The Role of Cathepsins in the Growth of Primary and Secondary Neoplasia in the Bone


Department of Radiation Oncology, University of Florida, Gainesville, FL 32610, USA
*
Author to whom correspondence should be addressed.
Osteology 2021, 1(1), 3-28; https://0-doi-org.brum.beds.ac.uk/10.3390/osteology1010002
Submission received: 20 August 2020
/
Revised: 21 September 2020
/
Accepted: 23 September 2020
/
Published: 9 October 2020
Abstract
:The upregulation of proteolytic enzymes has been demonstrated to promote primary tumor development and metastatic bone cancer. The secreted proteases increase tumor growth and angiogenesis, and potentiate neoplastic cell dissemination. This article reviews the role and mechanisms of cathepsins in normal physiology, cancer, bone remodeling, and the tumor–bone interface, with a specific focus on cathepsins B, D, H, G, L, and K. In this review, we highlight the role of cathepsins in primary bone cancer (i.e., osteosarcoma (OS)), as well as metastatic breast (BCa) and prostate (PCa) cancer. In addition, we discuss the clinical utility and therapeutic potential of cathepsin-targeted treatments in primary and secondary bone cancers.
1. Introduction
Bone tumors can present as either primary or secondary cancers. Primary bone cancer originates within the bone microenvironment. Secondary bone cancer occurs as a result of tumor cells that metastasize to the bone. In this review, we focus on osteosarcoma (OS) for primary bone cancer, and metastatic breast (BCa) and prostate cancer (PCa) for secondary bone cancer.
1.1. Osteosarcoma
OS is considered a rare disease, accounting for 1% of the cancers diagnosed annually (~900 new cases) in the United States [1]. OS is the most common primary malignant bone tumor, and the third leading cause of cancer-related death in adolescents and young adults [2,3]. OS most often (75% of cases) arises within the metaphysis (growth plate) of long bones such as the femur and tibia, and is characterized by the hyper-proliferation of mesenchymal cells depositing immature osteoid matrix into the bone tissue [1,4,5,6]. The 5-year survival rate for patients that present with non-metastatic osteosarcoma is about 70%. At diagnosis, approximately 80% of patients present with clinical or subclinical metastases [4,7]. For the 10–20% of OS patients who present with clinical metastasis (commonly found within the lung), the 5-year survival rate is 20% [8,9]. Since the mid-1980s, when the combination of chemotherapy and surgery became the standard of care treatment, the 5-year survival rate for patients with metastatic OS has plateaued [4,10,11].
1.2. Bone Metastasis in Breast and Prostate Cancer
In the US, breast (BCa) and prostate (PCa) cancers have the highest cancer incidence, and are the second leading cause of death in women and men, respectively [12]. It is predicted that one in eight women and one in nine men will be diagnosed with BCa and PCa in their lifetime, respectively. It is estimated that over 260,000 BCa and 170,000 PCa cases will be diagnosed in 2019, and about 20% of patients will die from their disease [12]. Metastasis is the leading cause of death in these patients.
The seed and soil hypothesis proposed by Stephen Paget in 1889 states that the spread of tumor cells from a primary tumor, which serve as ‘seeds’, grow in fertile ‘soil’, i.e., the host microenvironment [13]. BCa and PCa cells have a high affinity for the bone microenvironment. BCa and PCa cells express bone-specific factors to increase their survival within the bone environment, through a process known as osteomimicry [14,15,16,17]. In BCa and PCa, bone metastasis accounts for approximately 70% of all metastases [12,18,19,20]. Over 80% of the women who die of BCa develop skeletal lesions [21,22], and the 5-year survival rate for PCa patients with metastatic bone cancer is 3% [23].
Bone metastases are associated with significant morbidity and mortality [20,24]. Clinically, bone metastases potentiate hypercalcemia, nerve compression syndromes, pathologic fractures, and severe bone pain [18,20,25,26]. Traditionally, metastatic BCa is considered to be osteolytic, while metastatic PCa is believed to cause osteoblastic lesions within the bone [27]. However, it is currently believed that BCa and PCa both present with a mix of osteoclastic and osteoblastic cell function [28,29,30]. This process of bone remodeling is highly-regulated, based on the tumor–stromal interaction within the bone microenvironment.
Currently, once patients are diagnosed with metastatic primary and secondary bone disease, there are limited options for curative therapy. Thus there is a significant need for novel therapies that target bone related tumors in order to improve the survival and quality of life of patients with primary and secondary bone cancer.
2. Proteolytic Enzyme Targeting in Cancer
Protease secretion by tumor cells is critical for several stages of oncogenesis and tumor progression [31]. More specifically, extracellular proteases contribute to three key hallmarks of cancer: (1) tumor-promoting growth and inflammation, (2) the induction of angiogenesis, and (3) the activation of metastasis [32,33]. There are several families of proteases that have been implicated in the study of tumor progression and metastasis, including urokinase-type plasminogen activator (uPA), matrix metalloproteinases (MMP), and cathepsins. The role of MMPs in cancer progression led to the development of MMP inhibitors (MMPIs). Unfortunately the initial class of MMPIs lacked-specificity, which resulted in off-target effects and toxicity. Interest in the further development of MMPIs subsequently waned due to disappointing clinical trial data demonstrating adverse side effects with little to no clinical benefit [34,35,36,37,38]. The broad-spectrum inhibition of MMPs failed to account for the complex pro- and anti-tumorigenic roles of MMPs, as well as the physiologic role of MMPs under normal conditions [31,36,39]. However, this research did lead to the understanding that the development of agents to target specific proteases may be significantly beneficial in the inhibition of cancer progression.
A class of proteases that have been of particular interest in the field of metastasis are the cathepsins, with cysteine cathepsins predominating in the field of cathepsin-related cancer research. Cathepsins not only play a direct, multifaceted role in tumor progression and metastasis but their secretion also contributes to the activation of other extracellular proteases, such as uPA, proheparanase, and MMPs [40,41,42]. This review will focus on the role of cathepsins in bone remodeling, and the therapeutic potential of targeting cathepsins as a novel therapeutic intervention that targets bone-related primary tumors and metastases.
2.1. The Physiologic Role of Cathepsins
Cathepsins have been recognized primarily for their roles in lysosomal protein turnover, which contributes to a plethora of physiologic processes, such as antigen presentation, bone remodeling, and epidermal homeostasis. At physiologic pHs, cathepsins have negligible enzymatic activity; for their optimal function, cathepsins require acidic pHs (6.0–6.3) [43], such as those found within lysosomes, where they are primarily responsible for protein degradation. We will provide a brief overview of the physiologic role of cathepsins B, D, G, H, K, and L due to their highlighted role in bone remodeling.
Cathepsin B [CatB] is a cysteine protease present in normal cells, with a molecular weight of 24–29 kDa, which exhibits both exo- and endo-peptidase cleavage activity [43,44]. Pro-cathepsin B is activated by urokinase-plasminogen activator (uPA) [45]. Activated CatB degrades extracellular structure proteins and basement membrane components [46,47,48].
Cathepsin D [CatD] is an aspartyl protease [49]. Pro-CatD (52 kD) is catalyzed to different active forms of CatD (48kD, 34kD, 14kD) [50]. CatD is capable of hydrolyzing protumorigenic factors, such as insulin-like growth factor (IGF) binding proteins, which allow for the release of IGFs and subsequent downstream signaling [51]. CatD participates in the regulation of proCatB and L, and degrades and inactivates their active forms [52].
Cathepsin G [CatG] is a serine protease (28kDa) that has been shown to play a pivotal role in the bone microenvironment and tumor stromal interactions by activating MMP9, degrading the extra-cellular matrix (ECM), inducing apoptosis, modulating preosteoclastic chemotaxis, and cleaving the Receptor Activator of Nuclear Factor-Kappa β (RANK) Ligand (RANKL), a member of the tumor necrosis factor (TNF) superfamily, to form sRANKL [49,53,54].
Cathepsin H [CatH] is a ubiquitously expressed cysteine protease (proCatH 41kDa, mature CatH 29kDa) with exo- and endo-peptidase functions [55,56,57].
Cathepsin K [CatK], a.k.a. Cathepsin O2), a cysteine protease (proCatK 38kDa and mature 27kDa) and an endopeptidase, is exclusively expressed at the ruffled border of actively-resorbing osteoclasts [58,59,60,61,62,63,64,65]. In mice, CatK deficiency leads to osteopetrosis due to impaired osteoclast function [66,67]. In humans, the loss of CatK function leads to pycnodysostsis, an autosomal recessive disorder characterized by bone sclerosis, dysmorphic facial features, increased bone fragility and a short stature [64,66,67,68,69,70].
Cathepsin L [CatL] is a ubiquitously-expressed cysteine protease (proform 43kDa; mature 32 kDa) that has been found to participate in normal tissue function as well as physiologic bone turnover. In the lysosome, CatL degrades intracellular and endocytosed proteins [71].
2.2. Role of Cathepsins in Bone Remodeling
The secretion of cathepsins into the extracellular milieu occurs during normal physiological processes such as bone remodeling (Figure 1) [72,73,74,75,76,77]. The mineralized bone matrix is known to represent a rich storehouse of growth factors that are mobilized by osteoclastic resorption and become active in the local microenvironment.
Cathepsins B, K, and L have been shown to be actively involved in the cleavage of bone matrix proteins. CatK, the primary cathepsin associated with bone degradation, is secreted by osteoclasts within the bone lacunae [58,78]. CatK degrades type I collagen in the bone by cleaving the triple helical domains at multiple sites and directly releasing the telopeptides [79]. Although it is primarily secreted by osteoclasts, the secretion of CatK from osteoblasts also has been reported [80]. In addition to CatK, CatL is also expressed in osteoclasts, and plays an important role in bone remodeling due to its strong collagenolytic activity [81,82,83]. CatL is involved in the proteolytic degradation of the bone matrix and cartilage matrix components, such as gelatin and type I, II, IV, IX, and XI collagens [84,85,86,87]. Goto et al. [88] examined the localization of CatB in the osteoclasts of femoral bones and demonstrated that CatB was detected along the bone resorption lacunae, suggesting that CatB directly participates in the degradation of the bone matrix [40]. CatB degrades ECM proteins, such as type I and IV collagen, laminin, fibronectin, and proteoglycans. CatB also activates type I pro-collagenase, pro-stromelysin, and the soluble and receptor-bound forms of pro-uPA [43,89,90,91,92,93,94].
Several reports have demonstrated that the inhibition of cathepsin by agents such as CA074 (CatB inhbitor) and JMP-OEt (pan cathepsin inhibitor) leads to an increase in osteoclastogenesis, osteoclast surface area, and average cell size [95,96]. This suggests that cathepsin inhibition may increase the proliferation rates of precursor cells, thereby increasing the number of fusion events or the rate of osteoclast fusion. The pan inhibition of cysteine cathepsins enhances the differentiation of multinucleated osteoclasts, highlighting the potential suppressive role of cysteine cathepsin in the fusion of osteoclast precursor cells [95]. Although CatK deficiency increases osteoclast numbers and size, their ability to resorb bone is impaired [97,98].
Contrary to these findings, there have also been reports that demonstrate that pharmacologic cathepsin inhibition through endogenous cysteine cathepsin inhibitor cystatin C—as well as CatB inhibitor, CA-074, CatB/CatK inhibitor E64 and CatL/CatK inhibitor KGP94—leads to the opposite effect; i.e., cathepsin inhibition results in a decrease in osteoclastogenesis and the inhibition of osteoclast function [87,99,100,101,102]. Edgington-Mitchell et al. also demonstrated that the cysteine protease, legumain (asparaginyl endopeptidase), inhibits osteoclastogenesis through the autocleavage of its C-terminal domain and activates CatL, suggesting that cysteine protease inhibition enhances osteoclastogenesis, which has been implicated in bone metastasis [95].
Typically, cell-to-cell contact between osteoblasts (bone forming cells) and osteoclasts (bone resorbing cells) is required in order to activate RANKL on the surface of osteoblasts and RANK on the surface of osteoclasts. However, CatG has been shown to cleave RANKL on osteoblasts generated in soluble RANKL (sRANKL), negating the cell-to-cell contact needed between the osteoblast and osteoclast. In turn, CatG-generated sRANKL induces osteoclast activation and enhances osteolysis. The CatG–regulated chemotaxis of osteoclast inducing monocytes is mediated by the proteolytic activation of protease-activated receptor-1 (PAR-1). PAR-1 is a G protein-coupled receptor (GPCR) that initiates chemotaxis signaling via the Gαi/o subunit–mediated mechanism [53]. This G protein–coupled receptor (GPCR) is activated when the NH2 terminus is cleaved by proteases generating a tethered ligand that can bind intramolecularly and initiate signaling [103]. The inhibition of CatG reduces the number of multinucleated CD11b+ osteoclasts at the tumor–bone interface. Targeting PAR-1 in osteoclasts as well as CatG reduces osteolysis by inhibiting the recruitment, differentiation and activation of osteoclast precursors [53].
The modulation of CatK activity by small-molecule inhibitors reduces the bone resorption activity of osteoclasts in vitro and in vivo [66,67]. Ishikawa et al., demonstrated that CatK antisense blocks osteoclast development and formation [104]. Additionally, several studies have shown that pharmacological CatK inhibitors not only inhibit bone resorption but also stimulate bone formation [105,106]. In comparison to bisphosphonate zoledronic acid (ZOL), the CatK inhibitor L-235 protects against focal osteolysis and cortical disruption with an increase in bone density [107]. Approximately 70% of the cortical bone was eroded in vehicle control animals. The preventative administration of L-235 preserved cortical bone and reduced the cortical disruption by to 36%, 17%, and 11%, respectively, versus the vehicle, whereas ZOL resulted in 25% cortical disruption [107]. In the treatment regimen, L-235 and ZOL showed 31% and 24% cortical disruption, respectively, compared with the control group, which showed 58% cortical disruption [107].
Cathepsins also are secreted by immune cells such as macrophages and neutrophils [108,109]. CatB is expressed in macrophages and osteoclasts, and increases 2–3 fold in parallel with the CatK during osteoclast differentiation [110,111]. Myeloid Derived Suppressor Cells (MDSCs) express and demonstrate abundant cysteine cathepsin activity [95]. As myeloid precursor cells transition into macrophages, the activity of CatB and CatL is strongly upregulated, whereas during the differentiation of macrophages to osteoclast cathepsins, their activity is downregulated [95]. MDSCs increase CatK production and inhibit Cat B and L production in favor of osteoclastogenesis [95]. The endogenous cathepsin inhibitor cystatin B effectively prevents bone resorption by down-regulating CatK activity, and cystatin C decreases the formation of osteoclasts in mouse bone marrow cultures [78]. CatK and CatL have also been found to be secreted by innate immune cells and additional stromal cells within the bone microenvironment [40,108].
3. The Role of Cathepsin in Cancer
Several clinical studies that have demonstrated that cathepsins have prognostic value in cancer patients [112,113,114,115,116,117] and, notably, elevated levels of cathepsins are associated with an increase in metastatic incidence [118].
The expression of cathepsins is significantly upregulated in many cancer types, including breast, lung, colorectal, prostate and melanoma [113,114,119,120,121,122,123,124]. Cathepsins are involved in multiple aspects of tumor growth and metastasis, including cell migration and invasion, and the induction of angiogenesis [125,126,127]. The secreted cathepsins contribute to the metastatic potential of cancer cells by degrading ECM and basement membrane proteins as well as E-cadherin. By degrading the ECM, cathepsins enable tumor cells to escape from the primary tumor and invade into the vasculature, which enables the formation of distant metastases. The cleavage of E-cadherin, a key cell adhesion protein, disrupts adherens junctions, and increases cell migration and invasion. Cathepsins cleave the extracellular domain of cell adhesion molecule (CAM) proteins and transmembrane receptors which stabilize cell–cell contact; the inactivation of CAM leads to an increase in the migratory capacity of neoplastic cells [128,129]. Cathepsins have also been shown to activate growth factors, MMPs, uPA and other cathepsins in order to promote the establishment of tumors at secondary sites [31,130].
Cathepsins can also affect neoplastic primary and secondary growth through their impact on the immune response. For example, immature immune cells promote metastasis by inducing osteoclastogenesis and mediating immunosuppression [131,132]. MDSCs produce a large percentage of active CatB and CatL from both neutrophilic (CD11b+/Ly6G+) and monocytic (CD11b+/Ly6C+/Ly6G-) subsets, which are correlated with increased metastasis [106]. BMMs secrete CatK, which appears to be critical for macrophage invasion and tumor growth [133,134]. In CatK knockout mice, there is a decrease in the number of F4/80-positive macrophages in comparison to the F4/80-positive macrophages found in tumors from wild-type mice; the macrophages from the tumors of CatK knockout mice were also less invasive than the macrophages from tumors of wild-type mice [96].
The Role of Cathepsins in Primary Bone Cancer and Metastases to the Bone
The bone microenvironment is hospitable to bone tumor development and growth, as well as being a favored site for the metastases of cancers such as prostate and breast. Cathepsins play a key role in the malignant progression at this site because their secretion within the bone matrix enables the expansion of neoplastic cells and leads to the release of active growth factors that promote cancer cell growth [24,135]. Referred to as the ’vicious cycle’, the secretion of proteolytic enzymes from neoplastic cells, and the resulting hyperactive remodeling and release of pro-tumorigenic factors from the bone matrix, is critical to tumor progression in the bone [20]. The release of these growth factors constitutes one of the key mechanisms in the bone’s remodeling, allowing the physiological coupling between osteoblasts and the osteoclasts [136]. Neoplastic cells also secrete bone-remodeling cytokines and growth factors such as PTH related peptide (PTHrP), and IL -1, -6,-8, and -11 into the bone microenvironment, which, in turn, act on osteoblastic stromal cells to enhance the production of osteoclast-activating factors such as RANKL [20,78,137,138,139]. Additional tumor-derived growth factors, such as transforming growth factor- β (TGF-β), IGF-I, and IGF-II, as well as collagen type I peptides, also serve to attract tumor cells to the bone matrix, induce mitosis in tumor cells, and promote bone formation and remodeling by enhancing osteoclastogenesis [14,15,24,25,137,140,141,142,143]. The endogenous cathepsin inhibitor, cystatin C, inhibits TGF-beta signaling in normal and neoplastic cells [144]. Stromal and tumor microenvironmental conditions, such as acidosis within the bone, has been shown to increase osteoclasts’ function and resorption pit formation, which results in the release of multiple proteases: specifically members of the cysteine cathepsin family, such as cathepsins B, D, K and L [58,145,146].
Several studies have shown the importance of cathepsins for the invasion and metastasis of OS cell lines [43,46,48]. In normal human osteoblast cells and the OS cell line MG-63, the active secretion and RNA/protein production of CatB is generated in the presence of bone-resorbing agents such as IL-1β and PTH [43]. In malignant bone tumors, CatB demonstrates activity at the tumor–bone interface with low levels of CatB detected around the bone [110]. CatB is most extensively found at the periphery of primary tumors, correlating to its role in tumor invasion and the degradation of the ECM [147]. The ability of CatB to degrade other matrix proteins and activate additional proteases is associated with neoplastic cell invasion and metastasis. Contrary to these findings, Husmann et al., discovered that—in comparison to the non-metastatic parental line Saos-2—the expression of the mature form of CatB in LM5 and LM7 cells was decreased by approximately 50% [55].
Intriguingly, CatB has been discovered to also demonstrate anticancer effects in the OS cell line MG-63, which is mediated through the activation of the DR-5/p53/Bax/caspase-9/-3 signaling pathway and the DR-5/FADD/caspase-8/lysosomal/cathepsin B/caspase-3 signaling pathway [148]. The release of CatB from the lysosome into the cytosol induces cell death in vitro [44]. Caspase-8 mediated Bid cleavage leads to the permeabilization of the mitochondrial outer membrane and the release of cytochrome C [149]. In the human OS cell line U2OS, CatB activity is activated after exposure to tumor necrosis factor-related apoptosis inducing ligand (TRAIL). CatB contributes to TRAIL-mediated apoptosis in human cells through Bid cleavage [149,150,151]. CatB activity is significantly increased following 1–2 h of exposure to TRAIL [149]. Triptolide inhibits cell viability and proliferation, and induced apoptosis in OS cell line MG-63 [148]. Previous studies by Owa et al., support this finding, citing that triptolide induces lysosomal-mediated programmed cell death in MCF-7 BCa cells through CatB and caspase-3 [152]. It is believed that the upregulation of CatB increases caspase 3 cleavage and induced apoptosis. During the early stages of apoptosis, cytosolic levels of CatB are increased [148]. Triptolide increases lysosomal membrane permeability, causing a leakage of CatB into the cytosol within the first 3 h of treatment, triggering the apoptotic cascade. Triptolide increases DR-5, Bax, p53, and CatB proteins, as well as caspase-3,-8, and-9 activity [148]. Clinically, elevated levels of CatB and CatL have been shown to be significant predictors of relapse and death for BCa patients [116,117,153,154]. Approximately 77% of BCa samples express CatB [116]. The expression of CatB was also associated with the expression of CatD and CatL in BCa patient samples [116]. Interestingly, high levels of CatB protein and activity were found in DU145, PC3, and LNCaP bone tumors in severe combined immunodeficiency (SCID) mice, despite the low CatB expression in PC3 and LNCaP cell lines in vitro [155]. The use of activity-based probes provides a non-invasive modality in order to monitor cathepsin activity and the therapeutic efficacy of cathepsin-targeted agents. In metastatic bone cancer, CatB demonstrated a 2-fold increase compared to normal fibroblasts [156]. CatB is higher in MDSCs isolated from the bone marrow of 4T1.2 tumor-bearing mice compared to those taken from lungs, which further suggests a differential regulation of MDSC-derived CatB within the bone microenvironment [106]. BMV109, a pan-cysteine cathepsin probe, monitors for CatB and CatL [157]. BMV109+ cells were expressed to similar extents in primary tumors derived from BCa cells 67NR (non-metastatic) and 4T1.2 (metastatic) [95]. However, in mice which developed bone metastasis, 4T1.2 tumors exhibited a strong increase in cathepsin expression/activity compared to mice bearing 67NR tumors, suggesting that cathepsin activity is upregulated during metastasis [95]. Withana et al. also demonstrated a decrease in CatB activity after treatment with CA-074 using the GB123 activity-based probe [157].
In patient samples, Gemoll et al. demonstrates [158] that in, comparison to fetal osteoblast, CatD immunostaining localized in the cytoplasm is overexpressed in OS and pulmonary metastases. Prognostically, CatD reached a sensitivity of 76.47% at 100% specificity, as well as a sensitivity of 100% at 100% specificity to predict OS and pulmonary metastasis, respectively [158]. In vitro, Spreafico et al. [159] observed that CatD is also upregulated in human OS cell line Saos-2 in comparison to mature osteoblasts (Figure 2). The mRNA and protein expression of Saos-2 and its metastatic sublines LM5 and LM7 demonstrate a 2.5-fold upregulation of CatD [55]. Contrarily, Arkona et al., demonstrates that CatD is not elevated in bone metastases [156].
Elevated CatD in BCa tissue has been correlated to poor prognoses, and is a strong prognostic marker in BCa [116,160,161,162,163]. CatD expression is also associated with the expression of CatG, L in BCa [116]. Using BM2 to detect tumor-associated glycoprotein TAG12, which is expressed by almost all BCa cells and the anti-CatD antibody, Solomayer et al. demonstrated that patients with CatD-positive cells in their bone marrow have a significantly shorter metastasis-free interval (38 months) compared to patients who are CatD negative (64.5 months) [164]. The CatD present in patient bone marrow was only found on tumor cells, and was not found on stromal cells in the bone marrow of patients with primary BCa [164], suggesting that CatD may potentially serve as a clinical biomarker for bone malignancies to further guide treatment regimens.
The secretion of cathepsins into the microenvironment may also contribute to therapeutic drug resistance [165] For example, secreted CatD attenuates apoptosis through the P13-Akt pathway, which leads to chemoresistance [166]. CatD production also is implicated in doxorubicin (dox) resistance in OS and BCa [165,167].
The presence of CatG within the tumor has been demonstrated to increase metastasis [168]. CatG is associated with the expression of cysteine proteases CatK and CatL in BCa patients [116]. CatG is up-regulated at the tumor–bone interface and increases osteoclast differentiation, thereby inducing osteolytic lesions [169]. CatG also activates pro-MMP9; the activated MMP9 in turn cleaves and releases active TGF- β, which promotes tumor growth and activated osteoclast and bone resorption [143]. Wilson et al. [143] also demonstrated that the inhibition of CatG in vivo via N-tosyl-l-phenylalanine chloromethyl ketone (TPCK; 50 mg/kg/d subcutaneous) significantly decreases MMP9 activity and reduces TGF- β signaling at the tumor–bone interface using the C166 adenocarcinoma cell line in BALB/c mice.
Rojinik et al. [56] reported that CatH regulates bone morphogenetic protein 4 (BMP-4) in mice, but not in human OS and PCa. Previous studies demonstrated that CatH and BMP-4 expression is related to lung branching in mice, and that the inhibition of CatH leads to an accumulation of BMP-4 in mouse lungs [170]. Contrary to the human OS cell line HOS, which does not express mature CatH, the PCa cell line PC-3 does expresses mature CatH [56]. The addition of mature CatH into the culture with HOS increased the mRNA expression of BMP-4 2-fold, and decreased BMP-4 mRNA levels 0.5-fold in PC-3. Confirming Rojnik et al.’s studies, Husmann et al., also showed that mature CatH is undetectable in the Saos-2 parental and metastatic sublines (LM5 and LM7) [55]. In OS, CatH does not appear to be correlated to metastasis because the protein expression of LM5 and LM7 are downregulated compared to Saos-2 [55]. CatH appears to influence the relative expression of the BMP family members by upregulating the mRNA expression of BMP-3, -6, and -7, and down regulating BMP-1, -4, -5, and -8. However, Rojnik et al. [56] demonstrated that, in the human OS (HOS) and PCa (PC-3) cell lines, CatH and BMP-4 do not co-localize, and the inhibition of CatH does not increase the expression of BMP-4 and has no direct impact on the processing or degradation of BMP-4. These findings suggest that CatH may indirectly regulate BMP-4, although the mechanism has not been elucidated [56].
CatK is overexpressed in human cancer, and is associated with primary tumor growth and the metastatic process [55,171]. The development of primary bone tumors, such as OS, is associated with a local enhancement of CatK expression and excretion resulting in pathological excessive bone resorption [78]. The CatK in primary and metastatic tumors is involved in tumor cell invasion via ECM degradation, osteolysis, and the modulation of cytokines and chemokines such as IL-1α and CCL2 [171]. In 63% of OS tissue samples, tumor-associated osteoclasts were found within the tumor mass [168]. The presence of osteoclasts in OS biopsies at diagnosis correlated with metastases in 50% of clinical cases, whereas 100% of patients with no osteoclasts present within the primary tumor did not have clinically-detectable metastases at diagnosis [168]. In 90% of OS cases with osteoclast infiltration, CatK mRNA was present [168]. Serum tartrate-resistant acid phosphatase 5b (TRACP 5b), a marker of bone resorption, has been significantly correlated to serum N-terminal telopeptide (NTx), a biomarker of bone turnover, and CatK mRNA in tumor tissues [168]. Neoplastic cells expressing CatK and multinucleated tartrate-resistant acid phosphatase (TRAcP) have direct contact with the bone trabeculae and bone marrow spaces near metastatic tumor cells [172]. TRACP 5b has been demonstrated to correlate with tumors’ aggressiveness in OS [168].
The mRNA and protein expression of Saos-2 and its metastatic sublines LM5 and LM7 demonstrate a 2-fold upregulation of CatK [55]. The osteoclast differentiation induced from co-cultured human OS cell line MG-63 with human macrophages results in the production of cytokines such as RANKL and colony stimulating factor-1 (CSF-1) [110]. CatK demonstrates activity at the tumor–bone interface, with no CatK levels detected away from the bone [124]. While it is absent in cells, the CatK activity on bone substrate increases several-fold during multinucleated differentiation [110]. No NF-k B sites occur near the CatK transcription site [173,174,175]; therefore, the specific osteoclast products involve unidentified transcription factors [110].
CatK has also been implicated in metastatic OS [55]. Approximately 15% of high grade non-metastatic OS patients and 53% of patients with high grade metastatic OS stained intensity for CatK [55]. Husmann et al. demonstrated that, in patients with metastatic high-grade OS and low CatK expression at the time of diagnosis, the survival was significantly better than in those patients with high CatK staining, demonstrating that CatK expression may be of predictive prognostic value for patients with high-grade metastatic tumors at diagnosis [55].
Leung et al., detected high CatK expression by immunohistochemistry in breast (45%, n = 88) and prostate (75%, n = 64) tumors [176]. CatK mRNA increased approximately 3-fold in metastatic tissue compared to primary tumor tissue [107]. In comparison to primary BCa and PCa samples, CatK is upregulated in bone metastases, and it is associated with increased tumor cell invasiveness [58,109].
In BCa, there is a statistical difference between healthy sex-matched controls and patients with primary or metastatic BCa [144]. Tumminello et al. reported that, although CatK serum levels are decreased (mean 1.2–1.9 pmol/L) in BCa patients in comparison to their sex-matched healthy controls (mean: 11.3–14.0 pmol/L), cystatin C levels were significantly higher in BCa patients (mean: 1.5–0.59 mg/L) than their controls (mean: 0.85–0.16 mg/L) [144]. Although cystatin C is a useful clinical marker for the differentiation of cancer patients from healthy controls, the cystatin C serum levels in primary and metastatic BCa and PCa patients are not significantly different [144].
CatK is positive in 12% of PCa tumor cell samples and 31% of stroma cells surrounding the tumor [177], but whereas cystatin C serum levels are significantly increased in PCa patients (1.7–0.65 mg/L), serum CatK levels are not [144]. The authors also noted that, in PCa, there is no statistical difference between the controls and cancer patients who have primary or metastatic lesions to the bone. Intense immunoreactivity for CatK was observed in osteoclasts, and elevated levels of serum NTx were detected in patients with bone metastases [109]. The administration of ZOL to patients with bone metastasis induces a significant increase of CatK and cystatin C serum levels in PCa patients [144]. These findings suggest that cystatin C may be regarded as a possible marker to monitor the therapeutic response to bisphosphonate treatment (i.e: ZOL). The understanding of the role of CatK in the bone microenvironment has led to a multitude of efforts to evaluate whether CatK inhibition is an effective therapeutic strategy for patients with BCa or PCa that metastasizes to the bone.
CatK secretion from PCa cell lines (LNCaP, DU145) and macrophages participates in local invasion by mediating extracellular degradation [109,177]. Liang et al. [178] reported that CatK expression occurs both in PCa cell lines (LNCaP, C4-2B, and PC3) and in PCa tissues. CatK protein and enzymatic activity has been detected in the human PCa cell lines by Western blot and a fluorogenic assay, respectively [109].
Tumor-bearing osteoclasts secrete large amounts of cysteine proteases, especially pro-CatL, leading to tumor-associated bone absorption i.e., bone pit formations and the release of bone calcium, [179]. Typically, CatL expression and secretion is associated with pro tumorigenic factors and osteoclastic cytokines, such as (IL-1, IL-6, and TNF-α), PTH, vascular endothelial growth factor (VEGF), and cellular-SRC [116,135,180,181,182,183].
Damiens et al. showed that, in human OS cell lines, MG-63 and Saos2 CatL activity was increased in the presence of IL-1, IL-6 and oncostatin M (hOSM), which is a cytokine of the IL-6 family. hOSM induces osteoblast cell proliferation, matrix protein synthesis, and IL-6 secretion in osteoblasts [184,185]. Human growth factor and IGF-1 decrease cathepsin activity in OS media. In human OS, there is a prevalence of CatL activity over CatB activity using the CatB and L substrate Z-Phe-Arg-AMC [186]. The 2-fold elevation of Z-Phe-Arg-AMC hydrolysis in comparison to fibroblasts in metastatic bone cancer suggests that CatL has an active function in the metastatic process [156]. These results differ from the findings of Aisa et al. [43], who demonstrated a prevalence of CatB activity using the CatB-specific substrate Z-Arg-Arg-AMC.
In 50% of primary bone tumor tissue and 100% of metastatic tumor samples, CatL mRNA is expressed [187]. Arkona et al. [156] observed an elevation of CatL, but not other cysteine cathepsins such as Cat D and CatH, suggesting a possibly unique role of CatL in metastasis. In vitro studies in OS indicate that CatL contributes to the metastatic potential of neoplastic cells [55,187]. The mRNA and protein expression of Saos-2 and its metastatic sublines LM5 and LM7 demonstrate an upregulation of CatL in metastasis; however, the mature active form of CatL increases in the metastatic OS cell line LM5, but not in the parental Saos-2 or metastatic subline LM7 [55].
The secretion of CatL in the microenvironment may also contribute to therapeutic drug resistance. CatL was associated with poor response to neoadjuvant chemotherapy, and CatL-positive patients achieved a lower complete remission rate compared to CatL-negative patients [108]. However, CatL does not influence event-free-survival (EFS) or overall survival (OS) [108].
4. Targeting Cathepsin in Primary Bone and Metastatic Bone Cancer
Several approaches have been developed to target cathepsin activity. Most of our understanding of cathepsins in primary and metastatic bone cancers comes from single gene manipulation and small molecular inhibitor studies in the laboratory or the clinic. The summary of the studies, below, highlights the effect of genetic and targeted small-molecule cathepsin inhibition on either primary or secondary bone tumors (Table 1).
4.1. Cathepsin Targeting in Primary Bone Tumors
The inhibition of CatB expression through antisense gene silencing and pharmacologic inhibition via CA-074 significantly decreases OS cell invasion [45]. Palladacycle (BPC) selectively inhibits CatB activity [188]. The extracellular inhibition of CatB through BPC reduces the metastatic potential of neoplastic cells; however, intracellular inhibition has been shown to increase the concentration of BPC in the lysosome, inducing lysosomal membrane permeabilization and releasing CatB into the cytosol [189].
The cell-permeable CatB inhibitor CA-074Me inhibits both TRAIL and anti-DR5 mediated apoptosis by delaying the cleavage of pre-apoptotic Bid in OS cells, U2OS [149]. Although the levels of Bid in the CA-074Me-treated cells appeared to be unchanged at 2 h, there was a significant decrease in the levels of Bid between 2 h and 4 h. These findings demonstrate that treatment with CA-074Me delays, but does not prevent, the cleavage of Bid, and that CatB is constitutively active even in the absence of TRAIL signaling. These findings also suggest that CatB functions upstream of Bid cleavage and plays an important role in the TRAIL pathways for U2OS cells. These reports are supported by studies from Bechara et al., who demonstrated that CatB inhibition by CA-074 reduced BPC induced cell death in the OS cell line Saos-2 [190].
CatK inhibition impairs tumor growth, progression in the bone, macrophage infiltration and inflammation [96]. CatK inhibition has also been demonstrated to not only affect osteoclast function but also interfere with BMM function and metastasis in PCa [96]. The genetic inhibition of CatK impaired bone degradation and intraosseous tumor growth, which suggest that the role CatK inhibition may be bone-specific [96]. CatK deficiency leads to increased osteoclast differentiation within the tumor–bone interface in wild-type tumors, and results in bone loss in the tumor microenvironment, which is correlated with increasing levels of CatK. In contrast, only low levels of CatK were detected in bone tumors in genetically-inhibited CatK, despite an increased number of osteoclasts [96].
CatL antisense oligonucleotides in the OS cell line MNNG/HOS significantly reduced CatL mRNA expression and reduced CatL protein expression by 50–85% in comparison to the control [191]. Although the adhesion to collagen I matrix was not affected, migration and invasion were inhibited by 35–75% using antisense oligonucleotides [191]. These studies demonstrate that CatL is a potential therapeutic target in OS.
CatL has also been demonstrated to induce chemotherapeutic resistance in BCa. Dox is a key chemotherapeutic agent for both OS and metastatic Bca treatment [4,192]. Zheng et al., demonstrated that the inhibition of CatL by the inhibitor napsul-Ile-Trp-CHO (iCL) and short interfering (si) RNA facilitates the reversal of dox resistance [193]. The targeted inhibition of CatL in the human OS cell line Saos-2 hindered the dox resistance in cells in vitro and in vivo through proteolytic cleavage and the elimination of drug targets. These data suggest that the inhibition of CatL both prevents and reverses the development of dox drug resistance. Although iCL does not independently suppress the proliferation of tumor cells, the combination of iCL with Dox suppressed the proliferation of drug resistant tumors in nude mice [193] (Figure 3) in the absence of enhanced host toxicity. The combination of iCL and dox does not affect non-resistant cells, which suggests that the efficacy of iCL with dox in resistance cell lines may be due to alterations in drug targets and/or drug availability. Zheng et al. postulated that iCL and CatL inhibition through siRNA led to an accumulation of topoisomerase II alpha expression in a multi-drug resistance protein 1 (MRP1)-independent manner. Alternatively, since the inhibition of CatL through iCL or siRNA forces cells to undergo senescence, as is evidenced through the up-regulation of the expression of the CatL substrate and the cell cycle inhibitor p21/WAF1 [194], it is possible that the induction of senescence through CatL inhibition may be sufficient to reverse drug resistance [194].
4.2. Cathepsin Targeting in Secondary Bone Tumors
In the tumor microenvironment, CatB secretion increases tumor cells’ metastatic potential by increasing cell migration and invasiveness [191]. The intraperitoneal administration of the CatB inhibitor CA-074 has been shown to reduce bone metastasis in the syngeneic 4T1.2 BCa cell line in Balb/c mice [147]. However, the pan cysteine cathepsin inhibitor JPM-OEt did not reduce metastasis in tumor bearing mice [147]. Interestingly, the effect of CatB on the inhibition of metastasis is not correlated to its role in cell proliferation or tumor growth [147]. Several studies have demonstrated that CatG plays a role in tumor-mediated bone remodeling. In the moderately metastatic BCa cell line Cl66, CatG is significantly upregulated at the tumor–bone interface [169]. When the RAW264.7 macrophages were treated with 25% C166-conditioned medium, a 30% increase in osteoclast differentiation was observed. TPCK significantly inhibited the osteoclastogenesis induced by C166-conditioned medium [53,169,195].
Odanacatib (MK·0822, MK-822) is an orally-administered, non-basic, nitrile, highly selective CatK inhibitor with a high metabolic half-life developed by Merck & Co. Inc. Odanacatib exhibited robust, sustained and reversible antiresorptive activity, with no demonstrable effect on off-target cathepsins, as observed with competing CatK inhibitors [196]. Furthermore, Odanacatib showed significantly higher selectivity in cellular assays than other competing CatK inhibitors [197,198]. This agent was primarily marketed as an anti-osteoporotic agent; however, several clinical studies have demonstrated that cathepsin inhibition could be useful for patients with bone metastases [199]. However, the occurrence of a skin rash and rarer incidences of morphea-like skin reactions [200], as well as an increased stroke risk in osteoporotic patients during phase III clinical trials [201], led to the discontinuation of further clinical development of Odanacatib and other competing CaK inhibitors [202].
Although the use of Odanacatib is inappropriate for osteoporosis treatment due to the greater adverse effects, these adverse effects might not preclude the use of these drugs in life-threatening diseases such as bone metastasis. Indeed, targeting cathepsins (or combinations of cathepsins) may be promising in an oncologic setting. Studies with Odanacatib demonstrated that CatK inhibition suppressed bone resorption in women with primary BCa and metastatic bone cancer [199]. Odanacatib suppressed markers of bone resorption, such as uNTx (77%) and deoxypyridinoline (uDPD), (30%) at rates similar to the effect observed with ZOL [199].
The reversible small molecule CatK inhibitor L-235 (with structural similarity to Odanacatib) reduces tumor-mediated osteolysis in a preclinical Bca metastasis model [107,197,203]. When administered both preventatively (10, 30, and 100 mg/kg, p.o., b.i.d; with dosing started on day-1) and as a treatment (30 mg/kg with dosing started day-7 after tumor cell injection) for established MDA-MB-231 BCa tumors in nude rats, L-235 significantly reduced intratibial tumor growth. However, when administered as a treatment, L-235 and ZOL did not inhibit tumor cell infiltration in the trabecular bone. Although treatment with L-235 (up to 10 mmol/L) did not change the proliferation rate of MDA-MB- 231 cells in vitro, L-235 effectively decreased cell invasion in comparison to ZOL [107]. In vivo treatment with L-235 in the prevention protocol at 30 and 100 mg/kg also significantly reduced the metastatic incidence by 38 and 51%, respectively [107]. In the established tumor treatment protocol, L-235 (30 mg/kg) reduced the metastasis incidence by 42%, and the skeletal and soft tissue BCa tumor burden by 60–80% [107]. In the intratibial model of MDA-MB-231 BCa cells in nude rats, histology showed that L-235 significantly reduced micrometastases locally between trabeculae and distally in diaphyseal bone marrow, and directly inhibited BCa cell invasion in vitro [176]. However, it is important to note that, although L-235 is highly selective to CatK (Ki 0.25 nmol/L), it is also >4000-fold selective against human CatB and CatL, which also can affect bone remodeling [203], thus confounding whether the observed effects are strictly due to CatK inhibition. Observations with a variety of other CatK inhibitors further support the possibility that CatK could be a therapeutic target for the treatment of bone metastases. For example, CatK inhibition though AFG-495 was reported to reduce the skeletal tumor burden and cancer-mediated osteolysis when CatK-expressing human BT474 BCa cells were injected into the tibia of nude mice [204] (Figure 4). In the same tumor model, twice daily treatment with the CatK inhibitor CKI (50 mg/kg) resulted in osteolytic lesions that were 79% smaller than those of tumor-bearing mice treated with the drug vehicle, and in a 62% reduction in the skeletal tumor burden [204]. More recently, this inhibitor was used to treat C4-2B prostate cancer cells in the tibiae of SCID mice; it significantly inhibited the tumor establishment, reduced the tumor growth in the bone, and decreased the serum prostate-specific antigen, likely due to the noted inhibition of bone resorption demonstrated in vitro with the CKI treatment [178]. Interestingly, the inhibitory effects of the CatK inhibitor were enhanced when it was administered in combination with ZOL [178].
Another important cathepsin in the metastatic process to the bone is CatL. This enzyme is increased in the MCF-7 BCa cell line as well as the PCa cell lines LNCaP and ARCaP-E when the Snail transcription factor is activated [205]. The Snail transcription factor has been shown to be crucial for the epithelial to mesenchymal transition, which is the key for metastasis. The Snail-mediated increase in CatL activity has been correlated to phosphorylated STAT-3 (pSTAT-3) [205]. Muscadine grape skin extract has been shown to decrease Snail, pSTAT-3 and Cat-L activity, which also correlates with an abrogation of cell motility and invasion [205]. Previous studies have shown that RANKL can be upregulated by Snail overexpression [206]. Snail expression is higher in tumor samples from aggressive and metastatic PCa patients, and it promotes osteoclastogenesis in vitro and in vivo [206]. STAT-3 inhibition in cells that overexpress Snail decreases Snail and CatL expression [205]. Muscadine grape skin extract abrogates the Snail-mediated increase of CatL osteoclastogenesis to inhibit bone turnover [205]. CatL inhibition via CLIK-148 also decreased levels of hypercalcemia and metastasis in metastatic bone cancer [179]. CLIK-148 is able to suppress TNF-alpha, RANKL, and m-CSF mediated pit formation in vitro [179].
Given the important roles of CatK and CatL in bone remodeling and tumor progression, dual targeted cathepsin therapy may be an advantageous therapeutic approach. The CatL/CatK inhibitor KGP94 (3-bromophenyl-3-hydroxyphenyl-ketone thiosemicarbazone) selectively impairs CatL and CatK proteolytic function by targeting their active sites [207,208]. Importantly, it targets secreted CatL and CatK, and does not interfere with the intracellular functions of these cathepsins. In the metastatic PCa cell subline PC3-ML, the daily administration of KGP94 (20 mg/kg) significantly inhibited the establishment and growth of intracardially-injected tumor cells [99] (Figure 5). Prior studies from Sudhan et al. [99,209,210] demonstrated that KGP94 overcomes the increased metastatic potential of tumor cells under aberrant physiologic conditions and blocks multiple facets of PCa and BCa metastasis.
5. Conclusions and Perspectives
Metastases are the primary cause of cancer-related deaths. In prostate and breast cancer, bone is the favored site of tumor cell dissemination, and skeletal metastases lead to a drastic worsening of the treatment outcome and a substantial reduction in the quality of life. Cathepsins are a class of proteolytic enzymes that are critical contributors to bone remodeling physiologically. In cancer, evolving evidence has identified this family of enzymes as key contributors to neoplastic growth and progression in the bone environment. The ability for cathepsins to influence tumor progression and metastatic aggressiveness has made them an attractive target for the development of anti-metastatic therapeutic agents. Indeed, substantial data exists to support the notion that targeting these enzymes can effectively impair the bone tumor burden. Furthermore, the observation that multiple cathepsins can impact the progression and dissemination of primary bone cancer and bone metastases suggests that dual or multi-cathepsin-targeted therapies may potentially be most effective; this concept has received only limited attention, and deserves further exploration as a means to enhance cancer treatments. Clearly, additional studies are needed in order to elucidate the mechanism of actions of the family of cathepsins systemically as well as locally, particularly in light the multimodal roles of cathepsins in the human body. Still, the growing body of preclinical evidence and our increased understanding of the role of cathepsins in the bone microenvironment suggest that adjuvant systemic therapy targeting cathepsins may be beneficial for patients who are at high risk of a poorer prognosis.
Author Contributions
H.O.F. and D.W.S. wrote and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This review was carried out with funding provided by National Center for Advancing Translational Sciences of the National Institutes of Health under University of Florida Clinical and Translational Science Awards TL1TR001428 and UL1TR001427. The content is solely the responsibility of the authors, and does not necessarily represent the official views of the National Institutes of Health. This review is supported in part by grants from the National Cancer Institute (US Public Health Service Grants R01 CA169300 and R01 CA197477) and the Florida Education Fund McKnight Doctoral Fellowship.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| BCa | Breast Cancer |
| BMP-4 | Bone Morphogenic Protein 4 |
| CAM | Cell Adhesion Molecule |
| Cat | Cathepsin |
| CCL2 | Macrophage Chemo Attractant Protein-1 |
| CCR2 | C-C Chemokine Receptor Type 2 |
| CSF | Colony Stimulating Factor |
| Dox | Doxorubicin |
| ECM | Extracellular Matrix |
| GPCR | G Protein Coupled Receptor |
| hOSM | Oncostatin M |
| IGF | Insulin-Like Growth Factor |
| IL | Interleukin |
| m-CSF | Macrophage Colony Stimulating Factor |
| MDSC | Myeloid-Derived Suppressor Cells |
| NF-κB | Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells |
| OS | Osteosarcoma |
| PAR-1 | Protease-Activated Receptor-1 |
| PCa | Prostate Cancer |
| PTH | Parathyroid Hormone |
| PTHrP | Parathyroid Hormone Related Peptide |
| RANKL | Receptor Activator of NF-Kappa-β Ligand |
| SCID | Severe Combined Immunodeficiency |
| sRANKL | Soluble Receptor Activator of NF-Kappa-β Ligand |
| TGF-β | Transforming Growth Factor-beta |
| TNFα | Tumor Necrosis Factor-alpha |
| TRAIL-DR5 | Tumor Necrosis Factor-Related Apoptosis- Inducing Ligand Death Receptor-5 |
| TRACP 5b | Tartrate-Resistant Acid Phosphatase 5b |
| uPA | Urokinase-Plasminogen Activator |
| VEGF | Vascular Endothelial Growth Factor |
| ZOL | Zoledronic Acid |
References
- Ottaviani, G.; Jaffe, N. The epidemiology of osteosarcoma. Cancer Treat Res. 2009, 152, 3–13. [Google Scholar] [PubMed]
- Ries, L.A.G. Cancer Incidence and Survival among Children and Adolescents: United States SEER Program 1975–1995; Smith, M.A., Gurney, J.G., Linet, M., Tamra, T., Young, J.L., Bunin, G.R., Eds.; National Cancer Institute (NCI): Bethesda, MA, USA, 1999. [Google Scholar]
- Linabery, A.M.; Ross, J.A. Childhood and adolescent cancer survival in the US by race and ethnicity for the diagnostic period 1975–1999. Cancer 2008, 113, 2575–2596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geller, D.S.; Gorlick, R. Osteosarcoma: A review of diagnosis, management, and treatment strategies. Clin. Adv. Hematol. Oncol. 2010, 8, 705–718. [Google Scholar] [PubMed]
- Theriault, R.L. Biology of bone metastases. Cancer Control 2012, 19, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Durfee, R.A.; Mohammed, M.; Luu, H.H. Review of Osteosarcoma and Current Management. Rheumatol. Ther. 2016, 3, 221–243. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, W.S.; Goorin, A.M. Current treatment of osteosarcoma. Cancer Investig. 2001, 19, 292–315. [Google Scholar] [CrossRef]
- Fuchs, B.; Pritchard, D.J. Etiology of osteosarcoma. Clin. Orthop Relat Res. 2002, 397, 40–52. [Google Scholar] [CrossRef]
- Marina, N.; Gebhardt, M.; Teot, L.; Gorlick, R. Biology and therapeutic advances for pediatric osteosarcoma. Oncologist 2004, 9, 422–441. [Google Scholar] [CrossRef]
- Smith, M.A.; Seibel, N.L.; Altekruse, S.F.; Ries, L.A.; Melbert, D.L.; O’Leary, M.; Smith, F.O.; Reaman, G.H. Outcomes for children and adolescents with cancer: Challenges for the twenty-first century. J. Clin. Oncol. 2010, 28, 2625–2634. [Google Scholar] [CrossRef]
- Ferrari, S.; Serra, M. An update on chemotherapy for osteosarcoma. Expert Opin. Pharmacother. 2015, 16, 2727–2736. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paget, S. The distribution of secondary growths in cancer of the breast. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar] [CrossRef] [Green Version]
- Awolaran, O.; Brooks, S.A.; Lavender, V. Breast cancer osteomimicry and its role in bone specific metastasis; an integrative, systematic review of preclinical evidence. Breast 2016, 30, 156–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rucci, N.; Teti, A. Osteomimicry: How tumor cells try to deceive the bone. Front. Biosci. 2010, 2, 907–915. [Google Scholar]
- Barnes, G.L.; Hebert, K.E.; Kamal, M.; Javed, A.; Einhorn, T.A.; Lian, J.B.; Stein, G.S.; Gerstenfeld, L.C. Fidelity of Runx2 activity in breast cancer cells is required for the generation of metastases-associated osteolytic disease. Cancer Res. 2004, 64, 4506–4513. [Google Scholar] [CrossRef] [Green Version]
- Croucher, P.I.; McDonald, M.M.; Martin, T.J. Bone metastasis: The importance of the neighbourhood. Nat. Rev. Cancer 2016, 16, 373–386. [Google Scholar] [CrossRef]
- Coleman, R.E.; Rubens, R.D. The clinical course of bone metastases from breast cancer. Br. J. Cancer 1987, 55, 61–66. [Google Scholar] [CrossRef]
- Kakhki, V.R.; Anvari, K.; Sadeghi, R.; Mahmoudian, A.S.; Torabian-Kakhki, M. Pattern and distribution of bone metastases in common malignant tumors. Nucl. Med. Rev. Cent. East Eur. 2013, 16, 66–69. [Google Scholar] [CrossRef] [Green Version]
- Mundy, G.R. Metastasis to bone: Causes, consequences and therapeutic opportunities. Nat. Rev. Cancer 2002, 2, 584–593. [Google Scholar] [CrossRef]
- Dhillon, S.; Lyseng-Williamson, K.A. Zoledronic acid: A review of its use in the management of bone metastases of malignancy. Drugs 2008, 68, 507–534. [Google Scholar] [CrossRef]
- Rubens, R.D. Metastatic breast cancer. Curr. Opin. Oncol. 1995, 7, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Nørgaard, M.; Jensen, A.; Jacobsen, J.B.; Cetin, K.; Fryzek, J.P.; Sørensen, H.T. Skeletal related events, bone metastasis and survival of prostate cancer: A population based cohort study in Denmark (1999 to 2007). J. Urol. 2010, 184, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Roodman, G.D. Mechanisms of bone metastasis. Discov. Med. 2004, 4, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.J.; Singh, R.K. Proteases as modulators of tumor-stromal interaction: Primary tumors to bone metastases. Biochim. Biophys. Acta 2008, 1785, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, R.E. Metastatic bone disease: Clinical features, pathophysiology and treatment strategies. Cancer Treat. Rev. 2001, 27, 165–176. [Google Scholar] [CrossRef]
- Onishi, T.; Hayashi, N.; Theriault, R.L.; Hortobagyi, G.N.; Ueno, N.T. Future directions of bone-targeted therapy for metastatic breast cancer. Nat. Rev. Clin. Oncol. 2010, 7, 641–651. [Google Scholar] [CrossRef]
- Kingsley, L.A.; Fournier, P.G.; Chirgwin, J.M.; Guise, T.A. Molecular biology of bone metastasis. Mol. Cancer Ther. 2007, 6, 2609–2617. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.J.; Saylor, P.J.; Smith, M.R. Treatment and prevention of bone complications from prostate cancer. Bone 2011, 48, 88–95. [Google Scholar] [CrossRef] [Green Version]
- Roato, I.; D’Amelio, P.; Gorassini, E.; Grimaldi, A.; Bonello, L.; Fiori, C.; Delsedime, L.; Tizzani, A.; De Libero, A.; Isaia, G.; et al. Osteoclasts are active in bone forming metastases of prostate cancer patients. PLoS ONE. 2008, 3, e3627. [Google Scholar] [CrossRef]
- Palermo, C.; Joyce, J.A. Cysteine cathepsin proteases as pharmacological targets in cancer. Trends Pharmacol. Sci. 2008, 29, 22–28. [Google Scholar] [CrossRef]
- Mason, S.D.; Joyce, J.A. Proteolytic networks in cancer. Trends Cell Biol. 2011, 21, 228–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coussens, L.M.; Fingleton, B.; Matrisian, L.M. Matrix metalloproteinase inhibitors and cancer: Trials and tribulations. Science 2002, 295, 2387–2392. [Google Scholar] [CrossRef] [PubMed]
- Fingleton, B. Matrix metalloproteinases as valid clinical targets. Curr. Pharm. Des. 2007, 13, 333–346. [Google Scholar] [CrossRef] [Green Version]
- Overall, C.M.; Kleifeld, O. Tumour microenvironment—opinion: Validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat. Rev. Cancer 2006, 6, 227–239. [Google Scholar] [CrossRef]
- Overall, C.M.; López-Otín, C. Strategies for MMP inhibition in cancer: Innovations for the post-trial era. Nat. Rev. Cancer 2002, 2, 657–672. [Google Scholar] [CrossRef]
- Zucker, S.; Cao, J.; Chen, W.T. Critical appraisal of the use of matrix metalloproteinase inhibitors in cancer treatment. Oncogene 2000, 19, 6642–6650. [Google Scholar] [CrossRef]
- Krüger, A.; Kates, R.E.; Edwards, D.R. Avoiding spam in the proteolytic internet: Future strategies for anti-metastatic MMP inhibition. Biochim. Biophys. Acta 2010, 1803, 95–102. [Google Scholar] [CrossRef]
- Everts, V.; Korper, W.; A Hoeben, K.; Jansen, I.D.; Brömme, D.; Cleutjens, K.B.; Heeneman, S.; Peters, C.; Reinheckel, T.; Saftig, P.; et al. Osteoclastic bone degradation and the role of different cysteine proteinases and matrix metalloproteinases: Differences between calvaria and long bone. J. Bone Miner. Res. 2006, 21, 1399–1408. [Google Scholar] [CrossRef]
- Abboud-Jarrous, G.; Atzmon, R.; Peretz, T.; Palermo, C.; Gadea, B.B.; Joyce, J.A.; Vlodavsky, I. Cathepsin L is responsible for processing and activation of proheparanase through multiple cleavages of a linker segment. J. Biol. Chem. 2008, 283, 18167–18176. [Google Scholar] [CrossRef] [Green Version]
- Goretzki, L.; Schmitt, M.; Mann, K.; Calvete, J.J.; Chucholowski, N.; Kramer, M.; Günzler, W.A.; Jänicke, F.; Graeff, H. Effective activation of the proenzyme form of the urokinase-type plasminogen activator (pro-uPA) by the cysteine protease cathepsin L. FEBS Lett. 1992, 297, 112–118. [Google Scholar] [CrossRef] [Green Version]
- Aisa, M.C.; Rahman, S.; Senin, U.; Maggio, D.; Russell, R.G.G. Cathepsin B activity in normal human osteoblast-like cells and human osteoblastic osteosarcoma cells (MG-63): Regulation by interleukin-1-beta and parathyroid hormone. Biochim. Biophys. Acta 1996, 1290, 29–36. [Google Scholar] [CrossRef]
- Repnik, U.; Stoka, V.; Turk, V.; Turk, B. Lysosomes and lysosomal cathepsins in cell death. Biochim. Biophys. Acta 2012, 1824, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Krueger, S.; Haeckel, C.; Buehling, F.; Roessner, A. Inhibitory effects of antisense cathepsin B cDNA transfection on invasion and motility in a human osteosarcoma cell line. Cancer Res. 1999, 59, 6010–6014. [Google Scholar]
- Sloane, B.F.; Rozhin, J.; Johnson, K.; Taylor, H.; Crissman, J.D.; Honn, K.V. Cathepsin B: Association with plasma membrane in metastatic tumors. Proc. Natl. Acad. Sci. USA 1986, 83, 2483–2487. [Google Scholar] [CrossRef] [Green Version]
- Haeckel, C.; Ayala, A.G.; Radig, K.; Raymond, A.K.; Roessner, A.; Czerniak, B. Protease expression in dedifferentiated parosteal osteosarcoma. Arch. Pathol. Lab. Med. 1999, 123, 213–221. [Google Scholar]
- Köppel, P.; Baici, A.; Keist, R.; Matzku, S.; Keller, R. Cathepsin B-like proteinase as a marker for metastatic tumor cell variants. Exp. Cell Biol. 1984, 52, 293–299. [Google Scholar]
- Rawlings, N.D.; Waller, M.; Barrett, A.J.; Bateman, A. MEROPS: The database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2014, 42, D503–D509. [Google Scholar] [CrossRef] [Green Version]
- Rochefort, H.; Capony, F.; Garcia, M. Cathepsin D: A protease involved in breast cancer metastasis. Cancer Metastasis Rev. 1990, 9, 321–331. [Google Scholar] [CrossRef]
- Conover, C.A.; Perry, J.E.; Tindall, D.J. Endogenous cathepsin D-mediated hydrolysis of insulin-like growth factor-binding proteins in cultured human prostatic carcinoma cells. J. Clin. Endocrinol. Metab. 1995, 80, 987–993. [Google Scholar]
- Van der Stappen, J.W.; Williams, A.C.; Maciewicz, R.A.; Paraskeva, C. Activation of cathepsin B, secreted by a colorectal cancer cell line requires low pH and is mediated by cathepsin D. Int. J. Cancer 1996, 67, 547–554. [Google Scholar] [CrossRef]
- Wilson, T.J.; Nannuru, K.C.; Singh, R.K. Cathepsin G recruits osteoclast precursors via proteolytic activation of protease-activated receptor-1. Cancer Res. 2009, 69, 3188–3195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafatian, N.; Karunakaran, D.; Rayner, K.J.; Leenen, F.H.; Milne, R.W.; Whitman, S.C. Cathepsin G deficiency decreases complexity of atherosclerotic lesions in apolipoprotein E-deficient mice. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1141–H1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husmann, K.; Muff, R.; Bolander, M.E.; Sarkar, G.; Born, W.; Fuchs, B. Cathepsins and osteosarcoma: Expression analysis identifies cathepsin K as an indicator of metastasis. Mol. Carcinog. 2008, 47, 66–73. [Google Scholar] [CrossRef]
- Rojnik, M.; Jevnikar, Z.; Mirković, B.; Janeš, D.; Zidar, N.; Kikelj, D.; Kos, J. Cathepsin H indirectly regulates morphogenetic protein-4 (BMP-4) in various human cell lines. Radiol. Oncol. 2011, 45, 259–266. [Google Scholar] [CrossRef]
- Ketterer, S.; Gomez-Auli, A.; Hillebrand, L.E.; Petrera, A.; Ketscher, A.; Reinheckel, T. Inherited diseases caused by mutations in cathepsin protease genes. FEBS J. 2017, 284, 1437–1454. [Google Scholar] [CrossRef] [Green Version]
- Littlewood-Evans, A.J.; Bilbe, G.; Bowler, W.; Farley, D.; Wlodarski, B.; Kokubo, T.; Inaoka, T.; Sloane, J.; Evans, D.B.; A Gallagher, J. The osteoclast-associated protease cathepsin K is expressed in human breast carcinoma. Cancer Res. 1997, 57, 5386–5390. [Google Scholar]
- Vääräniemi, J.; Halleen, J.M.; Kaarlonen, K.; Ylipahkala, H.; Alatalo, S.L.; Andersson, G.; Kaija, H.; Vihko, P.; Väänänen, H.K. Intracellular machinery for matrix degradation in bone-resorbing osteoclasts. J. Bone Miner. Res. 2004, 19, 1432–1440. [Google Scholar] [CrossRef]
- Tezuka, K.; Tezuka, Y.; Maejima, A.; Sato, T.; Nemoto, K.; Kamioka, H.; Hakeda, Y.; Kumegawa, M. Molecular cloning of a possible cysteine proteinase predominantly expressed in osteoclasts. J. Biol. Chem. 1994, 269, 1106–1109. [Google Scholar]
- Inaoka, T.; Bilbe, G.; Ishibashi, O.; Tezuka, K.; Kumegawa, M.; Kokubo, T. Molecular cloning of human cDNA for cathepsin K: Novel cysteine proteinase predominantly expressed in bone. Biochem. Biophys. Res. Commun. 1995, 206, 89–96. [Google Scholar] [CrossRef]
- Li, Y.P.; Alexander, M.; Wucherpfennig, A.L.; Yelick, P.; Chen, W.; Stashenko, P. Cloning and complete coding sequence of a novel human cathepsin expressed in giant cells of osteoclastomas. J. Bone Miner. Res. 1995, 10, 1197–1202. [Google Scholar] [CrossRef] [PubMed]
- Bossard, M.J.; Tomaszek, T.A.; Thompson, S.K.; Amegadzie, B.Y.; Hanning, C.R.; Jones, C.; Kurdyla, J.T.; McNulty, D.E.; Drake, F.H.; Gowen, M.; et al. Proteolytic activity of human osteoclast cathepsin K. Expression, purification, activation, and substrate identification. J. Biol. Chem. 1996, 271, 12517–12524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelb, B.D.; Shi, G.P.; Chapman, H.A.; Desnick, R.J. Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency. Science 1996, 273, 1236–1238. [Google Scholar] [CrossRef] [PubMed]
- Saftig, P.; Hunziker, E.; Wehmeyer, O.; Jones, S.; Boyde, A.; Rommerskirch, W.; Moritz, J.D.; Schu, P.; Von Figura, K. Impaired osteoclastic bone resorption leads to osteopetrosis in cathepsin-K-deficient mice. Proc. Natl. Acad. Sci. USA 1998, 95, 13453–13458. [Google Scholar] [CrossRef] [Green Version]
- Duong, L.T. Therapeutic inhibition of cathepsin K-reducing bone resorption while maintaining bone formation. Bonekey Rep. 2012, 1, 67. [Google Scholar] [CrossRef] [Green Version]
- Costa, A.G.; Cusano, N.E.; Silva, B.C.; Cremers, S.; Bilezikian, J.P. Cathepsin K: Its skeletal actions and role as a therapeutic target in osteoporosis. Nat. Rev. Rheumatol. 2011, 7, 447–456. [Google Scholar] [CrossRef]
- Everts, V.; Aronson, D.C.; Beertsen, W. Phagocytosis of bone collagen by osteoclasts in two cases of pycnodysostosis. Calcif. Tissue Int. 1985, 37, 25–31. [Google Scholar] [CrossRef]
- Everts, V.; Hou, W.S.; Rialland, X.; Tigchelaar, W.; Saftig, P.; Gelb, B.D.; Beertsen, W. Cathepsin K deficiency in pycnodysostosis results in accumulation of non-digested phagocytosed collagen in fibroblasts. Calcif. Tissue Int. 2003, 73, 380–386. [Google Scholar] [CrossRef]
- Turan, S. Current research on pycnodysostosis. Intractable Rare Dis Res. 2014, 3, 91–93. [Google Scholar] [CrossRef] [Green Version]
- Barrett, A.J.; Kirschke, H. Cathepsin B, cathepsin H, and cathepsin L. Methods Enzymol. 1981, 80, 535–561. [Google Scholar] [PubMed]
- Hsing, L.C.; Rudensky, A.Y. The lysosomal cysteine proteases in MHC class II antigen presentation. Immunol. Rev. 2005, 207, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Zavasnik-Bergant, T.; Turk, B. Cysteine cathepsins in the immune response. Tissue Antigens 2006, 67, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Brix, K.; Dunkhorst, A.; Mayer, K.; Jordans, S. Cysteine cathepsins: Cellular roadmap to different functions. Biochimie 2008, 90, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim. Biophys. Acta 2012, 1824, 68–88. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, Y.; Kaleta, J.; Brömme, D. The role of cathepsins in osteoporosis and arthritis: Rationale for the design of new therapeutics. Adv. Drug Deliv. Rev. 2005, 57, 973–993. [Google Scholar] [CrossRef]
- Salminen-Mankonen, H.J.; Morko, J.; Vuorio, E. Role of cathepsin K in normal joints and in the development of arthritis. Curr. Drug Targets 2007, 8, 315–323. [Google Scholar] [CrossRef]
- Georges, S.; Ruiz Velasco, C.; Trichet, V.; Fortun, Y.; Heymann, D.; Padrines, M. Proteases and bone remodelling. Cytokine Growth Factor Rev. 2009, 20, 29–41. [Google Scholar] [CrossRef]
- Garnero, P.; Ferreras, M.; Karsdal, M.; NicAmhlaoibh, R.; Risteli, J.; Borel, O.; Qvist, P.; Delmas, P.; Foged, N.; Delaissé, J.-M. The type I collagen fragments ICTP and CTX reveal distinct enzymatic pathways of bone collagen degradation. J. Bone Miner. Res. 2003, 18, 859–867. [Google Scholar] [CrossRef]
- Mandelin, J.; Hukkanen, M.; Li, T.-F.; Korhonen, M.; Liljeström, M.; Sillat, T.; Hanemaaijer, R.; Salo, J.; Santavirta, S.; Konttinen, Y.T. Human osteoblasts produce cathepsin K. Bone 2006, 38, 769–777. [Google Scholar] [CrossRef]
- Kimura, S.; Sato, Y.; Matsubara, H.; Adachi, I.; Yamaguchi, K.; Suzuki, M.; Suemasu, K.; Abe, K. A retrospective evaluation of the medical treatment of malignancy-associated hypercalcemia. Jpn. J. Cancer Res. 1986, 77, 85–91. [Google Scholar]
- Rubens, R.D. Bone metastases—The clinical problem. Eur. J. Cancer 1998, 34, 210–213. [Google Scholar] [CrossRef]
- Delmas, P.D.; Fontana, A. Bone loss induced by cancer treatment and its management. Eur. J. Cancer 1998, 34, 260–262. [Google Scholar] [CrossRef]
- Kakegawa, H.; Nikawa, T.; Tagami, K.; Kamioka, H.; Sumitani, K.; Kawata, T.; Drobnič-Kosorok, M.; Lenarčič, B.; Turk, V.; Katunuma, N. Participation of cathepsin L on bone resorption. FEBS Lett. 1993, 321, 247–250. [Google Scholar] [CrossRef] [Green Version]
- Leto, G.; Crescimanno, M.; Flandina, C.; Sepporta, M.V.; Tumminello, F.M. Cathepsin L in Normal and Pathological Bone Remodeling. Clin. Rev. Bone Miner. Metab. 2011, 9, 107–121. [Google Scholar] [CrossRef]
- Nakase, T.; Kaneko, M.; Tomita, T.; Myoui, A.; Ariga, K.; Sugamoto, K.; Uchiyama, Y.; Ochi, T.; Yoshikawa, H. Immunohistochemical detection of cathepsin D, K, and L in the process of endochondral ossification in the human. Histochem. Cell Biol. 2000, 114, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Brage, M.; Abrahamson, M.; Lindström, V.; Grubb, A.; Lerner, U.H. Different cysteine proteinases involved in bone resorption and osteoclast formation. Calcif. Tissue Int. 2005, 76, 439–447. [Google Scholar] [CrossRef]
- Goto, T.; Tsukuba, T.; Kiyoshima, T.; Nishimura, Y.; Kato, K.; Yamamoto, K.; Tanaka, T. Immunohistochemical localization of cathepsins B, D and L in the rat osteoclast. Histochemistry 1993, 99, 411–414. [Google Scholar] [CrossRef]
- Evans, P.; Etherington, D.J. Characterisation of cathepsin B and collagenolytic cathepsin from human placenta. Eur. J. Biochem. 1978, 83, 87–97. [Google Scholar] [CrossRef]
- Burleigh, M.C.; Barrett, A.J.; Lazarus, G.S. Cathepsin B1. A lysosomal enzyme that degrades native collagen. Biochem. J. 1974, 137, 387–398. [Google Scholar] [CrossRef] [Green Version]
- Buck, M.R.; Karustis, D.G.; Day, N.A.; Honn, K.V.; Sloane, B.F. Degradation of extracellular-matrix proteins by human cathepsin B from normal and tumour tissues. Biochem. J. 1992, 282, 273–278. [Google Scholar] [CrossRef] [Green Version]
- Murphy, G.; Ward, R.; Gavrilovic, J.; Atkinson, S. Physiological mechanisms for metalloproteinase activation. Matrix Suppl. 1992, 1, 224–230. [Google Scholar] [PubMed]
- Kobayashi, H.; Schmitt, M.; Goretzki, L.; Chucholowski, N.; Calvete, J.; Kramer, M.; Günzler, W.A.; Jänicke, F.; Graeff, H. Cathepsin B efficiently activates the soluble and the tumor cell receptor-bound form of the proenzyme urokinase-type plasminogen activator (Pro-uPA). J. Biol. Chem. 1991, 266, 5147–5152. [Google Scholar] [PubMed]
- Jiao, W.J.; Xu, J.; Pan, H.; Wang, T.Y.; Shen, Y. Effect of endothelin-1 in esophageal squamous cell carcinoma invasion and its correlation with cathepsin B. World J. Gastroenterol. 2007, 13, 4002–4005. [Google Scholar] [CrossRef] [Green Version]
- Edgington-Mitchell, L.E.; Rautela, J.; Duivenvoorden, H.M.; Jayatilleke, K.M.; Van Der Linden, W.A.; Verdoes, M.; Bogyo, M.; Parker, B.S. Cysteine cathepsin activity suppresses osteoclastogenesis of myeloid-derived suppressor cells in breast cancer. Oncotarget 2015, 6, 27008–27022. [Google Scholar] [CrossRef] [PubMed]
- Herroon, M.K.; Rajagurubandara, E.; Rudy, D.L.; Chalasani, A.; Hardaway, A.L.; Podgorski, I. Macrophage cathepsin K promotes prostate tumor progression in bone. Oncogene 2013, 32, 1580–1593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiviranta, R.; Morko, J.; Alatalo, S.L.; NicAmhlaoibh, R.; Risteli, J.; Laitala-Leinonen, T.; Vuorio, E. Impaired bone resorption in cathepsin K-deficient mice is partially compensated for by enhanced osteoclastogenesis and increased expression of other proteases via an increased RANKL/OPG ratio. Bone 2005, 36, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.R.; Peters, C.; Saftig, P.; Brömme, D. Cathepsin K activity-dependent regulation of osteoclast actin ring formation and bone resorption. J. Biol. Chem. 2009, 284, 2584–2592. [Google Scholar] [CrossRef] [Green Version]
- Sudhan, D.R.; Pampo, C.; Rice, L.; Siemann, D.W. Cathepsin L inactivation leads to multimodal inhibition of prostate cancer cell dissemination in a preclinical bone metastasis model. Int. J. Cancer 2016, 138, 2665–2677. [Google Scholar] [CrossRef] [Green Version]
- Brage, M.; Lie, A.; Ransjö, M.; Kasprzykowski, F.; Kasprzykowska, R.; Abrahamson, M.; Grubb, A.; Lerner, U. Osteoclastogenesis is decreased by cysteine proteinase inhibitors. Bone 2004, 34, 412–424. [Google Scholar] [CrossRef]
- Patel, N.; Nizami, S.; Song, L.; Mikami, M.; Hsu, A.; Hickernell, T.; Chandhanayingyong, C.; Rho, S.; Compton, J.; Caldwell, J.-M.; et al. CA-074Me compound inhibits osteoclastogenesis via suppression of the NFATc1 and c-FOS signaling pathways. J. Orthop. Res. 2015, 33, 1474–1486. [Google Scholar] [CrossRef]
- Strålberg, F.; Henning, P.; Gjertsson, I.; Kindlund, B.; Souza, P.P.C.; Persson, E.; Abrahamson, M.; Kasprzykowski, F.; Grubb, A.; Lerner, U. Cysteine proteinase inhibitors regulate human and mouse osteoclastogenesis by interfering with RANK signaling. FASEB J. 2013, 27, 2687–2701. [Google Scholar] [CrossRef] [PubMed]
- Traynelis, S.F.; Trejo, J. Protease-activated receptor signaling: New roles and regulatory mechanisms. Curr. Opin. Hematol. 2007, 14, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Kamiyama, M.; Tani-Ishii, N.; Suzuki, H.; Ichikawa, Y.; Hamaguchi, Y.; Momiyama, N.; Shimada, H. Inhibition of osteoclast differentiation and bone resorption by cathepsin K antisense oligonucleotides. Mol. Carcinog. 2001, 32, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Grabowskal, U.; Chambers, T.J.; Shiroo, M. Recent developments in cathepsin K inhibitor design. Curr. Opin. Drug Discov. Dev. 2005, 8, 619–630. [Google Scholar]
- Stoch, S.A.; Wagner, J.A. Cathepsin K inhibitors: A novel target for osteoporosis therapy. Clin. Pharmacol. Ther. 2008, 83, 172–176. [Google Scholar] [CrossRef]
- Duong, L.T.; Wesolowski, G.A.; Leung, P.; Oballa, R.; Pickarski, M. Efficacy of a cathepsin K inhibitor in a preclinical model for prevention and treatment of breast cancer bone metastasis. Mol. Cancer Ther. 2014, 13, 2898–2909. [Google Scholar] [CrossRef] [Green Version]
- Biswas, B.; Sharma, M.C.; Mridha, A.R.; Bakhshi, S. Expression of Cathepsin L in tumor cells and tumor-associated macrophages in patients with Ewing sarcoma family of tumors: A pilot study. Indian J. Pathol. Microbiol. 2015, 58, 170–174. [Google Scholar]
- Brubaker, K.D.; Vessella, R.L.; True, L.D.; Thomas, R.; Corey, E. Cathepsin K mRNA and protein expression in prostate cancer progression. J. Bone Miner. Res. 2003, 18, 222–230. [Google Scholar] [CrossRef] [Green Version]
- Blair, H.C.; Sidonio, R.F.; Friedberg, R.C.; Khan, N.N.; Dong, S.S. Proteinase expression during differentiation of human osteoclasts in vitro. J. Cell. Biochem. 2000, 78, 627–637. [Google Scholar] [CrossRef]
- Joyce, J.A.; Baruch, A.; Chehade, K.; Meyer-Morse, N.; Giraudo, E.; Tsai, F.-Y.; Greenbaum, D.C.; Hager, J.H.; Bogyo, M.; Hanahan, D. Cathepsin cysteine proteases are effectors of invasive growth and angiogenesis during multistage tumorigenesis. Cancer Cell 2004, 5, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Battula, V.L.; Nguyen, K.; Sun, J.; Pitner, M.K.; Yuan, B.; Bartholomeusz, C.; Hail, N.; Andreeff, M. IKK inhibition by BMS-345541 suppresses breast tumorigenesis and metastases by targeting GD2+ cancer stem cells. Oncotarget 2017, 8, 36936–36949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berdowska, I. Cysteine proteases as disease markers. Clin. Chim. Acta 2004, 342, 41–69. [Google Scholar] [CrossRef] [PubMed]
- Jedeszko, C.; Sloane, B.F. Cysteine cathepsins in human cancer. Biol. Chem. 2004, 385, 1017–1027. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Kohno, K.; Kawamata, T.; Morimitsu, K.; Kuwano, M.; Miyakawa, I. Increased cathepsin L levels in serum in some patients with ovarian cancer: Comparison with CA125 and CA72-4. Gynecol. Oncol. 1995, 56, 357–361. [Google Scholar] [CrossRef]
- Sun, T.; Jiang, D.; Zhang, L.; Su, Q.; Mao, W.; Jiang, C. Expression profile of cathepsins indicates the potential of cathepsins B and D as prognostic factors in breast cancer patients. Oncol. Lett. 2016, 11, 575–583. [Google Scholar] [CrossRef] [Green Version]
- Thomssen, C.; Schmitt, M.; Goretzki, L.; Oppelt, P.; Pache, L.; Dettmar, P.; Jänicke, F.; Graeff, H. Prognostic value of the cysteine proteases cathepsins B and cathepsin L in human breast cancer. Clin. Cancer Res. 1995, 1, 741–746. [Google Scholar]
- Kageshita, T.; Yoshii, A.; Kimura, T.; Maruo, K.; Ono, T.; Himeno, M.; Nishimura, Y. Biochemical and immunohistochemical analysis of cathepsins B, H, L and D in human melanocytic tumours. Arch. Dermatol. Res. 1995, 287, 266–272. [Google Scholar] [CrossRef]
- Campo, E.; Muñoz, J.; Miquel, R.; Palacín, A.; Cardesa, A.; Sloane, B.F.; Emmert-Buck, M.R. Cathepsin B expression in colorectal carcinomas correlates with tumor progression and shortened patient survival. Am. J. Pathol. 1994, 145, 301–309. [Google Scholar]
- Werle, B.; Ebert, W.; Klein, W.; Spiess, E. Cathepsin B in tumors, normal tissue and isolated cells from the human lung. Anticancer Res. 1994, 14, 1169–1176. [Google Scholar]
- Schweiger, A.; Staib, A.; Werle, B.; Krasovec, M.; Lah, T.T.; Ebert, W.; Turk, V.; Kos, J. Cysteine proteinase cathepsin H in tumours and sera of lung cancer patients: Relation to prognosis and cigarette smoking. Br. J. Cancer 2000, 82, 782–788. [Google Scholar] [CrossRef] [Green Version]
- Fröhlich, E.; Schlagenhauff, B.; Möhrle, M.; Weber, E.; Klessen, C.; Rassner, G. Activity, expression, and transcription rate of the cathepsins B, D, H, and L in cutaneous malignant melanoma. Cancer 2001, 91, 972–982. [Google Scholar] [CrossRef]
- Lah, T.T.; Calaf, G.; Kalman, E.; Shinde, B.G.; Somers, R.; Estrada, S.; Salero, E.; Russo, J.; Daskal, I. Cathepsins D, B, and L in transformed human breast epithelial cells. Breast Cancer Res Treat. 1996, 39, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Fernández, P.L.; Farré, X.; Nadal, A.; Fernández, E.; Peiró, N.; Sloane, B.F.; Shi, G.-P.; Chapman, H.A.; Campo, E.; Cardesa, A. Expression of cathepsins B and S in the progression of prostate carcinoma. Int. J. Cancer 2001, 95, 51–55. [Google Scholar] [CrossRef]
- Gocheva, V.; Zeng, W.; Ke, D.; Klimstra, D.; Reinheckel, T.; Peters, C.; Hanahan, D.; Joyce, J.A. Distinct roles for cysteine cathepsin genes in multistage tumorigenesis. Genes Dev. 2006, 20, 543–556. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, M.M.; Sloane, B.F. Cysteine cathepsins: Multifunctional enzymes in cancer. Nat. Rev. Cancer 2006, 6, 764–775. [Google Scholar] [CrossRef]
- Skrzydlewska, E.; Sulkowska, M.; Koda, M.; Sulkowski, S. Proteolytic-antiproteolytic balance and its regulation in carcinogenesis. World J. Gastroenterol. 2005, 11, 1251–1266. [Google Scholar] [CrossRef]
- Cairns, R.A.; Khokha, R.; Hill, R.P. Molecular mechanisms of tumor invasion and metastasis: An integrated view. Curr. Mol. Med. 2003, 3, 659–671. [Google Scholar] [CrossRef]
- Sobotič, B.; Vizovišek, M.; Vidmar, R.; Van Damme, P.; Gocheva, V.; Joyce, J.A.; Gevaert, K.; Turk, V.; Turk, B.; Fonović, M. Proteomic Identification of Cysteine Cathepsin Substrates Shed from the Surface of Cancer Cells. Mol. Cell. Proteom. 2015, 14, 2213–2228. [Google Scholar] [CrossRef] [Green Version]
- Lankelma, J.M.; Voorend, D.M.; Barwari, T.; Koetsveld, J.; Van Der Spek, A.H.; De Porto, A.P.; Van Rooijen, G.; Van Noorden, C.J. Cathepsin L, target in cancer treatment? Life Sci. 2010, 86, 225–233. [Google Scholar] [CrossRef]
- Sawant, A.; Ponnazhagan, S. Myeloid-derived suppressor cells as osteoclast progenitors: A novel target for controlling osteolytic bone metastasis. Cancer Res. 2013, 73, 4606–4610. [Google Scholar] [CrossRef] [Green Version]
- D’Amico, L.; Roato, I. The Impact of Immune System in Regulating Bone Metastasis Formation by Osteotropic Tumors. J. Immunol. Res. 2015, 2015, 143526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gocheva, V.; Wang, H.W.; Gadea, B.B.; Shree, T.; Hunter, K.E.; Garfall, A.L.; Berman, T.; Joyce, J.A. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev. 2010, 24, 241–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podgorski, I.; Linebaugh, B.E.; Koblinski, J.E.; Rudy, D.L.; Herroon, M.K.; Olive, M.B.; Sloabe, B.F. Bone marrow-derived cathepsin K cleaves SPARC in bone metastasis. Am. J. Pathol. 2009, 175, 1255–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mundy, G.R. Mechanisms of bone metastasis. Cancer 1997, 80 (Suppl. 8), 1546–1556. [Google Scholar] [CrossRef]
- Mundy, G.R.; Chen, D.; Zhao, M.; Dallas, S.; Xu, C.; Harris, S. Growth regulatory factors and bone. Rev. Endocr. Metab. Disord. 2001, 2, 105–115. [Google Scholar] [CrossRef]
- Bussard, K.M.; Gay, C.V.; Mastro, A.M. The bone microenvironment in metastasis; what is special about bone? Cancer Metastasis Rev. 2008, 27, 41–55. [Google Scholar] [CrossRef]
- Horwood, N.J.; Elliott, J.; Martin, T.J.; Gillespie, M.T. Osteotropic agents regulate the expression of osteoclast differentiation factor and osteoprotegerin in osteoblastic stromal cells. Endocrinology 1998, 139, 4743–4746. [Google Scholar] [CrossRef]
- Rodan, G.A. The development and function of the skeleton and bone metastases. Cancer 2003, 97 (Suppl. 3), 726–732. [Google Scholar] [CrossRef]
- Guise, T.A.; Yin, J.J.; Mohammad, K.S. Role of endothelin-1 in osteoblastic bone metastases. Cancer 2003, 97 (Suppl. 3), 779–784. [Google Scholar] [CrossRef]
- Akhtari, M.; Mansuri, J.; Newman, K.A.; Guise, T.M.; Seth, P. Biology of breast cancer bone metastasis. Cancer Biol. Ther. 2008, 7, 3–9. [Google Scholar] [CrossRef]
- Futakuchi, M.; Nannuru, K.C.; Varney, M.L.; Sadanandam, A.; Nakao, K.; Asai, K.; Shirai, T.; Sato, S.; Singh, R. Transforming growth factor-beta signaling at the tumor-bone interface promotes mammary tumor growth and osteoclast activation. Cancer Sci. 2009, 100, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.J.; Nannuru, K.C.; Singh, R.K. Cathepsin G-mediated activation of pro-matrix metalloproteinase 9 at the tumor-bone interface promotes transforming growth factor-beta signaling and bone destruction. Mol. Cancer Res. 2009, 7, 1224–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumminello, F.M.; Flandina, C.; Crescimanno, M.; Leto, G. Circulating cathepsin K and cystatin C in patients with cancer related bone disease: Clinical and therapeutic implications. Biomed. Pharmacother. 2008, 62, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Arnett, T. Regulation of bone cell function by acid-base balance. Proc. Nutr. Soc. 2003, 62, 511–520. [Google Scholar] [CrossRef] [Green Version]
- Vasiljeva, O.; Reinheckel, T.; Peters, C.; Turk, D.; Turk, V.; Turk, B. Emerging roles of cysteine cathepsins in disease and their potential as drug targets. Curr. Pharm. Des. 2007, 13, 387–403. [Google Scholar] [CrossRef] [PubMed]
- Withana, N.P.; Blum, G.; Sameni, M.; Slaney, C.; Anbalagan, A.; Olive, M.B.; Bidwell, B.N.; Edington, L.; Wang, L.; Moin, K.; et al. Cathepsin B inhibition limits bone metastasis in breast cancer. Cancer Res. 2012, 72, 1199–1209. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Zhang, Q.; Chen, L. Triptolide induces the cell apoptosis of osteosarcoma cells through the TRAIL pathway. Oncol. Rep. 2016, 36, 1499–1505. [Google Scholar] [CrossRef]
- Garnett, T.O.; Filippova, M.; Duerksen-Hughes, P.J. Bid is cleaved upstream of caspase-8 activation during TRAIL-mediated apoptosis in human osteosarcoma cells. Apoptosis 2007, 12, 1299–1315. [Google Scholar] [CrossRef]
- Nagaraj, N.S.; Vigneswaran, N.; Zacharias, W. Cathepsin B mediates TRAIL-induced apoptosis in oral cancer cells. J. Cancer Res. Clin. Oncol. 2006, 132, 171–183. [Google Scholar] [CrossRef]
- Cirman, T.; Oresić, K.; Mazovec, G.D.; Turk, V.; Reed, J.C.; Myers, R.M.; Salvesen, G.S.; Turk, B. Selective disruption of lysosomes in HeLa cells triggers apoptosis mediated by cleavage of Bid by multiple papain-like lysosomal cathepsins. J. Biol. Chem. 2004, 279, 3578–3587. [Google Scholar] [CrossRef] [Green Version]
- Owa, C.; Messina, M.E.; Halaby, R. Triptolide induces lysosomal-mediated programmed cell death in MCF-7 breast cancer cells. Int. J. Womens Health 2013, 5, 557–569. [Google Scholar] [PubMed] [Green Version]
- Maguire, T.M.; Shering, S.G.; Duggan, C.M.; McDermott, E.W.; O’Higgins, N.J.; Duffy, M.J. High levels of cathepsin B predict poor outcome in patients with breast cancer. Int. J. Biol. Markers 1998, 13, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Foekens, J.A.; Kos, J.; Peters, H.A.; Krasovec, M.; Look, M.P.; Cimerman, N.; Meijer-van Gelder, M.E.; Henzen-Logmans, S.C.; van Putten, W.L.; Klijn, J.G. Prognostic significance of cathepsins B and L in primary human breast cancer. J. Clin. Oncol. 1998, 16, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Podgorski, I.; Linebaugh, B.E.; Sameni, M.; Jedeszko, C.; Bhagat, S.; Cher, M.L.; Sloane, B.F. Bone microenvironment modulates expression and activity of cathepsin B in prostate cancer. Neoplasia 2005, 7, 207–223. [Google Scholar] [CrossRef] [Green Version]
- Arkona, C.; Wiederanders, B. Expression, subcellular distribution and plasma membrane binding of cathepsin B and gelatinases in bone metastatic tissue. Biol. Chem. 1996, 377, 695–702. [Google Scholar] [CrossRef]
- Verdoes, M.; Oresic Bender, K.; Segal, E.; van der Linden, W.A.; Syed, S.; Withana, N.P.; Sanman, L.E.; Bogyo, M. Improved quenched fluorescent probe for imaging of cysteine cathepsin activity. J. Am. Chem. Soc. 2013, 135, 14726–14730. [Google Scholar] [CrossRef] [Green Version]
- Gemoll, T.; Epping, F.; Heinrich, L.; Fritzsche, B.; Roblick, U.J.; Szymczak, S.; Hartwig, S.; Depping, R.; Bruch, H.; Thorns, C.; et al. Increased cathepsin D protein expression is a biomarker for osteosarcomas, pulmonary metastases and other bone malignancies. Oncotarget 2015, 6, 16517–16526. [Google Scholar] [CrossRef] [Green Version]
- Spreafico, A.; Frediani, B.; Capperucci, C.; Chellini, F.; Paffetti, A.; D’Ambrosio, C.; Bernardini, G.; Mini, R.; Collodel, G.; Scaloni, A.; et al. A proteomic study on human osteoblastic cells proliferation and differentiation. Proteomics 2006, 6, 3520–3532. [Google Scholar] [CrossRef]
- Rochefort, H. Cathepsin D in breast cancer: A tissue marker associated with metastasis. Eur. J. Cancer 1992, 28A, 1780–1783. [Google Scholar] [CrossRef]
- Pujol, P.; Maudelonde, T.; Daures, J.P.; Rouanet, P.; Brouillet, J.P.; Pujol, H.; Rochefort, H. A prospective study of the prognostic value of cathepsin D levels in breast cancer cytosol. Cancer 1993, 71, 2006–2012. [Google Scholar] [CrossRef]
- Schultz, D.C.; Bazel, S.; Wright, L.M.; Tucker, S.; Lange, M.K.; Tachovsky, T.; Longo, S.; Niedbala, S.; Alhadeff, J.A. Western blotting and enzymatic activity analysis of cathepsin D in breast tissue and sera of patients with breast cancer and benign breast disease and of normal controls. Cancer Res. 1994, 54, 48–54. [Google Scholar] [PubMed]
- Ravdin, P.M. Evaluation of cathepsin D as a prognostic factor in breast cancer. Breast Cancer Res. Treat. 1993, 24, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Solomayer, E.F.; Diel, I.J.; Meyberg, G.C.; Gollan, C.; Bode, S.; Wallwiener, D.; Bastert, G. Prognostic relevance of cathepsin D detection in micrometastatic cells in the bone marrow of patients with primary breast cancer. Breast Cancer Res. Treat. 1998, 49, 145–154. [Google Scholar] [CrossRef]
- Arai, K.; Sakamoto, R.; Kubota, D.; Kondo, T. Proteomic approach toward molecular backgrounds of drug resistance of osteosarcoma cells in spheroid culture system. Proteomics 2013, 13, 2351–2360. [Google Scholar] [CrossRef] [PubMed]
- Sagulenko, V.; Muth, D.; Sagulenko, E.; Paffhausen, T.; Schwab, M.; Westermann, F. Cathepsin D protects human neuroblastoma cells from doxorubicin-induced cell death. Carcinogenesis 2008, 29, 1869–1877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Chen, C.M.; Yu, K.D.; Yang, W.T.; Shao, Z.M. A prognostic model to predict outcome of patients failing to achieve pathological complete response after anthracycline-containing neoadjuvant chemotherapy for breast cancer. J. Surg. Oncol. 2012, 105, 577–585. [Google Scholar] [CrossRef]
- Avnet, S.; Longhi, A.; Salerno, M.; Halleen, J.M.; Perut, F.; Granchi, D.; Ferrari, S.; Bertoni, F.; Giunti, A.; Baldini, N. Increased osteoclast activity is associated with aggressiveness of osteosarcoma. Int. J. Oncol. 2008, 33, 1231–1238. [Google Scholar] [CrossRef] [Green Version]
- Wilson, T.J.; Nannuru, K.C.; Futakuchi, M.; Sadanandam, A.; Singh, R.K. Cathepsin G enhances mammary tumor-induced osteolysis by generating soluble receptor activator of nuclear factor-kappaB ligand. Cancer Res. 2008, 68, 5803–5811. [Google Scholar] [CrossRef] [Green Version]
- Lü, J.; Qian, J.; Keppler, D.; Cardoso, W.V. Cathespin H is an Fgf10 target involved in Bmp4 degradation during lung branching morphogenesis. J. Biol. Chem. 2007, 282, 22176–22184. [Google Scholar] [CrossRef] [Green Version]
- Verbovšek, U.; Van Noorden, C.J.; Lah, T.T. Complexity of cancer protease biology: Cathepsin K expression and function in cancer progression. Semin Cancer Biol. 2015, 35, 71–84. [Google Scholar] [CrossRef]
- Blouin, S.; Baslé, M.F.; Chappard, D. Interactions between microenvironment and cancer cells in two animal models of bone metastasis. Br. J. Cancer 2008, 98, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Gelb, B.D.; Shi, G.P.; Heller, M.; Weremowicz, S.; Morton, C.; Desnick, R.J.; Chapman, H.A. Structure and chromosomal assignment of the human cathepsin K gene. Genomics 1997, 41, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Rood, J.A.; Van Horn, S.; Drake, F.H.; Gowen, M.; Debouck, C. Genomic organization and chromosome localization of the human cathepsin K gene (CTSK). Genomics 1997, 41, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.P.; Chen, W. Characterization of mouse cathepsin K gene, the gene promoter, and the gene expression. J. Bone Miner. Res. 1999, 14, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Leung, P.; Pickarski, M.; Wesolowski, G.A.; Duong, L.T. Anti-resorptive and potential anti-invasion effects of a cathepsin K inhibitor in the treatment of breast cancer-induced bone metastasis. Cancer Treat. Rev. 2008, 34 (Suppl. 1), S58. [Google Scholar] [CrossRef]
- Munari, E.; Cima, L.; Massari, F.; Bertoldo, F.; Porcaro, A.B.; Caliò, A.; Riva, G.; Ciocchetta, E.; Ciccarese, C.; Modena, A.; et al. Cathepsin K expression in castration-resistant prostate carcinoma: A therapeutical target for patients at risk for bone metastases. Int. J. Biol. Markers 2017, 32, e243–e247. [Google Scholar] [CrossRef] [Green Version]
- Liang, W.; Wang, F.; Chen, Q.; Dai, J.; Escara-Wilke, J.; Keller, E.T.; Zimmerman, J.; Hona, N.; Lu, Y.; Zhang, J. Targeting cathepsin K diminishes prostate cancer establishment and growth in murine bone. J. Cancer Res. Clin. Oncol. 2019, 145, 1999–2012. [Google Scholar] [CrossRef] [Green Version]
- Katunuma, N.; Tsuge, H.; Nukatsuka, M.; Asao, T.; Fukushima, M. Structure-based design of specific cathepsin inhibitors and their application to protection of bone metastases of cancer cells. Arch. Biochem. Biophys. 2002, 397, 305–311. [Google Scholar] [CrossRef]
- Yoneda, T. Cellular and molecular mechanisms of breast and prostate cancer metastasis to bone. Eur. J. Cancer 1998, 34, 240–245. [Google Scholar] [CrossRef]
- Furuyama, N.; Fujisawa, Y. Distinct roles of cathepsin K and cathepsin L in osteoclastic bone resorption. Endocr. Res. 2000, 26, 189–204. [Google Scholar] [CrossRef]
- Furuyama, N.; Fujisawa, Y. Regulation of collagenolytic protease secretion through c-Src in osteoclasts. Biochem. Biophys. Res. Commun. 2000, 272, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, F.; Yamashita, U.; Tanaka, Y.; Watanaba, K.; Sato, K.; Haratake, J.; Fujihira, T.; Oda, S.; Eto, S. Production of bone-resorbing activity corresponding to interleukin-1 alpha by adult T-cell leukemia cells in humans. Cancer Res. 1988, 48, 4284–4287. [Google Scholar] [PubMed]
- Jay, P.R.; Centrella, M.; Lorenzo, J.; Bruce, A.G.; Horowitz, M.C. Oncostatin-M: A new bone active cytokine that activates osteoblasts and inhibits bone resorption. Endocrinology 1996, 137, 1151–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenfield, E.M.; Horowitz, M.C.; Lavish, S.A. Stimulation by parathyroid hormone of interleukin-6 and leukemia inhibitory factor expression in osteoblasts is an immediate-early gene response induced by cAMP signal transduction. J. Biol. Chem. 1996, 271, 10984–10989. [Google Scholar] [CrossRef] [Green Version]
- Damiens, C.; Grimaud, E.; Rousselle, A.V.; Charrier, C.; Fortun, Y.; Heymann, D.; Padrines, M. Cysteine protease production by human osteosarcoma cells (MG63, SAOS2) and its modulation by soluble factors. Cytokine 2000, 12, 539–542. [Google Scholar] [CrossRef]
- Park, I.C.; Lee, S.Y.; Jeon, D.G.; Lee, J.S.; Hwang, C.S.; Hwang, Y.G.; Lee, S.H.; Hong, W.S.; Hong, S.I. Enhanced expression of cathepsin L in metastatic bone tumors. J. Korean Med. Sci. 1996, 11, 144–148. [Google Scholar] [CrossRef] [Green Version]
- Bincoletto, C.; Tersariol, I.L.; Oliveira, C.R.; Dreher, S.; Fausto, D.M.; Soufen, M.A.; Nascimento, F.D.; Caires, A.C.F. Chiral cyclopalladated complexes derived from N,N-dimethyl-1-phenethylamine with bridging bis(diphenylphosphine)ferrocene ligand as inhibitors of the cathepsin B activity and as antitumoral agents. Bioorg. Med. Chem. 2005, 13, 3047–3055. [Google Scholar] [CrossRef]
- Barbosa, C.M.; Oliveira, C.R.; Nascimento, F.D.; Smith, M.C.; Fausto, D.M.; Soufen, M.A.; Sena, E.; Araújo, R.C.; Tersariol, I.L.S.; Bincoletto, C.; et al. Biphosphinic palladacycle complex mediates lysosomal-membrane permeabilization and cell death in K562 leukaemia cells. Eur. J. Pharmacol. 2006, 542, 37–47. [Google Scholar] [CrossRef]
- Bechara, A.; Barbosa, C.M.; Paredes-Gamero, E.J.; Garcia, D.M.; Silva, L.S.; Matsuo, A.L.; Nascimento, F.D.; Rodrigues, E.G.; Caires, A.C.F.; Smaili, S.S.; et al. Palladacycle (BPC) antitumour activity against resistant and metastatic cell lines: The relationship with cytosolic calcium mobilisation and cathepsin B activity. Eur. J. Med. Chem. 2014, 79, 24–33. [Google Scholar] [CrossRef]
- Krueger, S.; Kellner, U.; Buehling, F.; Roessner, A. Cathepsin L antisense oligonucleotides in a human osteosarcoma cell line: Effects on the invasive phenotype. Cancer Gene Ther. 2001, 8, 522–528. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.; Friedrichs, K.; Noel, D.; Pintér, T.; Van Belle, S.; Vorobiof, D.; Duarte, R.; Gil Gil, M.; Bodrogi, I.; Murray, E.; et al. Prospective randomized trial of docetaxel versus doxorubicin in patients with metastatic breast cancer. J. Clin. Oncol. 1999, 17, 2341–2354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Chu, F.; Chou, P.M.; Gallati, C.; Dier, U.; Mirkin, B.L.; Mousa, S.A.; Rebbaa, A. Cathepsin L inhibition suppresses drug resistance in vitro and in vivo: A putative mechanism. Am. J. Physiol. Cell. Physiol. 2009, 296, C65–C74. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Chou, P.M.; Mirkin, B.L.; Rebbaa, A. Senescence-initiated reversal of drug resistance: Specific role of cathepsin L. Cancer Res. 2004, 64, 1773–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selak, M.A.; Chignard, M.; Smith, J.B. Cathepsin G is a strong platelet agonist released by neutrophils. Biochem. J. 1988, 251, 293–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewiecki, E.M. Odanacatib, a cathepsin K inhibitor for the treatment of osteoporosis and other skeletal disorders associated with excessive bone remodeling. IDrugs 2009, 12, 799–809. [Google Scholar] [PubMed]
- Gauthier, J.Y.; Chauret, N.; Cromlish, W.; Desmarais, S.; Duong, L.T.; Falgueyret, J.P.; Kimmel, D.B.; Lamontagne, S.; Léger, S.; LeRiche, T.; et al. The discovery of odanacatib (MK-0822), a selective inhibitor of cathepsin K. Bioorg. Med. Chem. Lett. 2008, 18, 923–928. [Google Scholar] [CrossRef]
- Desmarais, S.; Massé, F.; Percival, M.D. Pharmacological inhibitors to identify roles of cathepsin K in cell-based studies: A comparison of available tools. Biol. Chem. 2009, 390, 941–948. [Google Scholar] [CrossRef]
- Jensen, A.B.; Wynne, C.; Ramirez, G.; He, W.; Song, Y.; Berd, Y.; Wang, H.; Mehta, A.; Lombardi, A. The cathepsin K inhibitor odanacatib suppresses bone resorption in women with breast cancer and established bone metastases: Results of a 4-week, double-blind, randomized, controlled trial. Clin. Breast Cancer 2010, 10, 452–458. [Google Scholar] [CrossRef]
- Peroni, A.; Zini, A.; Braga, V.; Colato, C.; Adami, S.; Girolomoni, G. Drug-induced morphea: Report of a case induced by balicatib and review of the literature. J. Am. Acad. Dermatol. 2008, 59, 125–129. [Google Scholar] [CrossRef]
- Mullard, A. Merck &Co. drops osteoporosis drug odanacatib. Nat. Rev. Drug Discov. 2016, 15, 669. [Google Scholar]
- Althoff, E. Novartis R&D Update Highlights Industry Leading Development Pipeline Including Potential Blockbusters and Advanced Therapy Platforms; Novartis Media Relations: London, UK, 2018. [Google Scholar]
- Palmer, J.T.; Bryant, C.; Wang, D.X.; Davis, D.E.; Setti, E.L.; Rydzewski, R.M.; Venkatraman, S.; Tian, Z.Q.; Burrill, L.C.; Mendonca, R.V.; et al. Design and synthesis of tri-ring P3 benzamide-containing aminonitriles as potent, selective, orally effective inhibitors of cathepsin K. J. Med. Chem. 2005, 48, 7520–7534. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, C.; Bellahcène, A.; Bonnelye, E.; Gasser, J.A.; Castronovo, V.; Green, J.; Zimmermann, J.; Clézardin, P. A cathepsin K inhibitor reduces breast cancer induced osteolysis and skeletal tumor burden. Cancer Res. 2007, 67, 9894–9902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, L.J.; Smith, B.A.; Smith, B.N.; Loyd, Q.; Nagappan, P.; McKeithen, D.; Wilder, C.L.; Platt, M.O.; Hudson, T.; Odero-Marah, V.A. Muscadine grape skin extract can antagonize Snail-cathepsin L-mediated invasion, migration and osteoclastogenesis in prostate and breast cancer cells. Carcinogenesis 2015, 36, 1019–1027. [Google Scholar] [CrossRef]
- Odero-Marah, V.A.; Wang, R.; Chu, G.; Zayzafoon, M.; Xu, J.; Shi, C.; Marshall, F.F.; Zhau, H.E.; Chung, L.W.K. Receptor activator of NF-kappaB Ligand (RANKL) expression is associated with epithelial to mesenchymal transition in human prostate cancer cells. Cell Res. 2008, 18, 858–870. [Google Scholar] [CrossRef] [Green Version]
- Kumar, G.D.; Chavarria, G.E.; Charlton-Sevcik, A.K.; Yoo, G.K.; Song, J.; Strecker, T.E.; Siim, B.G.; Chaplin, D.J.; Trawick, M.L.; Pinney, K.G. Functionalized benzophenone, thiophene, pyridine, and fluorene thiosemicarbazone derivatives as inhibitors of cathepsin L. Bioorg. Med. Chem. Lett. 2010, 20, 6610–6615. [Google Scholar] [CrossRef]
- Kishore Kumar, G.D.; Chavarria, G.E.; Charlton-Sevcik, A.K.; Arispe, W.M.; Macdonough, M.T.; Strecker, T.E.; Chen, S.; Siim, B.G.; Chapli, D.J.; Trawick, M.L.; et al. Design, synthesis, and biological evaluation of potent thiosemicarbazone based cathepsin L inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 1415–1419. [Google Scholar] [CrossRef]
- Sudhan, D.R.; Rabaglino, M.B.; Wood, C.E.; Siemann, D.W. Cathepsin L in tumor angiogenesis and its therapeutic intervention by the small molecule inhibitor KGP94. Clin. Exp. Metastasis 2016, 33, 461–473. [Google Scholar] [CrossRef] [Green Version]
- Sudhan, D.R.; Siemann, D.W. Cathepsin L inhibition by the small molecule KGP94 suppresses tumor microenvironment enhanced metastasis associated cell functions of prostate and breast cancer cells. Clin. Exp. Metastasis 2013, 30, 891–902. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Distinct role of secreted Cathepsins at the bone–tumor interface. Cathepsins are secreted from neoplastic cells, osteoblasts and osteoclasts. Each of these Cathepsins plays a unique role in bone tumor progression. Figure created by: Veronica Hughes, PhD.
Figure 1.
Distinct role of secreted Cathepsins at the bone–tumor interface. Cathepsins are secreted from neoplastic cells, osteoblasts and osteoclasts. Each of these Cathepsins plays a unique role in bone tumor progression. Figure created by: Veronica Hughes, PhD.

Figure 2.
Representative images and data of the CatD validation of the cytospins of all of the cell lines (A,B) and the tissue microarray validations (C). The CatD showed an increased staining in osteosarcoma and pulmonary metastasis compared to fetal osteoblasts (A, left) that reached significance between three groups (B). A customized (C) tissue microarray-based evaluation of the CTSD showed a strong overexpression in osteosarcomas and pulmonary metastasis compared to normal bone tissue. Based on the lack of representative fetal osteoblasts in the normal bone tissue, they chose isolated osteocytes known to be descended from matured osteoblasts as the reference control (**: 0.001 < p < 0.01; * 0.01 < p < 0.05). Modified from refs. [158].
Figure 2.
Representative images and data of the CatD validation of the cytospins of all of the cell lines (A,B) and the tissue microarray validations (C). The CatD showed an increased staining in osteosarcoma and pulmonary metastasis compared to fetal osteoblasts (A, left) that reached significance between three groups (B). A customized (C) tissue microarray-based evaluation of the CTSD showed a strong overexpression in osteosarcomas and pulmonary metastasis compared to normal bone tissue. Based on the lack of representative fetal osteoblasts in the normal bone tissue, they chose isolated osteocytes known to be descended from matured osteoblasts as the reference control (**: 0.001 < p < 0.01; * 0.01 < p < 0.05). Modified from refs. [158].
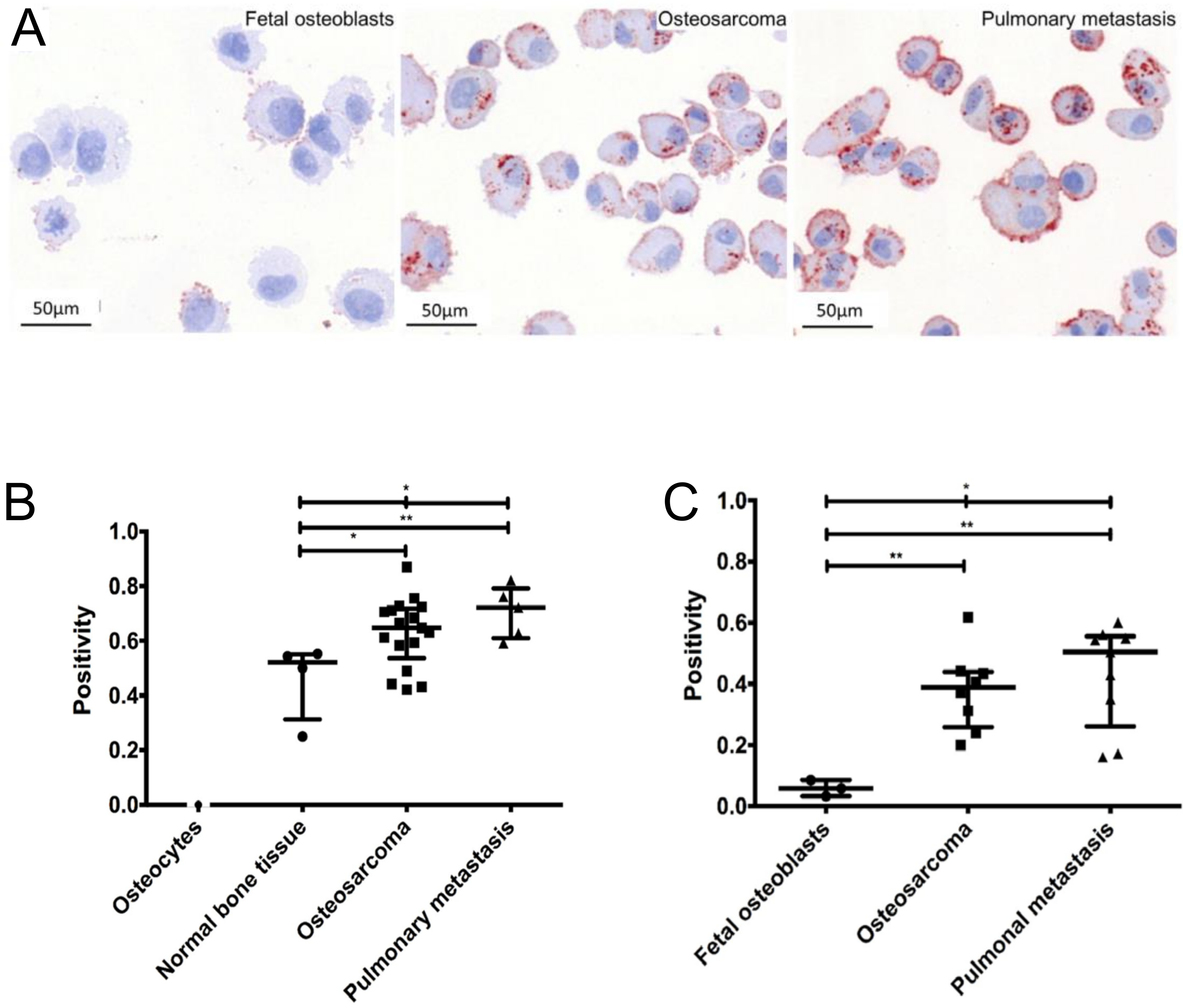
Figure 3.
Effect of CL inhibition on tumor growth in nude mice. The animals were injected with Dox-resistant SKN-SH/R cells (106 cells/100 L culture medium), and, after 10 days, they were assigned into the following four groups of seven animals: untreated (Ctl) mice, mice treated with Dox (1.5 mg/kg) alone, mice treated iCL (20 mg/kg) alone, and mice treated with the combination of both drugs (Dox iCL). The tumor volumes were measured every 3 days for up to 3 weeks. Tumor growth curves in the different animal groups. Data represent means of 7 determinations SE. *P < 0.001; **P < 0.05. Modified from refs. [193].
Figure 3.
Effect of CL inhibition on tumor growth in nude mice. The animals were injected with Dox-resistant SKN-SH/R cells (106 cells/100 L culture medium), and, after 10 days, they were assigned into the following four groups of seven animals: untreated (Ctl) mice, mice treated with Dox (1.5 mg/kg) alone, mice treated iCL (20 mg/kg) alone, and mice treated with the combination of both drugs (Dox iCL). The tumor volumes were measured every 3 days for up to 3 weeks. Tumor growth curves in the different animal groups. Data represent means of 7 determinations SE. *P < 0.001; **P < 0.05. Modified from refs. [193].

Figure 4.
Effect of cathepsin K inhibitor AFG-495 on the formation of experimental breast cancer bone metastasis. (A) Inhibitory effect of AFG-495 on the activity of RAW 264.7 osteoclastic cells in vitro. As shown by confocal microscopy, phalloidin-stained osteoclasts treated with the vehicle (panels i and ii) or AFG-495 (panels iii and iv) attach to the bone matrix and exhibit a typical sealing zone (white arrow), irrespective of the treatment. By contrast, AFG-495 inhibits the formation of resorption pits (panel iv) when compared with the vehicle (panel ii). (B) Effect of a preventive treatment with AFG-495 (50 mg/kg, twice daily) on experimental bone metastasis caused by human B02 breast cancer cells. Representative radiographs and a three-dimensional computed microtomographic reconstruction of the hind limbs from mice 32 days after the tumor cell inoculation are shown. There is a substantial reduction of bone destruction in the AFG-495-treated animals. Modified from ref. [204].
Figure 4.
Effect of cathepsin K inhibitor AFG-495 on the formation of experimental breast cancer bone metastasis. (A) Inhibitory effect of AFG-495 on the activity of RAW 264.7 osteoclastic cells in vitro. As shown by confocal microscopy, phalloidin-stained osteoclasts treated with the vehicle (panels i and ii) or AFG-495 (panels iii and iv) attach to the bone matrix and exhibit a typical sealing zone (white arrow), irrespective of the treatment. By contrast, AFG-495 inhibits the formation of resorption pits (panel iv) when compared with the vehicle (panel ii). (B) Effect of a preventive treatment with AFG-495 (50 mg/kg, twice daily) on experimental bone metastasis caused by human B02 breast cancer cells. Representative radiographs and a three-dimensional computed microtomographic reconstruction of the hind limbs from mice 32 days after the tumor cell inoculation are shown. There is a substantial reduction of bone destruction in the AFG-495-treated animals. Modified from ref. [204].

Figure 5.
CTSL ablation and KGP94 treatment reduces metastatic disease burden. (A) In vivo bone metastasis assay. Representative bioluminescence images of median mice inoculated with empty vector or CTSL shRNA transfected PC-3ML cells. (B) Measurement of bone metastasis burden based on bioluminescence intensity (photons per second); n10. Results are Mean ± S.E.M; analysis of variance. (C) Kaplan–Meier survival curves of mice inoculated with an empty vector of CTSL shRNA transfected PC-3ML cells; log-rank test. (D) In vivo bone metastasis assay. Representative bioluminescence images of median mice from control and KGP94-treated cohorts. (E) Bone metastases burden in control and KGP94 treated mice, measured based on photon flux (photons per second); n8. Results are Mean ± S.E.M, analysis of variance. (F) Kaplan– Meier survival curves of bone metastases-bearing mice treated with or without 20 mg/kg KGP94; log-rank test. (G) Representative Green Fluorescent Protein and hematoxylin and eosin images of bone metastases from each experimental group, scale bar = 1 mm. Modified from ref. [99].
Figure 5.
CTSL ablation and KGP94 treatment reduces metastatic disease burden. (A) In vivo bone metastasis assay. Representative bioluminescence images of median mice inoculated with empty vector or CTSL shRNA transfected PC-3ML cells. (B) Measurement of bone metastasis burden based on bioluminescence intensity (photons per second); n10. Results are Mean ± S.E.M; analysis of variance. (C) Kaplan–Meier survival curves of mice inoculated with an empty vector of CTSL shRNA transfected PC-3ML cells; log-rank test. (D) In vivo bone metastasis assay. Representative bioluminescence images of median mice from control and KGP94-treated cohorts. (E) Bone metastases burden in control and KGP94 treated mice, measured based on photon flux (photons per second); n8. Results are Mean ± S.E.M, analysis of variance. (F) Kaplan– Meier survival curves of bone metastases-bearing mice treated with or without 20 mg/kg KGP94; log-rank test. (G) Representative Green Fluorescent Protein and hematoxylin and eosin images of bone metastases from each experimental group, scale bar = 1 mm. Modified from ref. [99].


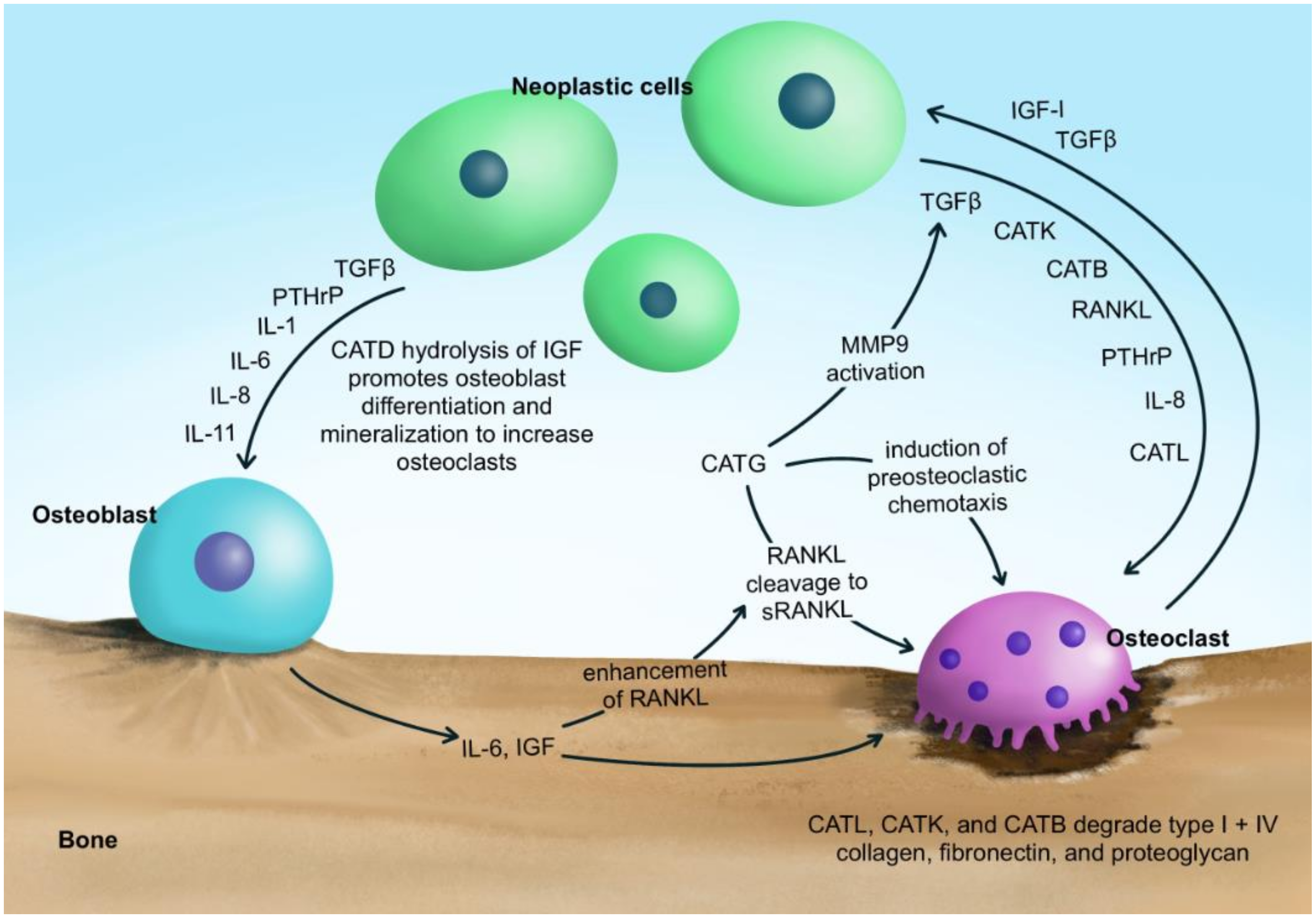{kind=link}
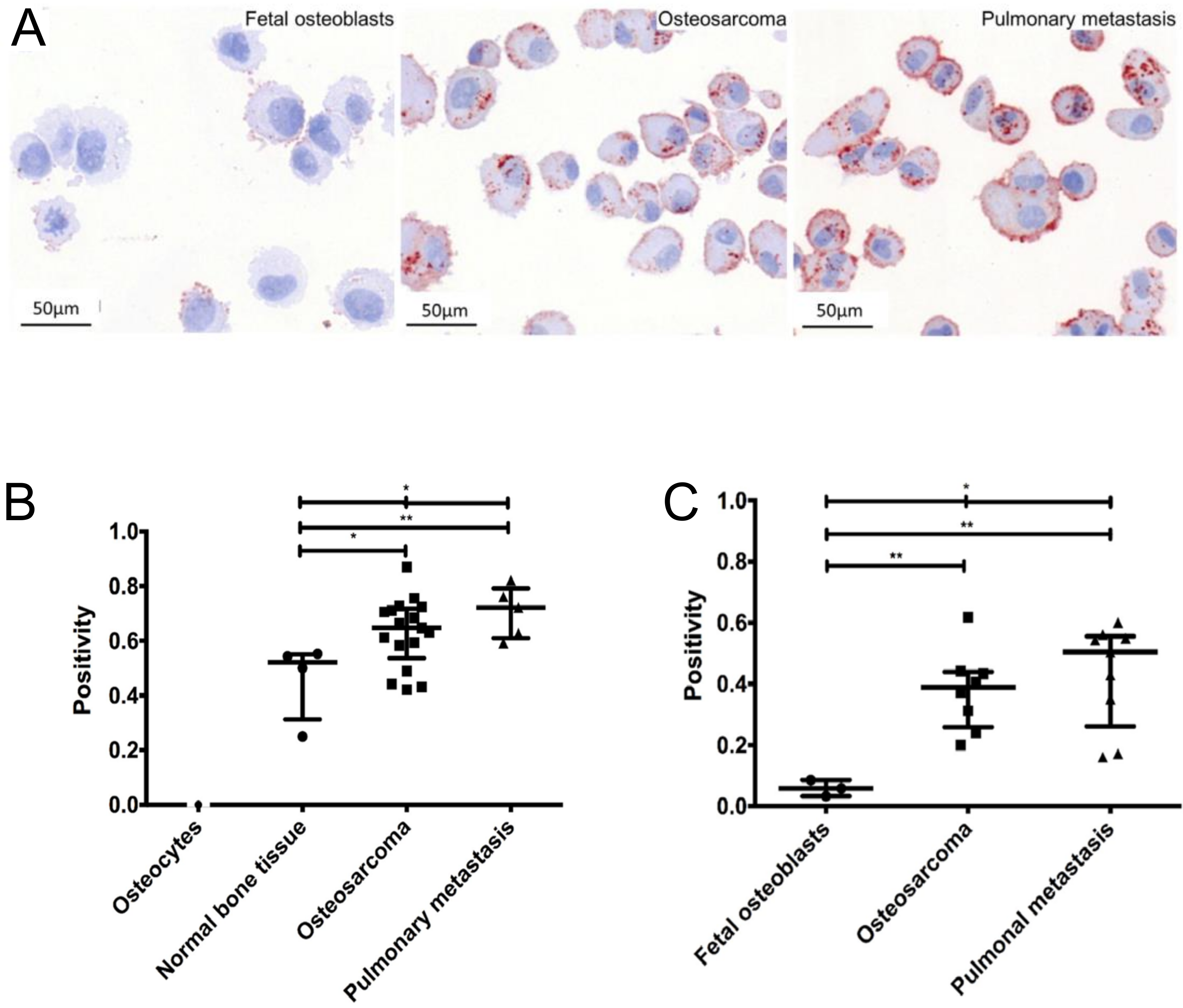{kind=link}
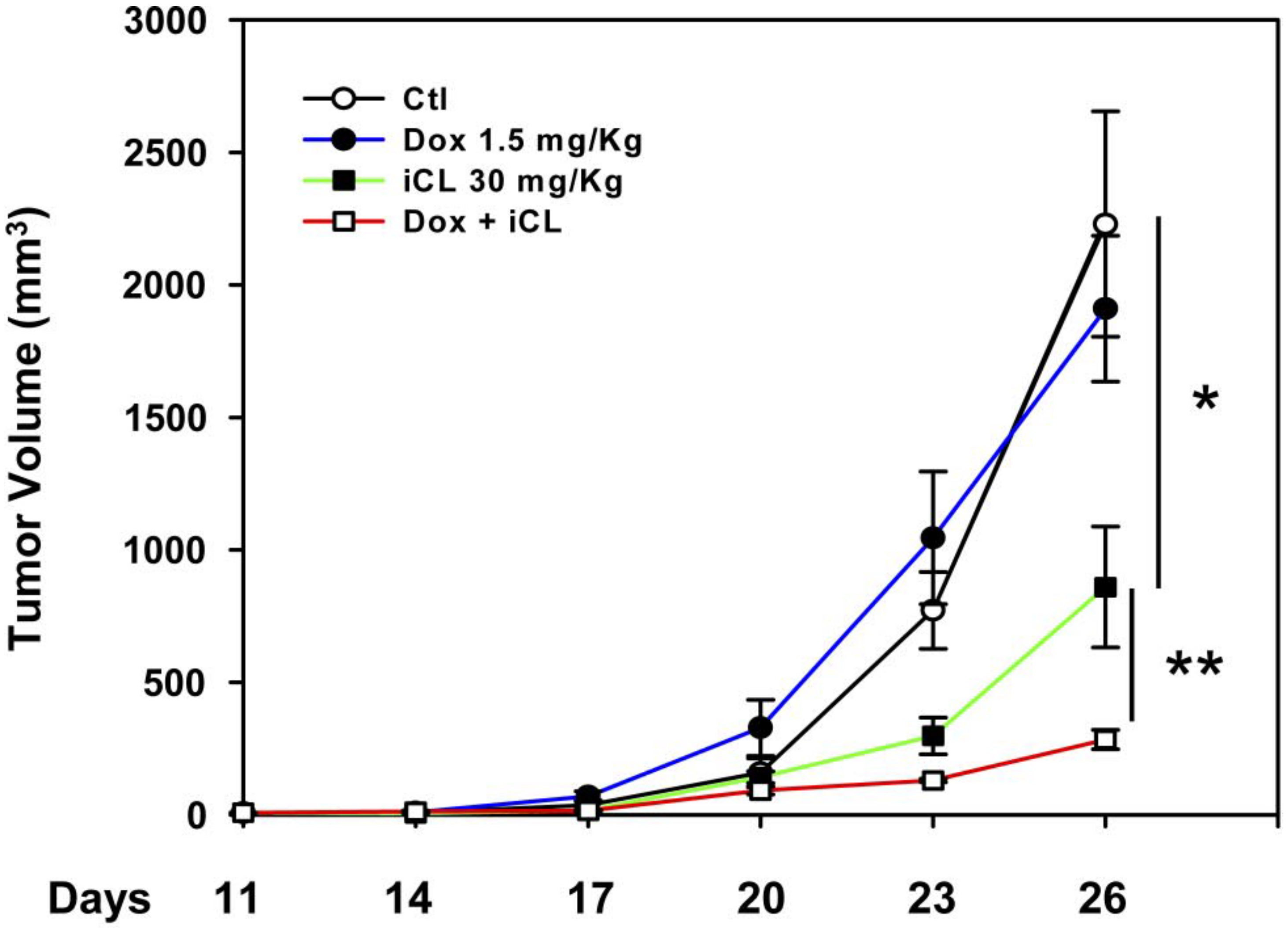{kind=link}
{kind=link}
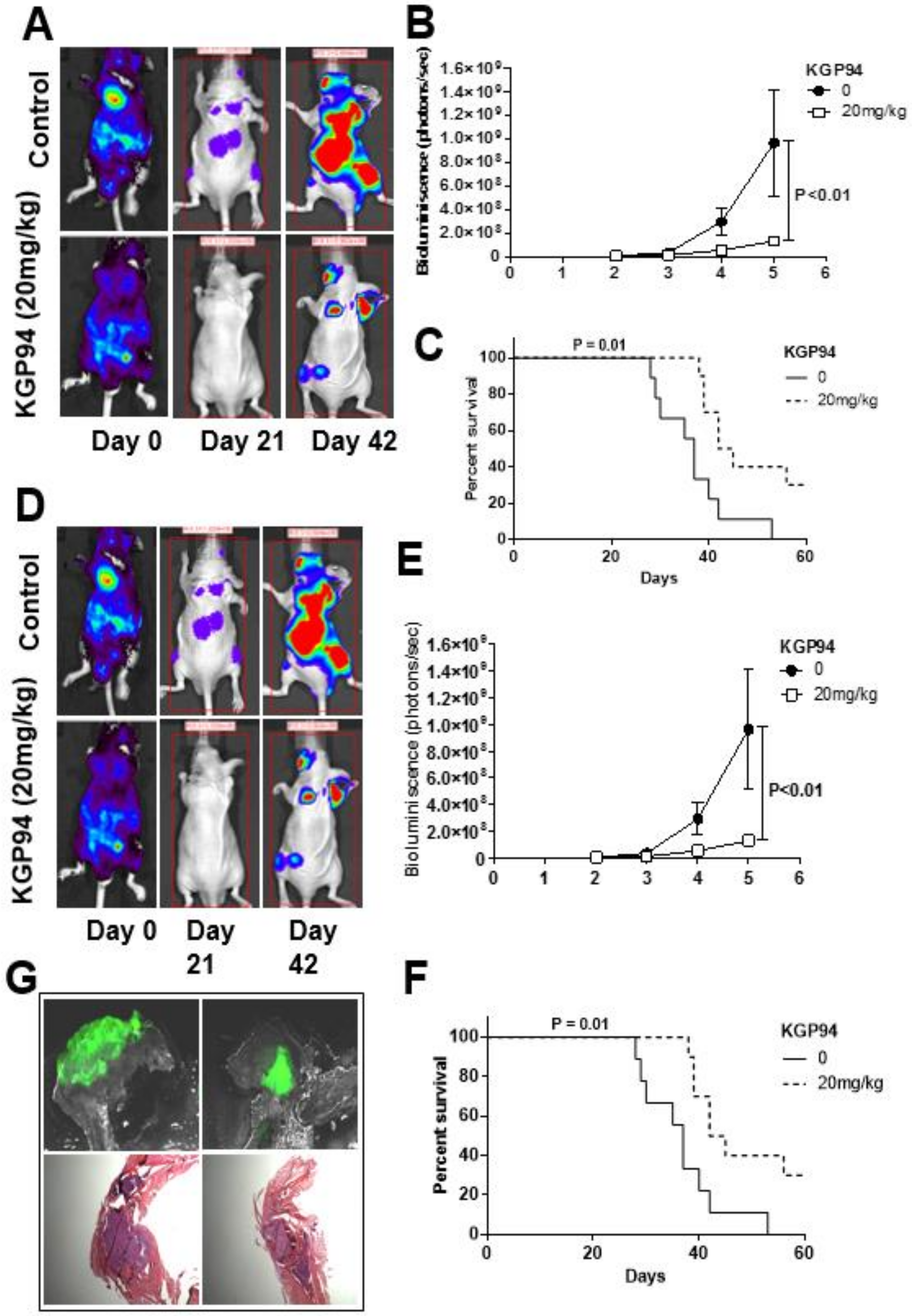{kind=link}
Table 1.
Summary of cathepsin-targeting agents in bone cancer.
| Cathepsin Target | Agent | Study Type | Target Approach | Effects | Side Effects |
|---|---|---|---|---|---|
| CatB | CA074Me | Preclinical | SMI |
| -- |
| BPC | Preclinical | SMI |
| -- | |
| CatG | TPCK | Preclinical | SMI |
| -- |
| CatK | Genetic inhibition | Preclinical | CatK null mice |
| -- |
| AFG-495 | Preclinical | SMI |
| -- | |
| CKI | Preclinical | SMI |
| -- | |
| L-235 |
| -- | |||
| Odanacatib | Preclinical/Clinical | SMI |
| -Increase risk of stroke | |
| -Skin rashes | |||||
| CatL | Genetic inhibition | Preclinical | Antisense oligonucleotides |
| -- |
| iCL | Preclinical | SMI |
| -- | |
| Muscadine grape skin extract | Preclinical |
| -- | ||
| CLIK-148 | Preclinical | SMI |
| -- | |
| CatB/CatK | E64 | Preclinical | SMI |
| -- |
| CatK/CatL | KGP94 | Preclinical | SMI |
| -- |
| Pan-cathepsin inhibitor | JMP-OEt | SMI |
| -- |
SMI = small molecule inhibitor.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Fasanya, H.O.; Siemann, D.W. The Role of Cathepsins in the Growth of Primary and Secondary Neoplasia in the Bone. Osteology 2021, 1, 3-28. https://0-doi-org.brum.beds.ac.uk/10.3390/osteology1010002
AMA Style
Fasanya HO, Siemann DW. The Role of Cathepsins in the Growth of Primary and Secondary Neoplasia in the Bone. Osteology. 2021; 1(1):3-28. https://0-doi-org.brum.beds.ac.uk/10.3390/osteology1010002
Chicago/Turabian StyleFasanya, Henrietta O., and Dietmar W. Siemann. 2021. "The Role of Cathepsins in the Growth of Primary and Secondary Neoplasia in the Bone" Osteology 1, no. 1: 3-28. https://0-doi-org.brum.beds.ac.uk/10.3390/osteology1010002