
Angiomatoid Fibrous Histiocytoma: Case Presentation with Review of Literature

 ,
,  , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Enzinger, F.M. Angiomatoid malignant fibrous histiocytoma: A distinct fibrohistiocytic tumor of children and young adults simulating a vascular neoplasm. Cancer 1979, 44, 2147–2157. [Google Scholar] [CrossRef]
- WHO. Soft Tissue and Bone Tumours WHO Classification of Tumours, 5th ed.; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Spatz, M.; Nussbaum, E.S.; Lyons, L.; Greenberg, S.; Kallmes, K.M.; Nussbaum, L.A. Primary intracranial angiomatoid fibrous histiocytoma: A case report and literature review. Br. J. Neurosurg. 2021, 35, 233–235. [Google Scholar] [CrossRef] [PubMed]
- Gunness, V.R.N.; Munoz, I.; González-López, P.; Alshafai, N.; Mikalkova, A.; Spears, J. Intracranial angiomatoid fibrous histiocytoma with Hodgkin lymphoma. Med. J. Malays. 2019, 74, 234–236. [Google Scholar]
- Bin Abdulqader, S.; Altuhaini, K.; Tallab, R.; AlTurkistani, A.; Alhussinan, M.; Alghamdi, S.; Al Saidi, K.; Almalki, S.; Alshakweer, W.; Alotaibi, F.E. Primary Intracranial Angiomatoid Fibrous Histiocytoma: Two Case Reports and Literature Review. World Neurosurg. 2020, 143, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Rani, D.; Gupta, A.; Gupta, A.; Rastogi, K. Cytological diagnosis of angiomatoid fibrous histiocytoma: Report of a case and review of literature. Diagn Cytopathol. 2021, 49, E36–E39. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Carreon, C.K.; Guerrero, J.; Putra, J. TLE-1 immunoreactivity in angiomatoid fibrous histiocytoma: A potential diagnostic pitfall. Pathology 2020, 52, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Abrahao-Machado, L.F.; Bacchi, L.M.; Fernandes, I.L.; Costa, F.D.; Bacchi, C.E. MUC4 Expression in Angiomatoid Fibrous Histiocytoma. Appl. Immunohistochem. Mol. Morphol. 2020, 28, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Cheah, A.L.; Zou, Y.; Lanigan, C.; Billings, S.D.; Rubin, B.P.; Hornick, J.L.; Goldblum, J.R. ALK Expression in Angiomatoid Fibrous Histiocytoma: A Potential Diagnostic Pitfall. Am. J. Surg. Pathol. 2019, 43, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Zhou, G.; Dong, Y.; Li, X.; Wang, L.; Guo, B.; Gao, C.; Xu, S.; Wang, F.; Sun, T. Angiomatoid fibrous histiocytoma in the spinal canal of T3-T4: A case report and literature review. Br. J. Neurosurg. 2020, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.Y.; Wang, L.C.; Hsu, H.S. Silent Angiomatoid Fibrous Histiocytoma of the Chest Wall. Ann. Thorac Surg. 2021, 111, e347–e348. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Kobayashi, E.; Yoshida, A.; Araki, Y.; Kubota, D.; Tanzawa, Y.; Kawai, A.; Yanagawa, T.; Takagishi, K.; Chuman, H. Angiomatoid fibrous histiocytoma: A series of seven cases including genetically confirmed aggressive cases and a literature review. BMC Musculoskelet. Disord. 2017, 18, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thway, K.; Fishern, C. Angiomatoid fibrous histiocytoma: The current status of pathology and genetics. Arch. Pathol. Lab. Med. 2015, 139, 674–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thway, K. Angiomatoid fibrous histiocytoma: A review with recent genetic findings. Arch. Pathol. Lab. Med. 2008, 132, 273–277. [Google Scholar] [CrossRef]
- Goldblum, J.; Weiss, S.; Folpe, A.L. Enzinger and Weiss’s Soft Tissue Tumors, 7th ed.; Elsevier: Amsterdam, The Netherlands.
- Albonico, G.; Pellegrino, G.; Maisano, M.; Africa, G.; Pedriali, M.; Nenci, I. Aneurysmatic fibrous histiocytoma: Case report and reivew of the literature. Pathologica 2001, 93, 136–138. [Google Scholar]
- Lee, H.S.; Kim, T.; Kim, J.S.; Lee, H.R.; Joo, M.; Park, J.Y.; Yi, S.Y. Angiomatoid fibrous histiocytoma as a second tumor in a young adult with testicular cancer. Cancer Res. Treat. 2013, 45, 239–243. [Google Scholar] [CrossRef]
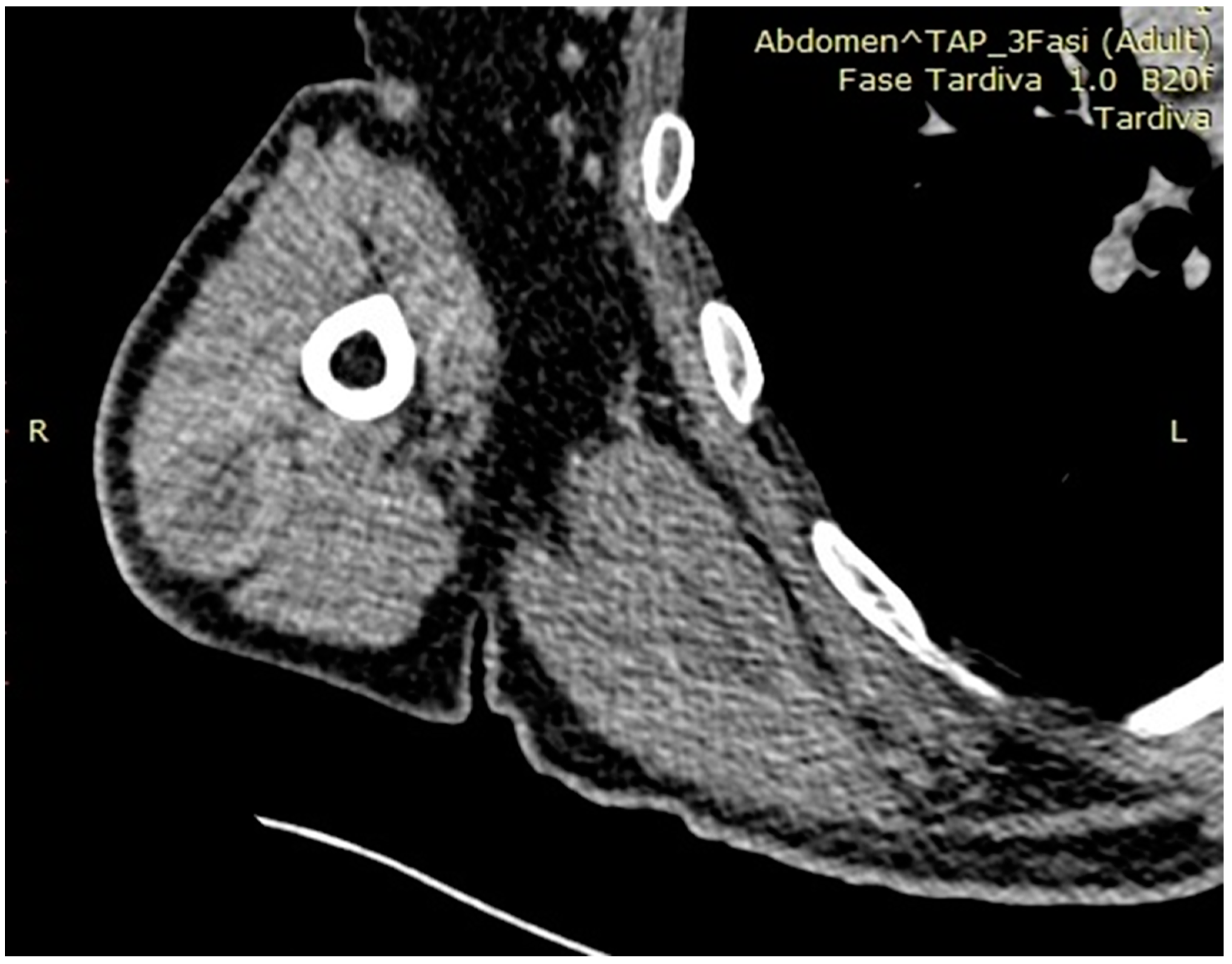

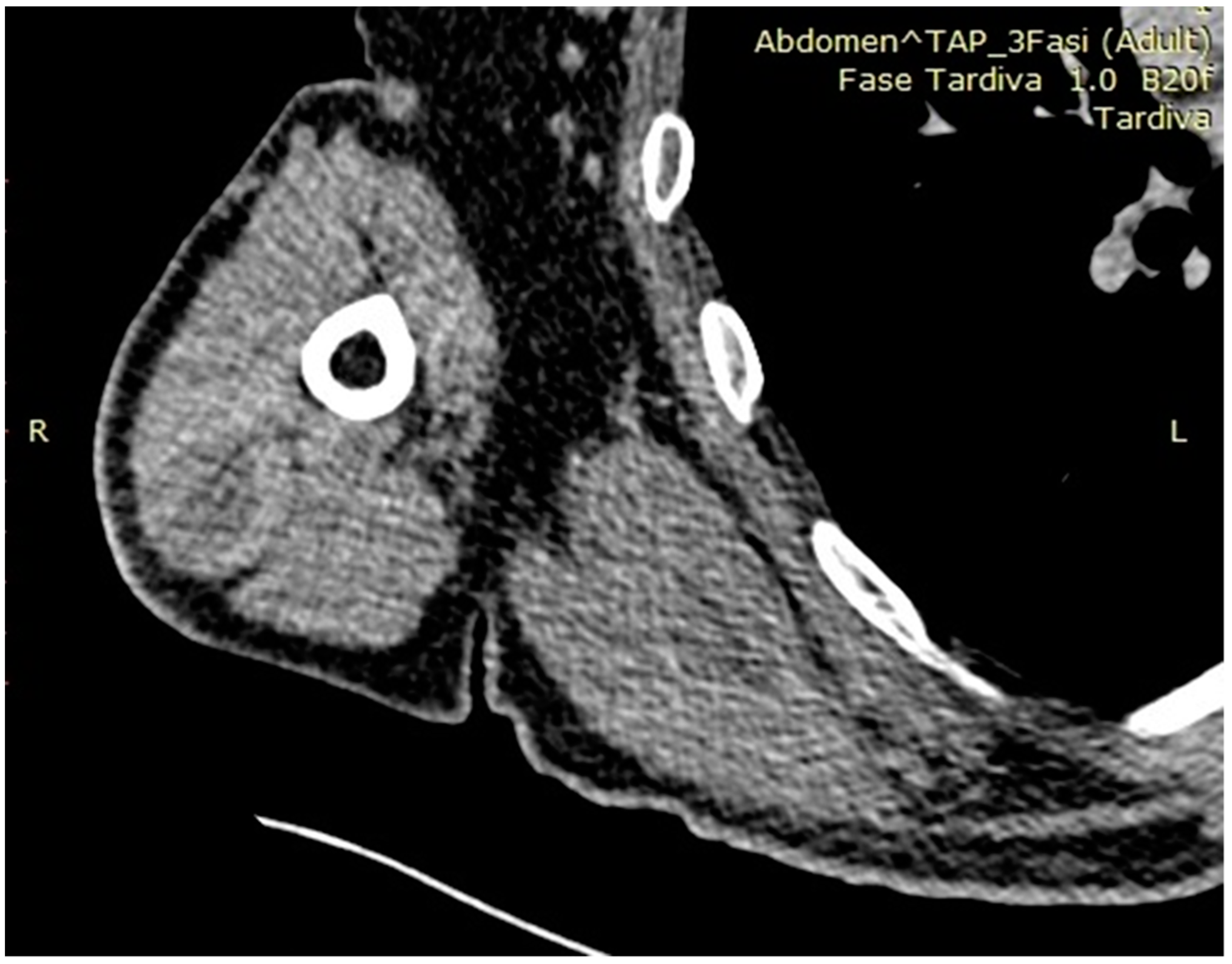{kind=link}
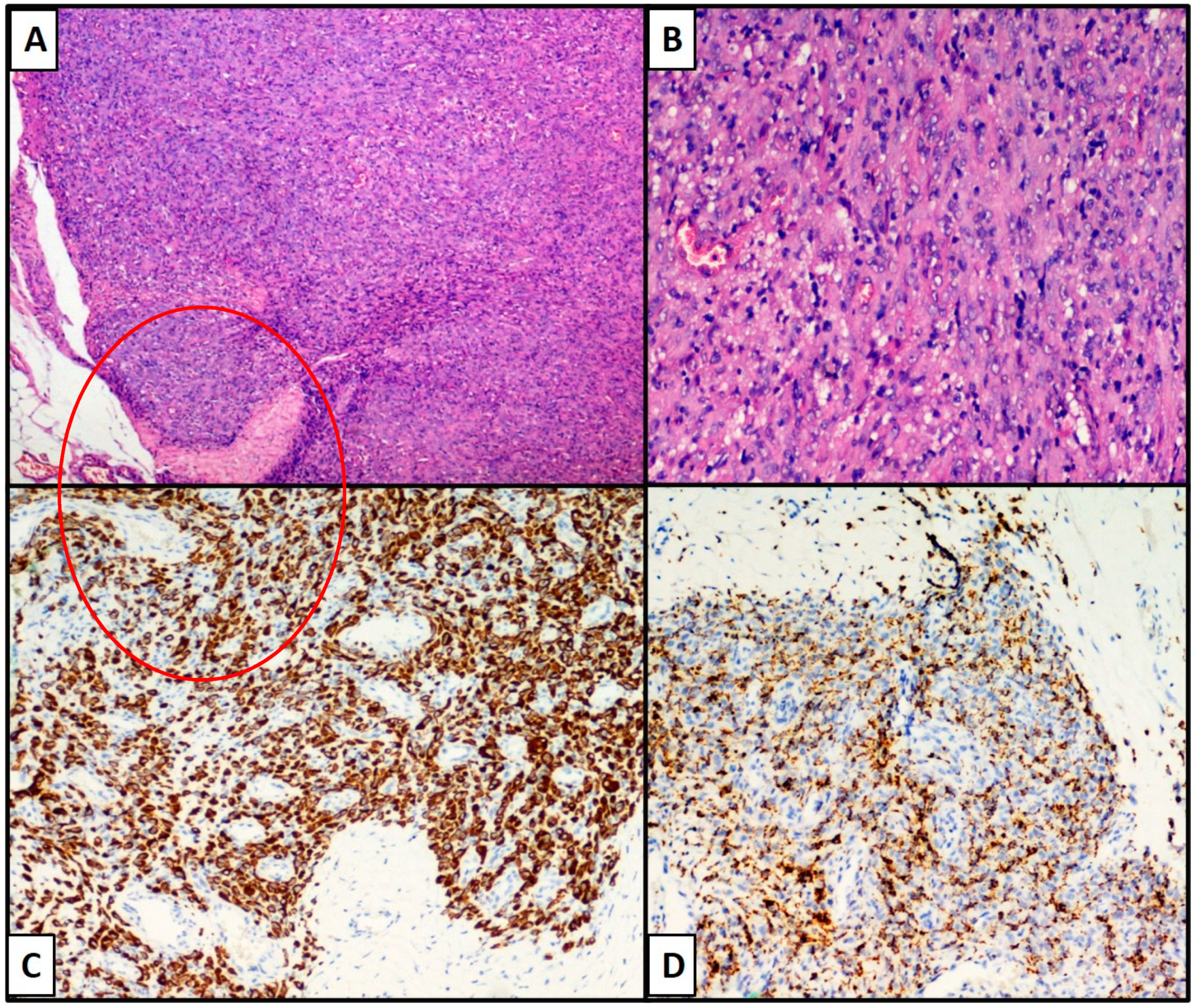
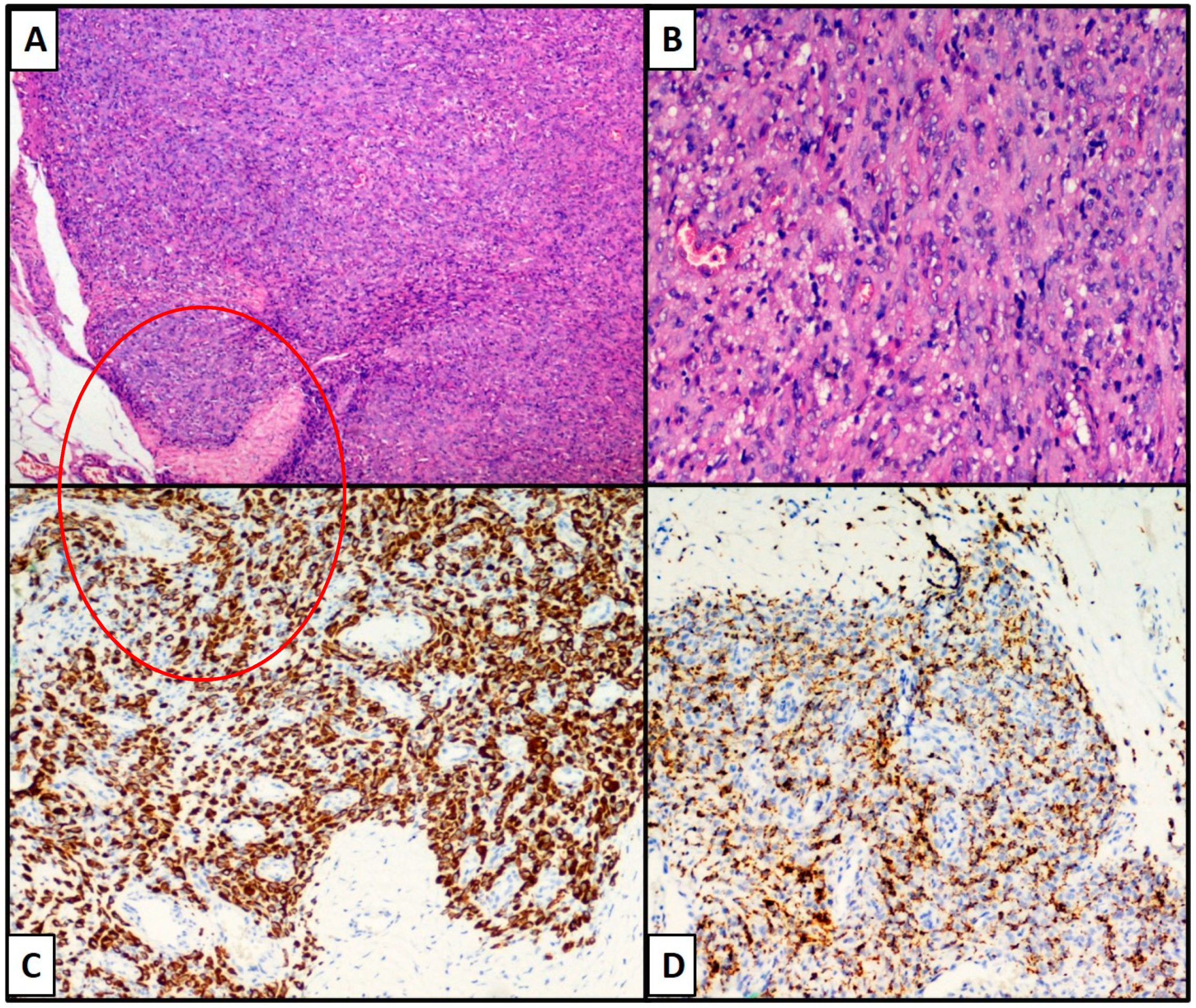{kind=link}
| Nature of Lesion | Fibrohistiocytic | Vascular | Neural/Melanocytic | Epithelial |
|---|---|---|---|---|
| Anti-CD68 (PG-M1; Dako, 1:800) | Anti-CD34 (QBEND/10, Dako, 1:100) | Anti-S-100 protein (Polyclonal, Dako, 1:500) | Anti-EMA (E29, Dako, 1:200) | |
| Anti-CD163 (Polyclonal, ThermoFisher 1:50) | Anti-CD31 (JC70A, Dako, 1:40) | Anti-SOX-10 (Polyclonal, ThermoFisher, 1:2000) | Anti-CK-Pool (M3515, Dako, 1:150) | |
| Immunohistochemical Markers (Clone, manufacturing company, dilution) | Anti-Desmin (PA5-16705, ThermoFisher, 1:200) | Anti-ERG (SP06-04, ThermoFisher, 1:100) | Anti-Melan-A (M2-7C10 + M2-9E3, ThermoFisher, 1:100) | Anti-CK 7 (Clone OV-TL 12/30, Dako, 1:100) |
| Anti-Podoplanin (D2-40, ThermoFisher, 1:40) | Anti-HMB-45 (ThermoFisher, 1:100) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cazzato, G.; Colagrande, A.; Cimmino, A.; Silecchia, M.; Lettini, T.; Resta, L.; Ingravallo, G. Angiomatoid Fibrous Histiocytoma: Case Presentation with Review of Literature. Osteology 2021, 1, 112-117. https://0-doi-org.brum.beds.ac.uk/10.3390/osteology1030012
Cazzato G, Colagrande A, Cimmino A, Silecchia M, Lettini T, Resta L, Ingravallo G. Angiomatoid Fibrous Histiocytoma: Case Presentation with Review of Literature. Osteology. 2021; 1(3):112-117. https://0-doi-org.brum.beds.ac.uk/10.3390/osteology1030012
Chicago/Turabian StyleCazzato, Gerardo, Anna Colagrande, Antonietta Cimmino, Mariella Silecchia, Teresa Lettini, Leonardo Resta, and Giuseppe Ingravallo. 2021. "Angiomatoid Fibrous Histiocytoma: Case Presentation with Review of Literature" Osteology 1, no. 3: 112-117. https://0-doi-org.brum.beds.ac.uk/10.3390/osteology1030012