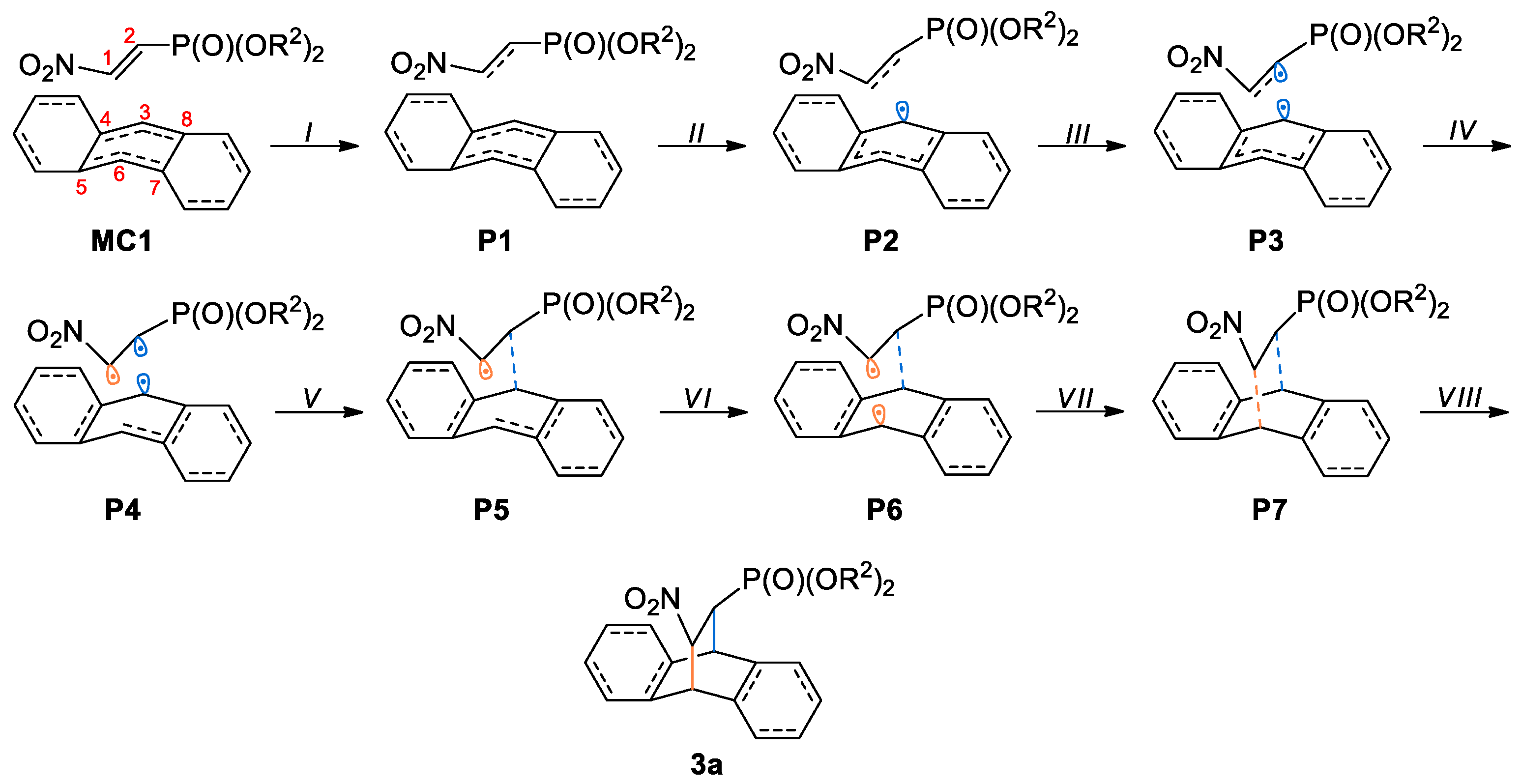
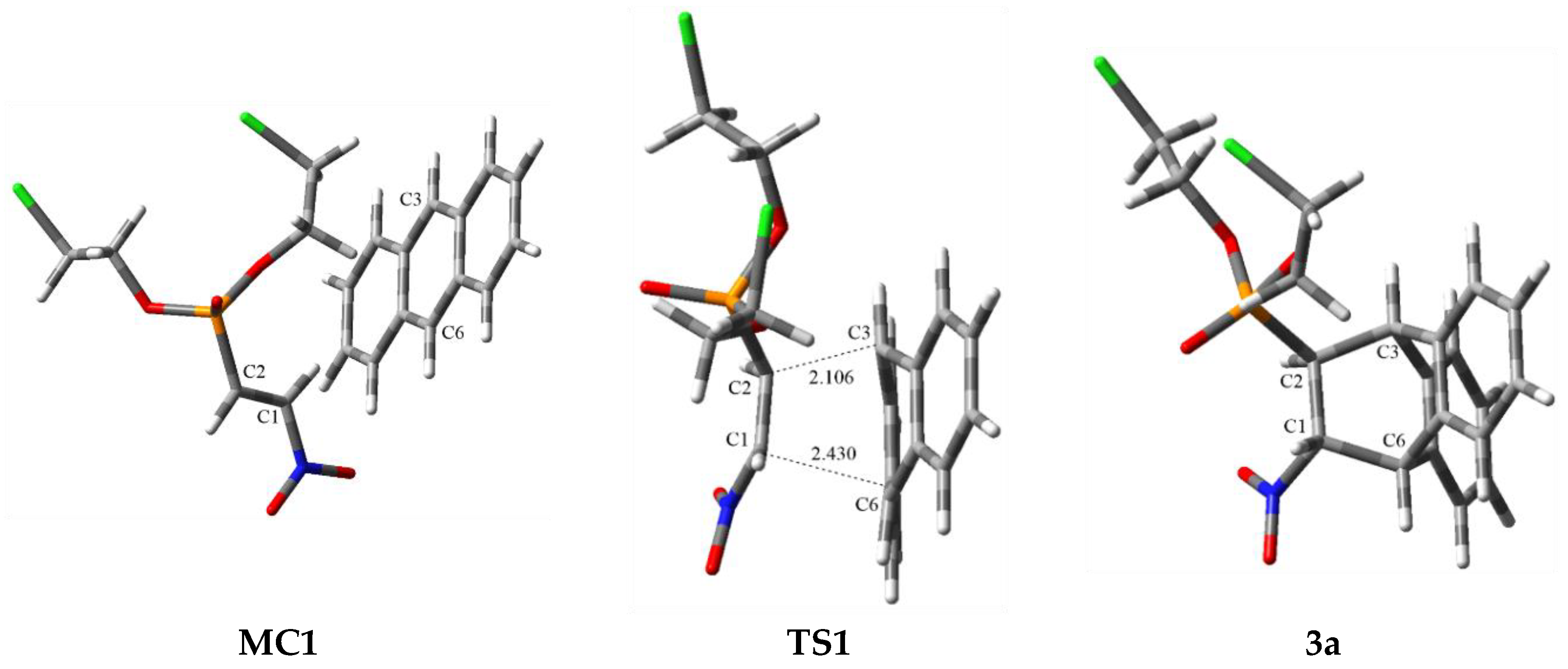
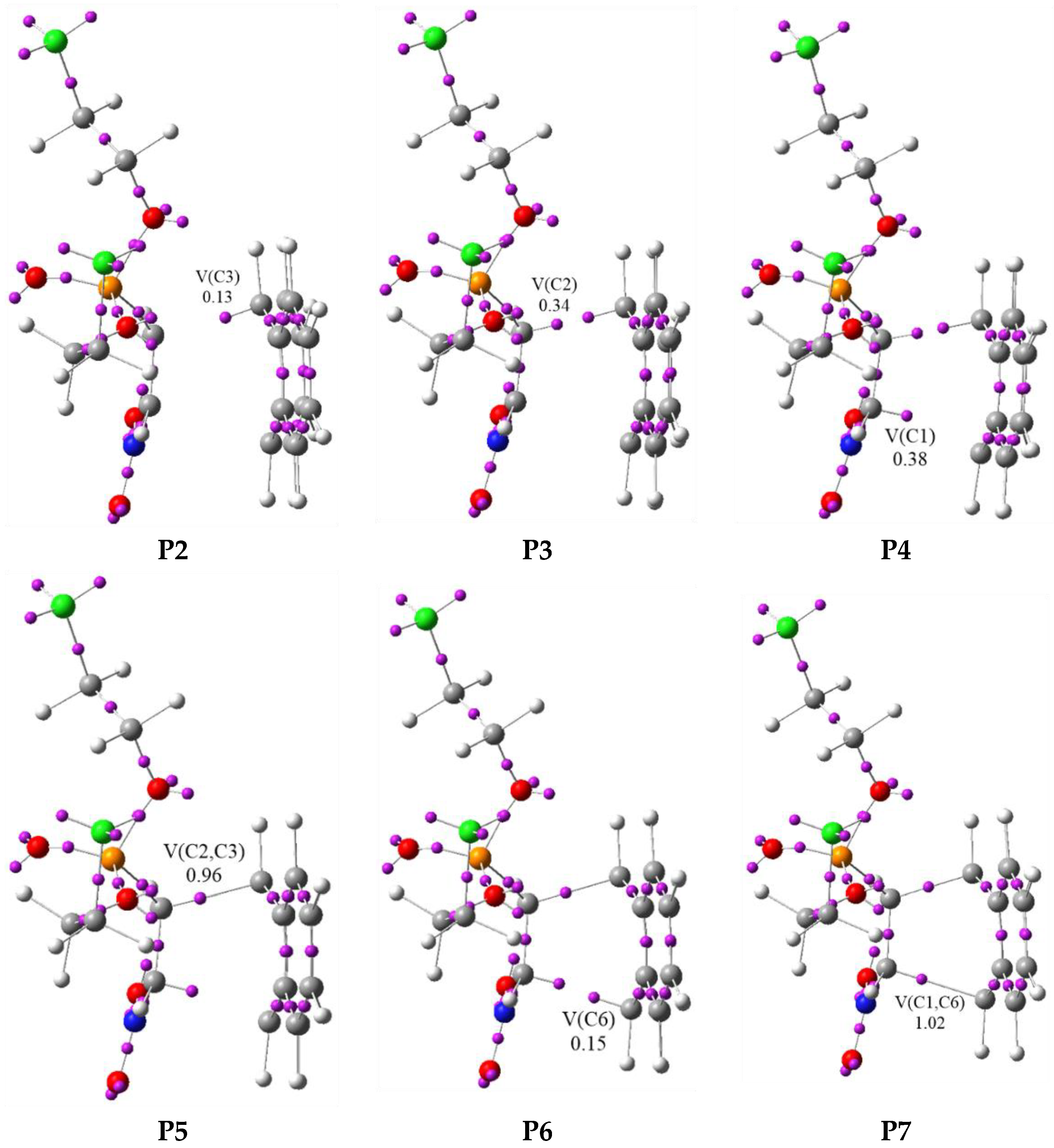
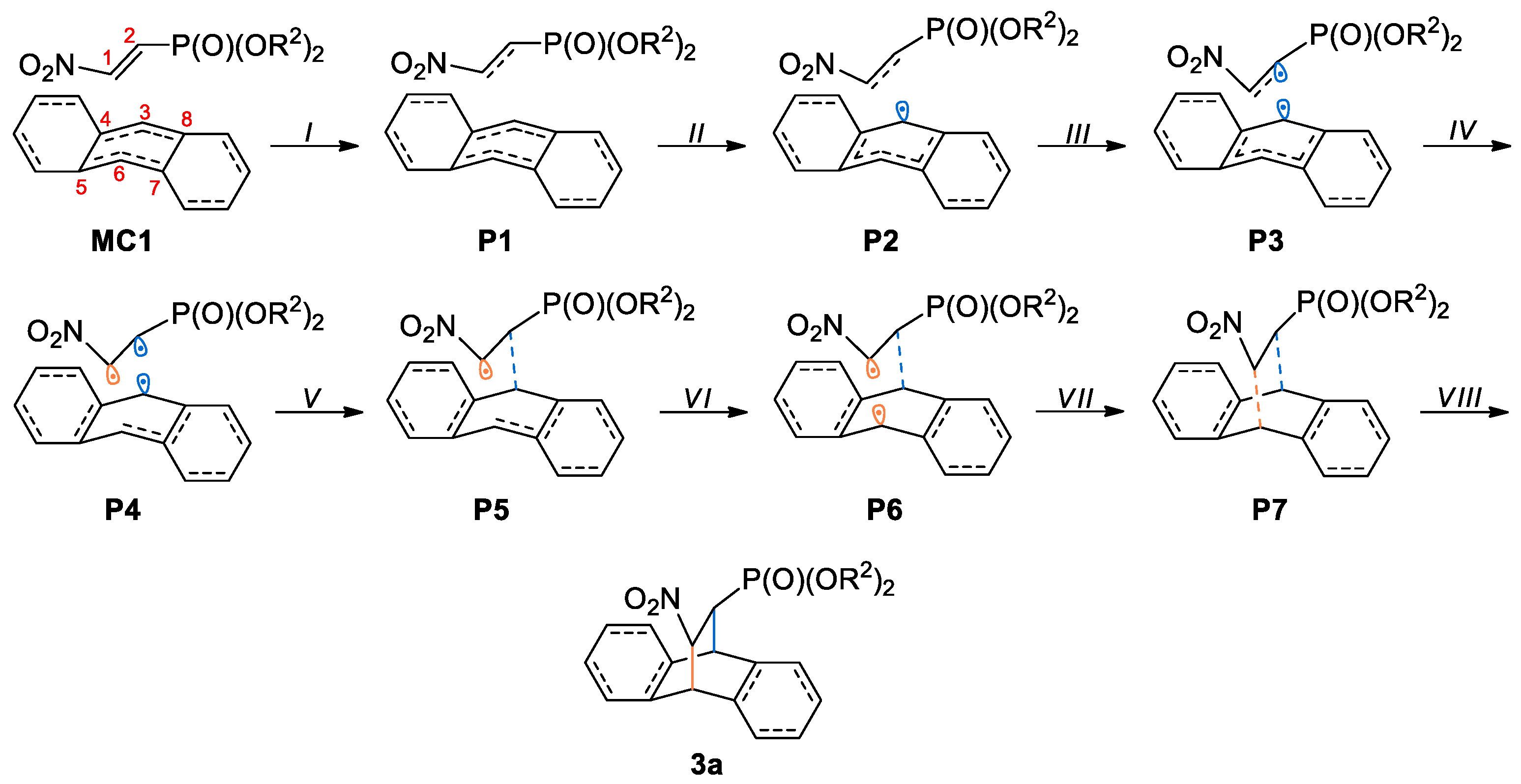
3.3. BET Study of the DA Reaction between bis(2-chloroethyl) 2-nitro-ethenylphosphonate 1a and Anthracene 2
4. Conclusions
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Coarey, E.J. Catalytic enantioselective Diels--Alder reactions: Methods, mechanistic fundamentals, pathways, and applications. Angew. Chem. Int. Ed. Engl. 2002, 17, 1650–1667. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Snyder, S.A.; Montagnon, T.; Vassilikogiannakis, G. The Diels–Alder Reaction in Total Synthesis. Angew. Chem. Int. Ed. Engl. 2002, 41, 1668–1698. [Google Scholar] [CrossRef]
- Heravi, M.M.; Ahmadi, T.; Ghavidel, M.; Heidari, B.; Himidi, H. Recent applications of the hetero Diels–Alder reaction in the total synthesis of natural products. RSC Adv. 2015, 5, 101999–102075. [Google Scholar] [CrossRef]
- Feuer, H. Nitrile Oxides, Nitrones and Nitronates in Organic Synthesis: Novel Strategies in Synthesis, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2008. [Google Scholar]
- Kącka-Zych, A.; Jasiński, R. Molecular mechanism of Hetero Diels-Alder reactions between (E)-1,1,1-trifluoro-3-nitrobut-2-enes and enamine systems in the light of Molecular Electron Density Theory. J. Mol. Graph. Model. 2020, 101, 107714. [Google Scholar] [CrossRef] [PubMed]
- Korotaev, V.Y.; Barkov, A.Y.; Slepukihin, P.A.; Kodess, M.I.; Sosnovskikh, V.Y. Diastereoselective reactions of 1,1,1-trichloro(trifluoro)-3-nitrobut-2-enes with 2-morpholinoalk-1-enes. Mendeleev Commun. 2011, 21, 112–114. [Google Scholar] [CrossRef]
- Korotaev, V.Y.; Barkov, A.Y.; Slepukihin, P.A.; Kodess, M.I.; Sosnovskikh, V.Y. Uncatalyzed reactions of α-(trihaloethylidene)nitroalkanes with push–pull enamines: A new type of ring–ring tautomerism in cyclobutane derivatives and the dramatic effect of the trihalomethyl group on the reaction pathway. Tetrahedron Lett. 2011, 52, 5764–5768. [Google Scholar] [CrossRef]
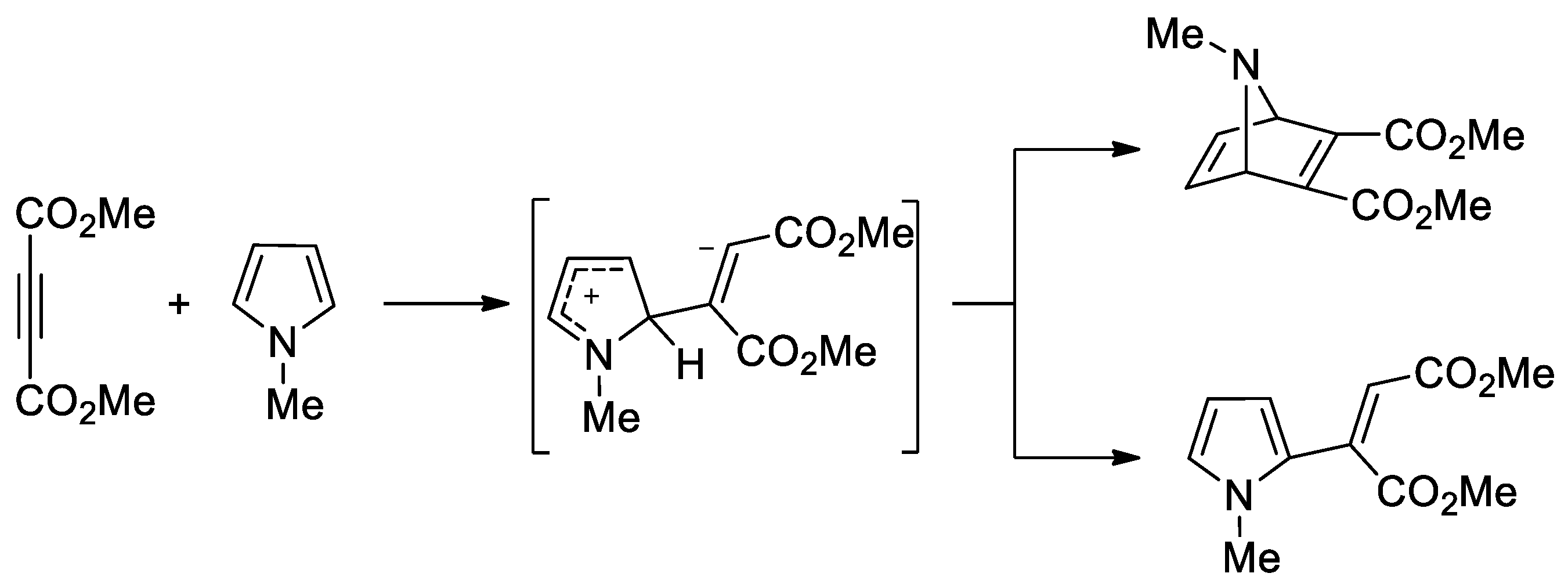
- Lee, C.K.; Hahn, C.S. Diels-Alder Reaction of Pyrrole with Dimethyl Acetylenedicarboxylate. J. Org. Chem. 1978, 43, 3727–3729. [Google Scholar] [CrossRef]
- Domingo, L.R.; Picher, M.T.; Zaragoza, R.J. Toward an Understanding of the Molecular Mechanism of the Reaction between 1-Methylpyrrole and Dimethyl Acetylenedicarboxylate. An ab Initio Study. J. Org. Chem. 1998, 63, 9183–9189. [Google Scholar] [CrossRef]
- Anisimova, N.A.; Kuzhaeva, A.A.; Berkova, G.A.; Deiko, L.I.; Berestovitskaya, V.M. Reactions of 2-Nitro- and 2-Bromo-2-nitroethenylphosphonates with Anthracene. Russ. J. Gen. Chem. 2005, 75, 689–693. [Google Scholar] [CrossRef]
- Domingo, L.R. Molecular Electron Density Theory: A Modern View of Reactivity in Organic Chemistry. Molecules 2016, 21, 1319. [Google Scholar] [CrossRef]
- Krokidis, X.; Noury, S.; Silvi, B. Characterization of elementary chemical processes by catastrophe theory. J. Phys. Chem. A 1997, 101, 7277–7283. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.; Vreven, T.J.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 16 Rev A.1; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Stephens, P.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Dresler, E.; Kącka-Zych, A.; Kwiatkowska, M.; Jasiński, R. Regioselectivity, stereoselectivity, and molecular mechanism of [3 + 2] cycloaddition reactions between 2-methyl-1-nitroprop-1-ene and (Z)-C-aryl-N-phenylnitrones: A DFT computational study. J. Mol. Model. 2018, 24, 329–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kącka-Zych, A. Understanding the molecular mechanism of the rearrangement of internal nitronic ester into nitronorbornene in light of the MEDT study. Molecules 2019, 24, 462. [Google Scholar] [CrossRef] [Green Version]
- Kącka-Zych, A.; Jasiński, R. Unexpected molecular mechanism of trimethylsilyl bromide elimination from 2-(trimethylsilyloxy)-3-bromo-3-methyl-isoxazolidines. Theor. Chem. Acc. 2019, 138, 81–86. [Google Scholar] [CrossRef] [Green Version]
- Kącka-Zych, A.; Ríos-Gutiérrez, M.; Domingo, L. A molecular electron density theory study of the Lewis acid–catalyzed decomposition reaction of nitroethyl benzoate using aluminum derivatives. J. Phys. Org. Chem. 2019, 32, e3938. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Unveiling the Lewis Acid Catalyzed Diels–Alder Reactions through the Molecular Electron Density Theory. Molecules 2020, 25, 2535. [Google Scholar] [CrossRef]
- Fukui, K. Formulation of the reaction coordinate. J. Phys. Chem. 1970, 74, 4161–4163. [Google Scholar] [CrossRef]
- Tapia, O. Solvent effect theories: Quantum and classical formalism and their applications in chemistry and biochemistry. J. Math. Chem. 1992, 10, 131–181. [Google Scholar] [CrossRef]
- Tomasi, J.; Perisco, M. Molecular interactions in solution: An overview of methods based on continuous distributions of the solvent. Chem. Rev. 1994, 94, 2017–2094. [Google Scholar] [CrossRef]
- Simkin, Y.; Sheikhet, I. Quantum Chemical and Statistical Theory of Solutions: A Computational Approach; Ellis Horwood: London, UK, 1995. [Google Scholar]
- Cances, E. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab initio study of solvated molecules: A new implementation of the polarizable continuum model. Chem. Phys. Chem. 1996, 225, 327–335. [Google Scholar] [CrossRef]
- Domingo, L.R. A new C–C bond formation model based on the quantum chemical topology of electron density. RSC Adv. 2014, 4, 32415–32428. [Google Scholar] [CrossRef] [Green Version]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Phys. Chem. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Greelings, P.; De Proft, F.; Langenaeker, W. Conceptual density functional theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the conceptual density functional theory indices to organic chemistry reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parr, R.G.; Szentpaly, L.V.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Domingo, L.R.; Chamorro, E.; Pérez, P. Understanding the Reactivity of Captodative Ethylenes in Polar Cycloaddition Reactions. A Theoretical Study. J. Org. Chem. 2008, 73, 4615–4624. [Google Scholar] [CrossRef]
- Domingo, L.R.; Pérez, P.; Sáez, J.A. Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv. 2013, 3, 1486–1494. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Noury, S.; Krokidis, X.; Fuster, F.; Silvi, B. Computational tools for the electron localization function topological analysis. Comput. Chem. 1999, 23, 597–604. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView Version 6.1; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Ahrens, J.; Geveci, B.; Law, C. ParaView: An End-User Tool for Large Data Visualization. In Visualization Handbook; Elsevier: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Ayachit, U. The ParaView Guide: A Parallel Visualization Application; Kitware: New York, NY, USA, 2015. [Google Scholar]
- Kącka-Zych, A. Push-pull nitronates in the [3+2] cycloaddition with nitroethylene: Molecular Electron Density Theory study. J. Mol. Graph. Model. 2020, 97, 107549. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Domingo, L.R.; Aurell, M.J.; Pérez, P.; Contreras, R. Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels–Alder reactions. Tetrahedron 2002, 58, 4417–4423. [Google Scholar] [CrossRef]
- Jaramillo, P.; Domingo, L.R.; Chamorro, E.; Pérez, P. A further exploration of a nucleophilicity index based on the gas-phase ionization potentials. J. Mol. Struct. THEOCHEM 2008, 865, 68–72. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| µ | η | ω | N | |
|---|---|---|---|---|
| 1a | −5.59 | 5.19 | 3.01 | 0.94 |
| 1b | −5.62 | 4.74 | 3.33 | 1.13 |
| 2 | −3.43 | 3.59 | 1.64 | 3.89 |
| Transition | ΔH353 | ΔG353 | ΔS353 |
|---|---|---|---|
| 1+2→MC1 | −2.1 | 7.9 | −33.7 |
| 1+2→TS1 | 21.8 | 36.8 | −50.4 |
| 1+2→3a | −13.6 | 1.5 | −50.7 |
| 1+2→MC2 | −3.6 | 4.8 | −28.0 |
| 1+2→TS2 | 18.7 | 35.2 | −55.3 |
| 1+2→3b | −15.0 | 3.8 | −63.0 |
| Structure | C2-C3 | C1-C6 | Δl | GEDT [e] | Imaginary Frequency [cm−1] | ||
|---|---|---|---|---|---|---|---|
| r [Å] | l | r [Å] | l | ||||
| MC1 | 6.816 | 3.925 | |||||
| TS1 | 2.106 | 0.664 | 2.430 | 0.448 | 0.22 | 0.30 | −399.48 |
| 3a | 1.576 | 1.566 | |||||
| MC2 | 7.256 | 5.187 | |||||
| TS2 | 1.958 | 0.762 | 2.805 | 0.203 | 0.56 | 0.36 | −332.75 |
| 3b | 1.581 | 1.561 | |||||
| Points | 1a | 2 | MC1 | P1 | P2 | P3 | P4 | P5 | P6 | P7 | 3a | TS1 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Phases | I | II | III | IV | V | VI | VII | VIII | ||||
| d(C2-C3) | 3.489 | 2.659 | 2.502 | 2.449 | 2.412 | 2.356 | 2.316 | 2.184 | 1.566 | 2.430 | ||
| d(C1-C6) | 3.643 | 2.429 | 2.208 | 2.132 | 2.081 | 2.005 | 1.956 | 1.821 | 1.576 | 2.106 | ||
| ΔE | 0.0 | 8.5 | 17.1 | 22.0 | 21.8 | 15.8 | 5.5 | −2.7 | −11.5 | 24.0 | ||
| V(C1,C2) | 1.74 | 1.75 | 3.32 | 3.19 | 2.95 | 2.69 | 2.55 | 2.45 | 2.22 | 1.99 | 2.87 | |
| V’(C1,C2) | 1.74 | 1.69 | ||||||||||
| V(C3,C4) | 2.71 | 2.81 | 2.65 | 2.58 | 2.55 | 2.53 | 2.49 | 2.45 | 2.33 | 2.05 | 2.54 | |
| V(C4,C5) | 2.47 | 2.45 | 2.59 | 2.60 | 2.61 | 2.62 | 2.65 | 2.66 | 2.70 | 2.75 | 2.61 | |
| V(C5,C6) | 2.71 | 2.79 | 2.65 | 2.50 | 2.40 | 2.35 | 2.29 | 2.23 | 2.16 | 2.05 | 2.37 | |
| V(C6,C7) | 2.93 | 2.83 | 2.69 | 2.53 | 2.46 | 2.41 | 2.34 | 2.31 | 2.22 | 2.07 | 2.44 | |
| V(C7,C8) | 2.47 | 2.48 | 2.54 | 2.60 | 2.61 | 2.62 | 2.64 | 2.65 | 2.69 | 2.77 | 2.61 | |
| V(C8,C3) | 2.93 | 2.81 | 2.76 | 2.69 | 2.64 | 2.61 | 2.61 | 2.47 | 2.33 | 2.06 | 2.61 | |
| V(C3) | 0.13 | 0.23 | 0.32 | 0.28 | ||||||||
| V(C2) | 0.34 | 0.45 | 0.40 | |||||||||
| V(C1) | 0.38 | 0.50 | 0.57 | |||||||||
| V(C2,C3) | 0.96 | 1.07 | 1.38 | 1.76 | ||||||||
| V(C6) | 0.15 | |||||||||||
| V(C1,C6) | 1.02 | 1.79 | ||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kącka-Zych, A. Participation of Phosphorylated Analogues of Nitroethene in Diels–Alder Reactions with Anthracene: A Molecular Electron Density Theory Study and Mechanistic Aspect. Organics 2020, 1, 36-48. https://0-doi-org.brum.beds.ac.uk/10.3390/org1010004
Kącka-Zych A. Participation of Phosphorylated Analogues of Nitroethene in Diels–Alder Reactions with Anthracene: A Molecular Electron Density Theory Study and Mechanistic Aspect. Organics. 2020; 1(1):36-48. https://0-doi-org.brum.beds.ac.uk/10.3390/org1010004
Chicago/Turabian StyleKącka-Zych, Agnieszka. 2020. "Participation of Phosphorylated Analogues of Nitroethene in Diels–Alder Reactions with Anthracene: A Molecular Electron Density Theory Study and Mechanistic Aspect" Organics 1, no. 1: 36-48. https://0-doi-org.brum.beds.ac.uk/10.3390/org1010004