
The Roles of Protein Structure, Taxon Sampling, and Model Complexity in Phylogenomics: A Case Study Focused on Early Animal Divergences



Abstract
:1. Introduction
2. Methods
3. Results
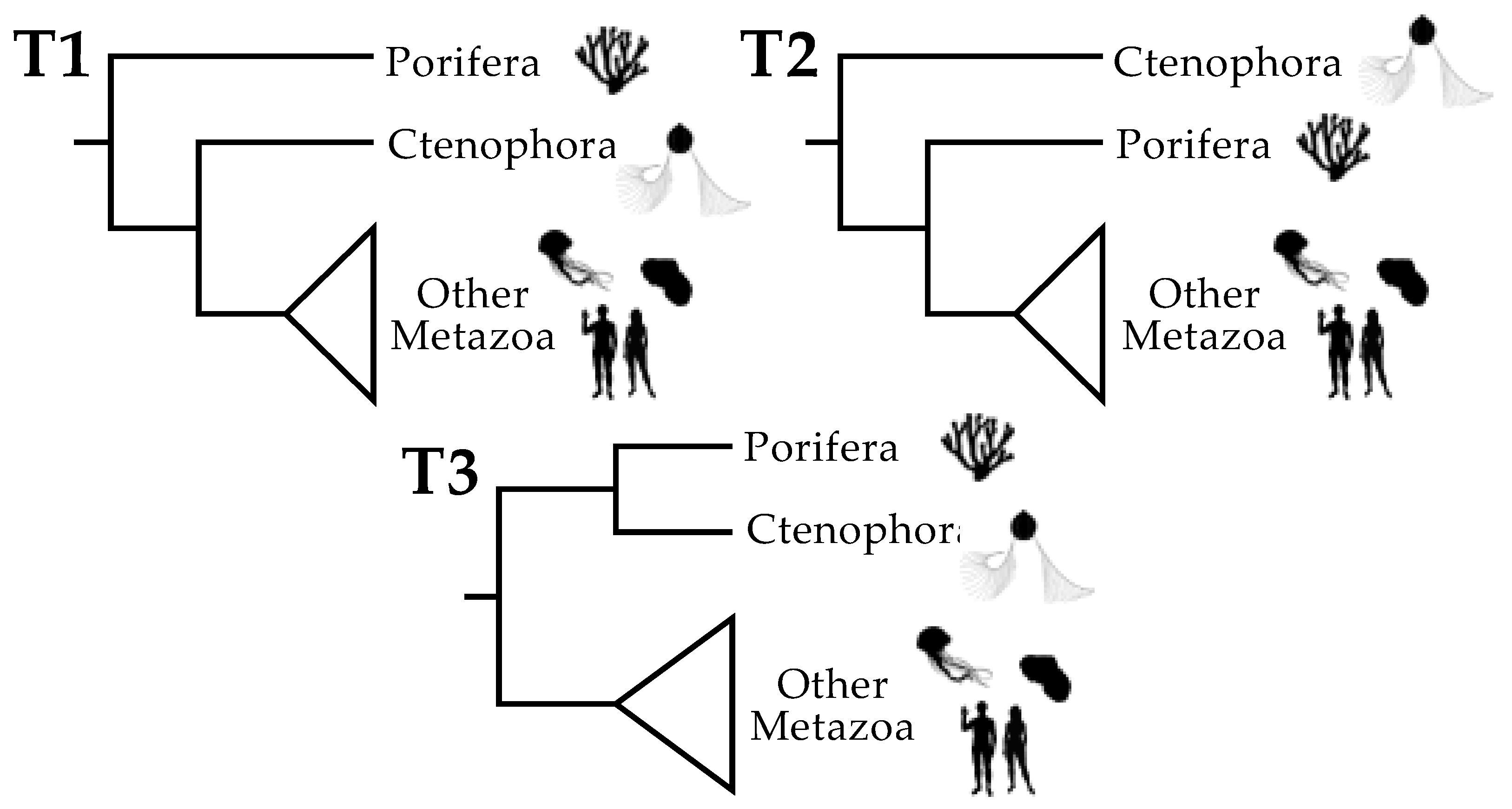
3.1. Tree Searches and the Use of Strongly Decisive Sites Lead to Similar Conclusions
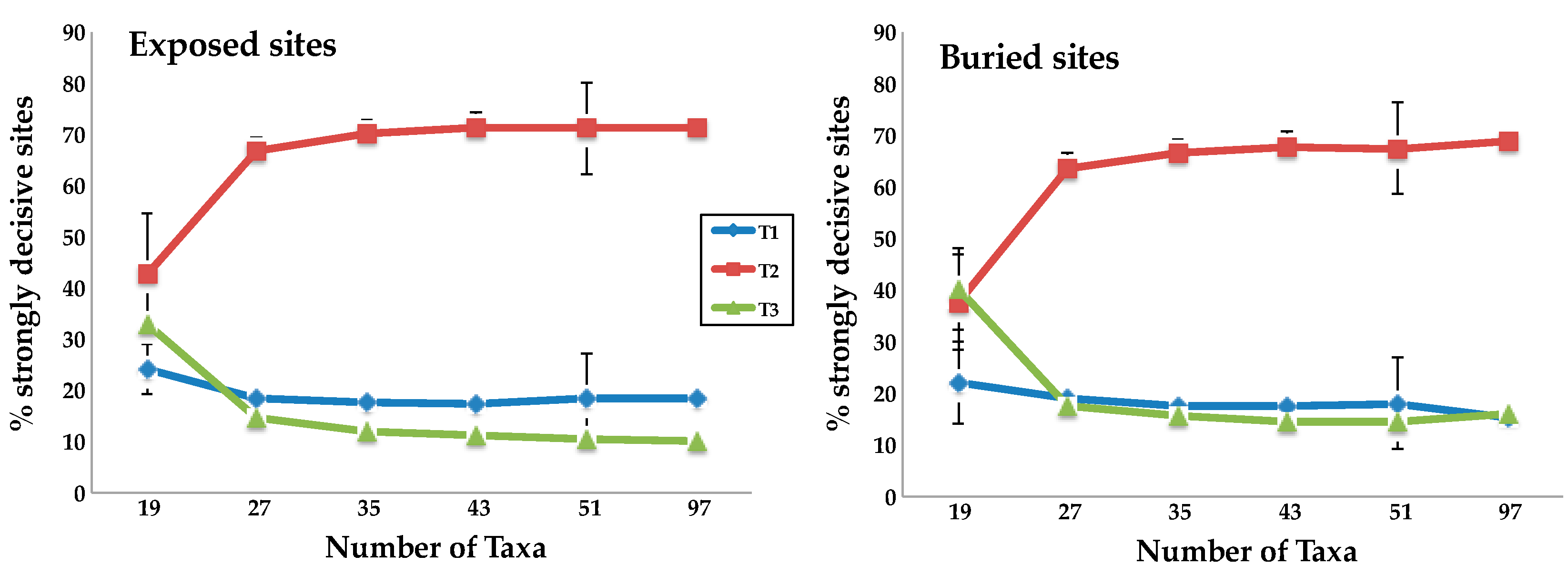
3.2. Taxon Addition Leads to Rapid Convergence in the Proportions of Strongly Decisive Sites Observed Using the Complete Taxon Sample
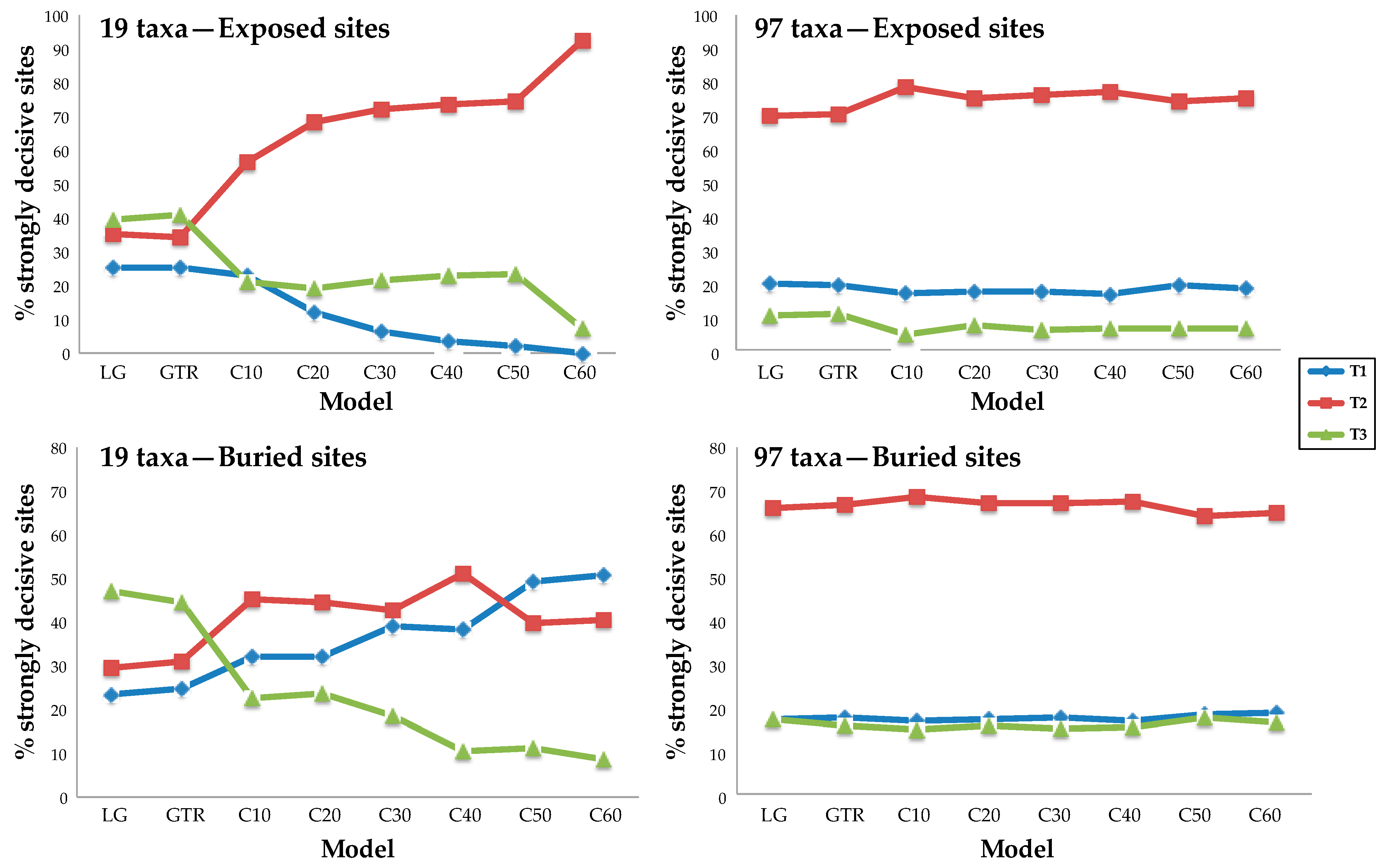
3.3. Rich Taxon Sampling Can Compensate for Low Model Complexity
4. Discussion
4.1. Taxon Sampling Has a Larger Impact Than Model Complexity
4.2. Strongly Decisive Sites, Phylogenetic Signal, and Protein Structure
5. Conclusions
- (1)
- There is gene tree–species tree discordance at the base of Metazoa, possibly reflecting ILS, combined with a structure-dependent bias. The bias leads to the observed asymmetry in the proportions of minority decisive site types in the relatively rapidly evolving solvent-exposed and helix sites.
- (2)
- There are processes of sequence evolution that generate conflicting strongly decisive sites without discordance among gene trees. Those processes result in equal proportions of minority decisive site types in low-rate structural environments and unequal proportions of minority decisive site types in higher rate structural environments.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zuckerkandl, E.; Pauling, L. Evolutionary divergence and convergence in proteins. In Evolving Genes and Proteins; Bryson, V., Vogel, H.J., Eds.; Elsevier: Amsterdam, The Netherlands, 1965; pp. 97–166. ISBN 9781483227344. [Google Scholar]
- Dickerson, R.E. The structures of cytochrome c and the rates of molecular evolution. J. Mol. Evol. 1971, 1, 26–45. [Google Scholar] [CrossRef]
- Alvarez-Ponce, D. Richard Dickerson, molecular clocks, and rates of protein evolution. J. Mol. Evol. 2020. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, J.-R. Determinants of the rate of protein sequence evolution. Nat. Rev. Genet. 2015, 16, 409–420. [Google Scholar] [CrossRef]
- Echave, J.; Spielman, S.J.; Wilke, C.O. Causes of evolutionary rate variation among protein sites. Nat. Rev. Genet. 2016, 17, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Gerstein, M.; Sonnhammer, E.L.; Chothia, C. Volume changes in protein evolution. J. Mol. Biol. 1994, 236, 1067–1078. [Google Scholar] [CrossRef]
- Goldman, N.; Thorne, J.L.; Jones, D.T. Assessing the impact of secondary structure and solvent accessibility on protein evolution. Genetics 1998, 149, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Illergård, K.; Ardell, D.H.; Elofsson, A. Structure is three to ten times more conserved than sequence--a study of structural response in protein cores. Proteins 2009, 77, 499–508. [Google Scholar] [CrossRef]
- Worth, C.L.; Gong, S.; Blundell, T.L. Structural and functional constraints in the evolution of protein families. Nat. Rev. Mol. Cell Biol. 2009, 10, 709–720. [Google Scholar] [CrossRef]
- Le, S.Q.; Gascuel, O. Accounting for solvent accessibility and secondary structure in protein phylogenetics is clearly beneficial. Syst. Biol. 2010, 59, 277–287. [Google Scholar] [CrossRef]
- Pandey, A.; Braun, E.L. Protein evolution is structure dependent and non-homogeneous across the tree of life. In Proceedings of the 11th ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics (ACM-BCB’20); ACM: New York, NY, USA, 2020; Article No.: 28, 11p. [Google Scholar] [CrossRef]
- Eisen, J.A. Phylogenomics: Improving functional predictions for uncharacterized genes by evolutionary analysis. Genome Res. 1998, 8, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Sjölander, K. Phylogenomic inference of protein molecular function: Advances and challenges. Bioinformatics 2004, 20, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Delsuc, F.; Brinkmann, H.; Philippe, H. Phylogenomics and the reconstruction of the tree of life. Nat. Rev. Genet. 2005, 6, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Dunn, C.W.; Hejnol, A.; Matus, D.Q.; Pang, K.; Browne, W.E.; Smith, S.A.; Seaver, E.; Rouse, G.W.; Obst, M.; Edgecombe, G.D.; et al. Broad phylogenomic sampling improves resolution of the animal tree of life. Nature 2008, 452, 745–749. [Google Scholar] [CrossRef]
- Hejnol, A.; Obst, M.; Stamatakis, A.; Ott, M.; Rouse, G.W.; Edgecombe, G.D.; Martinez, P.; Baguñà, J.; Bailly, X.; Jondelius, U.; et al. Assessing the root of bilaterian animals with scalable phylogenomic methods. Proc. R. Soc. B 2009, 276, 4261–4270. [Google Scholar] [CrossRef]
- Philippe, H.; Derelle, R.; Lopez, P.; Pick, K.; Borchiellini, C.; Boury-Esnault, N.; Vacelet, J.; Renard, E.; Houliston, E.; Quéinnec, E.; et al. Phylogenomics revives traditional views on deep animal relationships. Curr. Biol. 2009, 19, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Pick, K.S.; Philippe, H.; Schreiber, F.; Erpenbeck, D.; Jackson, D.J.; Wrede, P.; Wiens, M.; Alié, A.; Morgenstern, B.; Manuel, M.; et al. Improved phylogenomic taxon sampling noticeably affects nonbilaterian relationships. Mol. Biol. Evol. 2010, 27, 1983–1987. [Google Scholar] [CrossRef]
- Nosenko, T.; Schreiber, F.; Adamska, M.; Adamski, M.; Eitel, M.; Hammel, J.; Maldonado, M.; Müller, W.E.G.; Nickel, M.; Schierwater, B.; et al. Deep metazoan phylogeny: When different genes tell different stories. Mol. Phylogenet. Evol. 2013, 67, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J.F.; Pang, K.; Schnitzler, C.E.; Nguyen, A.-D.; Moreland, R.T.; Simmons, D.K.; Koch, B.J.; Francis, W.R.; Havlak, P.; NISC Comparative Sequencing Program; et al. The genome of the ctenophore Mnemiopsis leidyi and its implications for cell type evolution. Science 2013, 342, 1242592. [Google Scholar] [CrossRef]
- Moroz, L.L.; Kocot, K.M.; Citarella, M.R.; Dosung, S.; Norekian, T.P.; Povolotskaya, I.S.; Grigorenko, A.P.; Dailey, C.; Berezikov, E.; Buckley, K.M.; et al. The ctenophore genome and the evolutionary origins of neural systems. Nature 2014, 510, 109–114. [Google Scholar] [CrossRef]
- Borowiec, M.L.; Lee, E.K.; Chiu, J.C.; Plachetzki, D.C. Extracting phylogenetic signal and accounting for bias in whole-genome data sets supports the Ctenophora as sister to remaining Metazoa. BMC Genom. 2015, 16, 987. [Google Scholar] [CrossRef]
- Pisani, D.; Pett, W.; Dohrmann, M.; Feuda, R.; Rota-Stabelli, O.; Philippe, H.; Lartillot, N.; Wörheide, G. Genomic data do not support comb jellies as the sister group to all other animals. Proc. Natl. Acad. Sci. USA 2015, 112, 15402–15407. [Google Scholar] [CrossRef]
- Whelan, N.V.; Kocot, K.M.; Moroz, L.L.; Halanych, K.M. Error, signal, and the placement of Ctenophora sister to all other animals. Proc. Natl. Acad. Sci. USA 2015, 112, 5773–5778. [Google Scholar] [CrossRef] [PubMed]
- Feuda, R.; Dohrmann, M.; Pett, W.; Philippe, H.; Rota-Stabelli, O.; Lartillot, N.; Wörheide, G.; Pisani, D. Improved modeling of compositional heterogeneity supports sponges as sister to all other animals. Curr. Biol. 2017, 27, 3864–3870.e4. [Google Scholar] [CrossRef]
- Simion, P.; Philippe, H.; Baurain, D.; Jager, M.; Richter, D.J.; Di Franco, A.; Roure, B.; Satoh, N.; Quéinnec, É.; Ereskovsky, A.; et al. A large and consistent phylogenomic dataset supports sponges as the sister group to all other animals. Curr. Biol. 2017, 27, 958–967. [Google Scholar] [CrossRef] [PubMed]
- Whelan, N.V.; Kocot, K.M.; Moroz, T.P.; Mukherjee, K.; Williams, P.; Paulay, G.; Moroz, L.L.; Halanych, K.M. Ctenophore relationships and their placement as the sister group to all other animals. Nat. Ecol. Evol. 2017, 1, 1737–1746. [Google Scholar] [CrossRef] [PubMed]
- Laumer, C.E.; Fernández, R.; Lemer, S.; Combosch, D.; Kocot, K.M.; Riesgo, A.; Andrade, S.C.S.; Sterrer, W.; Sørensen, M.V.; Giribet, G. Revisiting metazoan phylogeny with genomic sampling of all phyla. Proc. R. Soc. B 2019, 286, 20190831. [Google Scholar] [CrossRef]
- Francis, W.R.; Canfield, D.E. Very few sites can reshape the inferred phylogenetic tree. PeerJ 2020, 8, e8865. [Google Scholar] [CrossRef]
- Kapli, P.; Telford, M.J. Topology-dependent asymmetry in systematic errors affects phylogenetic placement of Ctenophora and Xenacoelomorpha. Sci. Adv. 2020, 6, eabc5162. [Google Scholar] [CrossRef]
- Pandey, A.; Braun, E.L. Phylogenetic analyses of sites in different protein structural environments result in distinct placements of the metazoan root. Biology 2020, 9, 64. [Google Scholar] [CrossRef]
- Ryan, J.F.; Pang, K.; NISC Comparative Sequencing Program; Mullikin, J.C.; Martindale, M.Q.; Baxevanis, A.D. The homeodomain complement of the ctenophore Mnemiopsis leidyi suggests that Ctenophora and Porifera diverged prior to the ParaHoxozoa. Evodevo 2010, 1, 9. [Google Scholar] [CrossRef]
- Osigus, H.-J.; Rolfes, S.; Herzog, R.; Kamm, K.; Schierwater, B. Polyplacotoma mediterranea is a new ramified placozoan species. Curr. Biol. 2019, 29, R148–R149. [Google Scholar] [CrossRef]
- Nielsen, C. Early animal evolution: A morphologist’s view. R. Soc. Open Sci. 2019, 6, 190638. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.; Kimball, R.T.; Pandey, A.; Hosner, P.A.; Braun, M.J.; Hackett, S.J.; Han, K.-L.; Harshman, J.; Huddleston, C.J.; Kingston, S.; et al. Why do phylogenomic data sets yield conflicting trees? Data type influences the avian tree of life more than taxon sampling. Syst. Biol. 2017, 66, 857–879. [Google Scholar] [CrossRef]
- Braun, E.L.; Kimball, R.T. Data types and the phylogeny of Neoaves. Birds 2021, 2, 1–22. [Google Scholar] [CrossRef]
- Maddison, W.P. Gene trees in species trees. Syst. Biol. 1997, 46, 523–536. [Google Scholar] [CrossRef]
- Edwards, S.V. Is a new and general theory of molecular systematics emerging? Evolution 2009, 63, 1–19. [Google Scholar] [CrossRef]
- Philippe, H.; Brinkmann, H.; Lavrov, D.V.; Littlewood, D.T.J.; Manuel, M.; Wörheide, G.; Baurain, D. Resolving difficult phylogenetic questions: Why more sequences are not enough. PLoS Biol. 2011, 9, e1000602. [Google Scholar] [CrossRef]
- Warnow, T. Computational Phylogenetics (An Introduction to Designing Methods for Phylogeny Estimation), 1st ed.; Cambridge University Press: Cambridge, UK, 2017; p. 394. ISBN 978-1107184718. [Google Scholar]
- Pamilo, P.; Nei, M. Relationships between gene trees and species trees. Mol. Biol. Evol. 1988, 5, 568–583. [Google Scholar] [CrossRef]
- Le, S.Q.; Gascuel, O. An improved general amino acid replacement matrix. Mol. Biol. Evol. 2008, 25, 1307–1320. [Google Scholar] [CrossRef]
- Tavaré, S. Some probabilistic and statistical problems in the analysis of DNA sequences. Lect. Math. Life Sci. 1986, 17, 57–86. [Google Scholar]
- Dayhoff, M.O.; Schwartz, R.M.; Orcutt, B.C. A model of evolutionary change in proteins. In Atlas of Protein Sequence and Structure; Dayhoff, M.O., Ed.; National Biomedical Research Foundation: Silver Springs, MD, USA, 1978; Volume 5, pp. 345–352. [Google Scholar]
- Susko, E.; Roger, A.J. On reduced amino acid alphabets for phylogenetic inference. Mol. Biol. Evol. 2007, 24, 2139–2150. [Google Scholar] [CrossRef]
- Bruno, W.J. Modeling residue usage in aligned protein sequences via maximum likelihood. Mol. Biol. Evol. 1996, 13, 1368–1374. [Google Scholar] [CrossRef]
- Halpern, A.L.; Bruno, W.J. Evolutionary distances for protein-coding sequences: Modeling site-specific residue frequencies. Mol. Biol. Evol. 1998, 15, 910–917. [Google Scholar] [CrossRef]
- Melamed, D.; Young, D.L.; Gamble, C.E.; Miller, C.R.; Fields, S. Deep mutational scanning of an RRM domain of the Saccharomyces cerevisiae poly(A)-binding protein. RNA 2013, 19, 1537–1551. [Google Scholar] [CrossRef] [PubMed]
- Roscoe, B.P.; Bolon, D.N.A. Systematic exploration of ubiquitin sequence, E1 activation efficiency, and experimental fitness in yeast. J. Mol. Biol. 2014, 426, 2854–2870. [Google Scholar] [CrossRef] [PubMed]
- Starita, L.M.; Young, D.L.; Islam, M.; Kitzman, J.O.; Gullingsrud, J.; Hause, R.J.; Fowler, D.M.; Parvin, J.D.; Shendure, J.; Fields, S. Massively parallel functional analysis of BRCA1 RING domain variants. Genetics 2015, 200, 413–422. [Google Scholar] [CrossRef]
- Mighell, T.L.; Evans-Dutson, S.; O’Roak, B.J. A saturation mutagenesis approach to understanding PTEN lipid phosphatase activity and genotype-phenotype relationships. Am. J. Hum. Genet. 2018, 102, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Lartillot, N.; Philippe, H. A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol. Biol. Evol. 2004, 21, 1095–1109. [Google Scholar] [CrossRef]
- Le, S.Q.; Gascuel, O.; Lartillot, N. Empirical profile mixture models for phylogenetic reconstruction. Bioinformatics 2008, 24, 2317–2323. [Google Scholar] [CrossRef]
- Lartillot, N.; Brinkmann, H.; Philippe, H. Suppression of long-branch attraction artefacts in the animal phylogeny using a site-heterogeneous model. BMC Evol. Biol. 2007, 7, S4. [Google Scholar] [CrossRef]
- Whelan, N.V.; Halanych, K.M. Who let the CAT out of the bag? Accurately dealing with substitutional heterogeneity in phylogenomic analyses. Syst. Biol. 2017, 66, 232–255. [Google Scholar] [CrossRef]
- Wang, H.-C.; Minh, B.Q.; Susko, E.; Roger, A.J. Modeling site heterogeneity with posterior mean site frequency profiles accelerates accurate phylogenomic estimation. Syst. Biol. 2018, 67, 216–235. [Google Scholar] [CrossRef] [PubMed]
- Hillis, D.M. Inferring complex phytogenies. Nature 1996, 383, 130–131. [Google Scholar] [CrossRef] [PubMed]
- Hillis, D.M. Taxonomic sampling, phylogenetic accuracy, and investigator bias. Syst. Biol. 1998, 47, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Pollock, D.D.; Zwickl, D.J.; McGuire, J.A.; Hillis, D.M. Increased taxon sampling is advantageous for phylogenetic inference. Syst. Biol. 2002, 51, 664–671. [Google Scholar] [CrossRef]
- Zwickl, D.J.; Hillis, D.M. Increased taxon sampling greatly reduces phylogenetic error. Syst. Biol. 2002, 51, 588–598. [Google Scholar] [CrossRef]
- Hillis, D.M.; Pollock, D.D.; McGuire, J.A.; Zwickl, D.J. Is sparse taxon sampling a problem for phylogenetic inference? Syst. Biol. 2003, 52, 124–126. [Google Scholar] [CrossRef] [PubMed]
- Hedtke, S.M.; Townsend, T.M.; Hillis, D.M. Resolution of phylogenetic conflict in large data sets by increased taxon sampling. Syst. Biol. 2006, 55, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Braun, E.L.; Kimball, R.T. Examining basal avian divergences with mitochondrial sequences: Model complexity, taxon sampling, and sequence length. Syst. Biol. 2002, 51, 614–625. [Google Scholar] [CrossRef]
- Wiens, J.J. Can incomplete taxa rescue phylogenetic analyses from long-branch attraction? Syst. Biol. 2005, 54, 731–742. [Google Scholar] [CrossRef]
- Heath, T.A.; Hedtke, S.M.; Hillis, D.M. Taxon sampling and the accuracy of phylogenetic analyses. J. Syst. Evol. 2008, 46, 239–257. [Google Scholar] [CrossRef]
- Sullivan, J.; Swofford, D.L.; Naylor, G. The effect of taxon sampling on estimating rate heterogeneity parameters of maximum-likelihood models. Mol. Biol. Evol. 1999, 16, 1347–1356. [Google Scholar] [CrossRef]
- Pollock, D.D.; Bruno, W.J. Assessing an unknown evolutionary process: Effect of increasing site-specific knowledge through taxon addition. Mol. Biol. Evol. 2000, 17, 1854–1858. [Google Scholar] [CrossRef] [PubMed]
- Swofford, D.L.; Waddell, P.J.; Huelsenbeck, J.P.; Foster, P.G.; Lewis, P.O.; Rogers, J.S. Bias in phylogenetic estimation and its relevance to the choice between parsimony and likelihood methods. Syst. Biol. 2001, 50, 525–539. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ané, C.; Larget, B.; Ronquist, F. A Bayesian perspective on a non-parsimonious parsimony model. Syst. Biol. 2008, 57, 406–419. [Google Scholar] [CrossRef] [PubMed]
- Kimball, R.T.; Wang, N.; Heimer-McGinn, V.; Ferguson, C.; Braun, E.L. Identifying localized biases in large datasets: A case study using the avian tree of life. Mol. Phylogenet. Evol. 2013, 69, 1021–1032. [Google Scholar] [CrossRef]
- Tsirigos, K.D.; Peters, C.; Shu, N.; Käll, L.; Elofsson, A. The TOPCONS web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Res. 2015, 43, W401–W407. [Google Scholar] [CrossRef] [PubMed]
- Henikoff, S.; Henikoff, J.G. Position-based sequence weights. J. Mol. Biol. 1994, 243, 574–578. [Google Scholar] [CrossRef]
- Magnan, C.N.; Baldi, P. SSpro/ACCpro 5: Almost perfect prediction of protein secondary structure and relative solvent accessibility using profiles, machine learning and structural similarity. Bioinformatics 2014, 30, 2592–2597. [Google Scholar] [CrossRef]
- Maddison, D.R.; Swofford, D.L.; Maddison, W.P. NEXUS: An extensible file format for systematic information. Syst. Biol. 1997, 46, 590–621. [Google Scholar] [CrossRef]
- Swofford, D.L. PAUP* (*Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4.0b10; Sinauer Associates: Sunderland, MA, USA, 2002. [Google Scholar]
- Whelan, S.; Goldman, N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 2001, 18, 691–699. [Google Scholar] [CrossRef]
- Braun, E.L. An evolutionary model motivated by physicochemical properties of amino acids reveals variation among proteins. Bioinformatics 2018, 34, i350–i356. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Burnham, K.P.; Anderson, D.R. Model Selection and Multimodel Inference; Springer: New York, NY, USA, 2004; ISBN 978-0-387-95364-9. [Google Scholar]
- Kelchner, S.A.; Thomas, M.A. Model use in phylogenetics: Nine key questions. Trends Ecol. Evol. 2007, 22, 87–94. [Google Scholar] [CrossRef]
- Sanderson, M.J.; Kim, J. Parametric phylogenetics? Syst. Biol. 2000, 49, 817–829. [Google Scholar] [CrossRef]
- Gatesy, J. A tenth crucial question regarding model use in phylogenetics. Trends Ecol. Evol. 2007, 22, 509–510. [Google Scholar] [CrossRef]
- Graybeal, A. Is it better to add taxa or characters to a difficult phylogenetic problem? Syst. Biol. 1998, 47, 9–17. [Google Scholar] [CrossRef]
- Goldman, N. Phylogenetic information and experimental design in molecular systematics. Proc. R. Soc. B 1998, 265, 1779–1786. [Google Scholar] [CrossRef] [PubMed]
- Geuten, K.; Massingham, T.; Darius, P.; Smets, E.; Goldman, N. Experimental design criteria in phylogenetics: Where to add taxa. Syst. Biol. 2007, 56, 609–622. [Google Scholar] [CrossRef]
- Lanier, H.C.; Knowles, L.L. Applying species-tree analyses to deep phylogenetic histories: Challenges and potential suggested from a survey of empirical phylogenetic studies. Mol. Phylogenet. Evol. 2015, 83, 191–199. [Google Scholar] [CrossRef]
- Tamashiro, R.A.; White, N.D.; Braun, M.J.; Faircloth, B.C.; Braun, E.L.; Kimball, R.T. What are the roles of taxon sampling and model fit in tests of cyto-nuclear discordance using avian mitogenomic data? Mol. Phylogenet. Evol. 2019, 130, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Stiller, J.; Deng, Y.; Armstrong, J.; Fang, Q.; Reeve, A.H.; Xie, D.; Chen, G.; Guo, C.; Faircloth, B.C.; et al. Dense sampling of bird diversity increases power of comparative genomics. Nature 2020, 587, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Zoonomia Consortium. A comparative genomics multitool for scientific discovery and conservation. Nature 2020, 587, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Panova, M.; Aronsson, H.; Cameron, R.A.; Dahl, P.; Godhe, A.; Lind, U.; Ortega-Martinez, O.; Pereyra, R.; Tesson, S.V.M.; Wrange, A.-L.; et al. DNA extraction protocols for whole-genome sequencing in marine organisms. Methods Mol. Biol. 2016, 1452, 13–44. [Google Scholar] [CrossRef] [PubMed]
- Lakner, C.; Holder, M.T.; Goldman, N.; Naylor, G.J.P. What’s in a likelihood? Simple models of protein evolution and the contribution of structurally viable reconstructions to the likelihood. Syst. Biol. 2011, 60, 161–174. [Google Scholar] [CrossRef]
- Grau-Bové, X.; Torruella, G.; Donachie, S.; Suga, H.; Leonard, G.; Richards, T.A.; Ruiz-Trillo, I. Dynamics of genomic innovation in the unicellular ancestry of animals. eLife 2017, 6, e26036. [Google Scholar] [CrossRef] [PubMed]
- Rossberg, A.G.; Rogers, T.; McKane, A.J. Are there species smaller than 1 mm? Proc. R. Soc. B 2013, 280, 20131248. [Google Scholar] [CrossRef]
- Ewing, G.B.; Ebersberger, I.; Schmidt, H.A.; von Haeseler, A. Rooted triple consensus and anomalous gene trees. BMC Evol. Biol. 2008, 8, 118. [Google Scholar] [CrossRef]
- Arcila, D.; Ortí, G.; Vari, R.; Armbruster, J.W.; Stiassny, M.L.J.; Ko, K.D.; Sabaj, M.H.; Lundberg, J.; Revell, L.J.; Betancur-R, R. Genome-wide interrogation advances resolution of recalcitrant groups in the tree of life. Nat. Ecol. Evol. 2017, 1, 20. [Google Scholar] [CrossRef]
- Mirarab, S.; Warnow, T. ASTRAL-II: Coalescent-based species tree estimation with many hundreds of taxa and thousands of genes. Bioinformatics 2015, 31, i44–i52. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.J.; Macis, L.; Reitner, J.; Wörheide, G. A horizontal gene transfer supported the evolution of an early metazoan biomineralization strategy. Bmc Evol. Biol. 2011, 11, 238. [Google Scholar] [CrossRef]
- Boto, L. Horizontal gene transfer in the acquisition of novel traits by metazoans. Proc. R. Soc. B 2014, 281, 20132450. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.M.; Ryan, J.F. Horizontally transferred genes in the ctenophore Mnemiopsis leidyi. PeerJ 2018, 6, e5067. [Google Scholar] [CrossRef]
- Hehenberger, E.; Tikhonenkov, D.V.; Kolisko, M.; Del Campo, J.; Esaulov, A.S.; Mylnikov, A.P.; Keeling, P.J. Novel predators reshape holozoan phylogeny and reveal the presence of a two-component signaling system in the ancestor of animals. Curr. Biol. 2017, 27, 2043–2050.e6. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Cases in which parsimony or compatibility methods will be positively misleading. Syst. Biol. 1978, 27, 401–410. [Google Scholar] [CrossRef]
- Arenas, M.; Dos Santos, H.G.; Posada, D.; Bastolla, U. Protein evolution along phylogenetic histories under structurally constrained substitution models. Bioinformatics 2013, 29, 3020–3028. [Google Scholar] [CrossRef] [PubMed]
- Sharir-Ivry, A.; Xia, Y. Nature of long-range evolutionary constraint in enzymes: Insights from comparison to pseudoenzymes with similar structures. Mol. Biol. Evol. 2018, 35, 2597–2606. [Google Scholar] [CrossRef]
- Echave, J. Beyond stability constraints: A biophysical model of enzyme evolution with selection on stability and activity. Mol. Biol. Evol. 2019, 36, 613–620. [Google Scholar] [CrossRef]
- Wilke, C.O. Bringing molecules back into molecular evolution. PLoS Comput. Biol. 2012, 8, e1002572. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | T1 | T2 | T3 | SD Sites 1 | Total Sites |
|---|---|---|---|---|---|
| Exposed | 19.3 | 70.8 | 9.9 | 1420 | 161,897 |
| Buried | 16.6 | 66.1 | 17.3 | 1625 | 194,117 |
| Helix | 19.2 | 69.3 | 11.5 | 1290 | 161,117 |
| Sheet | 12.5 | 71.6 | 16.9 | 514 | 60,563 |
| Coil | 17.7 | 68 | 14.3 | 1194 | 134,334 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pandey, A.; Braun, E.L. The Roles of Protein Structure, Taxon Sampling, and Model Complexity in Phylogenomics: A Case Study Focused on Early Animal Divergences. Biophysica 2021, 1, 87-105. https://0-doi-org.brum.beds.ac.uk/10.3390/biophysica1020008
Pandey A, Braun EL. The Roles of Protein Structure, Taxon Sampling, and Model Complexity in Phylogenomics: A Case Study Focused on Early Animal Divergences. Biophysica. 2021; 1(2):87-105. https://0-doi-org.brum.beds.ac.uk/10.3390/biophysica1020008
Chicago/Turabian StylePandey, Akanksha, and Edward L. Braun. 2021. "The Roles of Protein Structure, Taxon Sampling, and Model Complexity in Phylogenomics: A Case Study Focused on Early Animal Divergences" Biophysica 1, no. 2: 87-105. https://0-doi-org.brum.beds.ac.uk/10.3390/biophysica1020008