
Design, Synthesis, and Utility of Defined Molecular Scaffolds


1
Graduate School of Pharmaceutical Sciences, Kyushu University, Fukuoka 812-8582, Japan
2
Department of Chemistry, North Carolina State University, Raleigh, NC 27695-8204, USA
3
Faculty of Pharmaceutical Sciences, Institute of Medical, Pharmaceutical, and Health Sciences, Kanazawa University, Kakuma-machi, Kanazawa 920-1192, Japan
*
Authors to whom correspondence should be addressed.
†
Equal contributions by both authors.
Organics 2021, 2(3), 161-273; https://0-doi-org.brum.beds.ac.uk/10.3390/org2030013
Submission received: 15 May 2021
/
Accepted: 18 June 2021
/
Published: 11 July 2021
(This article belongs to the Special Issue Feature Papers in Organics)
Abstract
:A growing theme in chemistry is the joining of multiple organic molecular building blocks to create functional molecules. Diverse derivatizable structures—here termed “scaffolds” comprised of “hubs”—provide the foundation for systematic covalent organization of a rich variety of building blocks. This review encompasses 30 tri- or tetra-armed molecular hubs (e.g., triazine, lysine, arenes, dyes) that are used directly or in combination to give linear, cyclic, or branched scaffolds. Each scaffold is categorized by graph theory into one of 31 trees to express the molecular connectivity and overall architecture. Rational chemistry with exacting numbers of derivatizable sites is emphasized. The incorporation of water-solubilization motifs, robust or self-immolative linkers, enzymatically cleavable groups and functional appendages affords immense (and often late-stage) diversification of the scaffolds. Altogether, 107 target molecules are reviewed along with 19 syntheses to illustrate the distinctive chemistries for creating and derivatizing scaffolds. The review covers the history of the field up through 2020, briefly touching on statistically derivatized carriers employed in immunology as counterpoints to the rationally assembled and derivatized scaffolds here, although most citations are from the past two decades. The scaffolds are used widely in fields ranging from pure chemistry to artificial photosynthesis and biomedical sciences.
1. Introduction
Molecular scaffolds defined here concern structures that serve as carriers of diverse covalently attached groups. The concept of carriers dates at least a century ago to studies in the early days of immunology [1]. Certain small molecules alone were found to be non-immunogenic; however, upon conjugation of multiple copies of the small molecules to a larger carrier, an immune response was elicited. From those studies originated a strong theme of research in immunology [2], and diverse carriers are now known to which small molecules (“haptens”) can be conjugated [3,4,5]. Such carriers include polysaccharides (e.g., dextran) [6], proteins (e.g., bovine serum albumin, keyhole limpet hemocyanin) [7,8], and dendrimers [9,10].
Valuable material entities for the development of scaffolds for diverse applications have been provided by peptides and proteins. The sequence specificity of proteins reflects an exquisite and compelling degree of molecular order, yet at the same time proteins present limitations toward derivatization. The limitations include the few distinct orthogonal reactive functional groups (e.g., amine, thiol) and the typical situation of statistical derivatization of multiple like functional groups leading to a heterogeneous mixture. Protein engineering has yielded towering accomplishments in molecular design, yet use of proteins as general scaffolds remains constrained due not only to the challenges of limited functional group availability but also the requirement for appropriate folding and avoidance of denaturation.
A growing theme in organic chemistry has been the development of molecular building blocks that can be assembled in a straightforward manner. Variation in the use of assorted building blocks enables access to distinct architectures. Building block chemistry enables de novo construction of target architectures, which can be used directly or derivatized further depending on the application. Accordingly, de novo synthesis with molecular building blocks (“bottom up”) is complementary to the use of intact, complex structures such as proteins as substrates for derivatization (“top down”).
In this paper, we focus on molecular scaffolds that are prepared by de novo synthesis. In the ideal case, a scaffold is associated with the following attributes: (1) the synthesis is scalable to gram-quantities; (2) the synthesis is rapid; (3) the scaffold sample is homogeneous (i.e., monodisperse); (4) the scaffold can be derivatized in a rational manner with regards to the number and type of attached units; (5) the derivatization of the scaffold can be done at an early or late-stage as desired; (6) the scaffold can be tailored with regards to polarity; (7) the scaffold is not susceptible to denaturation, or function does not depend on specific folding; and (8) the scaffold affords architectural control over the disposition of attached groups in the regime of 10–100 Å. The chief motivations for the preparation and use of scaffolds arise in the life sciences. Such applications are immensely diverse as illustrated herein; representative examples include molecular imaging and diagnostics, molecular brachytherapy, and therapeutic interventions [11,12,13,14]. Scaffolds enable preparation of multifunctional molecules, which are broadly defined as hybrid or conjugated agents comprised of two or more components [15].
The review is organized as follows. We first introduce and define the concept of a hub molecule, which is employed in the formation of scaffolds (Section 2). We then describe the use of graph theory to categorize the various scaffolds independent of their molecular composition (Section 3). We then describe the special features of polyethylene glycol (PEG) groups for aqueous solubilization given the wide use of the scaffolds in the life sciences (Section 4). We then launch into the core of the review, where illustrative scaffolds are described on the basis of the hubs (none, amino acids, triazine, benzene, nitrogen, carbon, and diverse functional entities; Section 5, Section 6, Section 7, Section 8, Section 9, Section 10 and Section 11). We finish with an overview of the various scaffolds (Section 12). Altogether, the review describes 30 hub molecules, 31 distinct molecular graphs, and 107 examples (scaffolds and derivatives thereof). To enable evaluation of synthetic accessibility, the syntheses of 19 scaffold examples are described. The review covers the period from inception to the present, although most citations concern the period 2000–2020. Taken together, the review aims to provide an in-depth assessment of diverse molecular scaffolds for facile construction of multifunctional molecules, the applications of which chiefly encompass the life sciences and material sciences.
2. Hub Molecules and Rational Chemistry
We define hub molecules as the constituents that make up a scaffold. A vast number of multifunctional molecules that utilize hub structures has been reported. Our focus is on hub units that fulfill the following criteria: (1) are small organic molecules, typically <400 Da, although hub molecules that provide an implicit function in addition to serving as a nexus may be larger; (2) are well-determined as a single compound by nuclear magnetic resonance (NMR) spectroscopy and mass spectrometry; (3) include provisions to conjugate ≥3 functional molecules, including ≥2 distinct molecular entities; (4) covalently anchor to linkers or functional molecules; and (5) support rational chemistry including formation of monodisperse, homogeneous scaffolds.
Rational chemistry entails exacting and successive introduction of different kinds of functional molecular entities. The counterpart to rational chemistry is statistical chemistry, where, for example, a set of reacting groups on a molecule is treated with a sub-stoichiometric quantity of derivatizing reagent, whereupon reaction proceeds in stochastic fashion. A distribution of product molecules results comprised of different numbers of derivatized groups and at distinct sites; for a given number of derivatized groups a large number of isomers is expected given the distinct sites of reaction. Separation of such a mixture is typically very challenging.
An example of statistical chemistry is shown in Figure 1. Consider a scaffold equipped with five identical sites that react equivalently and independently. Imagine the goal of preparing the target compound wherein only one site has been derivatized. Thus, treatment is carried out with one molar equivalent of a derivatization reagent. What is the outcome? This type of problem is described by the Poisson equation, and the resulting mixture is referred to as a Poisson distribution. The Poisson equation is derived from the binomial equation in the limit where the number of events becomes infinite and the probability of an event becomes infinitesimal. The Poisson and binomial distributions can be quite similar but the Poisson distribution can be more easily calculated. The Poisson equation is shown in Equation (1):
where m is the mean (here the loading number), k is the number of “hits” or sites that have undergone derivatization (k takes on integer values 0, 1, 2, 3, 4, etc.), and x is the expectation of a particular value of k [16].
The Poisson distribution for the mean number of derivatizations (“loading number”) equal to one is given in Figure 1; the probability of one and only one appended group is 36.8%, with 36.8% unreacted and 26.4% bearing two or more appended groups. Running the reaction with a higher loading of 1.5 gives 33.5% of the singly reacted scaffold and shifts material from unreacted (22.3%) to derivatized with ≥2 groups (44.2%). Moving in the opposite direction, with extreme sub-stoichiometric loading (0.3), the singly reacted scaffold is now 22.2%, and unreacted is 74.1%. There is no simple means to circumvent statistics in these situations.
The chemistry situation is actually substantially worse than these compositional statistics would indicate. For each product other than starting scaffold and the exhaustively derivatized scaffold, there are isomers. The number of isomers is given by the binomial equation. In this case for scaffold (S) with various number of appended groups (A), the number of isomers for A-Sn is shown in parentheses as follows: A-S0 (1), A-S1 (5), A-S2 (10), A-S3 (10), A-S4 (5), and A-S5 (1). The total number of distinct products is 32. For the reaction with mean loading of 1, the total yield of the singly derivatized scaffold A-S1 is 36.8%, and each of the five isomers therein is present at 7.36%. Even if one has available a method for separating the products that differ in number of appended groups, one still needs to separate the isomers for a given composition. The resulting yield of specific target compound is meager, indeed. We note also that the illustration given here concerns a simple case; a small protein such as myoglobin (~17 kDa) has 14 accessible amines (13 surface lysine ε-amines, and the α-amino group) [17], whereas an antibody (150 kDa) may have >50 accessible amines.
With the statistical example as a backdrop, it is worthwhile to point out the hubs and scaffolds that are excluded by the focus herein on rational chemistry. The following are omitted: dendrimers, synthetic or naturally derived polymers, solid nanoparticles, carbon nanomaterials, DNA nanostructures, metal–organic frameworks, proteins, dendrimers, polysaccharides, and multifunctional supramolecules (e.g., liposomes, micelles, host–guest complexes). We note that polysaccharides have been widely used as carrier molecules in the life sciences, and dendrimers provide more synthetically controlled architectures with defined numbers of substituents depending on the generation number (Figure 2). Regardless, the focus here concerns those scaffolds that support rational incorporation of ≥2 distinct groups. The hubs that underpin the scaffolds described herein are shown in Figure 3.
Subsequent sections delineate diverse scaffolds. For selected examples, the full synthesis is outlined. The compounds for which syntheses are displayed generally meet the following criteria:
- Published within the past 10–20 years.
- Represent the characteristics of the hub in molecular design and synthesis.
- Have an elaborate molecular structure and corresponding graph.
- Are prepared by a distinctive (sometimes non-obvious) synthesis.
- Are prepared by selective (convergent), unique, and efficient synthetic methods.
- Have reliable experimental data such as synthetic yield and characterization.
Before describing diverse scaffold architectures, we first turn to graph theory for abstract categorization of the various architectures.
3. Graph Theory in Design of Multifunctional Molecules
Graph theory is a branch of mathematics related to topology and combinatorics, and deals with the connectivity of plural objects [18]. Graph theory has been applied to chemistry to denote the connection network of atoms in terms of vertices and edges, which is known variously as a chemical graph, molecular graph, structural graph, or constitutional graph [19,20]. Inspired by this chemical graph theory, we describe multifunctional molecules in schematic tree diagrams.
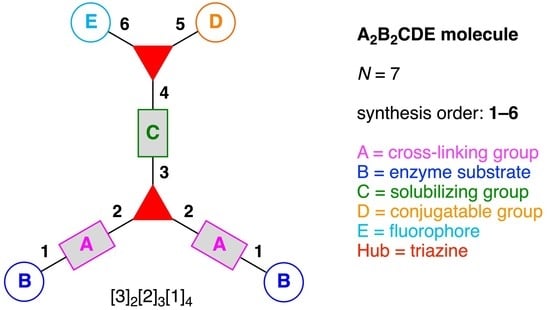
The present review encompasses 31 distinct trees. The trees are shown in Figure 4. The graph labeling is shown for each tree. In graph theory, a connected, undirected, and acyclic graph is termed a tree, which has >2 nodes (or vertices). Thus, the architecture of a multifunctional molecule may be regarded as “a tree” wherein each node is a molecular hub. In graph terminology, the number n of arms on a hub is denoted by [n] and the number of times m that the hub appears is given as a suffix, [n]m. We are aware that the denotation [n]m is a modification of common graph theory as required for molecular description. Distinct hubs are enumerated so that, for example, a tree with one four-arm hub and two three-arm hubs would be denoted by architecture [4]1[3]2, where the one can be omitted for clarity. The total number of groups attached to the scaffold are listed as a group regardless of identity. Thus, the tree with two three-arm hubs and one four-arm hub could have six substituents, in which case the tree would be denoted by architecture [4][3]2[1]6. Note that we use the terminology tree, graph, and architecture interchangeably here.
A shortcoming of the graph terminology is the absence of information concerning the number of distinct groups attached to the scaffold. For example, the aforementioned [4][3]2[1]6 tree could have six distinct groups or six identical groups or various combinations in between. To denote the richness of the various groups, we also use an abecedarian nomenclature. Thus, a [4][3]2[1]6 tree could be substituted with ABCDEF, A6, A2B2C2 groups, and so forth. Both the graph terminology to indicate the tree architecture and the abecedarian nomenclature to express the distinct derivatization of the scaffold are essential.
The scaffolds described here range from simple examples with tree designations such as architecture [3][1]3 to more complex examples such as architecture [3]2[2]3[1]4, as shown in Figure 4. Three molecular graphs have cyclic hubs, which are distinguished from the others by adding a prefix “c-” to the graph denotation such as architecture c-[4]6[1]12. Immense richness is available via derivatization of the various scaffolds with functional units (vide infra). The term “N” represents the number of the ABC groups. We now turn to delineate the graph terminology in the context of the specific types of hubs considered here. Additional terminology is described which conveys the sequence order for synthesis of the architecture including the scaffold and the functional substituents.
The multifunctional molecules in this review are composed of at least one hub that is tri- or tetra-armed (represented as a red triangle or rhombus). The ABCD functional units are attached to the scaffold via inert linkers or a functional linker. A functional unit is designated by an open circle containing an inscribed “X” (or ABCD). A linker that serves as a functional unit is displayed as a rectangle wherein “X” is inscribed. Hub molecules are rendered as a red triangle or rhombus. When the hub has a function other than a mere nexus, like that of ABCD molecules, an “X” is inscribed in the triangle or rhombus. This terminology fully describes the tree architecture and the distinctive composition of functional units. Note that there is not a 1:1 mapping of hubs (specific molecular entities) and trees (elaborated molecular constructs). One further terminology includes incorporating numbers alongside each functional unit in the graph to indicate the synthesis order of attachment on the hub or functional linker.
The following describes the steps to determine graphs and appropriate nomenclature for multifunctional molecules.
- Count all the edge-forming reaction steps of the graph. For a single hub, counting proceeds sequentially with each distinct (A, B, C, etc.) unit. An example is shown in Figure 5A. When there are ≥2 hubs, counting starts at the parent hub in linear sequential fashion. For a convergent synthesis step, wherein multiple identical groups become attached to one hub, the number of edge-forming reaction steps is tallied as one. If the edge has been formed in a commercially available building block, the counting for the edge is labeled as “0”. An example is shown in Figure 5B, which contains a scaffold composed of two hubs and A3BC substituents.
- Identify the parent hub. If the molecule contains ≥2 hubs, the number of edge-forming reaction steps starting at each hub is compared. The hub designated as the parent has the largest number of edge-forming reaction steps. If the steps are identical, four-arm hubs override three-arm hubs. If both the steps and the arm-numbers are identical, the parent hub is selected arbitrarily, considering the complexity of the molecular structures and the length of the synthetic process. An example is shown in Figure 5B, which contains two hubs in the scaffold, a three-arm and a four-arm hub. The three-arm hub is the parent because there are three distinct substituents (e.g., A, B and the four-arm hub) whereas the four-arm hub contains three identical substituents and the three-arm hub.
- Denote the synthesis order. The synthesis order is not unique to a particular tree but reflects the choices for a particular instantiation of a given tree to reach a particular target molecule. An example is provided in Figure 5C for the tree [3][2][1]3. Here, the three-arm hub is sequentially derivatized (Steps 1 and 2), the functional linker is derivatized with the functional unit Xd (Step 3), and the other terminus of the functional linker is joined to the third arm of the three-arm hub (Step 4). This terminology embodies the synthesis order, which for hubs, linkers, and functional units as molecular building blocks allows concise representation of a large quantity of information without expression of all the intricate details typical of a synthetic scheme.
- Assign the functional units. If the molecule contains ≥2 identical functional units, the units are named alphabetically in order of the number of the identical units. Thus, A2BC is preferred versus AB2C. If the number is identical, the units conjugated to the parent hub in the earliest step have the highest priority. For a subunit (a group of units), the unit with the least distance (edge number) from the parent hub has the highest priority. The example shown in Figure 6 is the tree with graph [3][2]3[1]3, functional units ABCDEF, and a synthesis that assembles A–E in a sequential manner to build the two arms of the three-arm hub and then attaches the functional unit F in the final step.
In summary, this terminology is not perfect but does embody a great deal of information in a shorthand way that concisely describes the architecture of the scaffold including the number of hubs, the number of distinct linkers and functional units, and the order of steps in the synthesis.
A multifunctional molecule is considered here as an assembly of ≥3 (bio)functional constituents attached to a hub unit, and each (bio)functional component is assigned with an abcedarian formula. For example, a multifunctional molecule with three distinct components is referred to as “ABC”. In another example, a multifunctional molecule that contains two identical units of A, one unit of B, and one unit of C is assigned as “A2BC”. Each so-named ABC… construct can be categorized by one of five chief functionalities as follows: (1) conjugatable group; (2) bioactive group; (3) imaging agent; (4) reactive group; and (5) other functionality. A bioactive group includes not only a ligand and drug that together act as a targeting entity, therapeutic agent, and molecular capture agent but also any enzyme substrate that engenders an enzyme responsive function. In the case of multifunctional molecules for cancer therapy, for example, representative bioactive groups are the Arg-Gly-Asp (RGD) motif for targeting, the Val-Cit (citrulline) peptide as a cathepsin B-cleavable sequence, and doxorubicin as the chemotherapy drug [21]. Imaging agents often include a fluorophore, quencher, metal chelator, and/or radioisotope [22,23,24,25]. Reactive groups such as light- or redox-responsive linkers as well as self-immolative linkers are used for not only chemical labeling probes but also stimuli-activatable probes for biological species [26,27,28,29]. Enzyme-responsible cleavable linkers (e.g., peptides cleaved by proteases) can serve as reactive groups or bioactive groups, but here were included solely in the latter category. Other functionalities include water-solubilizing tags, lipid tags, and ionic tags including, for example, a cell penetrating peptide.
4. Solubilizing Groups
Solubilizing groups can be appended to a hydrophobic structure or can be an integral constituent of a scaffold. Numerous groups have been employed as appendages to impart aqueous solubility to hydrophobic molecules. Examples of ionic solubilizing groups that are readily appended are pyridinium, ammonium, sulfonates, phosphonates, phosphates, and glucuronides, of which selected examples are displayed in Figure 7. The chief example of a non-ionic solubilizing group is provided by polyethylene glycol (PEG). Solubilizing groups often are incorporated or are integral to moieties that play a functional role. For example, self-immolative groups release a molecular cargo upon an appropriate trigger, such as enzymatic action or illumination. The chemistry concerning self-immolative linkers has developed immensely over the past few years. The reader is referred to excellent reviews on this topic [29,30,31].
Among various hydrophobic compounds, tetrapyrrole macrocycles may present one of the most significant challenges given the size of the disk-like macrocycle. The solubilization of tetrapyrroles has been examined with the full gamut of ionic and non-ionic solubilization motifs. Excellent reviews [32,33,34] concerning the solubilization of tetrapyrrole macrocycles have appeared, along with specific articles [35,36,37,38], and the various solubilization motifs will not be repeated here. Specific groups are described in the context of the 107 target molecules (vide infra). One group that does warrant explicit comment, paradoxically despite widespread use, is the PEG unit.
The PEG unit has been widely used for imparting aqueous solubility [36,39]. The synthetic chemistry for the preparation of PEG compounds is well developed. Indeed, monodisperse and heterotelechelic PEG reagents of specified length and bearing diverse handles are available, where monodisperse means all molecules have the same length (i.e., a homogeneous sample) and heterotelechelic means the two end groups are distinct [40,41,42,43]. The commercial availability of heterotelechelic monodisperse PEG reagents of specified length has facilitated widespread applications including in the biomedical arena [42,43,44].
One remarkable, unappreciated, and convenient aspect of PEG chemistry concerns solubility: PEG units are soluble both in water and in a variety of organic solvents [39]. On this account, a convenient purification tactic can be implemented. When hydrophobic compounds are PEGylated, purification often can be achieved by partitioning the product between aqueous and organic phases—the PEGylated compound partitions preferentially into the organic phase, whereas salts, polar solvents, and other polar entities (e.g., catalysts, reagents, byproducts) partition into the aqueous phase. Few if any other water-solubilization motifs exhibit this dual-solubility feature.
The PEG entity is typically drawn in the condensed form of –(CH2CH2O)n– with appropriate end groups, or is presented in extended fashion in a line drawing. Neither adequately conveys the features that impart water solubility. Some comments are warranted in this regard on the remarkable features of PEG groups in providing aqueous solubility to diverse hydrophobic molecules. Said differently, while the effects of PEG groups are widely exploited, just how unique such features are may be less appreciated. Begum and Matsuura (1997) address this issue as follows [45]:
“The fact that other polyethers such as poly(oxymethylene) (–OCH2–)m, poly(oxytrimethylene) (–OCH2CH2CH2–)m and poly(oxypropylene) [–OCH2CH(CH3)–]m are insoluble in water implies that the distance between the neighboring ether oxygen atoms along the chain and the hydrophobicity and size of the alkylene spacer between the ether oxygens are important factors of the water solubility. As the spatial distance between the ether oxygens depends directly on the conformation of the polymer chain, the phase behavior of the poly(oxyethylene)–water system should be closely related to the poly(oxyethylene) chain conformation.”
Begum and Matsuura further emphasize that two features: (1) stabilization of the gauche conformation of the O–C–C–O segment, and (2) the interaction of water, via hydrogen-bonding, with the –OCH2CH2– units—together give rise to a partly helical conformation and aqueous solubility [45].
The extent to which the PEG unit is unique in imparting aqueous solubility can perhaps be seen in the following comparison, expanding on the text by Begum and Matsuura. Polymers containing oxyalkyl repeat units are shown in Figure 8. The polymers contain the repeat unit (–OCH2–)m, (–OCH2CH2–)m (i.e., PEG) and (–OCH2CH2CH2–)m. Casual consideration might regard each to be water-soluble, extrapolating from the features of PEG, that the aqueous solubility of PEG group originates simply from the large number of oxygen atoms for hydrogen bonding with water. However, the (–OCH2–)m polymer (4a), which has twice the ratio of oxygen to carbon than –OCH2CH2– (PEG, 4b), is not water soluble, nor is the polymer composed of (–OCH2CH2CH2–)m units (4c). Among polyalkylethers, the structural requirements for aqueous solubility are sharply poised on the (–OCH2CH2–)m unit (PEG) and neither (–OCH2–)m nor (–OCH2CH2CH2–)m is water soluble.
5. Linear Functional Molecules, No Hubs (13)
Linear functional molecules that lack a hub come in considerable variety. Seven examples are shown in Figure 9, each of which has composition ABC and architecture [2][1]2. As a very compact example, an affinity-based probe (5a) for cell-based protein profiling and target validation was developed [46]. The probe is comprised of three functional components: (A) a diazirine linker as a photoaffinity labeling (PAL) agent; (B) an alkyne as a conjugatable group for visualization or enrichment of the labelling events, and (C) tankyrase1/2 inhibitor XAV-939 as a targeting agent for proteomic studies of the carcinogenic Wnt signaling pathway.
A trifunctional photoaffinity probe (5b) was developed for investigation of the potential target proteins of oleanolic acid (OA). The probe is comprised of three functional components: (A) a benzophenone linker as a PAL agent; (B) OA as a targeting agent for complementary proteins, and (C) a biotin tag [47].
An inflammation-guided redox-responsive cancer prodrug (5c) was developed [48]. The prodrug is comprised of three functional components: (A) a thioether linker cleaved by reducing (glutathione) or oxidative (H2O2) inputs likely to be present in physiological media and/or the tumor microenvironment; (B) a nonsteroidal anti-inflammatory drug both as a targeting agent for a cyclooxygenase enzyme (e.g., COX-2) that is overexpressed in various cancers and as an immunotherapeutic drug, and (C) topoisomerase-I inhibitor SN-38 as a chemotherapeutic drug.
An enzymatically activated prodrug of doxorubicin (5d) was developed that is comprised of three functional components: (A) a benzyloxycarbony group as a self-immolative linker; (B) galactose as a substrate sugar for β-galactosidase, and (C) doxorubicin as an anticancer drug [49]. A probe (5e) was developed for the in vivo detection and imaging of urokinase-like plasminogen activator (uPA) [50]. The probe undergoes Förster resonance energy transfer (FRET) [51,52,53,54] and is comprised of three functional components: (A) Ser-Gly-Arg-Ser-Ala-Asn-Ala heptapeptide linker cleaved by uPA; (B) a pentamethine cyanine dye (Cy5.0) as a fluorophore, and (C) a phenazinium-substituted azo dye referred to as Black Hole Quencher-3 (BHQ-3). A Kondrat’eva cycloaddition [55] of 5-alkoxyoxazole and maleimide reactants was utilized to couple the BHQ-3 moiety and the Cy5.0-heptapeptide unit.
A tumor-targeted agent (5f) was developed that is comprised of three functional components: (A) a GDEVDGSGK peptide as a pharmacokinetics-modifying linker to impart enhanced water solubility, better delivery efficiency, and decreased normal tissue toxicity; (B) a 64Cu-containing chlorophyll derivative, and (C) folate as a targeting agent for cancer cells wherein the FR is upregulated and over-expressed [56]. The compound was envisaged as a photosensitizer for photodynamic therapy (PDT) [57,58] and optical/positron emission tomography (PET) [59,60] imaging, although tetrapyrrole–copper chelates typically have short excited-state lifetimes [61]. As a general rule, long excited-state lifetimes are preferable for photoactivity, although PDT activity is the product of multiple factors, and counterexamples to this general rule are known [62].
A PAL probe (5g) was developed for the identification and characterization of molecular targets/domains in γ-secretase [63]. The probe is comprised of three functional components: (A) a phenyldiazirine linker as a PAL agent; (B) a biotin tag, and (C) a dibenzoazepine-type LY411575 analogue as a targeting agent for γ-secretase. In summary, each of the aforementioned seven compounds shown in Figure 9 constitutes an example of the architecture [2][1]2.
We now turn to linear molecules that contain more than three components. Six representative examples are shown in Figure 10. A matrix metalloproteinase (MMP)-activated prodrug (5h) was developed for cancer treatment [64]. The prodrug is comprised of four functional components: (A) a Leu residue for cleavage by intracellular or extracellular proteases; (B) a Hof-Tyr dipeptide for degradation by extracellular proteases, where Hof is homophenylalanine; (C) an Ac-Pro-Cit-Gly tripeptide for cleavage by MMP-2, MMP-9, and MMP-14 (which are frequently over-expressed in human tumors), and (D) doxorubicin as a cytotoxic drug. This construct (5h) has composition ABCD and architecture [2]2[1]2. The prodrug was cleaved in the middle of the hexapeptide by the presence of MMP-2, -9, and -14 to generate a Hof-Tyr-Leu–doxorubicin conjugate. The latter conjugate was subsequently degraded by extracellular proteases to release Leu–doxorubicin, which in turn may be converted by further proteolytic cleavage either inside or outside cells to give the cytotoxic doxorubicin.
A mitochondria-targeted DNA alkylating agent (5i) that is activated by hypoxia was developed for delivery to tumors and the mitochondria therein (Figure 10) [65]. The agent is comprised of four functional components: (A) rhodamine 123B as a fluorophore; (B) an azo linker as a reduction-cleavable quencher under hypoxic conditions typical of tumors [66]; (C) N,N-bis(2-chloroethyl)-1,4-benzenediamine as a DNA alkylating agent, and (D) triphenylphosphonium as a targeting agent for hyperpolarized mitochondria of cancer cells in solid tumors. This construct (5i) has composition ABCD and architecture [2]2[1]2.
A tumor-targeting prodrug (5j) for the precision therapy of non-small-cell lung cancer was developed (Figure 10) [67]. The prodrug is comprised of four functional components: (A) a disulfide linker cleaved by glutathione in tumors; (B) an aza-BODIPY as a fluorophore (the authors used the term azo-BODIPY but aza-BODIPY is employed here, where BODIPY refers to a difluoroboron-dipyrrin); (C) the drug Gefitinib for treatment of lung cancer, and (D) a polyamine derivative as a targeting agent for the polyamine uptake system. This construct (5j) has composition ABCD and architecture [2]2[1]2.
A theranostic dual-acting prodrug delivery probe (5k) also was developed for cancer therapy (Figure 10) [68]. The probe is comprised of four functional components: (A) a tetraphenylene derivative that exhibits aggregation-induced emission (AIE) [69,70] and serves as a donor in FRET; (B) cisplatin as an anticancer drug; (C) the cyclic peptide cyclo(-Arg-Gly-Asp-d-Phe-Pag-) as a targeting agent for αvβ3 integrin (where Pag refers to propargylglycine), which often is over-expressed in cancer cells, and (D) doxorubicin as a cytotoxic drug and FRET acceptor. This construct (5k) has composition ABCD and architecture [2]2[1]2.
A chemical probe (5l) was developed for labeling of galectin-3, which is a lectin associated with cancer (Figure 10). The probe is comprised of three functional components: (A) a benzophenone linker as a PAL agent; (B) an alkyne as a conjugatable group for post-labeling with a fluorophore, and (C) a thiodigalactoside as a targeting agent for galectin-3 [71]. This construct (5l) has composition A2B2C and architecture [2]3[1]2.
A cell-penetrating peptide bearing a H2O2-activatable cleavage site (5m) was developed (Figure 10) [72]. The agent is comprised of five functional components: (A) a phenylboronic acid linker, which is cleaved by H2O2; (B) a (D-Glu)9 peptide as a polyanionic linker; (C) a (D-Arg)9 peptide as a cell-penetrating peptide; (D) a pentamethine cyanine dye (Cy5) as a FRET acceptor, and (E) 5-carboxyfluorescein as a FRET donor. This construct (5m) has composition ABCDE and architecture [2]3[1]2. Oxidation of the phenyl boronic acid by H2O2 forms a phenolate that subsequently undergo a spontaneous 1,6-elimination, resulting in fragmentation of the agent and release of the cell-penetrating peptide domain. Visualization of the consequences of the oxidation with H2O2 was facilitated by fluorescent labeling of the two peptide domains.
6. Amino Acid and Peptide Hubs—General Features
Amino acid-based hubs provide versatile building blocks for construction of multifunctional molecules [73]. An amino acid has one amine (-NH2) and one carboxylic acid (-COOH), and many amino acids contain an additional functional group on the side chain, providing trifunctional hubs. The advantages of using an amino acid or peptide as a hub are as follows: (1) biocompatibility; (2) rich options for functionalizing the side chain (including non-natural amino acids); (3) seamless incorporation of peptide-based appendages; (4) expandability of amino acid hubs into peptides to give densely functionalized scaffolds, and (5) availability of well-developed reagents and protecting groups. Amino acids, as basic constituents of proteins, play a key role in virtually all biological processes [74]. Therefore, the biocompatibility of an amino acid or peptide core is ensured in principle, unless a non-natural amino acid is used. Native amino acids have up to eight distinct functional groups including amino, carboxy, hydroxy, carbamoyl, sulfanyl, amide, guanidino, and thiol groups as well as indole and imidazole rings. Moreover, a variety of non-natural azido or propargyl amino acids for click chemistry are now commercially available [75]. The components to be used in constructing the scaffold dictate the functionality of the side chains. Multifunctional molecules often contain one or more peptide components such as an enzymatically cleavable peptide, peptide ligand and inhibitor, or cell penetrating peptide [76,77,78,79]. When several peptide units are used as building blocks, it is possible to synthesize continuous linear peptide components via solid-phase peptide synthesis (SPPS), which allows for easy and fast (including automated) synthesis [80]. In addition, scaffolds that contain two or more amino acid hubs provide much functionality. New peptide coupling reagents and protecting groups have been extensively developed, and nowadays the desired peptide can be synthesized in high yield, in high purity, and in a scalable manner while minimizing side reactions [81,82].
On the other hand, amino acid-based chemistry is not without drawbacks and pitfalls, such as the following: (1) requirement for two or more orthogonal protecting groups; (2) ever-present possibility of racemization; (3) side reactions; (4) aggregation; and (5) low biostability. The various functional groups in amino acids make proper selection of protecting groups complicated. Hence, a promising synthetic strategy can be jeopardized if the corresponding protecting groups are not properly chosen. The protection of the C-terminal carboxylic acid is different in SPPS to liquid-phase peptide synthesis (LPPS). In SPPS, the C-terminus is commonly covalently bound to the resin as a solid support. Hence, C-terminal protection is not required because the resin acts both as a solid support and as a protecting group. For scaffold synthesis, where the number of peptide bonds may be few or the peptide may be elaborated as part of a non-peptide structure, it is common to use LPPS rather than the standard SPPS, which is common for preparing linear peptides of some length.
All native amino acids excepting glycine contain a chiral center at the α-carbon atom, and isoleucine and threonine have an additional chiral center at the β-carbon atom. The properties of amino acids and peptides crucially rely on maintaining the fidelity of the configuration of each chiral center. The inversion of a single chiral center in the peptide can affect its biological activity. To avoid epimerization, mild reaction conditions and appropriate coupling reagents are essential. The mechanisms of epimerization for a peptide (6a) upon activation of the carboxylic acid (6b) entail base-catalyzed direct enolization (Path A) to give the stereochemically altered product (6c) and formation of the oxazolone (6d) (Path B) (Figure 11A) [83]. Both side reactions can prevail when the carboxylic acid moiety is converted to the active ester during the coupling process to give the peptide product (6e). A second significant, and less obvious, side reaction is formation of the 2,5-diketopiperazine (DKP). A linear dipeptide (6f) containing a leaving group at the C-terminus can readily cyclize to form the six-membered DKP (6g) (Figure 11B) [84]. Additional unwanted reactions are many and typically entail involvement of side chains such as aspartimide and pyroglutamate formation, transference of cleaved protecting groups, guanidinylation, and so on [85,86,87]. Aggregation of nascent peptide chains during SPPS can lead to slow and incomplete reaction [88,89]. Aggregation propensity depends on the peptide sequence, which is difficult to predict accurately in advance [90]. A peptide consisting exclusively of natural amino acids (L-configuration) is easily hydrolyzed by proteases in vivo. Peptides containing D-amino acids or synthetic amino acids, or that exist as cyclic peptides, are often not recognized by proteases or by the immune system, and can exhibit high protease resistance (biostability) and perhaps lower immunogenicity.
Lysine, which has two amino and one carboxylic acid groups, is widely used for construction of ABC and A2B type multifunctional molecules. The requisite protecting groups for the two amines can be chosen from a variety of commercially available orthogonally protected lysine derivatives. Moreover, synthetic routes to azidolysine (2-amino-6-azidohexanoic acid) have been developed for the construction of the multifunctional molecules via click chemistry [91].
An amino acid used time and again as a hub is lysine, as described below. Yet glutamic acid, cysteine, serine and threonine are quite versatile and may be somewhat overlooked. Hence a short digression concerning these four amino acids, which have some subtle and perhaps non-obvious features, follows here.
Glutamic acid has two carboxylic acid groups and one amino group, and in that regard is the complement of lysine. The carboxylic acid can be preactivated as the isolable and storable ester of N-hydroxysuccinimide (NHS), providing a ready substrate for selective coupling with an amine. However, there are limitations on the use of Glu compared with Lys because the shorter side chain of Glu compared with Lys, often causing insertion of an appropriate linker to be mandatory. On the other hand, a common motif is Arg-Gly-Asp as part of a cyclic peptide (which can contain other amino acids but is designated as cRGD), which specifically binds to αvβ3 integrin (known to be overexpressed on cancer cells [92]). When the cRGD is used for targeting, the presence of a Lys in the cyclic peptide can be used advantageously as a conjugatable residue for attachment to an Nα-protected Glu hub. Indeed, cRGD peptides including a lysine residue such as cyclic Arg-Gly-Asp-Xaa-Lys (where Xaa is D-Phe or D-Tyr) are broadly used as targeting agents for cancer imaging and therapy. Multiple cRGD peptides have been reported to not only enhance the affinity to αvβ3 integrin by multivalent and cluster effects but also prevent rapid washout from a target tumor [93]. In such multiple cRGD strategies, two Lys-containing cRGD peptides can be easily and simultaneously conjugated with a multifunctional molecule between the amine on the side chain of Lys in the cRGD peptide and two carboxylic acids on the Nα-protected-Glu. In this case, a linker between Glu and cRGD peptide is not always necessary because the Lys side chain acts as the linker.
Cysteine can serve as an intrinsically trifunctional hub molecule given the presence of the nucleophilic thiol group in the side chain. The reaction of the thiol group in Cys with maleimide is a highly selective reaction that has been widely employed in the construction of multifunctional molecules [94]. In addition, disulfide bonds have been widely used to develop reduction-responsive drug delivery systems (DDS) for cancer therapy because cancer cells simultaneously overproduce reactive oxygen species (ROS) and glutathione, leading to a redox-heterogeneous intracellular environment [95]. The protection of Cys is essential because the thiol group can undergo or participate in several side reactions (beyond oxidation), including the following: (1) epimerization; (2) reaction with carbocations; and (3) β-elimination [96]. Cys is highly prone to racemize during peptide couplings, the extent of which depends on the protecting groups and coupling methods [97]. When Cys is anchored to a hydroxyl resin (e.g., the Wang resin in SPPS), epimerization can occur during the repetitive base treatments to remove the α-amino 9-fluorenylmethoxycarbonyl (Fmoc) group [98]. Use of the 2-chlorotrityl resin can minimize this side reaction. The most used protecting groups for the Fmoc/tBu strategy are trityl (Trt) groups to obtain the free thiol [99]. Cys can react with highly reactive carbocations during deprotection or cleavage of the peptide from the resin. In the case of deprotection of the tert-butyloxycarbonyl (Boc) and tBu groups, S-tert-butylated Cys has been observed. Concerning cleavage from the resin, a resin-bound carbocation generated during acidolysis can react with unprotected or protected Cys, leading to reattachment of the peptide to the resin [100].
β-Elimination can take place when the protected Cys is exposed to strong basic or acidic conditions, particularly when a peptide containing a C-terminal cysteine is synthesized by the Fmoc/tBu strategy [101]. Base-catalyzed β-elimination of protected Cys (6h) followed by addition of piperidine to the resulting dehydroalanine (6i) can occur during Fmoc deprotection to generate 3-(1-piperidinyl)alanine (6j) (Figure 12) [22]. The sterically hindered trityl protecting group can minimize β-elimination [102]. The aforementioned side reactions illustrate why incorporation of Cys is preferred in later stages of synthesis.
Serine and threonine each have a hydroxyl group in the side chain, and like cysteine, can serve as an intrinsically trifunctional hub molecule. However, they are rarely used as hub molecules because the hydroxyl group is less nucleophilic than that of an amine or thiol, leading to less selective reactions. Chief reactions with the hydroxyl group of Ser or Thr are as follows: (1) ester formation with an activated carboxylic acid, and (2) ether formation with an alkyl halide. The former has severe limitations owing to the electrophilicity of the required acyl halide or active ester [103] and the general limitations of sensitive groups that stem from the reagents required to prepare an acyl chloride, such as thionyl chloride, oxalyl chloride, or phosphorus chloride [104]. As an alternative to the side reactions of acid chlorides (hydrolysis and racemization under basic conditions through the ketene intermediate), acyl fluorides have been developed, which counterintuitively are less sensitive and can give diminished racemization and side reaction problems [105]. One of the most commonly used sets of conditions for the formation of an ester employs a carbodiimide and 4-(N,N-dimethylamino)pyridine (DMAP) [106]. In this reaction the corresponding urea byproduct is generated. In the case of activation by N,N′-dicyclohexylcarbodiimide (DCC), the insoluble N,N′-dicyclohexylurea (DCU) is formed. Analogues such as 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and the urea analogue are both water-soluble, providing advantages for coupling of biomolecules to multifunctional scaffolds. However, some side reaction such as acetyl transfer followed by formation of the unreactive N-acylurea are observed [107]. DMAP reacts faster than the competing acyl transfer and generates an active intermediate to avoid such side reactions [108].
7. Lysine Hubs (21)
Lysine has long been used as a nucleus for elaboration of amide-based architectures. The popularity stems from the availability of lysine in large quantities as well as the established protocols for amidation and functional group protection and deprotection. In this section, 21 distinct examples are reviewed.
An imaging agent (7a) for the bimodal features of PET and fluorescence imaging was developed (Figure 13) [109]. The agent is comprised of a single Lys as a hub and three functional components: (A) an NT-20.3 peptide agonist for targeting the neurotensin receptor 1, which is overexpressed in pancreatic adenocarcinomas and high-grade precancerous lesions; (B) the cyanine dye Cy5 as a fluorophore, and (C) the coordination complex [68Ga]-1,4,7-triazacyclononane,1-glutaric acid-4,7-acetic acid as a PET imaging agent. This construct (7a) has composition ABC and architecture [3][1]3.
A lysine-based heterofunctional cross-linking reagent for bioconjugation of drugs (7b) was developed (Figure 13) [110]. The reagent is comprised of a single Lys as a hub and two functional components: (A) doxorubicin as an anticancer drug, and (B) 2-pyridyl disulfide for thiol-disulfide exchange. This construct (7b) has composition A2B and architecture [3][1]3.
A bivalent small molecule-drug conjugate (7c) directed against carbonic anhydrase IX (CAIX) was developed for the delivery of therapeutic agents into neoplastic masses (Figure 13) [111]. The conjugate is comprised of a single Lys as a hub and three functional components: (A) acetazolamide as a targeting agent for CAIX, which is expressed in many solid tumors; (B) a disulfide bond between Cys and the drug moiety, to be cleaved by glutathione, and (C) DM1 as a potent cytotoxic drug. This construct (7c) has composition A2BC and architecture [3][2][1]3.
A photolabile fluorescently quenched probe (7d) was developed to assess an organelle-targeted light-mediated release strategy for controlling biological activity (Figure 13) [112]. The probe is comprised of a single Lys as a hub and four functional components: (A) a nitrobenzyl derivative as a photo-cleavable linker; (B) an Ac-(Cha-d-Arg-Cha-Lys)2 tetrapeptide (Cha = cyclohexylalanine) as a targeting agent for cell permeable mitochondrial localization; (C) tetramethylrhodamine (TAMRA) as a fluorophore, and (D) the quencher dye QSY-7. This construct (7d) has composition A2BC and architecture [3][2][1]3. The probe (7d) is readily absorbed by mitochondria and subsequently can be cleaved in a light-dependent fashion, leading to subcellular mitochondrial imaging.
A FRET-based theranostic probe (7e) was developed (Figure 13) [113]. The probe is comprised of a single Lys as a hub and four functional components: (A) Arg8 as a cell penetrating peptide; (B) Black Hole Quencher-2 (BHQ-2); (C) the tetrapeptide GFLG, which is cleaved by cathepsin B, and (D) doxorubicin as a fluorophore and anticancer drug. This construct (7e) has composition ABCD and architecture [3][2][1]3. When the probe (which is non-fluorescent prior to cellular entry) is subjected to cellular uptake and cathepsin B cleavage, doxorubicin is released thus resulting in fluorescence while also acting as a therapeutic agent.
Peptidic multifunctional molecules are of growing interest for medicinal applications such as diagnosis, imaging, or therapeutics, in which case efficient and versatile strategies are desired for peptide modification. An element-tagged activity-based photo-cleavable biotinylated chemical “hub” (7f) was prepared for absolute targeted-protease quantification and identification using inductively coupled plasma mass spectrometry (ICP-MS) and electrospray ionization mass spectrometry (ESI-MS) (Figure 14) [114]. The chemical “hub” is comprised of a single azidoLys bearing four functional components: (A) Eu-loaded 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) to produce ICP-MS signals for protein quantification; (B) an o-nitrobenzyl ether carbamate linker as a photocleavable linker; (C) a biotin tag, and (D) sulfonyl fluoride as a labeling agent for the hydroxyl group of the serine residue at the active site of serine proteases. This construct (7f) has composition ABCD and architecture [3][2][1]3. Upon addition of the chemical “hub” to a complex biological sample, targeted serine proteases are tagged due to sulfonyl fluoride, and the tagged proteases can be quantified by ICP-MS. Beyond this quantification, the tagged proteases containing the biotin tag are selectively captured by streptavidin-coated beads, followed by release of the targeted proteases from the beads by photocleavage of the o-nitrobenzyl ether linker upon ultraviolet (UV) irradiation. The released targeted proteases can be identified by ESI-MS.
A quenched ligand-directed tosylate (Q-LDT) reagent (7g) was developed for preparing fluorescently labeled proteins (Figure 14) [115]. The reagent is comprised of a single Lys hub and four functional components: (A) a 4-dimethylaminophenylazobenzene-4-carboxylic acid (Dabcyl) as a quencher; (B) a tosylate linker for protein labeling; (C) 7-diethylaminocoumarin (DEAC) as a fluorophore, and (D) a benzenesulfonamide for targeting carbonic anhydrase II (CAII). This construct (7g) has composition ABCD and architecture [3][2][1]3. Upon binding of the benzenesulfonamide moiety to the CAII, the DEAC is covalently transferred to the CAII through an SN2-type reaction between a nucleophilic amino acid located on the CAII surface and the tosylate-substituted linker. The quencher-tethered benzenesulfonamide remains noncovalently bound to the ligand-binding pocket of the CAII, thereby still allowing efficient FRET quenching of the fluorescence. The fluorescence can then be efficiently turned-on in the presence of the specific analyte ethoxzolamide by expelling the quencher-tethered benzenesulfonamide from the binding pocket of the CAII.
A multifunctional fluorescent probe utilizing a new “On–On” strategy (7h) was developed on the basis of the assembly and disassembly of fluorescein isothiocyanate nanoparticles (FITC-NPs) (Figure 14) [116]. The objective for the probe was for sequential detection of glutathione and caspase-3. The probe is comprised of a single Lys hub and five functional components: (A) a DEVD peptide cleaved by caspase-3; (B) Cys as a cross-linking group with 2-cyanobenzothiazole (CBT); (C) the StBu group as a reduction-cleavable group; (D) CBT as a cross-linking group with Cys, and (E) FITC as a fluorophore. This construct (7h) has composition ABCDE and architecture [3][2]2[1]3. Upon cleavage of the StBu group by glutathione-mediated reduction, the resulting Cys and CBT in the probe were immediately cross-linked. The cross-linking gave rise to assembly into nanoparticles (NPs) in the cells accompanied by moderate fluorescent emission. Subsequent hydrolysis of the peptide substrates on the NPs by intracellular caspase-3 led to enhanced fluorescence.
A branched fluorescent probe (7i) was developed as a model linker for an antibody–drug conjugate (ADC), where the ADC bears multiple payload molecules (Figure 14) [117]. The probe for a FRET assay was comprised of a single Lys hub and three functional components: (A) a Val-Cit dipeptide cleaved by cathepsin B; (B) 2,4-dinitrophenylethylenediamine as a quencher, and (C) Trp as a fluorophore. This construct (7i) has composition A2B2C and architecture [3][2]2[1]3.
Oriana and coworkers designed a multifunctionalized MMP2 FRET probe (7j) built on a peptide scaffold (Figure 14) [118]. The FRET probe is comprised of a single Lys hub and four functional components: (A) a substrate peptide cleaved by MMP2; (B) a PEG tag; (C) a FRET donor, and (D) a FRET acceptor. This construct (7j) has composition ABCD and architecture [3][2][1]3. The PEG tag in the probe improves biocompatibility and increases the half-life in the systemic circulation. In addition, the PEG unit engenders enhanced permeation and retention (known as the EPR effect), which can lead to significant accumulation of PEGylated compound in inflammatory tissues [119]. MMP2 is a zinc-dependent protease with activity related to diseases such as atherosclerosis and cancer [120,121].
The synthesis of 7j is representative of the construction of compounds that rely on use of Lys as a hub, and is outlined here. As an overview, a single Lys as hub was attached at the N-terminus of the substrate peptide, which provided a branch point for attachment of the FRET donor (coumarin). A Cys was attached to the C-terminus of the substrate peptide for attachment of the FRET acceptor. The design enables use of SPPS to create the peptide bearing Lys and Cys at the respective termini. Classical thiol–maleimide Michael addition, coupling using an NHS ester, and α-ketoacid–hydroxylamine (KAHA) ligation were respectively employed for introducing three functional molecules (PEG tag, FRET donor, and FRET acceptor) into the resulting peptide. These steps are expanded upon as follows. KAHA ligation employs an ε-(N-hydroxylamino) Lys and a trifluoroacetyl oxazolone derivative, and in so doing, enables chemoselective and late-stage modification of unprotected peptides [118]. KAHA ligation is reliable and offers the following advantages for constructing peptidic multifunctional molecules: (1) proceeds under mild and aqueous conditions; (2) is applied with an unprotected peptide without formation of noticeable byproducts; and (3) proceeds without a coupling reagent [122,123]. In this study, the KAHA ligation was advantageously used for attaching the FRET donor to the N-terminal lysine of the peptide for the detection of MMP2 activity.
The Rink amide 4-methylbenzhydrylamine (MBHA) resin (7j-1) was used in standard Fmoc SPPS to carry out the synthesis. Prior to the coupling of the Cys residue (to be used for the Michael addition), an Ala residue was loaded onto the resin to prevent the side reaction and racemization derived from the C-terminal Cys residue [124,125]. The desired peptide intermediate (7j-2) bearing the N-terminal Fmoc-protected γ-aminobutyric acid (GABA) was prepared and then extended with the target N-terminal lysine to give fully protected peptide 7j-3 (Figure 15). The amidation process was carried out with O-(1H-6-chlorobenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HCTU) in the presence of N-methylmorpholine (NMM) in the solvent N,N-dimethylformamide (DMF). A key building block was lysine bearing Fmoc protection at the Nα-site and both Boc and OBz protection of the Nε amine. The structure is shown in the insert in Figure 15. The lysine building block enabled selective elaboration at the Nα terminus (attachment of the PEG group), and subsequent elaboration at the Lys Nε site following cleavage of the peptide from the resin. Thus, after attachment of the PEG tag at the N-terminus using the corresponding NHS ester to give resin-bound peptide 7j-4, all of the protecting groups (except the Nε OBz group) and the resin were simultaneously removed under acidic conditions to obtain the peptide 7j-5. In parallel, ε-Boc-protected lysine (H-Lys(Boc)-OH, 7j-6) was treated with trifluoroacetic anhydride (TFAA) to give trifluoroacetyl oxazolone derivative 7j-7, which upon cleavage with trifluoroacetic acid (TFA) gave the free ε-amino lysine; the latter was derivatized with the FRET donor 7-methoxycoumarinyl-4-acetic acid (MCA) to give 7j-8. MCA-bearing trifluoroacetyl oxazolone derivative 7j-8 was initially hydrolyzed under acidic conditions to provide the non-cyclic α-ketoacid in situ, and the peptide 7j-5 was then added to the α-ketoacid solution. The KAHA ligation proceeded in the presence of unprotected functional groups such as the guanidino group and even a free thiol in the reaction mixture containing the reducing agent tris(2-carboxyethyl)phosphine (TCEP) to generate 7j-9 in 15% yield. Finally, the FRET acceptor, dinitrophenylaniline, was incorporated onto the side chain of the Cys residue by Michael addition with DNP maleimide in the presence of dimethylsulfoxide (DMSO) at room temperature (rt) to obtain the FRET probe 7j.
The probe 7j was incubated in the presence of MMP2 in Tris buffer (pH 7.5) at 37 °C, and the corresponding hydrolyzate could then be observed at 200 min upon analysis by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF-MS). Moreover, a significant increase in fluorescence emission derived from hydrolyzed 7j was observed after incubation of 7j with MMP2, while suppressed fluorescence emission was found in the presence of both MMP2 and its inhibitor. In this manner, MMP2 activity in vitro was successfully detected by using the fluorogenic probe 7j.
This study demonstrated that the KAHA ligation between an N-hydroxylamine Lys derivative and a trifluoroacetyl oxazolone derivative is suitable for constructing a multifunctional probe that contains Lys as a hub molecule. The ligation proceeded under mild and aqueous conditions in the presence of free functional groups. Accordingly, this strategy should be applicable for the synthesis of more elaborate probes such as those containing near infrared (NIR) dyes and proteins. In addition, this technique is potentially suitable for constructing multihub molecules.
An integrin α5β1-targeted probe (7k) for single-photon emission computed tomography (SPECT)/computed tomography (CT) imaging [126,127] was developed containing an albumin binder (Figure 16) [128]. The probe is comprised of two hubs (Lys and Glu) and three functional components: (A) the cyclic peptide cyclo[-d-Phg-isoAsp-Gly-Arg-d-Arg-] as a targeting agent for integrin α5β1, which displays high expression associated with glioma (where Phg is phenylglycine); (B) 4-(p-iodophenyl)butyric acid for conjugating with albumin to improve pharmacokinetics in blood, and (C) 99mTc-hydrazine-nicotinamide [tricine/trisodium triphenylphosphine-3,3′,3″-trisulfonate] as a SPECT/CT imaging agent. This construct (7k) has composition A2BC and architecture [3]2[1]4. The probe binds to serum proteins thereby affording prolonged residence time in the circulation and increased proclivity for tumor delivery.
A carboxyesterase (CES)-cleavable biotinylated NIR nanoprobe (7l) was developed for tumor dual-targeted imaging (Figure 16) [129]. The probe is comprised of two hubs (Lys and Ser) and six functional components: (A) biotin as a targeting agent for tumor cells that overexpress the biotin receptor; (B) a Cys for cross-linking with CBT; (C) the StBu group as a reduction-cleavable group; (D) CBT for cross-linking with Cys; (E) an ester bond between the Ser hub and the fluorophore to be cleaved by CES that is overexpressed in some tumor cells, and (F) the pentamethine cyanine dye Cy5.5 as a fluorophore. This construct (7l) has composition ABCDEF and architecture [3]2[2]2[1]4. Upon cleavage of the StBu group by glutathione-mediated reduction, the resulting Cys and CBT in the probe immediately cross-linked to yield biotinylated and fluorescence-quenched nanoparticles (NPs). The NPs target the biotin receptor-overexpressing tumor cells, and following translocation the fluorophore Cy5.5 is subsequently cleaved by intracellular CES, turning the NIR fluorescence signal “On”. The probe 7l has some functional similarity to 7h but the fluorophore is different, as is the molecular architecture.
A FRET-based theranostic probe (7m) was developed for simultaneous tumor targeting, PDT, and ratiometric imaging of the therapeutic effect (Figure 16) [130]. The probe is comprised of two hubs (both Lys) and five functional components: (A) the peptide Arg-Gly-Asp (RGD) as a targeting agent for the αvβ3 integrin, which is overexpressed in tumor cells; (B) the peptide DEVD, which is cleaved by caspase-3; (C) the quencher Dabcyl; (D) the fluorophore 5(6)-carboxyfluorescein; and (E) protoporphyrin IX (PpIX) as a photosensitizer and fluorophore (for use an internal fluorescence reference). This construct (7m) has composition ABCDE and architecture [3]2[2][1]4. The probe accumulated by RGD-mediated endocytosis in tumor cells where αvβ3 integrin was overexpressed. Upon light irradiation, ROS are generated via PpIX and activate apoptosis-related caspases, which cleave the DEVD peptide moiety in the probe. Thus, the turn-on of fluorescein fluorescence achieves ratiometric imaging of apoptosis.
A protein caging agent (7n) was developed that utilizes active-site recognition for propinquity labeling of a nonactive-site residue, thereby affording a light-activatable profluorescent protein (Figure 16) [131]. The agent is comprised of two hubs (Lys and Cys) and five functional components: (A) a 4-[4-(1-aminoethyl)-2-methoxy-5-nitrophenoxy] moiety as a photocleavable linker; (B) the peptide Ac-GRTGRRNAIHD (a protein kinase inhibitor, PKI) as a targeting agent for the active site of an adenosine monophosphate (AMP)-dependent protein kinase (PKA); (C) TAMRA as a fluorophore; (D) QSY-7 as a quencher, and (E) maleimide as a conjugatable group for the Cys residue. This construct (7n) has composition ABCDE and architecture [3]2[2][1]4. The agent inactivates the targeted enzyme PKA because the PKI peptide cages the Cys-199 at the base of the active site of PKA. The other proximate inactive site (Cys-343) of PKA is reacted with the maleimide to provide the photoactivatable profluorescent PKA. Photolysis of the photocleavable linker of the caging agent on covalently modified inactive PKA both restores enzymatic activity and simultaneously generates a fluorescent PKA (via release of the PKI on the active site and the appended quencher moiety).
A dual quenching probe (7o) was developed to detect intracellular furin activity with enhanced sensitivity (Figure 17) [132]. The probe was comprised of two hubs (both Lys) and six functional components: (A) a Cys for cross-linking with CBT; (B) an StBu as a reduction-cleavable group; (C) CBT for cross-linking with Cys; (D) a peptide substrate cleaved by furin; (E) the quencher Dabcyl, and (F) the fluorophore FITC. This construct (7o) has composition ABCDEF and architecture [3]2[2]2[1]4. Upon cleavage of the StBu group by glutathione-mediated reduction, the resulting Cys and CBT cross-link and self-assemble into dual quenched nanoparticles (NPs) in the cells. Subsequent hydrolysis of the peptide substrates on the NPs by the intracellular enzyme furin, which is a protease that is overexpressed in tumors, led to turning on the fluorescence with a high signal-to-noise ratio.
A paclitaxel/galactose-functionalized compound (7p) was developed to create fluorescent nanoparticles (NPs) for real-time imaging and cancer-cell targeting (Figure 17) [133]. The compound is comprised of two hubs (one Lys and one N,N-bis(carboxymethyl)-lysine) and three functional components: (A) galactose as a targeting agent for carbohydrate-binding proteins on the cell surface; (B) an Fmoc-protected amine as a conjugatable group, and (C) paclitaxel as an anticancer drug. This construct (7p) has composition A3BC and architecture [4][3][1]5.
A dendritic multivalent galactosyl carborane (7q) was developed as a potential cell-targeting agent in boron neutron capture therapy (BNCT) (Figure 17) [134]. The carborane derivative is comprised of double N,N-bis(carboxymethyl)lysines as hubs and two functional components: (A) galactose as a targeting agent for carbohydrate-binding proteins on cell surfaces, and (B) o-carborane for BNCT. This construct (7q) has composition A3B3 and architecture [4]2[1]6.
A cross-linkable molecular architecture (7r) was developed for possible in vivo immobilization and enzyme attachment in therapeutic applications (Figure 17) [135]. The molecular architecture is comprised of two hubs (two Lys) and four functional components: (A) indoxyl as a cross-linking agent; (B) glucose, to be cleaved by β-glucosidase thereby enabling enzymatically triggerable cross-linking; (C) the carbacephem antibiotic Loracarbef as a covalent labeling agent for a mutant β-lactamase, which as part of a fusion protein can bear another molecule (e.g., an enzyme) to elaborate the molecular function, and (D) maleimide for conjugation with a thiol group. This construct (7r) has composition A2B2CD and architecture [3]2[2]2[1]4. Upon cleavage of glucose by β-glucosidase, the resulting indoxyl moieties undergo spontaneous oxidative dimerization to form the corresponding indigoid product, which is insoluble and precipitates at or near the site of generation. This molecular scaffold, termed a “platform” or “nano-platform,” can be further functionalized via the maleimide and Loracarbef moieties.
The synthesis of 7r began with commercially available 5-benzyloxyindoxyl-3-carboxaldehyde (7r-1). Treatment with acetic anhydride afforded N-acetylation to give 7r-2, which upon Baeyer–Villiger oxidation (12-g scale) gave 7r-3. Glycosidation with acetobromo-α-d-glucose (7r-4) gave 7r-5 in 65% yield; hydrogenolysis removed the benzyl (Bn) group while leaving the N-acetyl group intact, affording a dihydroindole byproduct (7r-6) and the desired 7r-7 in 89% yield, albeit as an 8:1 mixture of β/α isomers (Figure 18).
Alkylation of 7r-7 (epimeric mixture) with benzyl α-bromoacetate (7r-8) enabled isolation of indoxyl-glycoside 7r-9 in stereoisomerically pure form (Figure 19). Subsequent hydrogenolysis to remove the benzyl group gave the indoxyl-glucoside bearing the free carboxylic acid (7r-10). DCC-mediated coupling of the latter with O-benzyl lysine (7r-11), which bears two free amines, gave the valuable, benzyl-protected bis(indoxyl-glucoside) 7r-12. Hydrogenolysis again liberated the free carboxylic acid, giving 7r-13.
DCC-mediated coupling of 7r-13 with α-Fmoc-O-benzyl lysine (7r-14) gave 7r-15, which upon removal of the Fmoc group with piperidine gave free amine 7r-16. Hydrogenolysis of the latter gave the amino acid 7r-17, and treatment with sodium methoxide caused global removal of the acetyl protecting groups from the glucoside (O-acetyl) and the indoxyl (N-acetyl) units to give 7r-18, containing the free amine and carboxylate groups (Figure 19).
Loracarbef provides an irreversible attachment motif (i.e., suicide linker) with a specially designed β-lactamase enzyme. Installation of the Loracarbef moiety required a protecting group for the Loracarbef 2-carboxylic acid group, taking into account the notoriously fickle nature of β-lactam chemistry. Thus, reaction of Loracarbef with Boc-anhydride gave the protected amine, which upon reaction with iodomethyl phenylacetate (7r-19) gave the doubly protected Loracarbef 7r-20 (Figure 20). Removal of the Boc group with TFA followed by an active ester of adipic acid (containing one NHS ester and one benzyl ester, 7r-21) afforded 7r-22, wherein Loracarbef bears an N-adipoyl tether. Hydrogenolysis of the benzyl group from the adipoyl tether followed by DCC-mediated coupling with NHS gave the active ester, 7r-23.
Bis(indoxyl-glucoside) 7r-18 was acidified to convert the carboxylate to the free acid, which upon reaction with Loracarbef derivative 7r-23 gave 7r-24. DCC-mediated activation of the latter with N-hydroxysuccinimide followed by reaction with N-(2-aminoethyl)maleimide gave the target compound 7r. Compound 7r (300 mg) contains two indoxyl-glucosides, a phenylacetoxymethyl-protected Loracarbef unit, and a maleimido unit for attachment to a biomolecule (Figure 20).
The synthesis of 7r required 17 steps beginning with commercially available reactants (Figure 20). The final structure contains 7 amide bonds: 6 as part of the peptide scaffold, and 1 as part of the Loracarbef structure. Loracarbef together with the mutant β-lactamase (not shown here) provides a new type of “click-chemistry”.
Fundamentally new strategies are needed to address cancer, particularly metastatic cancer, for metastasis is the source of the lethality of cancer. The cellular heterogeneity that underlies metastasis also stymies the efficacy of surgery, chemotherapy, and immunotherapy. Radiotherapy offers indiscriminate cell killing, which is attractive if targeted, but external beam radiotherapy causes too much collateral damage for use against metastases of unknown location. The ability to construct scaffolds in vivo may ultimately enable the localization of radiotherapeutic agents, affording a molecular brachytherapy [136], a theme which to date has been little explored.
A peptide-targeted Gd(III)-DOTA conjugate (7s) was developed for molecular imaging of prostate cancer (Figure 21) [137]. The conjugate is comprised of three hubs (all Lys) and two functional components: (A) Gd(III)-DOTA as a magnetic resonance imaging (MRI) agent, and (B) a cyclic CGLIIQKNEC peptide (CLT1) as a targeting agent for the clotted plasma proteins (fibrin–fibronectin complexes) that form in tumor stroma. Four Gd(III)-DOTA units are present. This construct (7s) has composition A4B and architecture [3]3[1]5.
A tris(carbohydrate) probe (7t) was developed for detecting cellular lectins (Figure 21) [138]. The probe is comprised of three hubs (two Lys, one Ser) and three functional components: (A) mannose as a targeting agent for the cell-surface lectins, (B) benzophenone (BP) as a PAL agent, and (C) the pentamethine cyanine dye Cy5 as a fluorophore. This construct (7t) has composition A3BC and architecture [3]3[1]5.
A driving force in the development of scaffolds has been the desire to display an array of carbohydrates in defined architectures for use in the life sciences. One approach creates dendrimers, but as outlined above, the desire to achieve greater specificity and functionality requires the modular synthetic amenability of scaffolds, which afford the capacity for irregular albeit rational derivatization. A chief rationale for creating arrays of carbohydrates is that interactions between carbohydrate ligands and carbohydrate-binding proteins play a key role in a broad range of biological and pathological processes. Such processes include fertilization, implantation, morphogenesis, development, differentiation, the immune response, cell migration, and cancer metastasis [139]. For example, such interactions initiate the first contact between pathogens (e.g., viruses and bacteria) and target cells during the infection process. While monovalent protein–glycan interactions show intrinsically weak affinity, multivalent and clustered glycans are displayed on the surface of the pathogens to enhance the binding affinity toward multimeric glycan-binding proteins on target cell surfaces such as lectin, which ultimately leads to infection [140]. This effect is termed “multivalency” or the “cluster-glycoside effect,” which is known to enhance overall ligand affinity and selectivity in lectin–carbohydrate interactions [141]. Hence, multivalent interactions have been a point of focus in the development of novel antimicrobial and antiviral interventions, and various heteroglycoclusters (hGCs), which imitate the heterogeneous expression of glycans at the cell surface, namely glycocalyx, have been synthesized [142,143]. Multivalent glyco(cyclo)peptides are currently used as ligands for carbohydrate-binding proteins due to the significant advantages mainly derived from cyclic peptide scaffolds as follows: (1) well-established synthetic procedures and extendability with regards to size and functionality; (2) imitability as native glycoprotein fragments; (3) conformational constraints and rigidity, and (4) non-immunogenicity and resistance against proteolytic degradation [144,145,146].
Toward these aims, a heteroglycocluster using a pentavalent cyclic decapeptide scaffold (7u) was created, which specifically is a fully synthetic multiantigenic vaccine candidate against tumors (Figure 21) [147]. The hGC in this study is comprised of a cyclic decapeptide scaffold containing five lysine hubs that respectively bear five distinct functional components: (A) a free amine group as a conjugatable group and (B–E) distinct carbohydrate motifs as targeting agents. This construct (7u) has composition ABCDE and architecture c-[3]5[1]5. The cyclic peptide scaffold displayed five functional groups (glyoxaldehyde, chloroacetyl, azide, alkene, and amine) by using five Lys residues, which allowed for bioorthogonal ligation of the five distinct functional units. The sophistication and elegance of the carbohydrate-appended cyclic decapeptide 7u is matched by the synthesis, which is described as follows.
Standard Fmoc SPPS using a super-acid sensitive resin (SASRIN) (7u-1) was employed to prepare a linear decapeptide (7u-2). The use of SASRIN affords facile cleavage under weak acidic conditions, thereby affording the protected peptide fragment 7u-3 (Figure 22). The decapeptide 7u-3 bears five Lys with orthogonal derivatization of the ε-amino group by use of cleavable protecting groups, the azide unit (which is carried through to the stage of carbohydrate attachment), and serine derivatization. The cleavable protecting groups include Boc, 1-(4,4-dimethyl-2,6-dioxocyclohexylidene)ethyl (Dde), and allyloxycarbonyl (Alloc). Head-to-tail cyclization of 7u-3 using the condensing agent benzotriazol-1-yltripyrrolidinophosphonium hexafluorophosphate (PyBOP) in DMF containing diisopropylethylamine (i-Pr2EtN, DIEA) afforded 7u-4, which was treated with hydrazine to remove the Dde group, followed by chloroacetylation of the resulting free amine to afford 7u-5.
An acidic deprotection cocktail comprised of TFA, triisopropylsilane (TIS), and water was then used to remove the Boc group of the Lys residue and the tBu group of the Ser residue. The resulting free Ser was then oxidized by sodium periodate to give the pentavalent scaffold 7u-6. The mild oxidation of N-terminal free Ser and Thr residues by periodate is known for preparing the corresponding aldehyde (glyoxylyl group), which can then be used for bioconjugation via oxime and hydrazone ligation (Figure 22) [148,149]. Scaffold 7u-6 bears a terminal alkene, an azide, an aldehyde, a chloroacetamide, and a primary amine (isolated as the TFA salt).
Scaffold 7u-6 was elaborated by sequential derivatization. Because the glyoxaldehyde is the most sensitive of the five functional groups, scaffold 7u-6 was initially reacted with a hydroxylamine derivative by oxime ligation [150]. Thus, β-glucosyl hydroxylamine 7u-7 was conjugated to 7u-6 under acidic conditions to obtain 7u-8 in 87% yield (Figure 23). Considering the chemical stability of the remaining four functional groups on the scaffold, photo-induced thiol–ene coupling, Cu-catalyzed click chemistry, and thiol–chloroacetyl coupling were carried out in succession. The N-acetylglucosamine thiol 7u-9 was reacted with the alkene 7u-8 in the presence of 2,2-dimethoxy-2-phenylacetophenone (DMPA) as a photoinitiator under UV irradiation (365 nm) to afford 7u-10 in 67% yield. To avoid a side reaction between 9 and the chloroacetyl group of 7u-8, the reaction was carried out for 30 min. Next, α-GalNAc propargyl 7u-11 underwent click reaction with 7u-10 in the presence of CuSO4, sodium ascorbate, and tris(3-hydroxypropyltriazolylmethyl)amine (THPTA) as a copper(I) stabilizing ligand in DMF and phosphate buffered saline (PBS) to obtain 7u-12 in 85% yield. Finally, α-Man thiol 7u-13 was reacted with the chloroacetyl group of 7u-12 in the presence of KI and DIEA to provide the heterofunctionalized glycosylated scaffold 7u in 84% yield. All compounds 7u-6, 7u-8, 7u-10, 7u-12, and 7u were handled as the TFA salt of the primary amine. The overall yield for the attachment of the carbohydrates was 42%.
Alternatively, sequential one-flask attachment of the carbohydrates was executed in the same order and under similar conditions with simple adjustments of both pH and solvent in the crude mixture. The reactivity and reaction efficiency were not dependent on the concentration used, and similar reaction times to the stepwise procedures were needed for complete conjugation via the sequential one-flask method. Final purification by reversed-phase high-performance liquid chromatography provided 7u in 47% overall yield, which showed that conjugation of the carbohydrate by a sequential one-flask process was somewhat preferable to the stepwise strategy.
This study demonstrated the utility of the pentavalent cyclic decapeptide scaffold for synthesizing a heterofunctionalized glycosylated architecture. The peptide scaffold 7u-6 was smoothly obtained by standard peptide synthesis methods, and four different carbohydrates could be conjugated to the cyclopeptide scaffold by sequential distinctive bioorthogonal ligations in a one-flask process with high efficiency. The hGC 7u may be useful for probing biologically significant multivalent protein–glycan interactions and exploring fully synthetic multi-antigenic synthetic vaccine candidates in a range of medical therapies [151]. In a display of expansive scope, a range of biologically relevant structures could be bound to the remaining conjugatable Lys side chain of 7u upon liberation of the amine from the TFA salt. The structures include biotin (7u-14), FITC (7u-15), poliovirus peptide fragment (7u-16), and a CpG oligodeoxynucleotide (7u-17) (Figure 24). In short, this synthetic strategy can be applied to the construction of various multifunctional molecules, affording modularity, versatility, and compatibility with not only biomolecules but also fluorescent dyes.
8. Other Amino Acid Hubs (17)
A fluorescence probe-labeled photoactivatable antiplasmodial naphthylisoquinoline (NIQ) derivative (8a) was developed for identifying possible molecular targets of NIQ within the malarial parasite Plasmodium falciparum (Figure 25) [152]. The derivative is comprised of a single Tyr hub and three functional components: (A) 5-(dimethylamino)naphthalene-1-sulfonyl (Dansyl) as a fluorophore; (B) BP as a PAL agent, and (C) dioncophylline A as a targeting agent for a protein in the malarial parasite. This construct (8a) has composition ABC and architecture [3][1]3.
A dual integrin and gastrin-releasing peptide receptor (GRPR)-binding heterodimeric peptide probe (8b) was developed for tumor imaging (Figure 25) [153]. The probe is comprised of one Glu hub and three functional components: (A) a bombesin peptide (BBN) as a targeting agent for GRPR, which is massively overexpressed in several human tumors; (B) the cyclic peptide cyclo(-Arg-Gly-Asp-d-Tyr-Lys-) as a targeting agent for integrin αvβ3, which is highly expressed on invasive tumors, and (C) a 4-18F-fluorobenzoate as a PET imaging agent. This construct (8b) has composition ABC and architecture [3][1]3.
A fluorophore-labeled cell-permeable peptide (8c) was developed that contains both lipophilic and positively charged side chains (Figure 25) [154]. The peptide is comprised of a single Ser as the hub and four functional components: (A) Lys3 provides a positively charged motif for cell permeation; (B) an ester bond to be cleaved by intracellular esterases and lipases; (C) a myristyl group as a hydrophobic tag for cell permeation and a lipid tag for lipase recognition, and (D) the fluorophore FITC. This construct (8c) has composition ABCD and architecture [3][2][1]3. The peptide displayed cellular uptake owing to the Lys3 tag and myristyl group, and the myristyl moiety was readily released on cellular internalization.
A small molecule probe (8d) was developed for tumor imaging (Figure 25) [155]. The probe is comprised of a single Glu hub and two functional components: (A) the cyclic peptide cyclo(-Arg-Gly-Asp-d-Phe-Lys-) as a targeting agent for integrin αvβ3, which is highly expressed on many tumor cells, new vessels, and tumor stromal cells, and (B) 64Cu-pyropheophorbide a as a PET imaging agent. This construct (8d) has composition A2B and architecture [3][1]3.
A trifunctional probe (8e) was developed that bears a carbene-generating photoaffinity probe, a biotin tag, and the antibiotic moenomycin A (Figure 25) [156]. The probe is comprised of single isoSer hub and three functional components: (A) aryl(trifluoromethyl)diazirine as the PAL agent; (B) biotin, and (C) moenomycin A as a targeting agent for the penicillin-binding protein 1b on bacterial cells. This construct (8e) has composition ABC and architecture [3][1]3.
An Asn-Gly-Arg (NGR)-based imaging probe (8f) was developed for noninvasive visualization of angiogenesis (Figure 26) [157]. The probe is comprised of single Cys hub and three functional components: (A) the cyclic peptide CNGRC as a targeting agent for aminopeptidase N (CD13), which is selectively expressed in angiogenic blood vessels; (B) biotin for immobilizing the probe on a carrier such as a nanoparticle (NP) via the avidin–biotin binding system, and (C) Gd(III)-diethylenetriamine pentaacetic acid as an MRI agent. This construct (8f) has composition ABC and architecture [3][1]3.
The challenge of exercising multimodal imaging has been a significant driving force for the development of scaffold molecules. The individual modalities include absorption/fluorescence, photoacoustics, positron emission, magnetic resonance, and beta or gamma emission, to name a few. For example, dual-modality optical/PET imaging probes that target a specific biomarker are considered as powerful tools for disease diagnosis and monitoring, and can enable improved therapy [11,158]. PET imaging allows for sensitive in vivo imaging without penetration limitations. On the other hand, optical imaging enables correlated fluorescent analysis such as fluorescent staining, immunohistochemical staining, and flow cytometry. Compared with the respective use of monomodality optical probes and PET probes, dual-modality optical/PET imaging probes intrinsically provide a better correlation between fluorescence and PET imaging experiments when the biological effect caused by the structural difference between mono-modality optical probes and the corresponding PET probes is not negligible [159]. On the other hand, multimodal imaging may well reside predominantly in the research domain, because clinical imaging, at least with the present single-modality instruments, would require use of multiple examinations of the patient, which is burdensome and expensive. While the latter considerations are very real at present, advances in instrumentation will require companion advances in the sophistication of the probes.
A dual-labeled cRGD peptide probe (8g) was developed for PET and optical imaging (Figure 26) [160]. The probe is comprised of a single Cys hub and three functional components: (A) cyclo(-Arg-Gly-Asp-d-Tyr-Lys-) as a targeting agent for integrin αvβ3 (which is expressed in a number of tumor types); (B) ZW800-1 as a fluorophore, and (C) 64Cu-DOTA as a PET imaging agent. This construct (8g) has composition ABC and architecture [3][1]3.
Dual-modality optical/PET imaging probes built around small peptides possess some desirable properties: (1) fast body clearance to reduce radio dose exposure time, and (2) easy-to-perform quality control to minimize batch-to-batch differences. However, the preparation often requires complicated multistep synthesis, use of orthogonal protecting groups, and chemoselective/bioorthogonal reactions, which restricts use in both research and clinical studies. A dual optical/PET peptide probe (8h) was developed for application to diagnosis and image-guided surgery (Figure 26) [161]. The probe is comprised of a single Cys hub and four functional components: (A) the cyclic peptide cyclo(-Arg-Gly-Asp-d-Phe-Lys-) as a tumor targeting peptide; (B) a 1,4-diazepine scaffold (AAZTA, 1,4-bis(carboxymethyl)-6-[bis(carboxymethyl)]amino-6-methylperhydro-1,4-diazepine) as a PET imaging reporter, and (C) the pentamethine cyanine dye Cy5.5 for optical imaging. This construct (8h) has composition ABC and architecture [3][1]3. The Cys-cyclo(-Arg-Gly-Asp-d-Phe-Lys-) unit was synthesized by SPPS and then conjugated to an AAZTA thioester by native chemical ligation, followed by Michael addition of a Cy5.5-maleimide to the remaining free thiol.
The process of native chemical ligation (NCL) [162,163], which entails reaction between a thioester moiety and N-terminal cysteine peptide to yield an amide, is well-established and enables the synthesis of long peptides and proteins. The approach also enables chemical labeling of N-terminal cysteine residues in peptides (Figure 27). The advantages of NCL are as follows: (1) the joining reaction proceeds at physiological pH under mild and aqueous conditions without any additives; (2) no additional protecting groups are required for any natural amino acids in the peptide chain, and (3) internal cysteine residues do not interfere with the reaction [164].
The cyclic RGD peptide unit of 8h was prepared by an on-resin cyclization technique. Thus, Wang ChemMatrix resin (8h-1) was initially attached onto the side chain of Boc-Asp-All (8h-2) (Figure 27), where All indicates the allyl group. Standard Fmoc SPPS was performed to obtain a fully protected linear RGD peptide (8h-3), whereupon the N-terminal Fmoc and C-terminal All groups were selectively deprotected. The coupling chemistry was carried out using N,N-diisopropylcarbodiimide (DIC) or O-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU). After coupling, any unreacted amines can be capped by treatment with acetic anhydride in N-methyl-2-pyrrolidine (NMP) in the presence of DIEA, then the Fmoc group is removed upon treatment with piperidine to begin a new cycle. The N-and C-terminal free peptide (8h-4) was cyclized on-resin through the use of PyBOP as a coupling reagent and DIEA in DMF at room temperature to yield the fully protected cyclic RGD peptide (8h-5). After selective deprotection of the 4-methyltrityl (Mtt) group on the side chain of Lys, the hub molecule Boc-Cys(Trt)-OH was linked to the site to give peptide 8h-6. Cleavage from the resin and global deprotection under acidic conditions gave the N-terminal cysteine peptide 8h-7 in 10% overall yield. Peptide 8h-7 constitutes one component for use in NCL.
Next, AAZTA precursor 8h-8 was activated as a thioester with sodium 2-mercaptoethanesulfonate (8h-9) and subsequent global deprotection in 38% yield to give AAZTA thioester 8h-10 (Figure 27). The thioester 8h-10 and N-terminal cysteine peptide 7 were conjugated by NCL to provide 8h-11 in 32% yield. The ligation was carried out in 0.2 M phosphate buffer (pH 7.4) including 5 M imidazole, 3 M guanidinium chloride, and 2 mM TCEP for 30 min at room temperature. Guanidinium chloride improves the solubility of the starting materials, and imidazole was used instead of a thiol (e.g., 4-mercaptophenylacetic acid) as a catalyst. The reducing agent TCEP inhibited formation of disulfide bonds, which can hinder the reaction. The optical reporter, Cy5.5-maleimide, was conjugated via Michael addition to the side chain of Cys in 8h-11, followed by treatment of 8h-12 with GaCl3 to obtain the optical/PET probe 8h.
Human glioblastoma U-87 MG cells, which overexpress αvβ3 integrin (that in turn can bind to the cyclic RGD peptide), were incubated in a medium containing different concentrations of the probe 8h-12 for 30 min at 0 °C. Flow cytometry experiments demonstrated that the probe 8h-12 bound, in a dose-dependent fashion, to the U-87 MG cells via the integrin receptor. This study demonstrated that NCL is a potent strategy for constructing a peptide-based dual-modal probe containing a fluorophore and a chelator. In an in vivo cell assay, the dual modality peptide probe 8h-12 showed affinity for the targeting molecule. The probe could be labeled with 68Ga. As such, the 68Ga-labelled probe 8h is a candidate for optical/PET imaging in diagnostic procedures and perhaps in image-guided surgery [165].
A cross-linking reagent with an isotope tag (8i) was developed for exploring protein interactions (Figure 28) [166]. The reagent is comprised of a single 2,3-diaminopropionic acid hub and four functional components: (A) BP as a PAL agent; (B) a deuterium-containing linker for protein identification via mass spectrometry; (C) a biotin tag, and (D) an ester of sulfo-N-hydroxysuccinimide (sulfo-NHS) for conjugation of a targeting molecule. This construct (8i) has composition ABCD and architecture [3][2][1]3.
A cleavable doxorubicin–octreotide macrocycle (8j) was developed for targeting overexpressed somatostatin receptors on tumor cells (Figure 28) [167]. The macrocycle is comprised of a single Glu hub and three functional components: (A) two disulfide linkers to be cleaved by intracellular glutathione; (B) doxorubicin as an anticancer drug, and (C) the peptide octreotide as a targeting agent for somatostatin receptor subtypes 2 and 5, which are highly expressed on various tumor cells and primary tumor tissues. This construct (8j) has composition A2BC and architecture [3][2]3[1]. The cyclic architecture is achieved by attachment of two aminoethylmercaptan units to the lysine hub. The octreotide bears two cysteines. Double disulfide bond formation between the dithiol-substituted lysine and the octreotide-bis(cysteine) gave the macrocycle.
A construct that contains two distinct enzymatically activated prodrugs (8k) was developed wherein the two prodrugs are anticancer agents (Figure 28) [168]. The construct is comprised of a single Glu hub and five functional components: (A) a Phe-Lys dipeptide to be cleaved by cathepsin B; (B) a p-aminobenzyloxycarbonyl spacer as a self-immolative linker; (C) paclitaxel as one anticancer drug; (D) a maleimide for conjugation to Cys or other thiol, and (E) doxorubicin as a second anticancer drug. This construct (8k) has composition A2B2CDE and architecture [3][2]4[1]3.
An optical imaging probe (8l) was developed that uses a chemically reactive azacyanine dye and is targeted for the integrin αvβ3 receptor (Figure 29) [169]. The probe is comprised of two Glu hubs and two types of functional components: (A) four copies of the cyclic peptide cyclo(-Arg-Gly-Asp-d-Phe-Lys-) as a targeting agent for integrin αvβ3, and (B) an azacyanine dye as a fluorophore. This construct (8l) has composition A4B and architecture [3]2[2][1]4.
A boron probe for total and specific molecular imaging probe (8m) was developed that relies on non-optical nanoscale secondary ion mass spectrometry (nanoSIMS [170]) (Figure 29) [171]. The probe is comprised of two hubs (Lys and Cys) and three functional components: (A) an o-carborane unit, which exhibits a strong signal in nanoSIMS; (B) Star635 as a fluorophore, and (C) a maleimide for conjugation with a Cys residue. This construct (8m) has composition A2BC and architecture [3]2[1]4. The carborane probe is initially coupled with an ectopic cysteine of a nanobody through a standard thiol–maleimide reaction. The probe-nanobody conjugate is then bound to target proteins on the cells via their epitopes to achieve cellular immunostaining, followed by nanoSIMS imaging of specific target proteins.
A photoaffinity multivalent glycoprobe (8n) was developed for the covalent capture of labile protein complexes via carbohydrate-mediated interactions (Figure 30) [172]. The glycoprobe contains two hubs, a single Glu and a 4-arm carbon unit, and three functional components: (A) galactose as a targeting agent for affinity binding to a lectin; (B) (trifluoromethyl)phenyldiazirine as a PAL agent, and (C) 3-fluorocyclooctyne as a conjugatable group for late-stage functionalization by strain-promoted [3 + 2]-azide–alkyne cycloaddition [173] (SPAAC). This construct (8n) has composition A3BC and architecture [4][3][1]5.
An amphiphilic biotin conjugate (8o) was developed for use in a targeted, triggered liposomal delivery system (Figure 30) [174]. The conjugate is comprised of two hubs, a single Ser and a three-arm carbon unit, and five functional components: (A) an octadecyl group; (B) a dansyl fluorophore; (C) a biotin tag; (D) a disulfide linker as a reduction-cleavable group, and (E) a quaternary ammonium salt as a water-solubilizing linker. This construct (8o) has composition A2BCDE and architecture [3]2[2]2[1]4.
A NIR fluorescent probe bearing tetrameric RGD peptides (8p) was developed for optical tumor imaging (Figure 31) [175]. The probe is comprised of three Glu hubs and two functional components: (A) the cyclic peptide cyclo(-Arg-Gly-Asp-d-Tyr-Lys-) as a targeting agent for integrin αvβ3, which is significantly up-regulated in activated endothelial cells and fast-growing solid tumor cells, and (B) the pentamethine cyanine dye Cy5.5 as a fluorophore. This construct (8p) has composition A4B and architecture [3]3[1]5.
A fully synthetic, pentavalent carbohydrate-based system (8q) was developed as an antitumor vaccine (Figure 31) [176]. The vaccine contains four 6-hydroxy norleucine (Nle(6-OH)) hubs and six functional components, of which five are antigens: (A) a Tn antigen; (B) a TF antigen; (C) a sialosyl-Tn antigen; (D) the Lewisy antigen, and (E) the Globo-H antigen, each of which is overexpressed on prostate and breast cancer cell surfaces. The sixth functional component, (F), is keyhole limpet hemocyanin [8] as an immunogenic carrier protein. This construct (8q) has composition ABCDEF and architecture [3]4[1]6.
9. Triazine Hubs (16)
The heterocycle 1,3,5-triazine has been widely used as a versatile scaffold for synthetic reagents, organic materials, bioactive compounds, and complex molecular architectures [177,178,179]. The popularity stems because the common synthetic precursor of these triazine compounds, 2,4,6-trichloro-1,3,5-triazine (cyanuric chloride, Figure 32A) [180], is inexpensive and capable of selective derivatization. The three chlorine atoms in cyanuric chloride can be substituted stepwise by O-, N-, S- and C-nucleophiles to afford a variety of 2,4,6-trisubstituted triazines. The triazine core provides a compact platform for assembly of three functional units. The stepwise substitution of cyanuric chloride is chiefly controlled by electronic effects independently of steric effects.
The reactivity of chloro-, alkoxy-, and amino-substituted triazines has been reported to be similar to that of acyl halides, esters, and amides, respectively [181]. On the other hand, to interpret reactivity, a trialkoxytriazine can be regarded as a formal trimer of the corresponding alkyl imidate [182]. In an extension of this analogy, cyanuric chloride can be considered as an electronically interactive trimer of carbonyl chlorides (Figure 32B). Substitution of the chlorine atom at one of the “carbonyl” groups causes electronic perturbation to affect (typically decrease) the reactivity of the others (Figure 32C). This progressive deactivation enables the stepwise substitution of the chlorines, which would not be expected among three isolated and identical carbonyl chlorides. Substitution of one chlorine generally does not sterically impede the reactivity of the others due to the 120° orientation angle; in other words, substituents at the “carbonyl” groups are spatially separated from each other. Thus, the steric demand of the first, the second, and the third chlorine substitution reactions at the triazine ring is nearly constant.
The substitution of cyanuric chloride, regardless of the nature of the nucleophile, results in displacement of chloride. For neutral nucleophiles, displacement liberates HCl. Accordingly, the nucleophilic displacement is typically carried out in the presence of a base. Over the years, a number of bases have been employed, of which the following organic amine bases are common: 1,10-phenanthroline, 2,6-lutidine, 2,4,6-collidine, triethylamine, pempidine (1,2,2,6,6-pentamethylpiperidine), and N,N-diisopropylethylamine. The base strengths range > 106-fold on the basis of the pKa value of the conjugate acid in water (vide infra). The roles and tactical implementation of the bases are described below.
A further distinctive feature of triazine chemistry resides in the nature of the nucleophiles. Bulky nucleophiles often react with chlorotriazines rather easily, unlike the typical profiles of nucleophiles in the nucleophilic aromatic substitution of benzene derivatives such as halonitrobenzenes. A plausible explanation for this is the absence of hydrogen atoms ortho to the reaction site that cause steric hindrance. For example, tertiary amines such as NMM and Et3N readily react with sufficiently reactive chlorotriazines to form the corresponding N-triazinylammonium salts, which undergo further substitution at the triazine or decomposition to (N,N-dialkylamino)triazines by N-dealkylation [183,184]. The role of tertiary amines as a nucleophilic catalyst for triazine substitution has been studied in the development of triazine-based condensing reagents [185].
In 1948, Moffatt and coworkers reported temperature-controlled, sequential substitution of cyanuric chlorides using alcohols, anilines, and amines as nucleophiles [186]. A general rule for the cyanuric chloride substitution (now known as Moffatt′s rule) was proffered: “In general, the first chlorine atom is replaced at 0 °C, the second at room temperature, and the third at 120–140 °C.” Although this statement highlights the tendency of cyanuric chloride substitution, the actual reaction rates depend on the reaction conditions, nucleophiles, and the substituents on the triazine. Thus, Moffat’s rule should be regarded as more of a heuristic than an irrefragable truism.
Kinetic experiments were performed to study substitution effects of chlorotriazines (Table 1) [187,188]. The reaction rate of chlorine substitution decreased upon introduction of substituents with low σm values. For example, 9a-1-Cl (Entry 1, Reaction A) reacted with benzylamine 150 or 17,000 times faster than 9a-1-OMe (Entry 4) or 9a-1-NMe2 (Entry 8) to afford 9a-2-OMe or 9a-2-NMe2, respectively. Similar results were observed in the reaction of chlorotriazine 9a-3-X with NaOMe to form 9a-4-X.
The reaction rate of 9a-3-Cl (Entry 1, Reaction B) was 190 or 75,000 times higher than that of 9a-3-OMe (Entry 4) or 9a-3-NMe2 (Entry 8), respectively. Owing to such stabilization effects, the chlorine atoms of cyanuric chloride and dichlorotriazines can be substituted stepwise with alcohols, phenols, amines, anilines, thiols, and carbon nucleophiles.
Amines and anilines are popular nucleophiles for cyanuric chloride. Melamine (triaminotriazine) and its derivatives have long been known as an important class of triazine compounds. Stepwise substitution of cyanuric chloride with naphthylamine [190] and ammonia and methylamine [191] was reported in the late 1800s. In the more recent era, Simanek and coworkers investigated the relative reactivity of a series of amines toward chloro(dimorpholino)triazine (Figure 33A) [192,193]. A cyclic secondary amine reacted considerably faster than primary amines. Reactivity of p-toluidine toward anilino(dichloro)triazine was 80 times lower than that of benzylamine [194]. Triazine-based dendrimers were synthesized efficiently using protection-free diamine linkers such as p-aminobenzylamine and 4-(aminomethyl)piperidine, the amino groups of which selectively react with chlorotriazine [195].
The question arises as to how to exercise orthogonal chemoselectivity with cyanuric chloride given a set of nucleophiles that differ in chemical composition—i.e., alcohols, phenols, thiols, and amines? Albericio and coworkers investigated the selective and stepwise substitution of cyanuric chloride using a set of nucleophiles including alcohols, phenol, thiols, and amines under mildly basic reaction conditions (i-Pr2EtN, CH2Cl2 or tetrahydrofuran (THF), −20 °C to 75 °C) [196,197,198]. Typically, the substitution of an alcohol (the weakest nucleophile) was carried out toward the highly electrophilic cyanuric chloride at the first step. Phenol and a thiol as well as an alcohol were required to be introduced to the triazine core before the amine substitution. These nucleophiles did not react with amino-substituted chlorotriazines because an amino group incorporated in the triazine ring strongly diminishes the electrophilicity (Table 1, Entry 8). Nevertheless, given the high nucleophilicity, an amine was able to react with such deactivated amino-substituted chlorotriazines. The high nucleophilicity of an amine toward chlorotriazines was demonstrated in competitive experiments (Figure 33B): treatment of chlorotriazine 9a-5 with an equimolar mixture of isopentyl alcohol, phenol, isopentyl mercaptan and isopentyl amine afforded only the corresponding amine-substituted product 9a-6 regardless of substituent X on the triazine [199]. In short, for reaction with cyanuric chloride, a substrate bearing a phenol and an alcohol reacts selectively at the phenol; a substrate with a phenol and an aromatic amine reacts selectively at the amine. The inherent distinctions in nucleophilicity enables some such substitution processes to be carried out sans protecting groups [200].
A distinct feature of amino-substituted triazines is hindered rotation of the exocyclic C−N single bond. The exocyclic carbon–nitrogen bond has partial double-bond character because of the conjugation of the lone pair on the sp2-hybridized amino nitrogen with the electron-deficient triazine ring. Unsymmetrically substituted triazine compounds (R1 ≠ R2, X1 ≠ X2) exist as a pair of rotamers (Ia and Ib, Figure 34A). Because of the rotamerism, decoalescence and broadening of NMR signals can occur depending on the nature of the substituents of the triazine and the temperature for the NMR measurement. Since one unsymmetrically substituted amino group can afford two triazine rotamers, in total eight rotamers are theoretically possible for an unsymmetric triaminotriazine [201]. The rotation barrier of the hindered exocyclic C−N bond in triaminotriazines was ~64 kJ/mol (coalescence temperature of ~32−42 °C), which is similar to the barrier toward C–N bond rotation in amides [202]. Chloro- or alkoxy-substituted diaminotriazines exhibit a higher barrier (>72 kJ/mol) to rotation around the C−N bond [203]. Thus, more obvious decoalescence of NMR signals is generally observed in these cases at room temperature versus that of triaminotriazines.
Several specific cases are germane here [200]:
- For a triazine that bears two identical substituents (X) and an alkylamino group (II), rotamers are not possible by symmetry (Figure 34B). Nonetheless, distinguishable signals in the NMR spectrum are possible because the two X substituents experience non-equivalent environments with respect to the orientation of the substituents on the attached nitrogen atom.
- For a triazine that bears two different substituents (X and Y) and one alkylamino group, a pair of rotamers (i.e., IIIa and IIIb) is possible (Figure 34C).
- For a triazine compound that bears two nonidentical alkylamino groups, four rotamers (IVa–IVd) are possible (Figure 34D).
- Possible tautomers IIa–c can in principle arise from Compound II (Figure 34E). Such tautomers are generally not detectable and ignored. Indeed, the tautomer equilibria between triazine and N-alkylimine species were excluded on the basis of different substituted triazines as reported in the literature. These types of equilibrium always shift towards triazine forms [201,204,205,206].
In all of the above cases, the presence of multiple peaks can complicate interpretation of the NMR spectra of aminotriazines, yet the underlying phenomenon is established and readily understandable.
On the basis of a vast amount of research over many decades, the following guidelines have emerged for the design and synthesis of the triazine hubs in multifunctional molecular architectures:
- Introduction of at least one amino group on the triazine hub is recommended. The amino group stabilizes the triazine hub and prevents undesired further nucleophilic substitution such as hydrolytic decomposition.
- The order of nucleophiles used toward cyanuric chloride should be as follows: alcohols first, phenols or thiols next, then anilines, and finally aliphatic amines.
- Alcohols and amines are preferred nucleophiles for the triazine hub. Building blocks containing hydroxy and amino groups are synthetically and commercially abundant. In particular, useful bifunctionalized linkers such as PEG linkers [41] are readily available with terminal alcohol and amine groups. Furthermore, unlike phenol- and aniline-based nucleophiles, aliphatic alcohols and amines do not contain a hydrophobic phenyl ring, which may impart water-solubility problems in biological applications.
- Cyanuric chloride (9a) undergoes successive substitution with an amine to give the monoamino (9a-7), diamino (9a-8), and triamino (9a-9) derivatives. A triaminotriazine (9a-9) is a representative triazine hub (Figure 35A, top). Each amino substitution reaction can be carried out in a straightforward manner by treating the chlorotriazine with the corresponding amine. However, base-labile functionalities could be damaged because the third amino substitution requires elevated temperature (typically >70 °C). If the first nucleophile is an alcohol, affording the monoalkoxy derivative (9a-10), the second substitution with an amine affords the monoalkoxy-monoamino derivative (9a-11), and a third substitution with an amine affords the monoalkoxy-diamino derivative (9a-12) (Figure 35A, middle). In this case, both the first alkoxy and second amino substitution occur at temperatures lower than room temperature. A general method for preparation of unsymmetric dialkoxychlorotriazines (9a-13) has not yet been established, precluding use of dialkoxy-aminotriazines (9a-14) as common hub designs (Figure 35A, bottom). Limited examples have adopted conditions at >100 °C for the second alkoxy substitution; the conditions entail 2,6-lutidine [207] or K2CO3/18-crown-6 [208] as a base, or solvolysis conditions in MeOH [209]. Note that primary alcohols are employed in Figure 35A. The alkoxy substitution with secondary alcohols proceeds slower than primary alcohols and thus was carried out under harsher heating conditions [210]. Tertiary alcohols generally require prior conversion to the corresponding metal alkoxide to react with cyanuric chloride [211].
- Substitution reactions of the chlorotriazine are typically carried out in the presence of an amine base. The most common bases are listed below along with the pKa value of the conjugate acid in water:The base strengths are solvent dependent; for example, the pKa value of 2,6-lutidine in H2O is 6.77 [213], whereas that in 50% EtOH-H2O is 5.77 [216]. Regardless, the trend shown here provides a general framework for selecting an appropriate base.The chloro-triazine substitutions are typically done in the presence of a hindered amine base such as Et3N and i-Pr2EtN. Since these tertiary amines are sufficiently basic, deactivation of the amine nucleophile by protonation with HCl is prevented, given that such protonation otherwise would occur during the chlorotriazine substitution. In contrast, Et3N is not employed for the cyanuric chloride substitution with an alcohol to prepare alkoxydichlorotriazines, due to a side reaction forming (N,N-diethyl)aminotriazine byproducts [217,218]. Instead, i-Pr2EtN is commonly used as the base to promote this alkoxy substitution at 0 °C, although the same type of side reactions could compete [210,219]. Alternatively, weaker bases such as 2,4,6-collidine [210] and 1,10-phenanthroline [207] can be used over the temperature range of 0 °C to room temperature to strictly suppress the side reaction forming the aminotriazine byproducts or the undesired second alkoxy substitution. The aromatic heterocyclic 1,10-phenanthroline is also an effective base for double-alkoxy substitution of cyanuric chloride to synthesize symmetric dialkoxychlorotriazines [209], although severe heating conditions (reflux in xylene) were employed. Recently, we introduced use of pempidine to this reaction [220]. Owing to the bulkiness and high basicity of this tertiary amine, the double-alkoxy substitution of cyanuric chloride proceeded at lower temperature (60 °C) without forming the (N,N-dialkyl)aminotriazine byproducts. Structures and the pKa values (in H2O) of amine bases employed in the chlorotriazine substitution are shown in Figure 35B.
- Rotamerism (vide supra) can present a nuisance for interpretation of NMR spectra, and the rotamers might potentially engender distinct features in biological systems. Rotamerism can be sidestepped through the use of cyclic amines. Thus, 4-substituted piperidinyl, 4-substituted piperazinyl, and 3-substituted azetidinyl groups are symmetric dialkylamino groups that do not give rise to triazine rotamers (Figure 35C). Such groups may serve as attractive linkers.
The use of triazine as a scaffold is exemplified in the following text. A bivalent antagonist (9b) for the nerve growth factor (NGF) receptor, TrkA, was developed (Figure 36) [221]. The antagonist is composed of a triazine hub and three functional units: (A) a monovalent TrkA agonist (a small molecule mimic of NGF), and (B) fluorescein. This construct (9b) has composition A2B and architecture [3][1]3.
A multifunctional chemical entity for the potential development of multimodal imaging agents (9c) was developed (Figure 36) [222]. The entity is composed of a triazine hub and three functional units: (A) two copies of a water-soluble sulfonated porphyrin for optical imaging, and (B) a DOTA-type ligand (DO3A) to chelate metal ions such as Gd3+ for use in MRI. This construct (9c) has composition A2B and architecture [3][1]3.
A compound targeting metal dysregulation and oxidative stress associated with Alzheimer’s disease (9d) was developed (Figure 36) [223]. The compound is composed of a triazine hub and three functional units: (A) two copies of cyclen for metal chelation, and (B) an antioxidant coumarin, which forms fluorescent hydroxycoumarins upon oxidation by ROS. This construct (9d) has composition A2B and architecture [3][1]2.
A fluorescent dimeric peptide ligand (9e) has been developed that targets 14-3-3σ, a protein regulating a wide range of signaling pathways (Figure 36) [224]. The peptide ligand is composed of a triazine hub and three functional units: (A) two copies of the peptide ligand (containing phosphoserine) for 14-3-3σ, and (B) fluorescein for detection. This construct (9e) has composition A2B and architecture [3][1]3.
A quinone-methide precursor for DNA alkylation (9f) has been developed (Figure 36) [225]. The pre-alkylator is composed of a triazine hub and three functional units: (A) two copies of cyclen to bind bivalent metals such as Zn2+, Ni2+, and Cd2+, and (B) a quinone-methide precursor containing a tert-butyldimethylsilyl (TBS) group, which can be deprotected by treatment with KF. This construct (9f) has composition A2B and architecture [3][1]3.
A triazine-based modular platform (9g) was developed for targeted PET imaging (Figure 36) [226]. The platform is composed of a triazine hub and three functional units: (A) two copies of a cyclo(RGDfK) peptide for targeting integrin αvβ3 receptors, and (B) DOTA for radiolabeling with 64Cu. This construct (9g) has composition A2B and architecture [3][1]3.
A trifunctional linker for “double-click” labeling (9h) has been developed (Figure 37) [227]. The linker is composed of a triazine hub and three functional units: (A) a terminal alkyne for Cu(I)-catalyzed azide–alkyne [3 + 2] cycloaddition (CuAAC, also referred to more casually as a copper-mediated click reaction or simply click chemistry) [228,229,230]; (B) tetrazine for Cu-free cycloaddition reaction with azides, and (C) an NHS ester as an amine-reactive group. This construct (9h) has composition ABC and architecture [3][1]3.
A β-lactam equipped bifunctional targeting module (9i) was developed for irreversible chemical programming of monoclonal aldolase antibody 38C2 (Figure 37) [231]. The module is composed of a triazine hub and three functional units: (A) an integrin inhibitor SCS-873 to target integrin; (B) luteinizing hormone releasing hormone (LHRH) containing a D-Lys6 modification to target a LHRH receptor, and (C) a β-lactam for covalent labeling of monoclonal antibody 38C2. This construct (9i) has composition ABC and architecture [3][1]3.
Toll-like receptor (TLR) agonists are used as adjuvants with antigens to control immune responses [232,233]. The covalent juxtaposition of three TLR agonists (9j) was constructed to serve as a novel vaccine adjuvant (Figure 37) [234]. The tri-agonist is composed of a triazine hub and three functional units: (A) a TLR4 agonist pyrimido[5,4-b]indole; (B) a TLR7 agonist loxoribine, and (C) a TLR9 agonist CpG-ODN1826. This construct (9j) has composition ABC and architecture [3][1]3.
A model system to mimic the photosynthetic antenna-–reaction center complex (9k) has been developed (Figure 37) [235]. The construct is composed of a triazine hub and three functional units: (A) a porphyrin as an energy-transfer acceptor and electron-transfer donor; (B) a BODIPY fluorescent dye as an energy-transfer donor, and (C) fullerene as an electron acceptor. The BODIPY dye serves as a light-harvesting antenna component whereas the porphyrin and fullerene serve as the reaction center. Note, however, that all three constituents are potent chromophores and hence provide light absorption. This construct (9k) has composition ABC and architecture [3][1]3.
A tumor-targeting theranostic drug conjugate (9l) was developed for SPECT imaging studies and therapy (Figure 38) [236]. The conjugate is composed of a triazine hub and four functional units: (A) a biotinyl group for tumor-targeting; (B) a disulfide-containing self-immolative linker; (C) the taxoid antitumor agent SB-T-1214, and (D) a dipyridyl-containing ligand for chelation of 99mTc. This construct (9l) has composition ABCD and architecture [3][2][1]3.
A tumor-targeting theranostic fluorescent probe (9m) was developed for internalization studies in vitro (Figure 38) [237]. The probe is composed of a triazine hub and four functional units: (A) biotin as a tumor-targeting agent; (B) a disulfide self-immolative linker for drug release; (C) the taxoid antitumor agent SB-T-1214, and (D) fluorescein for optical detection. This construct (9m) has composition ABCD and architecture [3][2][1]3.
A tumor-targeting dual-warhead conjugate (9n) was developed for a taxoid–camptothecin combination chemotherapy (Figure 38) [238]. The conjugate is composed of a triazine hub and five functional units: (A) two disulfide self-immolative linkers for release of both drugs; (B) biotin as a tumor-targeting agent, (C) the taxoid antitumor agent, SB-T-1214, and (D) the antitumor agent camptothecin. This construct (9n) has composition A2BCD and architecture [3][2]2[1]3.
A tetravalent platform (9o) was developed for theranostic applications (Figure 39) [239]. The target compound is composed of three triazine hubs (i.e., a generation-1 triazine dendron) and five functional units: (A) four copies of a thiol-reactive maleimide, and (B) DOTA as a nuclear medicine imaging agent. This construct (9o) has composition A4B and architecture [3]3[1]5.
One objective in molecular immunology has been to understand the response of TLR triagonists for both spatially organized and unorganized systems, where the three agonists are joined in a single molecular architecture or are more randomly available, respectively. A fluorescently labeled triagonist architecture (9p) was synthesized, built around two hubs (triazine and cysteine), to address this fundamental question. This construct (9p) has composition ABCD and architecture [3]2[1]4 (Figure 39) [240]. This compound is composed of three different TLR agonists: (A) a TLR4a (pyrimido-indole derivative); (B) a TLR1/2a (Pam3CSK4, Pam = palmitoyl), and (C) a TLR9a (oligodeoxynucleotide (ODN) 1826 CpG DNA containing a phosphorothioate backbone). The fourth component, (D), is a fluorescein molecule for detection and quantification.
The synthesis of 9p began with cyanuric chloride (9a). Standard temperature-controlled substitution of cyanuric chloride with three different amines in succession proceeded smoothly (72%, 99%, and 95% yield, respectively). Thus, reaction first with propargylamine afforded 9p-1, reaction next with a Boc-protected amine gave 9p-2, and final reaction with a piperidyl compound containing a protected maleimide (9p-3) gave the triaminotriazine 9p-4 (Figure 40). Removal of the Boc group and amidation with an Fmoc-protected amino-PEG12 linker (9p-5) gave 9p-6 in 81% yield. The amidation was carried out using O-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) and 1-hydroxybenzotriazole (HOBt, available as the monohydrate) in DMF containing collidine. Attachment of the first agonist unit (A) was achieved by amidation to afford 9p-7 in 41% yield. Deprotection of the maleimide followed by conjugation with the cysteine residue of 9p-8 bearing the agonist units (B) (protected form) and (C), and cleavage from the solid support, provided 9p-9 albeit in 2% yield. The cleavage process was carried out using a cocktail of TFA (for acidolysis), phenol and thiophenol (to scavenge carbocations), and ethanedithiol (EDT, to create a reducing milieu). Finally, the fluorescein unit (D) was attached by CuAAC to afford the desired ABCD compound 9p in 10% yield. The target compound 9p was purified via sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE) and gel extraction, characterized by ESI-MS, and quantified by fluorescence spectroscopy. Four other TLR triagonists were synthesized in a similar manner.
The ABCD compound 9p elicited drastically higher TH1-inducing cytokines than that of the unlinked counterparts in vitro, suggesting that spatial organization of TLR agonists with antigens enhances the immune response. Although no significant improvement of protection compared to a commercial vaccine was observed in an in vivo experiment (a live Coxiella burnetii aerosol challenge model in C56BL/6 mice) [241], the spatially organized adjuvant platform based on a triazine hub provides a concise architecture for further evaluation. Given the objective to elucidate the effects of spatial organization, it warrants comment that the reaction of a thiol and maleimide generates a stereocenter. Here, the TLR triagonist is comprised of a mixture of diastereomers owing to the ostensibly simple thiol–maleimide reaction. It warrants mention that in some systems, the thiol–maleimide reaction has been noted to undergo reversal (i.e., retro-Michael reaction), liberating the thiol over time [242].
We explored the formation of molecular scaffolds upon enzymatic action under physiological conditions. Such scaffolds can be utilized for diverse applications in the life sciences. To this end, we synthesized a compound (9q) composed of two 4,6-dibromoindoxyls (Unit A) each protected as a β-glucoside (Unit B), a sulfobetaine to improve water-solubility (Unit C), an azide as a conjugatable group (Unit D), and a coumarin dye for quantification (Unit E), all of which were built around two triazine hubs. This construct has composition A2B2CDE molecule and architecture [3]2[2]2[1]4 (Figure 39) [200].
The Units A–E were rapidly assembled utilizing triazine chemistry (Figure 41). Alcohol 9q-1 bearing the A unit (indoxyl) with attached B (O-acetyl-protected glucosyl group) provided the nucleophile for substitution of two chlorine atoms of cyanuric chloride (9a) in the presence of the base pempidine, affording the bis(glucosyl-indoxyl) triazine (9q-2). Removal of the indole N-acetyl groups with base followed by treatment with amino alcohol 9q-3 (containing Unit C) gave (dialkoxyamino)triazine 9q-4 in 29% yield from cyanuric chloride. The aliphatic hydroxy group in 9q-4 was used to attach to the second triazine hub. The reaction between 9q-4 and cyanuric chloride in the presence of the weak base 1,10-phenanthroline provided an (alkoxydichloro)triazine intermediate, wherein the two chlorine atoms were sequentially substituted by primary amines 9q-5 and 9q-6 bearing the Units D and E, respectively. As a result, 9q-7 was obtained in 39% yield from 9q-4. Global deprotection of the glucosyl O-acetyl groups in 9q-7 afforded the desired A2B2CDE compound 9q in quantitative yield. A minor point bearing on characterization is that the second triazine hub formed rotamers due to the unsymmetrically substituted amino groups. In contrast, rotamers were not formed on the first triazine hub given its symmetrical substitution pattern [200].
The glucosyl units (B) were designed to be removed upon treatment with β-glucosidase, thereby liberating the deprotected indoxyl units (A). The latter readily dimerize to form the indigoid species in the presence of O2. Given that 9q bears two indoxyl glucoside moieties, the enzymatic action gives rise to covalent cross-linking and thus affords a polymerized molecular scaffold containing the bioconjugatable azide group (Unit D) under physiological conditions. The progress of the cross-linking could be monitored by absorption spectroscopy owing to a chromogenic readout of the indigo (~630 nm). The coumarin in the molecule (Unit E) enabled ratiometric analysis, which is potentially valuable for studies in cells or tissues [200].
The A2B2CDE compound 9q (50 μM) underwent oligomerization to form a blue precipitate (~1 μm in size) by treatment with β-glucosidase (200 nM) in buffer (containing 5% DMF), as expected; however, the yield analyzed by absorption spectrometry was low (3%) [200]. In future studies, refinement of the molecular structure—through larger PEG groups or introduction of charged groups—is needed to improve water solubility and the yield of oligomerization.
10. Benzene Hubs (11)
The central importance of benzene in organic chemistry and the accompanying chemistry for introducing substituents has naturally led to the use of benzene as a hub in the design and synthesis of scaffolds. In this section, 11 examples are presented. A benzene hub was employed in a tri-functional compound (10a) for the fluorescence monitoring of DNA/RNA delivery via non-viral gene vectors (Figure 42) [243]. The tri-functional compound is comprised of a single 1,3,5-trisubstituted benzene hub and two types of functional components: (A) two copies of the triazamacrocycle [12]ane-N3 for binding to nucleic acids, and (B) a 1,8-naphthalimide derivative as a (yellow-green) fluorophore and hydrophobic tail. This construct (10a) has composition A2B and architecture [3][1]3. The tri-functional compound bound to, and caused condensation of, DNA/RNA, supporting the design for fluorescence monitoring of non-viral gene vectors.
Antibody-drug conjugates (ADCs) are of growing interest for targeted therapies [244,245]. In some cases, the linker bearing the drug to the antibody is cleavable to release the drug at the target site. A non-cleavable scaffold bearing two covalently attached drugs (10b) was developed for attachment to an antibody (Figure 42) [246]. The scaffold is comprised of a single 1,3,5-trisubstituted benzene hub and two types of functional components: (A) two copies of a pyrrolobenzodiazepine antitumor drug, and (B) maleimide for conjugation to the Cys of an antibody. This construct (10b) has composition A2B and architecture [3][1]3.
The trilactone, benzotrifuranone (10c), has C3h-symmetry and on inspection has three identical lactones (Figure 43) [247,248]. The three lactones, however, can be aminolyzed selectively: the relative reaction rate for opening of the first, second, and third ring was found to be 390:7.1:1.0 [248]. Thus, despite the symmetric structure, benzotrifuranone can be regarded as an ABC molecule (N = 3) bearing three different conjugatable groups around a benzene hub. This construct (10c) has composition ABC and architecture [3][1]3.
The inherent symmetry of benzotrifuranone enabled straightforward synthesis in four steps (Figure 43). Bromomethylation of 1,3,5-trimethoxybenzene (10c-1) provided 10c-2 in 37% yield [249]. Cyanide substitution to form 10c-3 (67%) was followed by hydrolysis and demethylation to afford 10c-4 (46%) [250]. Lactonization of 10c-4 by heating in polyphosphoric acid afforded the target benzotrifuranone (10c) in 45% yield. Sequential addition of furfurylamine, allylamine, and propargylamine to a solution of 10c in DMF at low temperature provided 10c-5 in 85% yield [247]. The origins of the unexpectedly efficient sequential aminolysis features of benzotrifuranone were rationalized through a synergy of ring strain and inductive effects. Although this scaffold has not yet been widely utilized, benzotrifuranone is a potential complement to cyanuric chloride (vide supra), affording sequential derivatization in a hub with three ostensibly equivalent reaction sites.
A synthetic antibody mimic of infliximab (10d) was developed with a tris(tertiary amine)-cyclotriveratrylene (TACTV) scaffold (Figure 44) [251]. The mimic is comprised of a single TACTV hub and three functional components: (A–C) complementarity determining regions (CDRs) that mimic the peptide loop of the monoclonal antibody infliximab. The latter is a clinically used biologic for treatment of human tumor necrosis factor alpha (hTNFα)-mediated autoimmune diseases and directly binds to hTNFα [252]. This construct (10d) has composition ABC and architecture [3][1]3. The TACTV derivative originally contained a triethylsilyl (TES)-protected alkyne, a triisopropylsilyl (TIPS)-protected alkyne, and a free alkyne. The first loop mimic (A) was initially introduced onto the free alkyne of the TACTV derivative via Cu-catalyzed click chemistry. After removal of the TES protecting group, the next CDR-mimicking peptide loop (B) was incorporated. Finally, the TIPS protecting group was removed, followed by the introduction of the CDR loop mimic (C) to obtain the synthetic antibody.
A tripodal scaffold bearing three distinct peptides (10e) was prepared wherein the three peptides on the scaffold were constructed by SPPS (Figure 44) [253]. The compound is comprised of a single 1,3,5-trisubstituted benzene hub and bears three dipeptides: (A) Ac-Ile-Asp; (B) Ac-Met-Lys, and (C) Ac-His-Trp. This construct (10e) has composition ABC and architecture [3][1]3. The scaffold originally contained a terminal carboxylic acid, an Fmoc-protected amine, a TES-protected alkyne, and a free alkyne. The carboxylic acid was initially anchored on an amide resin, and the first model peptide was then elongated from the Fmoc-protected amine. The second model peptide then was elaborated from the free alkyne, and the remaining TES-protected alkyne was finally used for introducing the third model peptide, followed by removal of the resin to yield the model peptide compound. This strategy elegantly combines the powerful methods of SPPS with a trifunctional, non-peptidic scaffold. At present, one limitation is the requirement to prepare the 1,3,5-trisubstituted benzene bearing three distinct groups from the symmetric trimesic acid (1,3,5-tricarboxybenzene).
A fluorescence probe containing building block for protein labeling and protein immobilization (10f) was developed (Figure 44) [254]. The building block is comprised of a 1,3,5-trisubstituted benzene hub and three functional components: (A) azide for conjugation to a protein via Staudinger ligation; (B) a protected Cys for conjugation to a protein via NCL, and (C) the cyanine dye Cy5 as a fluorophore. This construct (10f) has composition ABC and architecture [3][1]3. After deprotection of the Cys moiety, the resulting building block is bound to a thioester-bearing target protein via NCL. The derivatized protein can then be reacted with a phosphane-modified surface via Staudinger ligation to achieve protein immobilization.
A luminescent peptide–oligonucleotide model conjugate (10g) was developed on the basis of a heterotrifunctional benzenic scaffold equipped for multiple bioconjugations (Figure 44) [255]. The model conjugate is comprised of a 1,3,5-trisubstituted benzene hub and three functional components: (A) a model bioactive dodecapeptide; (B) Eu(III)-bis(pyridinyl)pyrazine as a luminescent complex, and (C) a 16-mer oligonucleotide as a model bioactive DNA fragment. This construct (10g) has composition ABC and architecture [3][1]3. The underlying scaffold contains three different and orthogonally bioconjugatable groups (aminooxy, azido and thiol). The aminooxy group was initially reacted with the model peptide bearing an aldehyde by oxime ligation. The thiol group was then reacted with a thiol-reactive Eu(III)-bis(pyridinyl)pyrazine-based chelate, and the click reaction between the azido group and the model nucleotide bearing the alkyne group was carried out to yield the luminescent peptide–oligonucleotide model conjugate.
Yoshida et al. first reported a tri-functionalized biphenyl hub bearing three azides [256]. The hub contains an unhindered aromatic azide, a primary aliphatic azide group, and a doubly sterically hindered aromatic azide. The azides could be coupled to the corresponding cycloaddition motifs with high selectivity to introduce a HaloTag ligand as labeling agent (Unit A), a BODIPY derivative as fluorophore (Unit B), and a biotin tag (Unit C) in sequence. The resulting trifunctionalized product (10h) is shown in Figure 44 [256]. The compound is reported to have a triazole with 1,5-disubstitution rather than the typical 1,4-disubstitution. This construct (10h) has composition ABC and architecture [3]1[1]3.
The synthesis of 10h began with 2,6-diisopropylaniline (10h-1), which underwent iodination at the 4-position to give 10h-2 [257]. Treatment with sodium azide gave the azide 10h-3, which upon Suzuki coupling with (Bpin)2 gave the p-Bpin derivative 10h-4 [256]. In parallel, ethyl 3-iodobenzoate (10h-5) was converted to the 3-Bpin derivative via iridium-based catalysis in the presence of 4,4′-di-tert-butyl-2,2′-bipyridyl (DTBPY), followed by installation of the azido group at the 5-position to give 10h-6. Reduction of the ester with diisobutylaluminum hydride (DIBAL-H) followed by treatment with diphenylphosphoryl azide (DPPA) gave 1-azido-3-(azidomethyl)-5-iodobenzene (10h-7) [258]. Suzuki coupling of 10h-4 and 10h-7 gave the triazide-containing biphenyl hub 10h-8 in a straightforward manner as shown in Figure 45.
Three subunits for 1,2,3-triazole formation were then synthesized following the routes in Figure 46. The chloro-amine 10h-9 underwent amidation with the NHS ester of 4-ethynylbenzoic acid to give 10h-10. The azido-PEG-amine 10h-11 was reacted with acetyl ketene to give PEG-azide 10h-12. The Cu-catalyzed click reaction between ethyne 10h-10 and PEG-azide 10h-12 in the presence of tris[(1-benzyl-1H-1,2,3-triazole-4-yl)methyl]amine (TBTA) gave the β-ketoamide-type HaloTag ligand 10h-13. In parallel, 5-((+)-biotinamido)pentylamine 10h-14 was reacted with the NHS ester of 4-ethynylbenzoic acid to give 10h-15, which upon click chemistry with azido-PEG-amine 10h-11 again in the presence of TBTA gave amino-biotin 10h-16. Reaction of the latter with the 4-nitrophenyl carbonate of the strained dibenzocyclooctyne (5,6-didehydro-11,12-dihydrodibenzo[a,e]cyclooctene, DIBO) gave cyclooctyne derivative 10h-17. In addition, in parallel, p-iodobenzaldehyde (10h-18) and 3-ethyl-2,4-dimethylpyrrole (10h-19) were converted via a four-step protocol [259] to the corresponding BODIPY 10h-20, which upon Sonogashira coupling with trimethylsilylethyne gave 10h-21 [260]. Standard cleavage of the trimethylsilyl (TMS) group with tetrabutylammonium fluoride (TBAF) gave the ethynyl-substituted BODIPY 10h-22 [260].
With all segments ready, the tri-azide hub 10h-8 was treated with labeling unit precursor 10h-13 to give mono-triazole 10h-23, then with fluorophore unit precursor 10h-22 with the reagent Cp*Ru(PPh3)Cl2 (where Cp* is pentamethylcyclopentadienyl) to give di-triazole 10h-24 (wherein the triazole contains 1,5-disubstitution rather than the typical 1,4-disubstitution) and then with biotin tag precursor 10h-17 to give tri-triazole 10h (Figure 47). The three couplings are restricted in sequence and must be carried out on the basis of steric and electronic differentiation among the three azido groups to give the trifunctional construct 10h.
As a result of extended study of the selectivity of various triazole-forming reactions, Yoshida and coworkers recently reported a tetra-azido platform (10i-1) for consecutive assembly of four modules (Figure 48) [261]. In addition to the aforementioned three different types of azido groups, the authors took advantage of the distinct selectivity of the 1-adamantyl azido group as the fourth joint and also introduced a branched oligoglycerol moiety as a water-solubilizing group (Unit D). Thus, 10i-1 bears four alkyl and aryl azide moieties with different degrees of steric hindrance: phenyl azide, benzyl azide, 2,6-diisopropylphenyl azide, and 1-adamantyl azide. The four azides in this order selectively formed the corresponding 1,2,3-triazole under appropriate reaction conditions (Figure 48) [261]. First, the phenyl azide in 10i-1 reacted with dicarbonyl compound 10i-2 in the presence of K2CO3. Second, the benzyl azide underwent ruthenium-catalyzed azide-alkyne cycloaddition with terminal alkyne 10i-3, affording a triazole with 1,5-disubstitution rather than the typical 1,4-disubstitution (analogous to 10h and precursors). Third, the 2,6-diisopropylphenyl azide reacted with cyclooctyne 10i-4 by strain-promoted azide-alkyne cycloaddition (SPAAC). Last, the 1-adamantyl azide reacted with terminal alkyne 10i-5 by CuAAC. The product tetrakis(triazole) 10i is composed of two benzene hubs and four functional units: (A) a HaloTag as a labeling agent; (B) a BODIPY as a fluorescent dye; (C) a biotin tag, and (D) a branched oligoglycerol for water-solubilization. This construct (10i) has composition ABCD and architecture [3]2[1]4. The work demonstrated an efficient route to assemble motifs onto tri-/tetra-azido platforms by the selection of the correct cycloaddition counterparts and reaction sequence, which points to a bright future for branched molecular design and synthesis.
A probe for detection of the emerging cancer marker galectin-3 (10j) was developed (Figure 49) [262]. The probe is comprised of a 1,3,5-trisubstituted benzene hub and three functional components: (A) lactose as a targeting agent for galectin-3, which is correlated with cancer aggressiveness, metastasis, and apoptosis; (B) BP as a PAL agent, and (C) an alkyne as a conjugatable group to introduce the fluorescent reporter molecule via click chemistry after carrying out photoaffinity labeling of galectin-3. This construct (10j) has composition A2B2C and architecture [3][2]2[1]3.
An A2BC compound (10k) was developed for forming chemically self-assembled nanoring (CSAN) structures that are endocytosed into T-leukemia cells and then visualized (Figure 49) [263]. This A2BC molecule is built around two hubs, a 1,3,5-trisubstituted benzene and a tertiary amine bearing three primary amines. The functional units include two methotrexate (MTX) units, which undergo dimerization in support of self-assembly (A), a green fluorescent protein (GFP) as protein cargo and fluorophore (B), and an azido- TAMRA fluorophore for imaging (C). This construct (10k) has composition A2BC and architecture [3]2[1]4. This triorthogonal-protein-labeling construct was treated with a dihydrofolate reductase-anti-CD3 fusion protein for macromolecular protein self-assembly. The resulting anti-CD3 CSANs were endocytosed into T-leukemia cells and then visualized.
The synthesis of 10k proceeded as follows. The versatile starting material 3,5-dihydroxybenzaldehyde (10k-1) was converted in three steps via bis(triflation) to give 10k-2, statistical mono-ethynylation to give 10k-3, and treatment with base to remove the triflate and TMS protecting groups and form 10k-4, a triorthogonal 3-ethynyl-5-hydroxybenzaldehyde (Figure 50). In parallel, geraniol (10k-5) was protected as the 2-tetrahydropyranyl (THP) ether (10k-6) by reaction with 3,4-dihydro-2H-pyran (DHP) with catalysis by p-toluenesulfonic acid (PTSA), which upon hydroperoxide treatment gave allylic hydroxylation of the terminal methyl group (10k-7), and the latter upon Mitsunobo reaction gave the terminal bromide (10k-8) [264,265]. The nucleophilic phenol of 10k-4 was alkylated with THP-protected hydoxyalkenyl bromide 10k-8 to give 10k-9, which upon cleavage of the THP group with the mild acid pyridinium p-toluenesulfonate (PPTS) gave ethyne-containing hydroxyalkenylbenzaldehde 10k-10. Phosphorylation of 10k-10 gave 10k-11, the first hub, which also constitutes a substrate for protein farnesyl transferase (PFTase).
The second hub was prepared beginning with 1,2-bis(2-aminoethoxy)ethane (10k-12). Boc protection of one amine followed by dialkylation with 5-bromovaleronitrile gave the dinitrile 10k-13. Hydrogenation gave the mono-Boc-protected triamine 10k-14, which was acylated with the side chain of N-protected glutamic acid O-methyl ester to give 10k-15; removal of the N-protecting group by hydrogenolysis followed by diacylation with MTX (10k-16) gave 10k-17 [266]. Treatment of 10k-17 to Boc removal under acid, coupling with 5-azidopropionic acid in the presence of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC), and saponification of the glutamic acid O-methyl ester gave the intermediate scaffold 10k-18 bearing two MTX groups and one azide (Figure 51).
Assembling the A2BC compound relied on coupling of Unit B, a GFP containing tetrapeptide sequence (10k-19, denoted as a CaaX box, CVIA in this case) at its C terminal, and the PFTase-ready substrate 10k-11 (Figure 52). The reaction was carried out in the presence of the water-soluble reducing agent 1,4-dithio-d-threitol (DTT). The reaction successfully incorporated both the aldehyde and alkyne groups to give the bifunctional (formyl, ethynyl) prenylated protein 10k-20. Co-incubation of 10k-20, aminooxy-TAMRA, and azido-bis(MTX) 10k-18 in the presence of m-phenylenediamine (mPDA) for 15 h gave the final target compound 10k. This work suggested that a protein bearing a C-terminal CaaX-box fusion is suitable for this strategy [263]. The triorthogonal reagent 10k-11 provides the protein with a hub for coupling with two orthogonal groups, which can be achieved in a one-flask procedure. In summary, this strategy expanded the property and function of the protein cargo by attachment with other functional groups for specific labeling.
11. Nitrogen Hubs (12)
The versatile properties of amines as nucleophiles has given rise to a rich variety of trialkylamine-containing scaffolds. In this section, a dozen such examples are presented. As a first example, a ratiometric time-resolved probe (11a) was prepared for bioanalytical applications (Figure 53) [267]. The probe is composed of a nitrogen hub and three functional units: (A) two copies of pyrene, which forms an excimer and serves as a FRET donor, and (B) fluorescein as a FRET acceptor. This construct (11a) has composition A2B and architecture [3][1]3. One attraction of the pyrene excimer is the long-lived excited state, which allows acquisition of the observed fluorescence in a time regime beyond the occurrence of cellular autofluorescence.
A PAL probe (11b) was prepared for derivatization of carbonic anhydrase IX (Figure 53) [14]. The probe is composed of a nitrogen hub and three functional units: (A) a terminal alkyne to conjugate a fluorescent dye after photoaffinity labeling; (B) phenyltrifluoromethyldiaziridine as a PAL agent, and (C) an arylsulfonamide as a targeting agent for carbonic anhydrase IX. This construct (11b) has composition ABC and architecture [3][1]3.
A PAL probe containing a mixed isotope label (11c) was prepared to facilitate identification of binding proteins by mass spectrometry (Figure 53) [268]. The probe is composed of a nitrogen hub and three functional units: (A) a deuterated benzophenone as an isotope-containing PAL agent; (B) a biotin tag for purification by affinity chromatography, and (C) cyclosporin A as a targeting agent. This construct (11c) has composition ABC and architecture [3][1]3.
A heterobivalent peptidic ligand (11d) was prepared for PET/CT breast cancer imaging (Figure 53) [269]. The ligand contains a nitrogen hub and three functional units: (A) 1,4,7-triazacyclononane-triacetic acid (NOTA) to chelate 68Ga; (B) a PEG4-BBN7–14 targeting the GRPR, where BBN7–14 (Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2) is one-half of the BBN peptide, and (C) [Lys4,Tyr4,Nle7]BVD15, which targets the neuropeptide Y receptor subtype 1 [NPY(Y1)R]. This construct (11d) has composition ABC and architecture [3][1]3.
A triple-click conjugation scaffold (11e) was developed that allows one-flask four-component coupling. The scaffold is built around a nitrogen hub with functional units including: (A) an alkyl azide; (B) a 1,2,4-triazine, and (C) a diazo group. This construct (11e) has composition ABC and architecture [3][1]3 (Figure 53) [270].
The synthesis began with 3-acetylaniline (11e-1), which upon reaction with 1,6-dibromohexane afforded the mono N-alkylated product 11e-2 in 40% yield (Figure 54). Subsequent acylation with bromoacetyl bromide followed by α-bromination with PhMe3NBr3 and sodium azide in succession gave triazide 11e-3 in 71% yield [271]. Treatment of 11e-3 with hydroxylamine and TBAF under optimized basic reaction conditions selectively transformed the alkyl azide adjacent to the ketone to the corresponding oxime 11e-4 in 65% yield. Elaboration of the oxime in 3-steps gave the 1,2,4-triazine moiety (11e-5) in 84% yield. The alkyl azide adjacent to the amide was then selectively converted to the diazo group (Unit C) to afford the ABC target compound 11e in 40% yield. The ABC compound 11e was sequentially treated in aqueous solution at room temperature with a pyrene-acrylate (11e-6), dibenzocyclooctyne (11e-7), and a trans-cyclooctene bearing a tethered biotin moiety (11e-8), affording selective conjugation at Units C, A, and B, respectively, and giving 11e-9 in 44% yield. The method may be utilized in site-selective assembly of multiple functional components onto a single molecular scaffold, thus facilitating the development of functional materials and biochemical tools.
A tetrafunctional photoaffinity probe bearing a m-nitroalkoxy substituted phenyltrifluoromethyldiazirine unit (11f) was developed (Figure 55) [272]. The probe is composed of a nitrogen hub and four functional units: (A) a biotin tag for affinity purification; (B) a m-nitrophenyl ether for cleavage by irradiation to simplify mass spectrometric analysis of labeled products; (C) phenyltrifluoromethyldiazirine as a PAL agent, and (D) a moenomycin derivative as a protein targeting agent. This construct (11f) has composition ABCD and architecture [3][2][1]3.
A sequence-defined oligothioether-amide structure (11g) was developed that contains heterotrifunctional cross-linkers with two orthogonal internal cleavage sites (Figure 55) [273]. The structure is built around a nitrogen hub and contains five functional units: (A) an azido group for click chemistry; (B) an oxidation-sensitive cleavable linker; (C) a reduction-sensitive cleavable linker; (D) rhodamine as a FRET acceptor, and (E) BODIPY as a FRET donor. This construct (11g) has composition ABCDE and architecture [3][2]2[1]3. Cleavage of the linkers can be monitored owing to the presence of the FRET fluorophores.
An amphiphile containing four steroidal heads and a polycationic tail (11h) was developed for delivery of small interfering RNA (siRNA) through cell membranes (Figure 56) [274]. The amphiphile is composed of a nitrogen hub, two lysine hubs, and six functional units: (A) four copies of cholic acid to construct a largely hydrophobic face; (B) a disulfide as a reduction-sensitive cleavable linker, and (C) a polycationic octaarginine peptide for binding to siRNA. This construct (11h) has composition A4BC and architecture [3]3[2][1]5.
An alkyne-functionalized scaffold bearing orthogonal protecting groups (11i) was developed (Figure 56) [275]. The scaffold is composed of three cyclic nitrogen hubs and three functional units: (A) a terminal alkyne; (B) a TIPS-protected terminal alkyne, and (C) a TES-protected terminal alkyne. This construct (11i) has composition ABC and architecture c-[3]3[1]3.
A compact T-shaped compound (11j) bearing three functional groups was developed (Figure 57) [276]. The synthesis is facile. The reaction of two molecules of o-mercaptobenzaldehyde and a primary amine occurs in aqueous solution at room temperature to afford a bicyclic [3.3.1] framework, a process somewhat reminiscent of the Robinson synthesis of the tropane skeleton [277,278]. The T-shaped compound is composed of a cyclic nitrogen hub and three functional units: (A) two copies of a terminal alkyne for conjugation, and (B) a model peptide bearing several nucleophilic amino acid residues (N-terminal proline, methionine, and histidine). This construct (11j) has composition A2B and architecture [3][1]3.
Selective chemical labeling and/or modification of proteins, which refers to installation of bioconjugatable handles onto defined protein surface locations, is an important strategy for understanding biological processes as well as pursuing applications in drug delivery, tissue engineering, and disease diagnosis and therapy [279,280]. Orthogonal dual modification of proteins, which enables the introduction of multiple modules on proteins, can significantly expand the scope of application in chemical biology; however, significant challenges remain concerning selectivity, efficiency, biostability, and compatibility with aqueous systems [281,282]. Though residue-specific protein labeling based on the functionalities of the natural amino acids—especially thiols and amines—is a classical and relatively straightforward strategy, the development of simple and robust methodologies that enable the installation of two specific modifications at a single site on a protein, especially when one handle can be used for bioorthogonal ligation in cells, is an area of prominent interest [283,284].
A method for chemoselective ligation of thiol- and amino-peptides was developed as exemplified by the compound 11k (Figure 57) [285]. The core scaffold is a bromomaleimide bearing a carboxylic acid tethered to the maleimido nitrogen (11k-1). The compound prepared is composed of a cyclic nitrogen hub and three functional units: (A) a first model peptide serves an amine nucleophile (on a solid phase) for coupling with the carboxylic acid of 11k-1; (B) the second model peptide serves as a thiol nucleophile, which causes displacement of the bromide and regeneration of the maleimide, and (C) the third model peptide serves as an amine nucleophile, which is attached via Michael addition. This construct (11k) has composition ABC and architecture [3][1]3.
An azabicyclic vinyl sulfone scaffold (11l) was developed for cysteine-specific dual labeling of proteins (Figure 57) [286]. The scaffold is comprised of a single [2.2.1]azabicyclic vinyl sulfone hub and three functional components: (A) an alkene as a bioconjugatable group; (B) a biotin tag, and (C) the C2A domain of Synaptotagmin-I (C2Am) mutant, which has a single Cys and binds to the phosphatidylserine on the surface of apoptotic cells, as a targeting agent. This construct (11l) has composition ABC and architecture [3][1]3.
The chemistry underlying the orthogonal reactivity of 11l warrants comment. Michael addition of a thiol group to a vinyl sulfone (11l-1) is an attractive methodology for protein labeling/modification because of the following features: (1) the reaction proceeds under mild conditions; (2) the reaction proceeds in almost quantitative fashion (hence without by-products); (3) the vinyl sulfone function is water-stable, and (4) the resulting thioether bond (11l-2) is stable (Figure 58A) [287]. Moreover, strained [2.2.1] bicyclic systems such as the norbornene-based structure (11l-3) react rapidly and specifically with tetrazine (11l-4) in inverse electron-demand Diels–Alder (iEDDA) cycloadditions to form stable adducts (11l-5). Consequently, this iEDDA reaction system is an attractive strategy for rapid and site-specific protein labeling (Figure 58B) [288]. According to these findings, a 7-azanorbornene structure was incorporated with a vinyl sulfone functionality to develop a trifunctionalized azanorbornadiene hub (11l-6) that first undergoes amidation to give the functionalized bridging nitrogen compound (11l-7), Michael addition with the vinyl sulfone to give the Michael adduct (11l-8), and iEDDA bioconjugation at the intrinsic double bond to give 11l-9 (Figure 58C).
A representative synthesis is shown in Figure 58D. The TMS group of 11l-10 was deprotected by NaF, followed by the reaction of the resulting ethynyl p-tolyl sulfone (11l-11) with N-Boc-pyrrole to afford the bicyclic 11l-12 in 71% yield over two steps. Upon deprotection of the Boc group under acidic conditions, the free azanorbornadiene (11l-13) was treated with a biotin unit bearing an acyl chloride tether (11l-14) to obtain 11l-15 in 75% yield. Finally, 11l-15 underwent Michael addition with a thiol of the C2Am mutant protein (11l-16) to provide the azabicyclic vinyl sulfone reagent 11l.
The binding ability of reagent 11l toward a phosphatidylserine phospholipid on the surface of apoptotic cells was investigated. Apoptotic HeLa cells were incubated with 11l, followed by incubation with streptavidin bearing the fluorescent dye Alexa555. Fluorescent microscopy images showed labeling of only apoptotic cells owing to the incubation with biotinylated 11l and streptavidin-Alexa555, which demonstrated that the C2Am unit in 11l worked as a targeting agent for the apoptotic cells. Finally, the labeling of 11l via iEDDA under physiological conditions was carried out. Apoptotic HeLa cells were incubated with 11l followed by treatment with 6-methyl-tetrazine-sulfo-Cy3 (Tz-Cy3) to functionalize the remaining double bond by iEDDA. Fluorescent imaging results showed intact labeling of the double bond in 11l with Tz-Cy3 by iEDDA in a cellular environment.
This study demonstrated the utility of a novel azabicyclic vinyl sulfone scaffold for cysteine-specific dual-protein labeling. The [2.2.1] azabicyclic vinyl sulfone hub 11l-12, which enables tri-functionalization via a bridging nitrogen, vinyl sulfone, and intrinsic double bond, could be further functionalized with biotin and a mutant protein to obtain the labeling reagent 11l. The sequence of transformations entails TFA-mediated acidolysis of the Boc group of 11l-12 to form 11l-13, acylation of the free amine in the latter with biotin-acid chloride 11l-14 to give 11l-15, which upon Michael addition at the vinyl sulfone with C2Am-SH (11l-16) in DMF containing aqueous sodium phosphate (NaPi) to give the target 11l. This mutant protein (C2-Am) worked as a pre-targeting agent for apoptotic cells, and post-labeling with biomolecules (e.g., streptavidin-Alexa555 or 6-methyl-tetrazine-sulfo-Cy3) via a biotin tag or iEDDA bioconjugations on cell surfaces could be achieved. This azanorbornadiene scaffold may find application for modification of target proteins and antibodies with two different functional units such as drugs, fluorophores, and tags for targeting identification.
12. Carbon Hubs (5)
Heterofunctional scaffolds, which contain a constellation of distinct (bio)conjugatable groups, are required for the rational construction of multifunctional molecules in service of bioconjugation, diagnosis, therapy and functionalization. Many reliable reactions for orthogonally linking two specific functional groups have been developed and applied to the synthesis of multifunctional molecules: the nucleophilic addition of amines on activated esters (e.g., NHS) ester, thiol–maleimide Michael addition, CuAAC, Cu-free strain-promoted azide–alkyne cycloaddition, tetrazine-based iEDDA with strained alkenes, and so on [289,290]. In this section, several all-carbon hubs are described that afford sequential couplings.
A multivalent and multimodal imaging probe (12a) was developed for PET/computed tomography (CT) tumor imaging by using a bifunctional chelator-based scaffold (Figure 59) [291]. The probe is comprised of single 3-arm carbon hub and three functional components: (A) 68Cu-1,4,7-NOTA as a PET/CT imaging agent; (B) AE105 peptide as a targeting agent for the uPA receptor, which is highly expressed in many human cancers, and (C) the cyclic peptide cyclo(-Arg-Gly-Asp-d-Tyr-Lys-) as a targeting agent for the tumor-associated integrin αvβ3. This construct (12a) has composition ABC and architecture [3][1]3.
A diazirine-based bis(glucose) derivative (12b) was developed for PAL of a GLUT (Figure 59) [292]. The derivative is comprised of a three-arm carbon hub and four functional components: (A) glucose as a targeting agent for GLUT; (B) a trifluoromethyl diazirine as a PAL agent; (C) a disulfide bond as a reduction-cleavable linker, and (D) a biotin tag. This construct (12b) has composition A2BCD and architecture [3][2]2[1]3.
An artificial tris(catecholate) siderophore–ampicillin conjugate (12c) was developed (Figure 59) [293]. The conjugate is comprised of a four-arm carbon hub and two functional components: (A) an acetylated catecholate as a ferric ion chelator, which provides bacterial membrane permeability via iron uptake pathways upon chelation with iron ion, and (B) aminopenicillin as an antibacterial drug. This construct (12c) has composition A3B and architecture [4][1]4.
A trifunctional compound (12d) built on a norbornene hub was developed (Figure 59) [294]. This compound was suitable for conducting three orthogonal click reactions in a one-flask process stemming from the presence of three conjugatable groups: a strained alkene from norbornene for iEDDA click chemistry [295,296] (Unit A), an alkyne for CuAAC (Unit B), and an electron-deficient maleimide group for thiol-Michael addition (Unit C). This construct (12d) has composition ABC and architecture [3]1[1]3.
The key design of 12d was based on the requirement of three different functional groups wherein each corresponding click reaction can be performed without any cross-reaction. As shown in Figure 60, maleic anhydride (12d-1) was first treated with cyclopentadiene [297] to give 12d-2 followed by propargyl alcohol to give the ring-opened difunctionalized intermediate 12d-3. In a separate route, 12d-1 was reacted with furan to give the Diels–Alder product 12d-4, which upon reaction with 2-aminoethanol gave 12d-5, which underwent thermal retro-Diels–Alder to give furan N-2-hydroxyethylmaleimide 12d-6 [298]. The final attachment was completed by esterification to join 12d-3 and 12d-6. The resulting scaffold 12d is amenable to one-flask reaction of the three aforementioned click reactions without any further protection technology or extra purification steps, which hints at attractive future designs as long as the reactions conditions and sequence are duly considered. Three different macromolecular segments now can be introduced simultaneously to impart complexity in the target molecule.
An oxazolone (also known as an azlactone) ring has potential as a powerful multifunctional scaffold because the ring-opening reaction with good nucleophiles such as amines is clean, specific and facile in a broad range of organic solvents as well as in aqueous solution at room temperature without any catalysts (Figure 61, inset) [299,300,301]. An oxazolone-based heteromultifunctional scaffold (12e) was developed for click chemistry (Figure 61) [302]. The scaffold is comprised of a single oxazolone hub and three functional components: (A) an alkene for conjugation in a thiol-ene radical reaction, which constitutes a type of click chemistry; (B) an alkyne for conjugation with an azide via Cu-catalyzed click reaction, and (C) azlactone for reaction with an amine via an oxazolone ring-opening reaction. This construct (12e) has composition A2BC and architecture [4][1]4.
The synthesis of 12e is shown in Figure 61. A TMS-protected alkyne was introduced to benzoic acid 12e-1 via Sonogashira cross-coupling, followed by conversion to the corresponding acylcyanohydrin 12e-2 in 75% yield. Treatment with an allylzinc reagent afforded the corresponding hydroxyamide 12e-3 in quantitative yield. A two-step sequence including 2-iodoxybenzoic acid (IBX) oxidation and Pinnick (Kraus) oxidation converted the alcohol 12e-3 into the corresponding carboxylic acid 12e-4 in 79% yield. After removal of the TMS group, intramolecular cyclization via mixed anhydride activation of the carboxylic acid 12e-4 by ethyl chloroformate gave the tri-functionalized oxazolone 12e in 98% yield.
The orthogonal reactivity of oxazolone 12e toward sequential aminolysis, Cu-catalyzed click chemistry, and radical alkene hydrothiolation was investigated with model benzyl substrates (Figure 61). Aminolysis and CuAAC are sequential reactions that proceeded in excellent yield regardless of the reaction order. The sequential coupling of an amine with 12e proceeded in 97% yield to give 12e-5, which then reacted quantitatively with an azide to give 12e-6. In addition, the simultaneous coupling of the amine and azide with 12e in a one-flask procedure provided 12e-6 in 95% yield, which demonstrates the orthogonality between the alkyne and oxazolone reactivities. The order of the amidation and reaction with azide could be reversed as shown by the intermediacy of 12e-7. In the case of aminolysis and the thiol-ene click reaction under UV irradiation (365 nm), the stepwise conjugation could also be achieved in satisfactory yield regardless of the reaction order to obtain 12e-8. On the contrary, the CuAAC reaction (12e → 12e-7) must be performed before the thiol-ene reaction (12e-6 → 12e-8 or 12e-7 → 12e-9) to avoid the undesired hydrothiolation of the alkyne. The CuAAC reaction was carried out in the presence of the ligand N,N,N′,N″,N″-pentamethyldiethylenetriamine (PMDETA), whereas the thiol-ene addition was carried out in the presence of the photoinitiator DMPA. Finally, the oxazolone scaffold 12e was successfully subjected to a one-flask orthogonal conjugation with three substrates in 85% yield without any purification (Figure 61).
This study demonstrated the utility of the new oxazolone-based heteromultifunctional scaffold for click chemistry reactions. The oxazolone scaffold 5, which contains azlactone, alkene, and alkyne groups, underwent stepwise orthogonal modifications with three model substrates (amine, azide, and thiol) to provide 12e-8 in high yield although the CuAAC reaction must be carried out before the thiol-ene reaction. Furthermore, the simultaneous one-flask conjugation with three model substrates on 12e was achieved in remarkable yield. These results strongly support use of this oxazolone-based scaffold 12e for bioconjugation or surface functionalization to construct multifunctional molecules.
13. Functional Hubs (12)
The members of the class described here have features in common with one or more of the aforementioned classes, but are distinguished by the role of the hub more than solely the composition of the hub. An enzymatically degradable cross-linking agent (13a) was developed for quantitation and visualization of hydrogel degradation (Figure 62) [303]. The enzyme of choice was MMP. The cross-linking agent contains two hubs, an enzymatically recognized Lys residue in a substrate peptide (B) cleaved by MMP and an independent Lys, and four functional components: (A) an acryloyl group as a cross-linking agent; (C) 2,4-dinitrofluorobenzene as a fluorescence quencher, and (D) MCA as a fluorophore. This construct (13a) has composition A2BCD and architecture [3]2[1]4.
A compound built around a triazine hub (13b) was developed as a new class of affinity glycoprobes for specific covalent labeling of carbohydrate-binding proteins (Figure 62) [304]. This affinity glycoprobe is composed of four units: (A) a triazine hub as a labeling agent; (B) an alkyne for CuAAC; (C) a furan for Diels–Alder conjugation to maleimide derivatives, and (D) a chitin pentaose as a targeting agent for carbohydrate-binding proteins. This construct (13b) has composition ABCD and architecture [3][1]3.
Treatment of cyanuric chloride (9a) with propargyl alcohol (Unit B) in the presence of i-Pr2EtN afforded mono-substitution to give dichlorotriazine 13b-1 in 80% yield (Figure 63). The subsequent reaction with furfurylamine (Unit C), also in the presence of i-Pr2EtN, provided diamino-substituted monochlorotriazine 13b-2 in 89% yield. In the next step, 13b-2 was treated with NMM and unprotected chitin pentaose (13b-3, an anomeric mixture) in aqueous solution to afford the desired ABCD molecule 13b (with α-anomeric configuration) in 45% yield. Most likely, NMM added to the reaction mixture functioned as a nucleophilic catalyst by forming a triazinylammonium intermediate. The selective substitution is attributed to the high acidity of the anomeric hydroxy group in chitin pentaose.
Compound 13b was used for affinity labeling of a chitin pentaose-binding protein, wheat germ agglutinin. Triazinylation of a nucleophilic residue in the protein was proposed to occur with release of chitin pentaose as a leaving group. The labeling of the protein was detected after treatment with azido-fluorescein, which attaches fluorescein to the alkyne (Unit B). Further study is necessary to quantify the protein labeling and to determine the position of the labeled amino acid residue(s).
Self-immolative linkers are valuable to enable release of a drug at a target site, particularly upon enzymatic triggering [305,306]. A compound (13c) containing a self-immolative linker that releases (upon enzymatic action) a phenol-containing drug was developed for use in ADCs (Figure 64) [307]. The linker is comprised of a single self-immolative hub (A) and four functional components: (B) a β-glucuronide that is cleaved by β-glucuronidase present in lysosomes and the tumor interstitium; (C) N,N′-dimethylethylenediamine (DMED) as a self-immolative linker; (D) psymberin as a cytotoxic drug, and (E) maleimide for conjugation to an antibody. This construct (13c) has composition ABCDE and architecture [3][2][1]3. Drug release arises upon enzymatic deglucuronidation, 1,6-elimination, decarboxylation, and cyclization of the DMED carbamate to liberate the free phenol moiety.
A highly efficient mitochondria-targeting antitumor theranostic prodrug (13d) was developed (Figure 64) [308]. The prodrug is comprised of two self-immolative hubs (A) and four functional components: (B) a phenylboronic ester linker that is cleaved by ROS such as H2O2, which is abundant in the mitochondria of tumor cells; (C) biotin for targeting biotin receptor-positive tumor cells; (D) 5-fluorouracil (5-FU) as a cytotoxic drug, and (E) ethidium as a fluorophore and targeting agent by intercalation with double-stranded RNA or DNA. This construct (13d) has composition A2B2C2D2E and architecture [3]2[2]3[1]4. In the presence of H2O2, the phenylboronic ester moiety is oxidized and hydrolyzed, and the self-immolative hubs are subsequently eliminated to release 5-FU and ethidium to the tumor cells, thereby providing both cytotoxicity and strong fluorescence.
Alsarraf et al. synthesized a dendritic folate-drug conjugate (13e) built around a combination of two self-immolative linkers (Figure 64) [309]. The folate-drug conjugate (13e) is composed of two monomethylauristatin E (MMAE) as anti-tumor drugs (Unit A), two self-immolative hubs (Units B and D), galactose as an enzymatic-cleavable trigger (Unit C) and folate as a FR targeting agent (Unit E). This construct (13e) has composition A2BCDE and architecture [3]2[1]4. This dendritic folate-drug conjugate can recognize the FR at the surface of cancer cells and was able to be endocytosed. Upon selective internalization, enzymatic cleavage of the glycosidic bond by lysosomal β-galactosidase followed by 1,6-elimination and decarboxylation leads to an aniline intermediate bearing two copies of the anti-neoplastic agent MMAE. The intermediate will later go through successive 1,6- and 1,4-elimination to release both MMAE units to kill cancer cells.
The synthesis is outlined in Figure 65. The alkylation of 3-nitro-4-hydroxybenzaldehyde (13e-1) with propargyl bromide afforded 13e-2, which upon glycosylation with acetobromo-β-d-galactose in the presence of 1,1,4,7,10,10-hexamethyltriethylenetetramine (HMTTA) afforded 13e-3. Carbonylation with p-nitrophenyl chloroformate afforded the key intermediate 13e-4 equipped with the self-immolative Unit B and sugar Unit C [310]. The synthesis of the self-immolative hub began by protection of bis(1,3-hydroxymethyl)benzene (13e-5) with silyl groups (13e-6), which upon nitration afforded nitroarene 13e-7. Catalytic transfer hydrogenation [311,312] gave aniline 13e-8, which upon treatment with fluoride gave the dimethanol aniline derivative 13e-9 [313]. It warrants mention that 13e-9 can be masked by two distinct protecting groups, which in later designs may enable conveyance of two distinct components by selective deprotection, whereas two identical MMAE motifs are used in this case.
The double self-immolative hub was then assembled by treating carbonate 13e-4 with aniline 13e-9 in the presence of HOBt to give the diol intermediate 13e-10 (Figure 65) [314]. Similar activation (via carbonylation) afforded active intermediate 13e-11, which upon nucleophilic substitution with MMAE afforded 13e-12. Full deprotection of the sugar hydroxy groups was then carried out and followed by copper-catalyzed click reaction with azido-folate to provide the final construct 13e. This work shows the elegance of installation of both targeting and enzyme-cleavable triggers onto an anti-tumor pro-drug. The chemical amplifier Unit D also provides a multi-fold drug releasing property, which is of great interest in the area of experimental therapeutics.
Fluorescence imaging is an attractive strategy for non-invasive visualization of cellular and biological processes that are involved in specific diseases, and has been widely used for disease diagnosis and treatment as well as clarification of complicated physiological events [315,316]. In particular, small molecule-based fluorescent probes for reporting in vivo enzymatic activities are a powerful tool for the detection and imaging of enzymatic activities in living systems owing to the following advantages: (1) rational designability and (in principle) well-defined synthesis; (2) high sensitivity; (3) fast and easy analysis; (4) cell permeability, and (5) real-time detection [24,317,318,319]. Although myriad small molecule-based fluorescent probes for in vivo detection have been developed, application to high-resolution imaging of enzyme activity and localization has been limited because these probes are usually subject to rapid diffusion from the reaction site [320]. Caspase is an attractive cysteine protease for imaging, because the caspase family is associated with human diseases such as cancer, degenerative diseases, and inflammatory disorders [321,322]. Therefore, the development of small molecule-based fluorescent probes for detecting and imaging the locale of caspase activity in the cells has been a sharp focus.
A fluorescence-quenched activity-based probe (qABP) based on quinolimine methide chemistry (13f) was developed for imaging live mammalian cells where caspase activities are localized (Figure 64) [323]. The qABP in this study is comprised of a functional quinolimine methide-based hub (A), a Lys hub, and four functional components: (B) Dabcyl as a quencher; (C) a biotin tag; (D) a substrate peptide cleaved by caspase, and (E) 5-carboxyfluorescein as a fluorophore. This construct (13f) has composition ABCDE and architecture [3]2[1]4.
Quinolimine (or quinone) methide-based structures have been used as functional scaffolds for constructing chemical imaging probes due to the self-immolative feature as well as the subsequent reactivity toward nucleophiles [324,325,326,327]. In this study, this quinolimine methide-based chemistry was advantageously applied to the development of the qABP. The specific compound (13f) designed includes the elimination of a peptide linked to a p-aminomandelic acid moiety via an amide bond to generate the reactive quinolimine methide species, which then reacts with a nucleophile. Proteolytic cleavage of the substrate peptide of qABP by caspase in the cell affords an intermediate (13f-1) that undergoes self-immolative 1,6-elimination of the quencher moiety to provide the reactive fluorescent quinolimine methide intermediate 13f-2 (Figure 66). The latter is electrophilic and was estimated to subsequently undergo three different labeling processes (Figure 66): (1) quenching by bulk water to generate the undesired hydrated product 13f-3, which could be easily washed away; (2) covalent binding to caspase to give 13f-4, and (3) reaction with a nearby nucleophilic species to give 13f-5 and likely accumulate in the cell. These immediate labeling reactions avoid the undesired rapid diffusion of the probe from the cell.
The synthesis of the quinolimine methide precursor (13f) began as is shown in Figure 67. Commercially available p-nitrophenylacetic acid (13f-6) was converted to the tert-butyl ester (13f-7) and subjected to benzylic bromination with N-bromosuccinimide (NBS) to give 13f-8. α-Acetoxylation gave p-nitromandelic acid (13f-9), which was subjected to cleavage of the tert-butyl group and condensation with O-allyl protected glycine (destined to serve as a linker) to give 13f-10. Removal of the acetyl group gave 13f-11, which upon treatment to dissolving metal reduction gave aniline 13f-12. Elaboration at the anilino nitrogen was then carried out by introduction of the first amino acid of the substrate peptide (Fmoc-Asp(OtBu)-OH) to obtain 13f-13. After activation of the hydroxyl group of 13f-13 by p-nitrophenyl chloroformate to give 13f-14, the Dabcyl moiety was coupled to yield 13f-15. Finally, deprotection of the allyl group by Pd catalysis afforded the p-aminomandelic acid building block 13f-16. Thus, in broad overview, preparation of the qABP relies on derivatization of the p-aminomandelic acid core 13f-16 with two amino acids (Fmoc-Asp(OtBu) and Gly) and the quencher. Building block 13f-16 was readily applicable to standard Fmoc SPPS because the structure is similar to that of an N-Fmoc-protected amino acid.
Building block 13f-16 and the other amino acids were used for constructing the probe by standard Fmoc SPPS. Consecutive Fmoc-deprotection and coupling reactions on the Rink amide AM resin 13f-17 successfully proceeded via intermediates 13f-18–20 to give 13f-21 (Figure 68). The couplings proceeded with various standard cocktails of reagents including use of HATU and 1-hydroxy-7-azabenzotriazole (HOAt) in DMF containing DIEA. Selective deprotection of the Mtt group of the Lys residue under acidic conditions, reaction of di-O-acetylfluorescein NHS ester with the resulting amino group, and removal of the di-O-acetyl protection under basic conditions gave 13f-22. Finally, removal of the resin and protecting groups by treatment with TFA provided the desired qABP 13f.
The substrate specificity of qABP 13f toward the caspase family was investigated in an in vitro enzymatic assay. The fluorescence results in the standard microplate-based enzymatic assay showed that the probe 13f was mainly hydrolyzed by caspase-3 and caspase-7. On the other hand, cathepsin B, L, and S as well as legumain, which are the most abundant cysteine proteases in mammalian cells, did not cleave 13f. In kinetic assays against caspase-3 and -7, probe 13f displayed 10-fold higher catalytic efficiency compared with the conventional coumarin-based caspase-3 and -7 substrate N-acetyl-Asp-Glu-Val-Asp-7-amido-4-trifluoromethylcoumarin; such results demonstrate the excellent substrate specificity of qABP 13f toward the intended enzyme targets (i.e., caspases).
The imaging ability of the qABP 13f for enzyme localization in cells was evaluated next with an in vivo assay. Apoptotic mammalian cells, where caspase-3 and -7 are overexpressed, were incubated with 13f, and the lysates were analyzed by in-gel fluorescence scanning. Various non-target proteins labeled with the quinolimine methide intermediate were observed (Pathway 3 in Figure 66). In contrast, any species labeled with the reactive intermediate were not observed in the case of the lysates of normal HeLa cells in which caspases were not expressed. This result indicated that caspase activities in the cells induced release of the reactive quinolimine methide intermediate, and the intermediate was predominantly trapped by other proteins in the vicinity. Finally, to demonstrate that the aforementioned labeling process provides sufficient spatial resolution to accurately report localization of caspases in the cell, normal and apoptotic HeLa cells treated with 13f were examined by confocal fluorescence microscopy. The apoptotic HeLa cells treated with 13f clearly showed strong fluorescence after washing off the fluorescent contaminants including the hydrated intermediate (Pathway 1 in Figure 66); in contrast, no fluorescence appeared in the normal HeLa cells treated with 13f. These results demonstrated that non-specific labeling with qABP 13f enables bioimaging of the live mammalian cells where caspase-3 and -7 are localized.
In conclusion, a new class of qABPs based on quinolimine methide chemistry for imaging of caspase activities in live mammalian cells was successfully synthesized from the amino acid-like quinolimine methide building block 13f-16. The in vitro caspase assay exhibited excellent substrate specificity and catalytic efficiency of qABP 13f against caspase-3 and -7 compared with that of the conventional coumarin-based probe. The fluorescence microscopic studies demonstrated that 13f could be used for bioimaging apoptotic cells through caspase activity. The biotin tag in 13f can be replaced with other functional molecules such as a cell-penetrating peptide, illustrating the versatility of this probe design. The modularity of this probe suggests applications to bioimaging of a variety of other endogenous enzymes.
A universal dark quencher (UDQ) (13g) was developed by using a trivalent nitro-Dabcyl quencher (Figure 69) [328]. The UDQ is comprised of a single nitro-Dabcyl as both hub and quencher (A) and four functional components: (B) a carboxylic acid as a conjugatable group; (C) BHQ-3 as a first quencher; (D) BHQ-1 as a second quencher, and (E) a sulfobetaine as a water-solubilizing group. This construct (13g) has composition ABCDE and architecture [3][2][1]3.
A molecular renal probe (13h) with high renal clearance efficiency was developed for in vivo optical imaging of drug-induced acute kidney injury (Figure 69) [329]. The probe is comprised of a single cyanine dye as a hub and fluorophore (B) and three functional components: (A) (2-hydroxypropyl)-β-cyclodextrin as a renal clearance moiety, which provides dramatic renal clearance efficiency of the probe; (C) a phenoxy-dioxetane as a chemiluminophore, and (D) a trifluoromethanesulfonyl (or triflyl) group cleaved by the superoxide anion (O2·−), which is a biomarker for acute kidney injury. This construct (13h) has composition A2BCD and architecture [3][2][1]3. The probe emits NIR fluorescence but becomes chemiluminescent only after the cleavage of the trifluoromethanesulfonyl group by the superoxide anion. The probe enables simultaneous monitoring of superoxide anion and clearance of the probe via chemiluminescent and NIR fluorescence signals respectively, providing a direct comparison of the time between upregulation of the superoxide anion and changes in glomerular filtration after nephrotoxic exposure in living animals.
A glycoporphyrin-based theranostic agent (13i) was developed for multimodal imaging and PDT towards cancer (Figure 69) [330]. The agent is comprised of a single Zn-porphyrin (as a combined hub, fluorophore, and photosensitizer, unit B) and two other components: (A) glucose as a targeting agent for the GLUT proteins on the cancer cells and (C) 2-fluoro-2-deoxy glucose (FDG) as a potential PET imaging agent. This construct (13i) has composition A3BC and architecture [4][1]4.
A porphyrin-based photosensitizer (13j) was developed that integrates MRI and fluorescence diagnostics with mitochondrial-targeted PDT in cancer theranostics (Figure 69) [331]. The photosensitizer is comprised of a single Mn-porphyrin (as a hub, fluorophore, photosensitizer, and MRI agent, B) and two functional components: (A) transplatin as a photosensitizer and (C) triphenylphosphonium cation as a targeting agent for mitochondria. This construct (13j) has composition A3BC and architecture [4][1]4.
The synthesis of 13j is shown in Figure 70. A mixed-aldehyde condensation [332] under Adler conditions with pyrrole (13j-1), 4-pyridylcarboxaldehyde (13j-2) and 4-hydroxybenzaldehyde (13j-3) in refluxing propionic acid containing acetic anhydride afforded a mixture of porphyrins. This one-flask procedure is ostensibly simple but chromatography in some instances can be prohibitively taxing given the formation of up to six porphyrins in addition to the inevitable numerous byproducts. For an overall yield of porphyrins of 20%, and the use of a 3:1 ratio of A and B aldehydes to favor the desired A3B-porphyrin (expected ~41% of the total porphyrins on the basis of statistics), the anticipated yield of the desired A3B-porphyrin would be about 8%. Losses on chromatography would of course degrade the yield further. Statistical reactions can be beguiling in this way [333]. (Here, the A3B terminology refers to the porphyrin substituents and not the abcedarian nomenclature of the graphs described throughout the text.) Chromatographic purification gave the desired tripyridylporphyrin (13j-4), albeit bearing the propionate ester of the lone phenol substituent. Saponification of the latter gave the free phenol (13j-5), which was alkylated with 4-bromobutyltriphenylphosphonium bromide (13j-6) to give the free base tripyridylporphyrin (13j-7). Metalation with manganese acetate gave metalloporphyrin 13j-8, which upon treatment with transplatin and silver nitrate gave, following removal of silver chloride, the tri-platinated porphyrin Pt-MnPor-PPh3 (13j) [334].
A cancer theranostic agent (13k) that contains a fluorophore and a cytotoxic drug was developed (Figure 69) [335]. Here, a BODIPY provides a functional hub. The constituents include a multifunctionalizable BODIPY as an optical imaging fluorophore (Unit A), a glucose derivative as a targeting agent (Unit B), a phosphonium as solubilizing group (Unit C), and an Au(I) complex as a cytotoxic drug (Unit D). This construct (13k) has composition ABCD and architecture [3]1[1]3.
The synthesis of 13k relies on synthesis of a tri-functionalizable BODIPY hub [336]. Thus, treatment of methyl 4-formylbenzoate (13k-1) with excess pyrrole [337] afforded the corresponding dipyrromethane 13k-2, which upon reaction with N-chlorosuccinimide (NCS) gave selective α-pyrrole chlorination yielding the 1,9-dichlorodipyrromethane 13k-3. Treatment with p-chloranil gave dipyrrin 13k-4, which upon exposure to BF3·O(Et)2 in the presence of triethylamine [259] gave the corresponding BODIPY 13k-5; this procedure is quite concise to convert an aryl aldehyde to the BODIPY [336] (Figure 71). Reaction with strong acid gave the free carboxylic acid 13k-6. The assembly of the following units was restricted by the fact that a second functionalization on the BODIPY pyrrole core ring was allowed only when a primary aliphatic amine was employed as the first nucleophile. On the basis of this reason, 13k-6 was treated with β-thioglucose tetraacetate to give nucleophilic substitution of the α-chloropyrrole and form 13k-7, which upon subsequent nucleophilic substitution of the remaining of the α-chloropyrrole with the amine of 2-(triphenylphosphonium)ethylammonium dibromide in the presence of NaHCO3 gave the difunctionalized intermediate 13k-8. Final introduction of the Au(I) complex was achieved by amidation of the p-carboxy group with 2-(diphenylphosphino)ethylamine-aurichloride to form the target 13k [335].
This work takes the advantage of the BODIPY as a simple tri-functionalizable platform as well as an optical imaging agent for tracking, which confirms the availability for rapid synthesis of a large library of compounds. Other analogues related to 13k also were prepared by Pliquett and coworkers through derivatization of this versatile BODIPY scaffold [335]. The well-established step-by-step synthetic protocol provides a promising path towards potential biological molecular designs.
Developing spatially defined architectures is an essential undertaking in the field of artificial photosynthesis, as photoinduced electron transfer and energy transfer are influenced by the three-dimensional arrangements of interacting pigments. We developed a one-flask synthesis of shape-persistent macrocycles [338] that incorporate six porphyrins. A representative example (13l) is shown in Figure 72. The one-flask synthesis relies on the Sonogashira coupling of a diethynylporphyrin (zinc chelate, 13l-1) and a diiodoporphyrin (free base, 13l-2) in the presence of a tripyridyl template (13l-3) (Figure 73). The template coordinates to the zinc porphyrins and guides the assembly to give the templated product (13l-4) [338]. This synthetic approach was applied to prepare eight cyclic hexamers of type 13l that differ in the nature of the substituents at the nonlinking positions (Figure 74) [338,339,340,341]. Yields range from 0.7–9.6% depending on the nature of the nonlinking substituents.
To call attention to the metalation states of the various porphyrins, the nomenclature in the published literature is employed here. Four types of cyclic hexamers, all of type 13l, have been prepared:
Type I cyclic hexamers (cyclo-Zn3Fb3-1a–c) employ two trans-A2B2-porphyrins wherein the two non-linking substituents on a given porphyrin are identical with each other. (Here, “Fb” stands for free base, indicating the absence of a centrally coordinated metal, and “Zn” indicates the presence of the zinc(II) chelate. For additional porphyrin nomenclature, see [332,342]). Here, all nonlinking substituents in the cyclic hexamer are identical with each other.
Type II cyclic hexamers (cyclo-Zn3Fb3-1d,e) employ two trans-A2B2-porphyrins wherein the two non-linking substituents on a given porphyrin are identical with each other. Here, the nonlinking substituents on the zinc porphyrin differ from those on the free base porphyrin [341].
Type III cyclic hexamers (cyclo-Zn3Fb3-1f–h) employ two trans-AB2C-porphyrins wherein the two non-linking substituents on a given porphyrin are not identical with each other. Here, the nonlinking substituents on the zinc porphyrin are identical with those on the free base porphyrin. Note that while the display shows all R substituents and R′ substituents on opposite edges of the macrocycle, the porphyrins can rotate about the trans axis, enabling randomization of the positions with respect to the two edges [341]. This construct (cyclo-Zn3Fb3-1f–h) has composition A2B2C2D2E2F2 and molecular architecture c-[4]6[1]12.
Type IV cyclic hexamers (cyclo-Zn2FbZn2Fb and cyclo-Zn2FbZn2Mg) employ two trans-A2B2-porphyrins wherein the two non-linking substituents on a given porphyrin are identical with each other. Here, all nonlinking substituents in the cyclic hexamer are identical with each other, and “Mg” indicates the presence of the magnesium(II) chelate. The synthesis of the two Type IV cyclic hexamers proceeded via a stepwise route wherein an acyclic pentamer of porphyrins was reacted with the sixth porphyrin, or two acyclic trimers of porphyrins were reacted to give the cyclic hexamer, with final macrocyclization yields of 10% and 13.6% yield, respectively. Such directed syntheses were essential to create the distinct metalation motifs [340].
The various metalation motifs enabled docking of a seventh porphyrin and channeling of excited-state energy among the porphyrins in the “wheel” of the cyclic hexamer (13l) and from such porphyrins to the docked porphyrin in the “spoke” position. The supramolecular architecture provides a platform for study of the flow of excited-state energy and electron transfer among the pigments [338,339,343,344,345]. The shape-persistent nature of the architectures is a key feature underpinning such studies [346,347]. While valuable for studies of excited-state energy flow among porphyrins following absorption of light, use of such large, shape-persistent compounds as versatile scaffolds to address broader issues in the energy sciences remains fallow.
A general synthetic route to ABCD-porphyrins is shown in Figure 75. A 1,9-diacyldipyrromethane (13l-5) is treated to reduction with sodium borohydride, which affords the corresponding dipyrromethane-1,9-dicarbinol (13l-6). The latter is partially purified and condensed with a dipyrromethane (13l-7) to give the resulting porphyrinogen (13l-8, a hexa-hydroporphyrin). Subsequent treatment with DDQ affords the porphyrin (13l-9). The route can be employed to access porphyrins with up to four distinct meso-substituents (i.e., ABCD-porphyrins) [342]. Yields of 20–30% are routine for this process, but more important than yield is the absence of acidolytic scrambling of substituents [333]. Conditions have been devised to enable high fidelity conversion of the substituents without such acidolysis and rearrangement of the substituents [348]. Other routes to such porphyrins also are available [349].
An example of the route is provided in Figure 75. Condensation of an aldehyde (13l-10) in the presence of excess pyrrole (as solvent and reactant) affords the dipyrromethane (13l-7a–f). The examples shown were prepared using TFA [341], although other acids can be employed [350]. A given dipyrromethane (13l-7b) undergoes acylation at the 1-position upon treatment with a Grignard reagent followed by an S-2-pyridyl thioate (13l-11) to afford the 1-acyldipyrromethane (13l-12); subsequent acylation at the 9-position is achieved with a Grignard reagent and an acid chloride (13l-13) to afford the 1,9-diacyldipyrromethane (13l-14). An alternative to the sequential acylation process is a one-flask process, wherein the Grignard reagent and an acid chloride (13l-13) are used in excess. Treatment of 1,9-diacyldipyrromethane (13l-14) with NaBH4 followed by short workup and subsequent reaction with a dipyrromethane (13l-7a) under suitable acidic conditions followed by oxidation afforded the corresponding porphyrin. Given a free base porphyrin, metalation with zinc acetate smoothly affords the zinc porphyrin. Thus, metalation with zinc acetate gave porphyrin 13l-15, which here contains two ethynes, one protected carboxylic acid, and one S-acetylthioether.
The successful preparation of 13l-15 illustrates the broad scope of the synthetic route [342]. While the route to trans-AB2C-porphyrins can be applied to prepare trans-A2B2-porphyrins, the latter also can be access via simpler routes including those that do not entail acylation of a dipyrromethane [342]. The upshot is that porphyrins can be prepared by rational synthetic methods to give hubs of composition up to ABCD. Such porphyrins can be elaborated in a rational manner to give large, shape-persistent macrocycles equipped with diverse functional groups.
14. Outlook
The de novo synthesis of molecular scaffolds enables rich opportunities in fields ranging from pure chemistry to materials science and the life sciences. Scaffolds serve as a nexus for juxtaposition of multiple groups. The thrust of the present review has been to illustrate the wide diversity of scaffolds that have been reported in recent years, chiefly the past two decades. The ability to prepare and derivatize scaffolds in a rational manner provides a valuable alternative to the traditional statistical derivatization of multifunctional carriers; the former requires more work but affords a homogeneous product; the latter is quicker but affords an intractably complex mixture.
Scaffolds have been categorized here with regards to composition (distinct appended groups ABCDE) and architecture (molecular graphs [M]x[N]y[O]z) to allow analysis apart from the chemical framework and functional groups. Index numbers provided along with the graphical abstract in each case highlight the sequence of couplings for assembly and derivatization of a given scaffold. Altogether, the review describes 30 hub molecules, 31 distinct molecular graphs, 107 examples (scaffolds and derivatives thereof), and 19 illustrative syntheses. A recurring theme in the development of scaffolds chemistry has been the adoption of techniques from bioconjugate chemistry and peptide synthesis, exemplified by use of click chemistry and the hub lysine. The survey here shows alternative hubs (e.g., cyanuric chloride, ABC; a [2.2.1] azabicyclic vinyl sulfone, ABC; benzotrifuranone, ABC; oxazolone, A2BC; porphyrin, ABCD) and companion reactions that would appear to be comparable to the superb features offered by extant click chemistry.
The demands for macromolecular conjugates a century ago could only be met with naturally occurring carriers and derivatization via statistical reactions. The advent of diverse scaffolds for rational chemistry points to a new era of chemistry that fulfills needs unmet by the powerful capabilities of macromolecular synthesis (access to large molecular weight polymers, albeit polydisperse), protein engineering (exquisite and versatile molecular structures with genetic expression, but often overly complex and susceptible to errant folding), and three-dimensional printing (top-down access to intricate structures, albeit not yet with the desired molecular scale or functional group specificity). A next challenge is the development of scaffolds for rational chemistry of mesoscale dimensions (few 10s–100s of nm) thereby offering a true and compelling complement to the traditional macromolecular carriers.
Author Contributions
Conceptualization, H.F. and J.S.L.; formal analysis, D.S., Z.W. and H.F.; writing—original draft preparation, D.S., Z.W. and H.F.; writing—review and editing, D.S., Z.W., H.F. and J.S.L.; funding acquisition, J.S.L. All authors have read and agreed to the published version of the manuscript.
Funding
The authors acknowledge funding from the Division of Chemical Sciences, Geosciences and Biosciences, Office of Basic Energy Sciences of the U.S. Department of Energy (DE-FG02-05ER15661).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data in the present review are from the cited papers or are derived herein.
Conflicts of Interest
The authors declare no competing financial interest.
Abbreviations
| AAZTA | 1,4-bis(carboxymethyl)-6-[bis(carboxymethyl)]amino-6-methylperhydro-1,4-diazepine |
| ADC | antibody–drug conjugate |
| 5-AVA | 5-aminovaleric acid |
| All | Allyl |
| Alloc | allyloxylcarbonyl |
| AMP | adenosine monophosphate |
| BBN | bombesin peptide |
| BHQ | black hole quencher |
| Bn | benzyl |
| BNCT | boron neutron capture therapy |
| Boc | tert-butyloxycarbonyl |
| BODIPY | boron-dipyrrin |
| BP | benzophenone |
| C2Am | C2A domain of synaptotagmin-I |
| CA | carbonic anhydrase |
| CBT | 2-cyanobenzothiazole |
| CDR | complementarity determining region |
| CES | carboxyesterase |
| Cha | cyclohexylalanine |
| Cit | citrulline |
| Cp* | pentamethylcyclopentadienyl |
| CSAN | chemically self-assembled nanoring |
| CT | computed tomography |
| CuAAC | copper-promoted [3 + 2]-azide–alkyne cycloaddition |
| Dabcyl | 4-dimethylaminophenylazobenzene-4-carboxylic acid |
| Dansyl | 5-(dimethylamino)naphthalene-1-sulfonyl |
| DCC | N,N′-dicyclohexylcarbodiimide |
| Dde | 1-(4,4-dimethyl-2,6-dioxocyclohexylidene)ethyl |
| DEAC | 7-diethylaminocoumarin |
| DHP | 3,4-dihydro-2H-pyran |
| DIBAL-H | diisobutylaluminum hydride |
| DIBO | 5,6-didehydro-11,12-dihydrodibenzo[a,e]cyclooctene |
| DIC | N,N′-diisopropylcarbodiimide |
| DIEA | diisopropylethylamine |
| DKP | 2,5-diketopiperazine |
| DMAP | 4-(N,N-dimethylamino)pyridine |
| DMED | N,N′-dimethylethylenediamine |
| DMF | N,N-dimethylformamide |
| DMPA | 2,2-dimethoxy-2-phenylacetophenone |
| DMSO | dimethylsulfoxide |
| DOTA | 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid |
| DPPA | diphenylphosphoryl azide |
| DTBPY | 4,4′-di-tert-butyl-2,2′-bipyridyl |
| DTT | 1,4-dithio-d-threitol |
| EDA | ethylenediamine |
| EDC | 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride |
| EDT | Ethanedithiol |
| EPR | enhanced permeation and retention |
| ESI-MS | electrospray ionization mass spectrometry |
| FITC | fluorescein isothiocyanate |
| Fmoc | 9-fluorenylmethoxycarbonyl |
| FR | folate receptor |
| FRET | Förster resonance energy transfer |
| 5-FU | 5-fluorouracil |
| GABA | γ-aminobutyric acid |
| GFP | green fluorescent protein |
| GLUT | glucose transporter |
| GRPR | gastrin-releasing peptide receptor |
| HATU | O-(1H-7-azabenzotriazol-1-yl)-1,1,3,3 -tetramethyluronium hexafluorophosphate |
| HBTU | O-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate |
| hGC | heteroglycocluster |
| HCTU | O-(1H-6-chlorobenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate |
| HMTTA | 1,1,4,7,10,10-hexamethyltriethylenetetramine |
| HOAt | 1-hydroxy-7-azabenzotriazole |
| HOBt | 1-hydroxybenzotriazole |
| Hof | homophenylalanine |
| IBX | 2-iodoxybenzoic acid |
| ICP-MS | Inductively coupled plasma mass spectrometry |
| iEDDA | inverse electron-demand Diels–Alder |
| KAHA | α-ketoacid–hydroxylamine |
| LHRH | luteinizing hormone releasing hormone |
| MALDI-TOF-MS | matrix-assisted laser desorption ionization time-of-flight mass spectrometry |
| MBHA | 4-methylbenzhydrylamine |
| MCA | 7-methoxycoumarinyl-4-acetic acid |
| MMAE | monomethylauristatin E |
| MMP | matrix metalloproteinase |
| mPDA | m-phenylenediamine |
| MRI | magnetic resonance imaging |
| Mtt | 4-methyltrityl |
| MTX | methotrexate |
| nanoSIMS | nanoscale secondary ion mass spectrometry |
| NaPi | sodium phosphate |
| NBS | N-bromosuccinimide |
| NCL | native chemical ligation |
| NCS | N-chlorosuccinimide |
| NGF | nerve growth factor |
| NHS | N-hydroxysuccinimide |
| NHS ester | N-hydroxysuccinimidyl ester |
| NIQ | naphthylisoquinoline |
| NIR | near infrared |
| NMM | N-methylmorpholine |
| NMP | N-methyl-2-pyrrolidone |
| NMR | Nuclear magnetic resonance |
| NOTA | 1,4,7-triazacyclononane-triacetic acid |
| NP | nanoparticle |
| NPY(Y1)R | neuropeptide Y receptor subtype 1 |
| OA | oleanolic acid |
| Pag | propargylglycine |
| PAL | photoaffinity labeling |
| Pam | palmitoyl |
| PBS | Phosphate-buffered saline |
| PDT | photodynamic therapy |
| PET | positron emission tomography |
| PFTase | protein farnesyl transferase |
| PKA | AMP-dependent protein kinase |
| PKI | protein kinase inhibitor |
| PMDETA | N,N,N′,N″,N″-pentamethyldiethylenetriamine |
| PpIX | protoporphyrin IX |
| PPTS | pyridinium p-toluenesulfonate |
| PTSA | p-toluenesulfonic acid |
| PyBOP | benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate |
| qABP | quenched activity-based probe |
| Q-LDT | quenched ligand-directed tosylate |
| RGD | Arg-Gly-Asp |
| ROS | reactive oxygen species |
| RT | room temperature |
| SASRIN | super acid sensitive resin |
| SDS-PAGE | sodium dodecylsulfate-polyacrylamide gel electrophoresis |
| siRNA | small interfering RNA |
| SPAAC | strain-promoted [3 + 2]-azide–alkyne cycloaddition |
| SPECT | single photon emission computed tomography |
| SPPS | solid-phase peptide synthesis |
| TACTV | tris-tertiary amine cyclotriveratrylene |
| TAMRA | tetramethylrhodamine |
| TBAF | tetrabutylammonium fluoride |
| TBS | tert-butyldimethylsilyl |
| TBTA | tris[(1-benzyl-1H-1,2,3-triazole-4-yl)methyl]amine |
| TCEP | tris(2-carboxyethyl)phosphine |
| TES | triethylsilyl |
| TFA | trifluoroacetic acid |
| TFAA | trifluoroacetic anhydride |
| THF | tetrahydrofuran |
| THP | 2-tetrahydropyranyl |
| THPTA | tris(3-hydroxypropyltriazolylmethyl)amine |
| TIPS | triisopropylsilyl |
| TIS | triisopropylsilane |
| TLR | Toll-like receptor |
| TMS | trimethylsilyl |
| Trt | trityl |
| Tz-Cy3 | 6-methyl-tetrazine-sulfo-Cy3 |
| UDQ | universal dark quencher |
| uPA | urokinase-like plasminogen activator |
| UV | ultraviolet |
References
- Landsteiner, K. The Specificity of Serological Reactions, Revised; Dover Publications: New York, NY, USA, 1962. [Google Scholar]
- King, T.P. Chemical and Biological Properties of Some Atopic Allergens. In Advances in Immunology Volume 23; Elsevier: Amsterdam, The Netherlands, 1976; pp. 77–105. [Google Scholar]
- Mayer, R.L. The Significance of Cross-Links in the Formation of Hapten-Carrier Complexes. Int. Arch. Allergy Immunol. 1956, 8, 115–129. [Google Scholar] [CrossRef] [PubMed]
- Plescia, O.J. The role of the carrier in antibody formation. Curr. Top. Microbiol. Immunol. 1969, 50, 78–106. [Google Scholar] [CrossRef] [PubMed]
- Kontiainen, S.; Makela, O.; Hurme, M. Immune responses to hapten conjugates in vitro. Q. Rev. Biophys. 1975, 8, 507–522. [Google Scholar] [CrossRef] [PubMed]
- King, T.P.; Kochoumian, L.; Ishizaka, K.; Lichtenstein, L.M.; Norman, P.S. Immunochemical studies of dextran coupled ragweed pollen allergen, antigen E. Arch. Biochem. Biophys. 1975, 169, 464–473. [Google Scholar] [CrossRef]
- Carter, J.M. Techniques for conjugation of synthetic peptides to carrier molecules. Methods Mol. Biol. 1994, 36, 155–191. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.R.; Markl, J. Keyhole limpet hemocyanin (KLH): A biomedical review. Micron 1999, 30, 597–623. [Google Scholar] [CrossRef]
- Bundle, D.R.; Paszkiewicz, E.; Elsaidi, H.R.H.; Mandal, S.S.; Sarkar, S. A Three Component Synthetic Vaccine Containing a betab-Mannan T-Cell Peptide Epitope and a beta-Glucan Dendritic Cell Ligand. Molecules 2018, 23, 1961. [Google Scholar] [CrossRef] [Green Version]
- Molina, N.; Martin-Serrano, A.; Fernandez, T.D.; Tesfaye, A.; Najera, F.; Torres, M.J.; Mayorga, C.; Vida, Y.; Montanez, M.I.; Perez-Inestrosa, E. Dendrimeric Antigens for Drug Allergy Diagnosis: A New Approach for Basophil Activation Tests. Molecules 2018, 23, 997. [Google Scholar] [CrossRef] [Green Version]
- Louie, A. Multimodality imaging probes: Design and challenges. Chem. Rev. 2010, 110, 3146–3195. [Google Scholar] [CrossRef] [Green Version]
- Beal, D.M.; Jones, L.H. Molecular scaffolds using multiple orthogonal conjugations: Applications in chemical biology and drug discovery. Angew. Chem. Int. Ed. 2012, 51, 6320–6326. [Google Scholar] [CrossRef]
- Kanfar, N.; Bartolami, E.; Zelli, R.; Marra, A.; Winum, J.Y.; Ulrich, S.; Dumy, P. Emerging trends in enzyme inhibition by multivalent nanoconstructs. Org. Biomol. Chem. 2015, 13, 9894–9906. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, J.; Ma, S.; Liu, Q.; Huang, L.; Chen, X.; Lou, K.; Wang, W. Recent developments in multimodality fluorescence imaging probes. Acta Pharm. Sin. B 2018, 8, 320–338. [Google Scholar] [CrossRef]
- Clave, G.; Boutal, H.; Hoang, A.; Perraut, F.; Volland, H.; Renard, P.Y.; Romieu, A. A novel heterotrifunctional peptide-based cross-linking reagent for facile access to bioconjugates. Applications to peptide fluorescent labelling and immobilisation. Org. Biomol. Chem. 2008, 6, 3065–3078. [Google Scholar] [CrossRef]
- Batschelet, E. Introduction to Mathematics for Life Scientists, 3rd ed.; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 1979; pp. 446–452. [Google Scholar]
- Jiang, J.; Chen, C.-Y.; Zhang, N.; Vairaprakash, P.; Lindsey, J.S. Polarity-Tunable and Wavelength-Tunable Bacteriochlorins Bearing a Single Carboxylic Acid or NHS Ester. Use in a Protein Bioconjugation Model System. New J. Chem. 2015, 39, 403–419. [Google Scholar] [CrossRef]
- Bondy, J.A.; Murty, U.S.R. Graph Theory with Applications; North Holland: New York, NY, USA, 1976. [Google Scholar]
- Garcia-Domenech, R.; Galvez, J.; de Julian-Ortiz, J.V.; Pogliani, L. Some new trends in chemical graph theory. Chem. Rev. 2008, 108, 1127–1169. [Google Scholar] [CrossRef] [PubMed]
- Amigo, J.M.; Galvez, J.; Villar, V.M. A review on molecular topology: Applying graph theory to drug discovery and design. Naturwissenschaften 2009, 96, 749–761. [Google Scholar] [CrossRef]
- Choi, K.Y.; Swierczewska, M.; Lee, S.; Chen, X. Protease-activated drug development. Theranostics 2012, 2, 156–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, S.; Zhang, H.; Huang, W.; Zhou, J.; Qian, H.; Chen, W. The application of an aryl hydrazine linker prevents beta-elimination side products in the SPPS of C-terminal cysteine peptides. J. Pept. Sci. 2010, 16, 309–313. [Google Scholar] [CrossRef]
- Price, E.W.; Orvig, C. Matching chelators to radiometals for radiopharmaceuticals. Chem. Soc. Rev. 2014, 43, 260–290. [Google Scholar] [CrossRef]
- Fu, Y.; Finney, N.S. Small-molecule fluorescent probes and their design. RSC Adv. 2018, 8, 29051–29061. [Google Scholar] [CrossRef] [Green Version]
- MacPherson, D.S.; Fung, K.; Cook, B.E.; Francesconi, L.C.; Zeglis, B.M. A brief overview of metal complexes as nuclear imaging agents. Dalton Trans. 2019, 48, 14547–14565. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, F.; Hyun, J.Y.; Wei, T.; Qiang, J.; Ren, X.; Shin, I.; Yoon, J. Recent progress in the development of fluorescent, luminescent and colorimetric probes for detection of reactive oxygen and nitrogen species. Chem. Soc. Rev. 2016, 45, 2976–3016. [Google Scholar] [CrossRef]
- Murale, D.P.; Hong, S.C.; Haque, M.M.; Lee, J.S. Photo-affinity labeling (PAL) in chemical proteomics: A handy tool to investigate protein-protein interactions (PPIs). Proteome Sci. 2017, 15, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Cheng, Y.; Zhao, X.; Luo, Y.; Chen, J.; Yuan, W.E. Advances in redox-responsive drug delivery systems of tumor microenvironment. J. Nanobiotechnol. 2018, 16, 74–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gisbert-Garzarán, M.; Manzano, M.; Vallet-Regí, M. Self-immolative chemistry in nanomedicine. Chem. Eng. J. 2018, 340, 24–31. [Google Scholar] [CrossRef]
- Alouane, A.; Labruere, R.; Le Saux, T.; Schmidt, F.; Jullien, L. Self-immolative spacers: Kinetic aspects, structure-property relationships, and applications. Angew. Chem. Int. Ed. 2015, 54, 7492–7509. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.E.; Green, O.; Gnaim, S.; Shabat, D. Dendritic, Oligomeric, and Polymeric Self-Immolative Molecular Amplification. Chem. Rev. 2016, 116, 1309–1352. [Google Scholar] [CrossRef]
- Dumoulin, F.; Durmuş, M.; Ahsen, V.; Nyokong, T. Synthetic pathways to water-soluble phthalocyanines and close analogs. Coord. Chem. Rev. 2010, 254, 2792–2847. [Google Scholar] [CrossRef]
- Pisarek, S.; Maximova, K.; Gryko, D. Strategies toward the synthesis of amphiphilic porphyrins. Tetrahedron 2014, 70, 6685–6715. [Google Scholar] [CrossRef]
- Luciano, M.; Bruckner, C. Modifications of Porphyrins and Hydroporphyrins for Their Solubilization in Aqueous Media. Molecules 2017, 22, 980. [Google Scholar] [CrossRef] [Green Version]
- Ogata, F.; Nagaya, T.; Maruoka, Y.; Akhigbe, J.; Meares, A.; Lucero, M.Y.; Satraitis, A.; Fujimura, D.; Okada, R.; Inagaki, F.; et al. Activatable Near-Infrared Fluorescence Imaging Using PEGylated Bacteriochlorin-Based Chlorin and BODIPY-Dyads as Probes for Detecting Cancer. Bioconjug. Chem. 2019, 30, 169–183. [Google Scholar] [CrossRef]
- Matsumoto, N.; Taniguchi, M.; Lindsey, J.S. Bioconjugatable synthetic chlorins rendered water-soluble with three PEG-12 groups via click chemistry. J. Porphyr. Phthalocyanines 2020, 24, 362–378. [Google Scholar] [CrossRef]
- Liu, R.; Liu, S.J.; Hu, G.F.; Lindsey, J.S. Aqueous solubilization of hydrophobic tetrapyrrole macrocycles by attachment to an amphiphilic single-chain nanoparticle (SCNP). New J. Chem. 2020, 44, 21293–21308. [Google Scholar] [CrossRef]
- Liu, S.; Rong, J.; Liu, R.; Lindsey, J.S. Single-Fluorophore Single-Chain Nanoparticle Undergoes Fluorophore-Driven Assembly with Fluorescence Features Retained in Physiological Milieu. ACS Appl. Polym. Mater. 2021, 3, 1767–1776. [Google Scholar] [CrossRef]
- Harris, J.M. Poly(ethylene glycol) Chemistry: Biotechnical and Biomedical Applications; Plenum Press: New York, NY, USA, 1992; pp. 1–14. [Google Scholar]
- Zalipsky, S.; Harris, J.M. (Eds.) Introduction to Chemistry and Biological Applications of Poly(ethylene glycol). In Poly(ethylene glycol) Chemistry and Biological Applications; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 1997; Volume 680, pp. 1–13. [Google Scholar]
- Thompson, M.S.; Vadala, T.P.; Vadala, M.L.; Lin, Y.; Riffle, J.S. Synthesis and applications of heterobifunctional poly(ethylene oxide) oligomers. Polymer 2008, 49, 345–373. [Google Scholar] [CrossRef] [Green Version]
- Larson, N.; Ghandehari, H. Polymeric conjugates for drug delivery. Chem. Mater. 2012, 24, 840–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herzberger, J.; Niederer, K.; Pohlit, H.; Seiwert, J.; Worm, M.; Wurm, F.R.; Frey, H. Polymerization of Ethylene Oxide, Propylene Oxide, and Other Alkylene Oxides: Synthesis, Novel Polymer Architectures, and Bioconjugation. Chem. Rev. 2016, 116, 2170–2243. [Google Scholar] [CrossRef] [Green Version]
- Kolate, A.; Baradia, D.; Patil, S.; Vhora, I.; Kore, G.; Misra, A. PEG—A versatile conjugating ligand for drugs and drug delivery systems. J. Control. Release 2014, 192, 67–81. [Google Scholar] [CrossRef]
- Begum, R.; Matsuura, H. Conformational properties of short poly(oxyethylene) chains in water studied by IR spectroscopy. J. Chem. Soc. Faraday Trans. 1997, 93, 3839–3848. [Google Scholar] [CrossRef]
- Lang, W.; Yuan, C.; Zhu, B.; Pan, S.; Liu, J.; Luo, J.; Nie, S.; Zhu, Q.; Lee, J.S.; Ge, J. Expanding the “minimalist” small molecule tagging approach to different bioactive compounds. Org. Biomol. Chem. 2019, 17, 3010–3017. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Y.; Dong, J.; Liu, J.; Zhang, L.; Sun, H. Design and synthesis of novel photoaffinity probes for study of the target proteins of oleanolic acid. Bioorg. Med. Chem. Lett. 2012, 22, 1036–1039. [Google Scholar] [CrossRef]
- Sharma, A.; Lee, M.G.; Won, M.; Koo, S.; Arambula, J.F.; Sessler, J.L.; Chi, S.G.; Kim, J.S. Targeting Heterogeneous Tumors Using a Multifunctional Molecular Prodrug. J. Am. Chem. Soc. 2019, 141, 15611–15618. [Google Scholar] [CrossRef] [PubMed]
- Devalapally, H.; Navath, R.S.; Yenamandra, V.; Akkinepally, R.R.; Devarakonda, R.K. Beta-galactoside prodrugs of doxorubicin for application in antibody directed enzyme prodrug therapy/prodrug monotherapy. Arch. Pharm. Res. 2007, 30, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Jouanno, L.A.; Chevalier, A.; Sekkat, N.; Perzo, N.; Castel, H.; Romieu, A.; Lange, N.; Sabot, C.; Renard, P.Y. Kondrat’eva ligation: Diels-Alder-based irreversible reaction for bioconjugation. J. Org. Chem. 2014, 79, 10353–10366. [Google Scholar] [CrossRef]
- van der Meer, B.W. Kappa-squared: From nuisance to new sense. J. Biotechnol. 2002, 82, 181–196. [Google Scholar] [CrossRef]
- Taniguchi, M.; Du, H.; Lindsey, J.S. PhotochemCAD 3: Diverse Modules for Photophysical Calculations with Access to Multiple Spectral Databases. Photochem. Photobiol. 2017, 94, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Qi, Q.; Taniguchi, M.; Lindsey, J.S. Heuristics from modeling of spectral overlap in forster resonance energy transfer (FRET). J. Chem. Inf. Model. 2019, 59, 652–667. [Google Scholar] [CrossRef]
- Lindsey, J.S.; Taniguchi, M.; Bocian, D.F.; Holten, D. The fluorescence quantum yield parameter in Förster resonance energy transfer (FRET)—Meaning, misperception, and molecular design. Chem. Phys. Rev. 2021, 2, 011302. [Google Scholar] [CrossRef]
- Jouanno, L.-A.; Renault, K.; Sabot, C.; Renard, P.-Y. 5-Alkoxyoxazole—A Versatile Building Block in (Bio)organic Synthesis. Eur. J. Org. Chem. 2016, 2016, 3264–3281. [Google Scholar] [CrossRef]
- Shi, J.Y.; Liu, T.W.B.; Chen, J.; Green, D.; Jaffray, D.; Wilson, B.C.; Wang, F.; Zheng, G. Transforming a Targeted Porphyrin Theranostic Agent into a PET Imaging Probe for Cancer. Theranostics 2011, 1, 363–370. [Google Scholar] [CrossRef] [Green Version]
- Benov, L. Photodynamic therapy: Current status and future directions. Med. Princ. Pract. 2015, 24, 14–28. [Google Scholar] [CrossRef]
- dos Santos, A.F.; de Almeida, D.R.Q.; Terra, L.F.; Baptista, M.S.; Labriola, L. Photodynamic therapy in cancer treatment—An update review. J. Cancer Metastasis Treat. 2019, 5, 25–44. [Google Scholar] [CrossRef] [Green Version]
- Ametamey, S.M.; Honer, M.; Schubiger, P.A. Molecular imaging with PET. Chem. Rev. 2008, 108, 1501–1516. [Google Scholar] [CrossRef] [PubMed]
- Mukai, H.; Watanabe, Y. Review: PET imaging with macro- and middle-sized molecular probes. Nucl. Med. Biol. 2021, 92, 156–170. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-Y.; Sun, E.; Fan, D.; Taniguchi, M.; McDowell, B.E.; Yang, E.; Diers, J.R.; Bocian, D.F.; Holten, D.; Lindsey, J.S. Synthesis and Physicochemical Properties of Metallobacteriochlorins. Inorg. Chem. 2012, 51, 9443–9464. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.; Diers, J.R.; Huang, Y.-Y.; Hamblin, M.R.; Lindsey, J.S.; Bocian, D.F.; Holten, D. Molecular Electronic Tuning of Photosensitizers to Enhance Photodynamic Therapy: Synthetic Dicyanobacteriochlorins as a Case Study. Photochem. Photobiol. 2013, 89, 605–618. [Google Scholar] [CrossRef] [Green Version]
- Fuwa, H.; Takahashi, Y.; Konno, Y.; Watanabe, N.; Miyashita, H.; Sasaki, M.; Natsugari, H.; Kan, T.; Fukuyama, T.; Tomita, T.; et al. Divergent synthesis of multifunctional molecular probes to elucidate the enzyme specificity of dipeptidic gamma-secretase inhibitors. ACS Chem. Biol. 2007, 2, 408–418. [Google Scholar] [CrossRef]
- Albright, C.F.; Graciani, N.; Han, W.; Yue, E.; Stein, R.; Lai, Z.; Diamond, M.; Dowling, R.; Grimminger, L.; Zhang, S.Y.; et al. Matrix metalloproteinase-activated doxorubicin prodrugs inhibit HT1080 xenograft growth better than doxorubicin with less toxicity. Mol. Cancer Ther. 2005, 4, 751–760. [Google Scholar] [CrossRef] [Green Version]
- Verwilst, P.; Han, J.; Lee, J.; Mun, S.; Kang, H.G.; Kim, J.S. Reconsidering azobenzene as a component of small-molecule hypoxia-mediated cancer drugs: A theranostic case study. Biomaterials 2017, 115, 104–114. [Google Scholar] [CrossRef] [PubMed]
- McKeown, S.R. Defining normoxia, physoxia and hypoxia in tumours-implications for treatment response. Br. J. Radiol. 2014, 87, 20130676. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Han, X.; Yu, F.; Zhang, X.; Chen, L.; Lv, C. Polyamine-Targeting Gefitinib Prodrug and its Near-Infrared Fluorescent Theranostic Derivative for Monitoring Drug Delivery and Lung Cancer Therapy. Theranostics 2018, 8, 2217–2228. [Google Scholar] [CrossRef]
- Yuan, Y.; Kwok, R.T.; Zhang, R.; Tang, B.Z.; Liu, B. Targeted theranostic prodrugs based on an aggregation-induced emission (AIE) luminogen for real-time dual-drug tracking. Chem. Commun. 2014, 50, 11465–11468. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Tang, B.Z. AIE Luminogens for Bioimaging and Theranostics: From Organelles to Animals. Chem 2017, 3, 56–91. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Lee, M.M.S.; Xu, W.; Kwok, R.T.K.; Lam, J.W.Y.; Tang, B.Z. Theranostics based on AIEgens. Theranostics 2018, 8, 4925–4956. [Google Scholar] [CrossRef] [PubMed]
- van Scherpenzeel, M.; Moret, E.E.; Ballell, L.; Liskamp, R.M.; Nilsson, U.J.; Leffler, H.; Pieters, R.J. Synthesis and evaluation of new thiodigalactoside-based chemical probes to label galectin-3. ChemBioChem 2009, 10, 1724–1733. [Google Scholar] [CrossRef]
- Weinstain, R.; Savariar, E.N.; Felsen, C.N.; Tsien, R.Y. In vivo targeting of hydrogen peroxide by activatable cell-penetrating peptides. J. Am. Chem. Soc. 2014, 136, 874–877. [Google Scholar] [CrossRef] [PubMed]
- Beal, D.M.; Albrow, V.E.; Burslem, G.; Hitchen, L.; Fernandes, C.; Lapthorn, C.; Roberts, L.R.; Selby, M.D.; Jones, L.H. Click-enabled heterotrifunctional template for sequential bioconjugations. Org. Biomol. Chem. 2012, 10, 548–554. [Google Scholar] [CrossRef]
- Akram, M.; Asif, H.M.; Uzair, M.; Akhtar, N.; Madni, A.; Shah, S.M.A.; ul Hasan, Z.; Ullah, A. Amino acids: A review article. J. Med. Plants Res. 2011, 5, 3997–4000. [Google Scholar] [CrossRef]
- Tang, W.; Becker, M.L. “Click” reactions: A versatile toolbox for the synthesis of peptide-conjugates. Chem. Soc. Rev. 2014, 43, 7013–7039. [Google Scholar] [CrossRef]
- Liu, S.P.; Zhou, L.; Lakshminarayanan, R.; Beuerman, R.W. Multivalent Antimicrobial Peptides as Therapeutics: Design Principles and Structural Diversities. Int. J. Pept. Res. Ther. 2010, 16, 199–213. [Google Scholar] [CrossRef] [Green Version]
- He, R.; Finan, B.; Mayer, J.P.; DiMarchi, R.D. Peptide Conjugates with Small Molecules Designed to Enhance Efficacy and Safety. Molecules 2019, 24, 1855. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Mazo, M.M.; Skaalure, S.C.; Thomas, M.R.; Schultz, S.R.; Stevens, M.M. Activatable cell-biomaterial interfacing with photo-caged peptides. Chem. Sci. 2019, 10, 1158–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poreba, M. Protease-activated prodrugs: Strategies, challenges, and future directions. FEBS J. 2020, 287, 1936–1969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovalova, A.; Pohl, R.; Vrabel, M. Stepwise triple-click functionalization of synthetic peptides. Org. Biomol. Chem. 2018, 16, 5960–5964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaradat, D.M.M. Thirteen decades of peptide synthesis: Key developments in solid phase peptide synthesis and amide bond formation utilized in peptide ligation. Amino Acids 2018, 50, 39–68. [Google Scholar] [CrossRef] [PubMed]
- Martin, V.; Egelund, P.H.G.; Johansson, H.; Thordal Le Quement, S.; Wojcik, F.; Sejer Pedersen, D. Greening the synthesis of peptide therapeutics: An industrial perspective. RSC Adv. 2020, 10, 42457–42492. [Google Scholar] [CrossRef]
- Al-Warhi, T.I.; Al-Hazimi, H.M.A.; El-Faham, A. Recent development in peptide coupling reagents. J. Saudi Chem. Soc. 2012, 16, 97–116. [Google Scholar] [CrossRef] [Green Version]
- El-Faham, A.; Albericio, F. Peptide coupling reagents, more than a letter soup. Chem. Rev. 2011, 111, 6557–6602. [Google Scholar] [CrossRef]
- Montalbetti, C.A.G.N.; Falque, V. Amide bond formation and peptide coupling. Tetrahedron 2005, 61, 10827–10852. [Google Scholar] [CrossRef]
- Isidro-Llobet, A.; Alvarez, M.; Albericio, F. Amino acid-protecting groups. Chem. Rev. 2009, 109, 2455–2504. [Google Scholar] [CrossRef] [Green Version]
- Behrendt, R.; White, P.; Offer, J. Advances in Fmoc solid-phase peptide synthesis. J. Pept. Sci. 2016, 22, 4–27. [Google Scholar] [CrossRef] [Green Version]
- Coin, I.; Dolling, R.; Krause, E.; Bienert, M.; Beyermann, M.; Sferdean, C.D.; Carpino, L.A. Depsipeptide methodology for solid-phase peptide synthesis: Circumventing side reactions and development of an automated technique via depsidipeptide units. J. Org. Chem. 2006, 71, 6171–6177. [Google Scholar] [CrossRef]
- Huang, Y.C.; Guan, C.J.; Tan, X.L.; Chen, C.C.; Guo, Q.X.; Li, Y.M. Accelerated Fmoc solid-phase synthesis of peptides with aggregation-disrupting backbones. Org. Biomol. Chem. 2015, 13, 1500–1506. [Google Scholar] [CrossRef]
- Paradís-Bas, M.; Tulla-Puche, J.; Albericio, F. The road to the synthesis of “difficult peptides”. Chem. Soc. Rev. 2016, 45, 631–654. [Google Scholar] [CrossRef]
- Pícha, J.; Buděšínský, M.; Macháčková, K.; Collinsová, M.; Jiráček, J. Optimized syntheses of Fmoc azido amino acids for the preparation of azidopeptides. J. Pept. Sci. 2017, 23, 202–214. [Google Scholar] [CrossRef] [Green Version]
- Weis, S.M.; Cheresh, D.A. Tumor angiogenesis: Molecular pathways and therapeutic targets. Nat. Med. 2011, 17, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Dou, X.; Nomoto, T.; Takemoto, H.; Matsui, M.; Tomoda, K.; Nishiyama, N. Effect of multiple cyclic RGD peptides on tumor accumulation and intratumoral distribution of IRDye 700DX-conjugated polymers. Sci. Rep. 2018, 8, 8126–8137. [Google Scholar] [CrossRef]
- Renault, K.; Fredy, J.W.; Renard, P.Y.; Sabot, C. Covalent Modification of Biomolecules through Maleimide-Based Labeling Strategies. Bioconjug. Chem. 2018, 29, 2497–2513. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Luo, C.; Yu, H.; Zhang, X.; Chen, Q.; Yang, W.; Wang, M.; Kan, Q.; Zhang, H.; Wang, Y.; et al. Disulfide Bond-Driven Oxidation- and Reduction-Responsive Prodrug Nanoassemblies for Cancer Therapy. Nano Lett. 2018, 18, 3643–3650. [Google Scholar] [CrossRef] [PubMed]
- Postma, T.M.; Albericio, F. Disulfide Formation Strategies in Peptide Synthesis. Eur. J. Org. Chem. 2014, 2014, 3519–3530. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Akaji, K.; Kiso, Y. Racemization-free synthesis of C-terminal cysteine-peptide using 2-chlorotrityl resin. Chem. Pharm. Bull. 1994, 42, 724–726. [Google Scholar] [CrossRef] [Green Version]
- Lukszo, J.; Patterson, D.; Albericio, F.; Kates, S.A. 3-(1-Piperidinyl)alanine formation during the preparation of C-terminal cysteine peptides with the Fmoc/t-Bu strategy. Lett. Pept. Sci. 1996, 3, 157–166. [Google Scholar] [CrossRef]
- Gongora-Benitez, M.; Mendive-Tapia, L.; Ramos-Tomillero, I.; Breman, A.C.; Tulla-Puche, J.; Albericio, F. Acid-labile Cys-protecting groups for the Fmoc/tBu strategy: Filling the gap. Org. Lett. 2012, 14, 5472–5475. [Google Scholar] [CrossRef]
- Stathopoulos, P.; Papas, S.; Pappas, C.; Mousis, V.; Sayyad, N.; Theodorou, V.; Tzakos, A.G.; Tsikaris, V. Side reactions in the SPPS of Cys-containing peptides. Amino Acids 2013, 44, 1357–1363. [Google Scholar] [CrossRef]
- Lelievre, D.; Terrier, V.P.; Delmas, A.F.; Aucagne, V. Native Chemical Ligation Strategy to Overcome Side Reactions during Fmoc-Based Synthesis of C-Terminal Cysteine-Containing Peptides. Org. Lett. 2016, 18, 920–923. [Google Scholar] [CrossRef]
- Tsuda, S.; Masuda, S.; Yoshiya, T. Epimerization-Free Preparation of C-Terminal Cys Peptide Acid by Fmoc SPPS Using Pseudoproline-Type Protecting Group. J. Org. Chem. 2020, 85, 1674–1679. [Google Scholar] [CrossRef] [PubMed]
- Albericio, F.; El-Faham, A. Choosing the Right Coupling Reagent for Peptides: A Twenty-Five-Year Journey. Org. Process Res. Dev. 2018, 22, 760–772. [Google Scholar] [CrossRef]
- Valeur, E.; Bradley, M. Amide bond formation: Beyond the myth of coupling reagents. Chem. Soc. Rev. 2009, 38, 606–631. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.; Bharathi, K.; Haseena, B.B. Applications of peptide coupling reagents—An update. Intl. J. Pharm. Sci. Rev. Res. 2011, 8, 108–119. [Google Scholar]
- Tsakos, M.; Schaffert, E.S.; Clement, L.L.; Villadsen, N.L.; Poulsen, T.B. Ester coupling reactions--an enduring challenge in the chemical synthesis of bioactive natural products. Nat. Prod. Rep. 2015, 32, 605–632. [Google Scholar] [CrossRef] [PubMed]
- Dunetz, J.R.; Magano, J.; Weisenburger, G.A. Large-Scale Applications of Amide Coupling Reagents for the Synthesis of Pharmaceuticals. Org. Process Res. Dev. 2016, 20, 140–177. [Google Scholar] [CrossRef]
- Sakakura, A.; Kawajiri, K.; Ohkubo, T.; Kosugi, Y.; Ishihara, K. Widely useful DMAP-catalyzed esterification under auxiliary base- and solvent-free conditions. J. Am. Chem. Soc. 2007, 129, 14775–14779. [Google Scholar] [CrossRef]
- Renard, E.; Dancer, P.A.; Portal, C.; Denat, F.; Prignon, A.; Goncalves, V. Design of Bimodal Ligands of Neurotensin Receptor 1 for Positron Emission Tomography Imaging and Fluorescence-Guided Surgery of Pancreatic Cancer. J. Med. Chem. 2020, 63, 2426–2433. [Google Scholar] [CrossRef] [PubMed]
- Lelle, M.; Peneva, K. An amino acid-based heterofunctional cross-linking reagent. Amino Acids 2014, 46, 1243–1251. [Google Scholar] [CrossRef] [PubMed]
- Krall, N.; Pretto, F.; Neri, D. A bivalent small molecule-drug conjugate directed against carbonic anhydrase IX can elicit complete tumour regression in mice. Chem. Sci. 2014, 5, 3640–3644. [Google Scholar] [CrossRef]
- Lee, H.M.; Priestman, M.A.; Lawrence, D.S. Light-mediated spatial control via photolabile fluorescently quenched peptide cassettes. J. Am. Chem. Soc. 2010, 132, 1446–1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lock, L.L.; Tang, Z.; Keith, D.; Reyes, C.; Cui, H. Enzyme-Specific Doxorubicin Drug Beacon as Drug-Resistant Theranostic Molecular Probes. ACS Macro Lett. 2015, 4, 552–555. [Google Scholar] [CrossRef]
- Yan, X.; Li, Z.; Liang, Y.; Yang, L.; Zhang, B.; Wang, Q. A chemical “hub” for absolute quantification of a targeted protein: Orthogonal integration of elemental and molecular mass spectrometry. Chem. Commun. 2014, 50, 6578–6581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukiji, S.; Wang, H.; Miyagawa, M.; Tamura, T.; Takaoka, Y.; Hamachi, I. Quenched ligand-directed tosylate reagents for one-step construction of turn-on fluorescent biosensors. J. Am. Chem. Soc. 2009, 131, 9046–9054. [Google Scholar] [CrossRef]
- Huang, R.; Wang, X.; Wang, D.; Liu, F.; Mei, B.; Tang, A.; Jiang, J.; Liang, G. Multifunctional fluorescent probe for sequential detections of glutathione and caspase-3 in vitro and in cells. Anal. Chem. 2013, 85, 6203–6207. [Google Scholar] [CrossRef]
- Anami, Y.; Xiong, W.; Gui, X.; Deng, M.; Zhang, C.C.; Zhang, N.; An, Z.; Tsuchikama, K. Enzymatic conjugation using branched linkers for constructing homogeneous antibody-drug conjugates with high potency. Org. Biomol. Chem. 2017, 15, 5635–5642. [Google Scholar] [CrossRef] [PubMed]
- Oriana, S.; Cai, Y.; Bode, J.W.; Yamakoshi, Y. Synthesis of tri-functionalized MMP2 FRET probes using a chemo-selective and late-stage modification of unprotected peptides. Org. Biomol. Chem. 2017, 15, 1792–1800. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Nayak, B.; Dey, R.K. PEGylation in anti-cancer therapy: An overview. Asian J. Pharm. Sci. 2016, 11, 337–348. [Google Scholar] [CrossRef] [Green Version]
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [Google Scholar] [CrossRef]
- Hu, J.; Van den Steen, P.E.; Sang, Q.X.; Opdenakker, G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat. Rev. Drug Discov. 2007, 6, 480–498. [Google Scholar] [CrossRef]
- Bode, J.W. Chemical Protein Synthesis with the alpha-Ketoacid-Hydroxylamine Ligation. Acc. Chem. Res. 2017, 50, 2104–2115. [Google Scholar] [CrossRef] [Green Version]
- Zuo, C.; Zhang, B.; Yan, B.; Zheng, J.S. One-pot multi-segment condensation strategies for chemical protein synthesis. Org. Biomol. Chem. 2019, 17, 727–744. [Google Scholar] [CrossRef]
- Arbour, C.A.; Kondasinghe, T.D.; Saraha, H.Y.; Vorlicek, T.L.; Stockdill, J.L. Epimerization-free access to C-terminal cysteine peptide acids, carboxamides, secondary amides, and esters via complimentary strategies. Chem. Sci. 2018, 9, 350–355. [Google Scholar] [CrossRef] [Green Version]
- Zuo, C.; Yan, B.J.; Zhu, H.Y.; Shi, W.W.; Xi, T.K.; Shi, J.; Fang, G.M. Robust synthesis of C-terminal cysteine-containing peptide acids through a peptide hydrazide-based strategy. Org. Biomol. Chem. 2019, 17, 5698–5702. [Google Scholar] [CrossRef]
- Mariani, G.; Bruselli, L.; Kuwert, T.; Kim, E.E.; Flotats, A.; Israel, O.; Dondi, M.; Watanabe, N. A review on the clinical uses of SPECT/CT. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 1959–1985. [Google Scholar] [CrossRef]
- Israel, O.; Pellet, O.; Biassoni, L.; De Palma, D.; Estrada-Lobato, E.; Gnanasegaran, G.; Kuwert, T.; la Fougere, C.; Mariani, G.; Massalha, S.; et al. Two decades of SPECT/CT—The coming of age of a technology: An updated review of literature evidence. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1990–2012. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.; Luo, C.; Yang, G.; Du, S.; Li, X.; Zhao, H.; Shi, J.; Wang, F. Improved in Vivo Targeting Capability and Pharmacokinetics of (99m)Tc-Labeled isoDGR by Dimerization and Albumin-Binding for Glioma Imaging. Bioconjug. Chem. 2019, 30, 2038–2048. [Google Scholar] [CrossRef]
- Chen, P.; Kuang, W.; Zheng, Z.; Yang, S.; Liu, Y.; Su, L.; Zhao, K.; Liang, G. Carboxylesterase-Cleavable Biotinylated Nanoparticle for Tumor-Dual Targeted Imaging. Theranostics 2019, 9, 7359–7369. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Y.; Cheng, H.; Xie, B.R.; Qiu, W.X.; Song, L.L.; Zhuo, R.X.; Zhang, X.Z. A ratiometric theranostic probe for tumor targeting therapy and self-therapeutic monitoring. Biomaterials 2016, 104, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.M.; Xu, W.; Lawrence, D.S. Construction of a photoactivatable profluorescent enzyme via propinquity labeling. J. Am. Chem. Soc. 2011, 133, 2331–2333. [Google Scholar] [CrossRef] [Green Version]
- Hai, Z.; Wu, J.; Saimi, D.; Ni, Y.; Zhou, R.; Liang, G. Smart Dual Quenching Strategy Enhances the Detection Sensitivity of Intracellular Furin. Anal. Chem. 2018, 90, 1520–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, C.H.; Chang, T.C.; Chuang, Y.J.; Tzou, D.L.; Lin, C.C. Stepwise orthogonal click chemistry toward fabrication of paclitaxel/galactose functionalized fluorescent nanoparticles for HepG2 cell targeting and delivery. Bioconjug. Chem. 2013, 24, 1698–1709. [Google Scholar] [CrossRef]
- Lai, C.H.; Lin, Y.C.; Chou, F.I.; Liang, C.F.; Lin, E.W.; Chuang, Y.J.; Lin, C.C. Design of multivalent galactosyl carborane as a targeting specific agent for potential application to boron neutron capture therapy. Chem. Commun. 2012, 48, 612–614. [Google Scholar] [CrossRef]
- Lee, D.L.; Chin, H.-L.M.; Knudsen, C.G.; Mayers, G.L.; Rose, D.S.; Skogstrom, R.K.; Palzkill, T.; Fujita, H.; Zhang, Y.; Wu, Z.; et al. Peptide-based scaffolds for in vivo immobilization and enzyme attachment in therapeutic applications. Proc. SPIE 2020, 11477, 1147708. [Google Scholar] [CrossRef]
- Yao, Z.; Borbas, K.E.; Lindsey, J.S. Soluble Precipitable Porphyrins for Use in Targeted Molecular Brachytherapy. New J. Chem. 2008, 32, 436–451. [Google Scholar] [CrossRef]
- Wu, X.; Burden-Gulley, S.M.; Yu, G.P.; Tan, M.; Lindner, D.; Brady-Kalnay, S.M.; Lu, Z.R. Synthesis and evaluation of a peptide targeted small molecular Gd-DOTA monoamide conjugate for MR molecular imaging of prostate cancer. Bioconjug. Chem. 2012, 23, 1548–1556. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.R.; Jung, D.W.; Williams, D.; Shin, I. Efficient solid-phase synthesis of trifunctional probes and their application to the detection of carbohydrate-binding proteins. Org. Lett. 2005, 7, 5477–5480. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Garcia, M.; Benito, J.M.; Rodriguez-Lucena, D.; Yu, J.X.; Chmurski, K.; Ortiz Mellet, C.; Gutierrez Gallego, R.; Maestre, A.; Defaye, J.; Garcia Fernandez, J.M. Probing secondary carbohydrate-protein interactions with highly dense cyclodextrin-centered heteroglycoclusters: The heterocluster effect. J. Am. Chem. Soc. 2005, 127, 7970–7971. [Google Scholar] [CrossRef]
- Jimenez Blanco, J.L.; Ortiz Mellet, C.; Garcia Fernandez, J.M. Multivalency in heterogeneous glycoenvironments: Hetero-glycoclusters, -glycopolymers and -glycoassemblies. Chem. Soc. Rev. 2013, 42, 4518–4531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Cuesta, M.; Ortiz Mellet, C.; Garcia Fernandez, J.M. Carbohydrate supramolecular chemistry: Beyond the multivalent effect. Chem. Commun. 2020, 56, 5207–5222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, C.; Despras, G.; Lindhorst, T.K. Organizing multivalency in carbohydrate recognition. Chem. Soc. Rev. 2016, 45, 3275–3302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyard, D.; Thomas, B.; Gillon, E.; Imberty, A.; Renaudet, O. Heteroglycoclusters With Dual Nanomolar Affinities for the Lectins LecA and LecB From Pseudomonas aeruginosa. Front. Chem. 2019, 7, 666–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galan, M.C.; Dumy, P.; Renaudet, O. Multivalent glyco(cyclo)peptides. Chem. Soc. Rev. 2013, 42, 4599–4612. [Google Scholar] [CrossRef]
- Pifferi, C.; Berthet, N.; Renaudet, O. Cyclopeptide scaffolds in carbohydrate-based synthetic vaccines. Biomater. Sci. 2017, 5, 953–965. [Google Scholar] [CrossRef] [Green Version]
- Richard, M.; Ariztia, J.; Lamandé-Langle, S.; Pellegrini Moïse, N. Sugar γ-Amino Acids as Building Blocks for the Synthesis of Cyclic Neoglycopeptides. ChemistrySelect 2018, 3, 9121–9126. [Google Scholar] [CrossRef]
- Thomas, B.; Fiore, M.; Daskhan, G.C.; Spinelli, N.; Renaudet, O. A multi-ligation strategy for the synthesis of heterofunctionalized glycosylated scaffolds. Chem. Commun. 2015, 51, 5436–5439. [Google Scholar] [CrossRef]
- Chelius, D.; Shaler, T.A. Capture of peptides with N-terminal serine and threonine: A sequence-specific chemical method for peptide mixture simplification. Bioconjug. Chem. 2003, 14, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Spears, R.J.; Fascione, M.A. Site-selective incorporation and ligation of protein aldehydes. Org. Biomol. Chem. 2016, 14, 7622–7638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Mahdi, O.; Melnyk, O. alpha-Oxo aldehyde or glyoxylyl group chemistry in peptide bioconjugation. Bioconjug. Chem. 2013, 24, 735–765. [Google Scholar] [CrossRef]
- Mettu, R.; Chen, C.Y.; Wu, C.Y. Synthetic carbohydrate-based vaccines: Challenges and opportunities. J. Biomed. Sci. 2020, 27, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bringmann, G.; Gampe, C.M.; Reichert, Y.; Bruhn, T.; Faber, J.H.; Mikyna, M.; Reichert, M.; Leippe, M.; Brun, R.; Gelhaus, C. Synthesis and pharmacological evaluation of fluorescent and photoactivatable analogues of antiplasmodial naphthylisoquinolines. J. Med. Chem. 2007, 50, 6104–6115. [Google Scholar] [CrossRef]
- Liu, Q.H.; Yan, X.L.; Guo, J.C.; Wang, D.H.; Li, L.; Yan, F.Y.; Chen, L.G. Spectrofluorimetric determination of trace nitrite with a novel fluorescent probe. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2009, 73, 789–793. [Google Scholar] [CrossRef]
- Carrigan, C.N.; Imperiali, B. The engineering of membrane-permeable peptides. Anal. Biochem. 2005, 341, 290–298. [Google Scholar] [CrossRef]
- Fan, D.; Wang, K.; Gao, H.; Luo, Q.; Wang, X.; Li, X.; Tong, W.; Zhang, X.; Luo, C.; Yang, G.; et al. A (64)Cu-porphyrin-based dual-modal molecular probe with integrin αvβ3 targeting function for tumour imaging. J. Label. Compd. Radiopharm. 2020, 63, 212–221. [Google Scholar] [CrossRef]
- Ruhl, T.; Volke, D.; Stembera, K.; Hatanaka, Y.; Hennig, H.; Schumer, F.; Welzel, P. Isoserine-based biotinylated photoaffinity probes that interact with penicillin-binding protein 1b. Chem. Commun. 2002, 1630–1631. [Google Scholar] [CrossRef]
- Dijkgraaf, I.; Van de Vijver, P.; Dirksen, A.; Hackeng, T.M. Synthesis and application of cNGR-containing imaging agents for detection of angiogenesis. Bioorg. Med. Chem. 2013, 21, 3555–3564. [Google Scholar] [CrossRef]
- Seibold, U.; Wangler, B.; Schirrmacher, R.; Wangler, C. Bimodal imaging probes for combined PET and OI: Recent developments and future directions for hybrid agent development. BioMed Res. Int. 2014, 2014, 153741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Ding, J.; Xing, W.; Gai, Y.; Sheng, J.; Zeng, D. Novel Strategy for Preparing Dual-Modality Optical/PET Imaging Probes via Photo-Click Chemistry. Bioconjug. Chem. 2016, 27, 1200–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Guo, N.; Li, Q.; Ma, Y.; Jacboson, O.; Lee, S.; Choi, H.S.; Mansfield, J.R.; Niu, G.; Chen, X. Dynamic PET and Optical Imaging and Compartment Modeling using a Dual-labeled Cyclic RGD Peptide Probe. Theranostics 2012, 2, 746–756. [Google Scholar] [CrossRef] [Green Version]
- Hawala, I.; De Rosa, L.; Aime, S.; D’Andrea, L.D. An innovative approach for the synthesis of dual modality peptide imaging probes based on the native chemical ligation approach. Chem. Commun. 2020, 56, 3500–3503. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.S.; Sayers, J.; Premdjee, B.; Payne, R.J. Rapid and efficient protein synthesis through expansion of the native chemical ligation concept. Nat. Rev. Chem. 2018, 2, 0122–0147. [Google Scholar] [CrossRef]
- Agouridas, V.; El Mahdi, O.; Diemer, V.; Cargoet, M.; Monbaliu, J.M.; Melnyk, O. Native Chemical Ligation and Extended Methods: Mechanisms, Catalysis, Scope, and Limitations. Chem. Rev. 2019, 119, 7328–7443. [Google Scholar] [CrossRef]
- Thapa, P.; Zhang, R.Y.; Menon, V.; Bingham, J.P. Native chemical ligation: A boon to peptide chemistry. Molecules 2014, 19, 14461–14483. [Google Scholar] [CrossRef] [Green Version]
- Debie, P.; Hernot, S. Emerging Fluorescent Molecular Tracers to Guide Intra-Operative Surgical Decision-Making. Front. Pharmacol. 2019, 10, 510–529. [Google Scholar] [CrossRef] [Green Version]
- Trester-Zedlitz, M.; Kamada, K.; Burley, S.K.; Fenyo, D.; Chait, B.T.; Muir, T.W. A modular cross-linking approach for exploring protein interactions. J. Am. Chem. Soc. 2003, 125, 2416–2425. [Google Scholar] [CrossRef]
- Lelle, M.; Kaloyanova, S.; Freidel, C.; Theodoropoulou, M.; Musheev, M.; Niehrs, C.; Stalla, G.; Peneva, K. Octreotide-Mediated Tumor-Targeted Drug Delivery via a Cleavable Doxorubicin-Peptide Conjugate. Mol. Pharm. 2015, 12, 4290–4300. [Google Scholar] [CrossRef]
- Abu Ajaj, K.; Biniossek, M.L.; Kratz, F. Development of protein-binding bifunctional linkers for a new generation of dual-acting prodrugs. Bioconjug. Chem. 2009, 20, 390–396. [Google Scholar] [CrossRef]
- Sinisi, R.; Morales, A.R.M.; Dubikovskaya, E.A.; Singh, R. Azacyanine Dyes and Use Thereof. U.S. Patent US2018/0079906 A1, 22 March 2018. [Google Scholar]
- Nunez, J.; Renslow, R.; Cliff, J.B., 3rd; Anderton, C.R. NanoSIMS for biological applications: Current practices and analyses. Biointerphases 2018, 13, 03B301-301–303B301-326. [Google Scholar] [CrossRef] [Green Version]
- Kabatas, S.; Agui-Gonzalez, P.; Saal, K.A.; Jahne, S.; Opazo, F.; Rizzoli, S.O.; Phan, N.T.N. Boron-Containing Probes for Non-optical High-Resolution Imaging of Biological Samples. Angew. Chem. Int. Ed. 2019, 58, 3438–3443. [Google Scholar] [CrossRef]
- Chang, T.C.; Adak, A.K.; Lin, T.W.; Li, P.J.; Chen, Y.J.; Lai, C.H.; Liang, C.F.; Chen, Y.J.; Lin, C.C. A photo-cleavable biotin affinity tag for the facile release of a photo-crosslinked carbohydrate-binding protein. Bioorg. Med. Chem. 2016, 24, 1216–1224. [Google Scholar] [CrossRef]
- Kim, E.; Koo, H. Biomedical applications of copper-free click chemistry: In vitro, in vivo, and ex vivo. Chem. Sci. 2019, 10, 7835–7851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodersen, N.; Arbuzova, A.; Herrmann, A.; Egger, H.; Liebscher, J. Synthesis of novel amphiphilic conjugates with a biological recognition function for developing targeted triggered liposomal delivery systems. Tetrahedron 2011, 67, 7763–7774. [Google Scholar] [CrossRef]
- Cheng, Z.; Wu, Y.; Xiong, Z.; Gambhir, S.S.; Chen, X. Near-infrared fluorescent RGD peptides for optical imaging of integrin αvβ3 expression in living mice. Bioconjug. Chem. 2005, 16, 1433–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragupathi, G.; Koide, F.; Livingston, P.O.; Cho, Y.S.; Endo, A.; Wan, Q.; Spassova, M.K.; Keding, S.J.; Allen, J.; Ouerfelli, O.; et al. Preparation and evaluation of unimolecular pentavalent and hexavalent antigenic constructs targeting prostate and breast cancer: A synthetic route to anticancer vaccine candidates. J. Am. Chem. Soc. 2006, 128, 2715–2725. [Google Scholar] [CrossRef]
- Giacomelli, G.; Porcheddu, A.; De Luca, L. [1,3,5]-triazine: A versatile heterocycle in current applications of organic chemistry. Curr. Org. Chem. 2004, 8, 1497–1519. [Google Scholar] [CrossRef]
- Bretterbauer, K.; Schwarzinger, C. Melamine Derivatives—A Review on Synthesis and Application. Curr. Org. Synth. 2012, 9, 342–356. [Google Scholar] [CrossRef]
- Shinde, R.S. Review on Synthesis and Biological Study of Triazines Derivatives. United J. Chem. 2018, 1, 92–100. [Google Scholar]
- Blotny, G. Recent applications of 2,4,6-trichloro-1,3,5-triazine and its derivatives in organic synthesis. Tetrahedron 2006, 62, 9507–9522. [Google Scholar] [CrossRef]
- Thurston, J.T.; Schaefer, F.C.; Dudley, J.R.; Holmhansen, D. Cyanuric Chloride Derivatives. V. Reaction of Alkoxy-S-Triazines and Aryloxy-Ss-Triazines with Amines. J. Am. Chem. Soc. 1951, 73, 2992–2996. [Google Scholar] [CrossRef]
- Yamada, K.; Fujita, H.; Kunishima, M. A novel acid-catalyzed O-benzylating reagent with the smallest unit of imidate structure. Org. Lett. 2012, 14, 5026–5029. [Google Scholar] [CrossRef]
- Kunishima, M.; Kawachi, C.; Iwasaki, F.; Terao, K.; Tani, S. Synthesis and characterization of 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride. Tetrahedron Lett. 1999, 40, 5327–5330. [Google Scholar] [CrossRef]
- Mibu, N.; Yokomizo, K.; Aki, H.; Ota, N.; Fujii, H.; Yuzuriha, A.; Saneyoshi, S.; Tanaka, A.; Koga, A.; Zhou, J.; et al. Synthesis and Antiviral Evaluation of Some C(3)-Symmetrical Trialkoxy-Substituted 1,3,5-Triazines and Their Molecular Geometry. Chem. Pharm. Bull. 2015, 63, 935–944. [Google Scholar] [CrossRef]
- Kunishima, M.; Ujigawa, T.; Nagaoka, Y.; Kawachi, C.; Hioki, K.; Shiro, M. Study on 1,3,5-triazine chemistry in dehydrocondensation: Gauche effect on the generation of active triazinylammonium species. Chem. Eur. J. 2012, 18, 15856–15867. [Google Scholar] [CrossRef]
- Cuthbertson, W.W.; Moffatt, J.S. 115. Contributions to the chemistry of synthetic antimalarials. Part VI. Some derivatives of 1:3:5-triazine. J. Chem. Soc. 1948, 561–564. [Google Scholar] [CrossRef]
- Goi, M. Reactivities of Cyanuric Chloride Derivatives. II Displacement Reactions of 2-Chloro-4-substituted-6-anilino-S-triazines with Benzylamine. J. Synth. Org. Chem. Jpn. 1960, 18, 332–336. [Google Scholar] [CrossRef] [Green Version]
- Fukushima, Y.; Hashida, Y.; Matsui, K. Substituent Effects in 1,3,5-Triazine Derivatives. Nippon. Kagaku Kaishi 1972, 629–634. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Taft, R.W. A Survey of Hammett Substituent Constants and Resonance and Field Parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Fries, H.H. XXXI.—Contributions to a knowledge of cyanuric derivatives. J. Chem. Soc. Trans. 1886, 49, 314–316. [Google Scholar] [CrossRef] [Green Version]
- Diels, O. Zur Kenntniss der Cyanurverbindungen. Ber. Dtsch. Chem. Ges. 1899, 32, 691–702. [Google Scholar] [CrossRef]
- Steffensen, M.B.; Simanek, E.E. Chemoselective building blocks for dendrimers from relative reactivity data. Org. Lett. 2003, 5, 2359–2361. [Google Scholar] [CrossRef]
- Simanek, E.E.; Abdou, H.; Lalwani, S.; Lim, J.; Mintzer, M.; Venditto, V.J.; Vittur, B. The 8 year thicket of triazine dendrimers: Strategies, targets and applications. Proc. R. Soc. A Math. Phys. Eng. Sci. 2010, 466, 1445–1468. [Google Scholar] [CrossRef]
- Goi, M. Reactivities of Cyanuric Chloride Derivatives II Displacement Reactions of 2-Amino-4,6-dichloro-S-triazines with Aromatic Amines. J. Synth. Org. Chem. Jpn. 1960, 18, 337–342. [Google Scholar] [CrossRef]
- Moreno, K.X.; Simanek, E.E. Identification of Diamine Linkers with Differing Reactivity and their Application in the Synthesis of a Melamine Dendrimers. Tetrahedron Lett. 2008, 49, 1152–1154. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; El-Faham, A.; de la Torre, B.G.; Albericio, F. Exploring the Orthogonal Chemoselectivity of 2,4,6-Trichloro-1,3,5-Triazine (TCT) as a Trifunctional Linker With Different Nucleophiles: Rules of the Game. Front. Chem. 2018, 6, 516–526. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Sheyi, R.; Kumar, A.; El-Faham, A.; de la Torre, B.G.; Albericio, F. Investigating Triorthogonal Chemoselectivity. Effect of Azide Substitution on the Triazine Core. Org. Lett. 2019, 21, 7888–7892. [Google Scholar] [CrossRef]
- Maes, B.; Sheyi, R.; Sharma, A.; Kumar, A.; El-Faham, A.; Torre, B.G.d.l.; Albericio, F. 1,3,5-Triazine as core for the preparation of dendrons. Arkivoc 2020, 2020, 64–73. [Google Scholar] [CrossRef]
- Sharma, A.; Kumar, A.; El-Faham, A.; de la Torre, B.G.; Albericio, F. Exploiting azido-dichloro-triazine as a linker for regioselective incorporation of peptides through their N, O, S functional groups. Bioorg. Chem. 2020, 104, 104334. [Google Scholar] [CrossRef]
- Fujita, H.; Zhang, Y.; Wu, Z.; Lindsey, J.S. Chromogenic agents built around a multifunctional double-triazine framework for enzymatically triggered cross-linking under physiological conditions. New J. Chem. 2020, 44, 3856–3867. [Google Scholar] [CrossRef]
- Bourguet, E.; Correia, I.; Dorgeret, B.; Chassaing, G.; Sicsic, S.; Ongeri, S. Synthesis and conformational studies of pseudopeptides containing an unsymmetrical triazine scaffold. J. Pept. Sci. 2008, 14, 596–609. [Google Scholar] [CrossRef]
- Amm, M.; Platzer, N.; Guilhem, J.; Bouchet, J.P.; Volland, J.P. Structural and conformational study of substituted triazines by N-15 NMR and x-ray analysis. Magn. Reson. Chem. 1998, 36, 587–596. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Ghiviriga, I.; Steel, P.J.; Oniciu, D.C. Restricted rotations in 4,6-bis- and 2,4,6-tris-(N,N-dialkylamino)-s-triazines. J. Chem. Soc. Perk. Trans. 2 1996, 443–447. [Google Scholar] [CrossRef]
- Birkett, H.E.; Harris, R.K.; Hodgkinson, P.; Carr, K.; Charlton, M.H.; Cherryman, J.C.; Chippendale, A.M.; Glover, R.P. NMR studies of exchange between triazine rotamers. Magn. Reson. Chem. 2000, 38, 504–511. [Google Scholar] [CrossRef]
- Birkett, H.E.; Cherryman, J.C.; Chippendale, A.M.; Hazendonk, P.; Harris, R.K. Molecular modelling studies of side-chain rotation in substituted triazine rings. J. Mol. Struct. 2002, 602, 59–70. [Google Scholar] [CrossRef]
- Birkett, H.E.; Cherryman, J.C.; Chippendale, A.M.; Evans, J.S.O.; Harris, R.K.; James, M.; King, I.J.; McPherson, G.J. Structural investigations of three triazines: Solution-state NMR studies of internal rotation and structural information from solid-state NMR, plus a full structure determination from powder x-ray diffraction in one case. Magn. Reson. Chem. 2003, 41, 324–336. [Google Scholar] [CrossRef]
- Kunishima, M.; Asao, R.; Yamada, K.; Kitamura, M.; Fujita, H. Development of acid-catalyzed fluorous benzylating reagents based on a triazinedione core. J. Fluor. Chem. 2016, 190, 68–74. [Google Scholar] [CrossRef]
- Menicagli, R.; Malanga, C.; Peluso, P. Selective Mono- or Dialkoxylation of 2,4,6-Trichloro-1,3,5-triazine in Solid-Liquid Phase Transfer Conditions. Synth. Commun. 1994, 24, 2153–2158. [Google Scholar] [CrossRef]
- Fujita, H.; Hayakawa, N.; Kunishima, M. Study of the Reactivities of Acid-Catalyzed O-Benzylating Reagents Based on Structural Isomers of 1,3,5-Triazine. J. Org. Chem. 2015, 80, 11200–11205. [Google Scholar] [CrossRef] [PubMed]
- Mibu, N.; Yokomizo, K.; Koga, A.; Honda, M.; Mizokami, K.; Fujii, H.; Ota, N.; Yuzuriha, A.; Ishimaru, K.; Zhou, J.R.; et al. Synthesis and Antiviral Activities of Some 2,4,6-Trisubstituted 1,3,5-Triazines. Chem. Pharm. Bull. 2014, 62, 1032–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, H.; Kakuyama, S.; Fukuyoshi, S.; Hayakawa, N.; Oda, A.; Kunishima, M. Triazine-Based Cationic Leaving Group: Synergistic Driving Forces for Rapid Formation of Carbocation Species. J. Org. Chem. 2018, 83, 4568–4580. [Google Scholar] [CrossRef]
- Robb, W.; Nicholson, C.G. Kinetics of Exchange-Reactions of Ethylenediamine with a Series of Cationic Rhodium(I)Complexes. S. Afr. J. Chem. 1978, 31, 1–5. [Google Scholar] [CrossRef]
- Clarke, K.; Rothwell, K. A Kinetic Study of the Effect of Substituents on the Rate of Formation of Alkylpyridinium Halides in Nitromethane Solution. J. Chem. Soc. 1960, 1885–1895. [Google Scholar] [CrossRef]
- Hall, H.K. Steric Effects on the Base Strengths of Cyclic Amines. J. Am. Chem. Soc. 1957, 79, 5444–5447. [Google Scholar] [CrossRef]
- Fujii, T.; Nishida, H.; Abiru, Y.; Yamamoto, M.; Kise, M. Studies on Synthesis of the Antibacterial Agent Nm441. II. Selection of a Suitable Base for Alkylation of 1-Substituted Piperazine with 4-(Bromomethyl)-5-methyl-1,3-dioxol-2-one. Chem. Pharm. Bull. 1995, 43, 1872–1877. [Google Scholar] [CrossRef] [Green Version]
- Brown, H.C.; Kanner, B. Preparation and Reactions of 2,6-Di-t-butylpyridine and Related Hindered Bases. A Case of Steric Hindrance toward the Proton. J. Am. Chem. Soc. 1966, 88, 986–992. [Google Scholar] [CrossRef]
- Kober, E.; Ratz, R. Reaction of Tertiary Amines with Halo-S-Triazines and Halopyrimidines. J. Org. Chem. 1962, 27, 2509–2514. [Google Scholar] [CrossRef]
- Kolesinska, B.; Kaminski, Z.J. The umpolung of substituent effect in nucleophilic aromatic substitution. A new approach to the synthesis of N,N-disubstituted melamines (triazine triskelions) under mild reaction conditions. Tetrahedron 2009, 65, 3573–3576. [Google Scholar] [CrossRef]
- Mannion, J.C.; Dax, S.L.; Colder, F.J.; Macintyre, D.E.; Mcleod, J.; Ozola, V.; Suna, E.; Shubin, K.; Mencel, J.J.; Peng, S.X. Novel Orally Bioavailable Breathing Control Modulating Compounds, and Methods of Using Same. International Patent Application No WO2014078575A2, 22 May 2014. [Google Scholar]
- Fujita, H.; Dou, J.; Matsumoto, N.; Wu, Z.; Lindsey, J.S. Enzymatically triggered chromogenic cross-linking agents under physiological conditions. New J. Chem. 2020, 44, 719–743. [Google Scholar] [CrossRef]
- Brahimi, F.; Liu, J.; Malakhov, A.; Chowdhury, S.; Purisima, E.O.; Ivanisevic, L.; Caron, A.; Burgess, K.; Saragovi, H.U. A monovalent agonist of TrkA tyrosine kinase receptors can be converted into a bivalent antagonist. Biochim. Biophys. Acta 2010, 1800, 1018–1026. [Google Scholar] [CrossRef] [Green Version]
- Calvete, M.J.F.; Pinto, S.M.A.; Burrows, H.D.; Castro, M.M.C.A.; Geraldes, C.F.G.C.; Pereira, M.M. Multifunctionalization of cyanuric chloride for the stepwise synthesis of potential multimodal imaging chemical entities. Arabian J. Chem. 2020, 13, 2517–2525. [Google Scholar] [CrossRef]
- Gonzalez, P.; Pota, K.; Turan, L.S.; da Costa, V.C.P.; Akkaraju, G.; Green, K.N. Synthesis, Characterization, and Activity of a Triazine Bridged Antioxidant Small Molecule. ACS Chem. Neurosci. 2017, 8, 2414–2423. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Chung, B.; Ko, D.; Lim, H.S. A solid-phase method for synthesis of dimeric and trimeric ligands: Identification of potent bivalent ligands of 14-3-3sigma. Bioorg. Chem. 2019, 91, 103141. [Google Scholar] [CrossRef]
- Lönnberg, T.; Hutchinson, M.; Rokita, S. Selective Alkylation of C-Rich Bulge Motifs in Nucleic Acids by Quinone Methide Derivatives. Chem. Eur. J. 2015, 21, 13127–13136. [Google Scholar] [CrossRef]
- Li, H.; Zhou, H.; Krieger, S.; Parry, J.J.; Whittenberg, J.J.; Desai, A.V.; Rogers, B.E.; Kenis, P.J.; Reichert, D.E. Triazine-based tool box for developing peptidic PET imaging probes: Syntheses, microfluidic radiolabeling, and structure-activity evaluation. Bioconjug. Chem. 2014, 25, 761–772. [Google Scholar] [CrossRef]
- Zong, H.; Goonewardena, S.N.; Chang, H.N.; Otis, J.B.; Baker, J.R., Jr. Sequential and parallel dual labeling of nanoparticles using click chemistry. Bioorg. Med. Chem. 2014, 22, 6288–6296. [Google Scholar] [CrossRef] [PubMed]
- Haldón, E.; Nicasio, M.C.; Perez, P.J. Copper-catalysed azide-alkyne cycloadditions (CuAAC): An update. Org. Biomol. Chem. 2015, 13, 9528–9550. [Google Scholar] [CrossRef]
- Meldal, M.; Diness, F. Recent Fascinating Aspects of the CuAAC Click Reaction. Trends Chem. 2020, 2, 569–584. [Google Scholar] [CrossRef]
- Fantoni, N.Z.; El-Sagheer, A.H.; Brown, T. A Hitchhiker’s Guide to Click-Chemistry with Nucleic Acids. Chem. Rev. 2021. [Google Scholar] [CrossRef] [PubMed]
- Gavrilyuk, J.I.; Wuellner, U.; Salahuddin, S.; Goswami, R.K.; Sinha, S.C.; Barbas, C.F., 3rd. An efficient chemical approach to bispecific antibodies and antibodies of high valency. Bioorg. Med. Chem. Lett. 2009, 19, 3716–3720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnjatic, S.; Sawhney, N.B.; Bhardwaj, N. Toll-like receptor agonists: Are they good adjuvants? Cancer J. 2010, 16, 382–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirtland, M.E.; Tsitoura, D.C.; Durham, S.R.; Shamji, M.H. Toll-Like Receptor Agonists as Adjuvants for Allergen Immunotherapy. Front. Immunol. 2020, 11, 599083. [Google Scholar] [CrossRef] [PubMed]
- Tom, J.K.; Dotsey, E.Y.; Wong, H.Y.; Stutts, L.; Moore, T.; Davies, D.H.; Felgner, P.L.; Esser-Kahn, A.P. Modulation of Innate Immune Responses via Covalently Linked TLR Agonists. ACS Cent. Sci. 2015, 1, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Kuhri, S.; Charalambidis, G.; Angaridis, P.A.; Lazarides, T.; Pagona, G.; Tagmatarchis, N.; Coutsolelos, A.G.; Guldi, D.M. A new approach for the photosynthetic antenna-reaction center complex with a model organized around an s-triazine linker. Chem. Eur. J. 2014, 20, 2049–2057. [Google Scholar] [CrossRef]
- Wang, T.; Vineberg, J.G.; Honda, T.; Ojima, I. Design and synthesis of tumor-targeting theranostic drug conjugates for SPECT and PET imaging studies. Bioorg. Chem. 2018, 76, 458–467. [Google Scholar] [CrossRef]
- Vineberg, J.G.; Wang, T.; Zuniga, E.S.; Ojima, I. Design, synthesis, and biological evaluation of theranostic vitamin-linker-taxoid conjugates. J. Med. Chem. 2015, 58, 2406–2416. [Google Scholar] [CrossRef] [Green Version]
- Vineberg, J.G.; Zuniga, E.S.; Kamath, A.; Chen, Y.J.; Seitz, J.D.; Ojima, I. Design, synthesis, and biological evaluations of tumor-targeting dual-warhead conjugates for a taxoid-camptothecin combination chemotherapy. J. Med. Chem. 2014, 57, 5777–5791. [Google Scholar] [CrossRef]
- Lee, C.; Ji, K.; Simanek, E.E. Functionalization of a Triazine Dendrimer Presenting Four Maleimides on the Periphery and a DOTA Group at the Core. Molecules 2016, 21, 335. [Google Scholar] [CrossRef] [Green Version]
- Albin, T.J.; Tom, J.K.; Manna, S.; Gilkes, A.P.; Stetkevich, S.A.; Katz, B.B.; Supnet, M.; Felgner, J.; Jain, A.; Nakajima, R.; et al. Linked Toll-Like Receptor Triagonists Stimulate Distinct, Combination-Dependent Innate Immune Responses. ACS Cent. Sci 2019, 5, 1137–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilkes, A.P.; Albin, T.J.; Manna, S.; Supnet, M.; Ruiz, S.; Tom, J.; Badten, A.J.; Jain, A.; Nakajima, R.; Felgner, J.; et al. Tuning Subunit Vaccines with Novel TLR Triagonist Adjuvants to Generate Protective Immune Responses against Coxiella burnetii. J. Immunol. 2020, 204, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.J.; Omarjee, S.; Galloway, W.; Kwan, T.T.; Sore, H.F.; Parker, J.S.; Hyvonen, M.; Carroll, J.S.; Spring, D.R. A general approach for the site-selective modification of native proteins, enabling the generation of stable and functional antibody-drug conjugates. Chem. Sci. 2019, 10, 694–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.G.; Huangfu, S.Y.; Patil, S.; Tang, Q.; Sun, W.; Li, Y.; Lu, Z.L.; Qian, A. [12]aneN3-based multifunctional compounds as fluorescent probes and nucleic acids delivering agents. Drug Deliv. 2020, 27, 66–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody-Drug Conjugates: A Comprehensive Review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchikama, K.; An, Z. Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell 2016, 9, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Tiberghien, A.C.; Gregson, S.J.; Masterson, L.A.; Levy, J.-N.; Kemp, G.C.; Adams, L.R.; Patel, N.V.; Howard, P.W. An optimised synthesis of SG3376, a non-cleavable antibody-drug conjugate pyrrolobenzodiazepine drug-linker. Tetrahedron Lett. 2017, 58, 4363–4366. [Google Scholar] [CrossRef]
- Baker, M.B.; Ghiviriga, I.; Castellano, R.K. Molecular multifunctionalization via electronically coupled lactones. Chem. Sci. 2012, 3, 1095–1099. [Google Scholar] [CrossRef]
- Baker, M.B.; Ferreira, R.B.; Tasseroul, J.; Lampkins, A.J.; Al Abbas, A.; Abboud, K.A.; Castellano, R.K. Selective and Sequential Aminolysis of Benzotrifuranone: Synergism of Electronic Effects and Ring Strain Gradient. J. Org. Chem. 2016, 81, 9279–9288. [Google Scholar] [CrossRef]
- Li, H.; Homan, E.A.; Lampkins, A.J.; Ghiviriga, I.; Castellano, R.K. Synthesis and self-assembly of functionalized donor-sigma-acceptor molecules. Org. Lett. 2005, 7, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lampkins, A.J.; Baker, M.B.; Sumpter, B.G.; Huang, J.; Abboud, K.A.; Castellano, R.K. Benzotrifuranone: Synthesis, structure, and access to polycyclic heteroaromatics. Org. Lett. 2009, 11, 4314–4317. [Google Scholar] [CrossRef] [PubMed]
- Longin, O.; van de Langemheen, H.; Gannon, S.; Ward, D.; Liskamp, R.M.J. Synthesis of tris-tertiary amine CycloTriVeratrilene (TACTV) derivatives as water soluble pre-organized three aromatic ring containing molecular scaffolds for the construction of protein mimics. Tetrahedron Lett. 2019, 60, 151245–151249. [Google Scholar] [CrossRef]
- Longin, O.; Hezwani, M.; van de Langemheen, H.; Liskamp, R.M.J. Synthetic antibody protein mimics of infliximab by molecular scaffolding on novel CycloTriVeratrilene (CTV) derivatives. Org. Biomol. Chem. 2018, 16, 5254–5274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pícha, J.; Fabre, B.; Buděšínský, M.; Hajduch, J.; Abdellaoui, M.; Jiráček, J. Tri-Orthogonal Scaffolds for the Solid-Phase Synthesis of Peptides. Eur. J. Org. Chem. 2018, 2018, 5180–5192. [Google Scholar] [CrossRef]
- Watzke, A.; Gutierrez-Rodriguez, M.; Kohn, M.; Wacker, R.; Schroeder, H.; Breinbauer, R.; Kuhlmann, J.; Alexandrov, K.; Niemeyer, C.M.; Goody, R.S.; et al. A generic building block for C- and N-terminal protein-labeling and protein-immobilization. Bioorg. Med. Chem. 2006, 14, 6288–6306. [Google Scholar] [CrossRef]
- Viault, G.; Dautrey, S.; Maindron, N.; Hardouin, J.; Renard, P.Y.; Romieu, A. The first “ready-to-use” benzene-based heterotrifunctional cross-linker for multiple bioconjugation. Org. Biomol. Chem. 2013, 11, 2693–2705. [Google Scholar] [CrossRef]
- Yoshida, S.; Kanno, K.; Kii, I.; Misawa, Y.; Hagiwara, M.; Hosoya, T. Convergent synthesis of trifunctional molecules by three sequential azido-type-selective cycloadditions. Chem. Commun. 2018, 54, 3705–3708. [Google Scholar] [CrossRef] [Green Version]
- Hospital, A.; Gibard, C.; Gaulier, C.; Nauton, L.; Thery, V.; El-Ghozzi, M.; Avignant, D.; Cisnetti, F.; Gautier, A. Access to functionalised silver(I) and gold(I) N-heterocyclic carbenes by [2 + 3] dipolar cycloadditions. Dalton Trans. 2012, 41, 6803–6812. [Google Scholar] [CrossRef]
- Yoshida, S.; Misawa, Y.; Hosoya, T. Formal C-H-Azidation—Based Shortcut to Diazido Building Blocks for the Versatile Preparation of Photoaffinity Labeling Probes. Eur. J. Org. Chem. 2014, 2014, 3991–3995. [Google Scholar] [CrossRef]
- Wagner, R.W.; Lindsey, J.S. Boron-Dipyrromethene Dyes for Incorporation in Synthetic Multi-Pigment Light-Harvesting Arrays. Pure Appl. Chem. 1996, 68, 1373–1380. [Google Scholar] [CrossRef]
- Azov, V.A.; Schlegel, A.; Diederich, F. Functionalized Calix[4]resorcinarene Cavitands. Versatile Platforms for the Modular Construction of Extended Molecular Switches. Bull. Chem. Soc. Jpn. 2006, 79, 1926–1940. [Google Scholar] [CrossRef]
- Yoshida, S.; Sakata, Y.; Misawa, Y.; Morita, T.; Kuribara, T.; Ito, H.; Koike, Y.; Kii, I.; Hosoya, T. Assembly of four modules onto a tetraazide platform by consecutive 1,2,3-triazole formations. Chem. Commun. 2021, 57, 899–902. [Google Scholar] [CrossRef] [PubMed]
- Ballell, L.; van Scherpenzeel, M.; Buchalova, K.; Liskamp, R.M.; Pieters, R.J. A new chemical probe for the detection of the cancer-linked galectin-3. Org. Biomol. Chem. 2006, 4, 4387–4394. [Google Scholar] [CrossRef]
- Rashidian, M.; Kumarapperuma, S.C.; Gabrielse, K.; Fegan, A.; Wagner, C.R.; Distefano, M.D. Simultaneous dual protein labeling using a triorthogonal reagent. J. Am. Chem. Soc. 2013, 135, 16388–16396. [Google Scholar] [CrossRef] [Green Version]
- Rashidian, M.; Song, J.M.; Pricer, R.E.; Distefano, M.D. Chemoenzymatic reversible immobilization and labeling of proteins without prior purification. J. Am. Chem. Soc. 2012, 134, 8455–8467. [Google Scholar] [CrossRef] [Green Version]
- Gaon, I.; Turek, T.C.; Distefano, M.D. Farnesyl and geranylgeranyl pyrophosphate analogs incorporating benzoylbenzyl ethers: Synthesis and inhibition of yeast protein farnesyltransferase. Tetrahedron Lett. 1996, 37, 8833–8836. [Google Scholar] [CrossRef]
- Fegan, A.; Kumarapperuma, S.C.; Wagner, C.R. Chemically self-assembled antibody nanostructures as potential drug carriers. Mol. Pharm. 2012, 9, 3218–3227. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.X.; Zhang, X.B.; Li, J.B.; Zhang, C.C.; Liang, H.; Mao, G.J.; Zhou, L.Y.; Tan, W.; Yu, R.Q. Bispyrene-fluorescein hybrid based FRET cassette: A convenient platform toward ratiometric time-resolved probe for bioanalytical applications. Anal. Chem. 2014, 86, 10389–10396. [Google Scholar] [CrossRef]
- Lamos, S.M.; Krusemark, C.J.; McGee, C.J.; Scalf, M.; Smith, L.M.; Belshaw, P.J. Mixed isotope photoaffinity reagents for identification of small-molecule targets by mass spectrometry. Angew. Chem. Int. Ed. 2006, 45, 4329–4333. [Google Scholar] [CrossRef]
- Vall-Sagarra, A.; Litau, S.; Decristoforo, C.; Wangler, B.; Schirrmacher, R.; Fricker, G.; Wangler, C. Design, Synthesis, In Vitro, and Initial In Vivo Evaluation of Heterobivalent Peptidic Ligands Targeting Both NPY(Y(1))- and GRP-Receptors–An Improvement for Breast Cancer Imaging? Pharmaceuticals 2018, 11, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoi, T.; Ueda, T.; Tanimoto, H.; Morimoto, T.; Kakiuchi, K. Site-selective conversion of azido groups at carbonyl alpha-positions into oxime groups leading triazide to a triple click conjugation scaffold. Chem. Commun. 2019, 55, 1891–1894. [Google Scholar] [CrossRef]
- Yokoi, T.; Tanimoto, H.; Ueda, T.; Morimoto, T.; Kakiuchi, K. Site-Selective Conversion of Azido Groups at Carbonyl alpha-Positions to Diazo Groups in Diazido and Triazido Compounds. J. Org. Chem. 2018, 83, 12103–12121. [Google Scholar] [CrossRef]
- Daghish, M.; Hennig, L.; Findeisen, M.; Giesa, S.; Schumer, F.; Hennig, H.; Beck-Sickinger, A.G.; Welzel, P. Tetrafunctional photoaffinity labels based on Nakanish’s m-nitroalkoxy-substituted phenyltrifluoromethyldiazirine. Angew. Chem. Int. Ed. 2002, 41, 2293–2297. [Google Scholar] [CrossRef]
- Sorkin, M.R.; Walker, J.A.; Brown, J.S.; Alabi, C.A. Versatile Platform for the Synthesis of Orthogonally Cleavable Heteromultifunctional Cross-Linkers. Bioconjug. Chem. 2017, 28, 907–912. [Google Scholar] [CrossRef]
- Cline, L.L.; Janout, V.; Fisher, M.; Juliano, R.L.; Regen, S.L. A molecular umbrella approach to the intracellular delivery of small interfering RNA. Bioconjug. Chem. 2011, 22, 2210–2216. [Google Scholar] [CrossRef] [Green Version]
- Werkhoven, P.R.; van de Langemheen, H.; van der Wal, S.; Kruijtzer, J.A.; Liskamp, R.M. Versatile convergent synthesis of a three peptide loop containing protein mimic of whooping cough pertactin by successive Cu(I)-catalyzed azide alkyne cycloaddition on an orthogonal alkyne functionalized TAC-scaffold. J. Pept. Sci. 2014, 20, 235–239. [Google Scholar] [CrossRef]
- Dai, Y.; Weng, J.; George, J.; Chen, H.; Lin, Q.; Wang, J.; Royzen, M.; Zhang, Q. Three-Component Protein Modification Using Mercaptobenzaldehyde Derivatives. Org. Lett. 2019, 21, 3828–3833. [Google Scholar] [CrossRef]
- Haley, C.A.C.; Maitland, P. Organic Reactions in Aqueous Solution at Room Temperature. Part 1. The Influence of pH on Condensations Involving the Linking of Carbon to Nitrogen and of Carbon to Carbon. J. Chem. Soc. 1951, 3155–3174. [Google Scholar] [CrossRef]
- Medley, J.W.; Movassaghi, M. Robinson’s landmark synthesis of tropinone. Chem. Commun. 2013, 49, 10775–10777. [Google Scholar] [CrossRef] [PubMed]
- Boutureira, O.; Bernardes, G.J. Advances in chemical protein modification. Chem. Rev. 2015, 115, 2174–2195. [Google Scholar] [CrossRef]
- Shadish, J.A.; DeForest, C.A. Site-Selective Protein Modification: From Functionalized Proteins to Functional Biomaterials. Matter 2020, 2, 50–77. [Google Scholar] [CrossRef]
- Maruani, A.; Richards, D.A.; Chudasama, V. Dual modification of biomolecules. Org. Biomol. Chem. 2016, 14, 6165–6178. [Google Scholar] [CrossRef]
- Tessier, R.; Ceballos, J.; Guidotti, N.; Simonet-Davin, R.; Fierz, B.; Waser, J. “Doubly Orthogonal” Labeling of Peptides and Proteins. Chem 2019, 5, 2243–2263. [Google Scholar] [CrossRef]
- Baslé, E.; Joubert, N.; Pucheault, M. Protein chemical modification on endogenous amino acids. Chem. Biol. 2010, 17, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Spicer, C.D.; Davis, B.G. Selective chemical protein modification. Nat. Commun. 2014, 5, 4740–4753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramesh, S.; Cherkupally, P.; Govender, T.; Kruger, H.G.; Albericio, F.; de la Torre, B.G. Highly chemoselective ligation of thiol- and amino-peptides on a bromomaleimide core. Chem. Commun. 2016, 52, 2334–2337. [Google Scholar] [CrossRef]
- Gil de Montes, E.; Jimenez-Moreno, E.; Oliveira, B.L.; Navo, C.D.; Cal, P.; Jimenez-Oses, G.; Robina, I.; Moreno-Vargas, A.J.; Bernardes, G.J.L. Azabicyclic vinyl sulfones for residue-specific dual protein labelling. Chem. Sci. 2019, 10, 4515–4522. [Google Scholar] [CrossRef] [Green Version]
- Morales-Sanfrutos, J.; Lopez-Jaramillo, J.; Ortega-Munoz, M.; Megia-Fernandez, A.; Perez-Balderas, F.; Hernandez-Mateo, F.; Santoyo-Gonzalez, F. Vinyl sulfone: A versatile function for simple bioconjugation and immobilization. Org. Biomol. Chem. 2010, 8, 667–675. [Google Scholar] [CrossRef]
- Lang, K.; Davis, L.; Torres-Kolbus, J.; Chou, C.; Deiters, A.; Chin, J.W. Genetically encoded norbornene directs site-specific cellular protein labelling via a rapid bioorthogonal reaction. Nat. Chem. 2012, 4, 298–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mushtaq, S.; Yun, S.J.; Jeon, J. Recent Advances in Bioorthogonal Click Chemistry for Efficient Synthesis of Radiotracers and Radiopharmaceuticals. Molecules 2019, 24, 3567. [Google Scholar] [CrossRef] [Green Version]
- Koniev, O.; Wagner, A. Developments and recent advancements in the field of endogenous amino acid selective bond forming reactions for bioconjugation. Chem. Soc. Rev. 2015, 44, 5495–5551. [Google Scholar] [CrossRef] [Green Version]
- Gai, Y.; Xiang, G.; Ma, X.; Hui, W.; Ouyang, Q.; Sun, L.; Ding, J.; Sheng, J.; Zeng, D. Universal Molecular Scaffold for Facile Construction of Multivalent and Multimodal Imaging Probes. Bioconjug. Chem. 2016, 27, 515–520. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, M.; Hatanaka, Y.; Yang, J.; Dhesi, J.; Holman, G.D. Synthesis of biotinylated bis(D-glucose) derivatives for glucose transporter photoaffinity labelling. Carbohydr. Res. 2001, 331, 119–127. [Google Scholar] [CrossRef]
- Ji, C.; Miller, P.A.; Miller, M.J. Iron transport-mediated drug delivery: Practical syntheses and in vitro antibacterial studies of tris-catecholate siderophore-aminopenicillin conjugates reveals selectively potent antipseudomonal activity. J. Am. Chem. Soc. 2012, 134, 9898–9901. [Google Scholar] [CrossRef] [Green Version]
- Knall, A.C.; Hollauf, M.; Saf, R.; Slugovc, C. A trifunctional linker suitable for conducting three orthogonal click chemistries in one pot. Org. Biomol. Chem. 2016, 14, 10576–10580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knall, A.C.; Slugovc, C. Inverse electron demand Diels-Alder (iEDDA)-initiated conjugation: A (high) potential click chemistry scheme. Chem. Soc. Rev. 2013, 42, 5131–5142. [Google Scholar] [CrossRef] [Green Version]
- Pagel, M. Inverse electron demand Diels-Alder (IEDDA) reactions in peptide chemistry. J. Pept. Sci. 2019, 25, e3141. [Google Scholar] [CrossRef] [Green Version]
- Citron, C.A.; Wickel, S.M.; Schulz, B.; Draeger, S.; Dickschat, J.S. A Diels-Alder/Retro-Diels-Alder Approach for the Enantioselective Synthesis of Microbial Butenolides. Eur. J. Org. Chem. 2012, 6636–6646. [Google Scholar] [CrossRef]
- Gramlich, W.M.; Robertson, M.L.; Hillmyer, M.A. Reactive Compatibilization of Poly(l-lactide) and Conjugated Soybean Oil. Macromolecules 2010, 43, 2313–2321. [Google Scholar] [CrossRef]
- Ho, H.T.; Levere, M.E.; Fournier, D.; Montembault, V.; Pascual, S.; Fontaine, L. Introducing the Azlactone Functionality into Polymers through Controlled Radical Polymerization: Strategies and Recent Developments. Aust. J. Chem. 2012, 65, 970–977. [Google Scholar] [CrossRef]
- de Castro, P.P.; Carpanez, A.G.; Amarante, G.W. Azlactone Reaction Developments. Chem. Eur. J. 2016, 22, 10294–10318. [Google Scholar] [CrossRef]
- Yu, X.; Herberg, A.; Kuckling, D. Micellar Organocatalysis Using Smart Polymer Supports: Influence of Thermoresponsive Self-Assembly on Catalytic Activity. Polymers 2020, 12, 2265. [Google Scholar] [CrossRef]
- Pantin, M.; Caillé, J.; Boeda, F.; Fontaine, L.; Pearson-Long, M.S.M.; Bertus, P. Heteromultifunctional Oxazolones as Versatile Linkers for Click Chemistry Reactions. Eur. J. Org. Chem. 2019, 2019, 7359–7366. [Google Scholar] [CrossRef]
- Moss, J.A.; Stokols, S.; Hixon, M.S.; Ashley, F.T.; Chang, J.Y.; Janda, K.D. Solid-phase synthesis and kinetic characterization of fluorogenic enzyme-degradable hydrogel cross-linkers. Biomacromolecules 2006, 7, 1011–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masselin, A.; Petrelli, A.; Donzel, M.; Armand, S.; Cottaz, S.; Fort, S. Unprecedented Affinity Labeling of Carbohydrate-Binding Proteins with s-Triazinyl Glycosides. Bioconjug. Chem. 2019, 30, 2332–2339. [Google Scholar] [CrossRef]
- Blencowe, C.A.; Russell, A.T.; Greco, F.; Hayes, W.; Thornthwaite, D.W. Self-immolative linkers in polymeric delivery systems. Polym. Chem. 2011, 2, 773–790. [Google Scholar] [CrossRef] [Green Version]
- Gonzaga, R.V.; do Nascimento, L.A.; Santos, S.S.; Machado Sanches, B.A.; Giarolla, J.; Ferreira, E.I. Perspectives About Self-Immolative Drug Delivery Systems. J. Pharm. Sci. 2020, 109, 3262–3281. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, S.C.; De Brabander, J.; Miyamoto, J.; Senter, P.D. Expanded Utility of the beta-Glucuronide Linker: ADCs That Deliver Phenolic Cytotoxic Agents. ACS Med. Chem. Lett. 2010, 1, 277–280. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Han, J.; Lim, H.J.; Ren, W.X.; Lim, J.Y.; Kim, J.H.; Kim, J.S. Mitochondrial induced and self-monitored intrinsic apoptosis by antitumor theranostic prodrug: In vivo imaging and precise cancer treatment. J. Am. Chem. Soc. 2014, 136, 17836–17843. [Google Scholar] [CrossRef] [PubMed]
- Alsarraf, J.; Peraudeau, E.; Poinot, P.; Tranoy-Opalinski, I.; Clarhaut, J.; Renoux, B.; Papot, S. A dendritic beta-galactosidase-responsive folate-monomethylauristatin E conjugate. Chem. Commun. 2015, 51, 15792–15795. [Google Scholar] [CrossRef]
- Legigan, T.; Clarhaut, J.; Tranoy-Opalinski, I.; Monvoisin, A.; Renoux, B.; Thomas, M.; Le Pape, A.; Lerondel, S.; Papot, S. The first generation of beta-galactosidase-responsive prodrugs designed for the selective treatment of solid tumors in prodrug monotherapy. Angew. Chem. Int. Ed. 2012, 51, 11606–11610. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhou, X.-Y.; Wu, H.; Lei, Y.-Z.; Li, J.-H. Highly efficient reduction of nitro compounds: Recyclable Pd/C-catalyzed transfer hydrogenation with ammonium formate or hydrazine hydrate as hydrogen source. Synth. Commun. 2018, 48, 2475–2484. [Google Scholar] [CrossRef]
- Wiener, H.; Blum, J.; Sasson, Y. Studies on the Mechanism of Transfer Hydrogenation of Nitroarenes by Formate Salts Catalyzed by Pd/C. J. Org. Chem. 1991, 56, 4481–4486. [Google Scholar] [CrossRef]
- Erez, R.; Shabat, D. The azaquinone-methide elimination: Comparison study of 1,6- and 1,4-eliminations under physiological conditions. Org. Biomol. Chem. 2008, 6, 2669–2672. [Google Scholar] [CrossRef] [PubMed]
- Grinda, M.; Legigan, T.; Clarhaut, J.; Peraudeau, E.; Tranoy-Opalinski, I.; Renoux, B.; Thomas, M.; Guilhot, F.; Papot, S. An enzyme-responsive system programmed for the double release of bioactive molecules through an intracellular chemical amplification process. Org. Biomol. Chem. 2013, 11, 7129–7133. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Ogawa, M.; Alford, R.; Choyke, P.L.; Urano, Y. New strategies for fluorescent probe design in medical diagnostic imaging. Chem. Rev. 2010, 110, 2620–2640. [Google Scholar] [CrossRef] [Green Version]
- Peterson, J.D. Paradigms in Fluorescence Molecular Imaging: Maximizing Measurement of Biological Changes in Disease, Therapeutic Efficacy, and Toxicology/Safety. Mol. Imaging Biol. 2019, 21, 599–611. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Ye, D. Molecular imaging of enzyme activity in vivo using activatable probes. Sci. Bull. 2016, 61, 1672–1679. [Google Scholar] [CrossRef]
- Liu, H.W.; Chen, L.; Xu, C.; Li, Z.; Zhang, H.; Zhang, X.B.; Tan, W. Recent progresses in small-molecule enzymatic fluorescent probes for cancer imaging. Chem. Soc. Rev. 2018, 47, 7140–7180. [Google Scholar] [CrossRef]
- Singh, H.; Tiwari, K.; Tiwari, R.; Pramanik, S.K.; Das, A. Small Molecule as Fluorescent Probes for Monitoring Intracellular Enzymatic Transformations. Chem. Rev. 2019, 119, 11718–11760. [Google Scholar] [CrossRef]
- Baruch, A.; Jeffery, D.A.; Bogyo, M. Enzyme activity—it’s all about image. Trends Cell Biol. 2004, 14, 29–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinon, F.; Tschopp, J. Inflammatory caspases: Linking an intracellular innate immune system to autoinflammatory diseases. Cell 2004, 117, 561–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Opdenbosch, N.; Lamkanfi, M. Caspases in Cell Death, Inflammation, and Disease. Immunity 2019, 50, 1352–1364. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Li, L.; Wu, H.; Su, Y.; Yang, P.Y.; Uttamchandani, M.; Xu, Q.H.; Yao, S.Q. Multicolor, one- and two-photon imaging of enzymatic activities in live cells with fluorescently Quenched Activity-Based Probes (qABPs). J. Am. Chem. Soc. 2011, 133, 12009–12020. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Girish, A.; Chattopadhaya, S.; Yao, S.Q. Developing novel activity-based fluorescent probes that target different classes of proteases. Chem. Commun. 2004, 1512–1513. [Google Scholar] [CrossRef] [PubMed]
- Sellars, J.D.; Landrum, M.; Congreve, A.; Dixon, D.P.; Mosely, J.A.; Beeby, A.; Edwards, R.; Steel, P.G. Fluorescence quenched quinone methide based activity probes—A cautionary tale. Org. Biomol. Chem. 2010, 8, 1610–1618. [Google Scholar] [CrossRef]
- Gnaim, S.; Shabat, D. Quinone-methide species, a gateway to functional molecular systems: From self-immolative dendrimers to long-wavelength fluorescent dyes. Acc. Chem. Res. 2014, 47, 2970–2984. [Google Scholar] [CrossRef]
- Gnaim, S.; Shabat, D. Activity-Based Optical Sensing Enabled by Self-Immolative Scaffolds: Monitoring of Release Events by Fluorescence or Chemiluminescence Output. Acc. Chem. Res. 2019, 52, 2806–2817. [Google Scholar] [CrossRef]
- Chevalier, A.; Hardouin, J.; Renard, P.Y.; Romieu, A. Universal dark quencher based on “clicked” spectrally distinct azo dyes. Org. Lett. 2013, 15, 6082–6085. [Google Scholar] [CrossRef]
- Huang, J.; Li, J.; Lyu, Y.; Miao, Q.; Pu, K. Molecular optical imaging probes for early diagnosis of drug-induced acute kidney injury. Nat. Mater. 2019, 18, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Arja, K.; Elgland, M.; Appelqvist, H.; Konradsson, P.; Lindgren, M.; Nilsson, K.P.R. Synthesis and Characterization of Novel Fluoro-glycosylated Porphyrins that can be Utilized as Theranostic Agents. ChemistryOpen 2018, 7, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Deng, J.; Guo, D.; Sun, Q.; Wang, Z.; Wang, K.; Wu, F. Mitochondria-targeting Pt/Mn porphyrins as efficient photosensitizers for magnetic resonance imaging and photodynamic therapy. Dyes Pigment. 2019, 166, 189–195. [Google Scholar] [CrossRef]
- Lindsey, J.S. Synthesis of meso-Substituted Porphyrins. In The Porphyrin Handbook; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; Academic Press: San Diego, CA, USA, 2000; Volume 1, pp. 45–118. [Google Scholar]
- Taniguchi, M.; Lindsey, J.S. Enumeration of Isomers of Substituted Tetrapyrrole Macrocycles: From Classical Problems in Biology to Modern Combinatorial Libraries. In Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific: Singapore, 2012; Volume 23, pp. 1–80. [Google Scholar]
- Yang, J.; Yang, Y.W. Metal-Organic Frameworks for Biomedical Applications. Small 2020, 16, e1906846. [Google Scholar] [CrossRef] [PubMed]
- Pliquett, J.; Amor, S.; Ponce-Vargas, M.; Laly, M.; Racoeur, C.; Rousselin, Y.; Denat, F.; Bettaieb, A.; Fleurat-Lessard, P.; Paul, C.; et al. Design of a multifunctionalizable BODIPY platform for the facile elaboration of a large series of gold(I)-based optical theranostics. Dalton Trans. 2018, 47, 11203–11218. [Google Scholar] [CrossRef] [PubMed]
- Laine, M.; Barbosa, N.A.; Kochel, A.; Osiecka, B.; Szewczyk, G.; Sarna, T.; Ziółkowski, P.; Wieczorek, R.; Filarowski, A. Synthesis, structural, spectroscopic, computational and cytotoxic studies of BODIPY dyes. Sens. Actuators B Chem. 2017, 238, 548–555. [Google Scholar] [CrossRef]
- Gryko, D.; Li, J.; Diers, J.R.; Roth, K.M.; Bocian, D.F.; Kuhr, W.G.; Lindsey, J.S. Studies Related to the Design and Synthesis of a Molecular Octal Counter. J. Mater. Chem. 2001, 11, 1162–1180. [Google Scholar] [CrossRef]
- Li, J.; Ambroise, A.; Yang, S.I.; Diers, J.R.; Seth, J.; Wack, C.R.; Bocian, D.F.; Holten, D.; Lindsey, J.S. Template-Directed Synthesis, Excited-State Photodynamics, and Electronic Communication in a Hexameric Wheel of Porphyrins. J. Am. Chem. Soc. 1999, 121, 8927–8940. [Google Scholar] [CrossRef]
- Ambroise, A.; Li, J.; Yu, L.; Lindsey, J.S. A Self-Assembled Light-Harvesting Array of Seven Porphyrins in a Wheel and Spoke Architecture. Org. Lett. 2000, 2, 2563–2566. [Google Scholar] [CrossRef]
- Yu, L.; Lindsey, J.S. Rational Syntheses of Cyclic Hexameric Porphyrin Arrays for Studies of Self-Assembling Light-Harvesting Systems. J. Org. Chem. 2001, 66, 7402–7419. [Google Scholar] [CrossRef]
- Tomizaki, K.-y.; Yu, L.; Wei, L.; Bocian, D.F.; Lindsey, J.S. Synthesis of Cyclic Hexameric Porphyrin Arrays. Anchors for Surface Immobilization and Columnar Self-Assembly. J. Org. Chem. 2003, 68, 8199–8207. [Google Scholar] [CrossRef]
- Lindsey, J.S. Synthetic Routes to meso-Patterned Porphyrins. Acc. Chem. Res. 2010, 43, 300–311. [Google Scholar] [CrossRef]
- Holten, D.; Bocian, D.F.; Lindsey, J.S. Probing Electronic Communication in Covalently Linked Multiporphyrin Arrays. A Guide to the Rational Design of Molecular Photonic Devices. Acc. Chem. Res. 2002, 35, 57–69. [Google Scholar] [CrossRef]
- Song, H.-e.; Kirmaier, C.; Schwartz, J.K.; Hindin, E.; Yu, L.; Bocian, D.F.; Lindsey, J.S.; Holten, D. Mechanisms, Pathways, and Dynamics of Excited-State Energy Flow in Self-Assembled Wheel-and-Spoke Light-Harvesting Architectures. J. Phys. Chem. B 2006, 110, 19121–19130. [Google Scholar] [CrossRef]
- Song, H.-e.; Kirmaier, C.; Schwartz, J.K.; Hindin, E.; Yu, L.H.; Bocian, D.F.; Lindsey, J.S.; Holten, D. Effects of Multiple Pathways on Excited-State Energy Flow in Self-Assembled Wheel-and-Spoke Light-Harvesting Architectures. J. Phys. Chem. B 2006, 110, 19131–19139. [Google Scholar] [CrossRef]
- Tiede, D.M.; Zhang, R.; Chen, L.X.; Yu, L.; Lindsey, J.S. Structural Characterization of Modular Supramolecular Architectures in Solution. J. Am. Chem. Soc. 2004, 126, 14054–14062. [Google Scholar] [CrossRef]
- Mardis, K.L.; Sutton, H.M.; Zuo, X.; Lindsey, J.S.; Tiede, D.M. Solution-State Conformational Ensemble of a Hexameric Porphyrin Array Characterized Using Molecular Dynamics and X-Ray Scattering. J. Phys. Chem. A 2009, 113, 2516–2523. [Google Scholar] [CrossRef] [Green Version]
- Geier, G.R., III; Callinan, J.B.; Rao, P.D.; Lindsey, J.S. A Survey of Acid Catalysts in Dipyrromethanecarbinol Condensations Leading to meso-Substituted Porphyrins. J. Porphyr. Phthalocyanines 2001, 5, 810–823. [Google Scholar] [CrossRef]
- Senge, M.O. Stirring the porphyrin alphabet soup–functionalization reactions for porphyrins. Chem. Commun. 2011, 47, 1943–1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laha, J.K.; Dhanalekshmi, S.; Taniguchi, M.; Ambroise, A.; Lindsey, J.S. A Scalable Synthesis of Meso-Substituted Dipyrromethanes. Org. Process Res. Dev. 2003, 7, 799–812. [Google Scholar] [CrossRef]
Figure 1.
Poisson distribution for m = 0.3, 1.0 and 1.5 (top panel). Scaffold S (equipped with five equal and independent reaction sites) undergoes derivatization here with mean loading of one to give a mixture of 32 species (bottom panel).
Figure 1.
Poisson distribution for m = 0.3, 1.0 and 1.5 (top panel). Scaffold S (equipped with five equal and independent reaction sites) undergoes derivatization here with mean loading of one to give a mixture of 32 species (bottom panel).
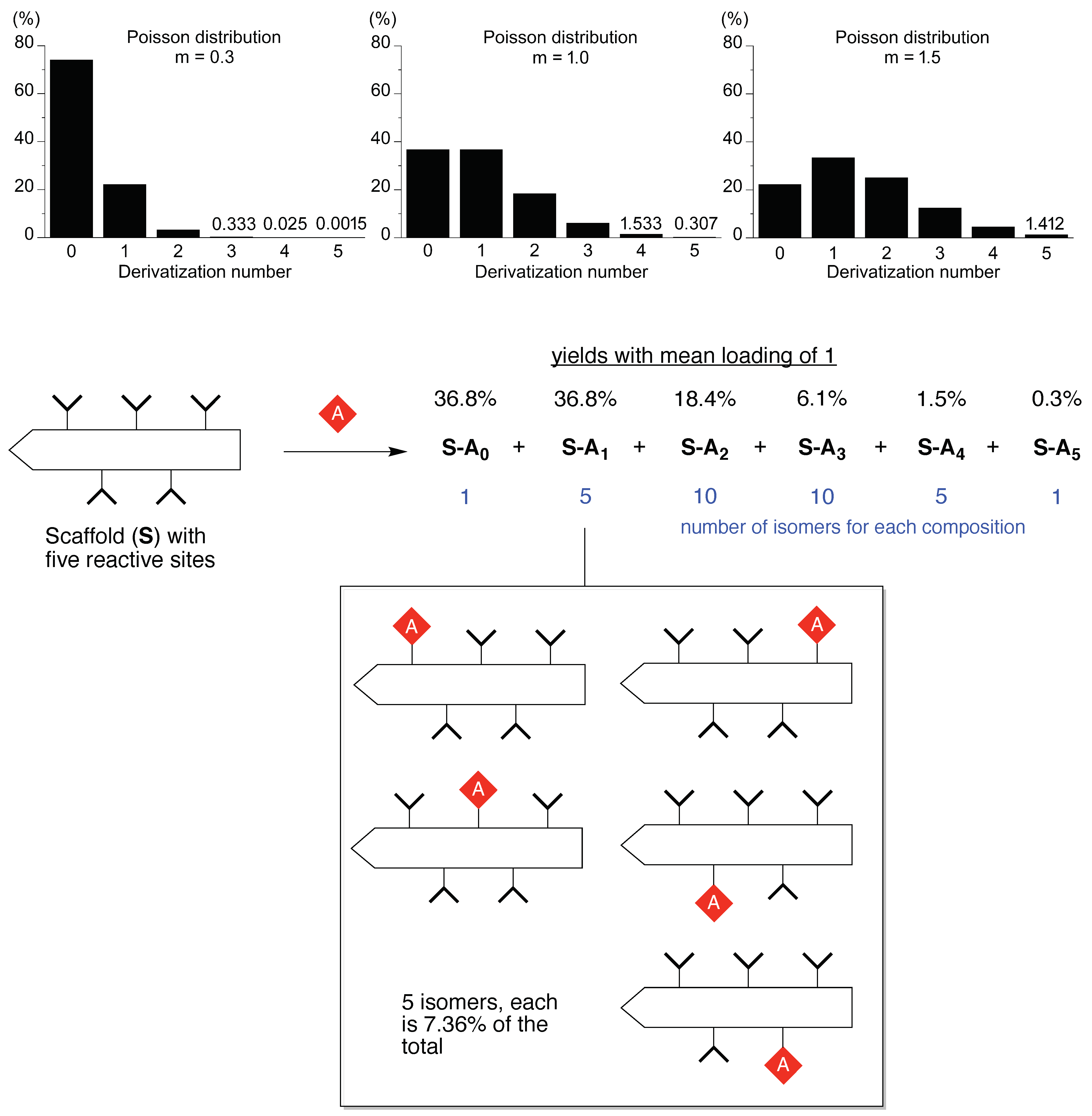
Figure 2.
Structures of natural polysaccharide carriers and a synthetic dendrimer.
Figure 3.
Hub molecules described herein.

Figure 4.
All graphs treated herein.

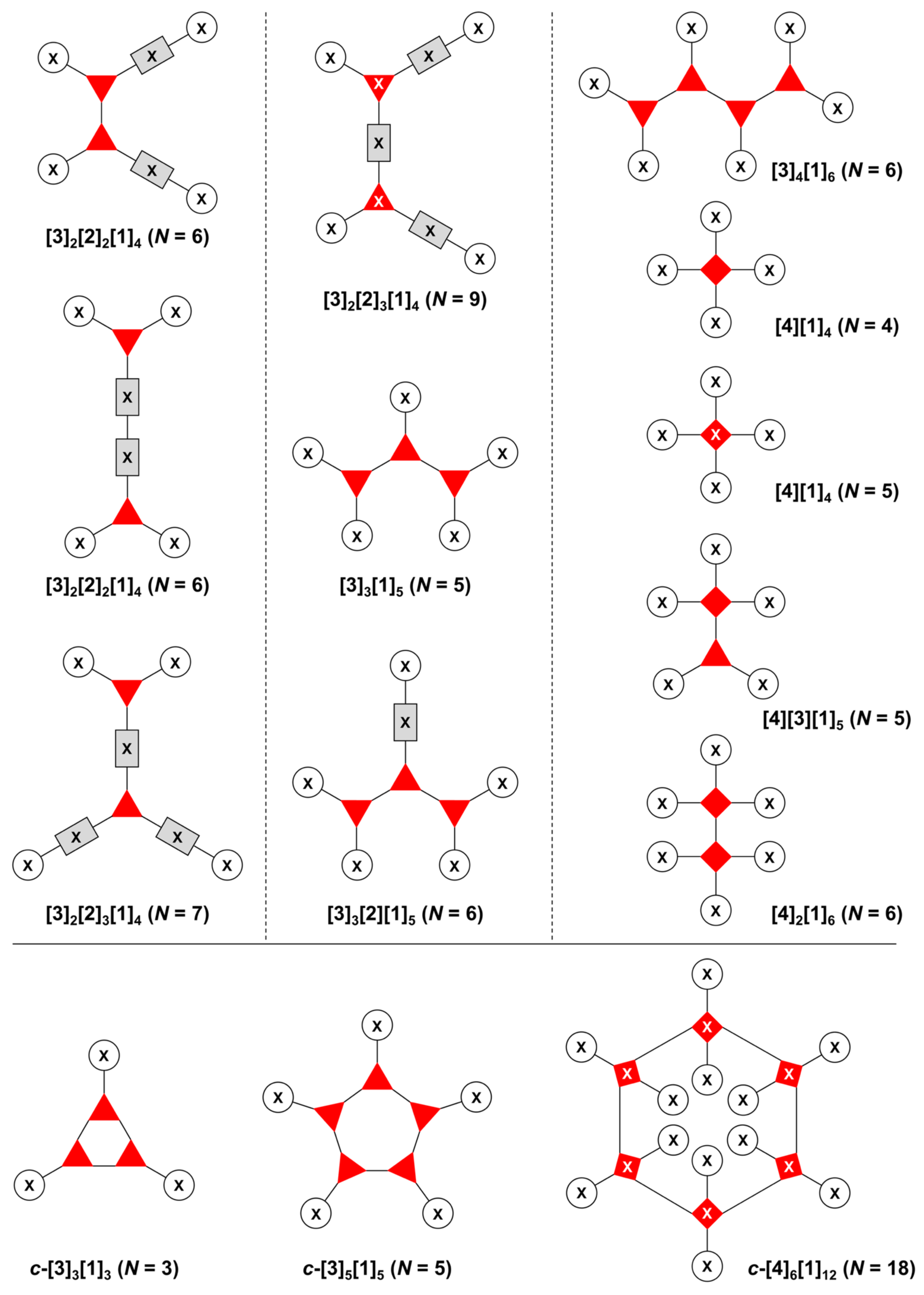
Figure 5.
Procedure for the composition of graphs to represent multifunctional molecules. (A) A single hub with architecture [3][1]3 and composition ABC. (B) Two hubs with architecture [4][3][1]5 and composition A3BC. (C) A single hub with a functional linker giving architecture [3][2][1]3 and composition ABCD. The synthesis step order (1–4) is given by numbers alongside the tree branches.
Figure 5.
Procedure for the composition of graphs to represent multifunctional molecules. (A) A single hub with architecture [3][1]3 and composition ABC. (B) Two hubs with architecture [4][3][1]5 and composition A3BC. (C) A single hub with a functional linker giving architecture [3][2][1]3 and composition ABCD. The synthesis step order (1–4) is given by numbers alongside the tree branches.

Figure 6.
Representative graph [3][2]3[1]3 (one three-arm hub, three two-arm hubs, and three appendages) with functional units ABCDEF and synthesis order 1–6.
Figure 6.
Representative graph [3][2]3[1]3 (one three-arm hub, three two-arm hubs, and three appendages) with functional units ABCDEF and synthesis order 1–6.
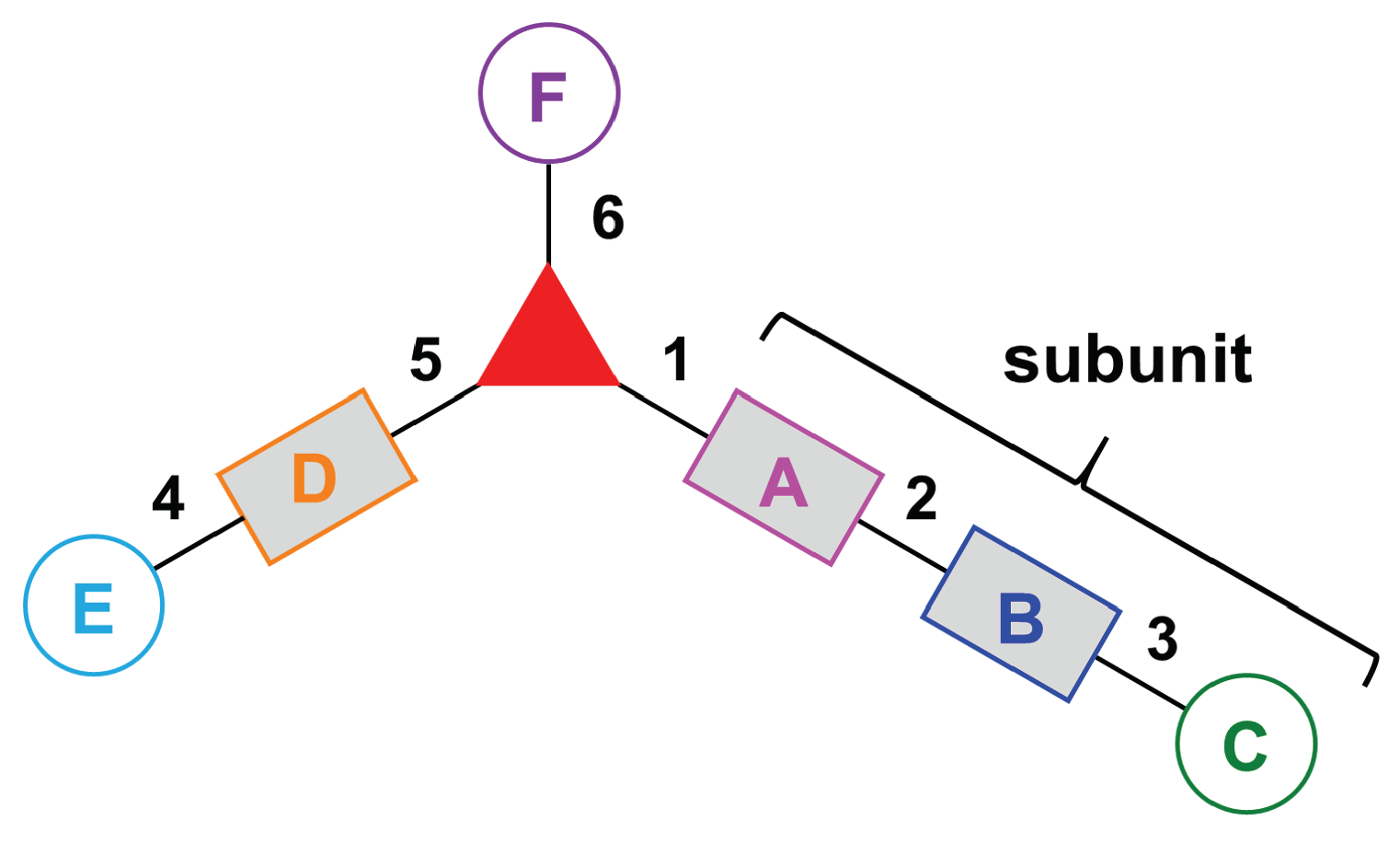
Figure 7.
Diverse groups for water solubilization.
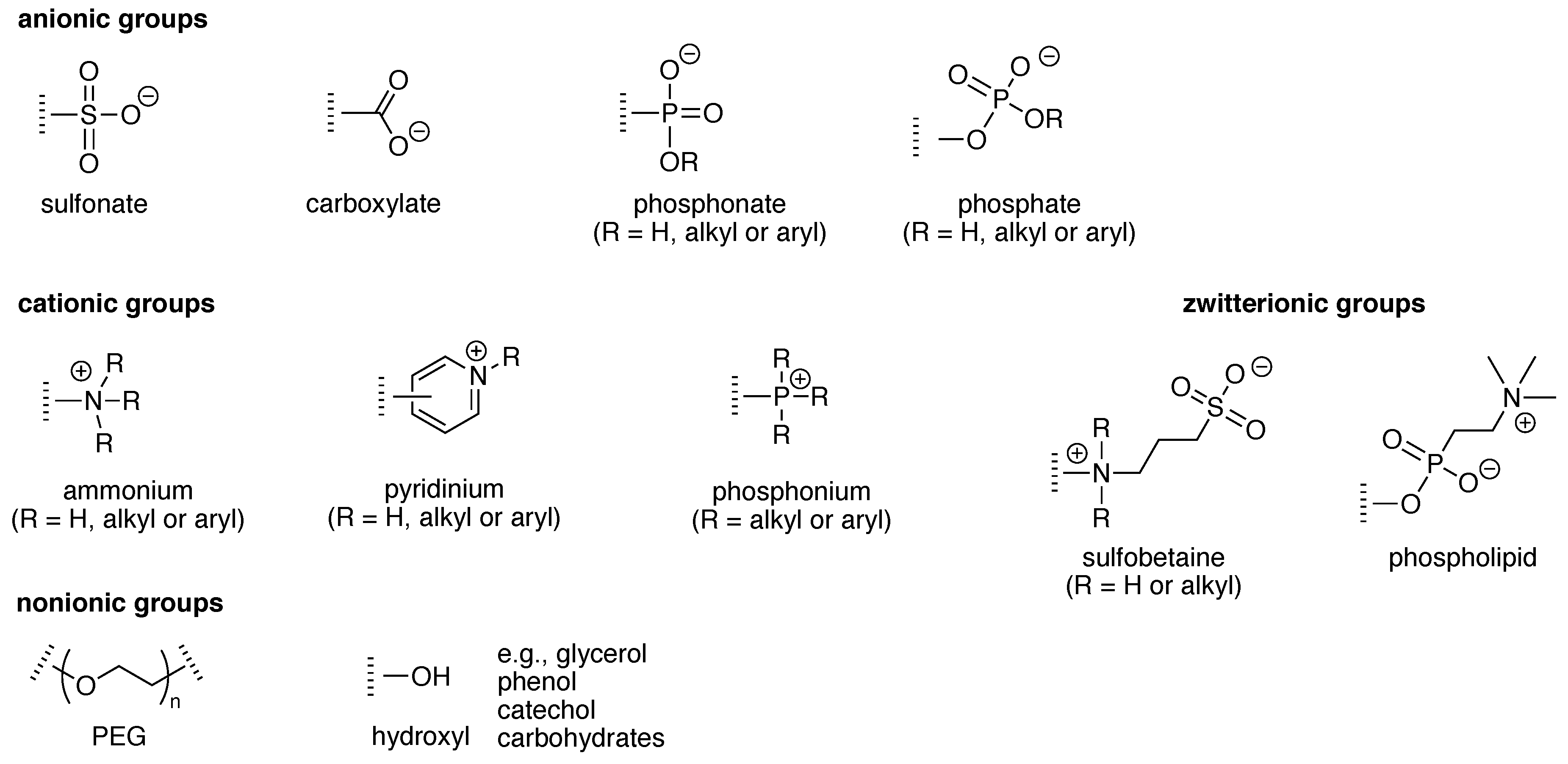
Figure 8.
Solubility of the PEG unit (4b) compared with two homologous oxyalkyl polymers (left panel), and the gauche conformational preference for PEG groups (right panel).
Figure 8.
Solubility of the PEG unit (4b) compared with two homologous oxyalkyl polymers (left panel), and the gauche conformational preference for PEG groups (right panel).
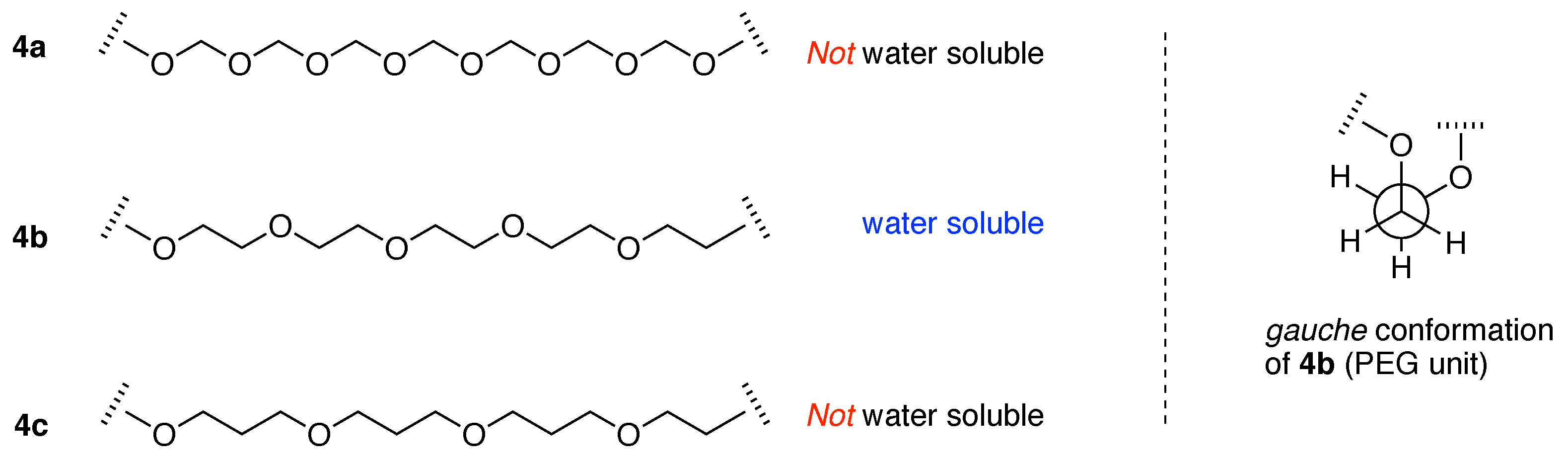
Figure 9.
Seven linear multifunctional molecules (no hubs) of graph [2][1]2.
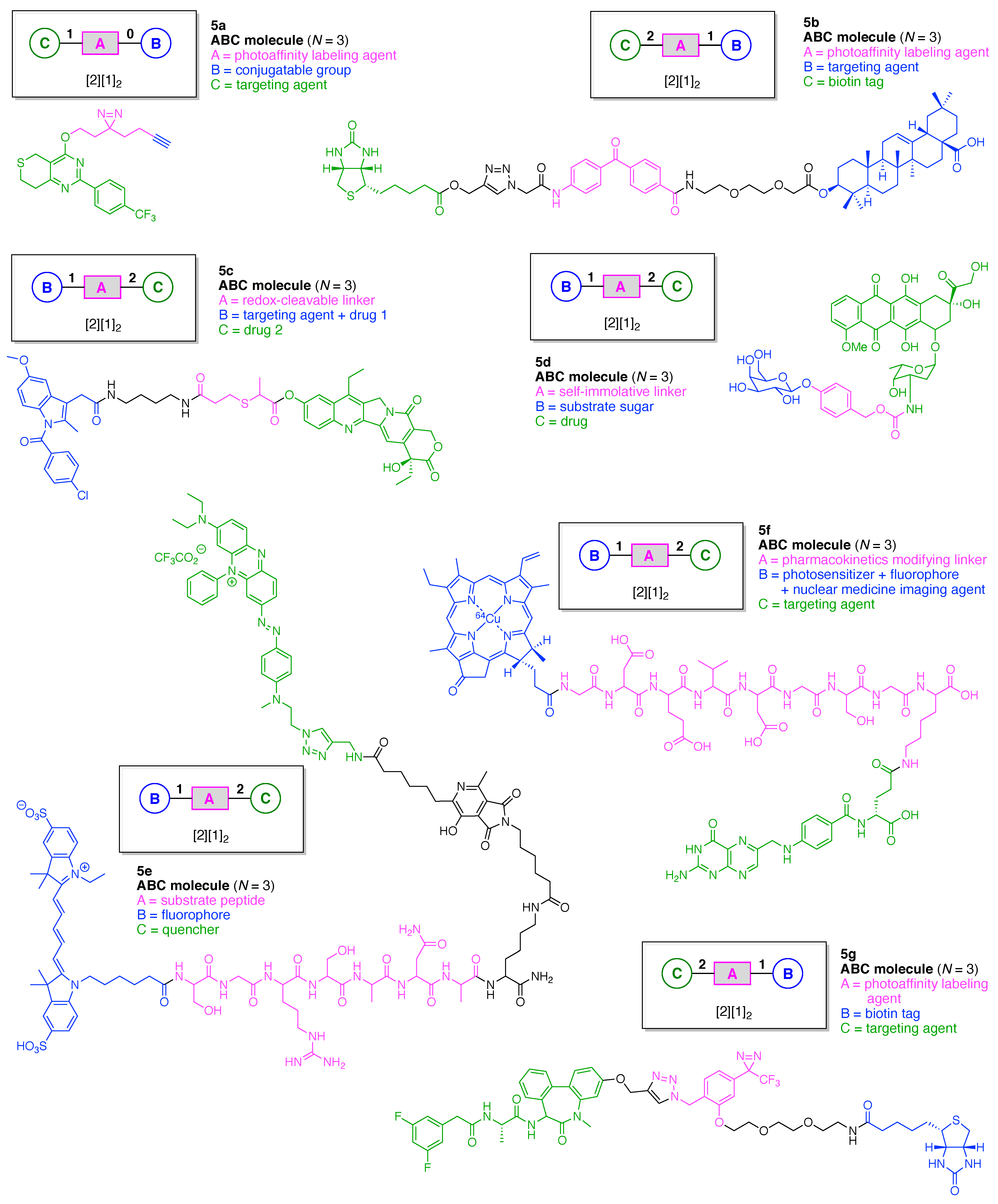
Figure 10.
Six linear multifunctional molecules (no hubs) of graphs [2]2[1]2 and [2]3[1]2.

Figure 11.
Side reactions during peptide synthesis including (A) epimerization and (B) DKP formation.
Figure 11.
Side reactions during peptide synthesis including (A) epimerization and (B) DKP formation.

Figure 12.
β-Elimination of Cys-Wang resin during Fmoc deprotection (with piperidine) on SPPS.

Figure 13.
Five examples each with a single Lys hub.

Figure 14.
Five examples each with a single Lys hub.
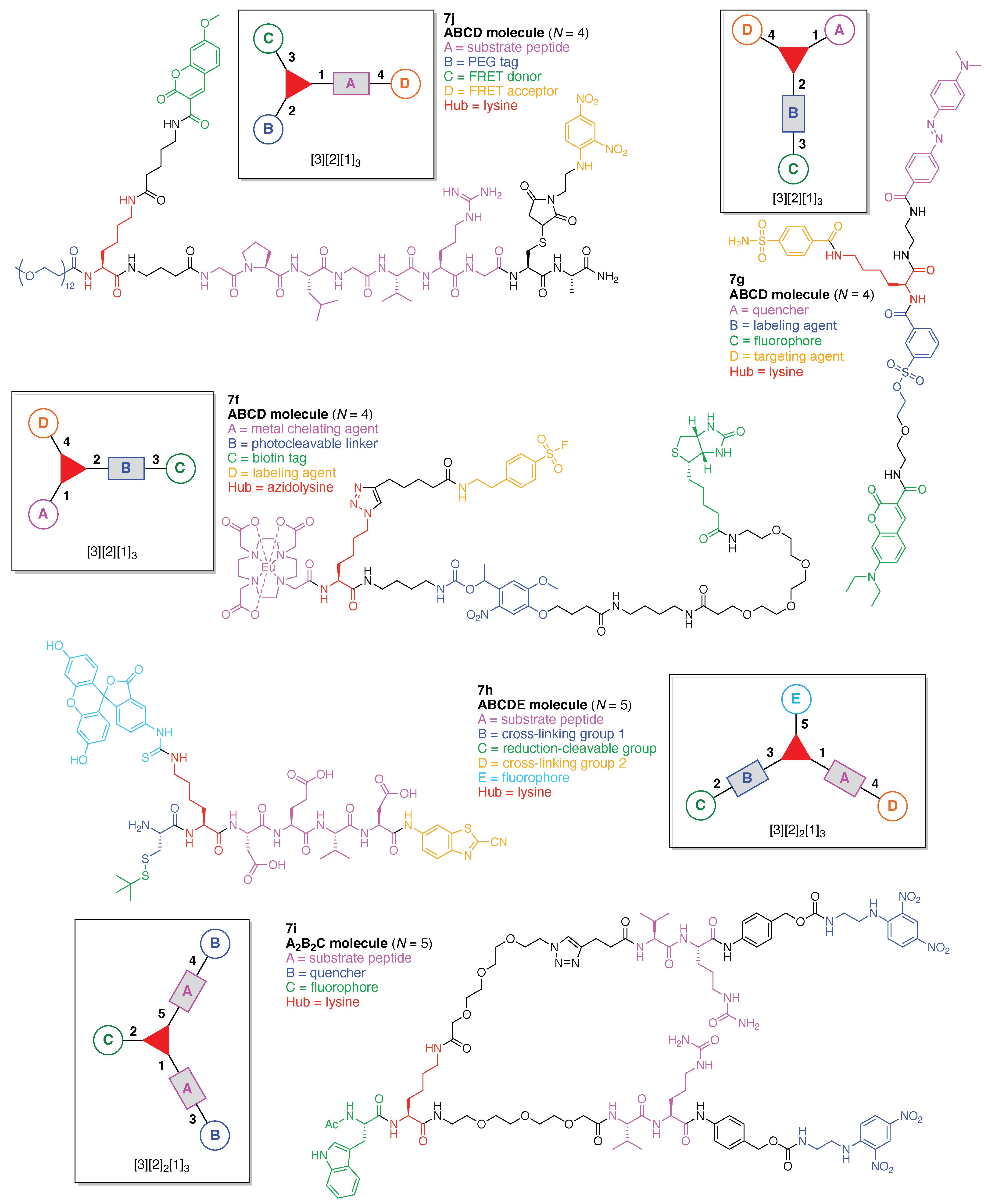
Figure 15.
Synthesis of an ABCD-peptide.
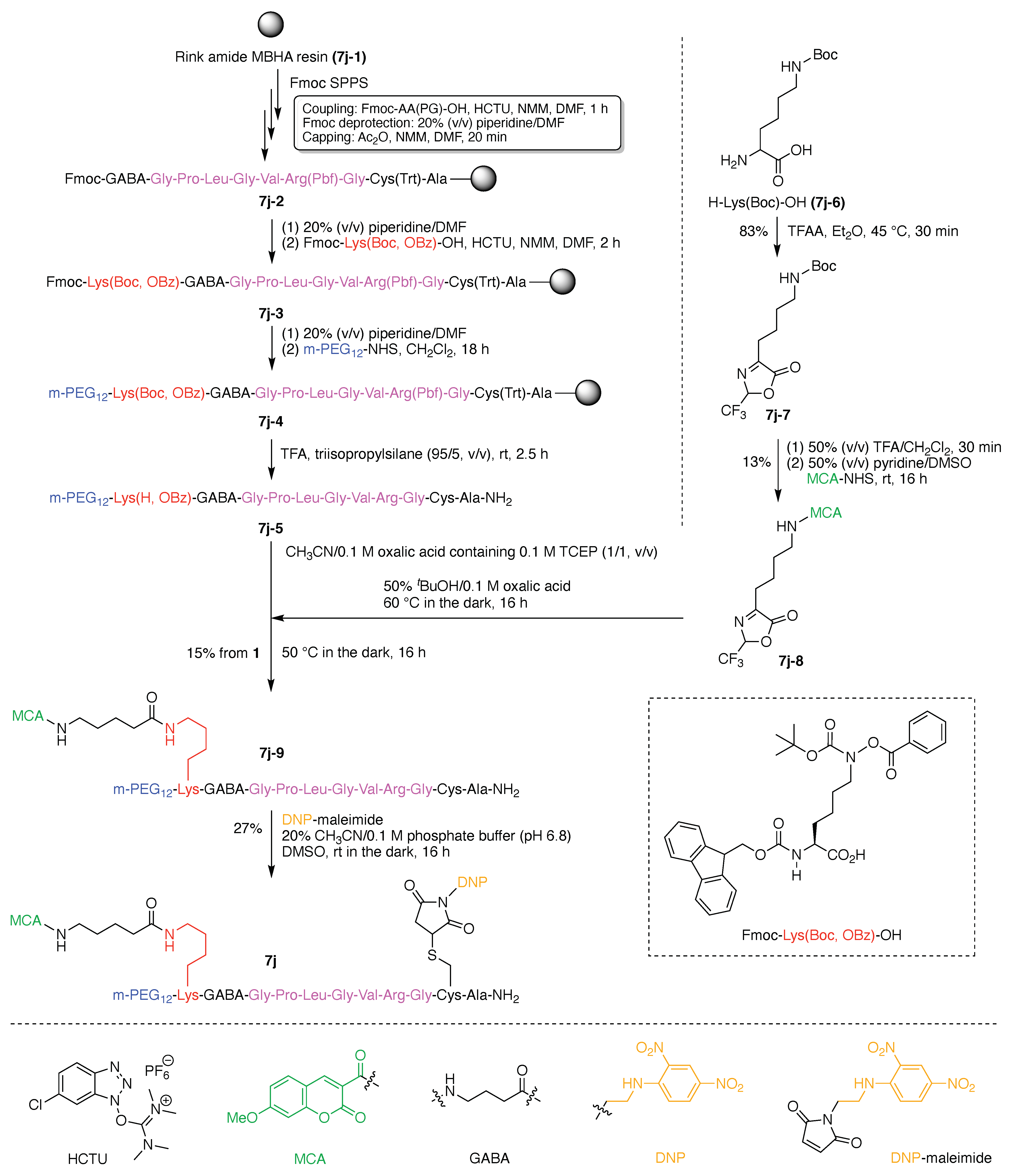
Figure 16.
Four examples each with one Lys hub and one additional amino acid hub.
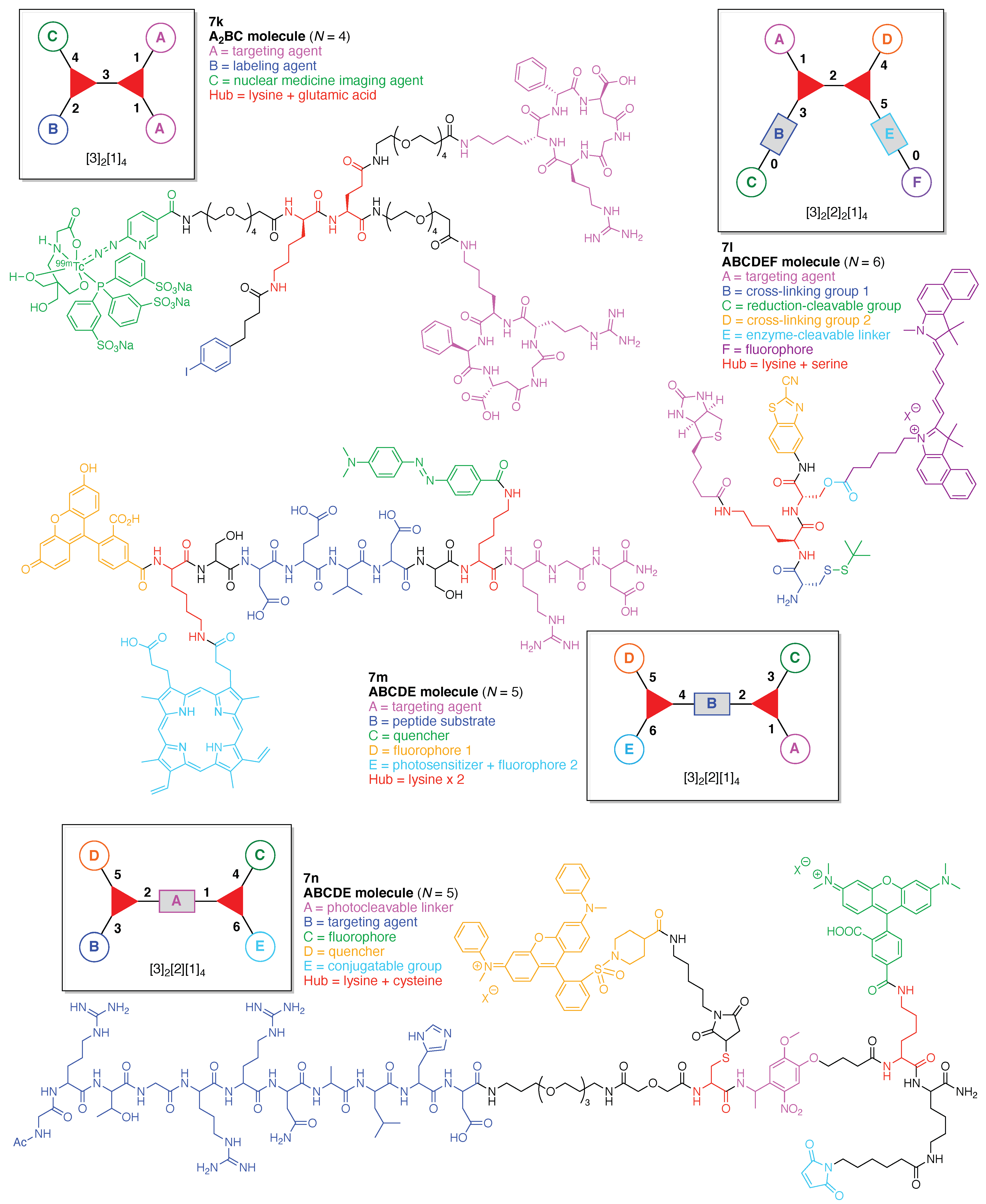
Figure 17.
Four examples each with two lysine or lysine-based hubs.
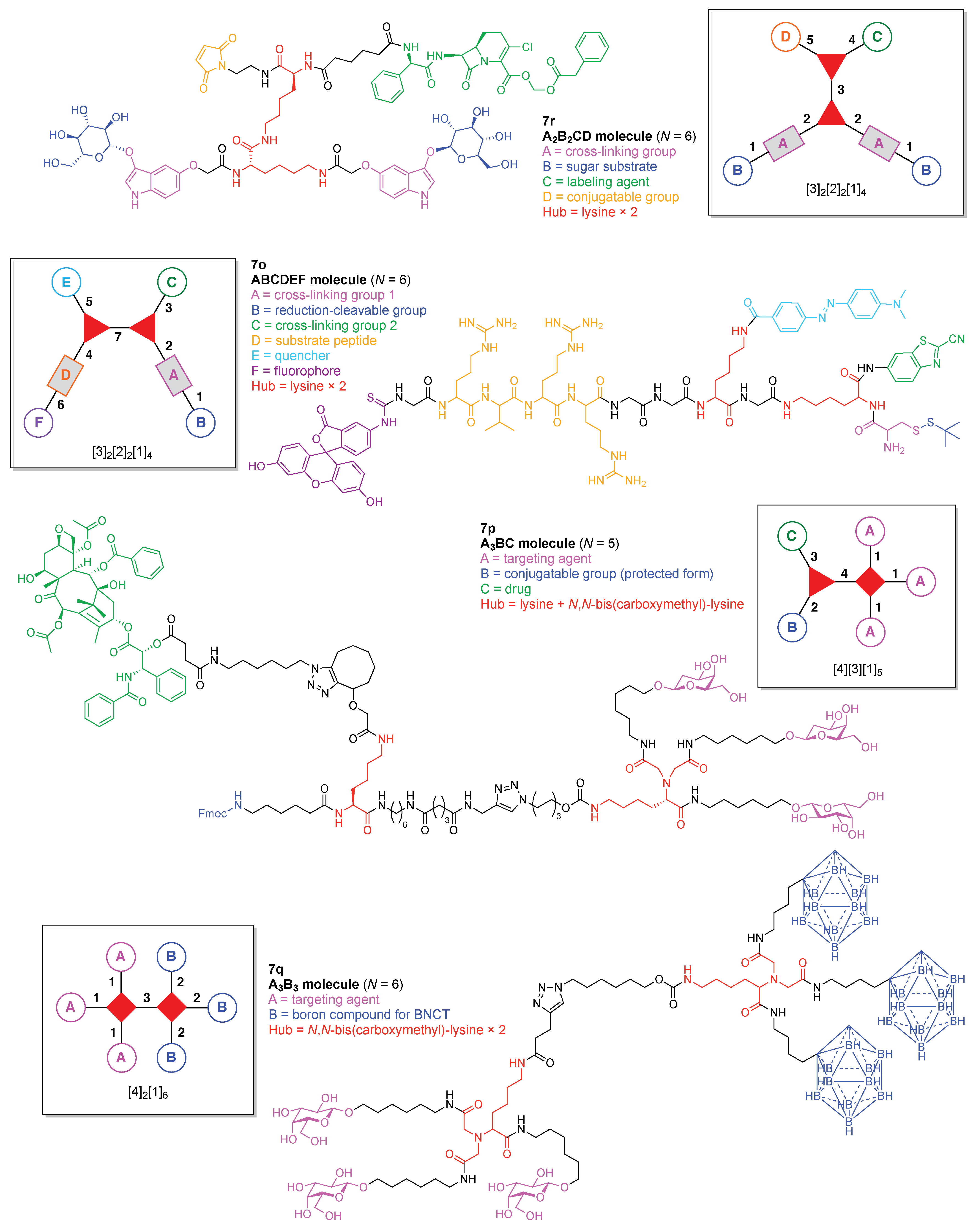
Figure 18.
Synthesis of β-indoxyl-glucoside.
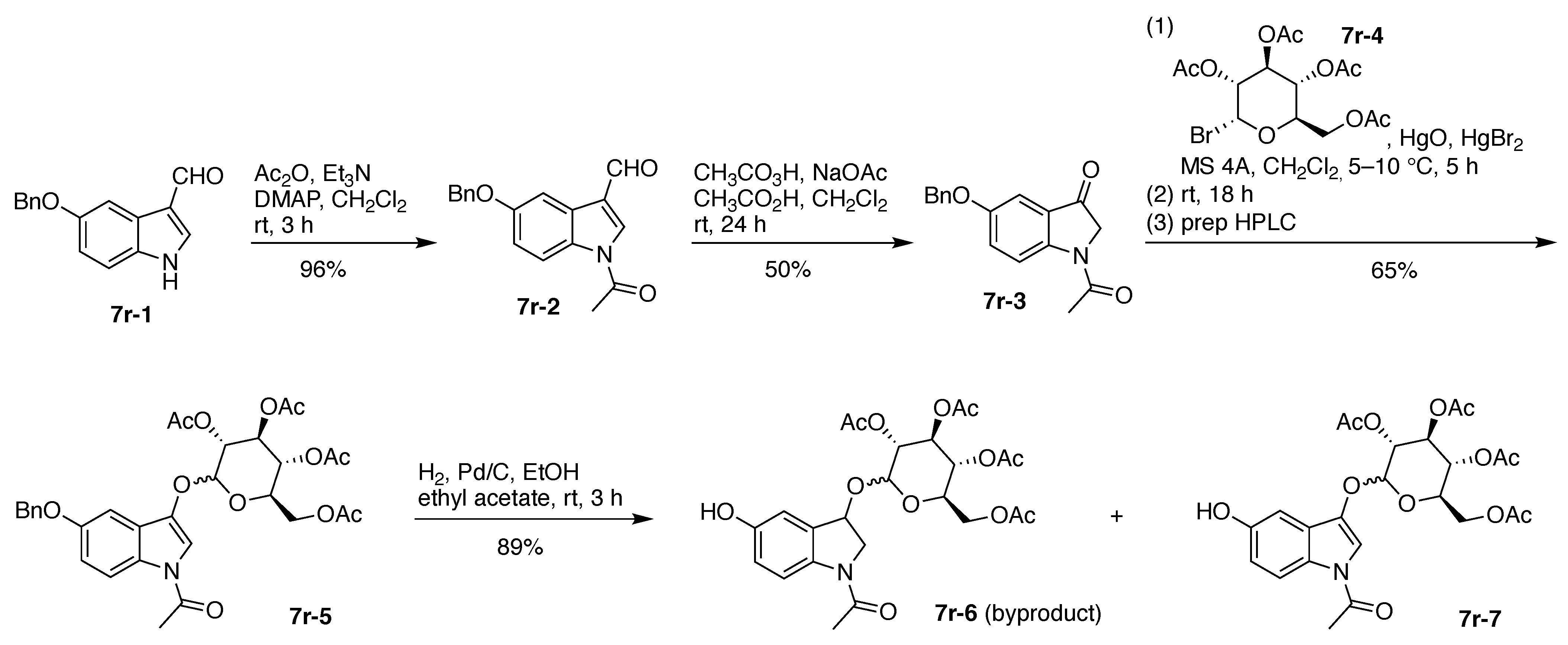
Figure 19.
Synthesis of a Lys hub bearing two protected glucosyl-indoxyl moieties and extension of a second Lys hub.
Figure 19.
Synthesis of a Lys hub bearing two protected glucosyl-indoxyl moieties and extension of a second Lys hub.
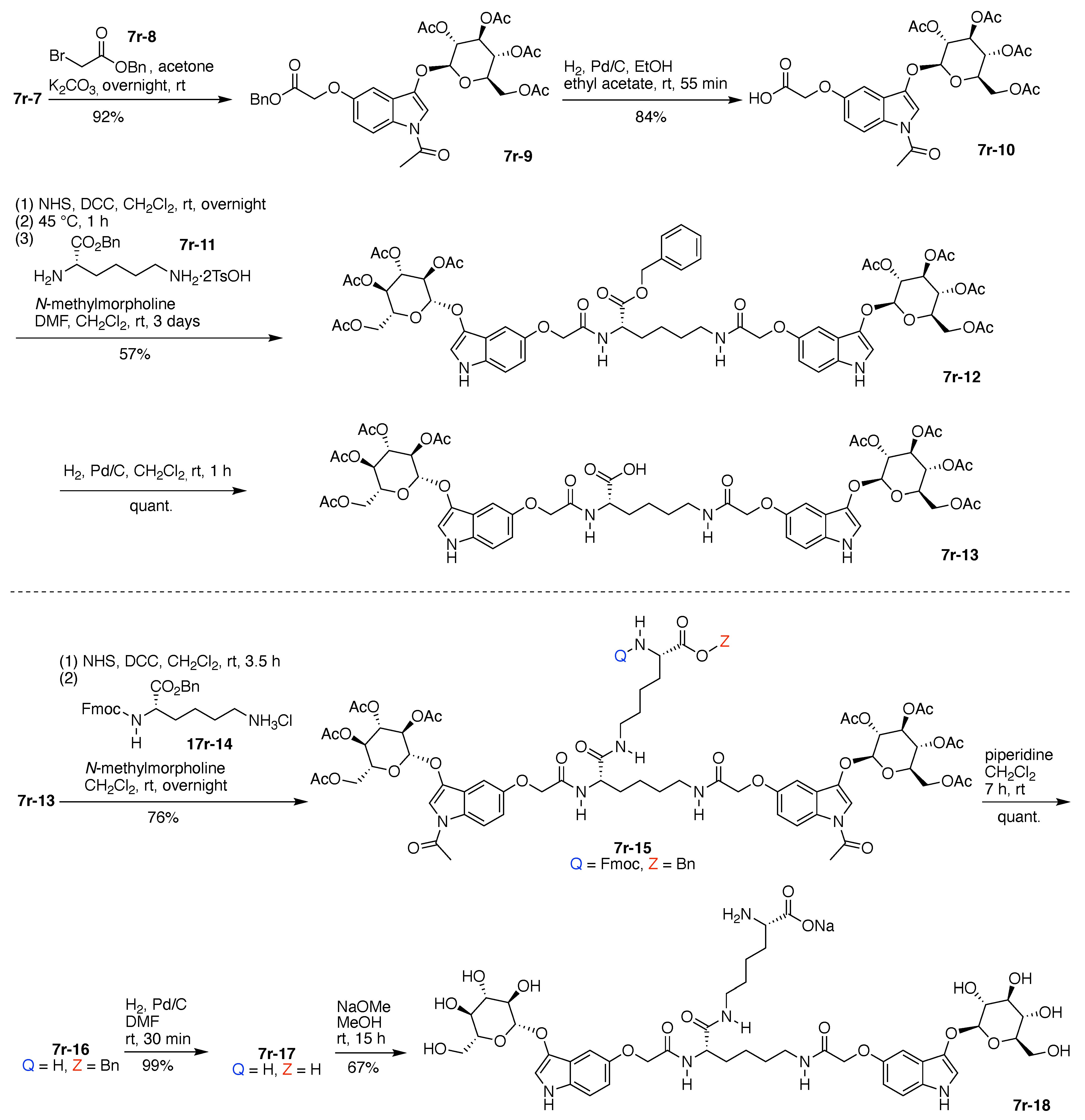
Figure 20.
Completion of the synthesis of 7r, with composition A2B2CD and architecture [3]2[2]2[1]4.
Figure 20.
Completion of the synthesis of 7r, with composition A2B2CD and architecture [3]2[2]2[1]4.

Figure 21.
Examples each with two or more Lys hubs.

Figure 22.
Synthesis of a cyclic decapeptide.
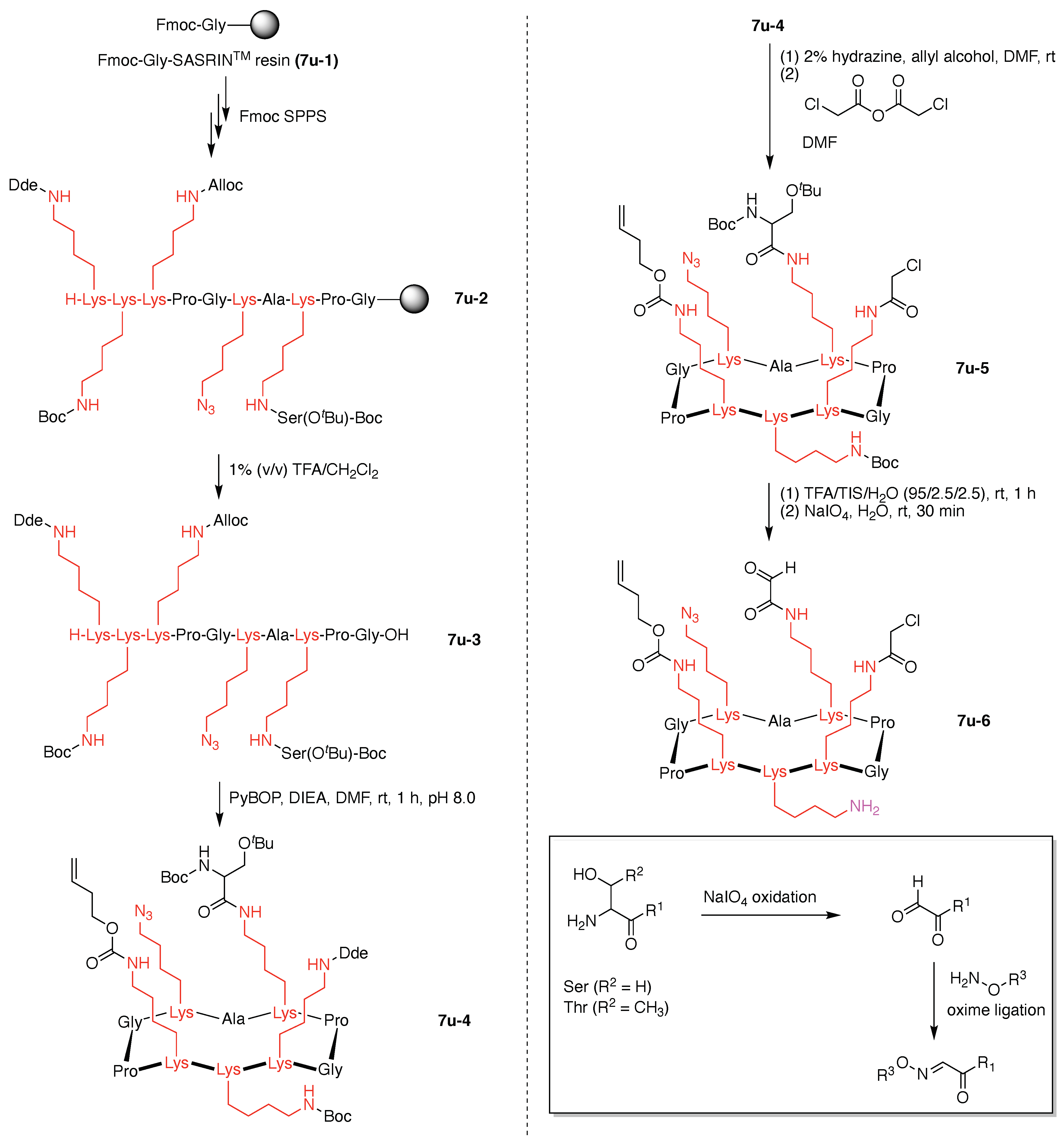
Figure 23.
Rational introduction of B, C, D, E units on a decapeptide scaffold.
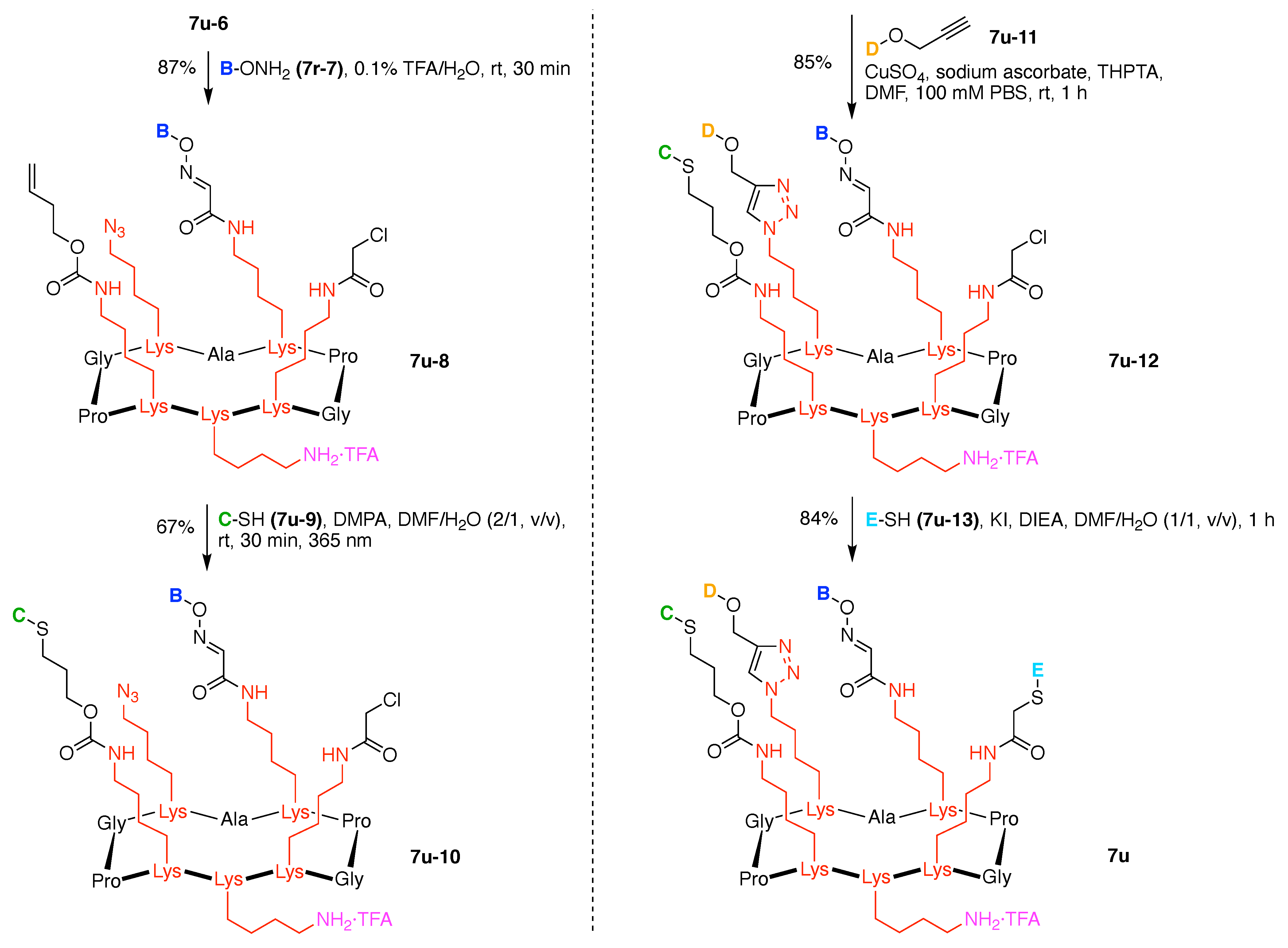
Figure 24.
Decapeptide scaffold with expansive scope.
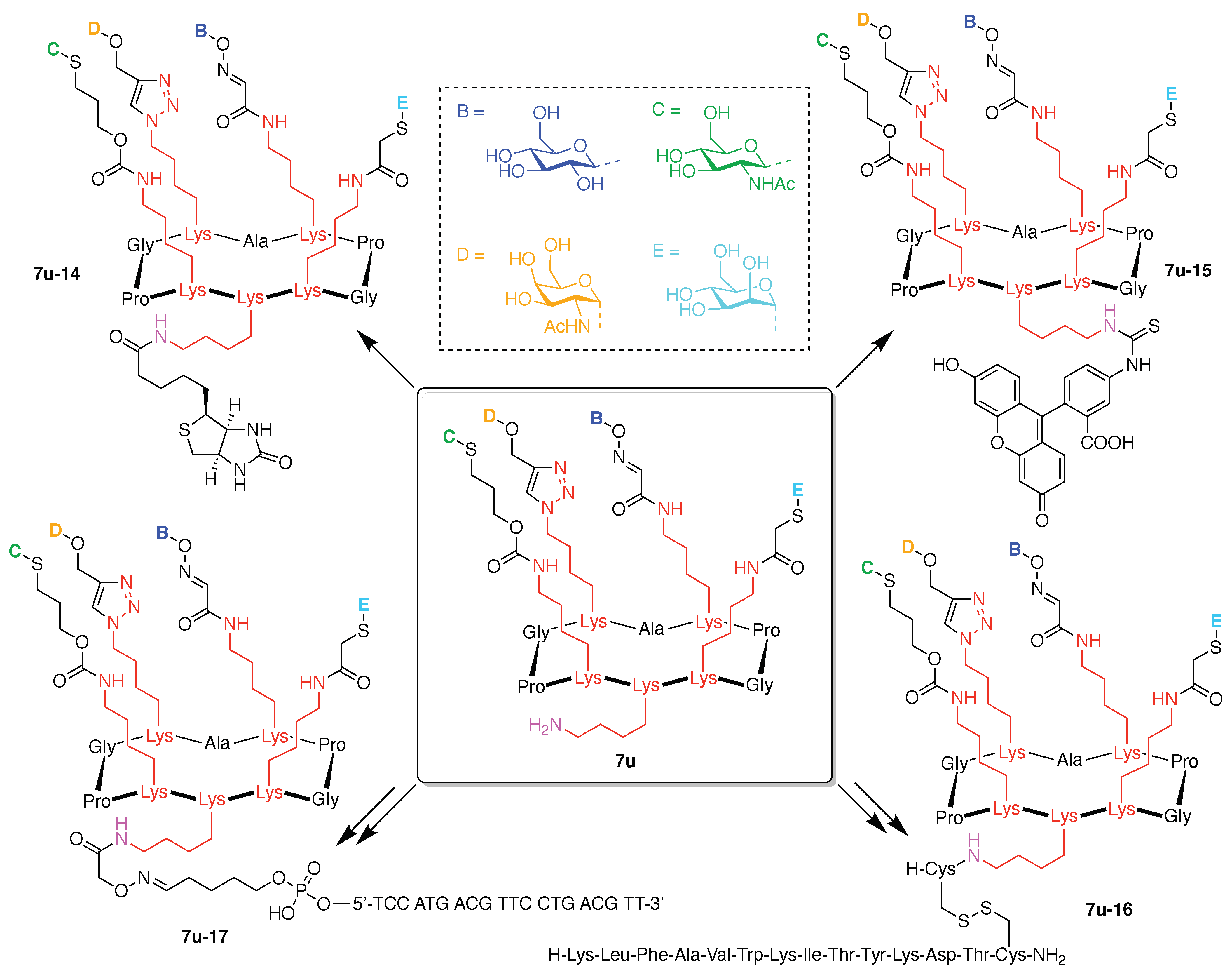
Figure 25.
Five examples each with one (non-lysine) amino acid hub.

Figure 26.
Three examples each with one cysteine hub.
Figure 27.
Labeling of cysteine in a cyclic peptide. Native chemical ligation is shown in the top panel.
Figure 27.
Labeling of cysteine in a cyclic peptide. Native chemical ligation is shown in the top panel.

Figure 28.
Three examples each with one (non-lysine) amino acid hub.
Figure 29.
Two examples each with two amino acid hubs.
Figure 30.
Two examples each with two hubs including one amino acid.

Figure 31.
Two examples each with several amino acid hubs.
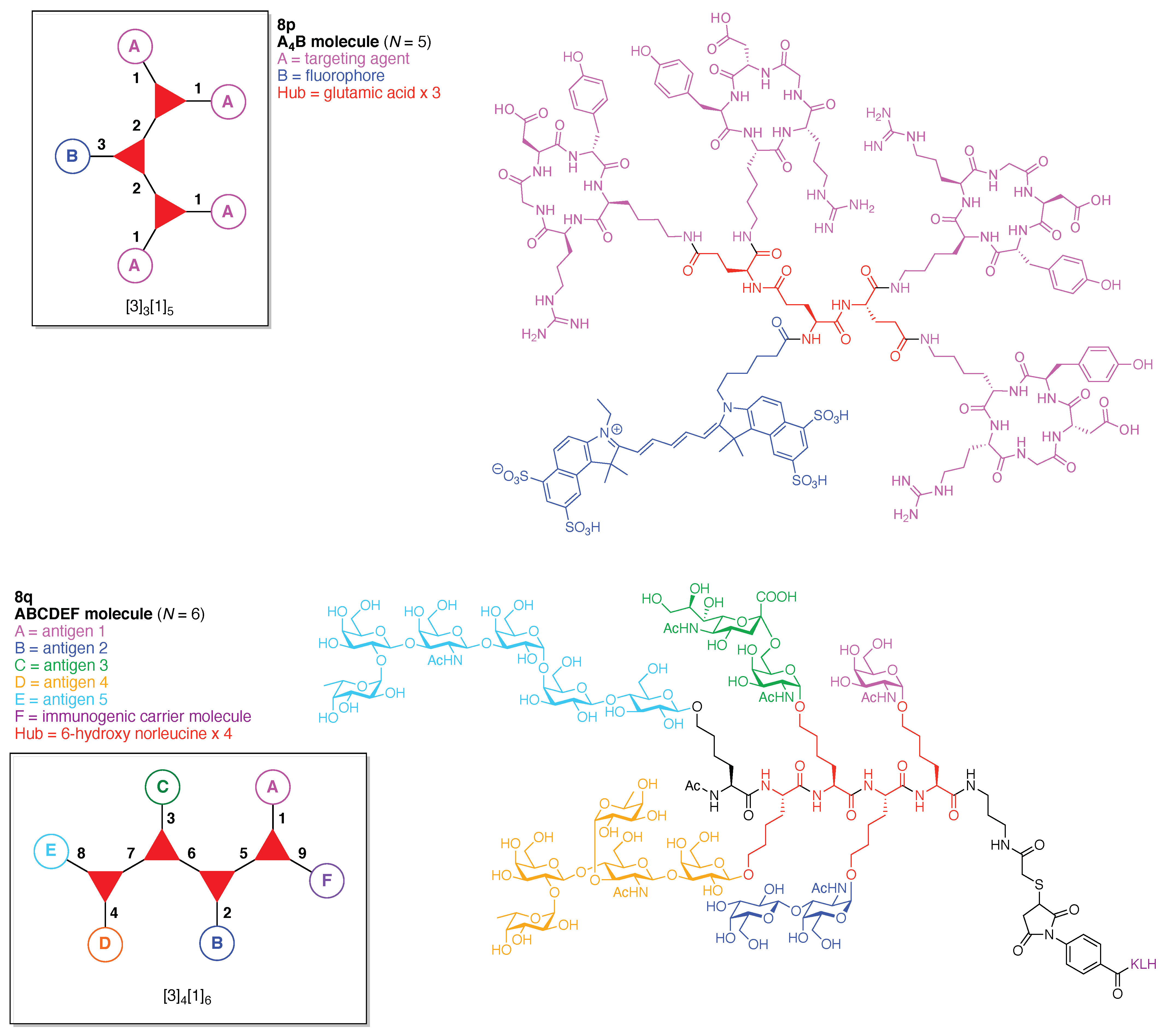
Figure 32.
(A) Structure of cyanuric chloride; (B) analogy of cyanuric chloride to a formal trimer of carbonyl chlorides; and (C) electronic and steric effects of the substituent X.
Figure 32.
(A) Structure of cyanuric chloride; (B) analogy of cyanuric chloride to a formal trimer of carbonyl chlorides; and (C) electronic and steric effects of the substituent X.
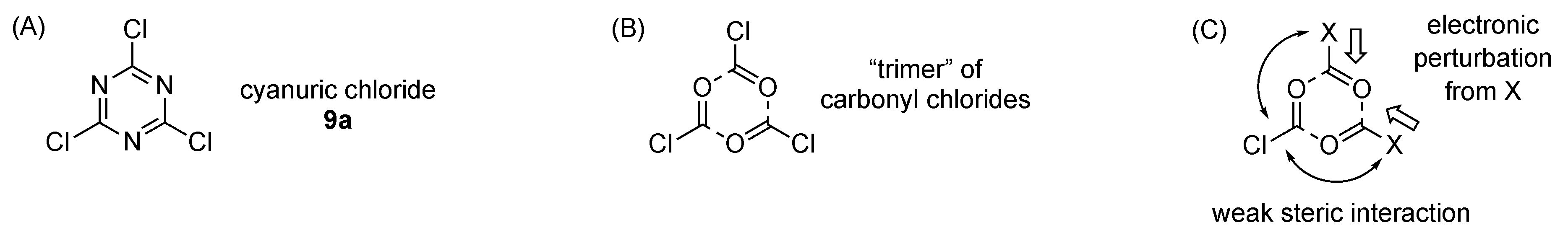
Figure 33.
Relative reactivity of amines in nucleophilic substitution of triazine chlorides (A). Competitive reactions of various nucleophiles for chlorotriazines (B).
Figure 33.
Relative reactivity of amines in nucleophilic substitution of triazine chlorides (A). Competitive reactions of various nucleophiles for chlorotriazines (B).
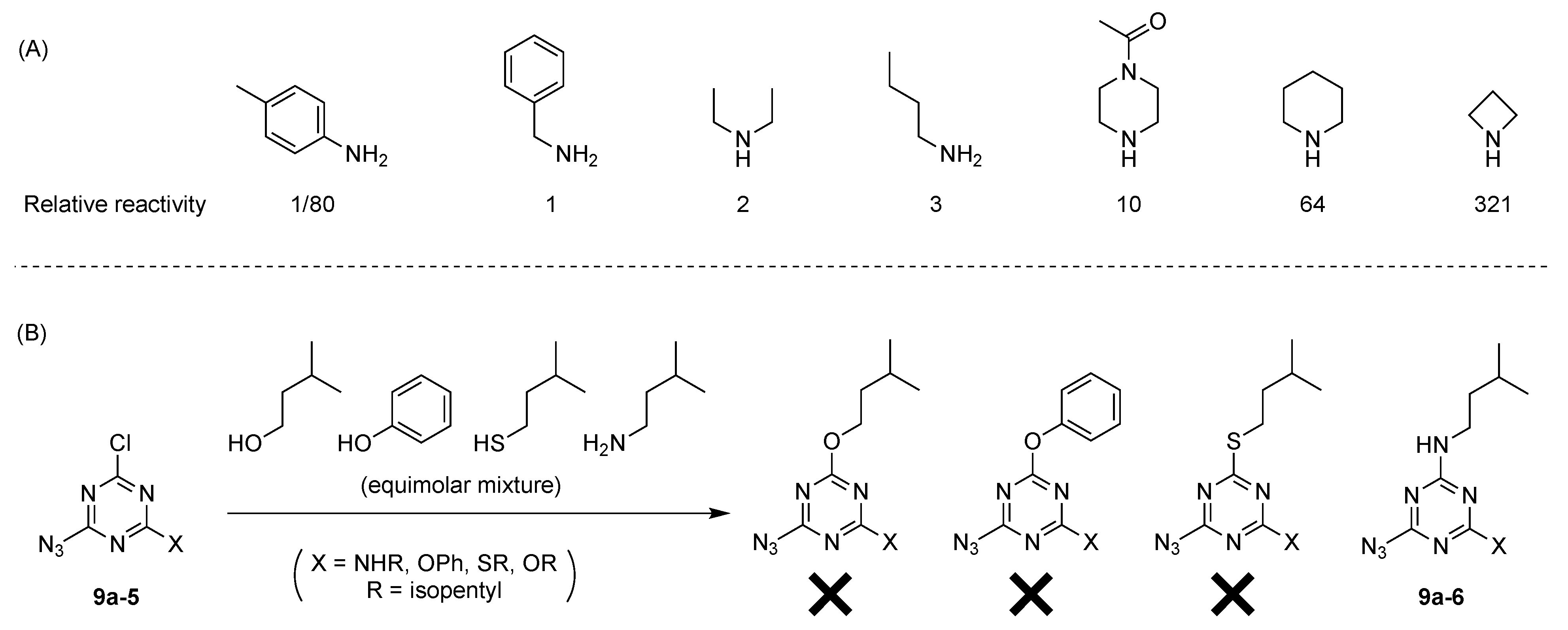
Figure 34.
Conformational interconversion due to hindered C–N bond rotation in substituted aminotriazines. (A) Rotamers Ia and Ib; (B) the two structures shown are identical (II) yet the two X groups have distinct environments, which may be manifest in the NMR spectrum; and (C) Rotamers IIIa and IIIb; (D) four rotamers of IV; (E) tautomers IIa–c of II.
Figure 34.
Conformational interconversion due to hindered C–N bond rotation in substituted aminotriazines. (A) Rotamers Ia and Ib; (B) the two structures shown are identical (II) yet the two X groups have distinct environments, which may be manifest in the NMR spectrum; and (C) Rotamers IIIa and IIIb; (D) four rotamers of IV; (E) tautomers IIa–c of II.

Figure 35.
(A) Synthesis of triazine hubs bearing amino or alkoxy groups: (top) triaminotriazine, (middle) alkoxydiaminotriazine, and (bottom) dialkoxyaminotriazine; (B) amine bases employed in chlorotriazine substitution with amines and alcohols. The pKa values (in H2O) are shown in parentheses; and (C) dialkylamino substituents avoiding formation of rotamers.
Figure 35.
(A) Synthesis of triazine hubs bearing amino or alkoxy groups: (top) triaminotriazine, (middle) alkoxydiaminotriazine, and (bottom) dialkoxyaminotriazine; (B) amine bases employed in chlorotriazine substitution with amines and alcohols. The pKa values (in H2O) are shown in parentheses; and (C) dialkylamino substituents avoiding formation of rotamers.
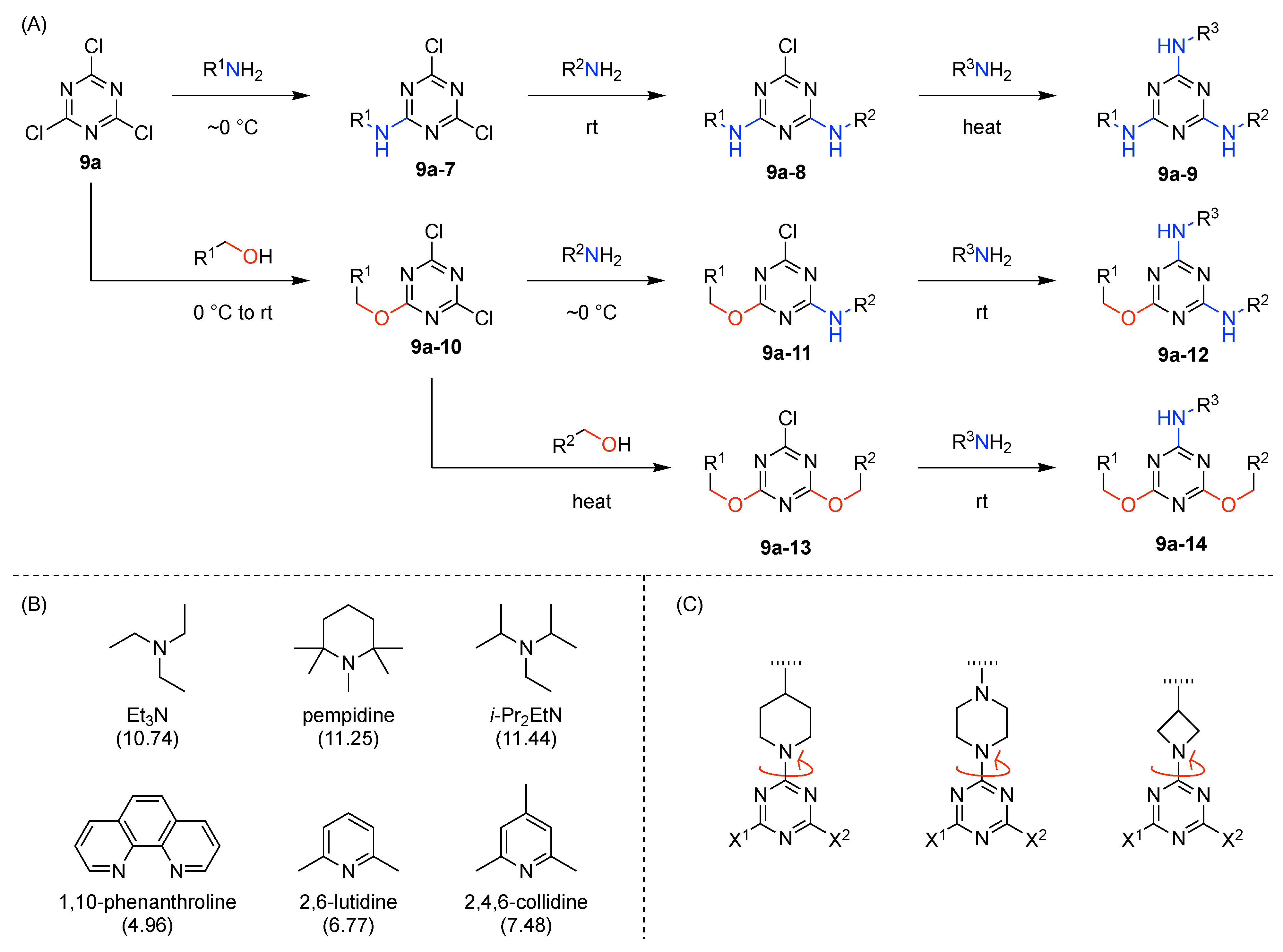
Figure 36.
Examples wherein each contains a single triazine hub with A2B substituents.
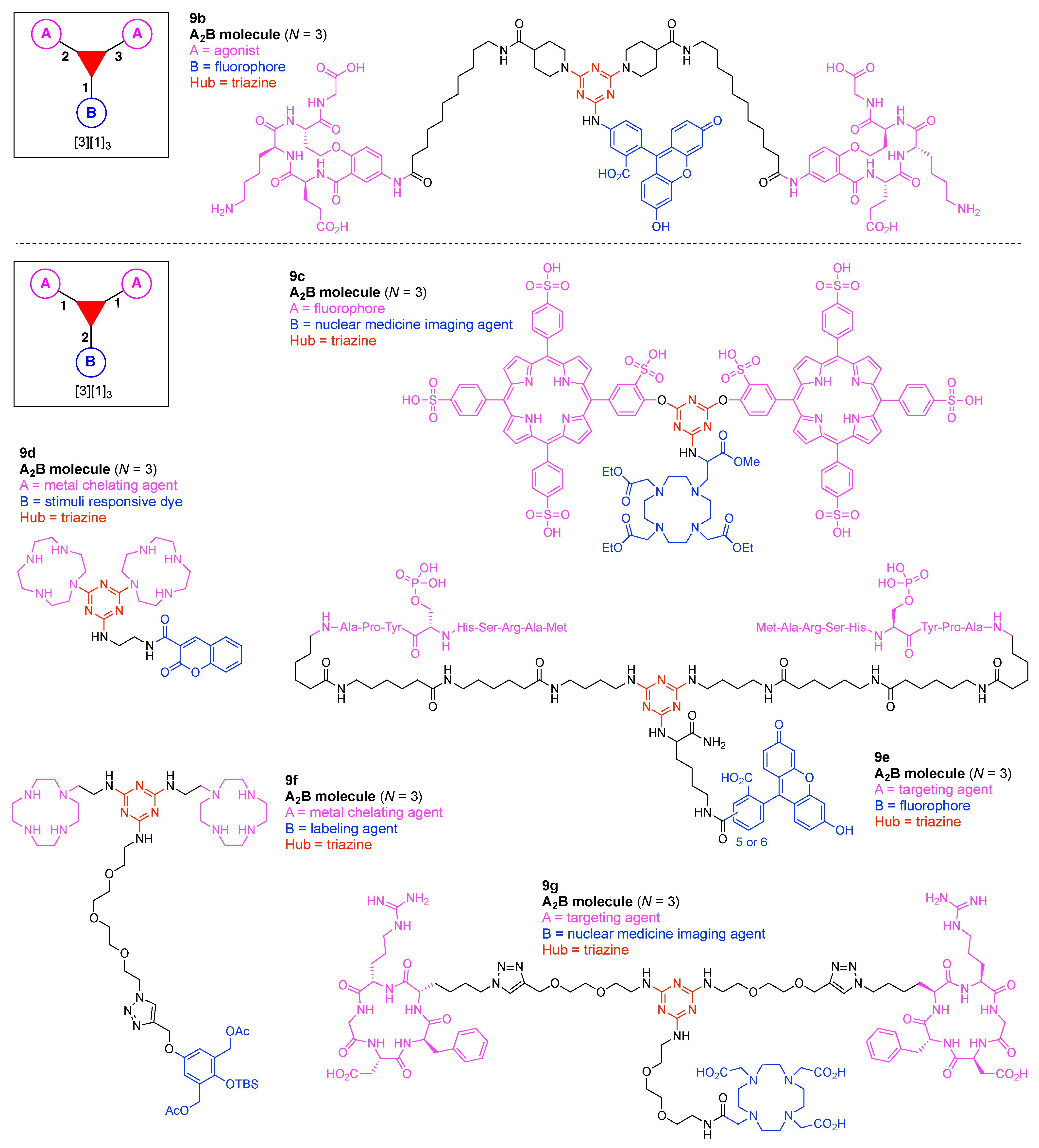
Figure 37.
Examples wherein each contains a single triazine hub with ABC substituents.

Figure 38.
Three examples each containing a single triazine hub.

Figure 39.
Examples wherein each contains at least one triazine hub.
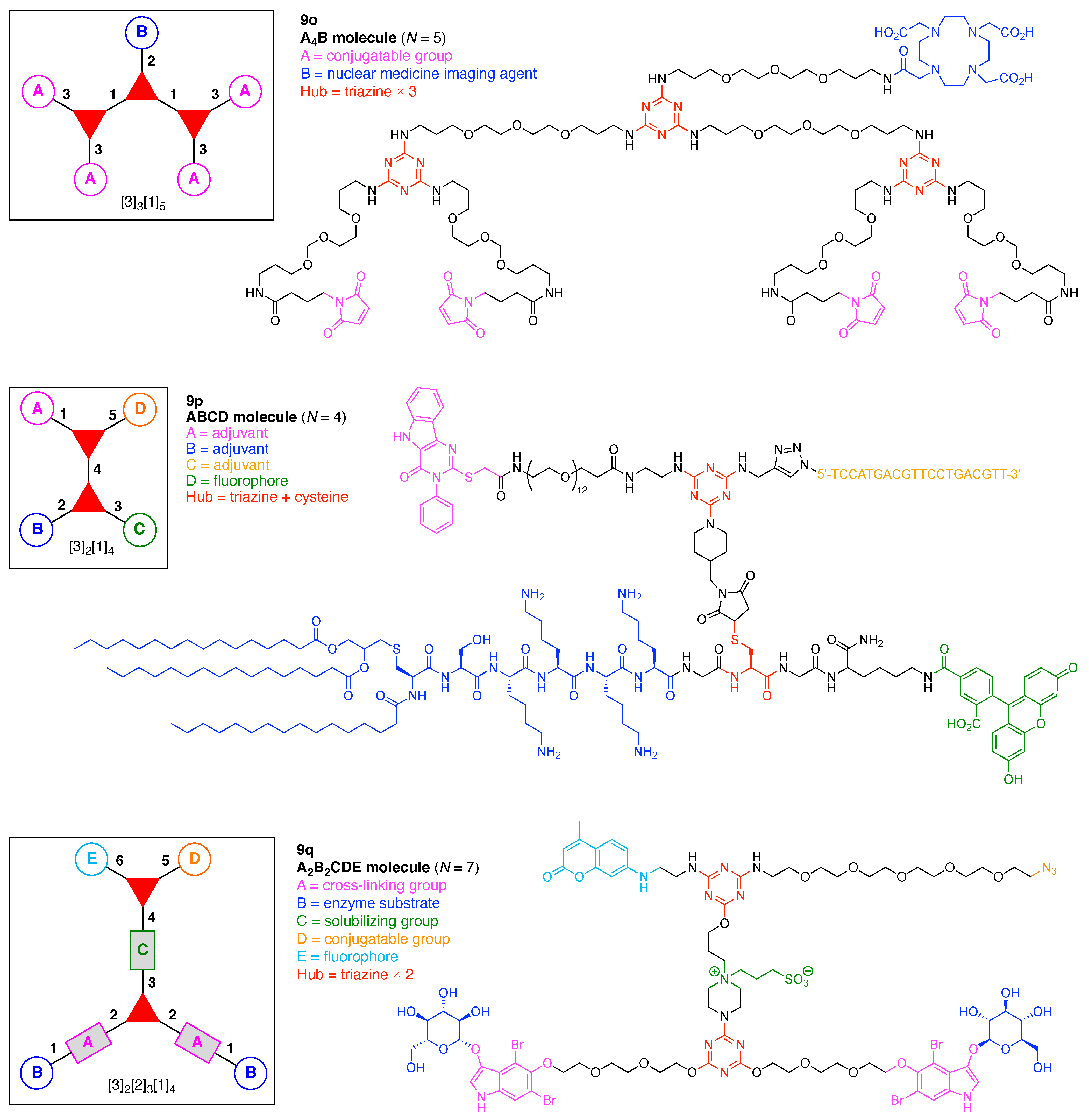
Figure 40.
Synthesis of a TLR triagonist connected to a fluorophore via a triazine hub.
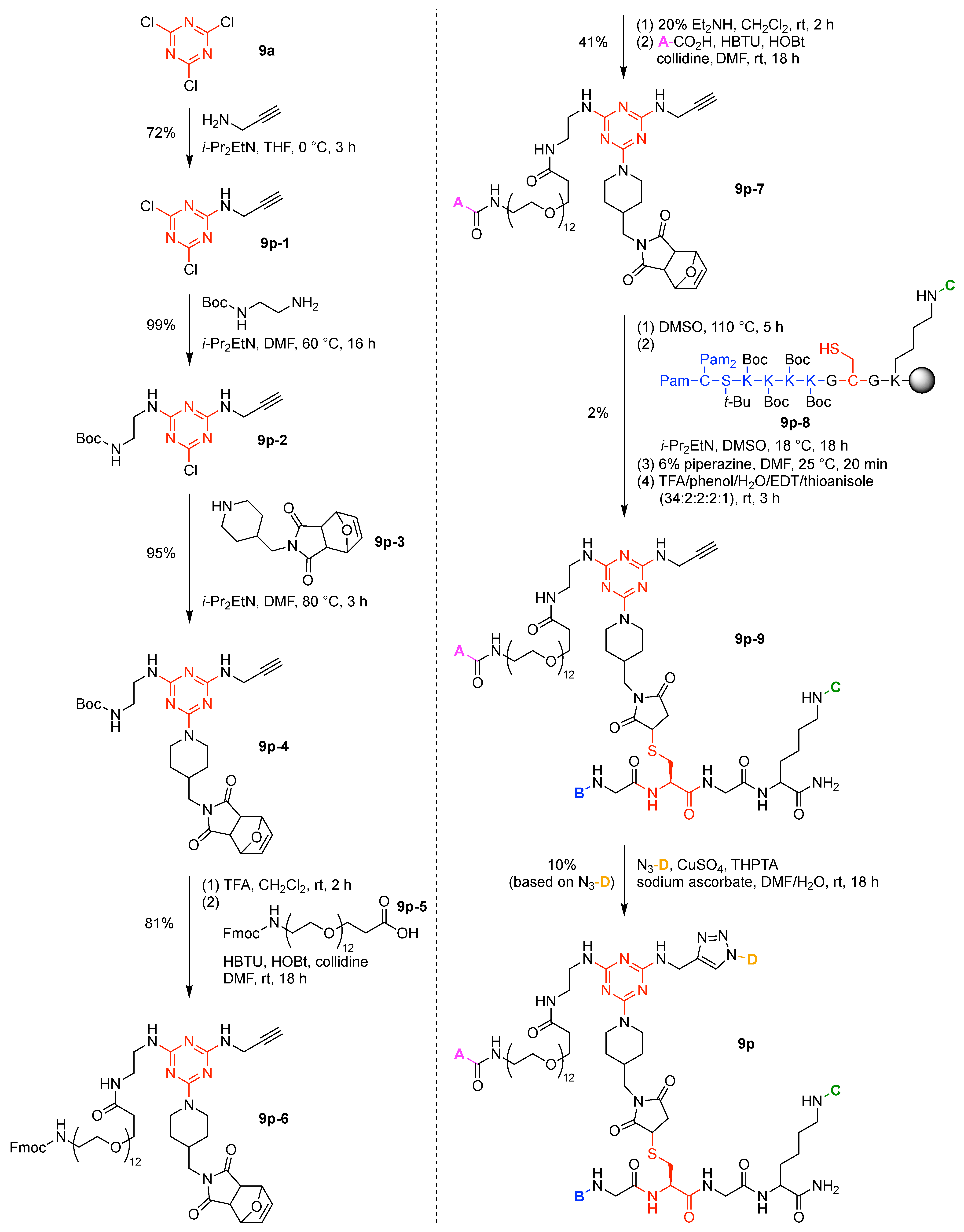
Figure 41.
Synthesis of a bioconjugatable compound, built around triazine hubs, that enables enzymatically triggered cross-linking chemistry.
Figure 41.
Synthesis of a bioconjugatable compound, built around triazine hubs, that enables enzymatically triggered cross-linking chemistry.
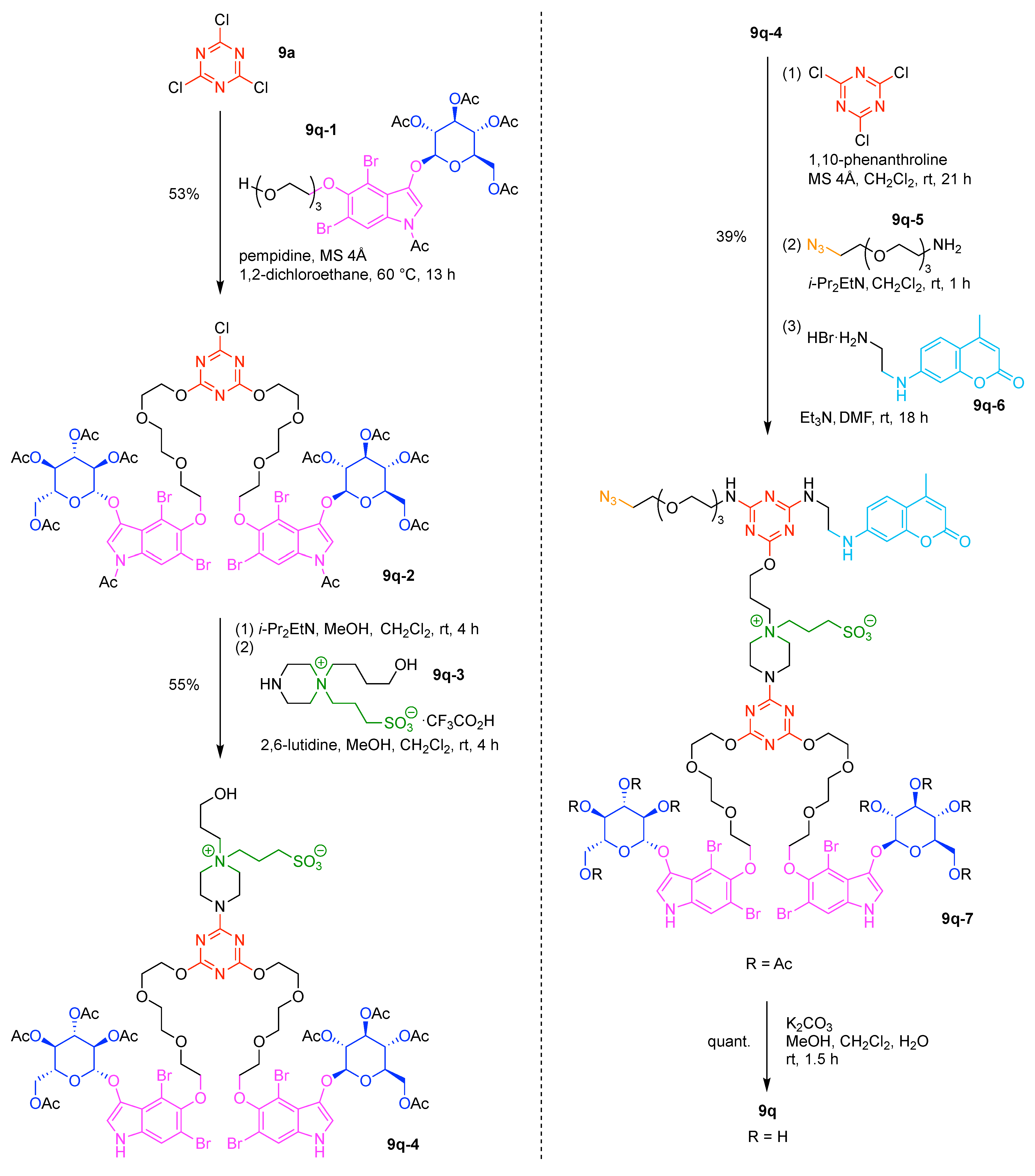
Figure 42.
Examples wherein each contains a single benzene hub.

Figure 43.
A benzotrifuranone scaffold affords sequential derivatization (top) and is formed via straightforward synthesis (bottom).
Figure 43.
A benzotrifuranone scaffold affords sequential derivatization (top) and is formed via straightforward synthesis (bottom).
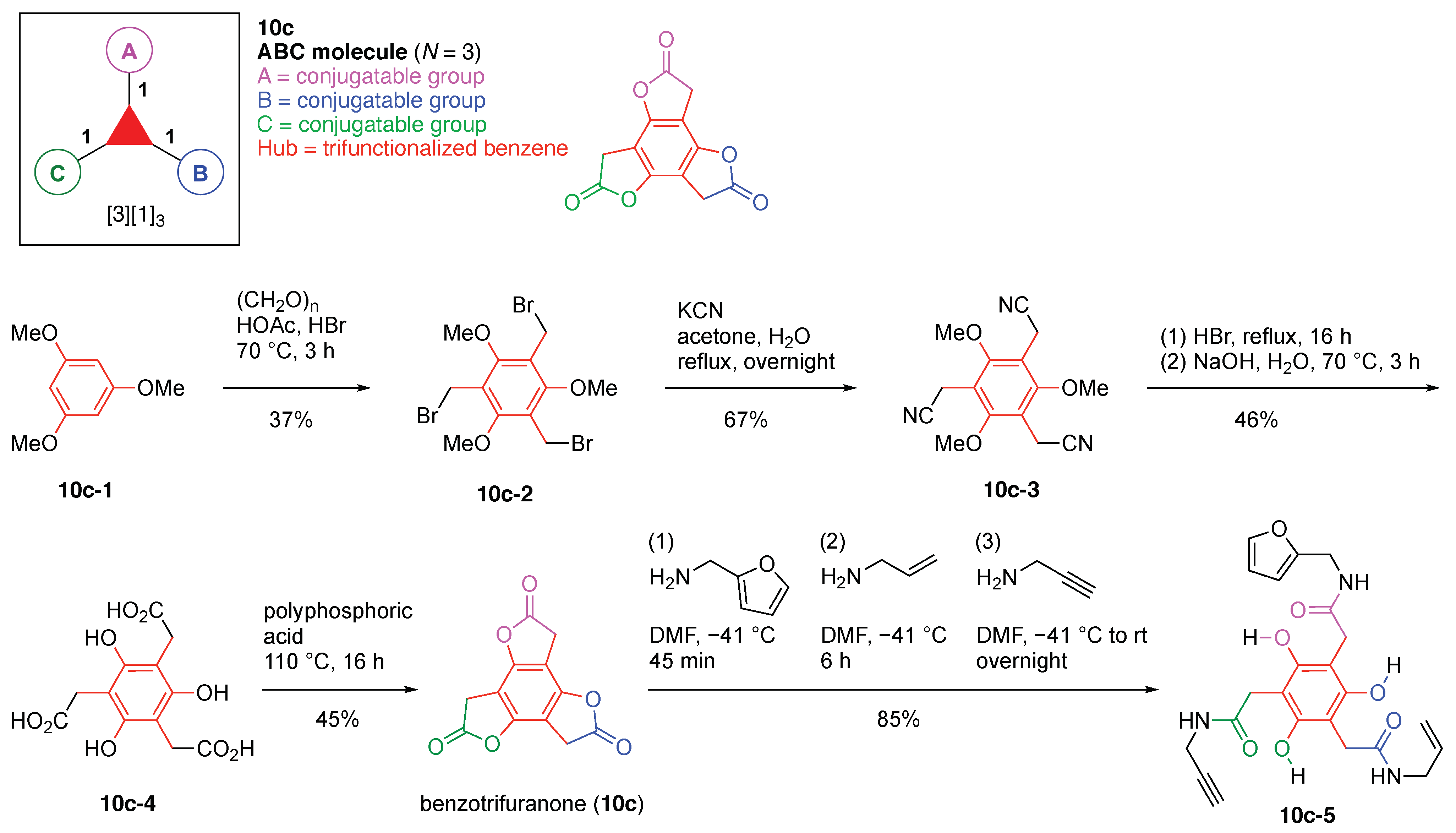
Figure 44.
Examples of single benzene hubs.

Figure 45.
Synthesis of a triazido hub.
Figure 46.
Synthesis of three appendages for the triazido hub.

Figure 47.
Sequential click chemistry with the triazido hub.

Figure 48.
A tetrafunctionalized scaffold built around two benzenes and elaborated by consecutive formation of 1,2,3-triazoles.
Figure 48.
A tetrafunctionalized scaffold built around two benzenes and elaborated by consecutive formation of 1,2,3-triazoles.
Figure 49.
Examples wherein each contains one or two benzene hubs.
Figure 50.
Synthesis of a triorthogonal scaffold comprised of a single benzene hub.
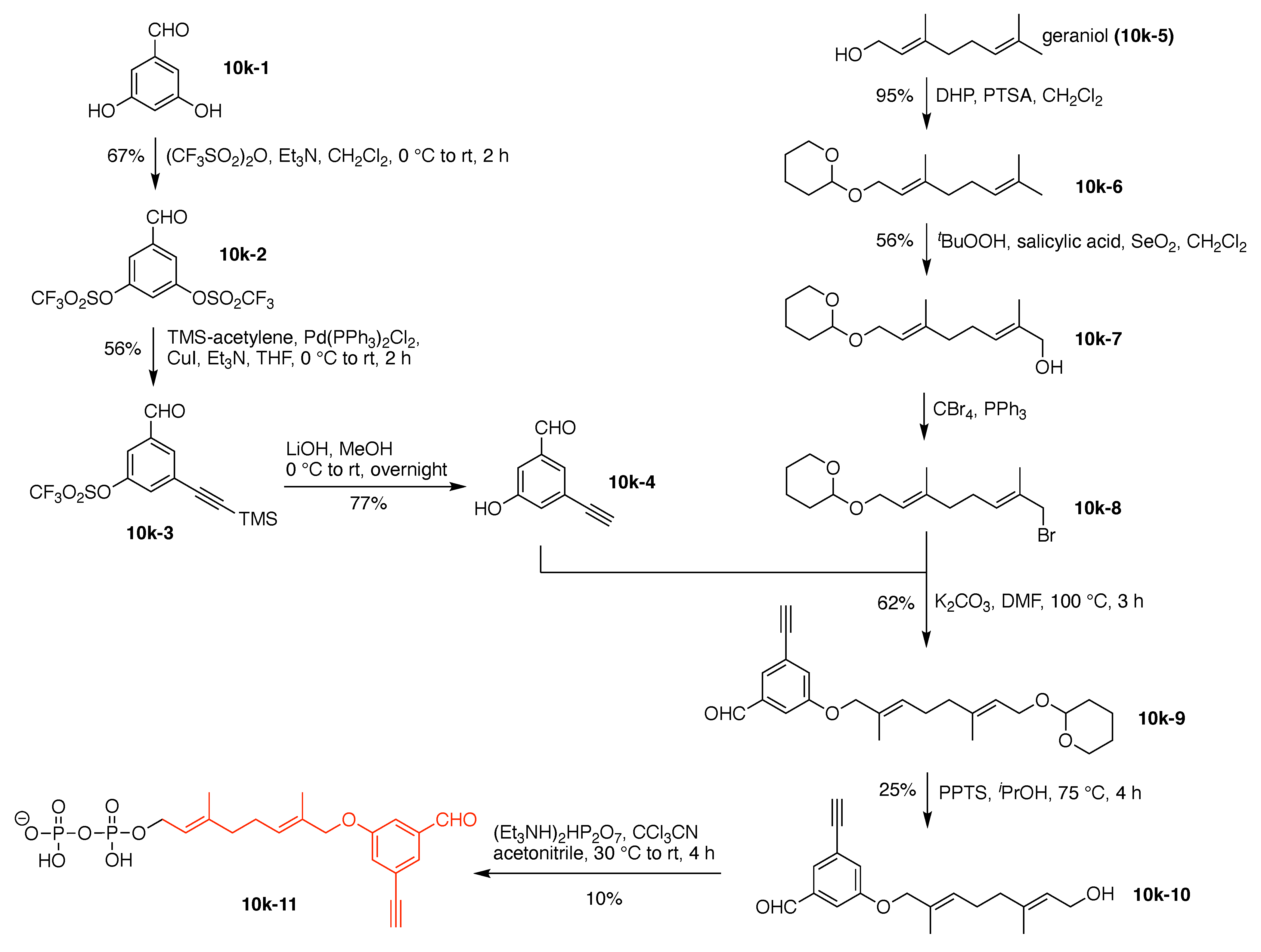
Figure 51.
Synthesis of a bis(MTX) construct bearing a single azide.

Figure 52.
Rational assembly of GFP, TAMRA and azido-bis(MTX) on a benzene scaffold.

Figure 53.
Examples wherein each contains a single nitrogen hub.

Figure 54.
Synthesis of a triple-click conjugation scaffold built around a single nitrogen hub.

Figure 55.
Two examples each containing a single nitrogen hub.
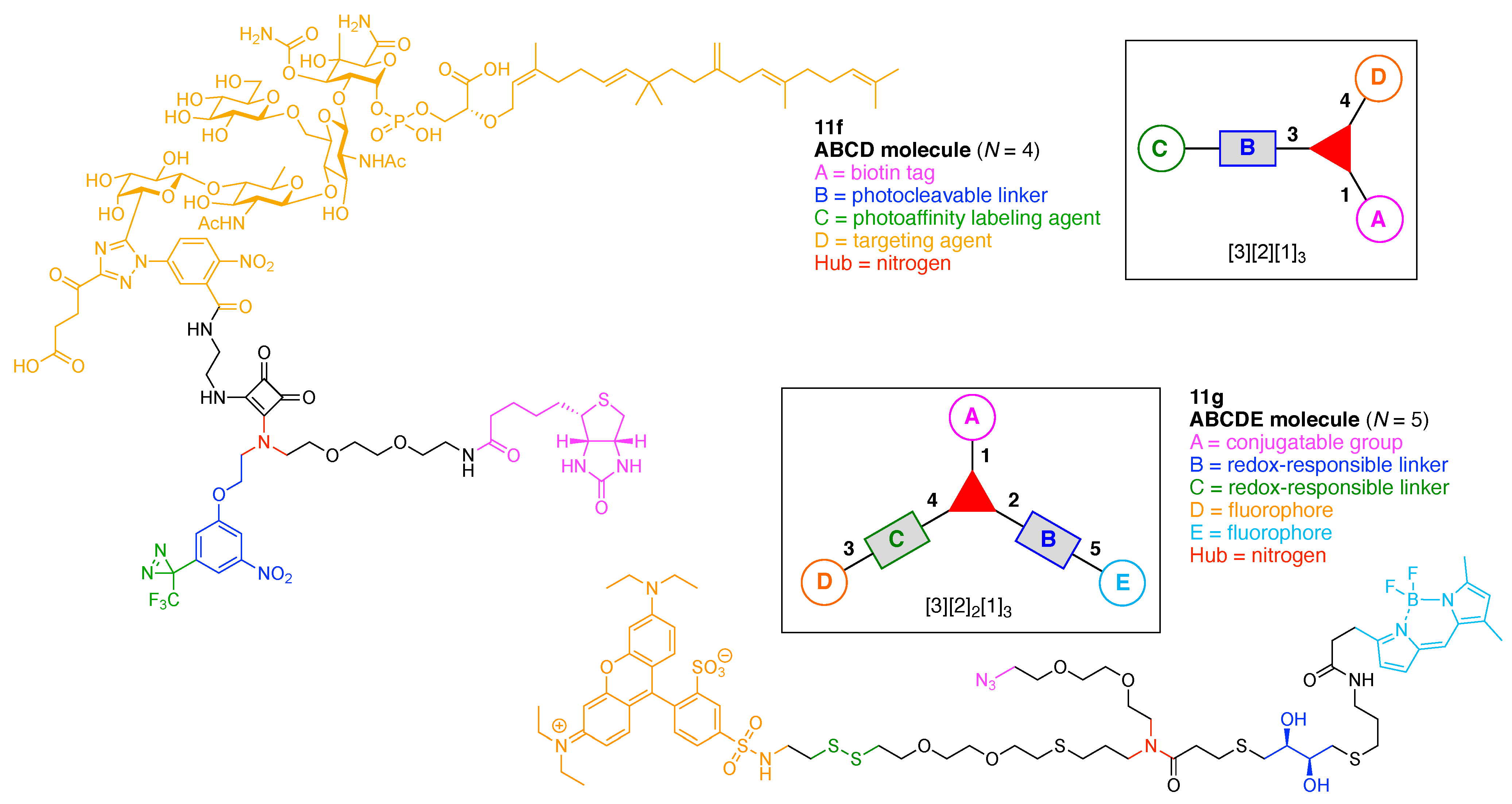
Figure 56.
Two examples each containing three nitrogen hubs.
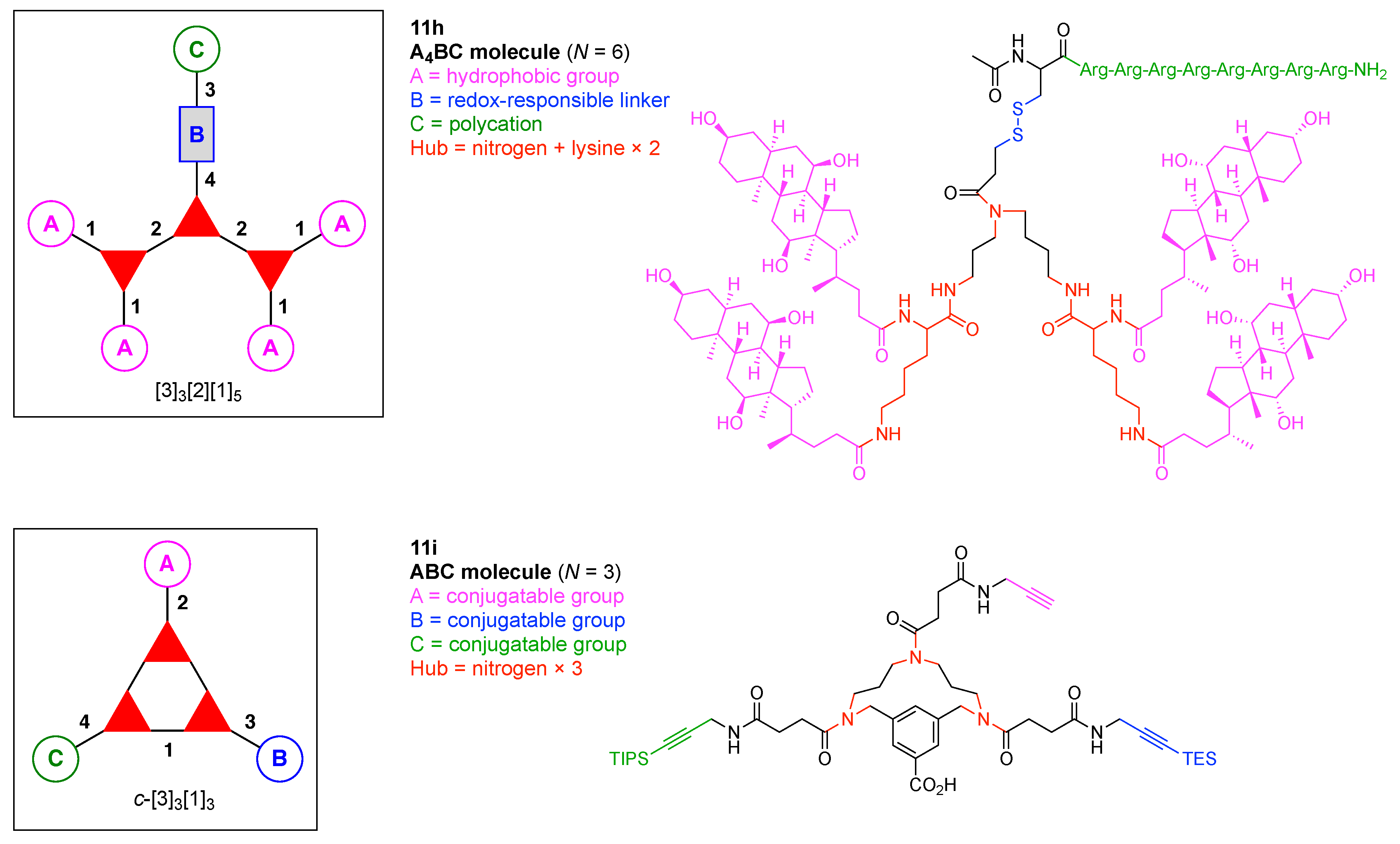
Figure 57.
Examples wherein each contains a single nitrogen hub.
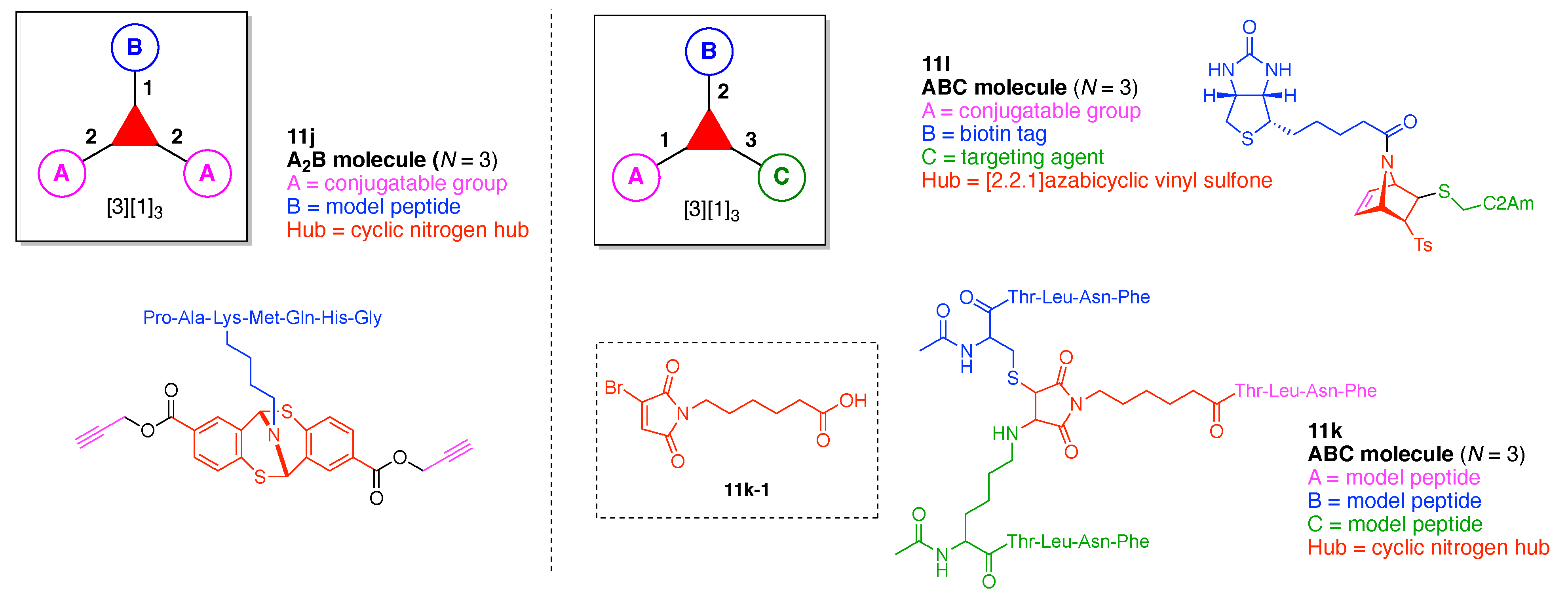
Figure 58.
Orthogonal bioconjugation reactions (A–C). Synthesis of the azanorbornadiene reagent for protein modification (D).
Figure 58.
Orthogonal bioconjugation reactions (A–C). Synthesis of the azanorbornadiene reagent for protein modification (D).

Figure 59.
Examples wherein each contains a single carbon hub.

Figure 60.
Synthesis of an alkynyl-maleimide hub.

Figure 61.
An oxazolone for multiple couplings (lower left). The ring-opening reaction of the oxazolone with a nucleophile (insert in box). Synthesis of a heteromultifunctional oxazolone scaffold (top). Reactivity of the scaffold towards aminolysis, Cu-catalyzed click reaction, and thiol-ene click reaction (bottom).
Figure 61.
An oxazolone for multiple couplings (lower left). The ring-opening reaction of the oxazolone with a nucleophile (insert in box). Synthesis of a heteromultifunctional oxazolone scaffold (top). Reactivity of the scaffold towards aminolysis, Cu-catalyzed click reaction, and thiol-ene click reaction (bottom).

Figure 62.
Two examples of enzymatically recognized hubs.

Figure 63.
Synthesis of an affinity labeling probe for carbohydrate-binding proteins.

Figure 64.
Four examples each containing a self-immolative hub.

Figure 65.
Synthesis of a tetra-substituted scaffold.

Figure 66.
Enzymatic activation of the qABP in this study via quinolimine methide chemistry, and possible subsequent reactions in an intracellular environment.
Figure 66.
Enzymatic activation of the qABP in this study via quinolimine methide chemistry, and possible subsequent reactions in an intracellular environment.
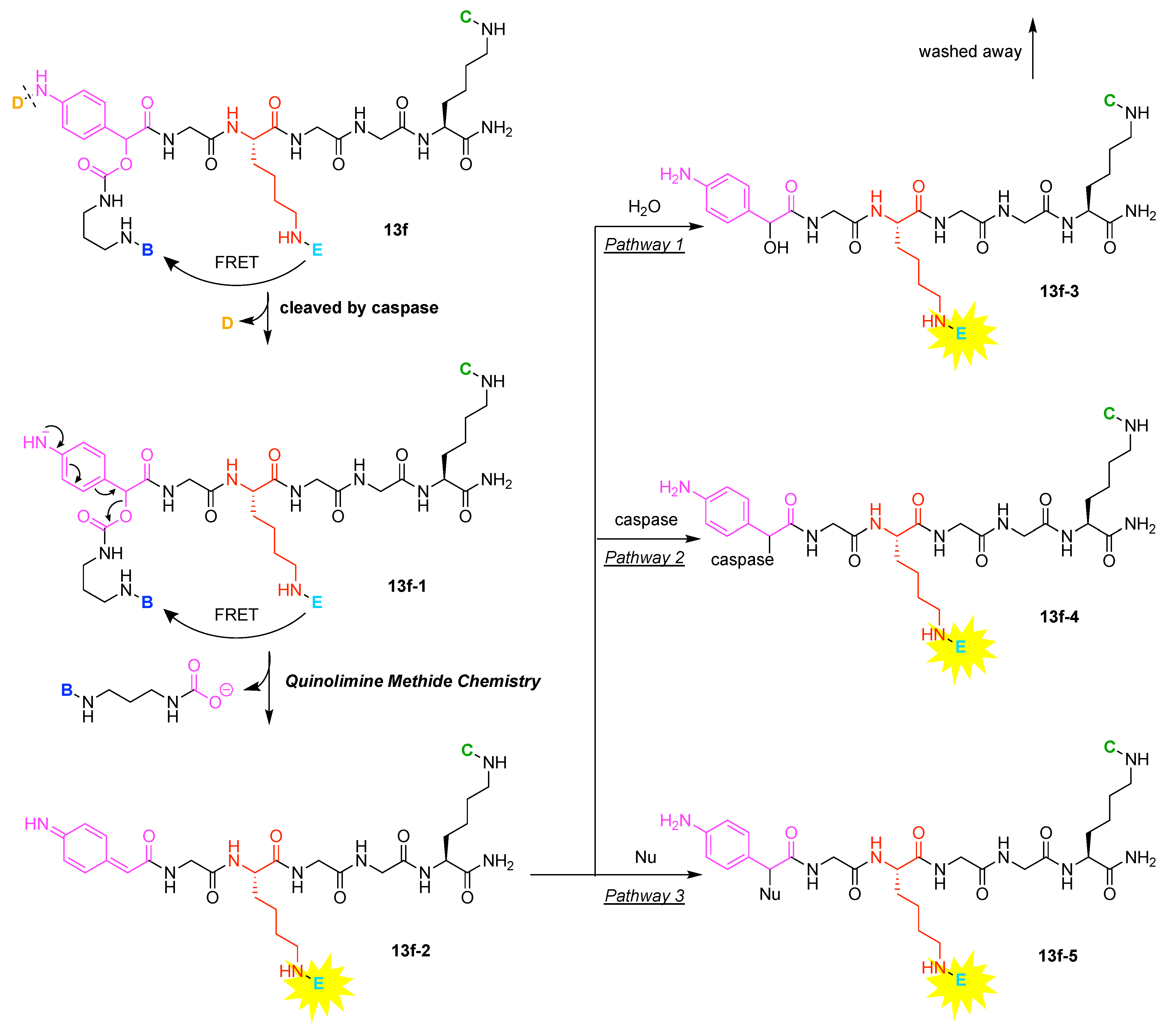
Figure 67.
Synthesis of a p-aminomandelic acid building block.
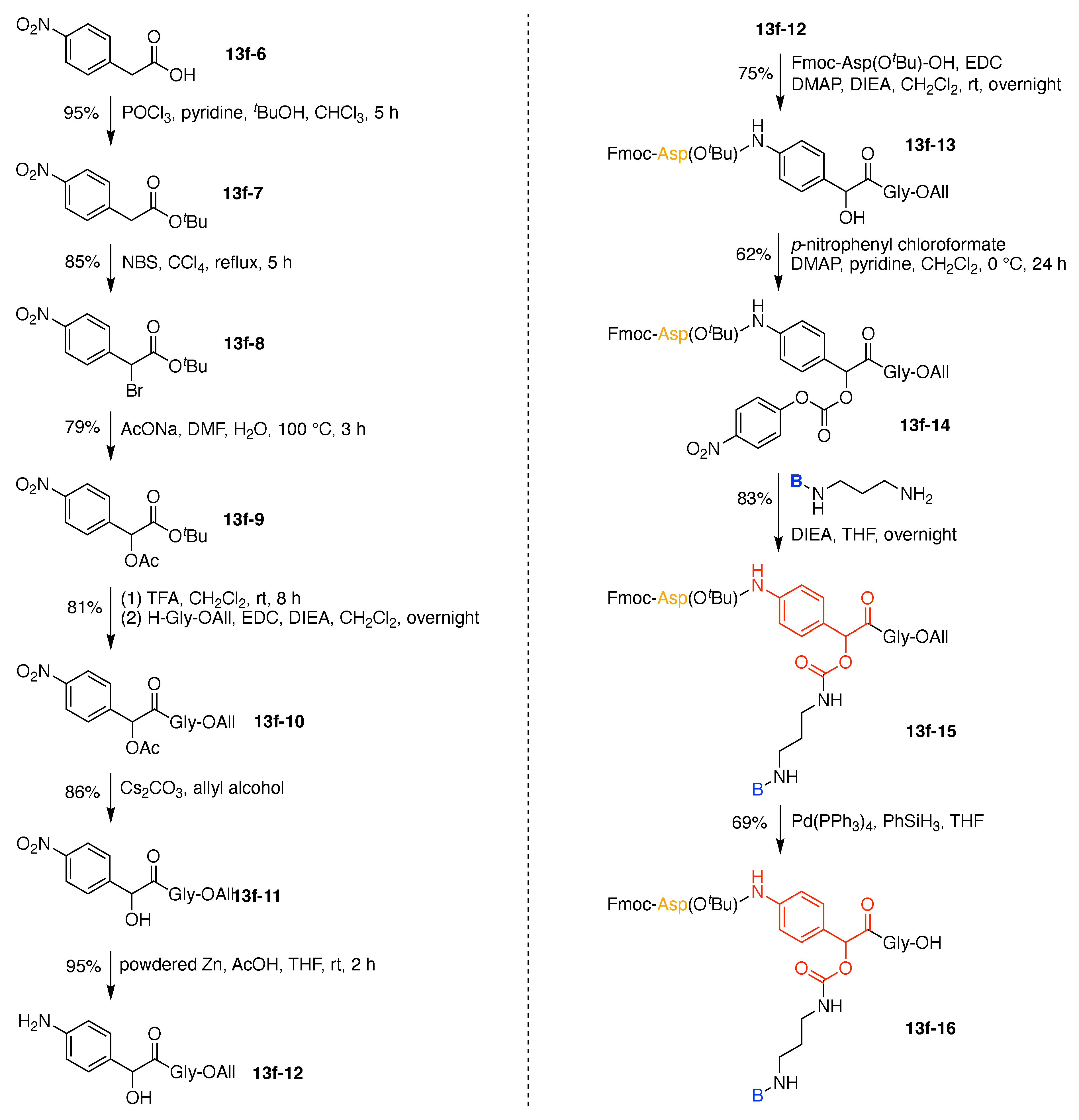
Figure 68.
Synthesis of the qABP unit by Fmoc SPPS.

Figure 69.
Five examples each containing a dye hub.
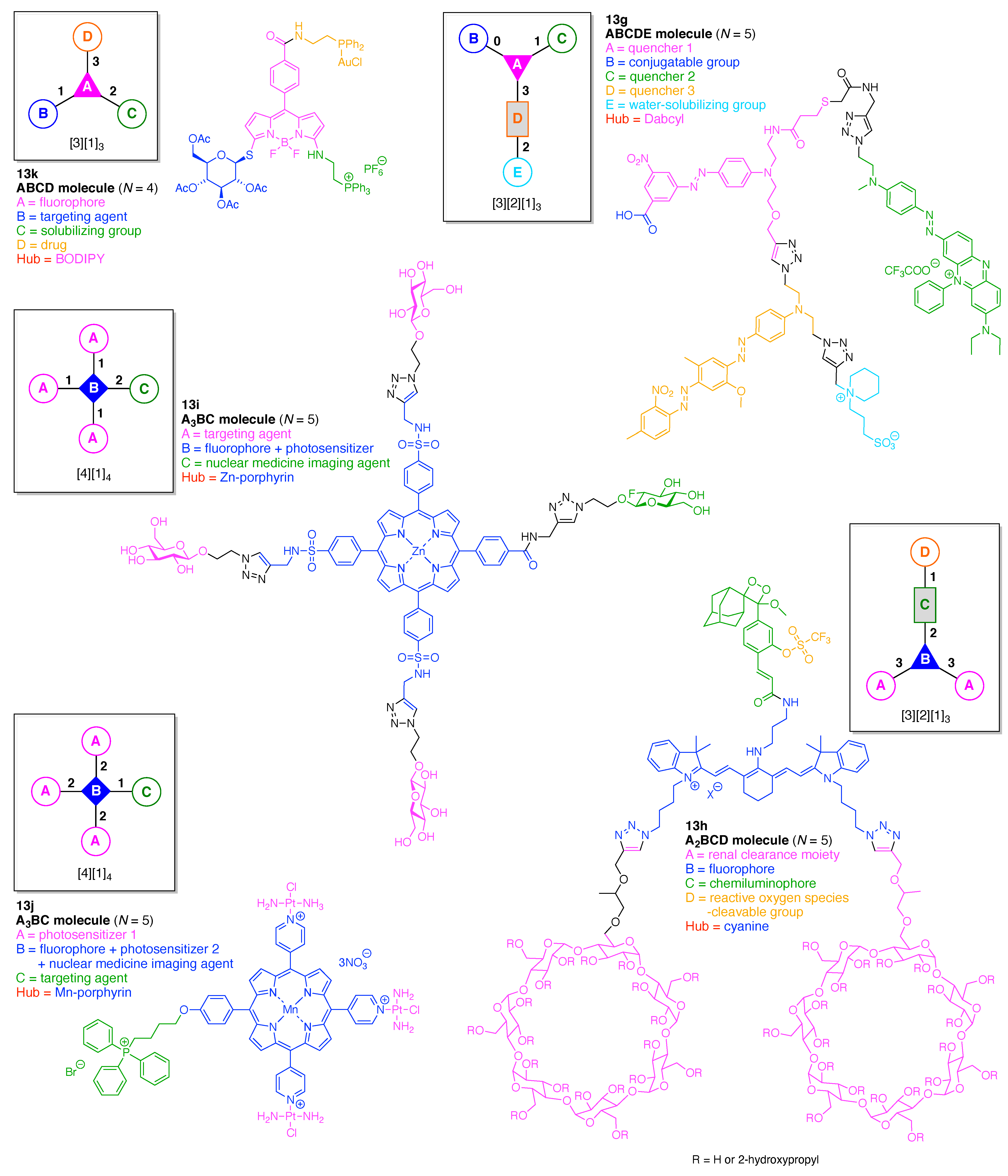
Figure 70.
Statistical synthesis of a porphyrin.

Figure 71.
Synthesis of a tri-functionalized BODIPY.
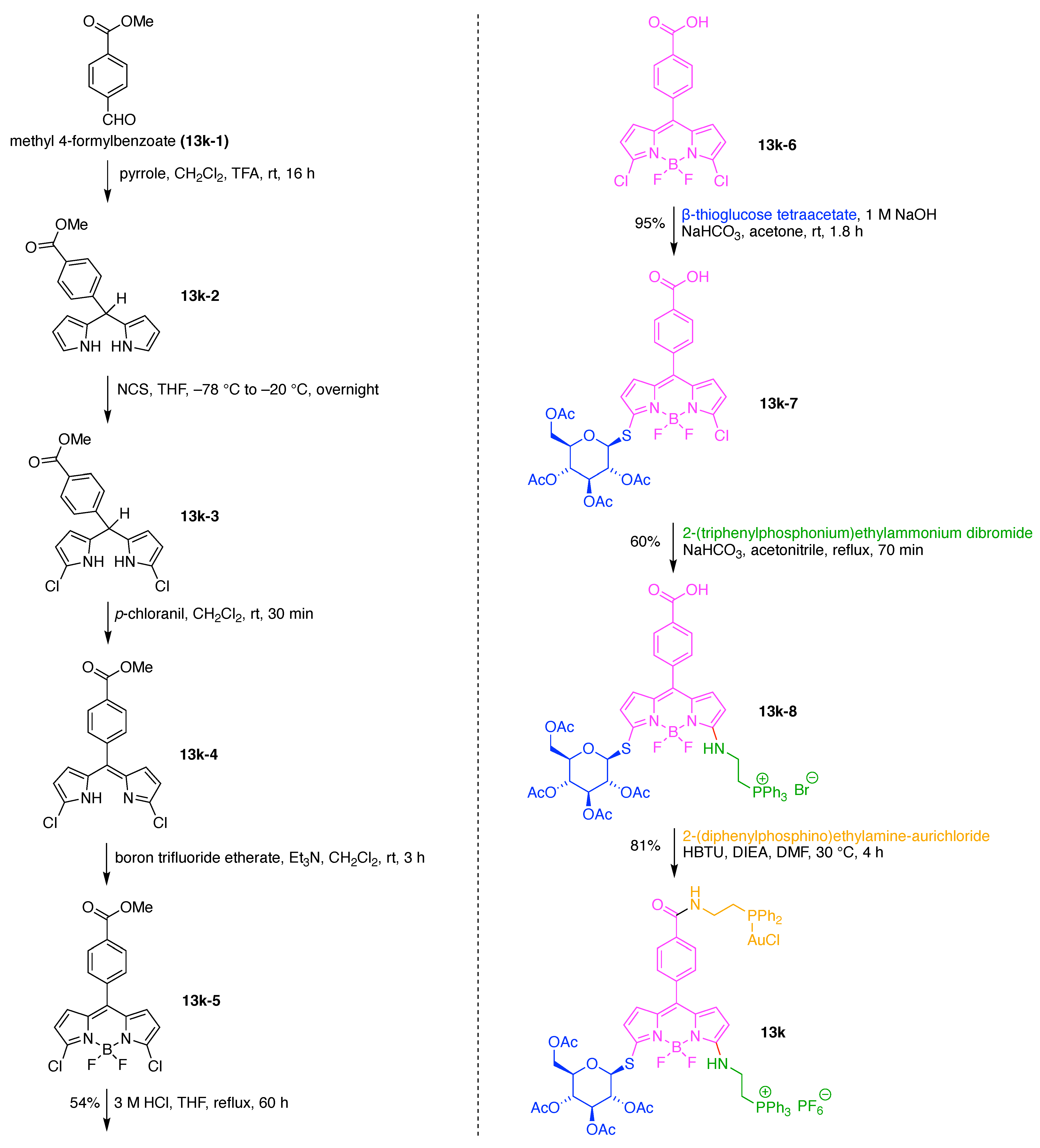
Figure 72.
A shape-persistent macrocycle with 12 derivatizable groups.

Figure 73.
One-flask synthesis of shape-persistent macrocycles encompassing six porphyrins.
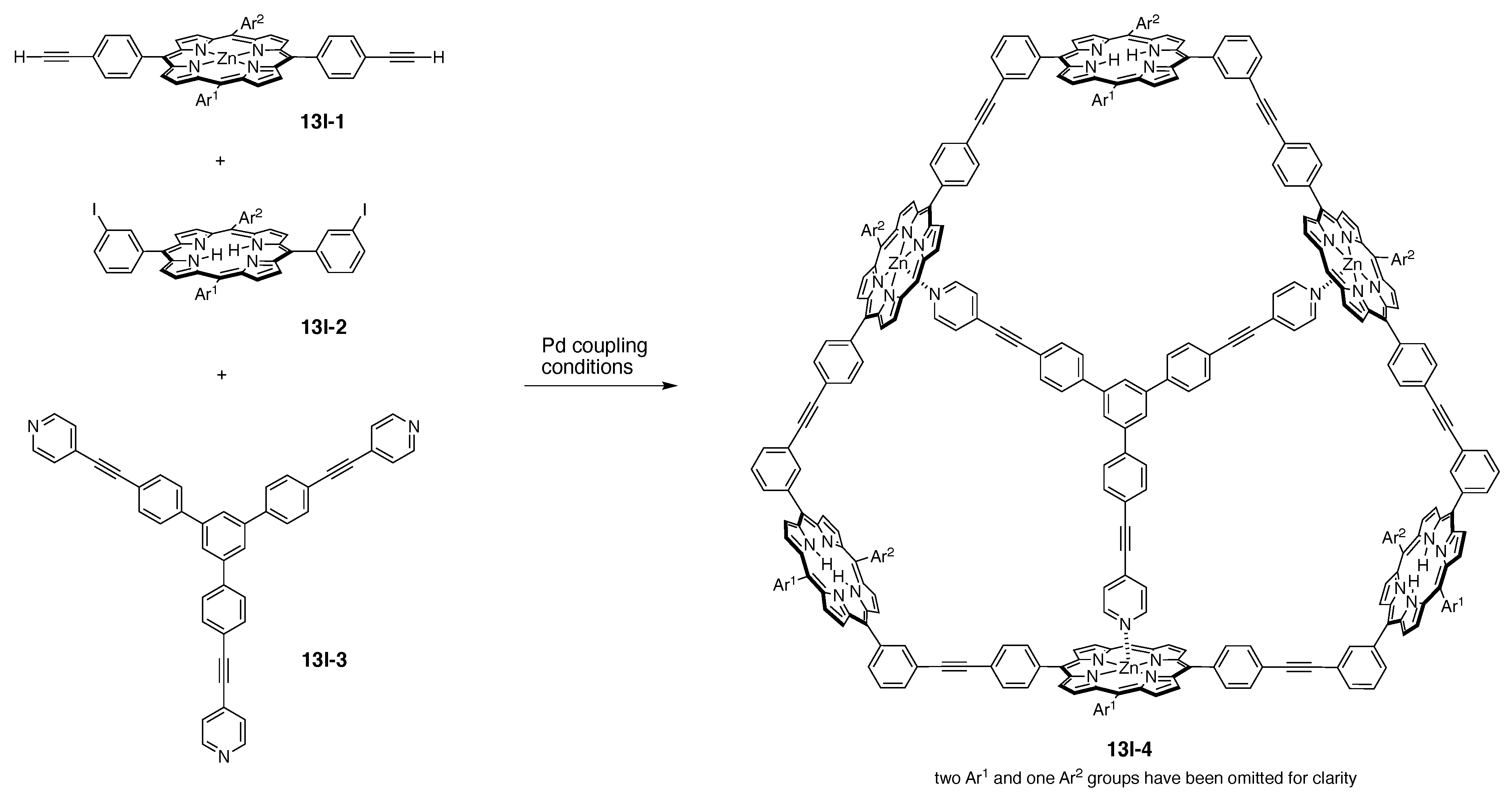
Figure 74.
Diverse shape-persistent macrocycles with control over metalation state and peripheral substituents.
Figure 74.
Diverse shape-persistent macrocycles with control over metalation state and peripheral substituents.
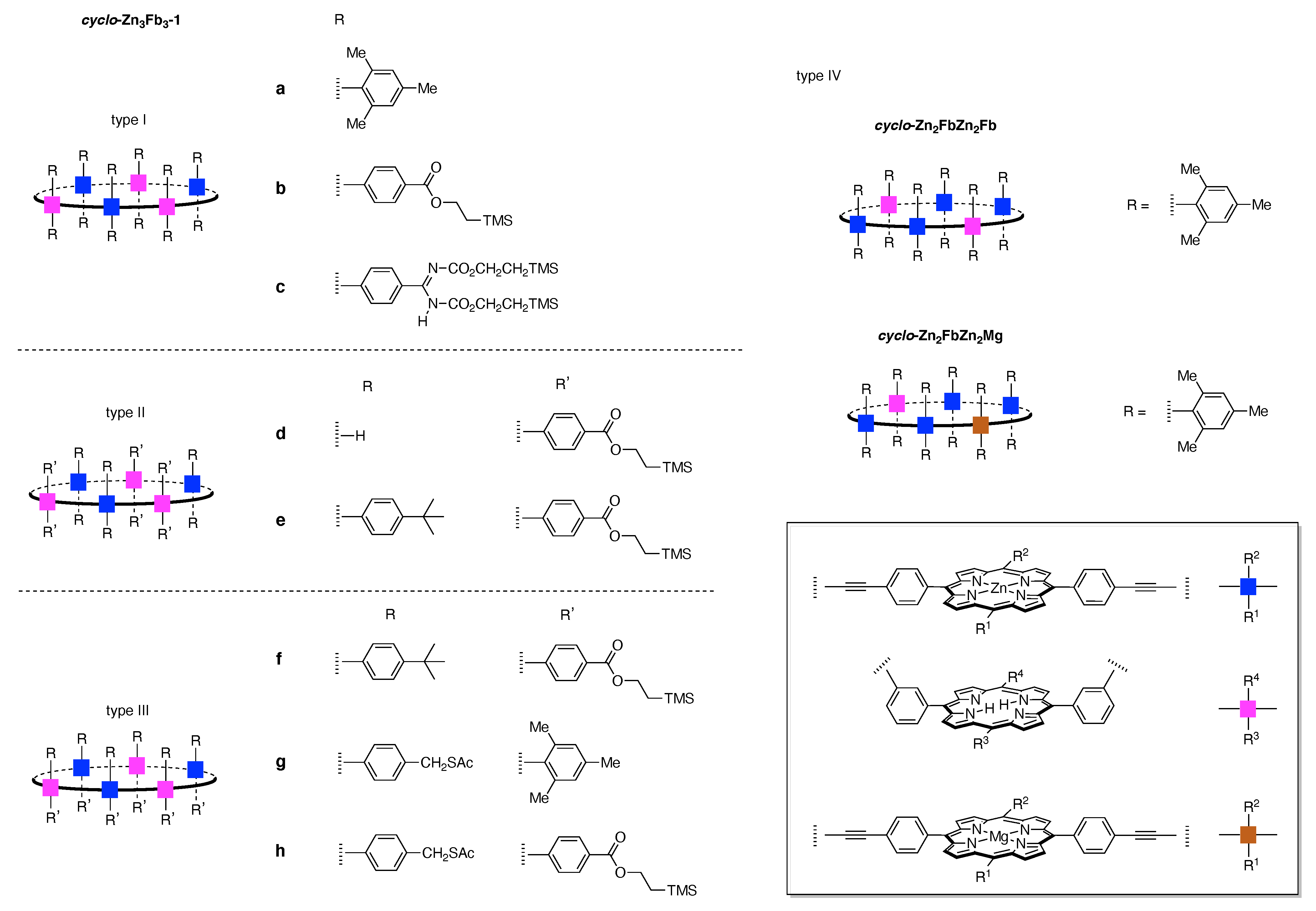
Figure 75.
Rational synthesis of porphyrins with four distinct meso-substituents (top panel). One-flask synthesis of dipyrromethanes (second panel). Synthesis of 1,9-diacyldipyrromethanes (third panel). Use of a 1,9-diacyldipyrromethane and a dipyrromethane to form a porphyrin bearing two ethynes, one protected ester, and one S-acetylthioether (bottom panel).
Figure 75.
Rational synthesis of porphyrins with four distinct meso-substituents (top panel). One-flask synthesis of dipyrromethanes (second panel). Synthesis of 1,9-diacyldipyrromethanes (third panel). Use of a 1,9-diacyldipyrromethane and a dipyrromethane to form a porphyrin bearing two ethynes, one protected ester, and one S-acetylthioether (bottom panel).
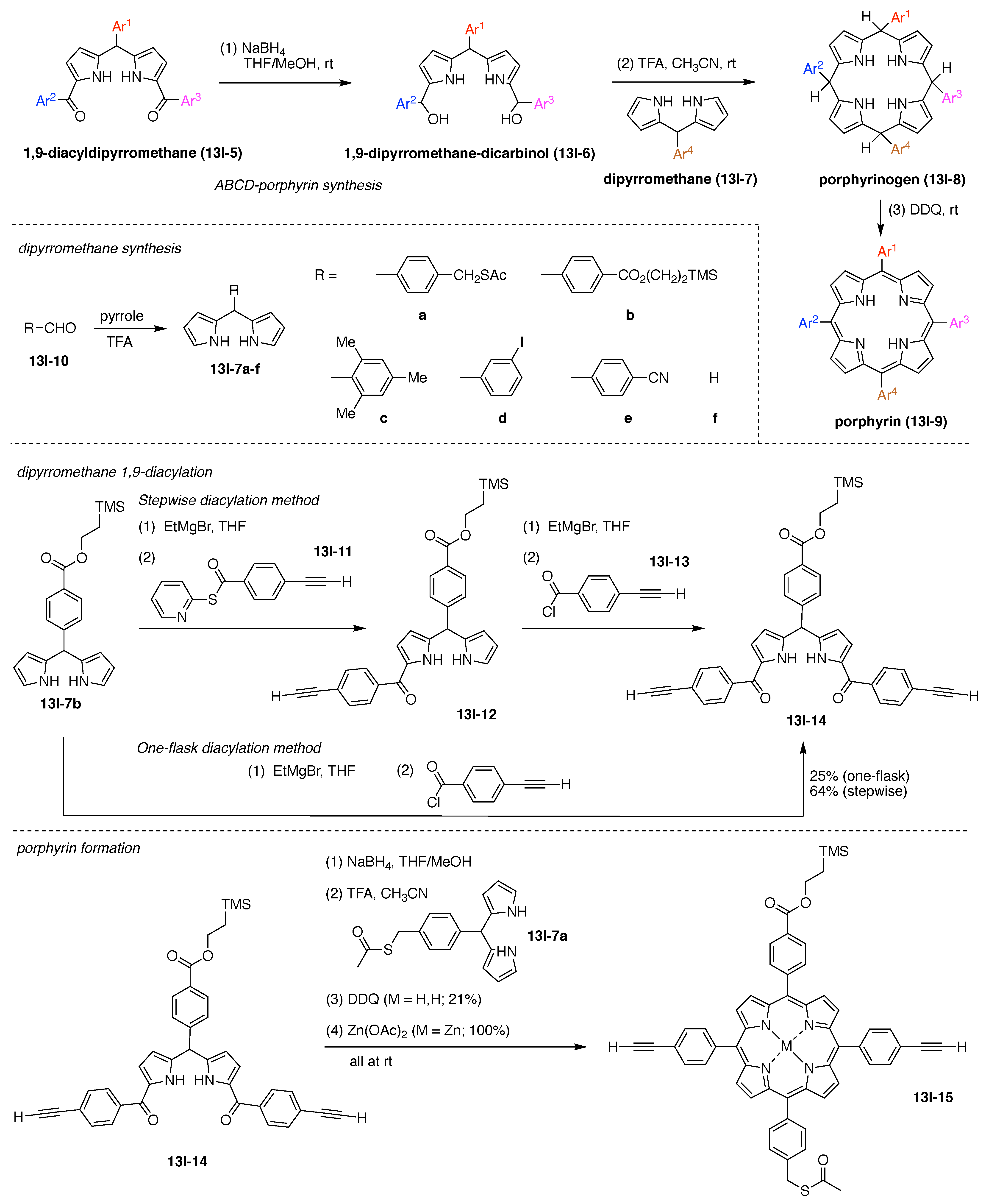

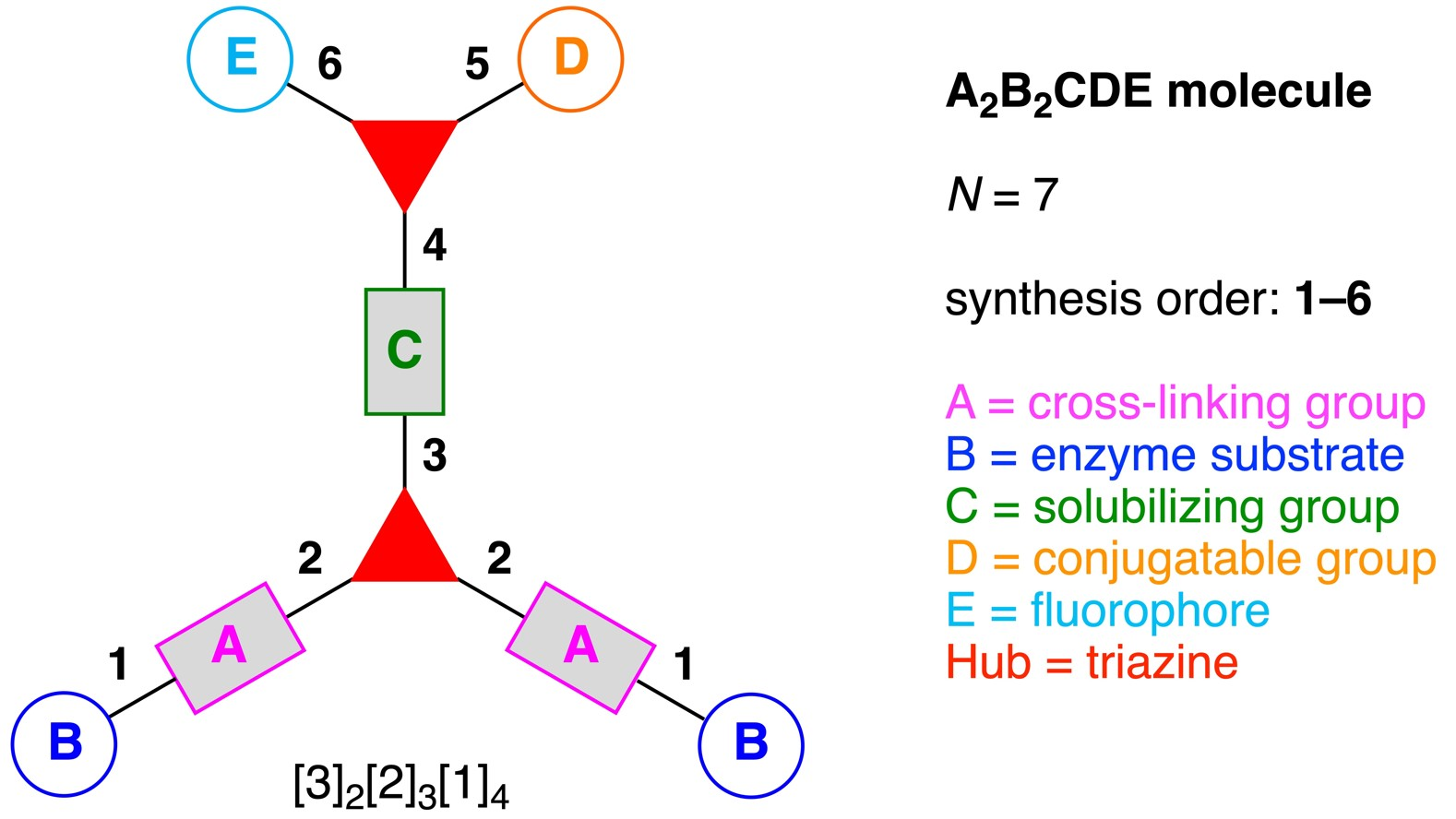{kind=link}
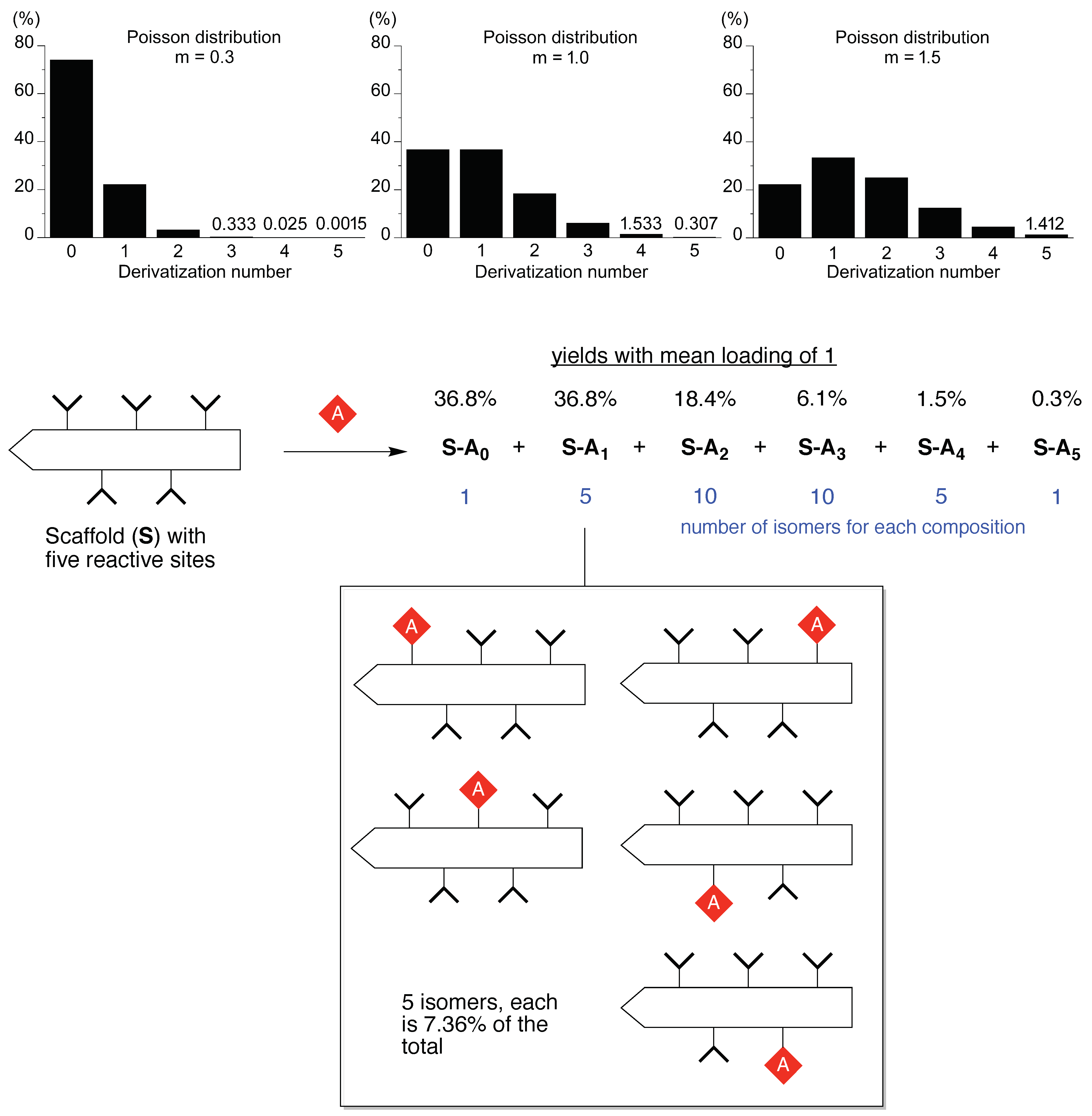{kind=link}
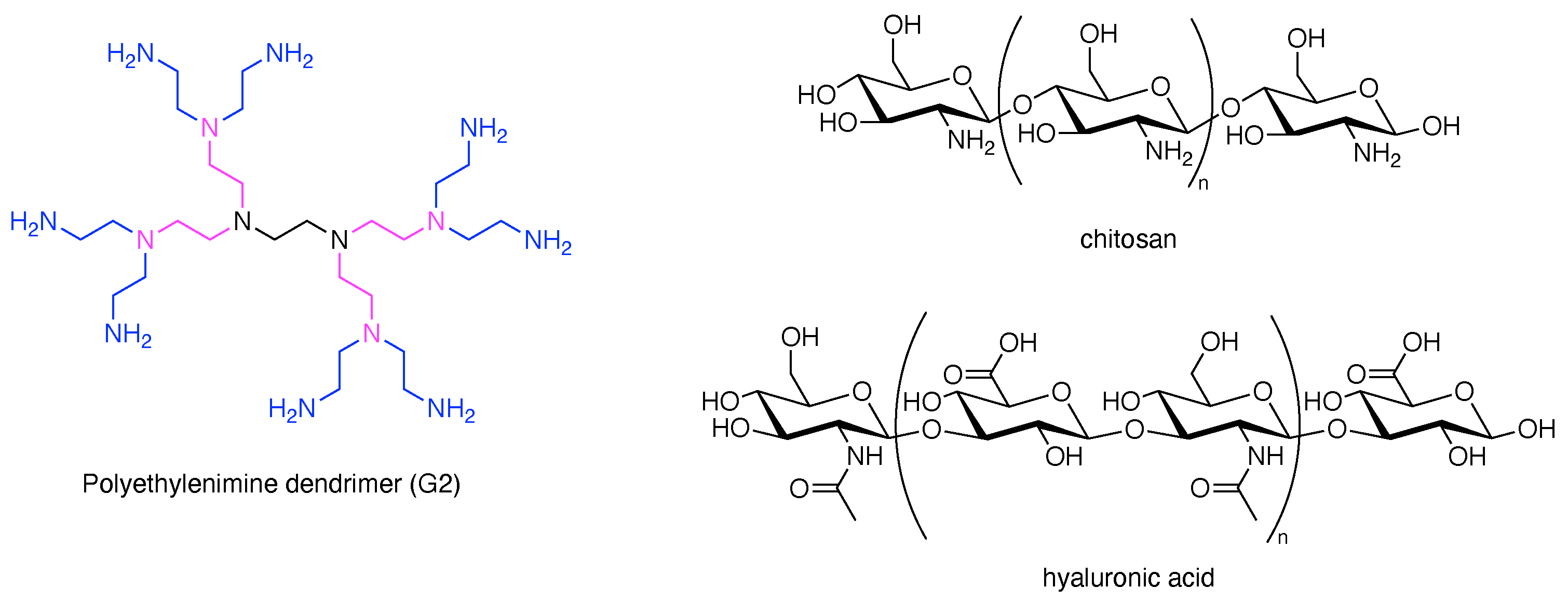{kind=link}
{kind=link}
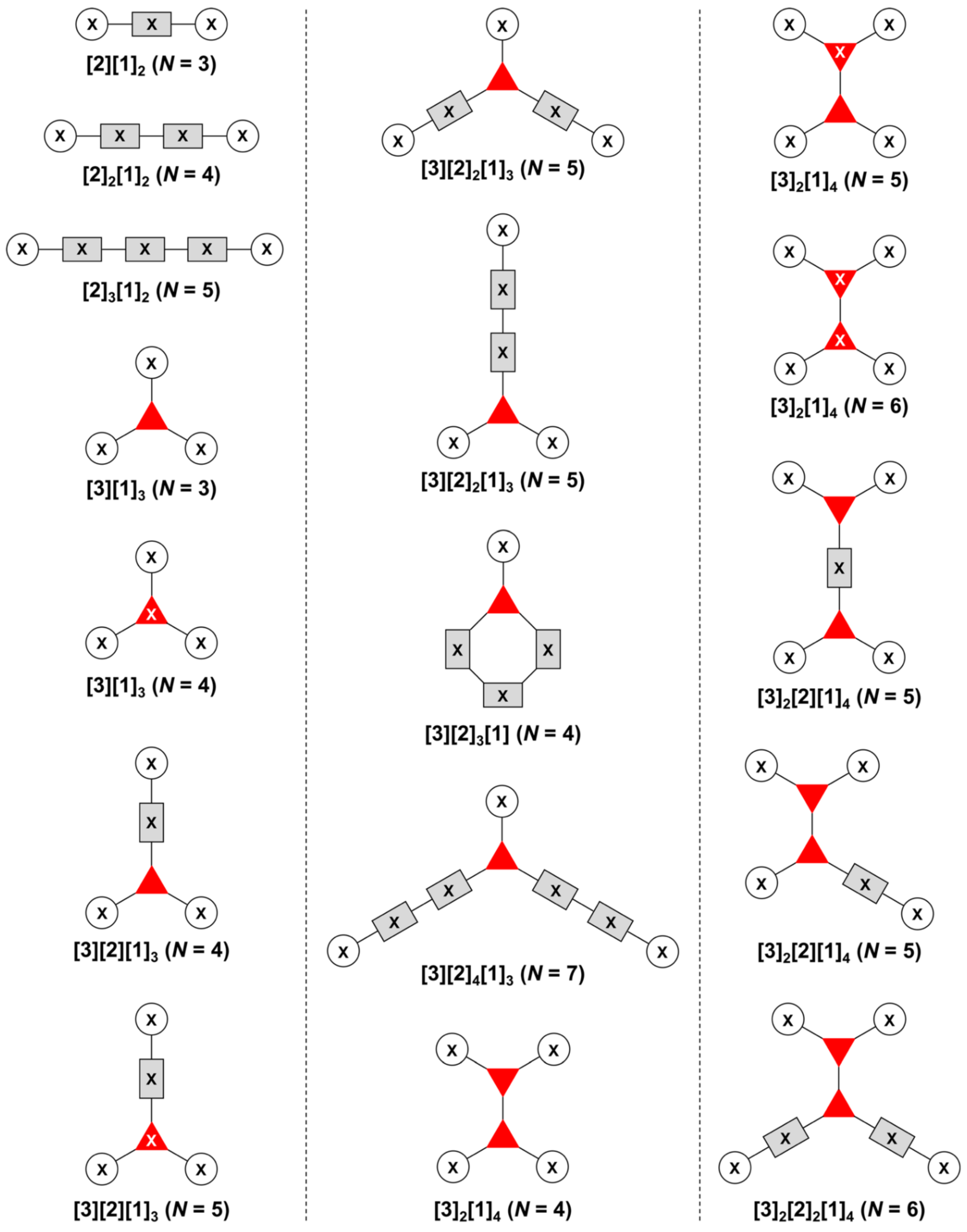{kind=link}
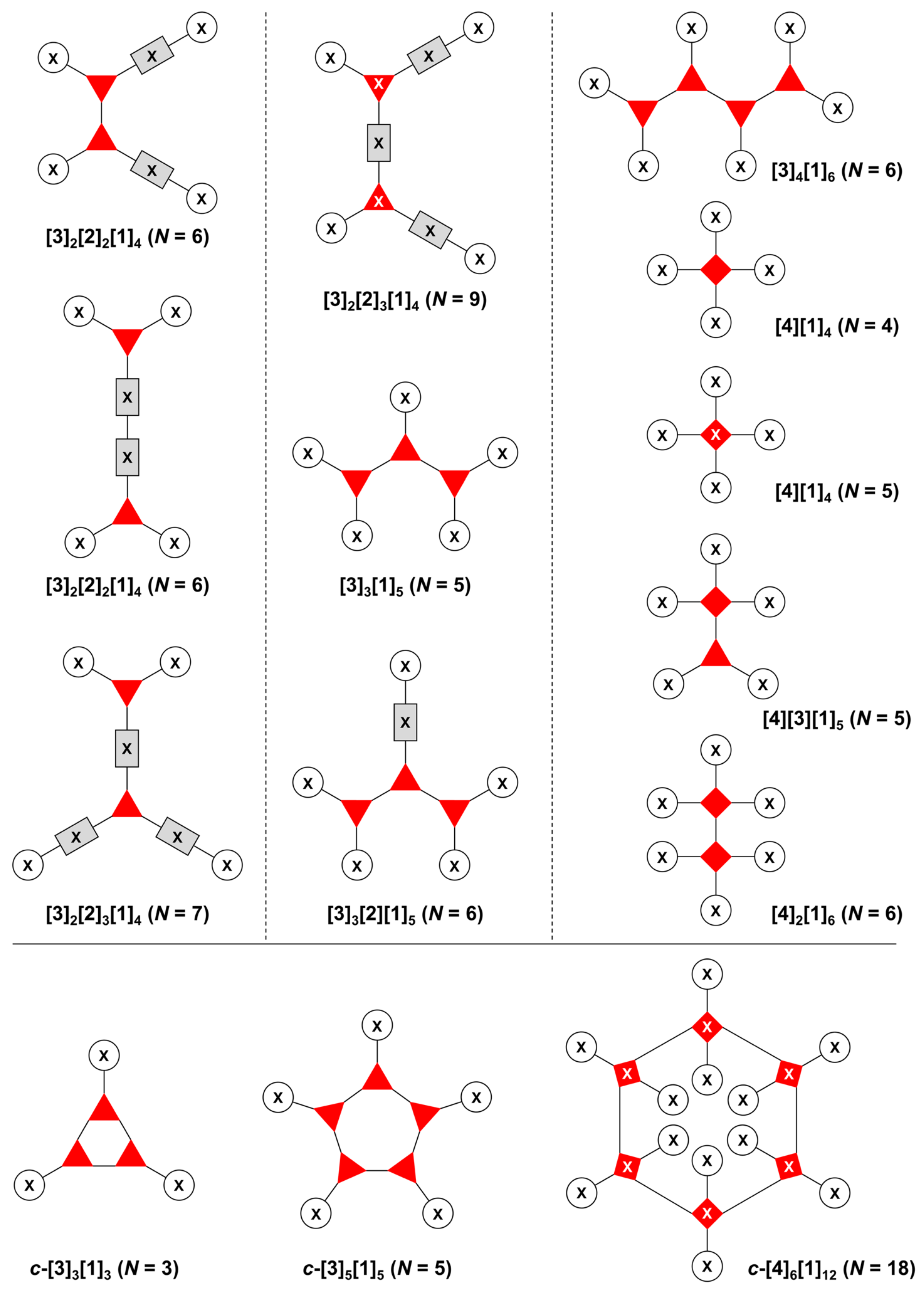{kind=link}
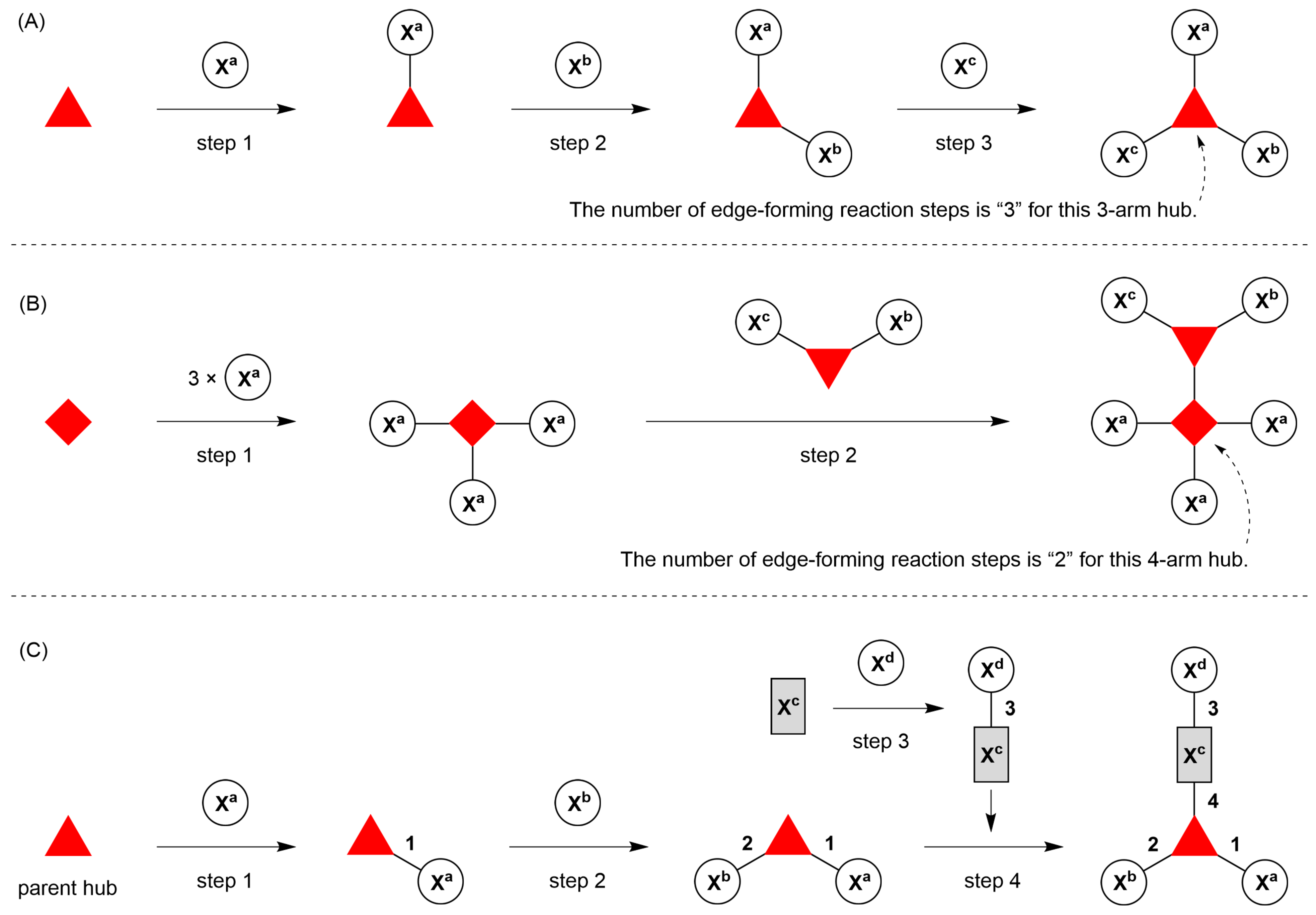{kind=link}
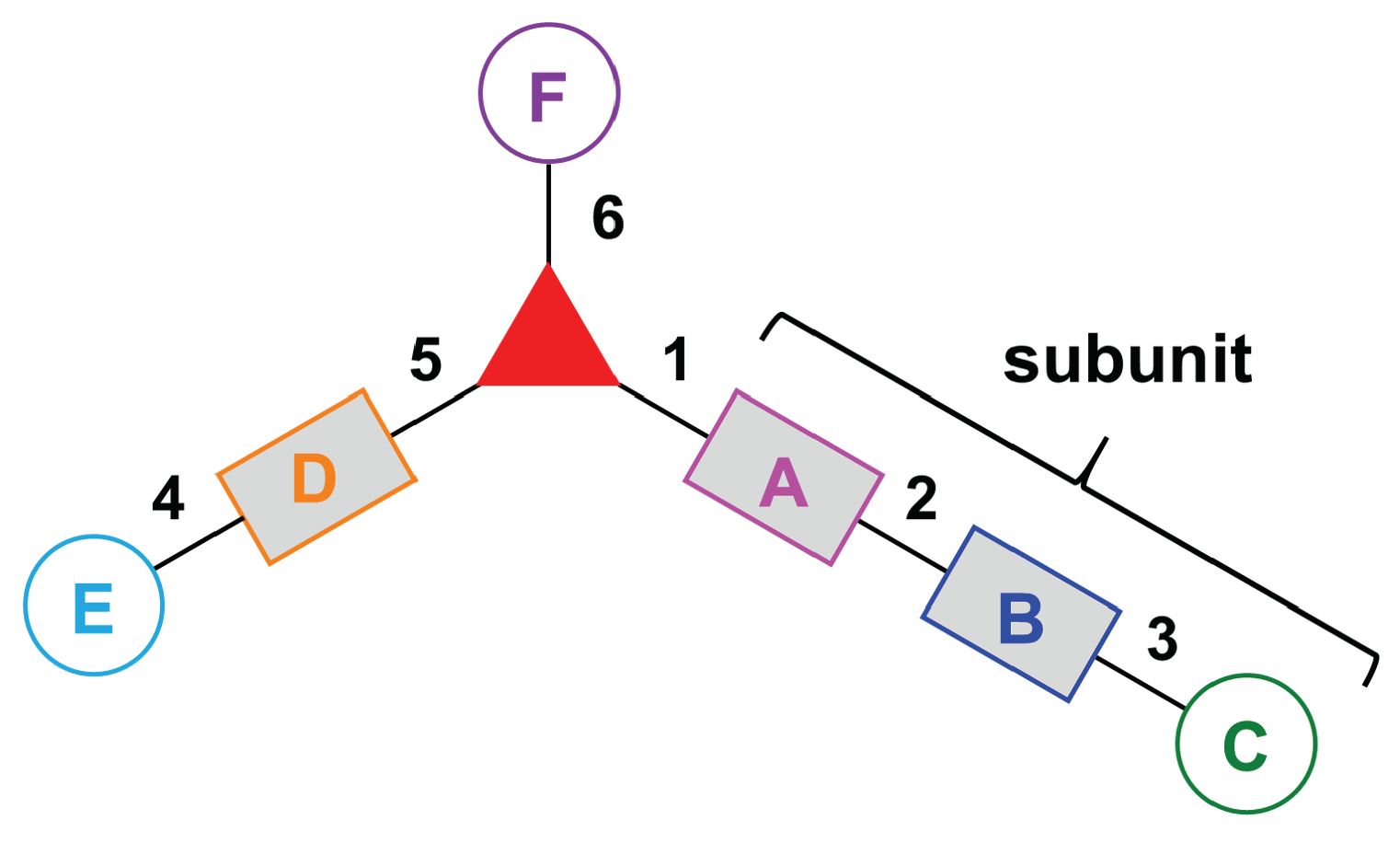{kind=link}
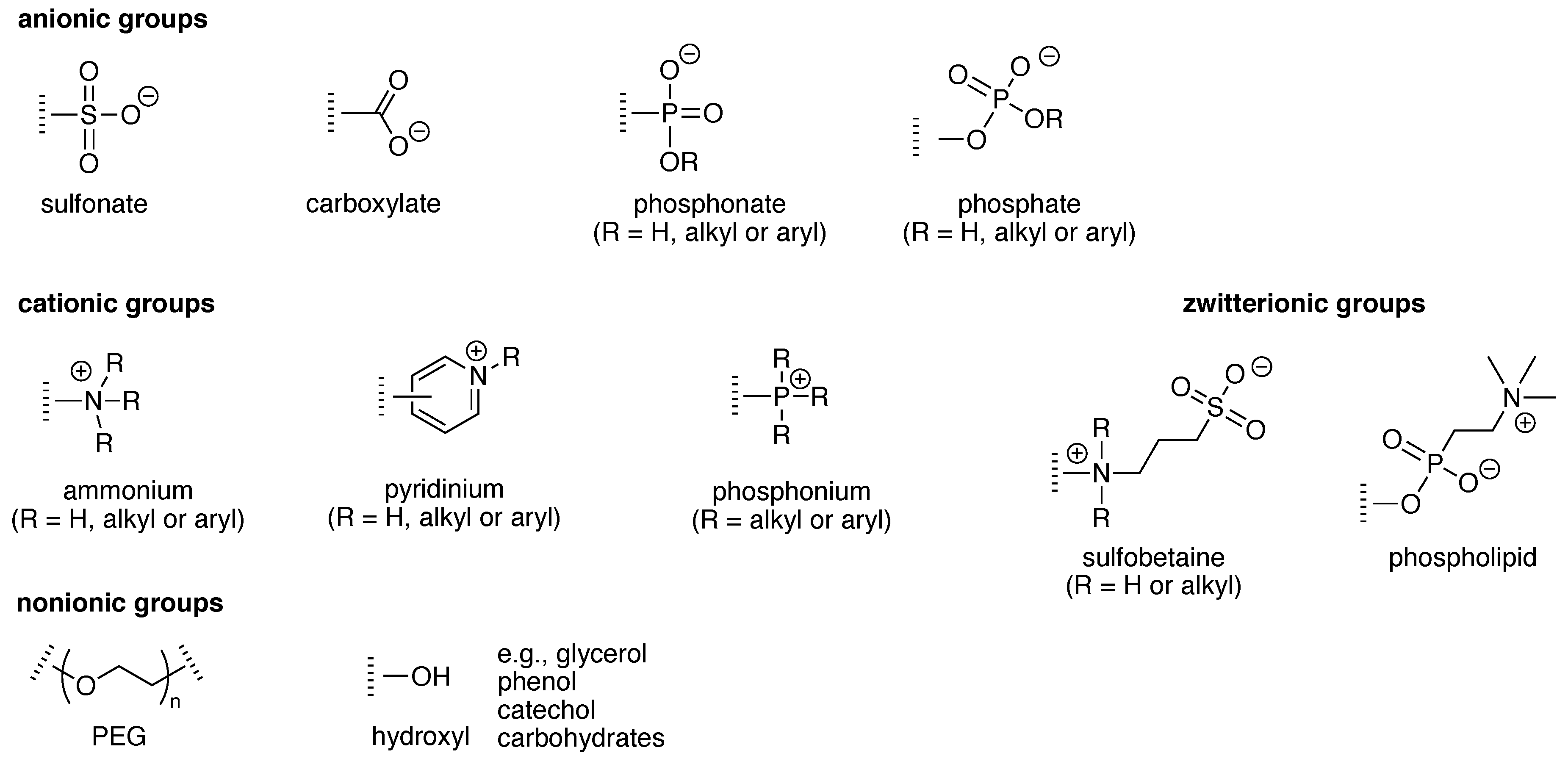{kind=link}
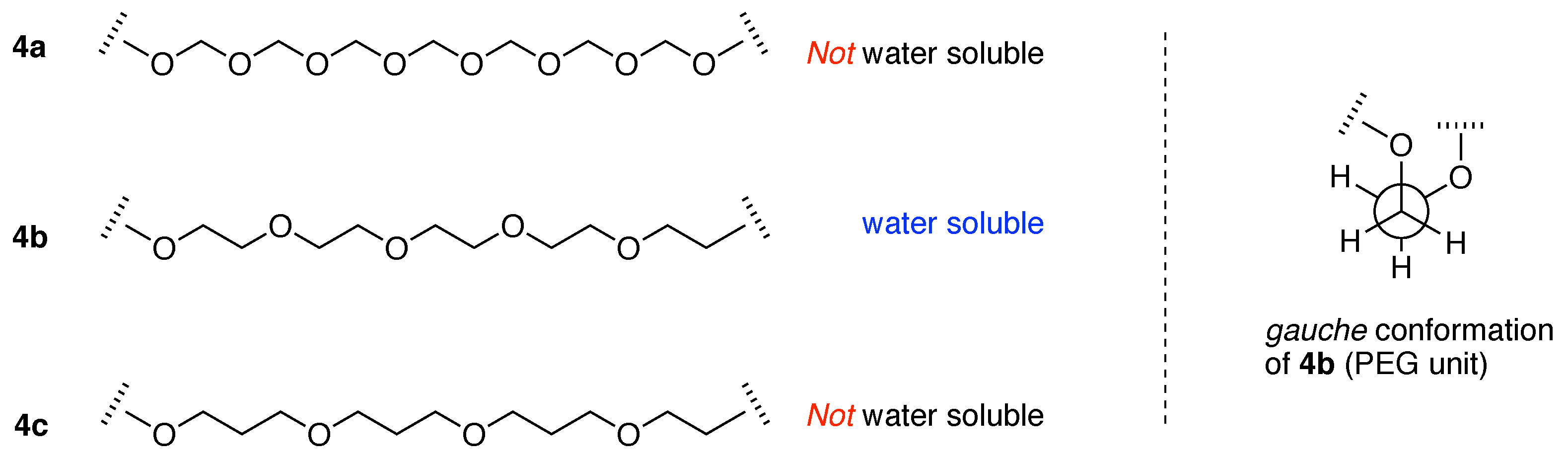{kind=link}
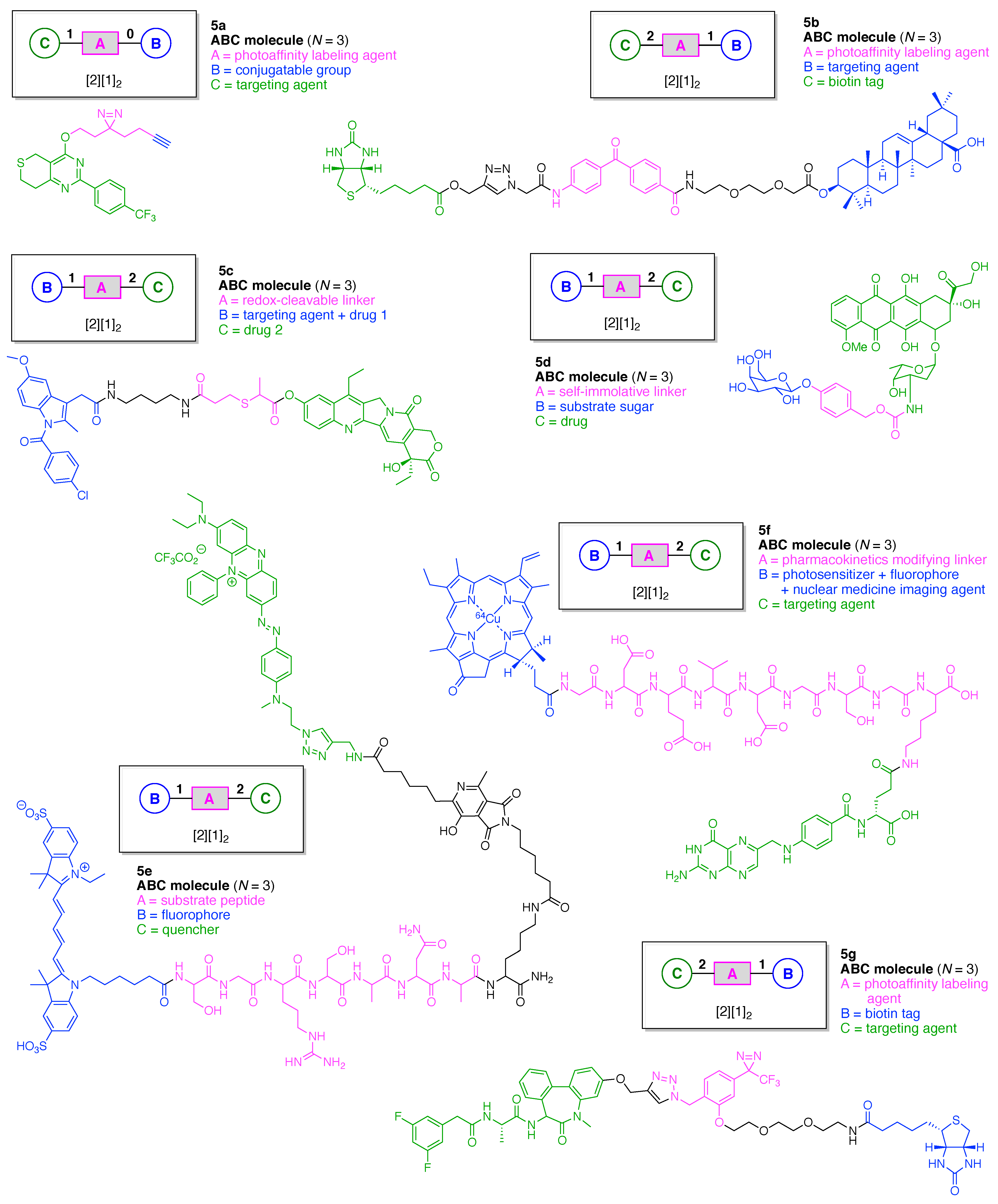{kind=link}
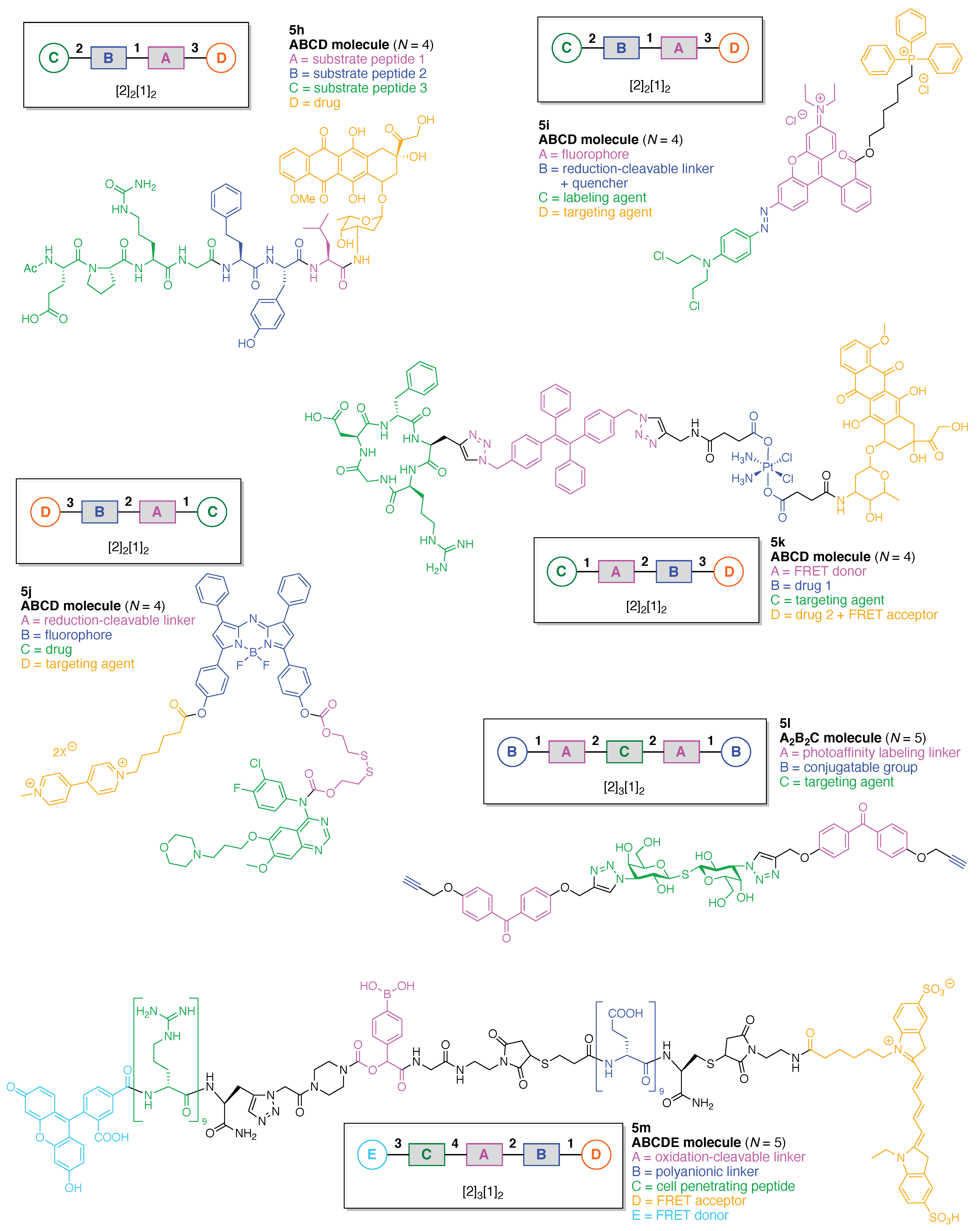{kind=link}
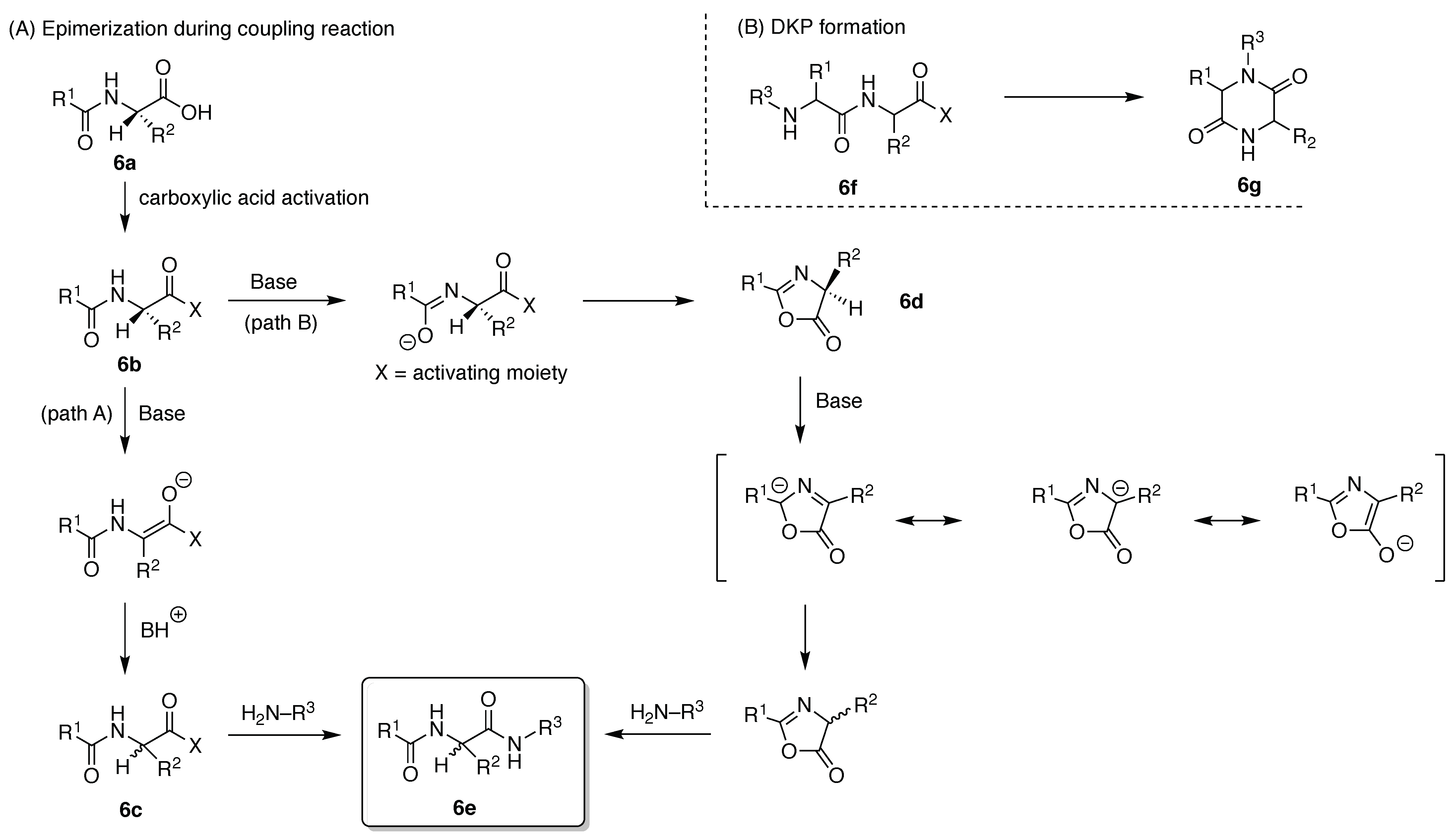{kind=link}
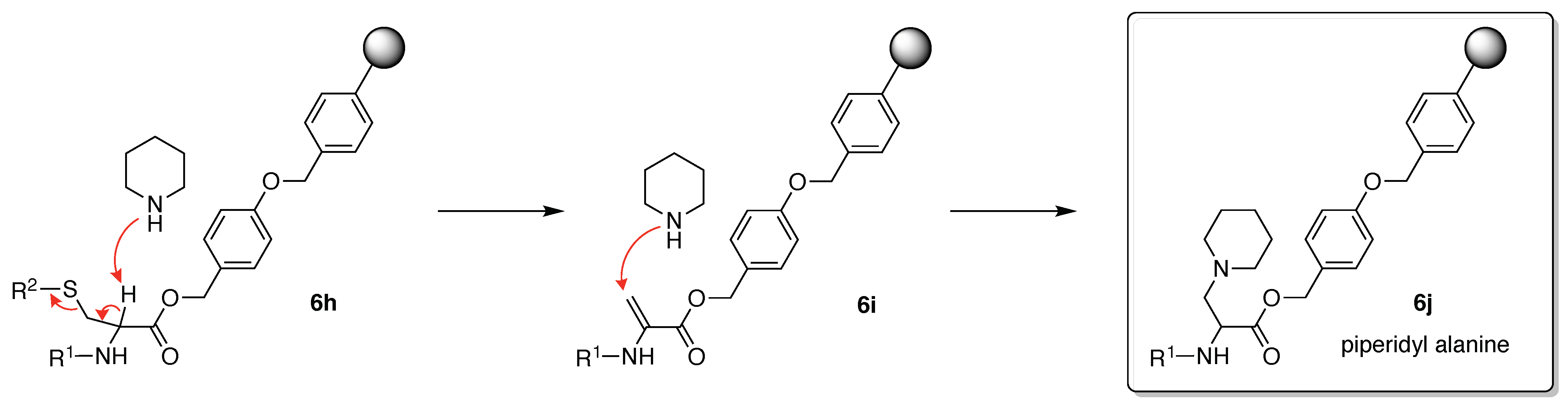{kind=link}
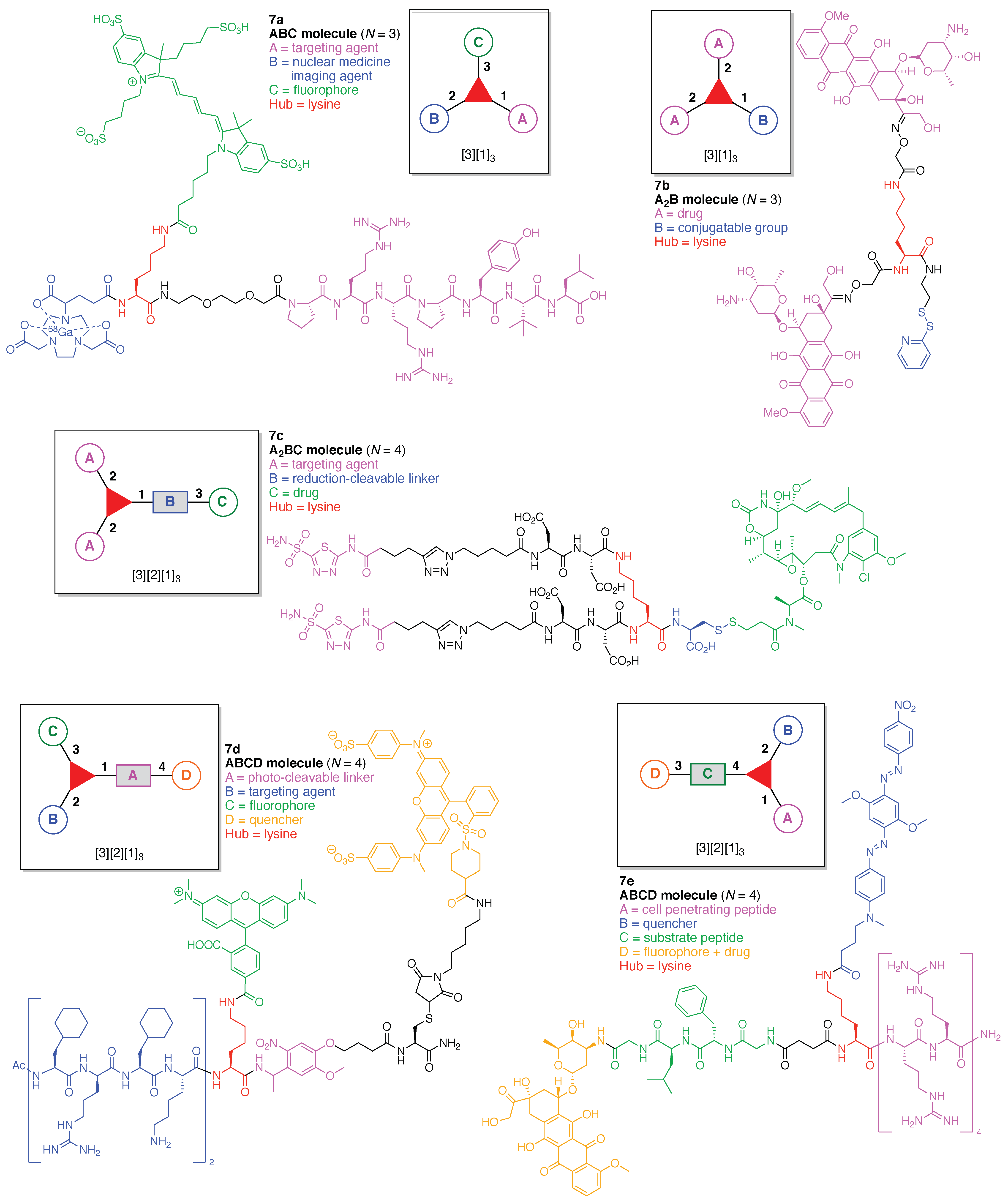{kind=link}
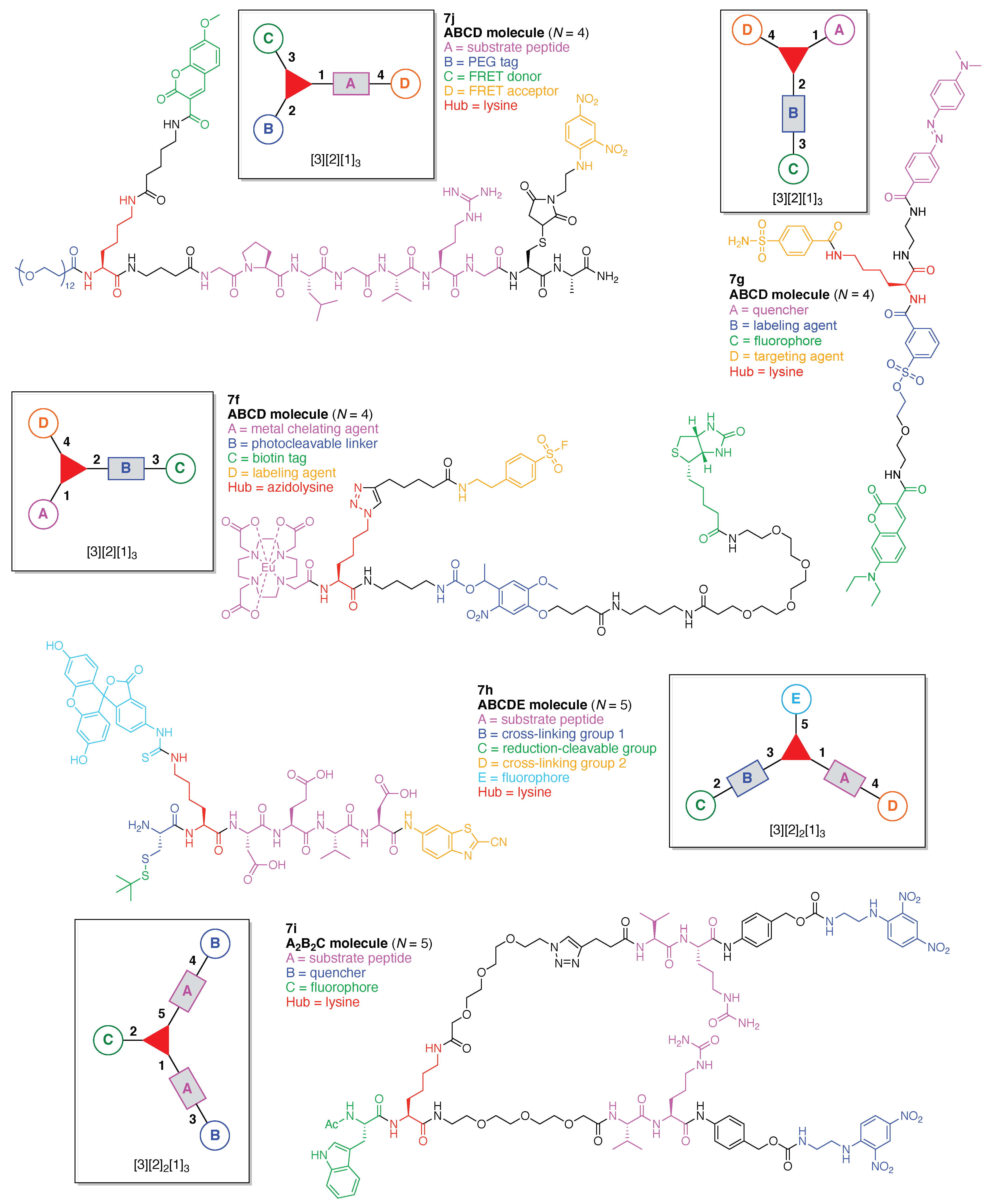{kind=link}
{kind=link}
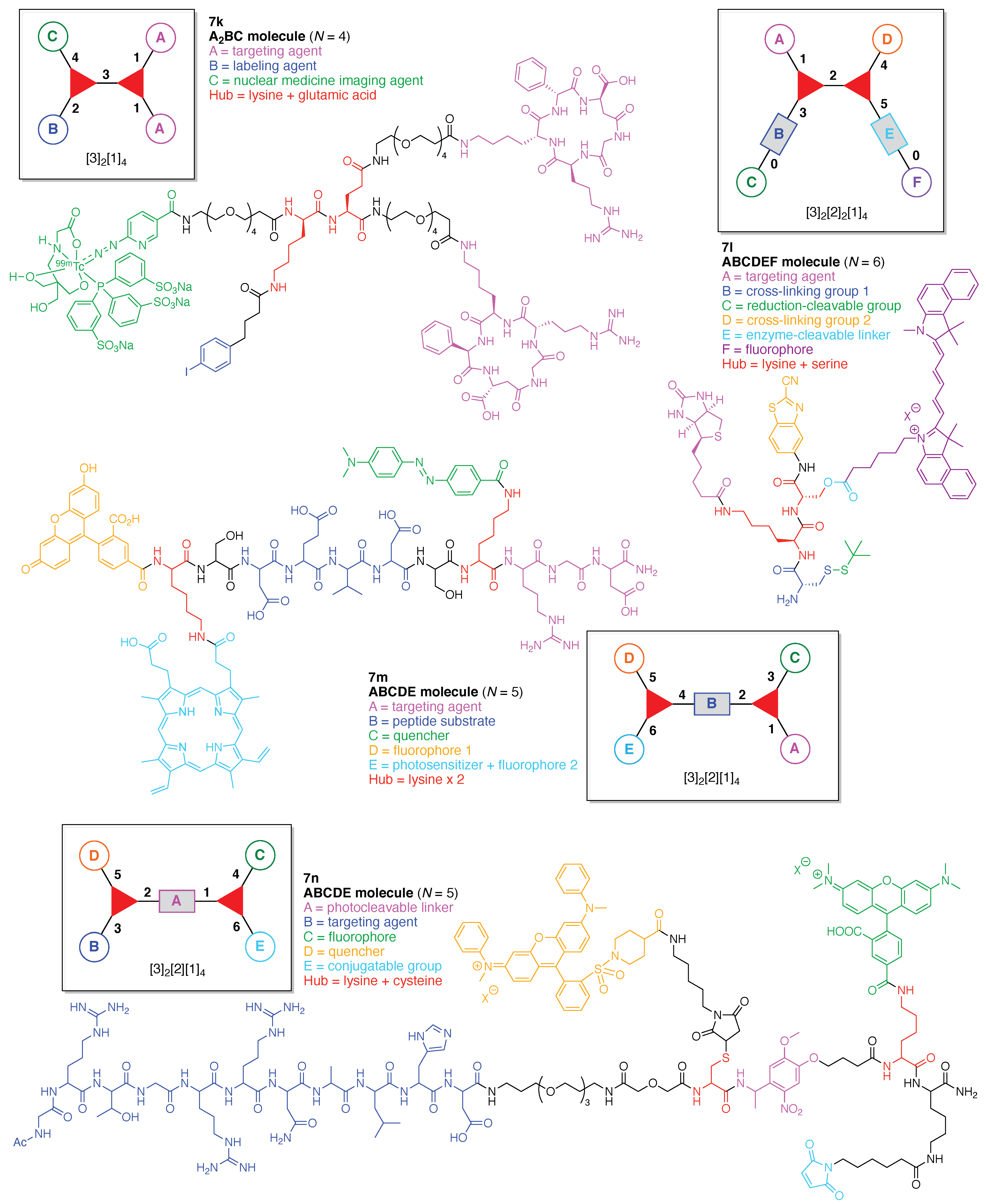{kind=link}
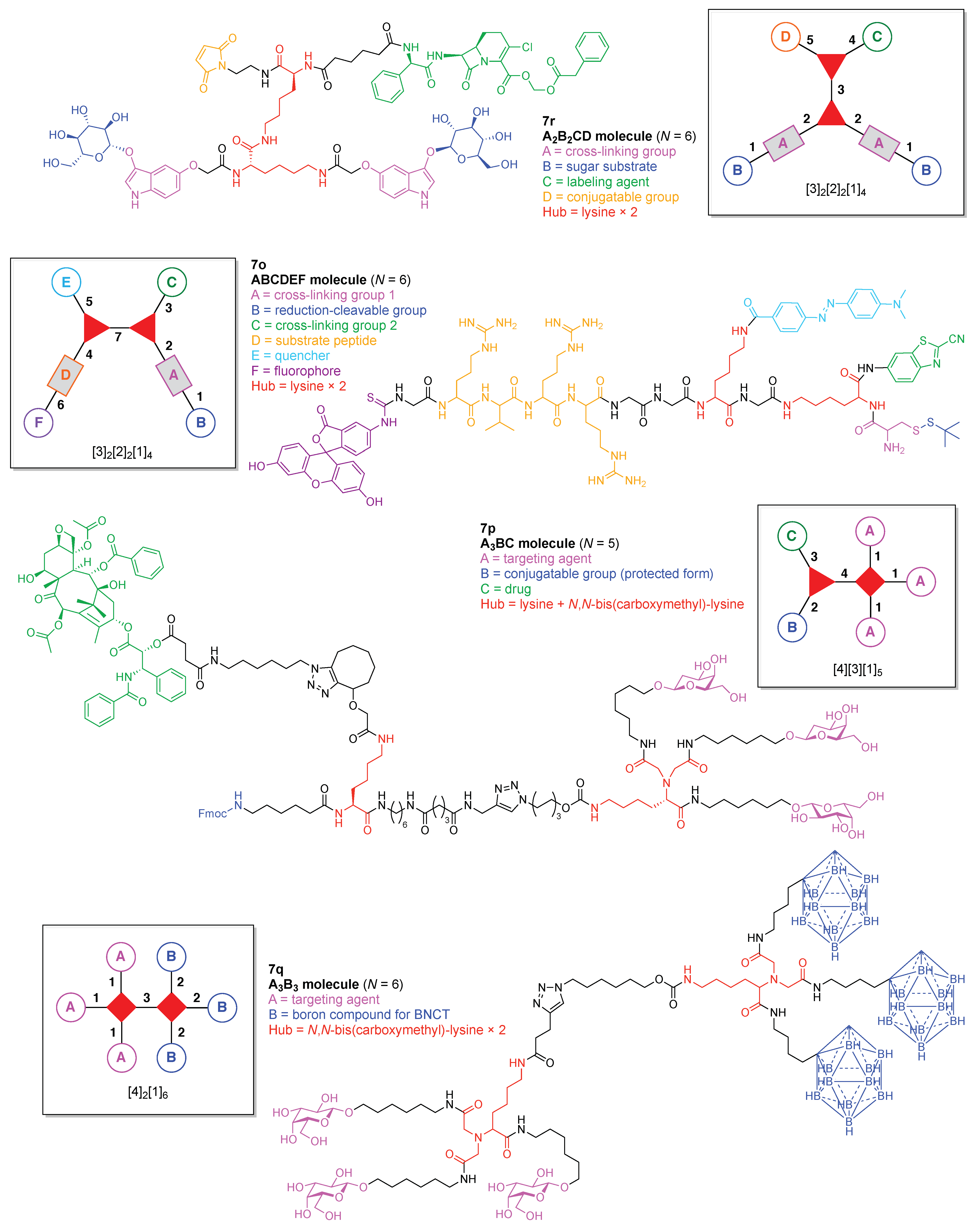{kind=link}
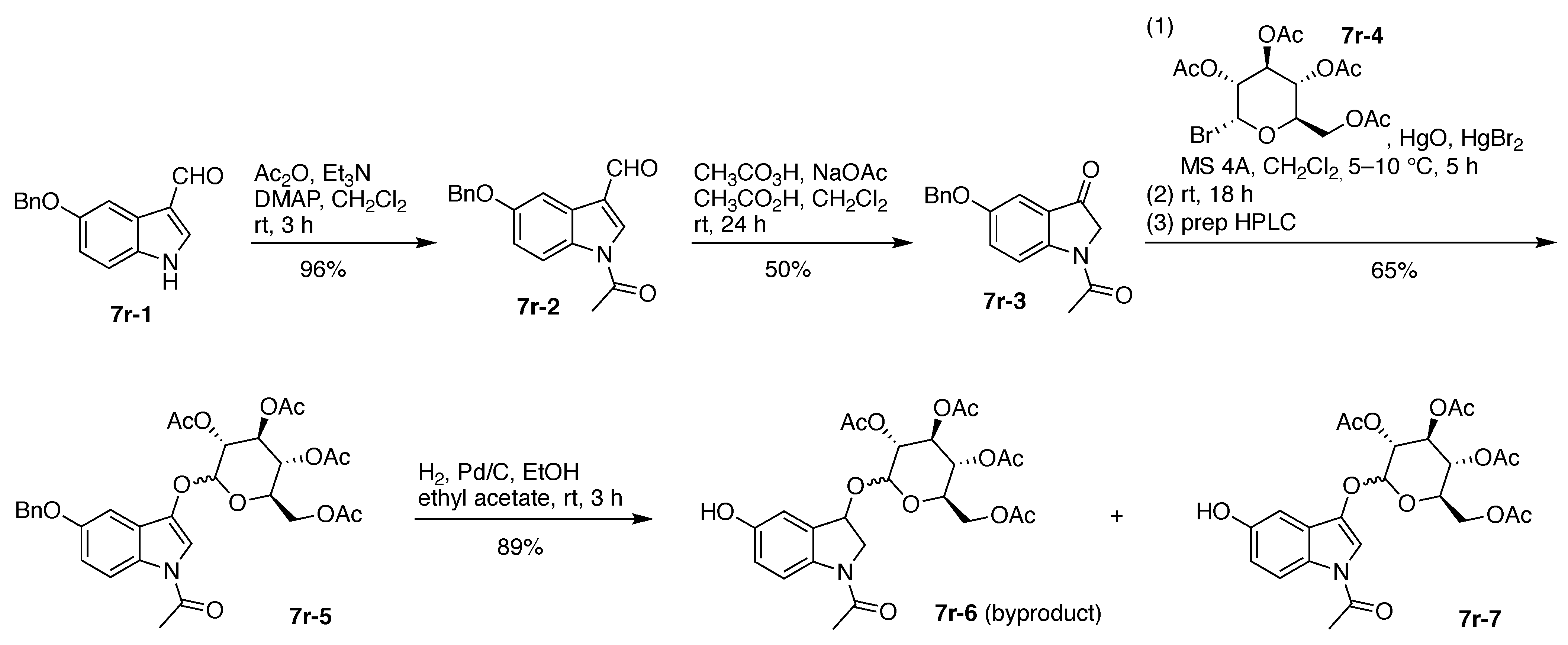{kind=link}
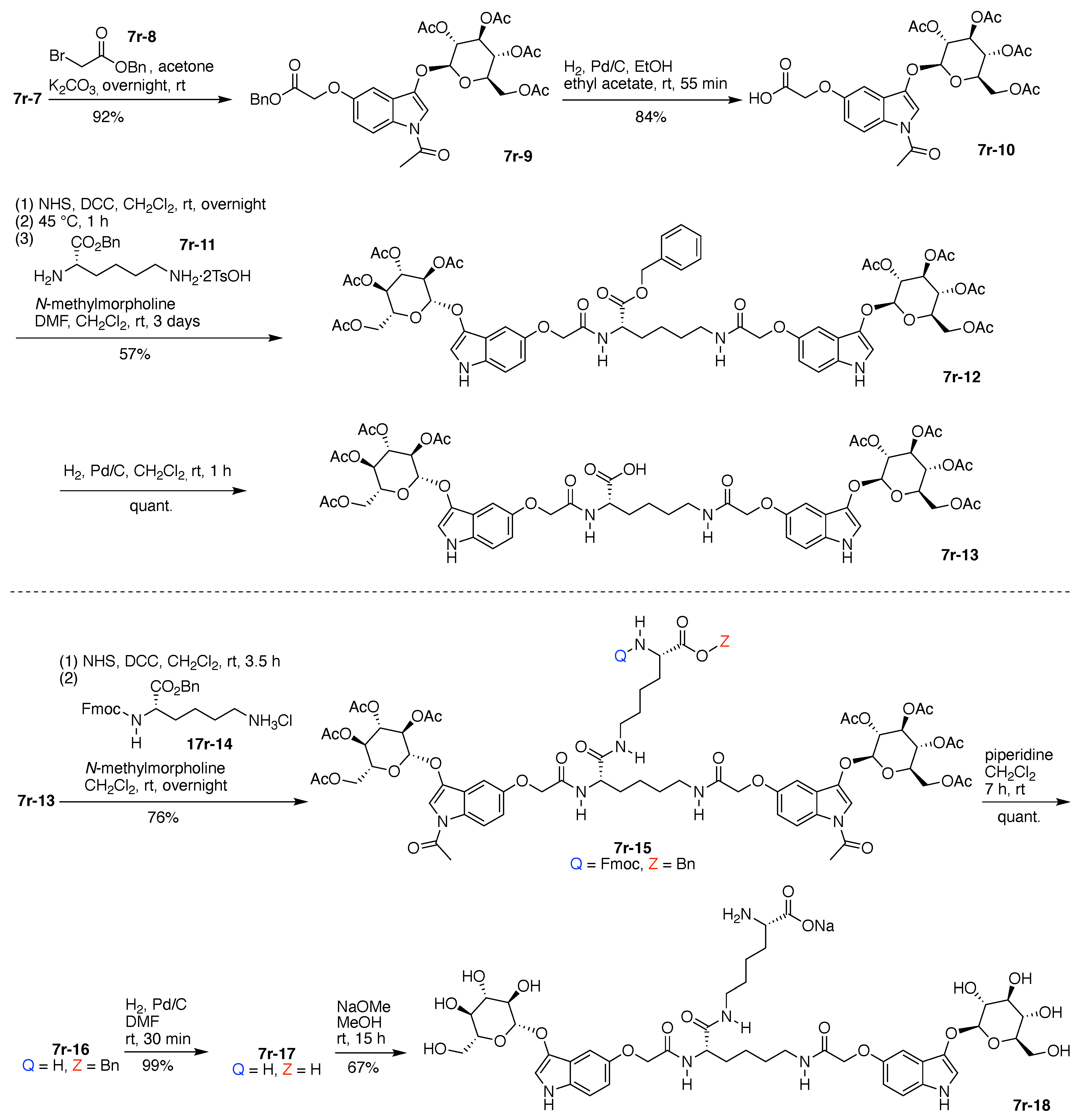{kind=link}
{kind=link}
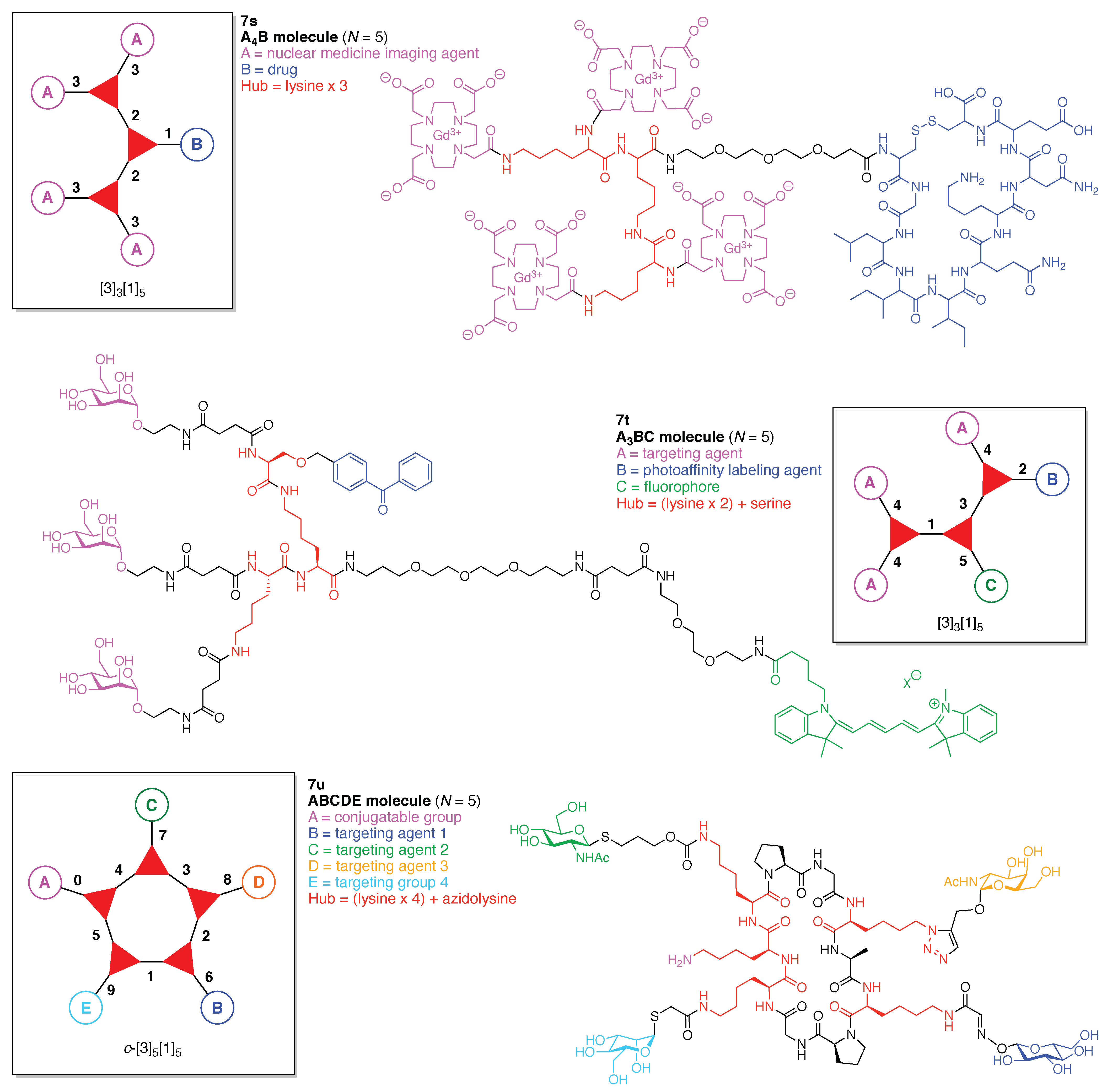{kind=link}
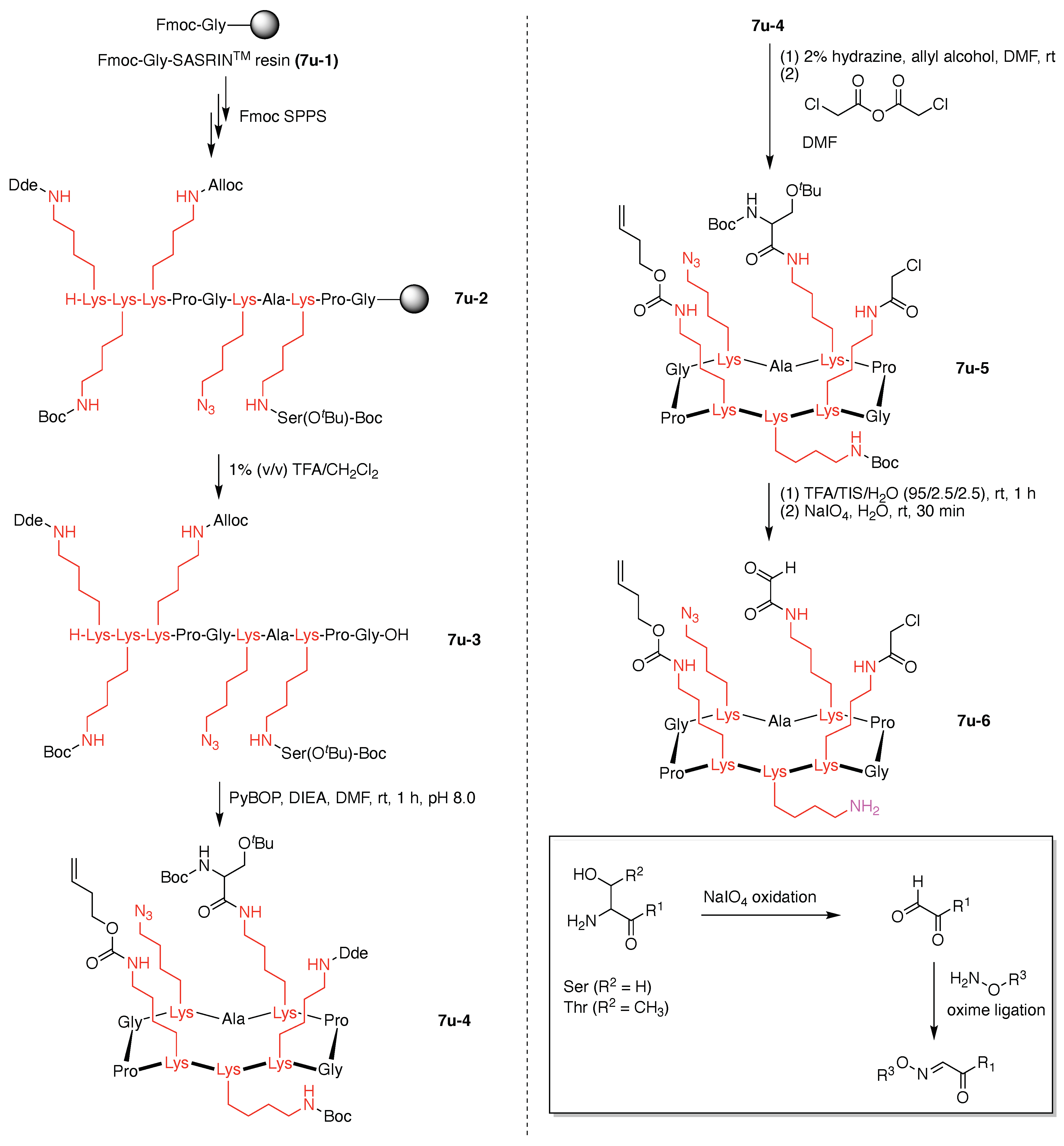{kind=link}
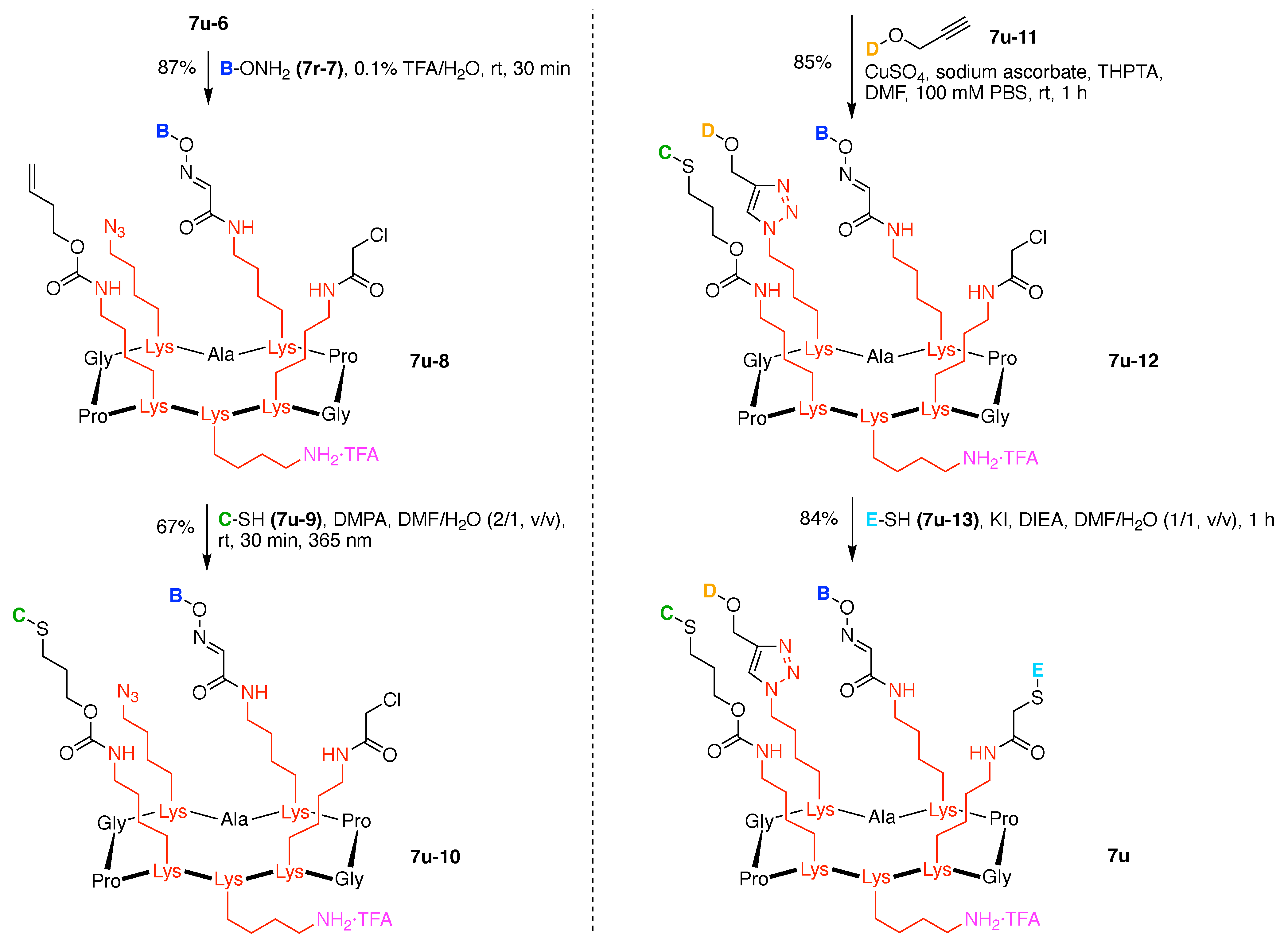{kind=link}
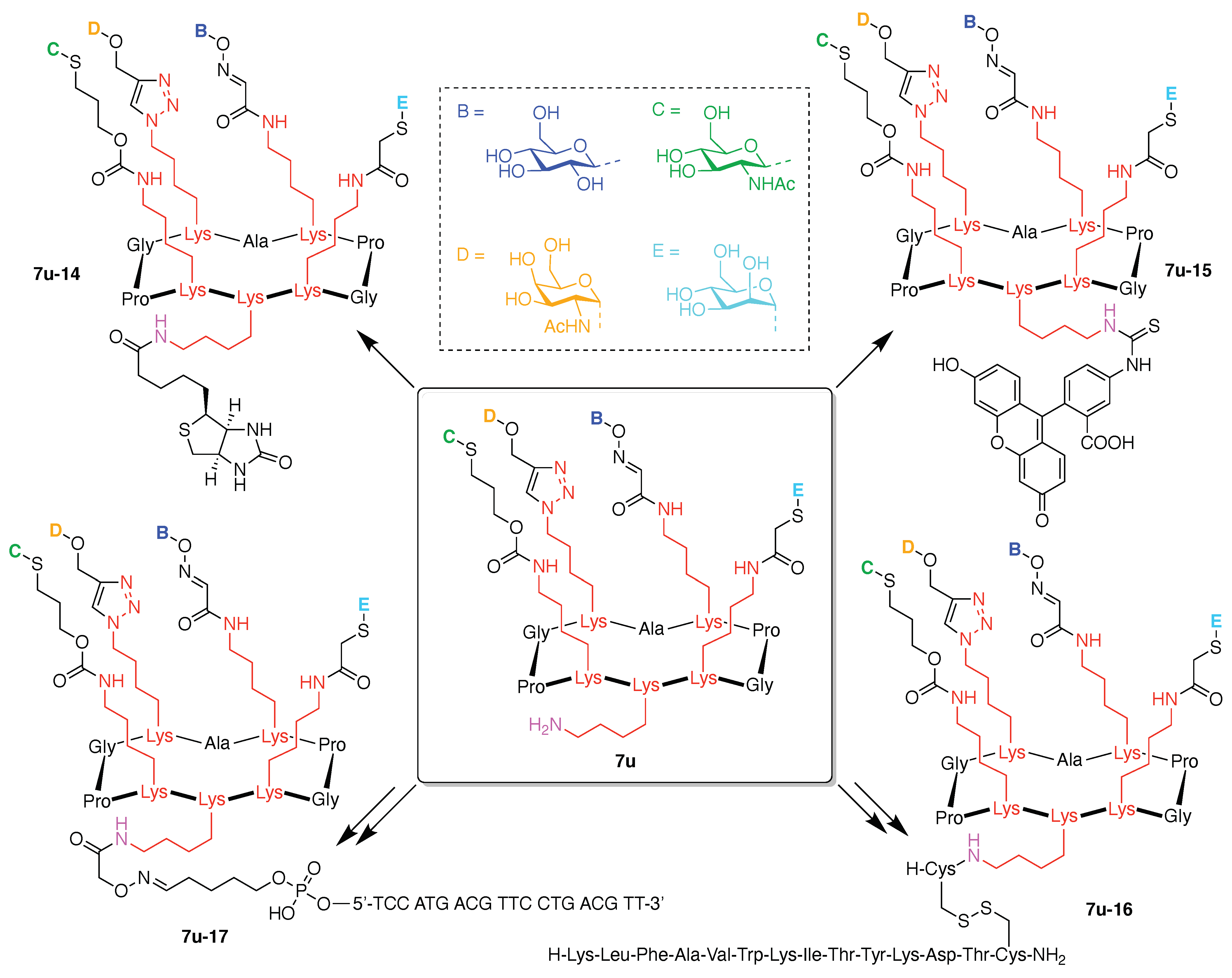{kind=link}
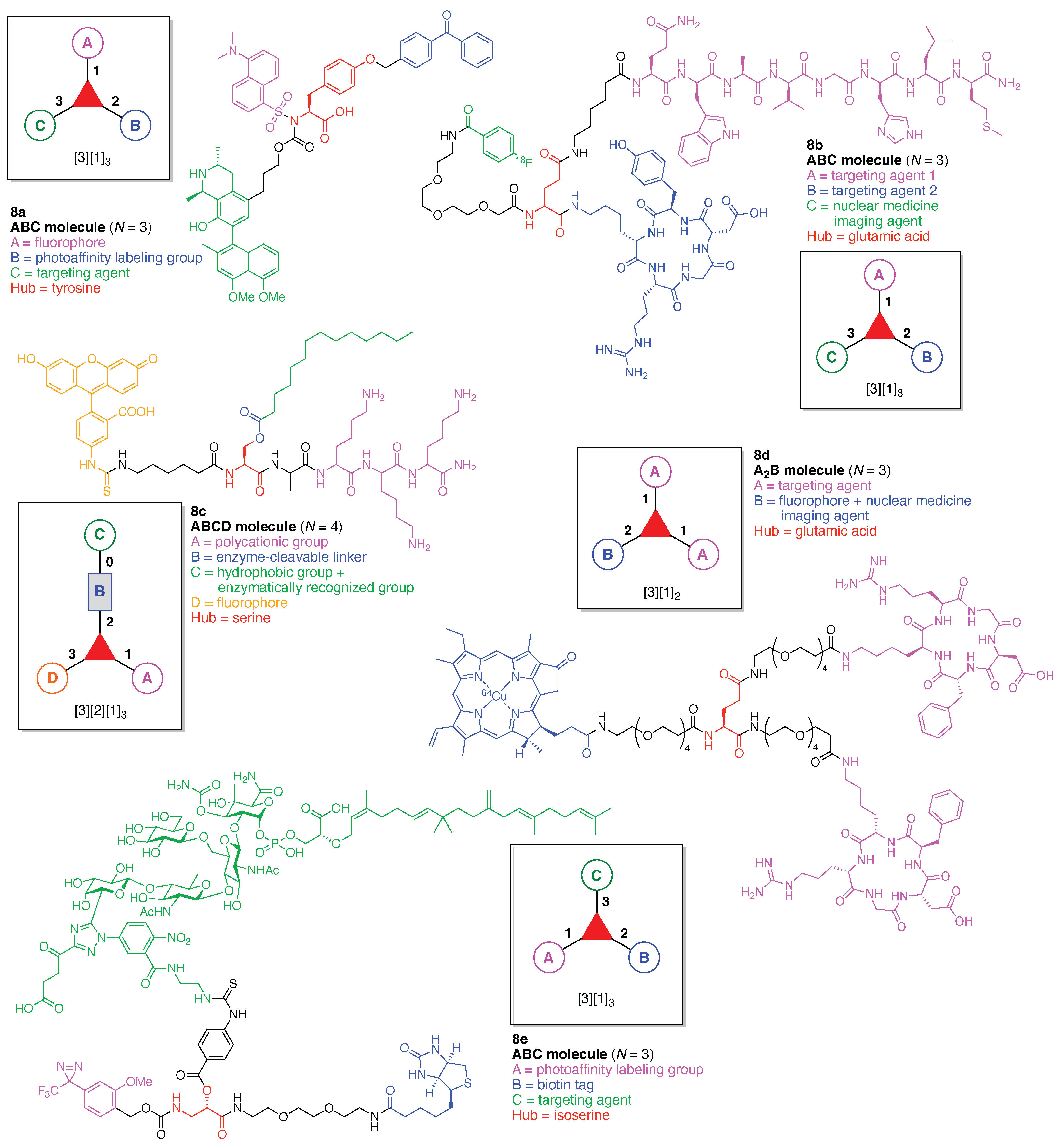{kind=link}
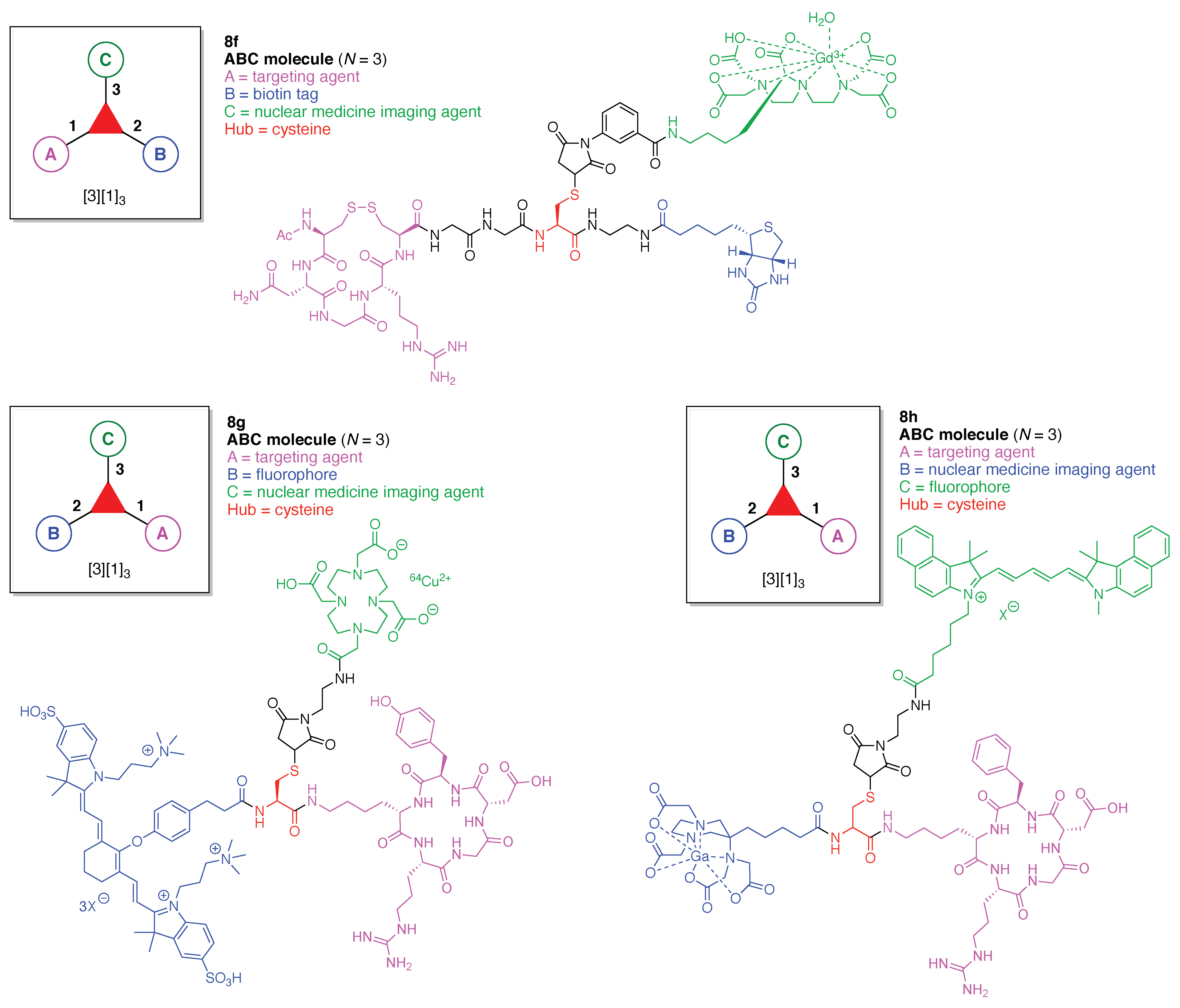{kind=link}
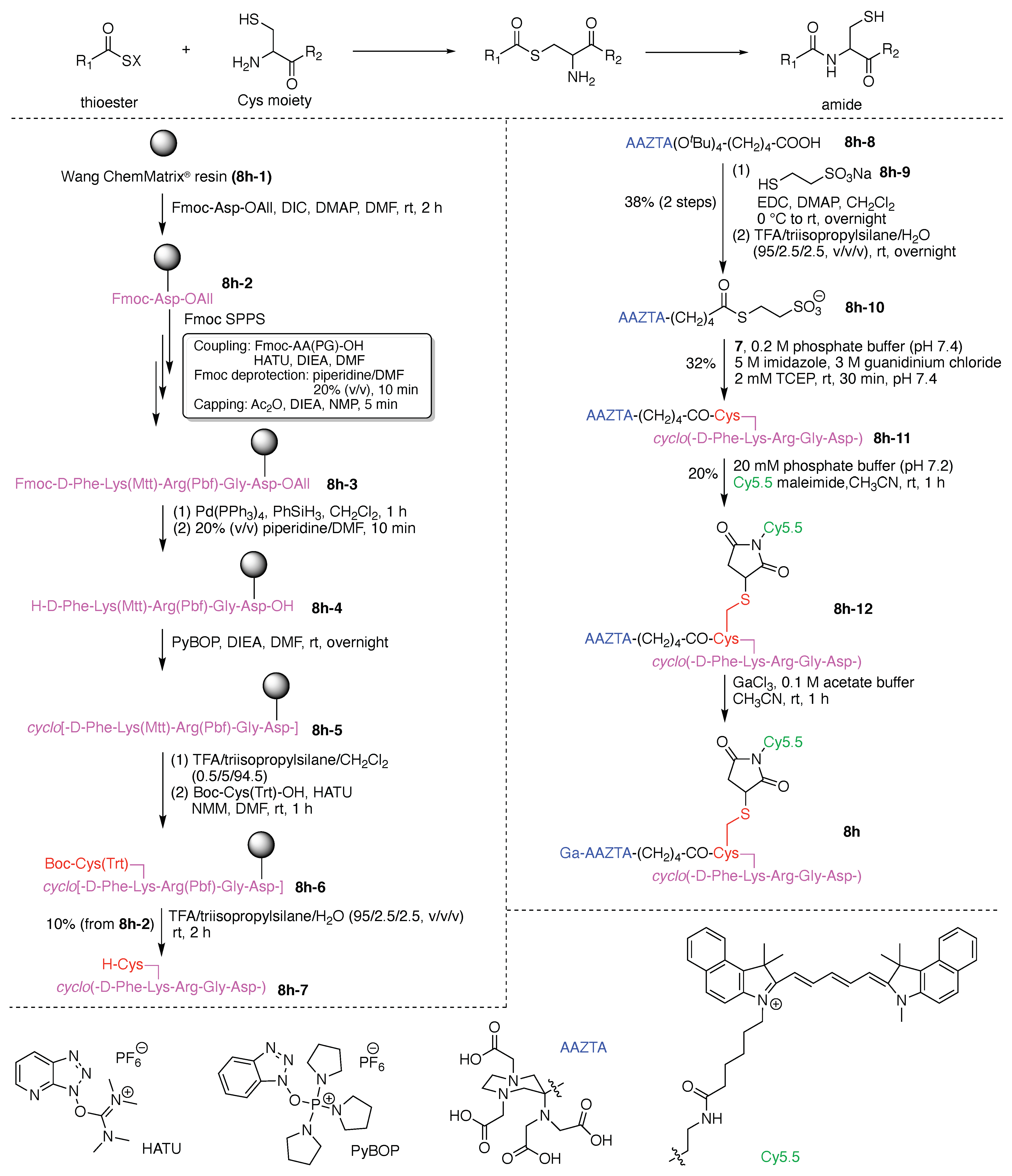{kind=link}
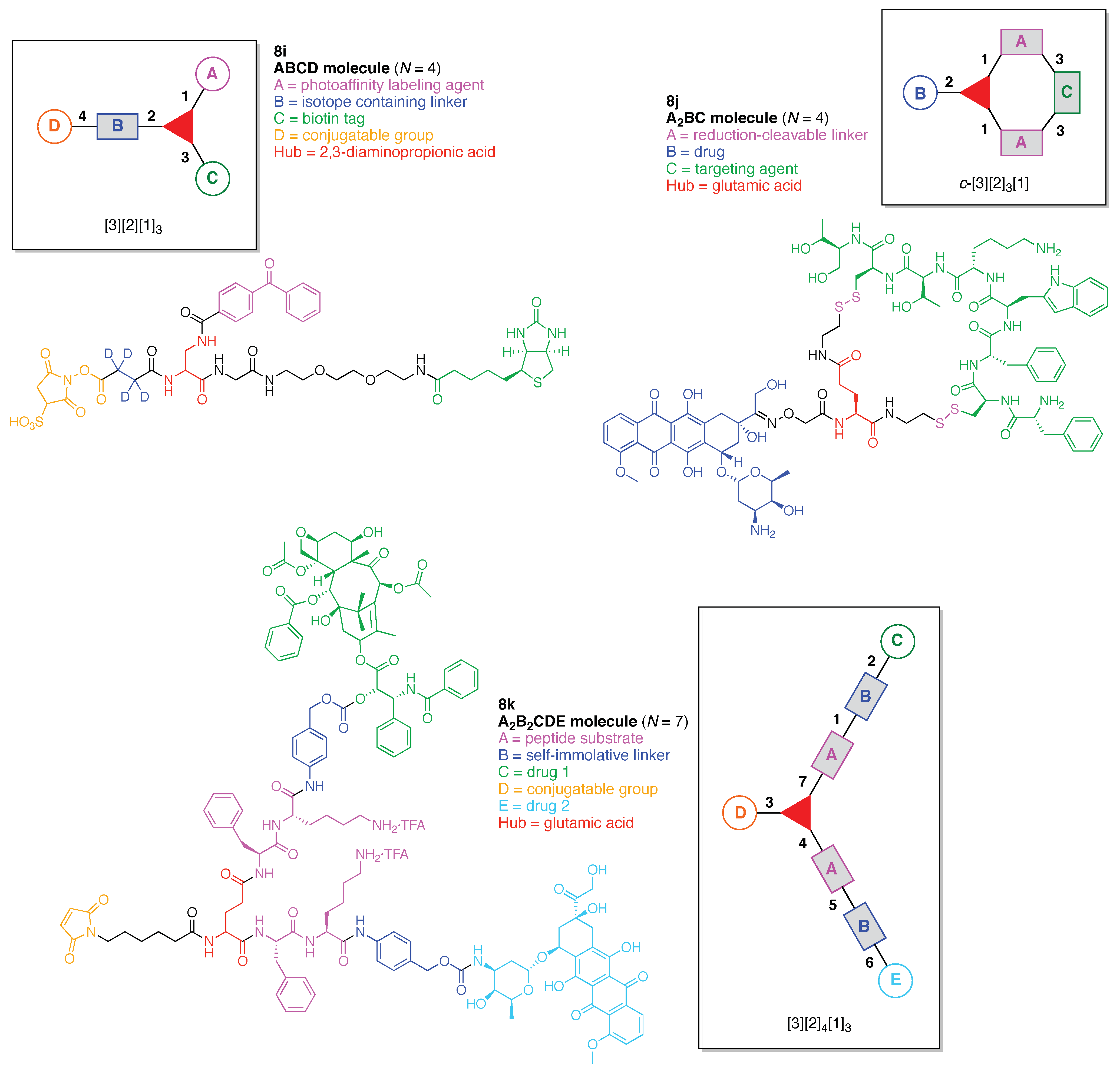{kind=link}
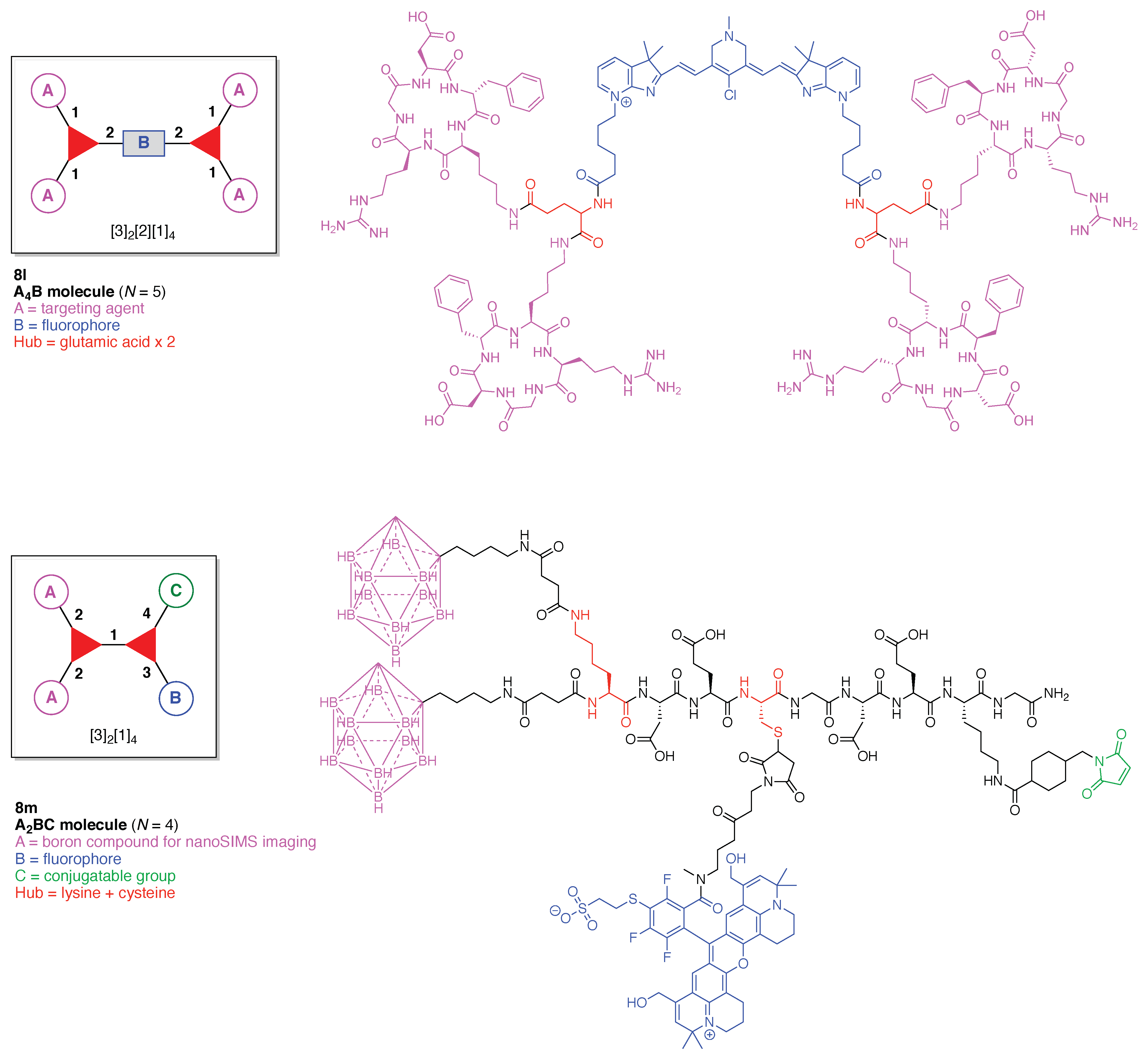{kind=link}
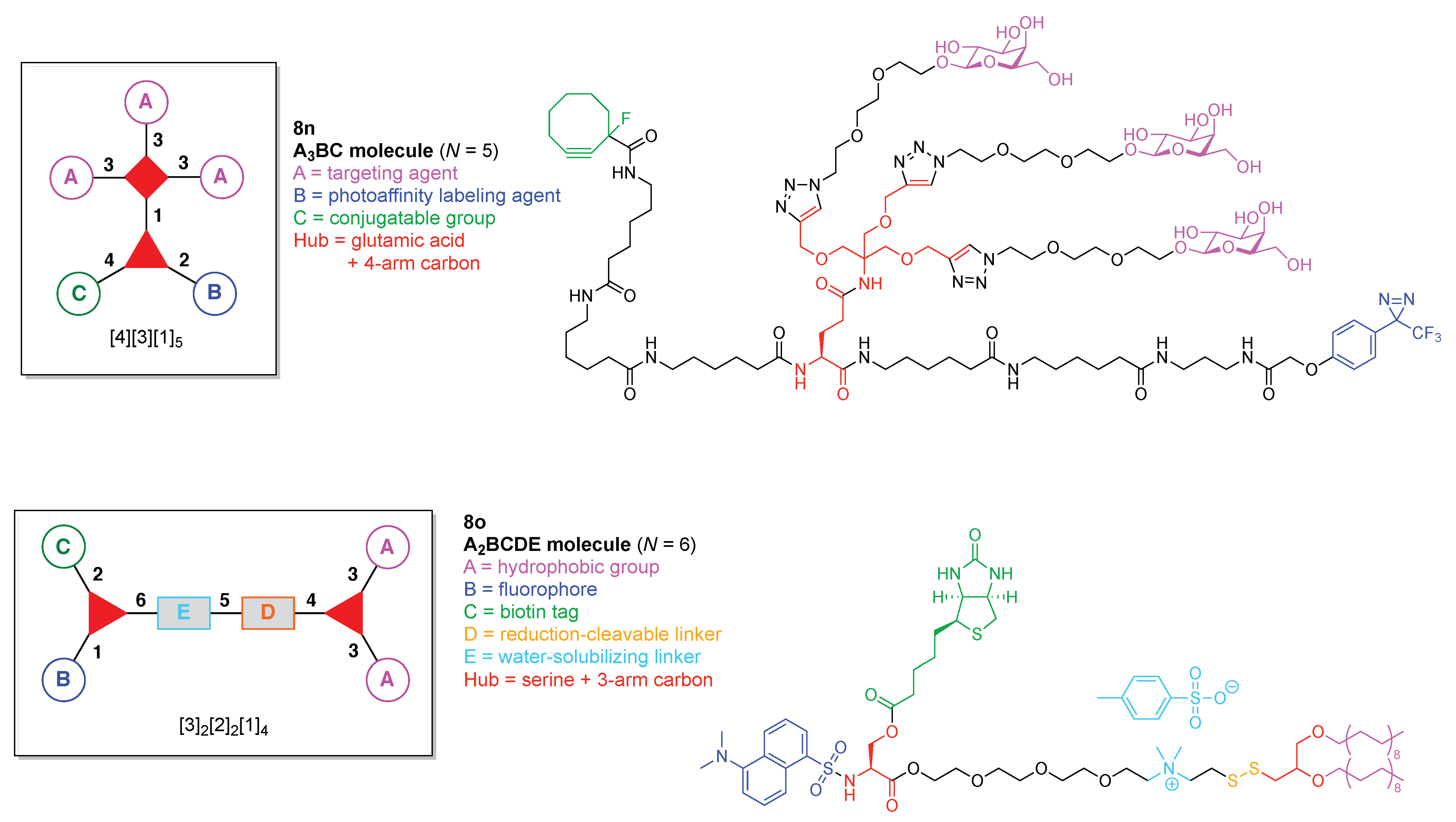{kind=link}
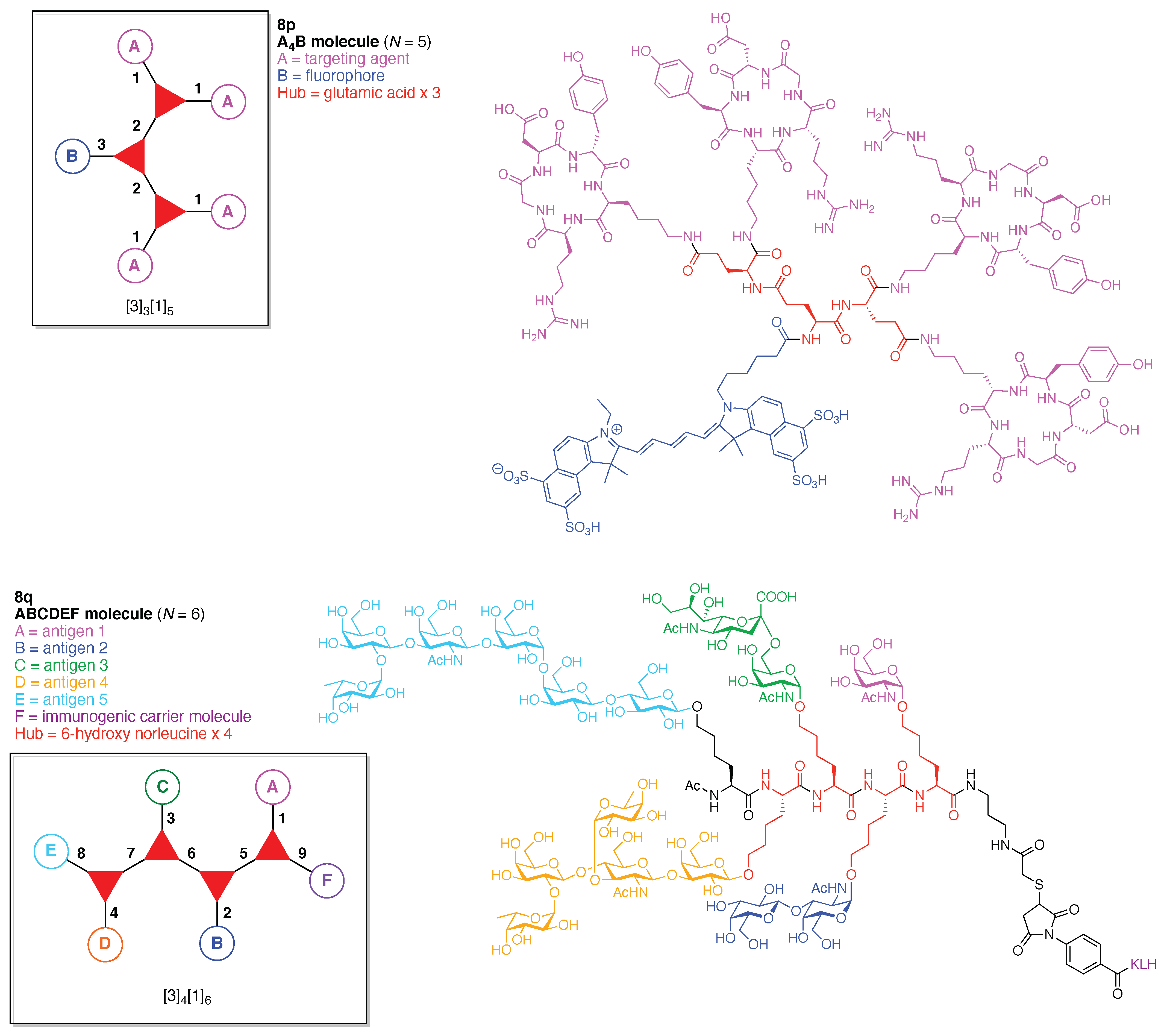{kind=link}
{kind=link}
{kind=link}
{kind=link}
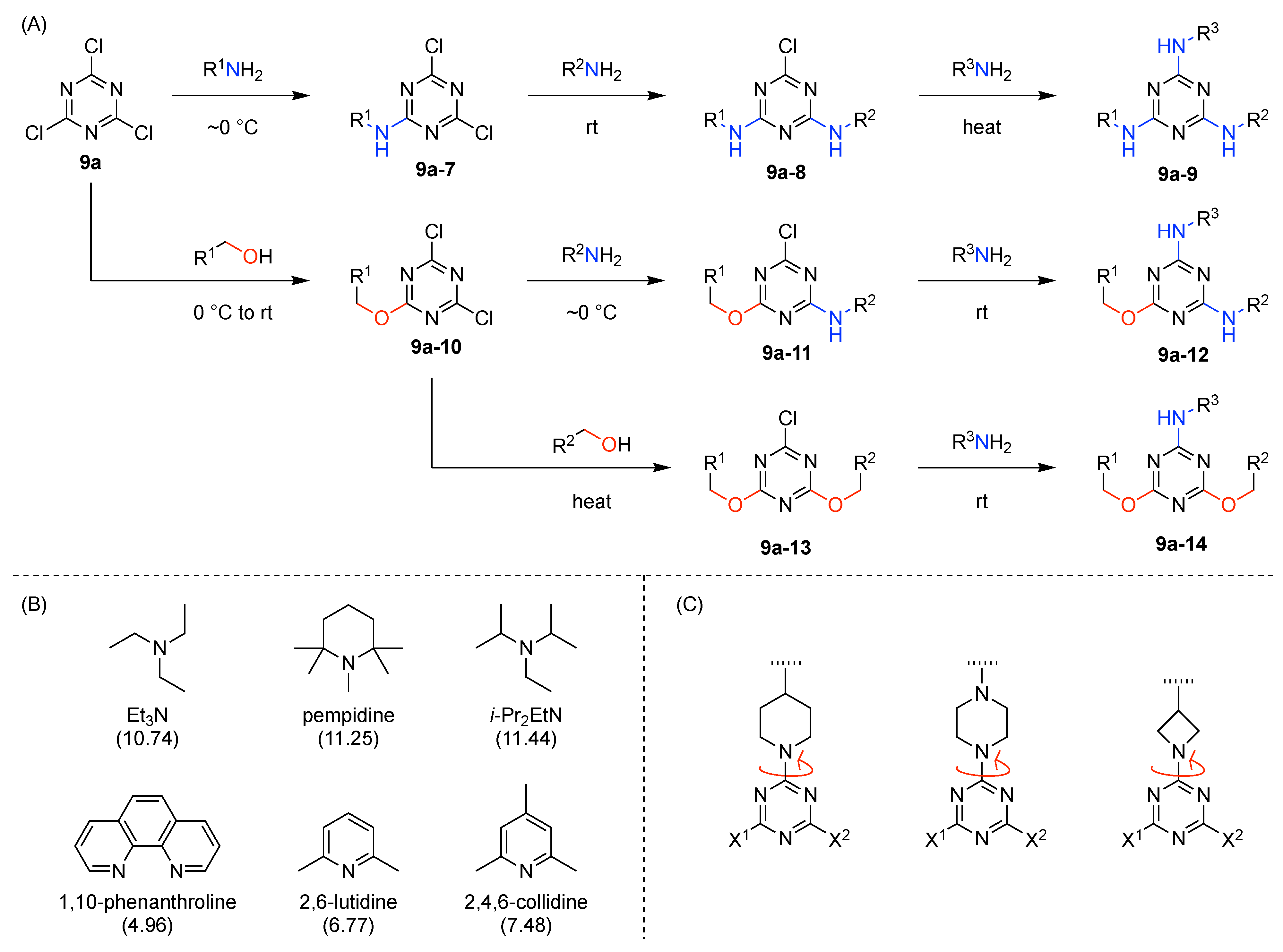{kind=link}
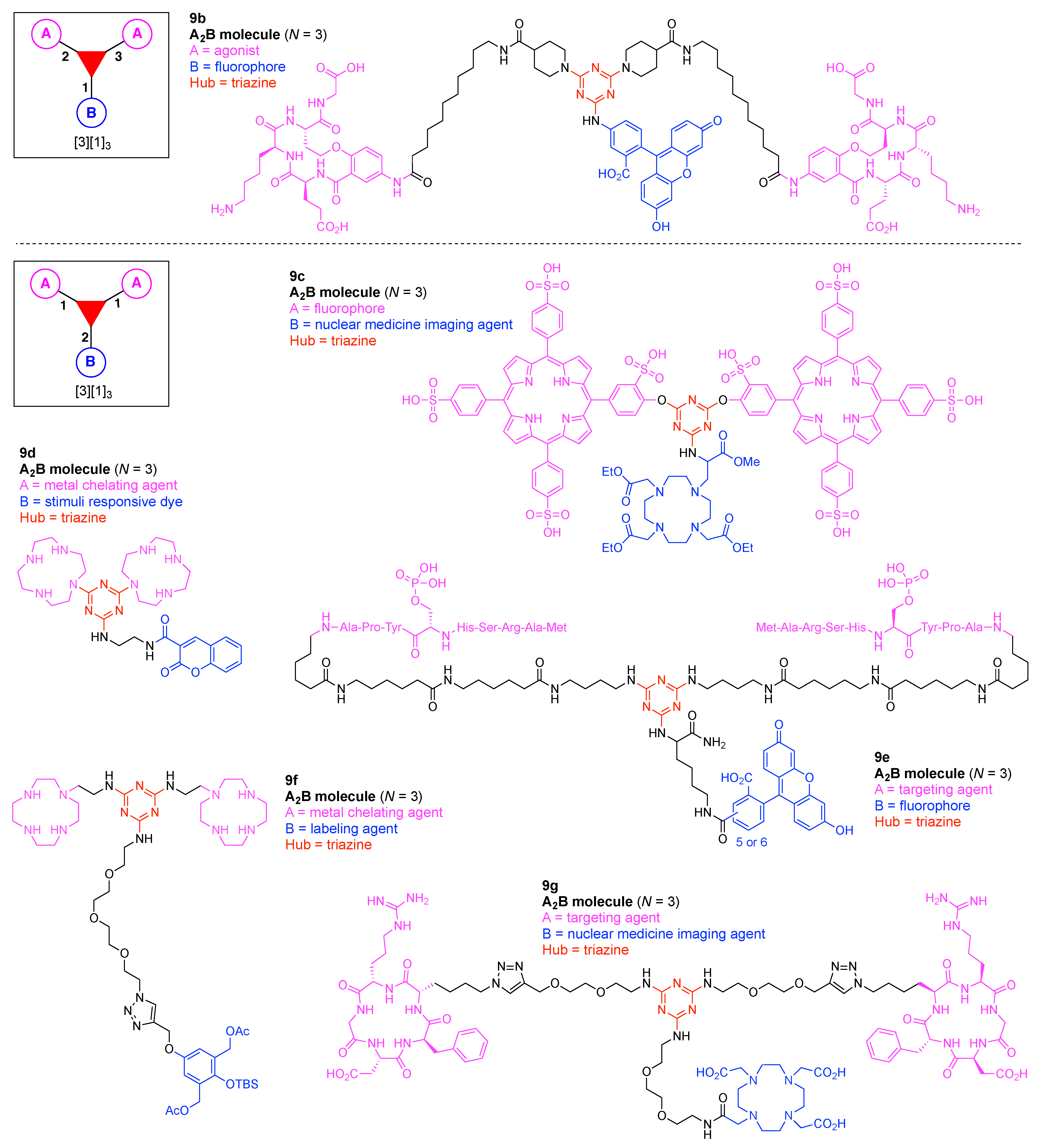{kind=link}
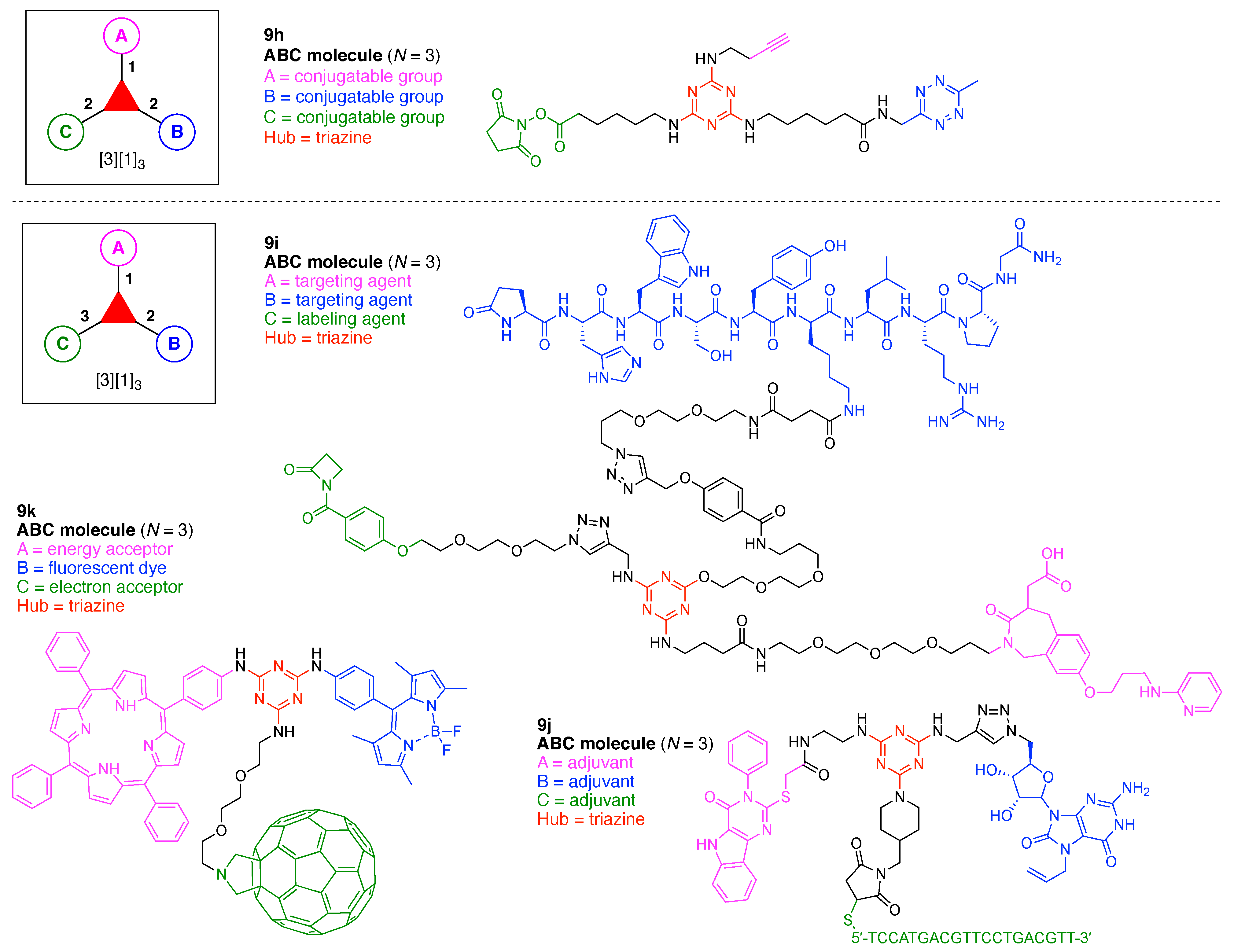{kind=link}
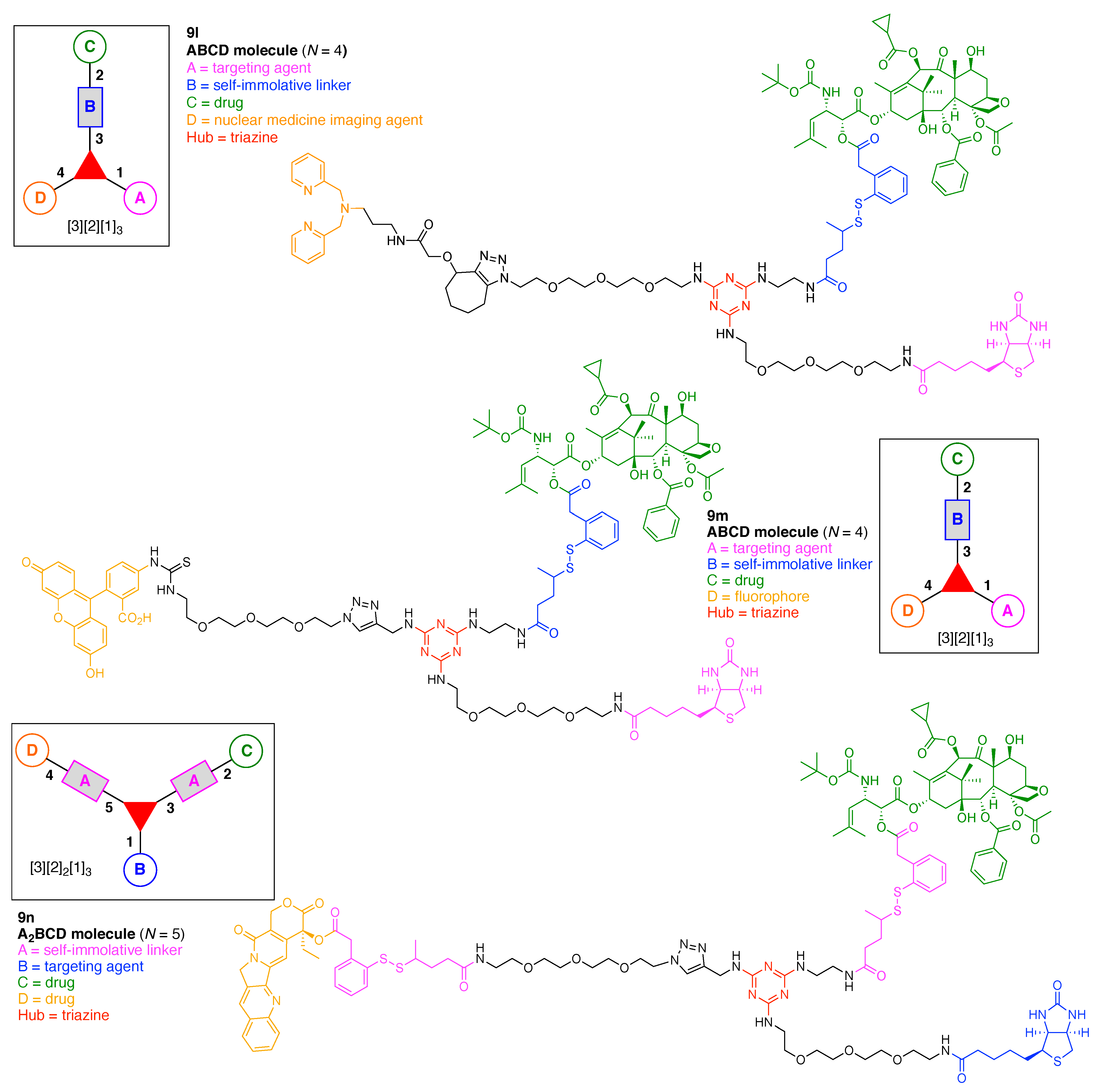{kind=link}
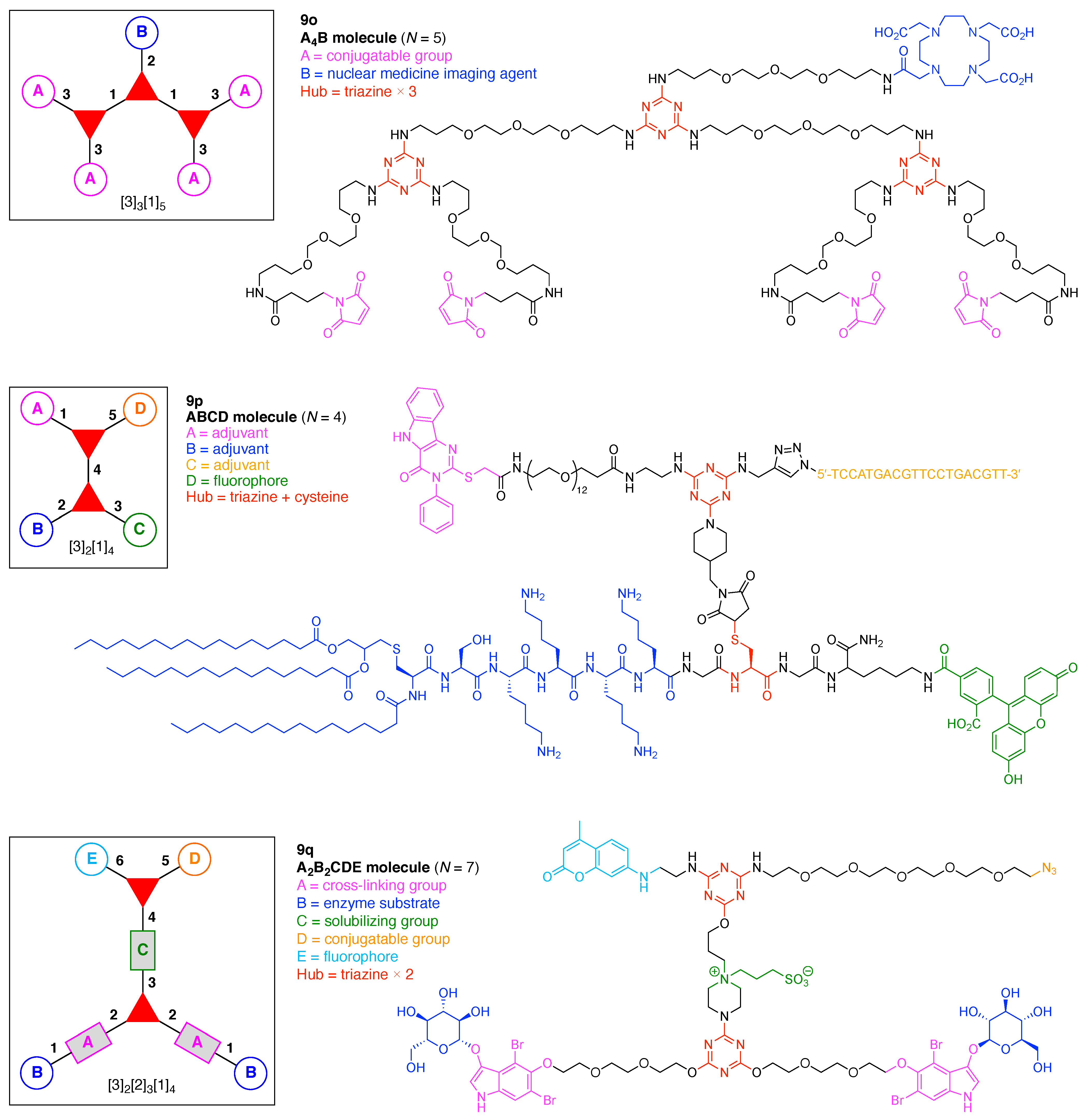{kind=link}
{kind=link}
{kind=link}
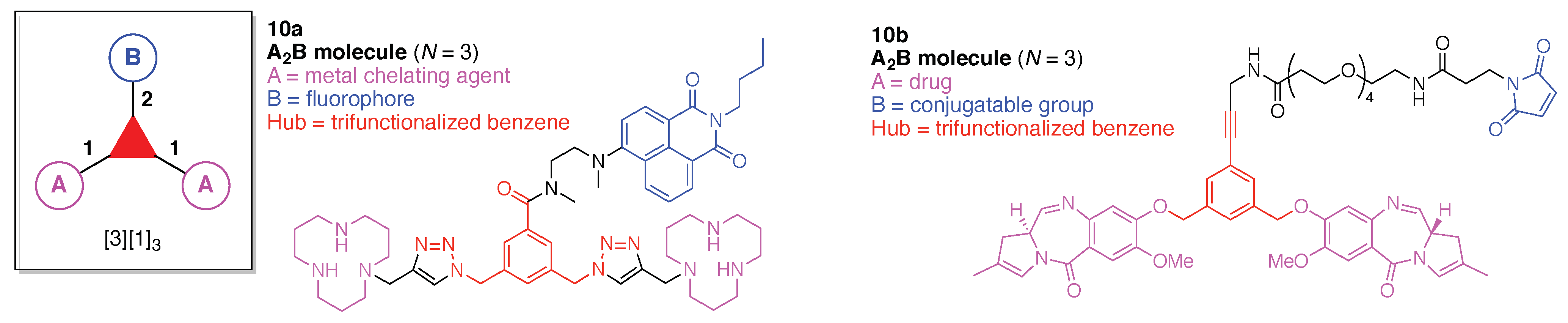{kind=link}
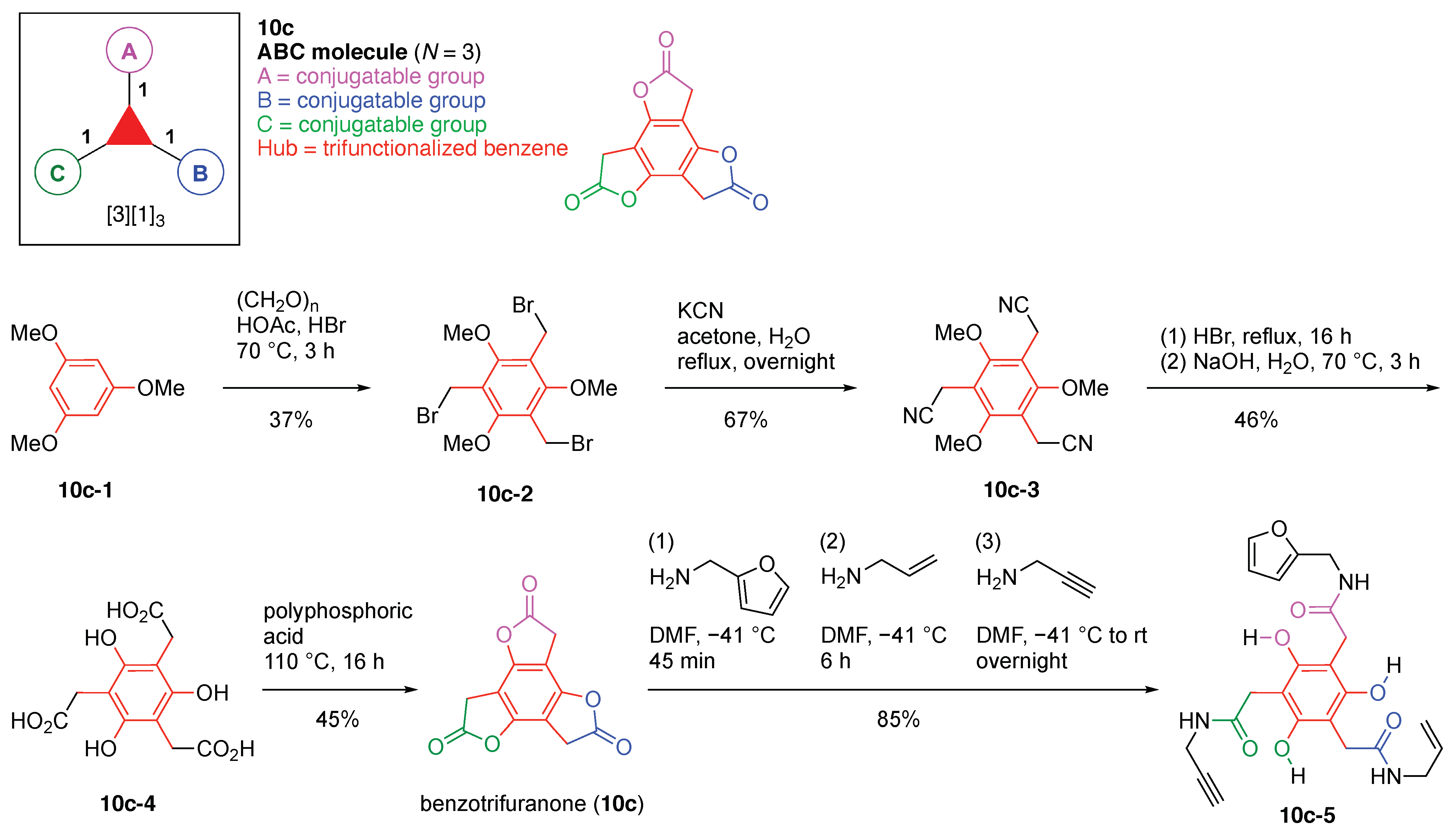{kind=link}
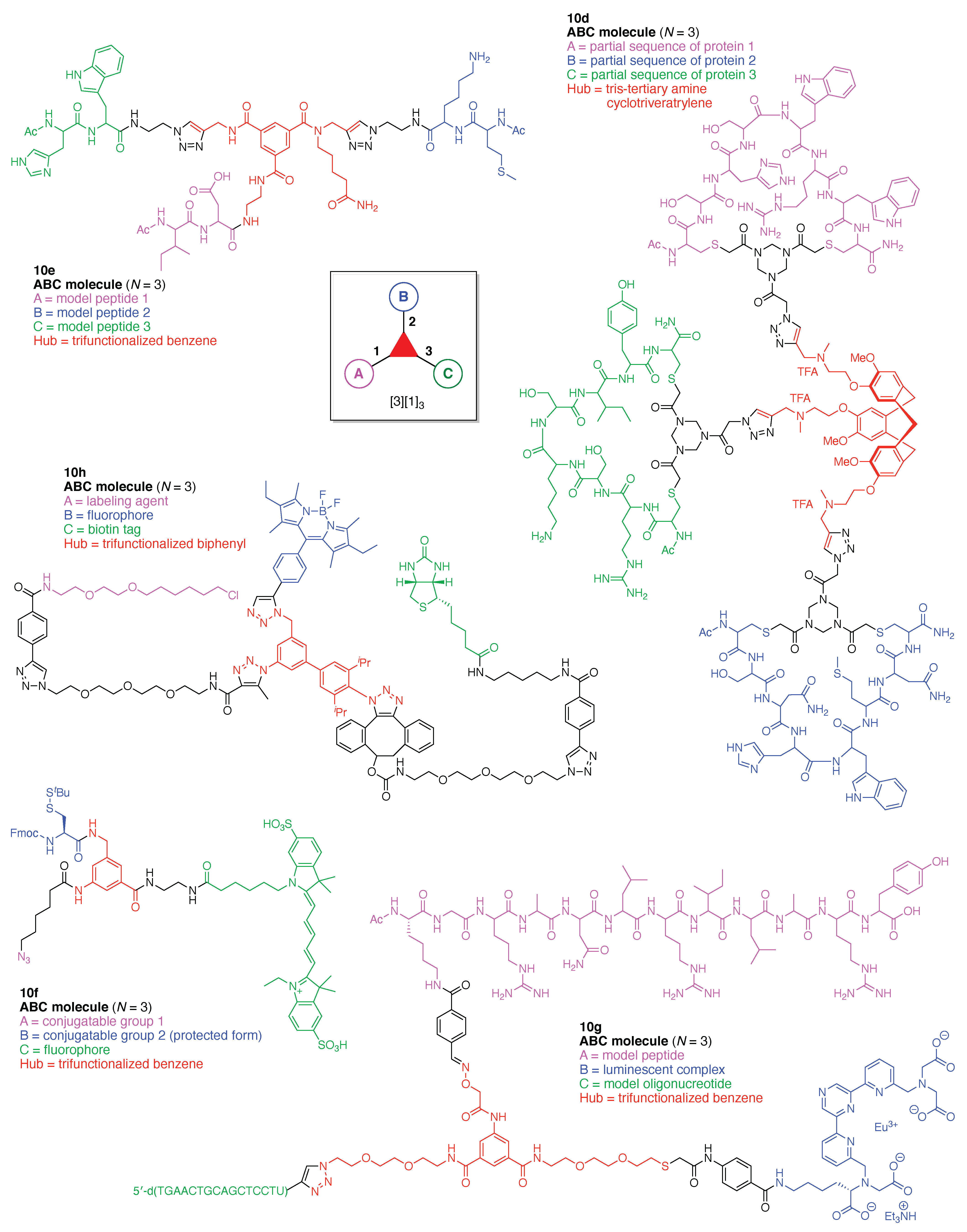{kind=link}
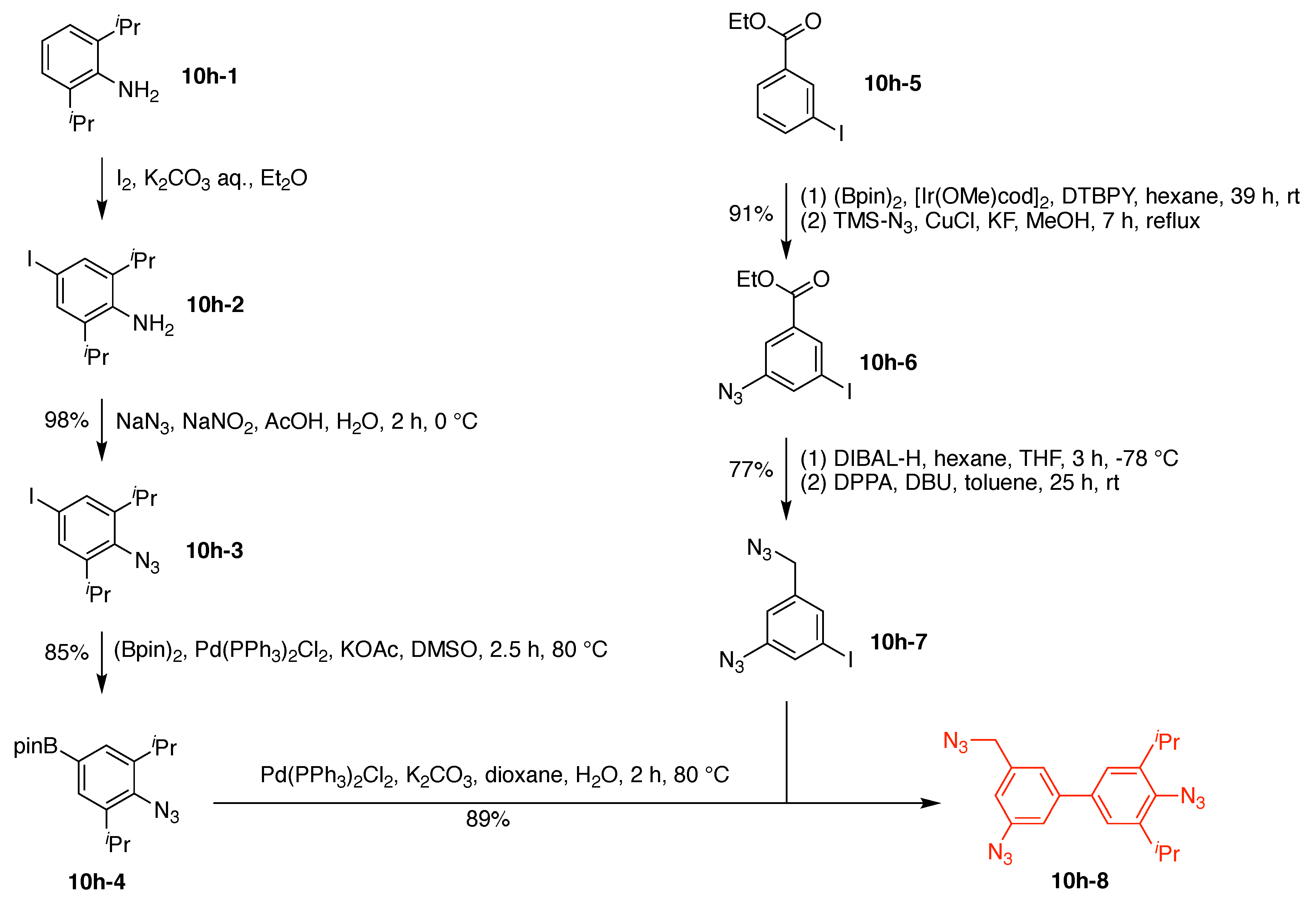{kind=link}
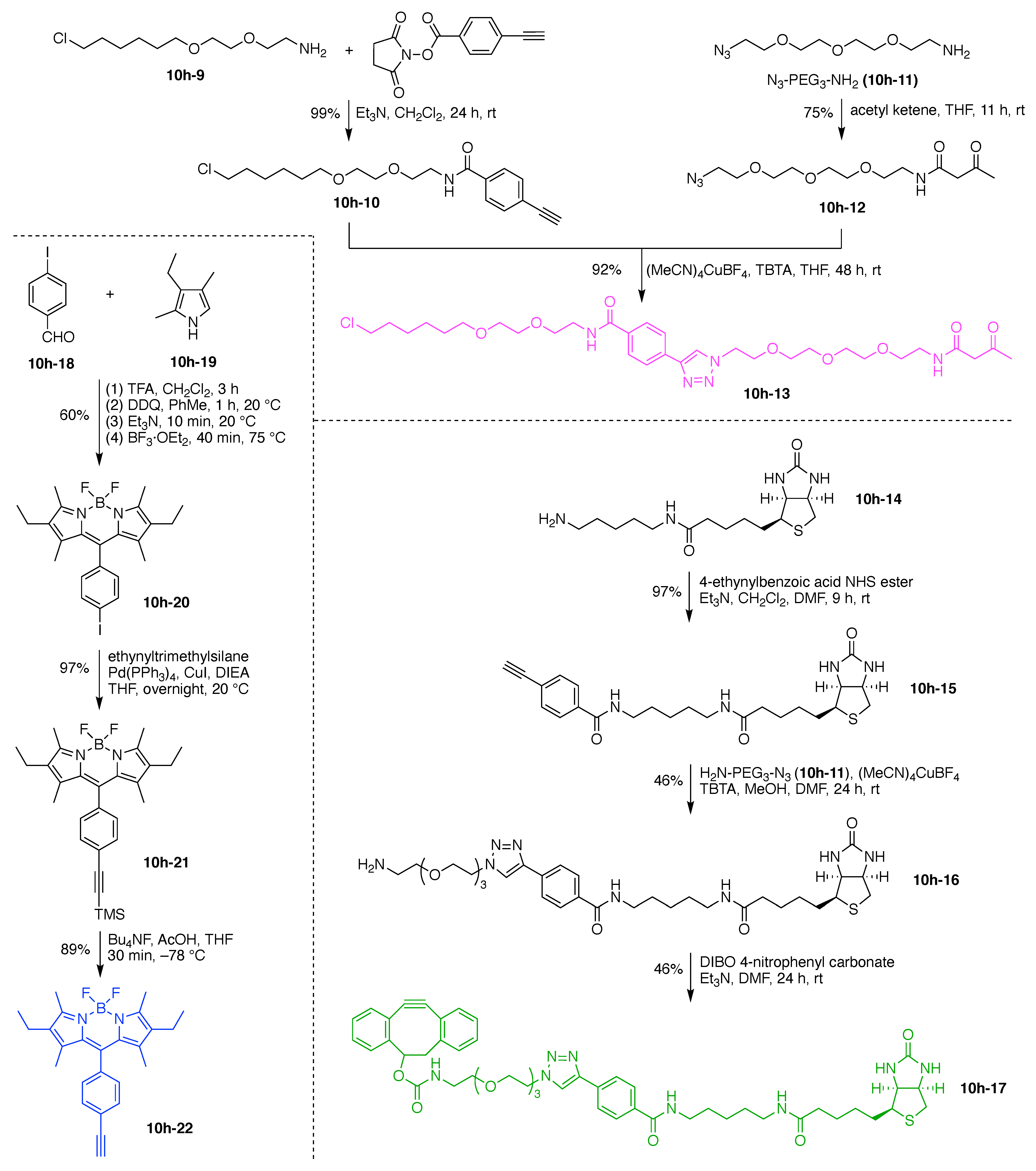{kind=link}
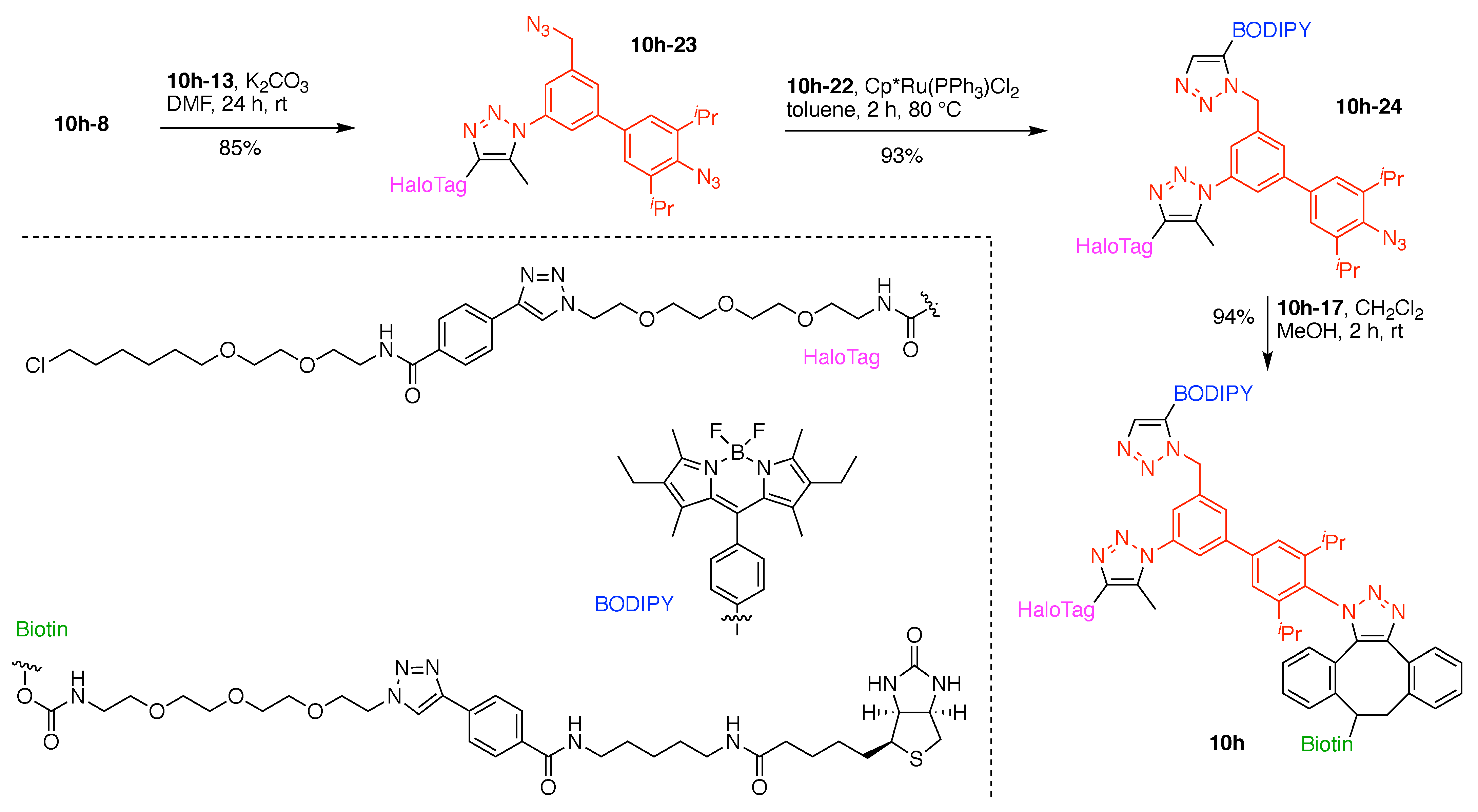{kind=link}
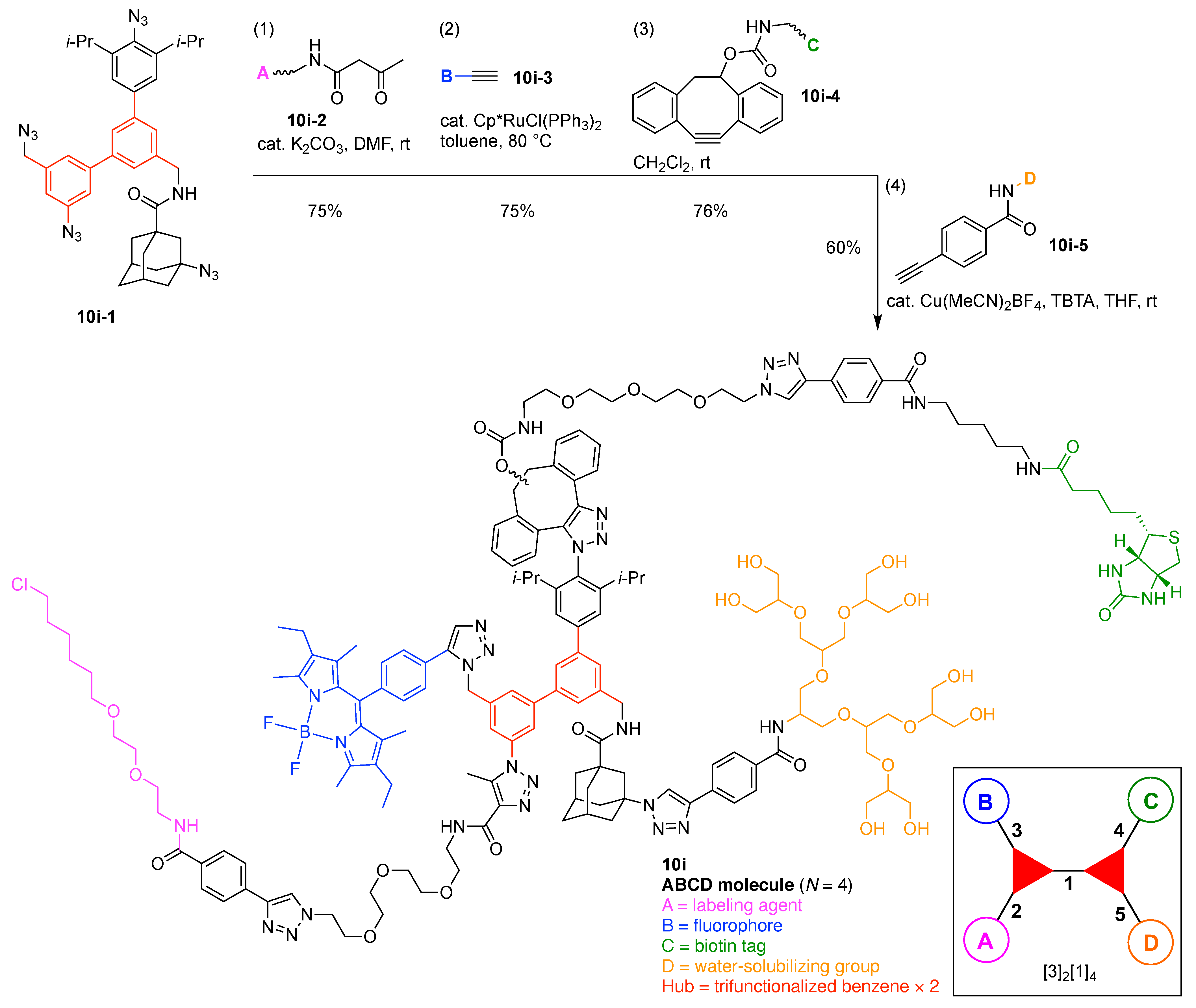{kind=link}
{kind=link}
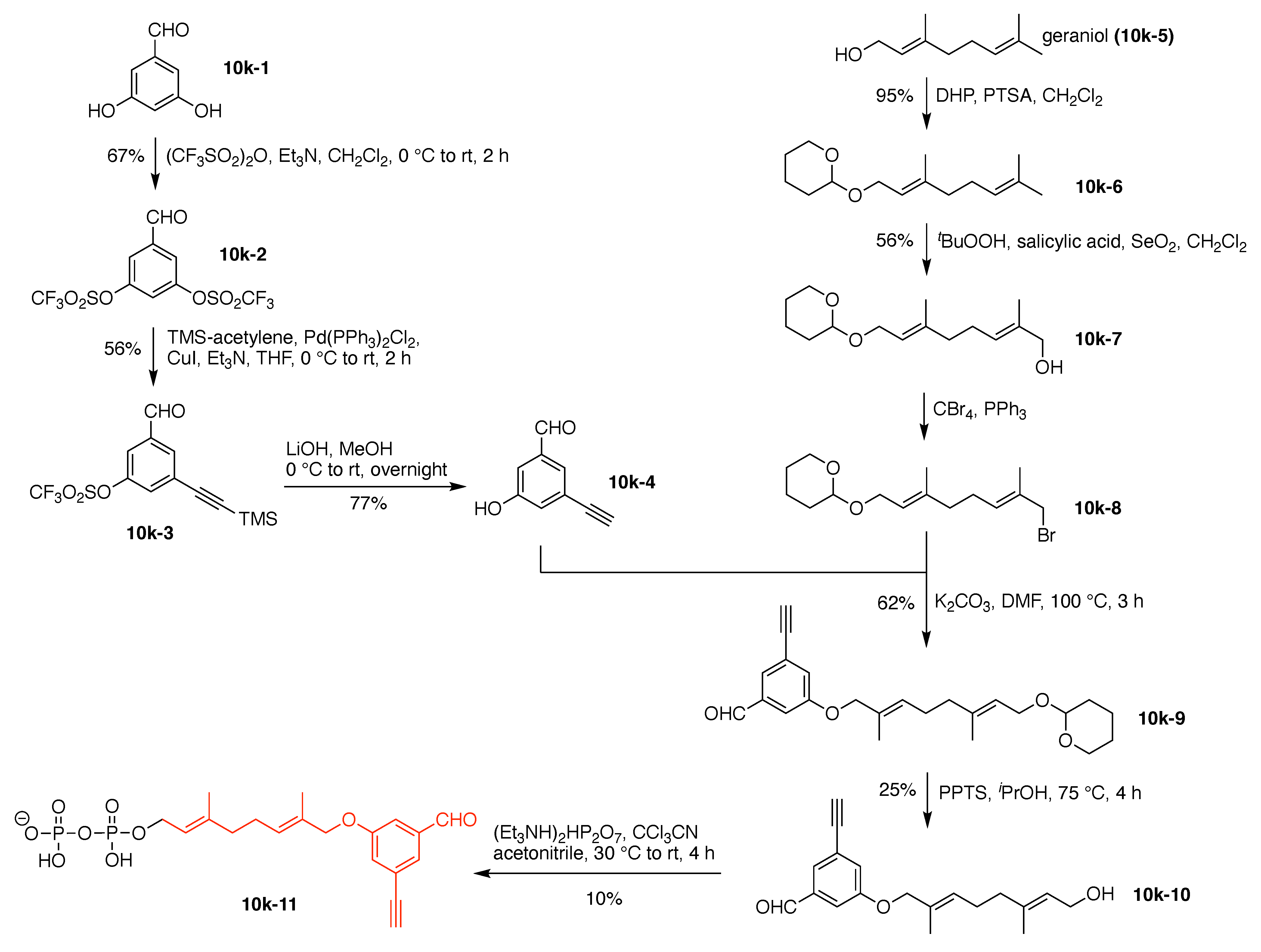{kind=link}
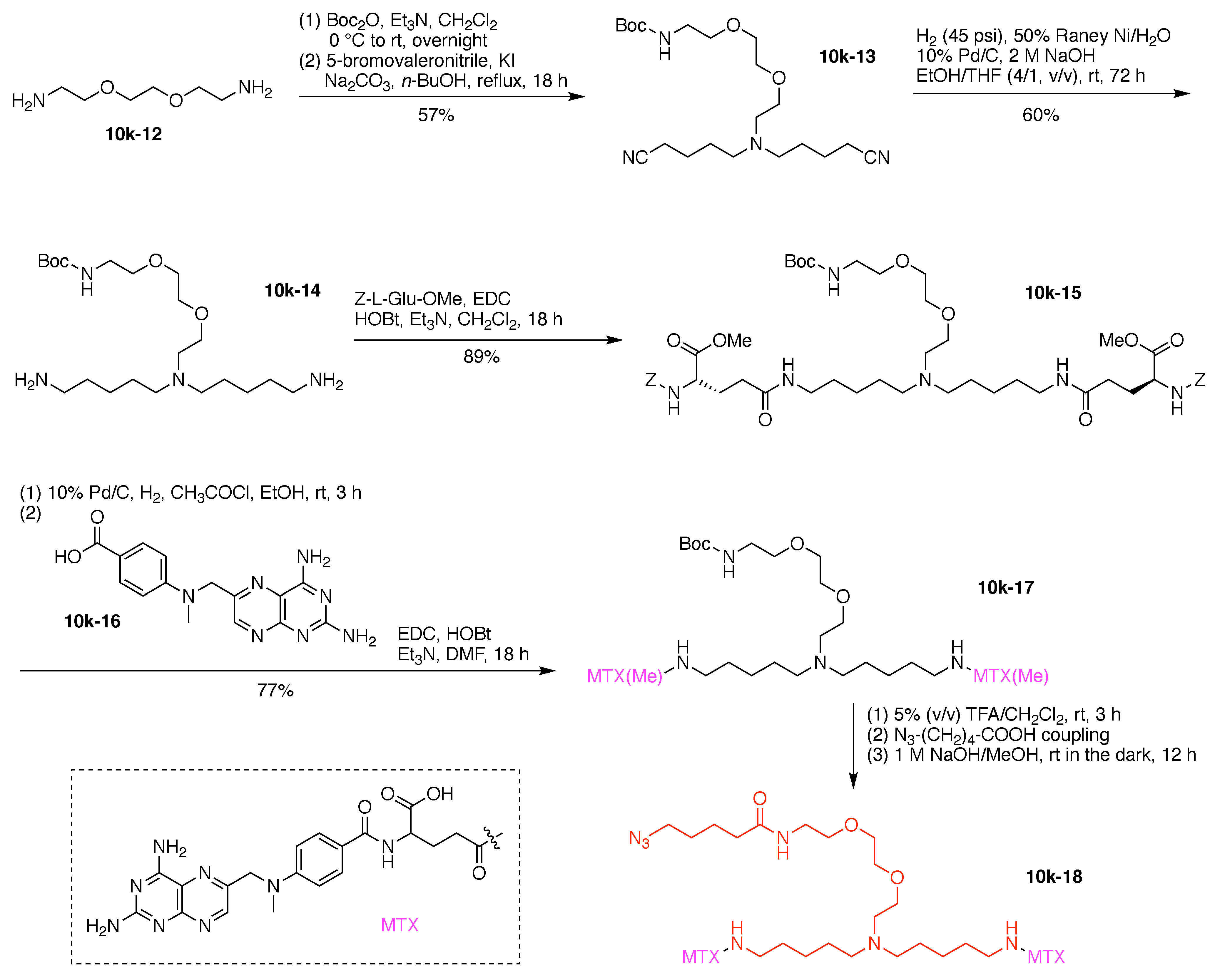{kind=link}
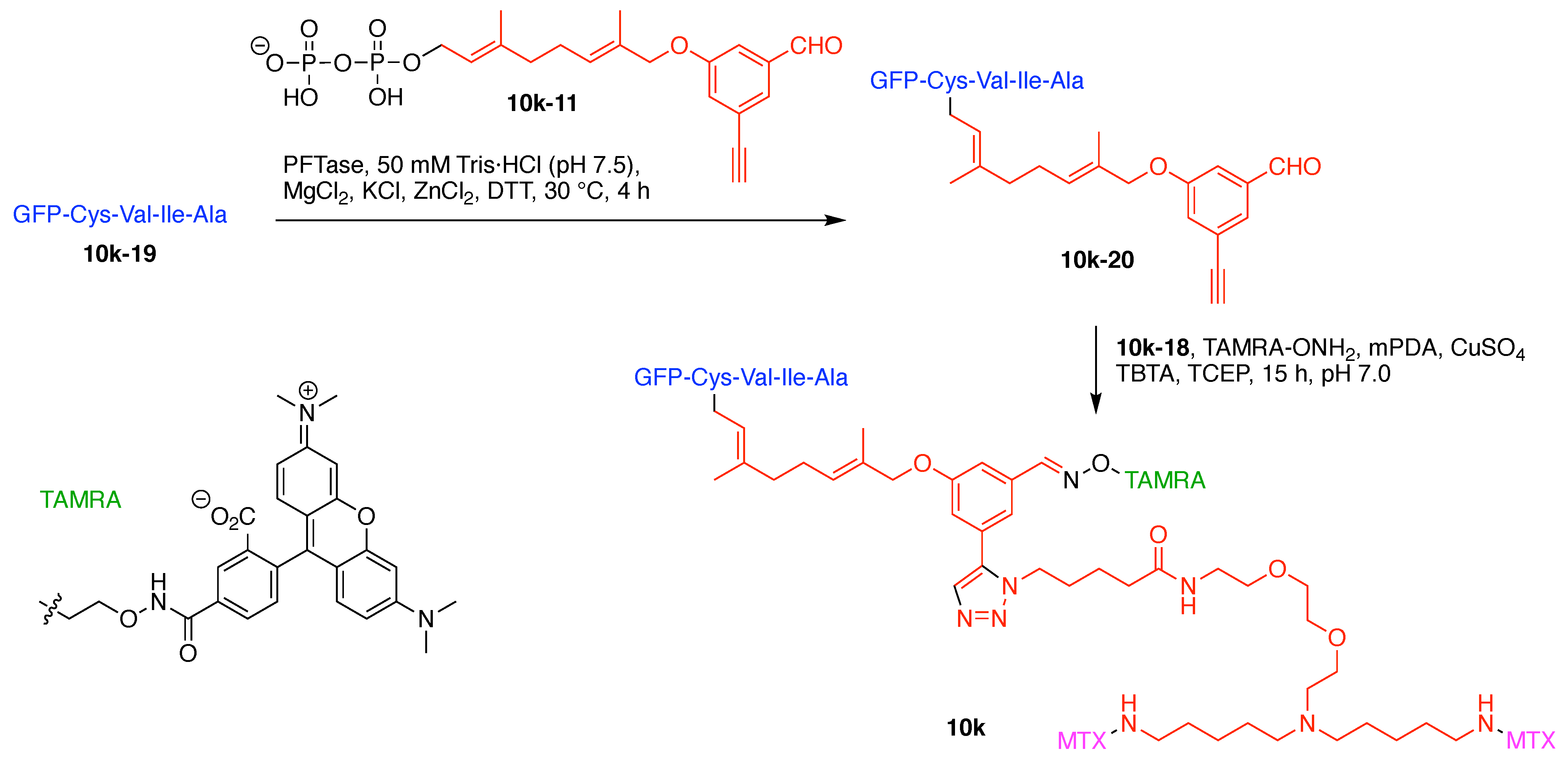{kind=link}
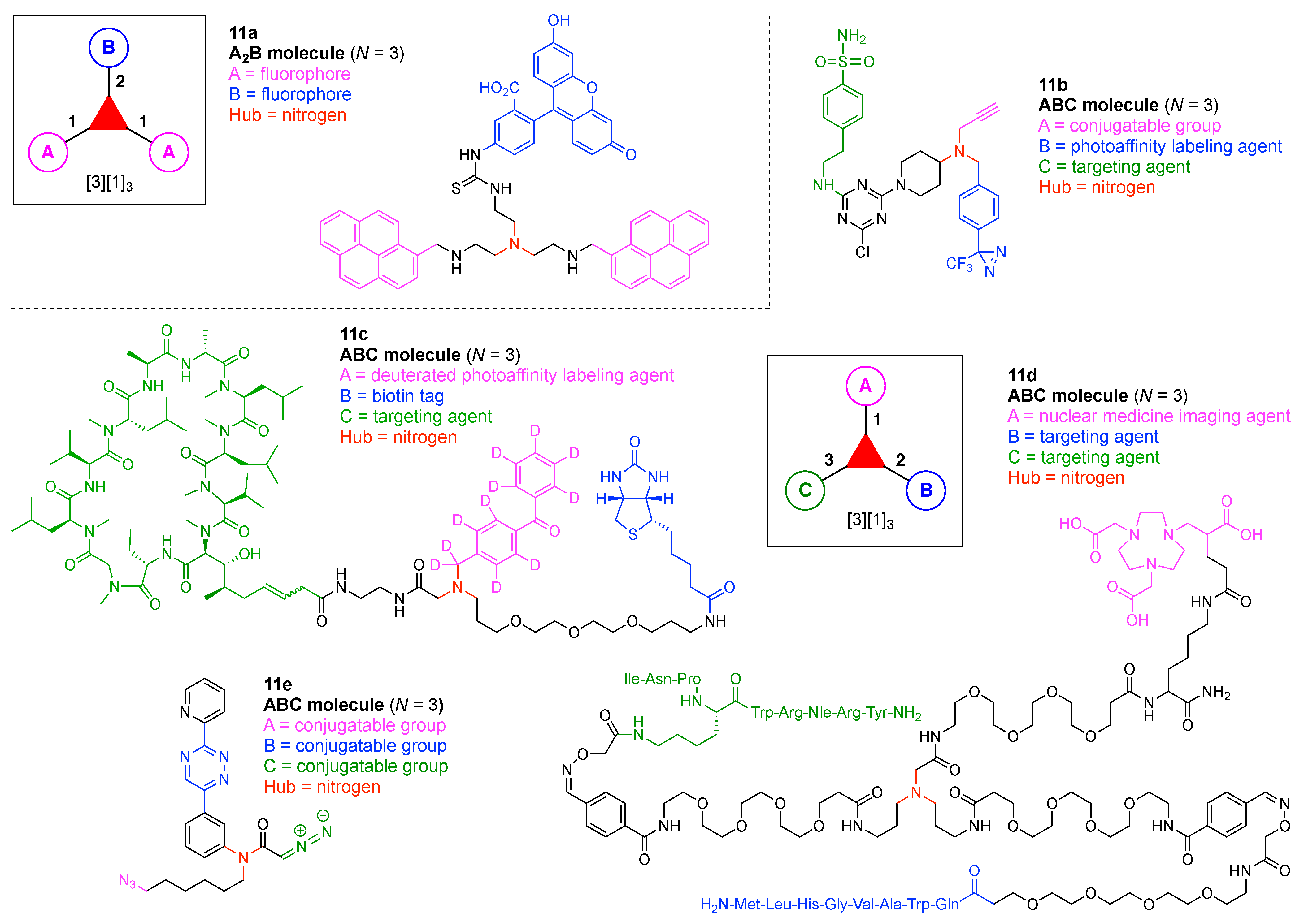{kind=link}
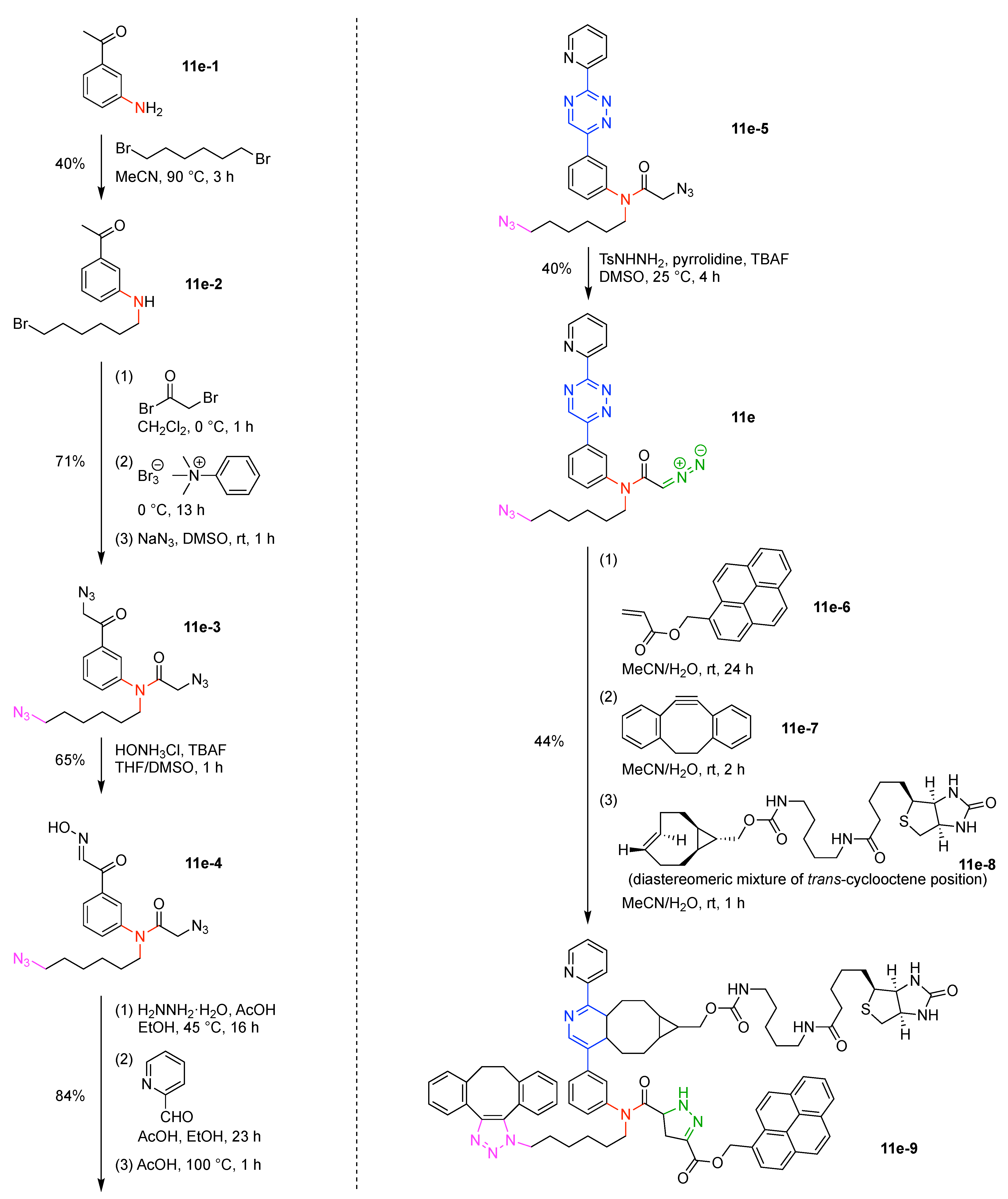{kind=link}
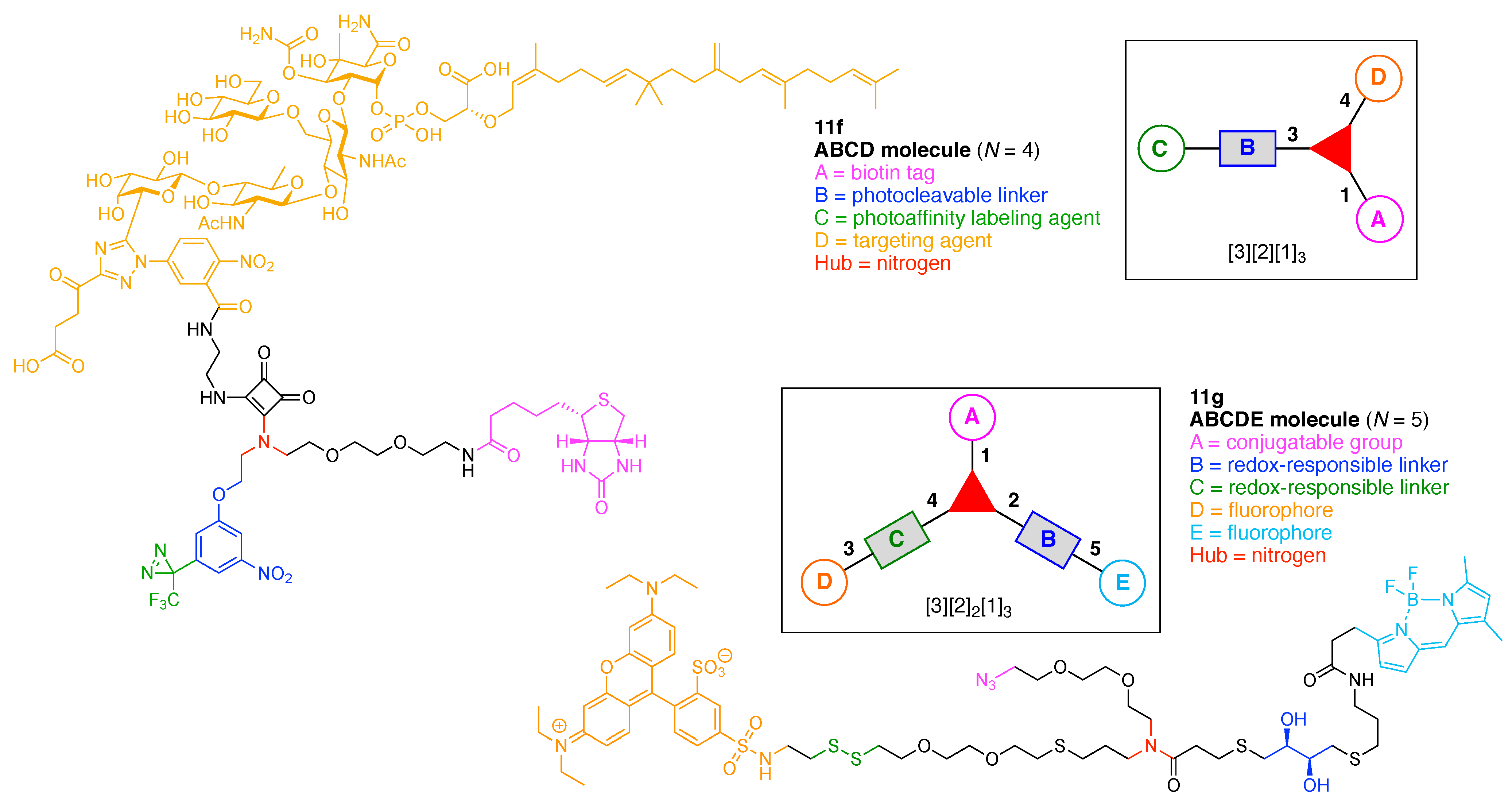{kind=link}
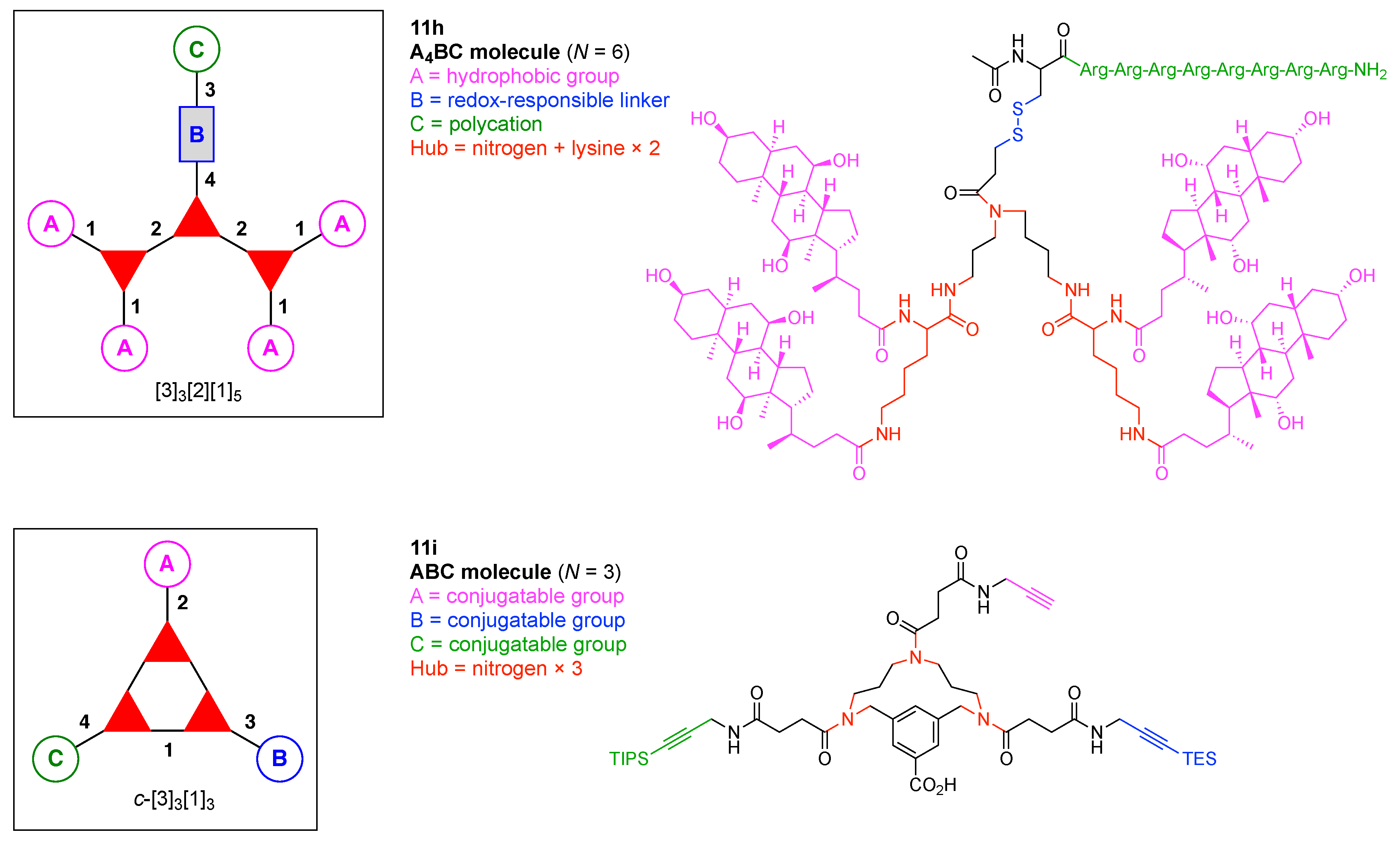{kind=link}
{kind=link}
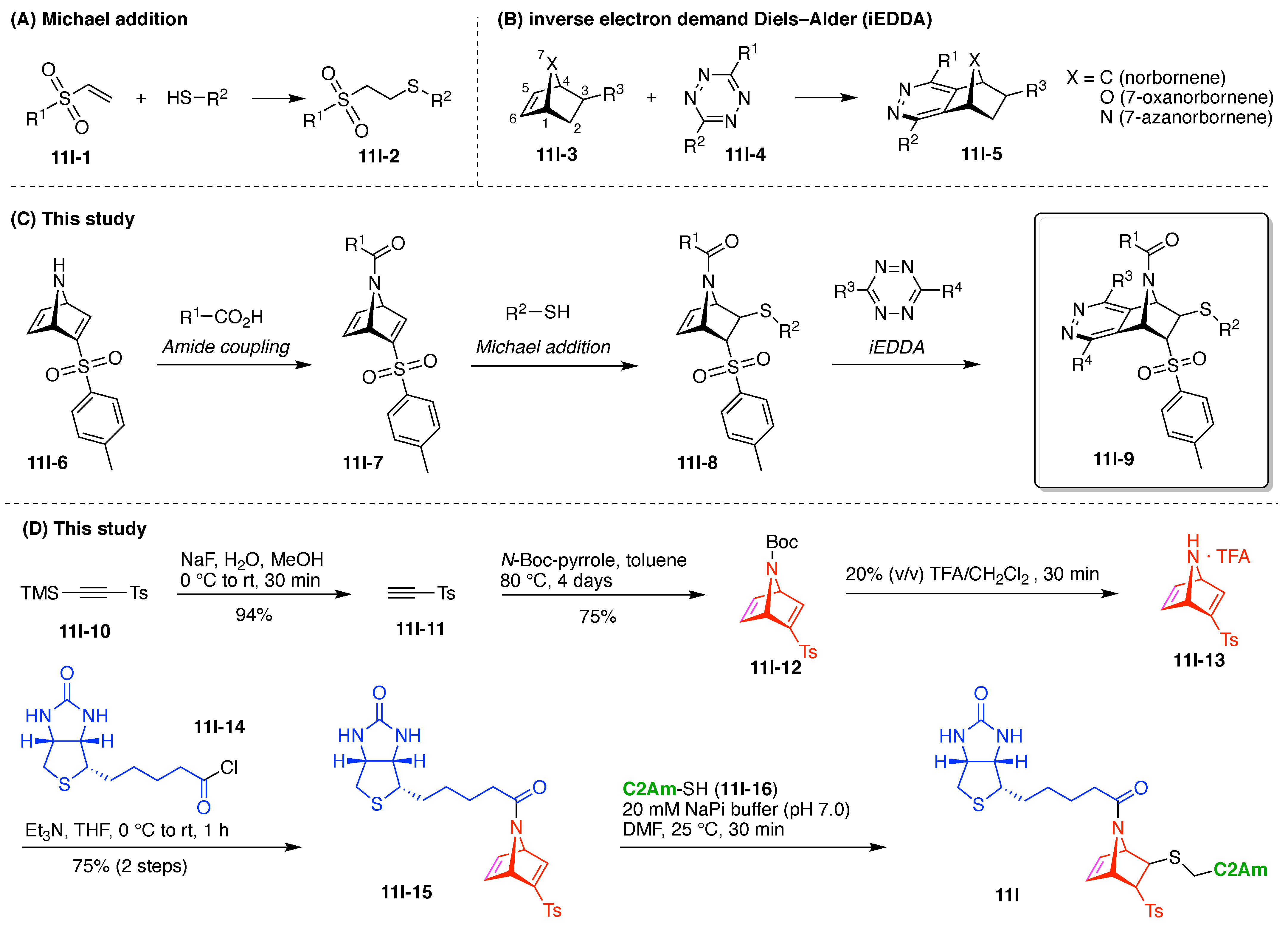{kind=link}
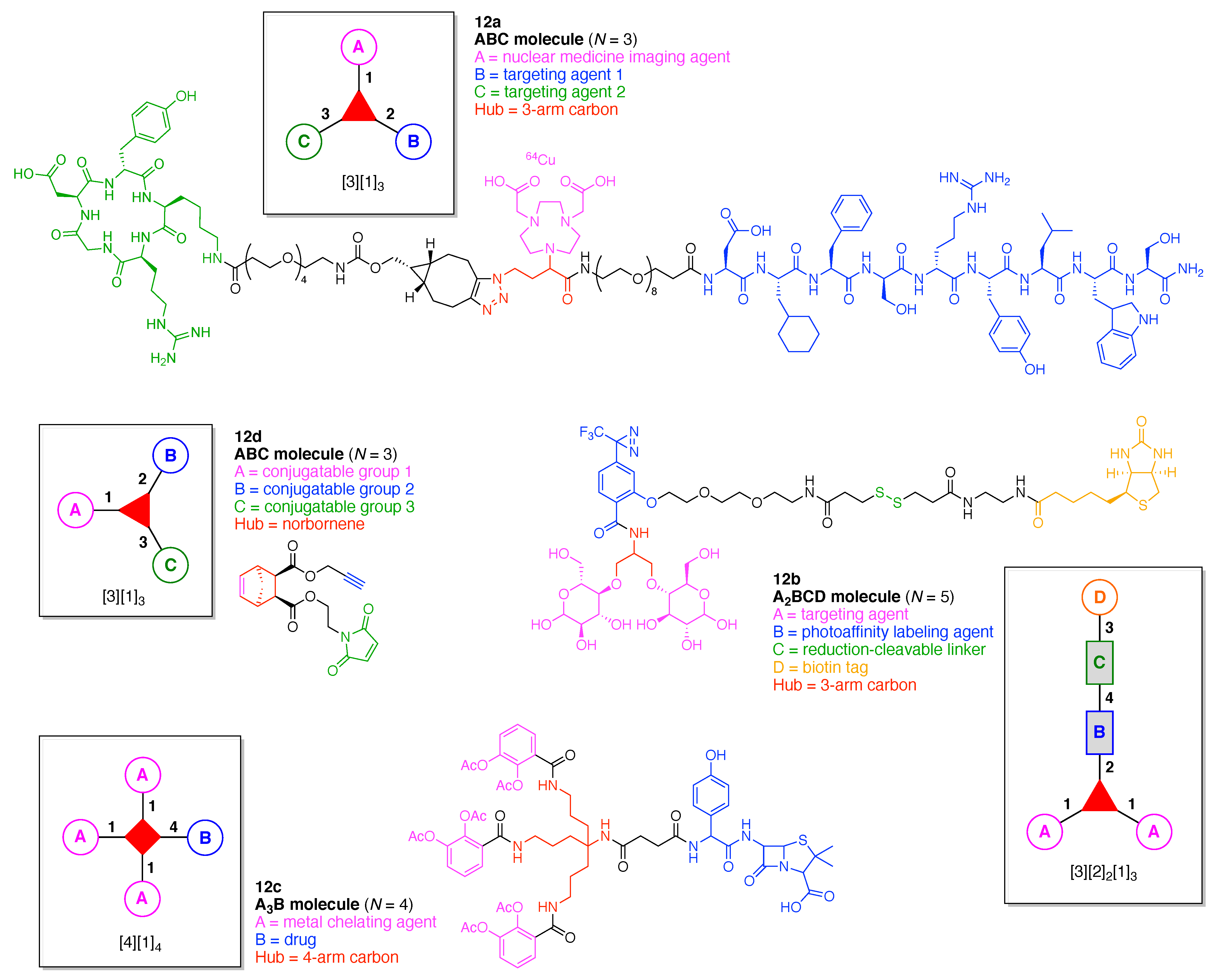{kind=link}
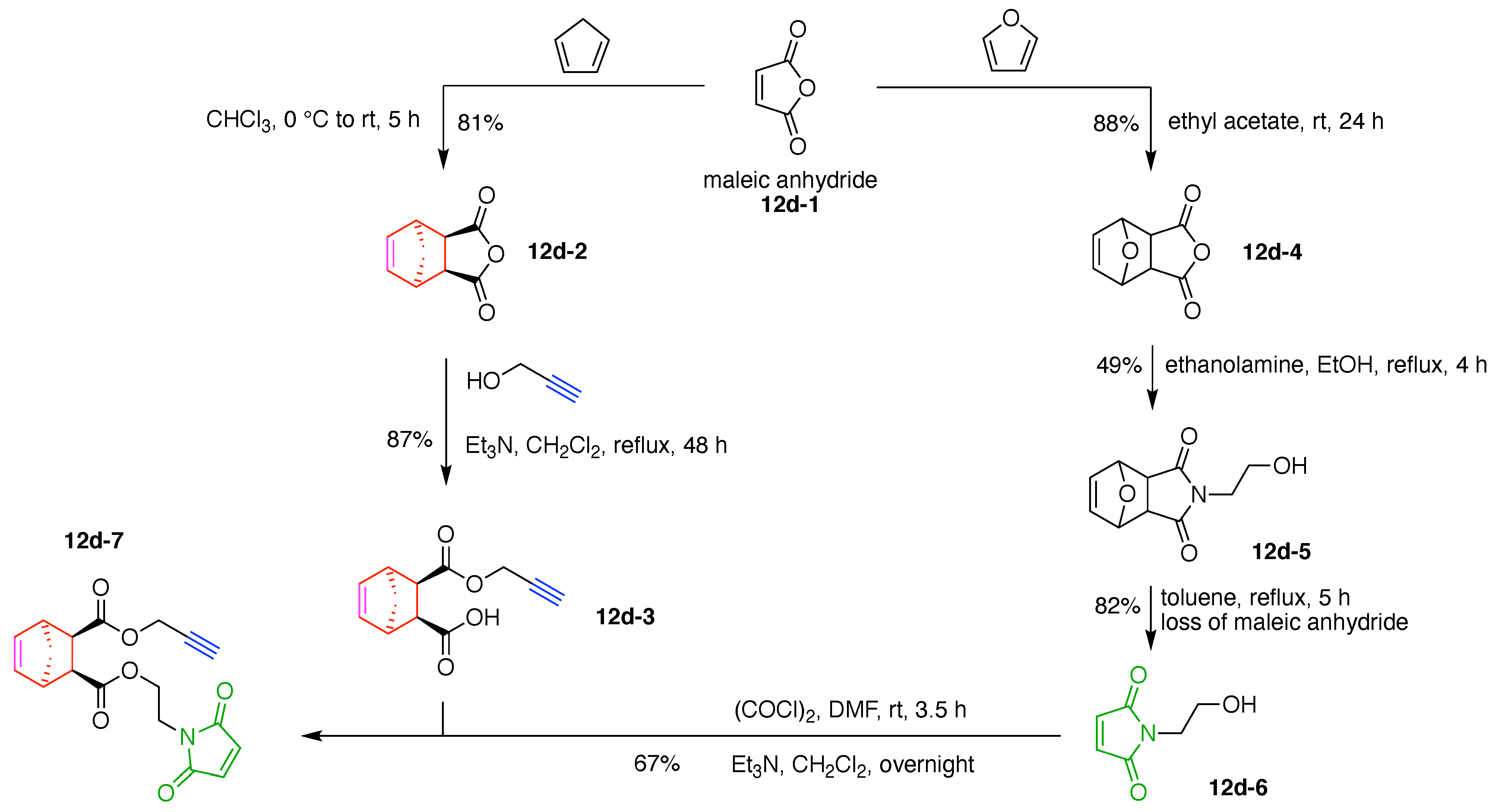{kind=link}
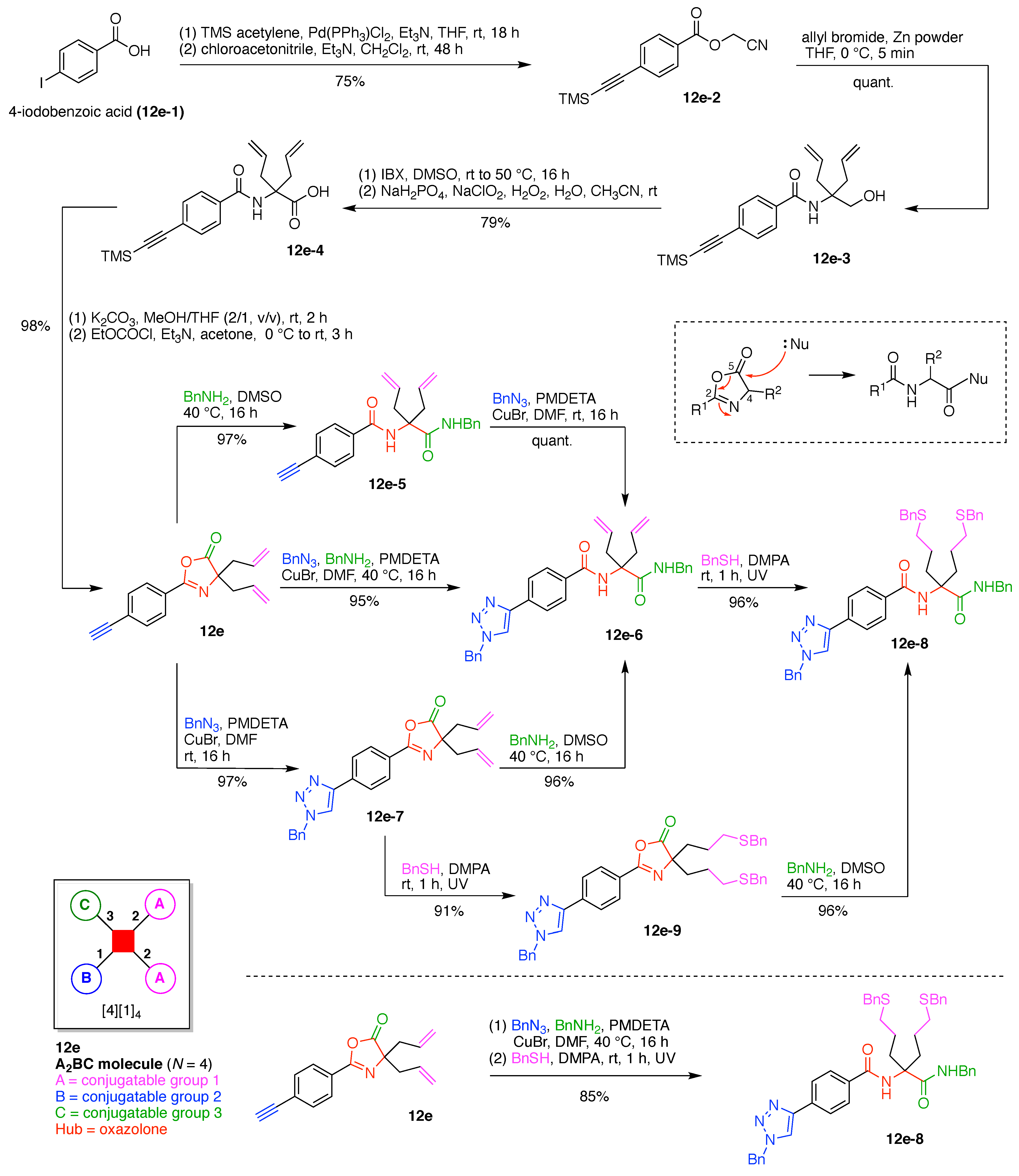{kind=link}
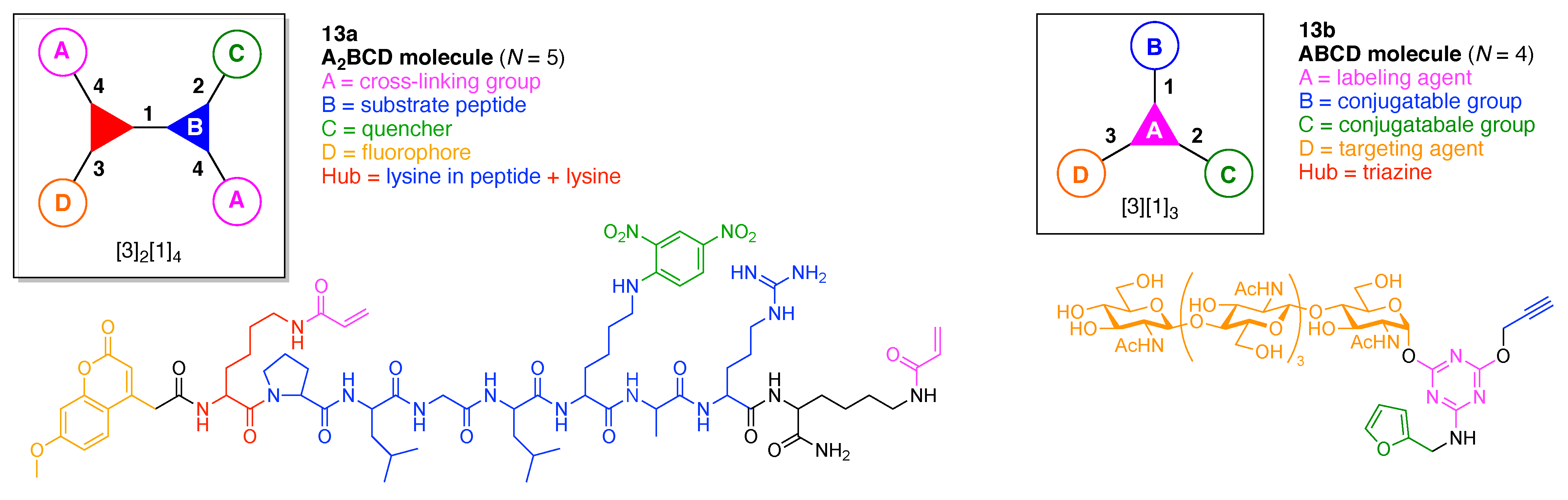{kind=link}
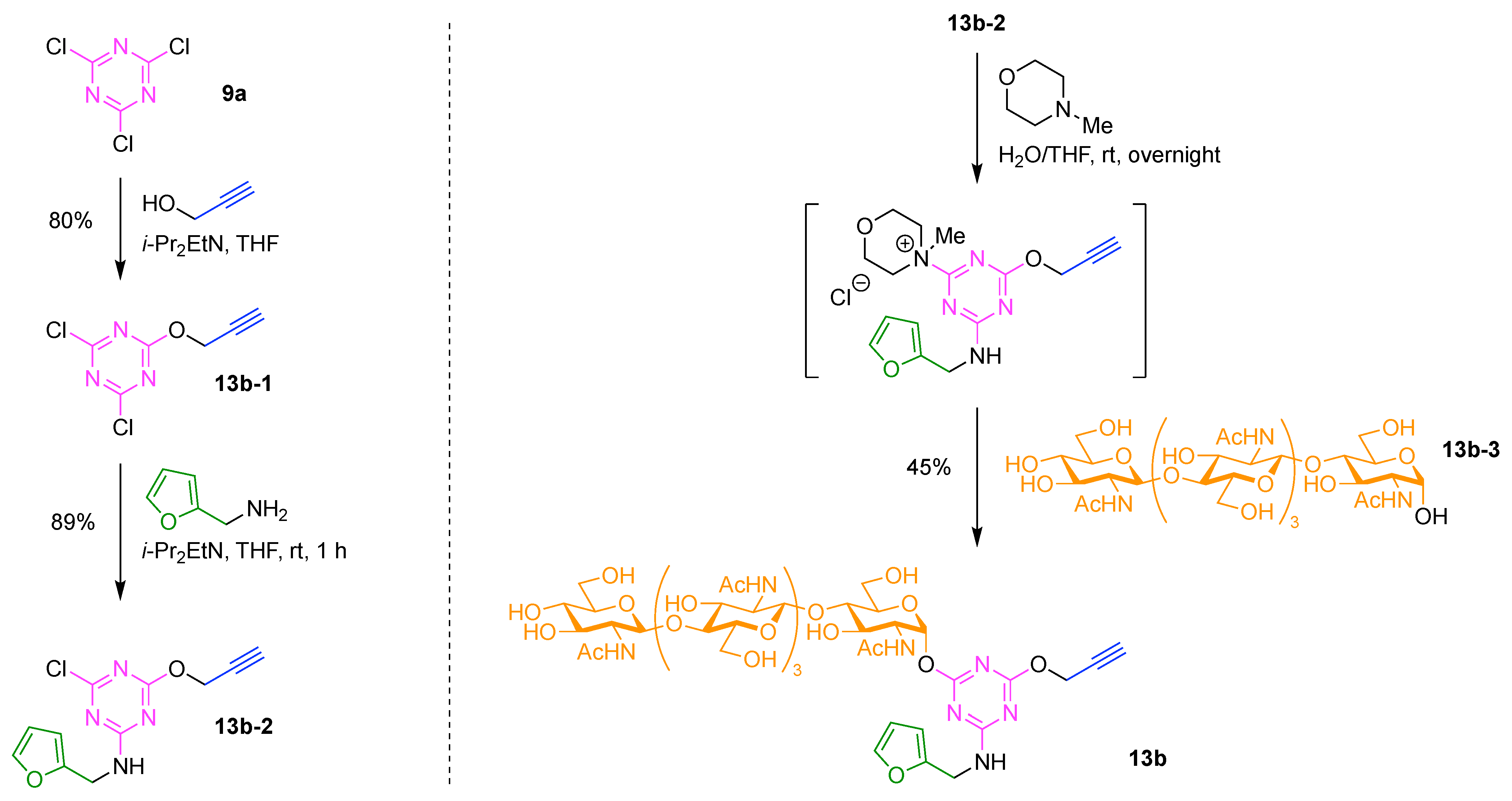{kind=link}
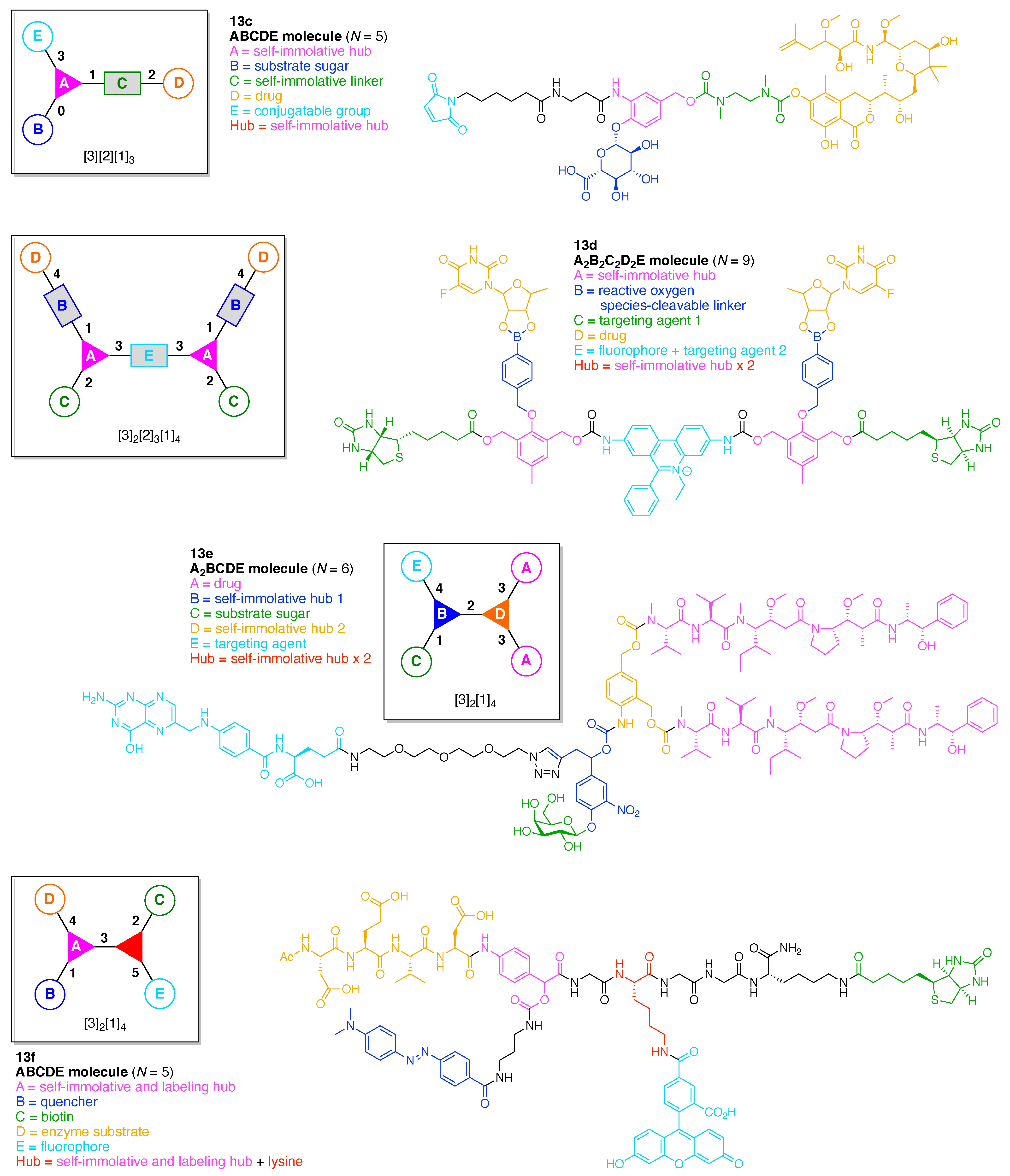{kind=link}
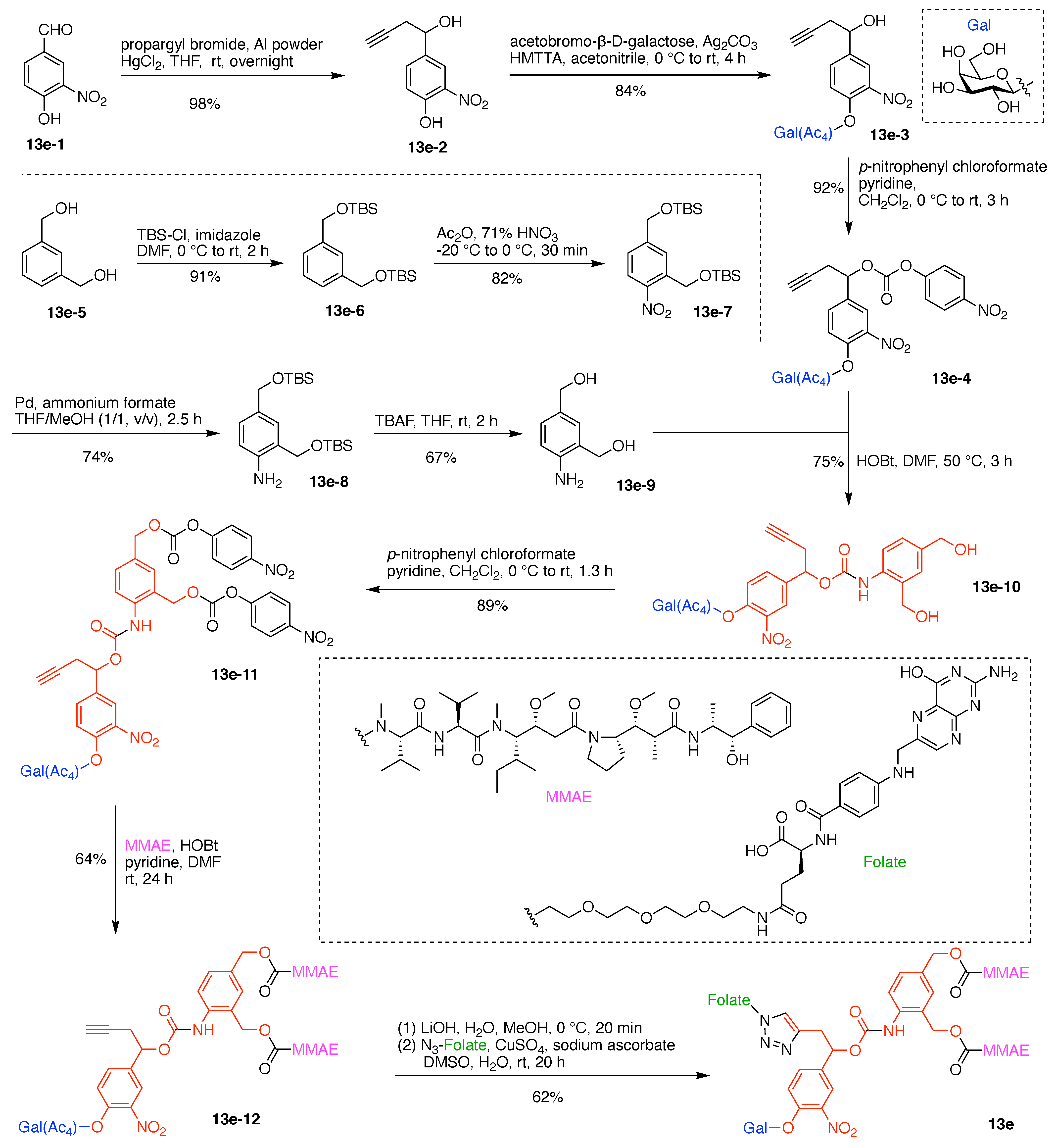{kind=link}
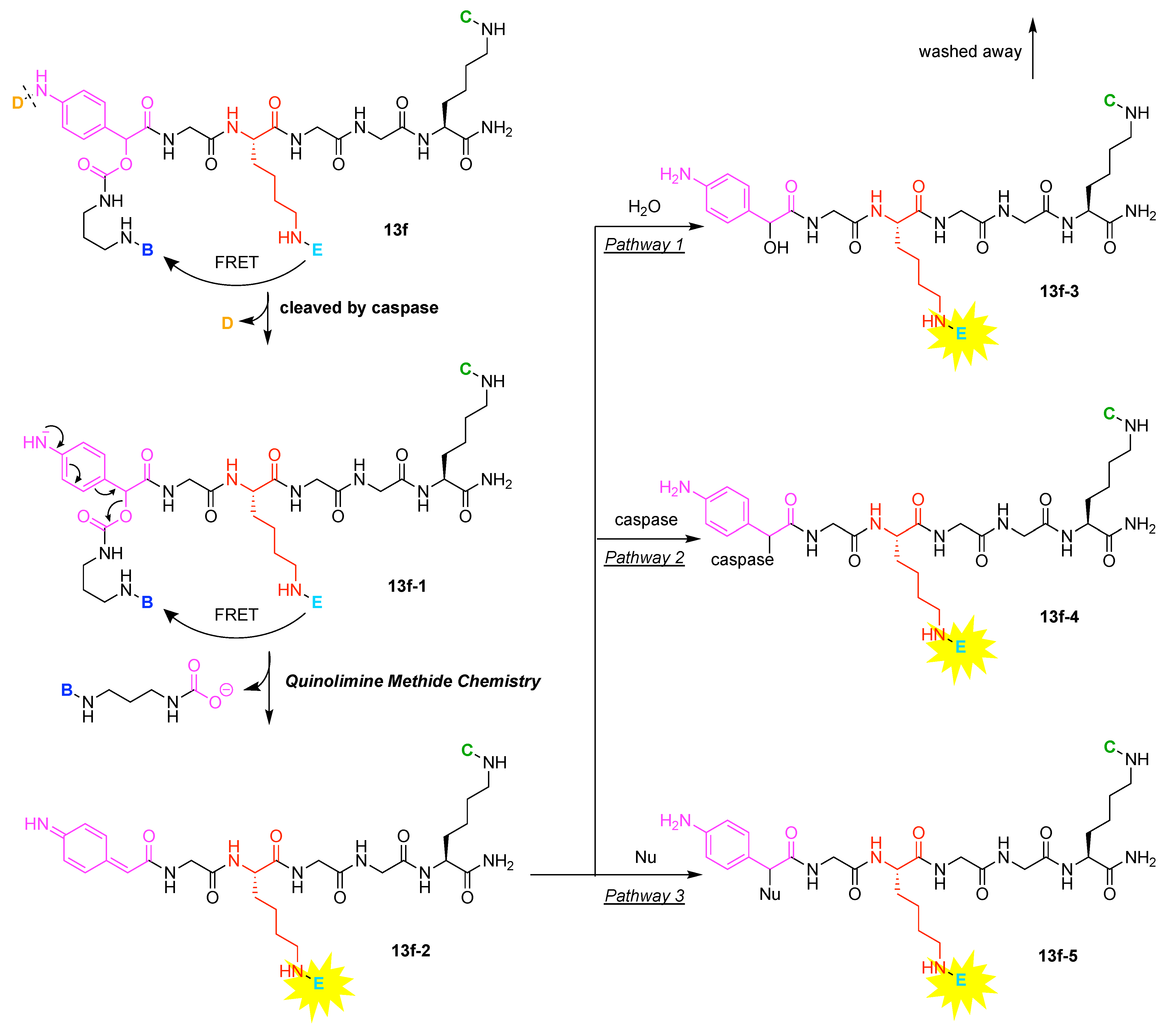{kind=link}
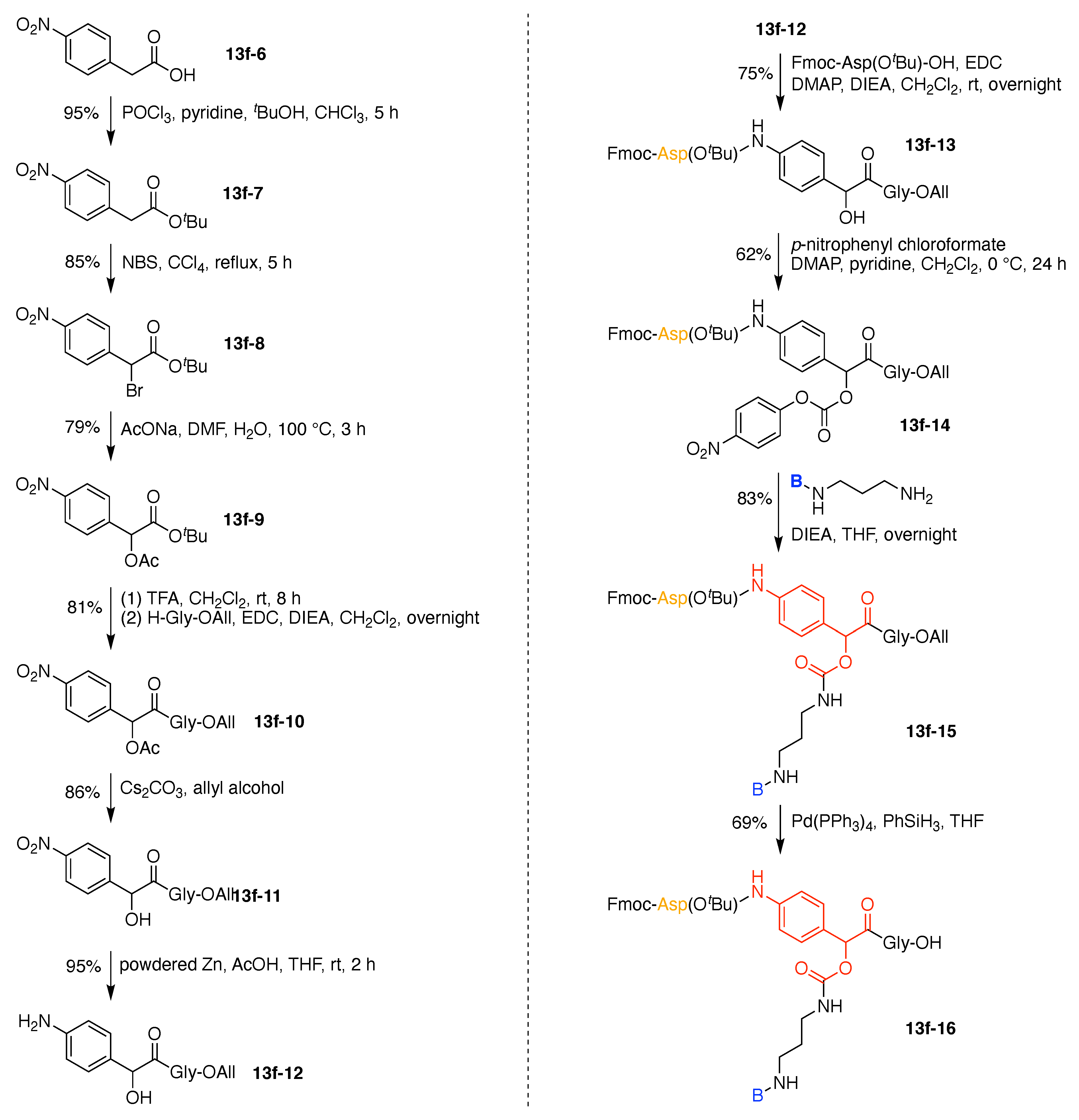{kind=link}
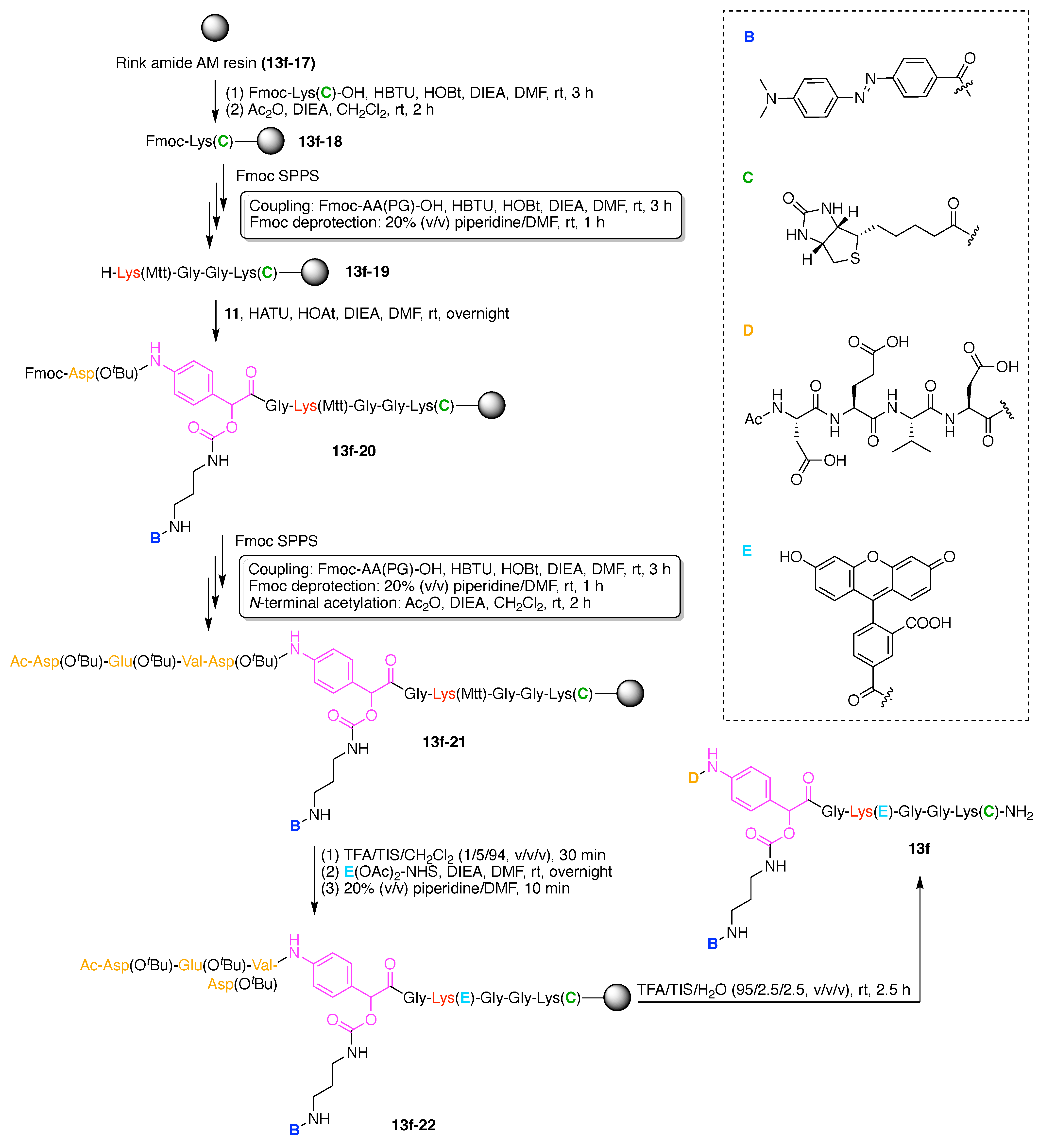{kind=link}
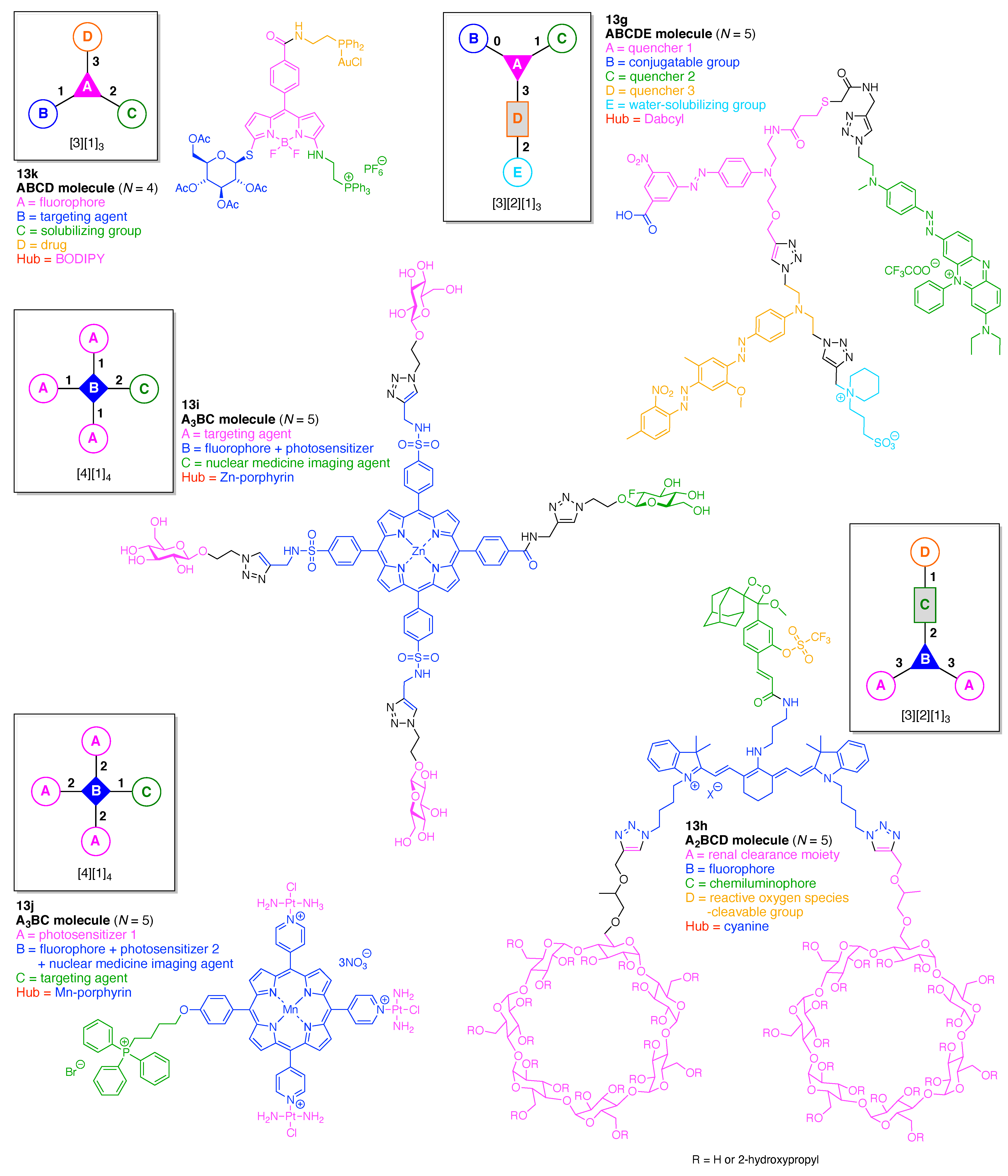{kind=link}
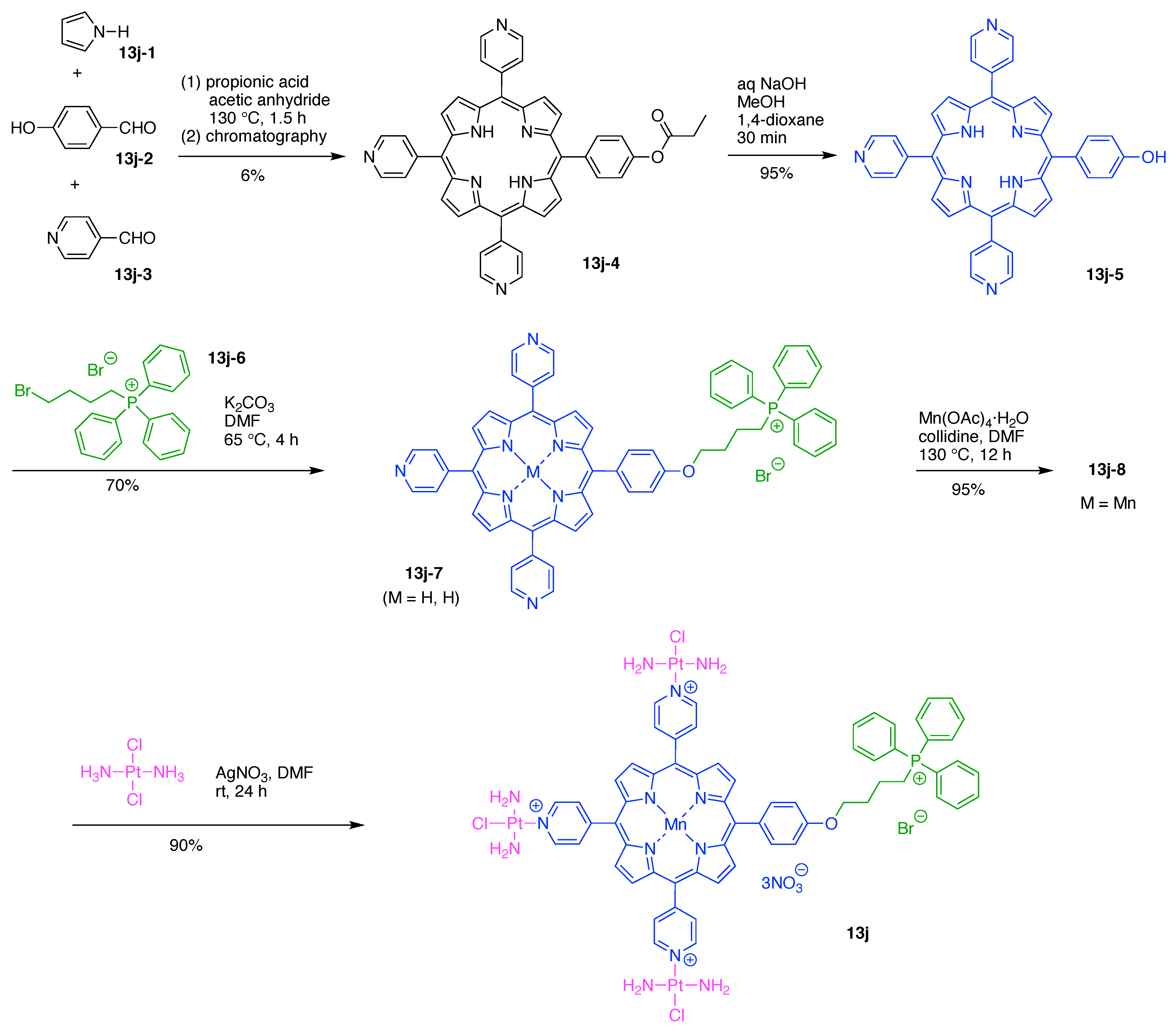{kind=link}
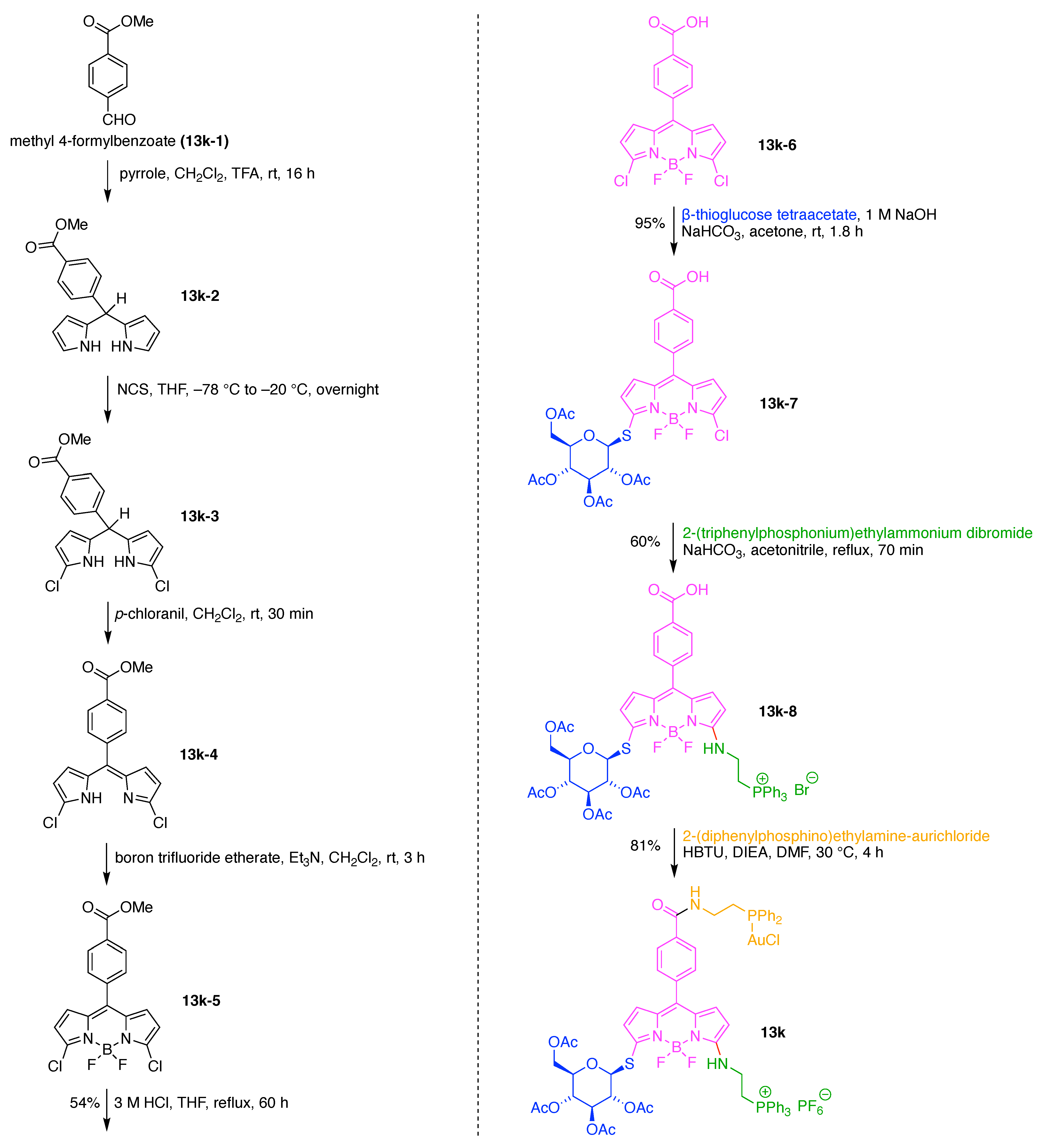{kind=link}
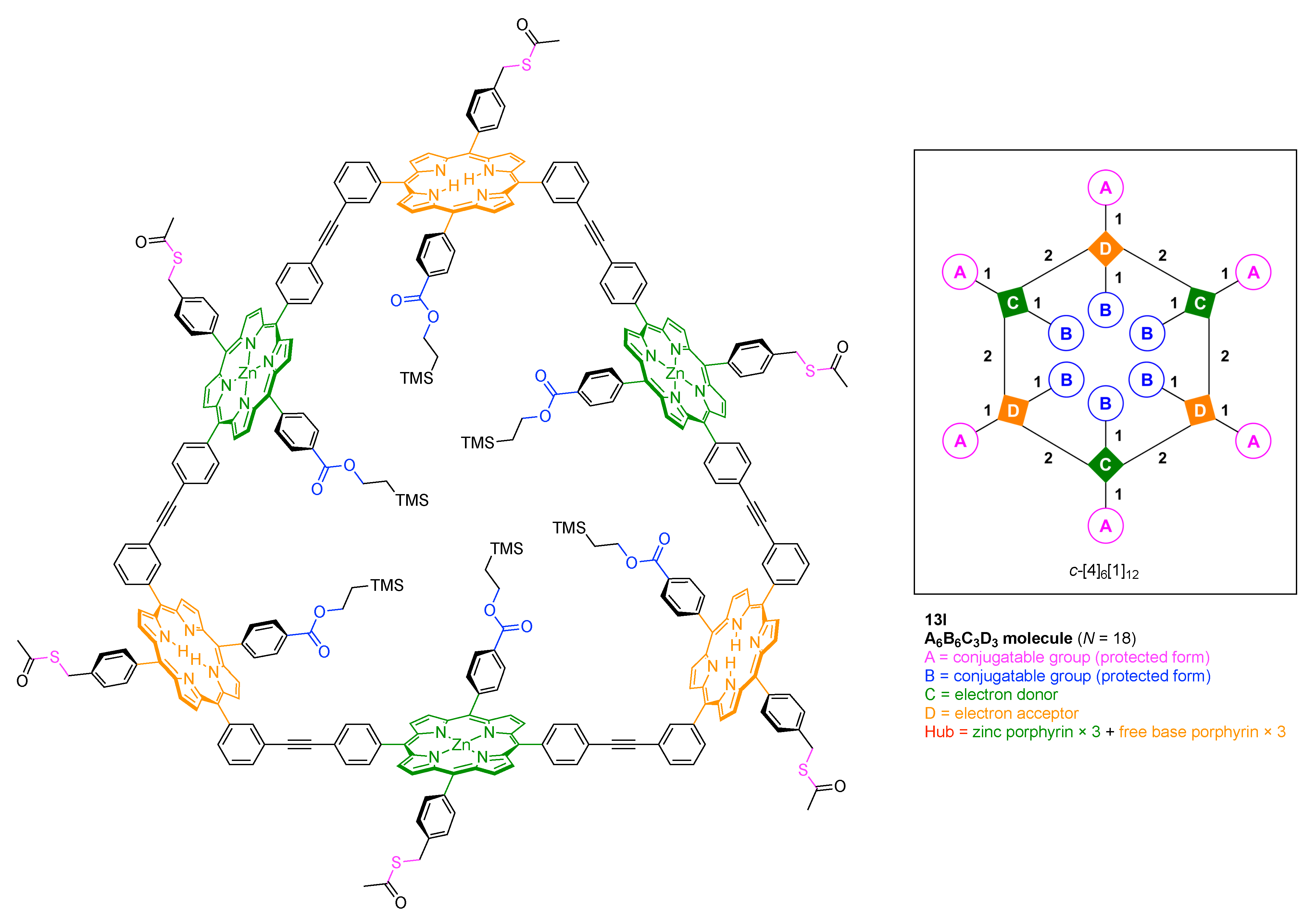{kind=link}
{kind=link}
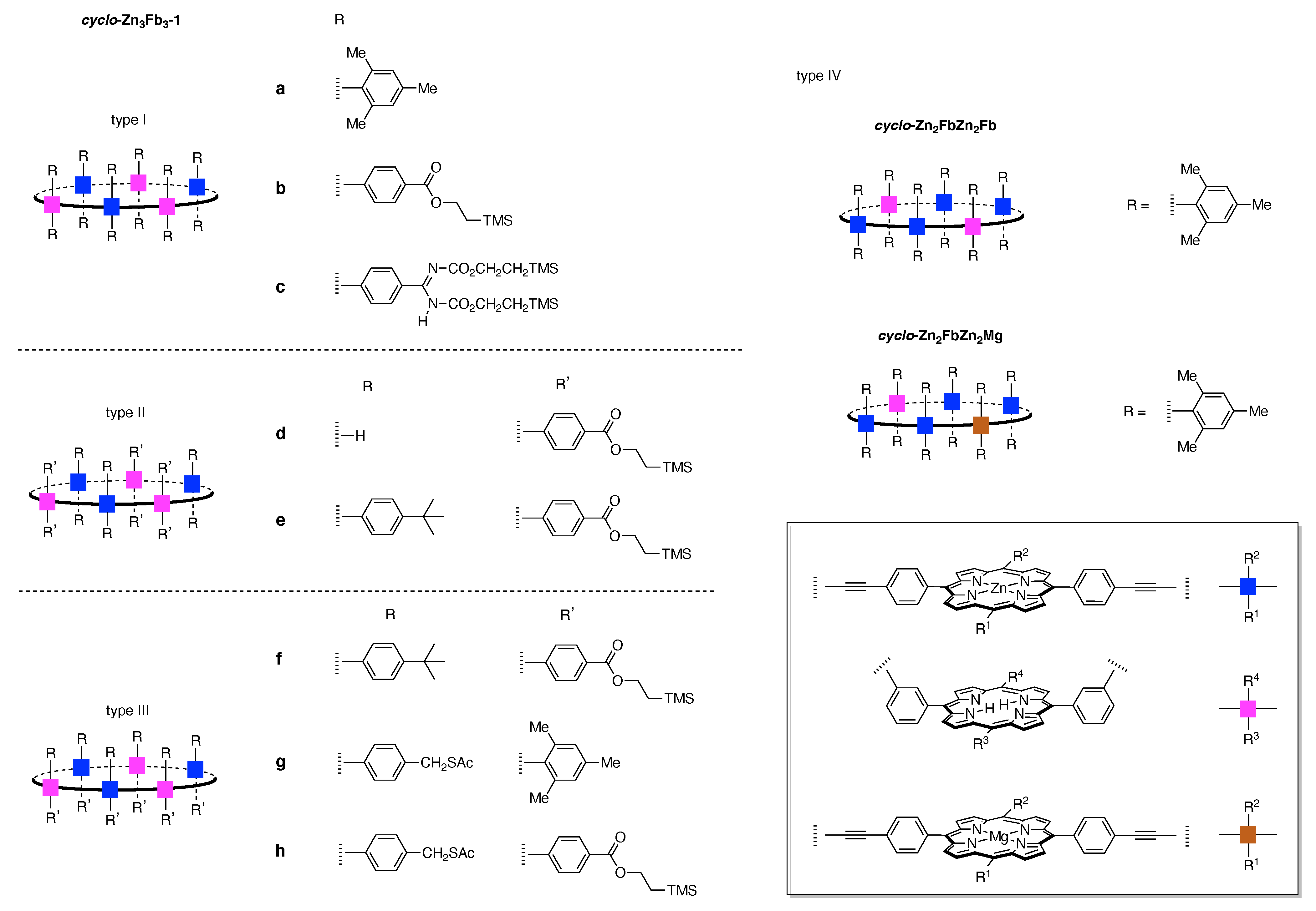{kind=link}
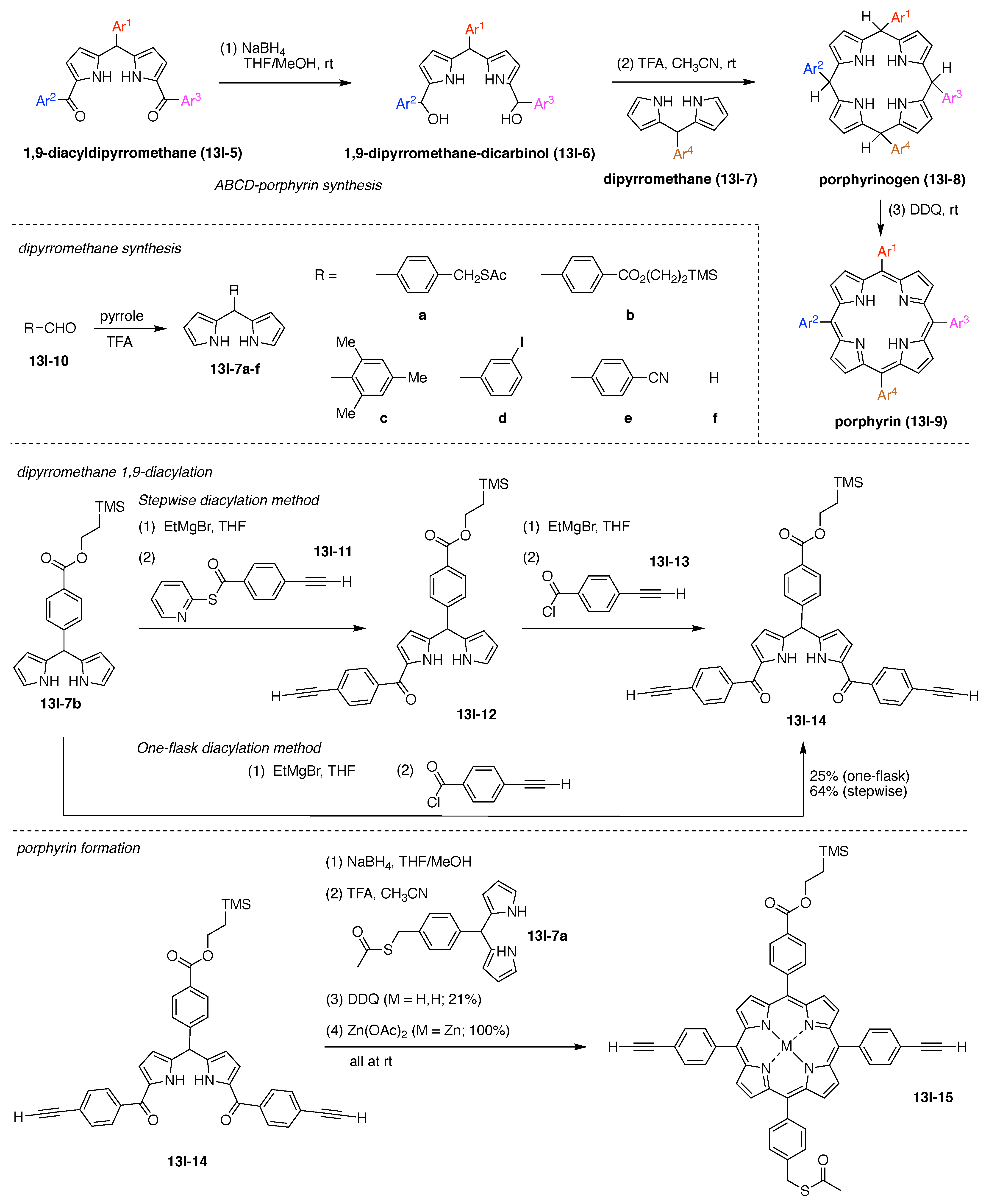{kind=link}
Table 1.
Substituent effects of chlorotriazines in nucleophilic aromatic substitution.

| Entry | X | σma | Relative Rate Constant | |
|---|---|---|---|---|
| Reaction A | Reaction B | |||
| 1 | –Cl | 0.37 | 1.0 b | 1.0 b |
| 2 | –OPh | 0.25 | 1/52 | |
| 3 | –SMe | 0.15 | 1/130 | |
| 4 | –OMe | 0.12 | 1/150 | 1/190 |
| 5 | –Ph | 0.06 | 1/290 | |
| 6 | –NHPh | −0.02 | 1/2900 | |
| 7 | –N-morpholino | 1/6000 | ||
| 8 | –NMe2 | −0.16 | 1/17,000 | 1/75,000 |
a Data from [189]. b The observed rate constant was halved to calculate the relative rate constant per one chloro group.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sato, D.; Wu, Z.; Fujita, H.; Lindsey, J.S. Design, Synthesis, and Utility of Defined Molecular Scaffolds. Organics 2021, 2, 161-273. https://0-doi-org.brum.beds.ac.uk/10.3390/org2030013
AMA Style
Sato D, Wu Z, Fujita H, Lindsey JS. Design, Synthesis, and Utility of Defined Molecular Scaffolds. Organics. 2021; 2(3):161-273. https://0-doi-org.brum.beds.ac.uk/10.3390/org2030013
Chicago/Turabian StyleSato, Daisuke, Zhiyuan Wu, Hikaru Fujita, and Jonathan S. Lindsey. 2021. "Design, Synthesis, and Utility of Defined Molecular Scaffolds" Organics 2, no. 3: 161-273. https://0-doi-org.brum.beds.ac.uk/10.3390/org2030013