
Exploring the Mechanism of Shikimate Kinase through Quantum Mechanical and Molecular Mechanical (QM/MM) Methods


High Performance Computing Center North (HPC2N), Umeå University, S-90187 Umeå, Sweden
Biophysica 2021, 1(3), 334-343; https://0-doi-org.brum.beds.ac.uk/10.3390/biophysica1030025
Submission received: 15 August 2021
/
Revised: 1 September 2021
/
Accepted: 2 September 2021
/
Published: 5 September 2021
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The chemical step of Shikimate Kinase Helicobacter pylori, involving the transfer of a phosphoryl group, has been studied by using quantum mechanical and molecular mechanical (QM/MM) methods. Understanding the mechanism of this chemical step, present in bacteria and other microorganisms but absent in humans, can lead to the development of novel drugs for the treatment of common diseases caused by those pathogenic organisms. Different mechanisms including associative, dissociative, and concerted have been proposed up to now but there is not a consensus on the type of pathway that the reaction follows. Herein, we found that the mechanism has features from the associative and concerted types. An analysis of the free energy landscape of the chemical step reveals that the reaction is a two-step process without a well-defined intermediate state.
1. Introduction
Shikimate kinase (SK) catalyses the conversion of ATP and shikimic acid (SKM) to ADP and shikimate-3-phosphate (S3P) by transferring the terminal phosphoryl group of ATP to SKM. This phosphorylation reaction is an essential step in the overall shikimate pathway whose purpose is to produce the chorismate intermediate, a compound that is further processed in a chain of enzymatic reactions, that results in the synthesis of aromatic residues. Because this catalytic reaction is only found in microorganisms such as bacteria [1], for instance, Helicobacter pylori, which is responsible for gastrointestinal ulcers [2], it makes this reaction pathway a good target for novel drugs to inactivate such organisms. Several crystal structures of SK have been published up to now in the reactant state (RS) or product state (PS) [3,4] but a structure containing a transition state (TS) or TS mimic, which is crucial for drug development, has not been reported yet.
Depending on the surroundings, the phosphorylation reaction can follow any continuous pathway which is bounded by two extreme cases [5,6], also known as associative and dissociative mechanisms. In the associative mechanism, the bond between the leaving oxygen atom and the leaving phosphorus atom (P-Olg) cleaves after the nucleophilic attack has started and the new bond between P and the nucleophilic group is formed (P-Onuc). In the dissociative mechanism, the P-Olg bond of the leaving group cleaves before the new bond P-Onuc has been formed, see Figure 1 for an explanation of these distances. The associative mechanism is then characterised by a pentavalent phosphorene intermediate while the dissociative mechanism is by a planar metaphosphate intermediate. In between these two mechanisms, one can find a special case called the concerted mechanism where bond cleaving and forming occur simultaneously and the associative/dissociative character depends on the bond orders in the transition state. The reaction mechanism of SK has been studied previously both experimentally [2] and theoretically [7], however, there is not a consensus about the actual pathway for this reaction. For instance, based on the long distances between reactants observed in molecular dynamics simulations, Prado et al. [2] suggested that the reaction involves a dissociative mechanism. This finding was also supported by the fact that the catalytic turnover was achieved in experiments with ADP/metaphosphoric acid (an ATP-mimetic) in the absence of the native reactant ATP. This showed that a tight bond between the leaving group and ADP was not necessary. Contrary to the previous result, Hartmann et al. [3] proposed previously an associative reaction mechanism based on a geometric distance analysis from a model structure obtained from a series of crystal structures along the catalytic step. Recently, Yao et al. [7] proposed a concerted one-step reaction with a loose TS, by using QM/MM simulations. This was the first time that a model for the TS was proposed.
The difference in the previous proposed results regarding the mechanism of the SK’s chemical step motivated us to further investigate it by using different levels of theory within the QM/MM [8,9,10] framework. They allowed us to compute free energies along a predefined reaction coordinate which in turn helped us to assess the type of mechanism involved with particular emphasis on the TS.
2. Materials and Methods
Helicobacter pylori SK in complex with shikimate-3-phosphate (S3P) and adenosine diphosphate (ADP), PDB ID 3MUF [4], was used as the initial structure of SK, S3P and ADP. One Mg2+ atom, present in other protein kinases [11], was manually added between ADP and S3P. In addition to this, four coordinating water molecules were added around the Mg2+ atom to achieve its six-fold coordination. CHARMM package (versions 40a2 and 43b1) [12] was used to build the initial system which contained the protein, ADP, S3P, Mg2+ ion, 59 crystal waters, 16 Na+ ions, 13 Cl− ions, and 5184 bulk water molecules resulting in a system of 18,447 total number of atoms. The system was simulated as a rhombic dodecahedron (RHDO) box with a length of 63.6 Å. The CHARMM27 [13,14,15] force field was employed to simulate the protein chain, waters and ions. Water molecules were simulated with the TIP3P [16] model. SHAKE [17] constraints were applied to all bonds that included hydrogens. Electrostatic interactions were solved with the 6th-order particle mesh Ewald (PME) summation technique [18] with a 10 Å real space cut-off. The van der Waals interactions were truncated with a switching function between 9 Å and 10 Å.
The MM part in our QM/MM simulations was performed with the CHARMM engine while the QM part was solved with either the MNDO97 [19,20] CHARMM module or the Gaussian 09 [21] package through the CHARMM interface [10]. The QM region included ADP, S3P, four coordinating waters, Mg2+, and the terminal atoms of the following residues: K14 (cut at Cδ atom), protonated D33 (cut at Cα), R57 (cut at Cγ atom), R107 (cut at Cγ atom), and R116 (cut at Cγ atom). The cutoff scheme for treating QM boundary atoms was the generalized hybrid orbital (GHO) method [22]. Although residues R57 and R107 are not close to the chemically important region, we included them in the QM region to zero out the total charge of it. The total number of QM atoms was 131. In the MNDO simulations, the Mg2+, S3P and ADP molecules were modelled with the AM1/d-PhoT [23] set of parameters. As for the Gaussian 09 simulations, the level of theory was B3LYP [24,25] with the 6-31+g(d) basis set. The extended Lagrangian formulation with dissipation in the Born-Oppenheimer approximation [26,27] was used in the MNDO simulations to speed up the convergence of the self-consistent procedure.
The initial structure (in the PS) was first minimised using the ABNR method [28] during 1000 steps while keeping the protein chain, ADP, S3P, and Mg2+ ion fixed (excluding all hydrogens). A subsequent minimisation of 500 steps was done where the constraints on the protein chain were relaxed. The generation of the initial pathway started with the latter structure and subsequent structures were obtained from the previous one by gradually reducing the value of the reaction coordinate (RC) which was computed as:
And whose initial value was µ = 2.5 Å. Each reduction of the RC was followed by a minimisation procedure of 5 and 500 steps for B3LYP and MNDO methods, respectively. Two pathways were generated by using B3LYP (500 windows) and the computationally cheaper semiempirical MNDO methods (50 windows). Free energy calculations were performed on the pathway obtained with the MNDO method by using the umbrella sampling [29] (US) technique. These finite temperature simulations were performed at 298.15 K in the constant pressure (CPT) ensemble with the Nosé–Hoover Langevin piston method [30,31].
3. Results
3.1. Details of the Initial Structure in the PS
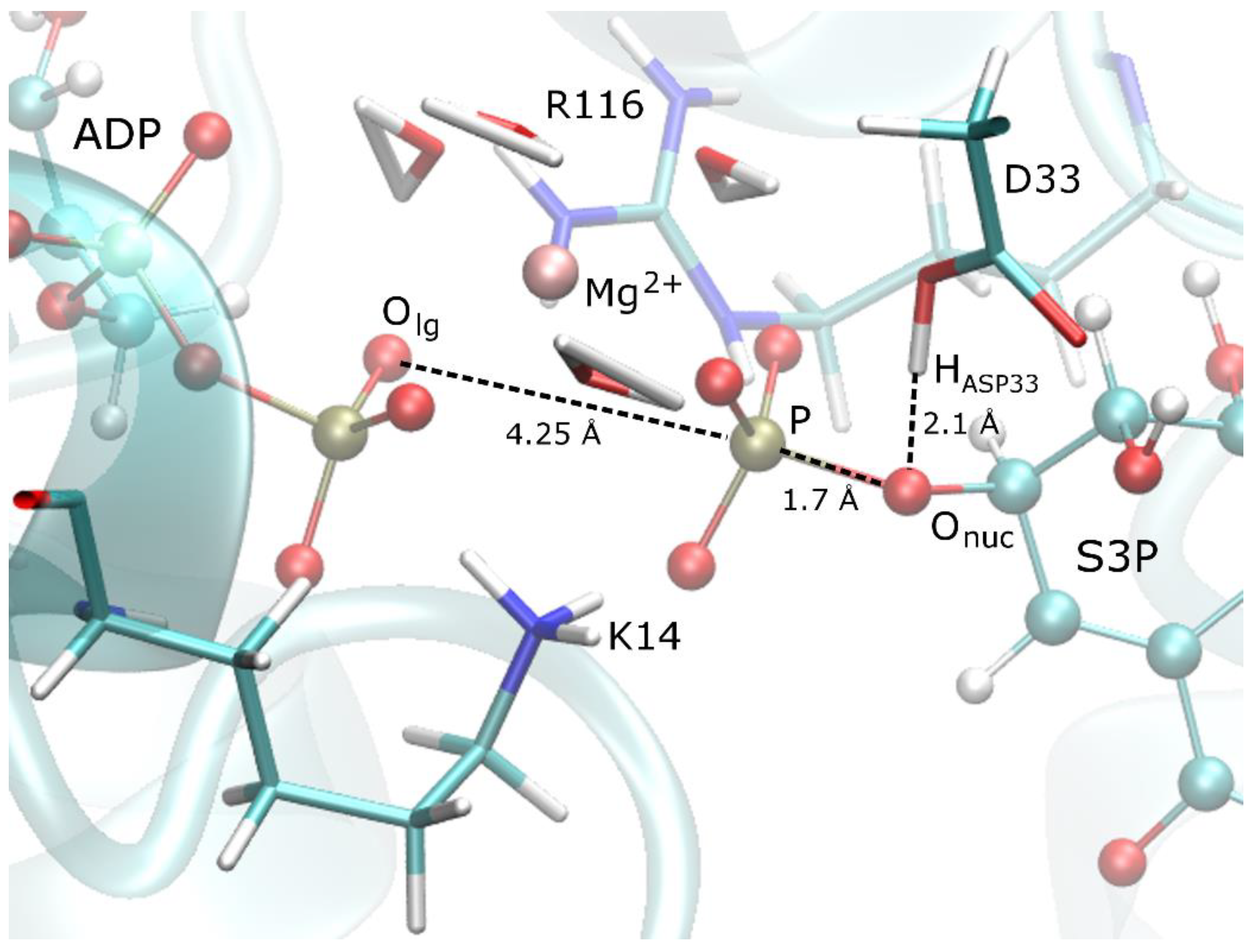
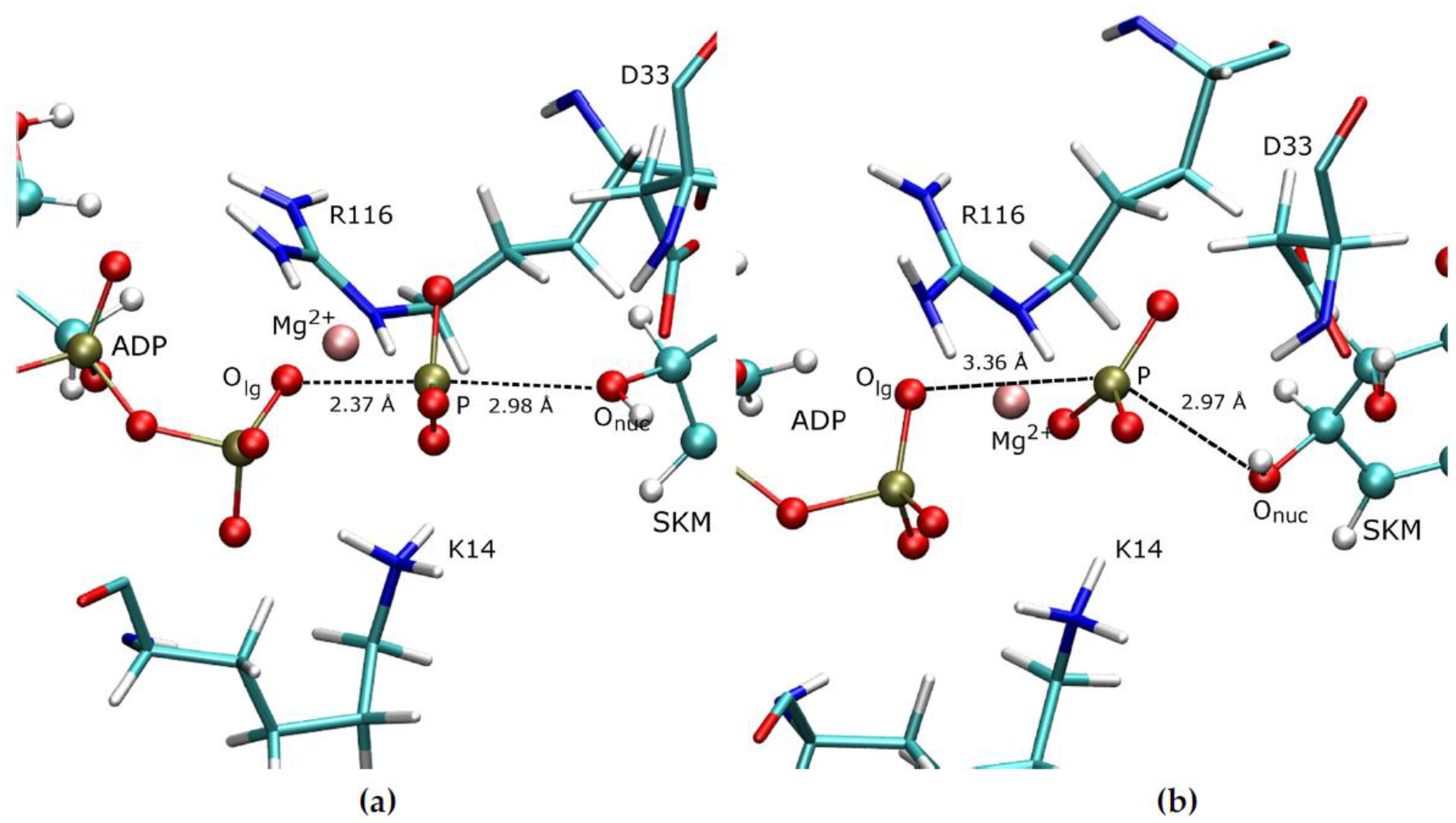
The binding site of the initially minimised structure with the MNDO method, see Figure 1, shows that the products ADP and S3P have a network of hydrogen bonds with the nearby residues K14 and R116 and therefore they function as a bridge between the products. This network seems to be essential for the catalytic efficiency of the enzyme as demonstrated by the dramatic reduction of activity upon mutations of these residues [4,35]. In this structure, D33 is protonated at the PS and forms a hydrogen bond with the nucleophilic oxygen (Onuc). The Mg2+ ion helps to stabilise the negatively charged leaving group and to keep the leaving group in place.
3.2. Reaction Mechanism of the Chemical Step
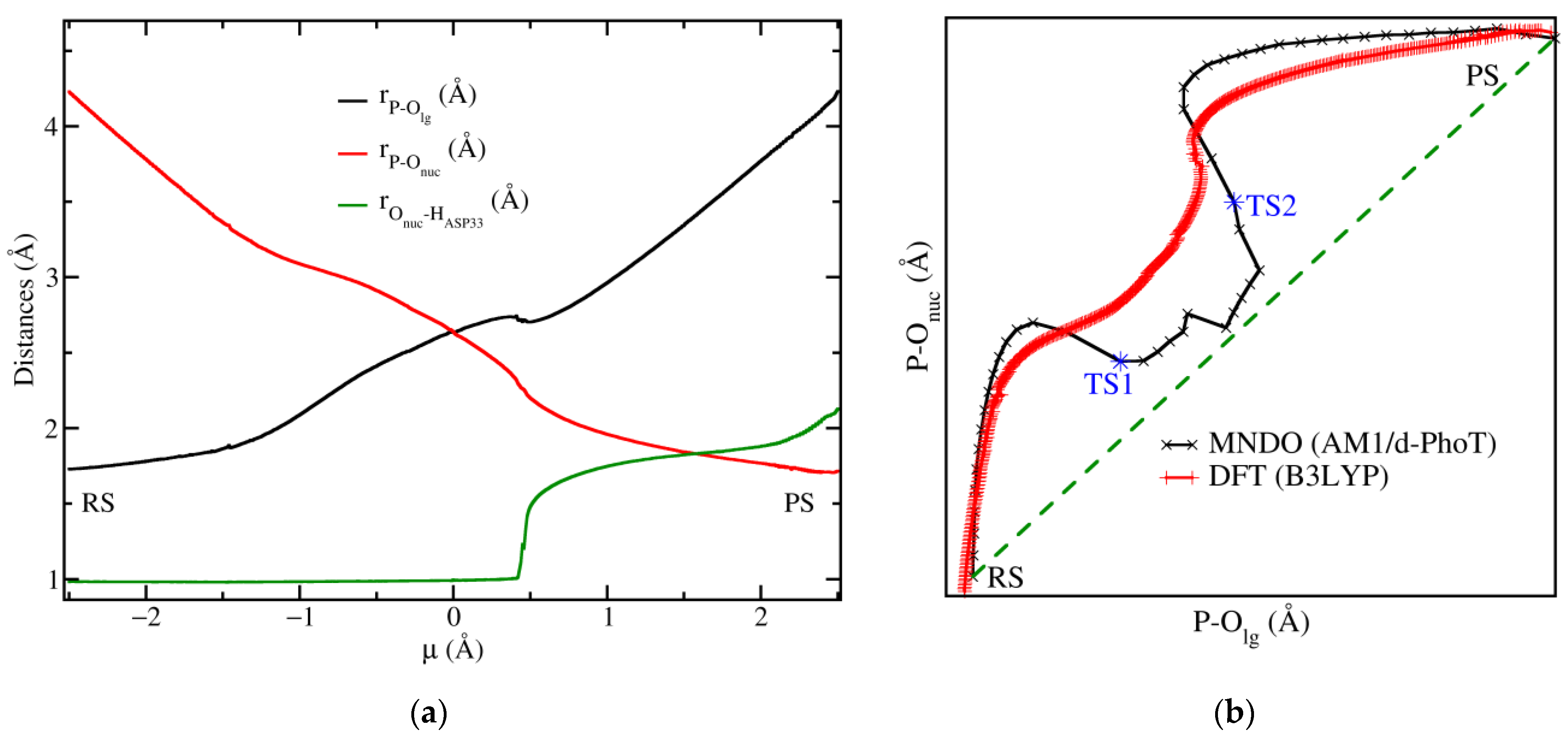
The B3LYP-QM/MM minimised pathway shows that the distances between P-Olg and P-Onuc gradually change as was found by Yao et al. [7] with the crossing point of the curves occurring at µ = 0 Å. The distances for P-Olg and P-Onuc at this point were both equal to 2.64 Å, while the ones reported in this reference had values of 2.37 Å and 2.57 Å, respectively, see Figure 2a. The application of Pauling’s formula to these sets of distances results in a dissociative character DC obtained from Equation (2) of 96.9%, in our case and a close result of 94.1% in Yao’s DFT calculations. We noticed that the dissociative character is in fact closer to our value in the case of Yao’s MP2 calculation because the bonds at the TS are slightly looser than in the DFT results cited above. We also noticed that although P-Olg and P-Onuc gradually change, the slope of the curves changes producing a bending feature near the crossing point.
This bending feature is better captured in a More O’Ferrall Jencks plot [36,37] as it can be observed in Figure 2b. A plot for the B3LYP (red line) result shows that starting from the RS the reaction follows an associative mechanism, but close to the crossing point µ = 0 Å the curvature of the P-Olg vs. P-Onuc curves changes. A similar pattern was observed with MNDO (black line) level of theory. This bending suggests that the mechanism is shifted towards a more concerted one depicted by the green dashed line in the same figure. It is interesting to notice that in the case of the MNDO result, the reaction pathway is even closer to the concerted mechanism than in the B3LYP case which is a consequence of the looser bonds obtained with the former method, for instance at the crossing point the P-Olg and P-Onuc distances were both equal to 2.64 Å and 2.9 Å for B3LYP and MNDO methods, respectively.
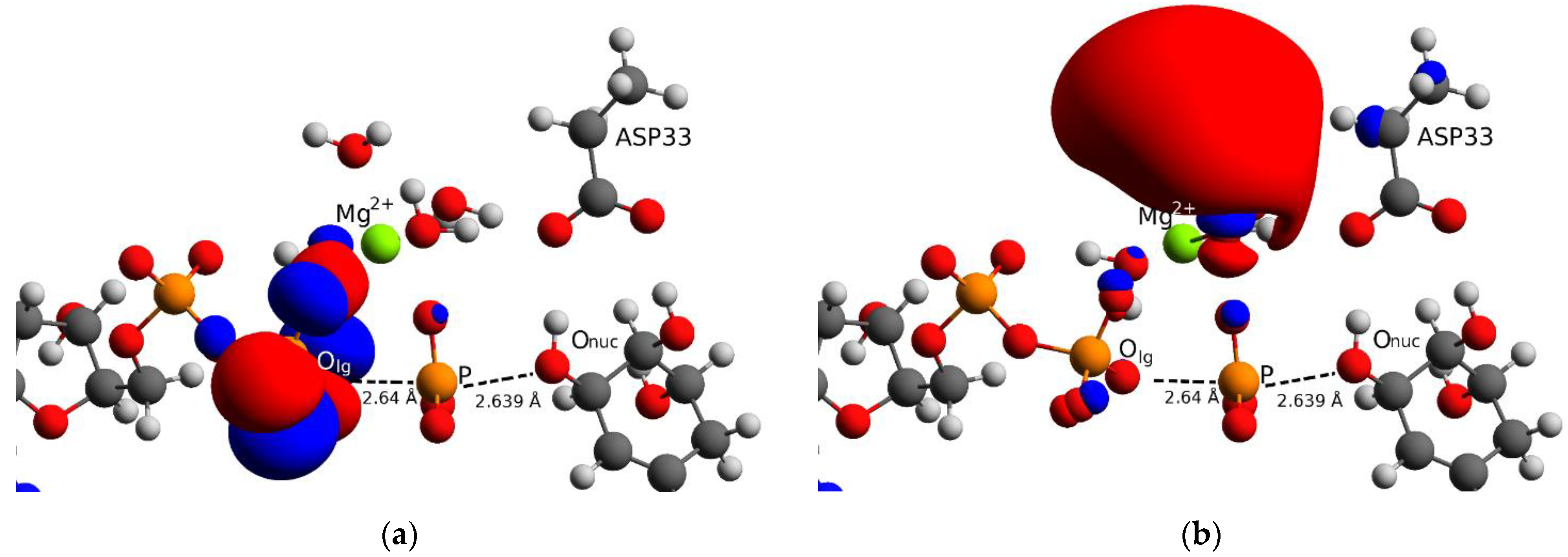
Regarding the crossing point, we performed a frontier molecular orbital analysis on the structure obtained with B3LYP method at µ = 0 Å, this can be seen in Figure 3. We found that the highest occupied molecular orbital (HOMO), Figure 3a, is localised in the β phosphate group of ADP but the expected location was at the level of Onuc as this region is supposed to donate the highest-energy electrons for attacking the leaving group. This is not the case in the present reaction mechanism because the proton of SKM is transferred to D33 beyond the crossing point (µ > 0.42 Å) and the attacking oxygen Onuc is not yet enabled, this is shown in Figure 2a (green line). The lowest unoccupied molecular orbital (LUMO), Figure 3b, is localised around the Mg2+ ion showing that this ion assists in the stabilisation of the negative charges (red blob) formed when reactants approach each other. All these findings suggest that the crossing point is not a real transition state in our case but only one intermediate in an ensemble of states which can be accessed due to the loose nature of the bonds. The latter can be assessed by turning on a thermostat (finite temperature) which will make the relevant bonds fluctuate and therefore prove the stability of the state at the crossing point.
3.3. Free Energy Landscape of the Chemical Step
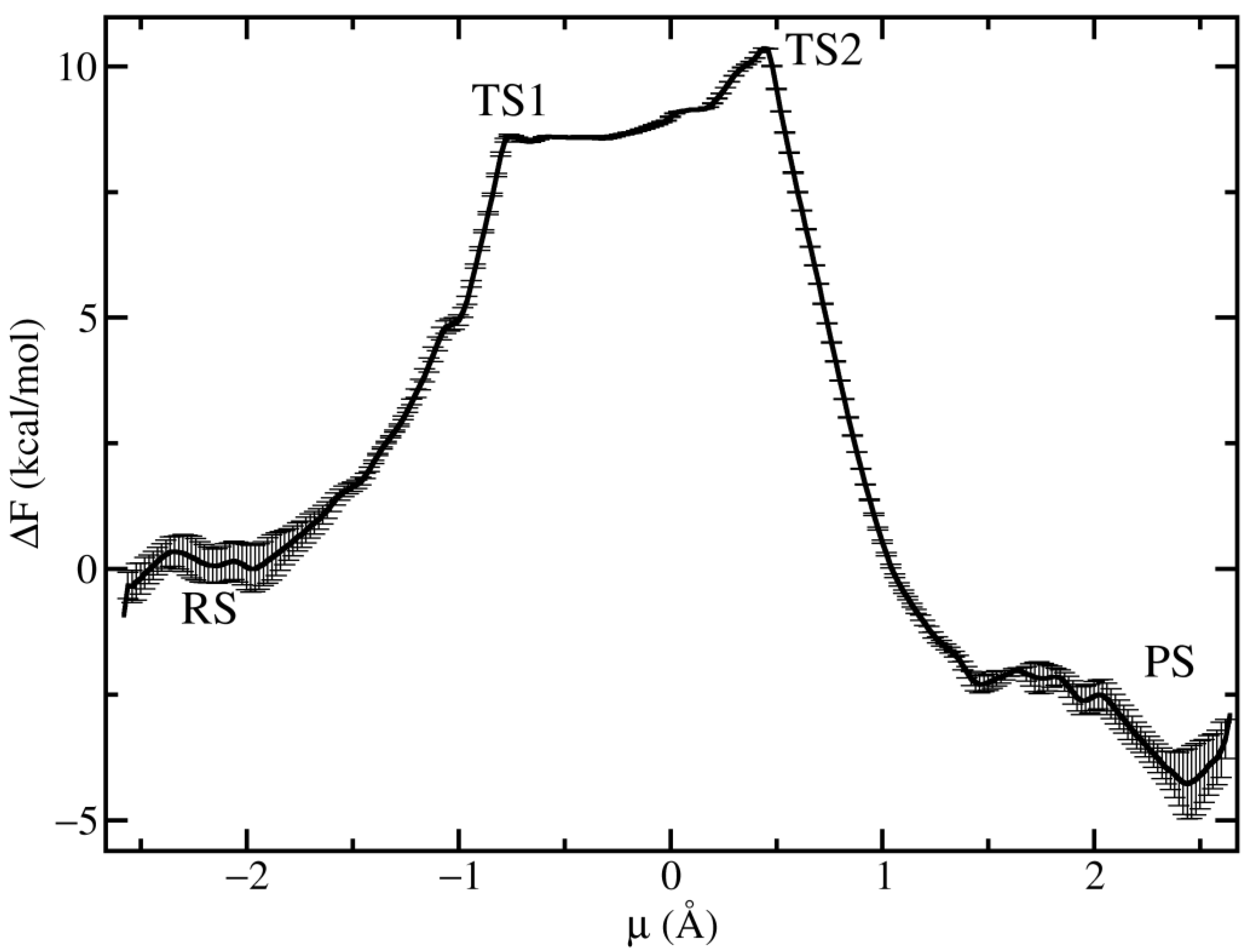
In order to explore the behaviour of the proposed reaction mechanism, with particular emphasis on the crossing point, we performed finite temperature simulations and computed the potential of mean force (PMF) along the reaction coordinate defined in Equation (1), the results are displayed in Figure 4. The PMF shows that the reaction proceeds through two transition states TS1 (µ = −0.75 Å) and TS2 (µ = 0.44 Å) without a clear stable intermediate state between them. This finding discards the fully associative/dissociative mechanisms where a stable phosphorene/methaphosphate intermediate is required. The role of TS1 is to decrease the overall barrier height by functioning as an intermediate state before the reactants climb to TS2, which is the rate-limiting step with a barrier height of 10.3 kcal/mol. This overall barrier height is lower than the experimental value of 15.3 kcal/mol derived from the kcat value reported previously [35] by using Eyring’s formula of transition state theory [38]. It is known that semiempirical methods tend to underestimate the energy barriers [7] which are usually corrected by applying a more accurate but also more computationally expensive level of theory. However, we have not attempted to correct energies in this work because our main interest is on the pathway rather than on the exact energies. The PMF together with the More O’Ferrall plot confirms the idea that the reaction mechanism shares some features of the associative and concerted mechanisms.
Representative structures of the transition state ensemble TS1 and TS2 are displayed in Figure 5a,b, respectively. This figure shows that in the TS1, the leaving group is closer to ADP than to SKM which is the opposite in TS2. The Mg2+ ion helps to keep in place the leaving group. In both TS1 and TS2, the proton at the nucleophilic oxygen has not been transferred yet which contributes to the bond elongations.
4. Discussion
The analysis of the P-Olg and P-Onuc distances along the reaction coordinates reveals that the reaction starts by following an associative mechanism. However, as it approaches the crossing point µ = 0 Å, the bond lengths become looser and the reaction tends to be shifted towards a more concerted mechanism, see Figure 2b. The crossing point showed a 96.9% dissociative character suggesting that the bonds at this point are loose. Thus, we claim that the reaction is neither fully associative nor concerted but that it has features from both types of mechanisms. These results can explain the findings from Prado et al. [2], that the catalytic turnover can proceed with the mimetic metaphosphoric acid, where a tight bond between the leaving phosphoryl group and ADP is not necessary. Our model can explain this finding because near the crossing point the bonds are loose and dissociative in nature. The previous finding of Hartmann [3], regarding the associative character of the reaction can be explained by our model because right at the beginning of the chemical step, the reaction follows an associative mechanism. Finally, the results of Yao [7], are also explained by our results as the reaction becomes concerted near the crossing point. Finite temperature simulations showed that the crossing point is not the TS of the reaction, but that it is separated into two TSs instead of the well-defined TS found in the previous reference.
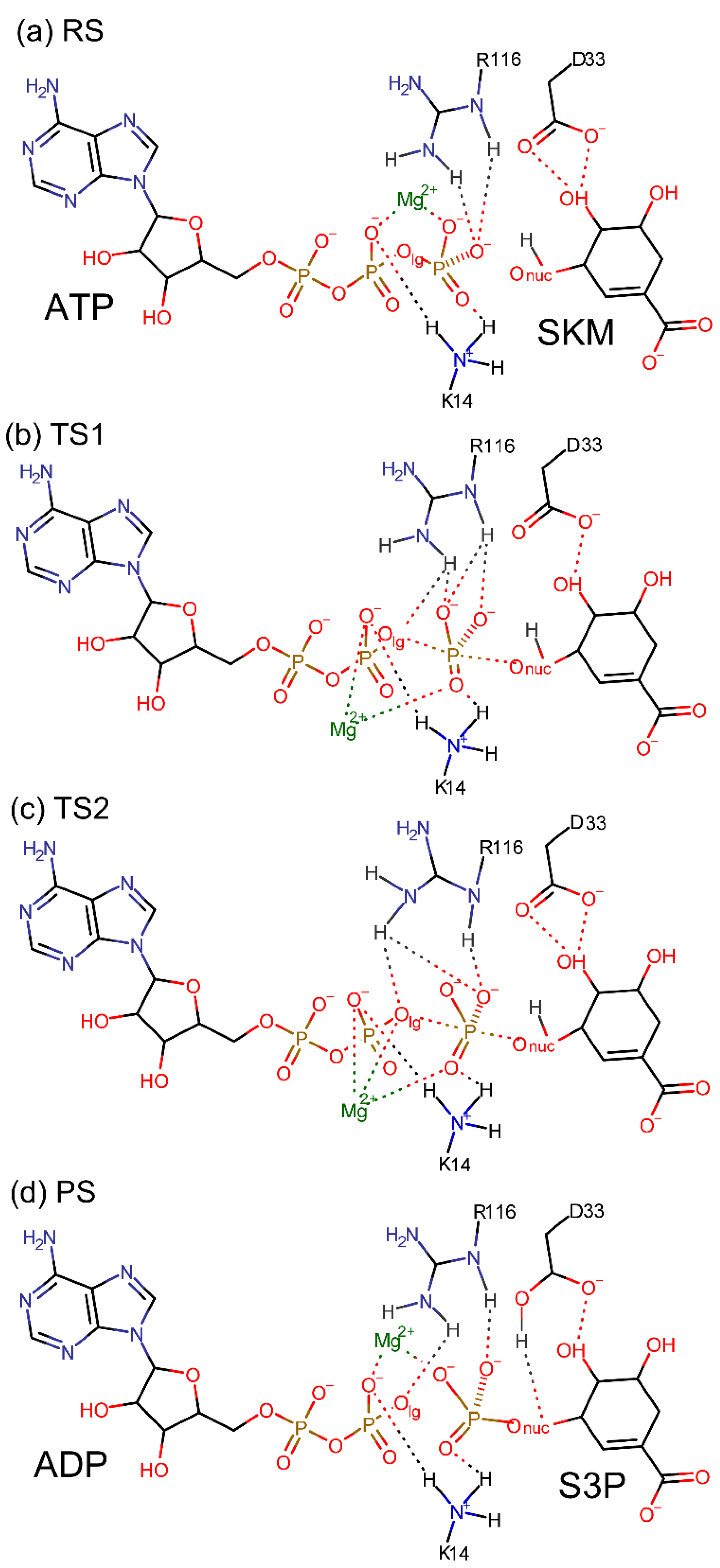
The proposed two-step mechanism for the SK-catalysed phosphoryl transfer reaction is presented in Figure 6a–d. Reactants are kept in place by the two long arm residues K14 and R116 and the Mg2+ ion. Upon reaching the two TSs, K14 and R116 show high flexibility which is reflected in the displacement of their terminal groups. The flexibility allows these residues to form and break hydrogen bonds with the reactants with a low energy cost. The Mg2+ ion also assists in the rearrangement of the hydrogen bond pattern by changing its coordination, with the leaving and nucleophilic groups’ oxygen atoms, depending upon the reaction state. In Figure 6d we also see that D33 is the acceptor of the proton released by SKM.
5. Conclusions
In the present work, we analysed the SK-catalysed phosphoryl transfer reaction through QM/MM simulations at different levels of QM theory. We found that the reaction mechanism lies between the fully associative and dissociative mechanisms. Analysis of distances through the More O’Ferrall Jencks plot and the dissociative character showed that the reaction turns into a concerted mechanism near the crossing point of the P-Olg and P-Onuc distance curves. We also found that the reaction proceeds through two TSs. Our work reconciles in a single model previous results that suggested three different possible mechanisms.
Funding
This research received no external funding.
Data Availability Statement
Not applicable.
Acknowledgments
This research was conducted using the resources of High-Performance Computing Center North (HPC2N) at the Swedish National Infrastructure for Computing (SNIC).
Conflicts of Interest
The author declares no conflict of interest.
References
- Coracini, J.D.; de Azevedo, W.F., Jr. Shikimate kinase, a protein target for drug design. Curr. Med. Chem. 2014, 21, 592–604. [Google Scholar] [CrossRef]
- Prado, V.; Lence, E.; Vallejo, J.A.; Beceiro, A.; Thompson, P.; Hawkins, A.R.; González-Bello, C. Study of the Phosphoryl-Transfer Mechanism of Shikimate Kinase by NMR Spectroscopy. Chem. Eur. J. 2016, 22, 2758–2768. [Google Scholar] [CrossRef]
- Hartmann, M.D.; Bourenkov, G.P.; Oberschall, A.; Strizhov, N.; Bartunik, H.D. Mechanism of Phosphoryl Transfer Catalyzed by Shikimate Kinase from Mycobacterium tuberculosis. J. Mol. Biol. 2006, 364, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.-C.; Chen, Y.-F.; Wang, H.-J.; Hsu, K.-C.; Lin, S.-C.; Chen, T.-J.; Yang, J.-M.; Wang, W.-C. Structures of Helicobacter pylori Shikimate Kinase Reveal a Selective Inhibitor-Induced-Fit Mechanism. PLoS ONE 2012, 7, e33481. [Google Scholar] [CrossRef] [Green Version]
- Kamerlin, S.C.L.; Florián, J.; Warshel, A. Associative Versus Dissociative Mechanisms of Phosphate Monoester Hydrolysis: On the Interpretation of Activation Entropies. ChemPhysChem 2008, 9, 1767–1773. [Google Scholar] [CrossRef] [PubMed]
- Barnes, J.A.; Williams, I.H.; Wilkie, J. Transition-state structural variation and mechanistic change. J. Chem. Soc. Faraday Trans. 1994, 90, 1709–1714. [Google Scholar] [CrossRef]
- Yao, J.; Wang, X.; Luo, H.; Gu, P. Understanding the Catalytic Mechanism and the Nature of the Transition State of an Attractive Drug-Target Enzyme (Shikimate Kinase) by Quantum Mechanical/Molecular Mechanical (QM/MM) Studies. Chem. Eur. J. 2018, 23, 16380–16387. [Google Scholar] [CrossRef]
- Warshel, A.; Levitt, M. Theoretical studies of enzymic reactions: Dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J. Mol. Biol. 1976, 103, 227–249. [Google Scholar] [CrossRef]
- Gao, J. Methods and Applications of Combined Quantum Mechanical and Molecular Mechanical Potentials. In Reviews in Computational Chemistry; Lipkowitz, K.B., Boyd, D.B., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1996; pp. 119–185. [Google Scholar]
- Field, M.J.; Bash, P.A.; Karplus, M. A combined quantum mechanical and molecular mechanical potential for molecular dynamics simulations. J. Comput. Chem. 1990, 11, 700–733. [Google Scholar] [CrossRef]
- Bellinzoni, M.; Haouz, A.; Graña, M.; Munier-Lehmann, H.; Shepard, W.; Alzari, P.M. The crystal structure of Mycobacterium tuberculosis adenylate kinase in complex with two molecules of ADP and Mg2+ supports an associative mechanism for phosphoryl transfer. Protein Sci. 2006, 15, 1489–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- MacKerell, A.D., Jr.; Feig, M.; Brooks, C.L. Improved Treatment of the Protein Backbone in Empirical Force Fields. J. Am. Chem. Soc. 2004, 126, 698–699. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Banavali, N.; Foloppe, N. Development and current status of the CHARMM force field for nucleic acids. Biopolymers 2000, 56, 257–265. [Google Scholar] [CrossRef]
- Foloppe, N.; MacKerell, J.A.D. All-atom empirical force field for nucleic acids: I. Parameter optimization based on small molecule and condensed phase macromolecular target data. J. Comput. Chem. 2000, 21, 86–104. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Nam, K.; Gao, J.; York, D.M. An Efficient Linear-Scaling Ewald Method for Long-Range Electrostatic Interactions in Combined QM/MM Calculations. J. Chem. Theory Comput. 2005, 1, 2–13. [Google Scholar] [CrossRef]
- Nam, K. Acceleration of Ab Initio QM/MM Calculations under Periodic Boundary Conditions by Multiscale and Multiple Time Step Approaches. J. Chem. Theory Comput. 2014, 10, 4175–4183. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Rev. E.01; Gaussian Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Gao, J.; Amara, P.; Alhambra, C.; Field, M.J. A Generalized Hybrid Orbital (GHO) Method for the Treatment of Boundary Atoms in Combined QM/MM Calculations. J. Phys. Chem. A 1998, 102, 4714–4721. [Google Scholar] [CrossRef]
- Nam, K.; Cui, Q.; Gao, J.; York, D.M. Specific Reaction Parametrization of the AM1/d Hamiltonian for Phosphoryl Transfer Reactions: H, O, and P Atoms. J. Chem. Theory Comput. 2007, 3, 486–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becke, A.D. Density-Functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niklasson, A.M.N.; Steneteg, P.; Odell, A.; Bock, N.; Challacombe, M.; Tymczak, C.J.; Holmström, E.; Zheng, G.; Weber, V. Extended Lagrangian Born–Oppenheimer molecular dynamics with dissipation. J. Chem. Phys. 2009, 130, 214109. [Google Scholar] [CrossRef] [Green Version]
- Zheng, G.; Niklasson, A.M.N.; Karplus, M. Lagrangian formulation with dissipation of Born-Oppenheimer molecular dynamics using the density-functional tight-binding method. J. Chem. Phys. 2011, 135, 044122. [Google Scholar] [CrossRef] [Green Version]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.; Karplus, M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Torrie, G.M.; Valleau, J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Pauling, L. Atomic Radii and Interatomic Distances in Metals. J. Am. Chem. Soc. 1947, 69, 542–553. [Google Scholar] [CrossRef]
- Li, Q.; Fan, S.; Li, X.; Jin, Y.; He, W.; Zhou, J.; Cen, S.; Yang, Z. Insights into the Phosphoryl Transfer Mechanism of Human Ubiquitous Mitochondrial Creatine Kinase. Sci. Rep. 2016, 6, 38088. [Google Scholar] [CrossRef]
- Mildvan, A.S. Mechanisms of signaling and related enzymes. Proteins Struct. Funct. Bioinform. 1997, 29, 401–416. [Google Scholar] [CrossRef]
- Krell, T.; Maclean, J.; Boam, D.J.; Cooper, A.; Resmini, M.; Brocklehurst, K.; Kelly, S.M.; Price, N.C.; Lapthorn, A.J.; Coggins, J.R. Biochemical and X-ray crystallographic studies on shikimate kinase: The important structural role of the P-loop lysine. Protein Sci. 2001, 10, 1137–1149. [Google Scholar] [CrossRef] [Green Version]
- Jencks, W.P. A primer for the Bema Hapothle. An empirical approach to the characterization of changing transition-state structures. Chem. Rev. 1985, 85, 511–527. [Google Scholar] [CrossRef]
- More, O.; Ferrall, R.A. Relationships between E2 and E1cB mechanisms of β-elimination. J. Chem. Soc. B 1970, 274, 77. [Google Scholar] [CrossRef]
- Rosenstock, H.M.; Wallenstein, M.B.; Wahrhaftig, A.L.; Eyring, H. Absolute Rate Theory for Isolated Systems and the Mass Spectra of Polyatomic Molecules. Proc. Natl. Acad. Sci. USA 1952, 38, 667–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
The binding site of SK showing the products ADP/S3P, the nearby residues K14, R116, and D33 and the Mg2+ ion with its coordinating waters.
Figure 1.
The binding site of SK showing the products ADP/S3P, the nearby residues K14, R116, and D33 and the Mg2+ ion with its coordinating waters.

Figure 2.
Plot of the P-Olg and P-Onuc along the reaction: (a) distances for the minimised pathway obtained with the B3LYP method. The proton distance is also displayed (green line); (b) More O’Ferrall plot for the MNDO (black line) and B3LYP (red line) results showing the curve bending feature and the tendency towards a concerted mechanism (green dashed line). The location of the transition states TS1 and TS2 is also shown (see main text).
Figure 2.
Plot of the P-Olg and P-Onuc along the reaction: (a) distances for the minimised pathway obtained with the B3LYP method. The proton distance is also displayed (green line); (b) More O’Ferrall plot for the MNDO (black line) and B3LYP (red line) results showing the curve bending feature and the tendency towards a concerted mechanism (green dashed line). The location of the transition states TS1 and TS2 is also shown (see main text).

Figure 3.
Frontier molecular orbitals for the crossing point (µ = 0 Å) structure obtained with the B3LYP method: (a) the HOMO shows that the highest energy electrons are localised on the ADP side; (b) LUMO shows that the negative charges formed when the reactants are close to each other are stabilised by the Mg2+ ion. Onuc is not involved in either HOMO and LUMO as it is still in the protonated and therefore it is not yet chemically reactive.
Figure 3.
Frontier molecular orbitals for the crossing point (µ = 0 Å) structure obtained with the B3LYP method: (a) the HOMO shows that the highest energy electrons are localised on the ADP side; (b) LUMO shows that the negative charges formed when the reactants are close to each other are stabilised by the Mg2+ ion. Onuc is not involved in either HOMO and LUMO as it is still in the protonated and therefore it is not yet chemically reactive.

Figure 4.
PMF for the chemical step showing the RS, PS as well as the TS1 and TS2. The reaction proceeds without a well-defined intermediate state.
Figure 4.
PMF for the chemical step showing the RS, PS as well as the TS1 and TS2. The reaction proceeds without a well-defined intermediate state.

Figure 5.
Representative structures of the (a) TS1 and (b) TS2 ensembles.

Figure 6.
Proposed two-step reaction mechanism for SK-catalysed phosphoryl transfer reaction proceeding through two TSs. (a) In the RS, SKM is protonated and forms hydrogen bonds with D33. ATP terminal phosphate groups form hydrogen bonds with K14 and R116. These hydrogen bonds assist in the correct positioning of the reactants. (b) TS1. (c) TS2. (d) PS containing ADP and S3P molecules. The residues K14 and R116 are highly flexible during the reaction as reflected in the different hydrogen bond patterns found in (a–d). Only the relevant hydrogen atoms are shown.
Figure 6.
Proposed two-step reaction mechanism for SK-catalysed phosphoryl transfer reaction proceeding through two TSs. (a) In the RS, SKM is protonated and forms hydrogen bonds with D33. ATP terminal phosphate groups form hydrogen bonds with K14 and R116. These hydrogen bonds assist in the correct positioning of the reactants. (b) TS1. (c) TS2. (d) PS containing ADP and S3P molecules. The residues K14 and R116 are highly flexible during the reaction as reflected in the different hydrogen bond patterns found in (a–d). Only the relevant hydrogen atoms are shown.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ojeda-May, P. Exploring the Mechanism of Shikimate Kinase through Quantum Mechanical and Molecular Mechanical (QM/MM) Methods. Biophysica 2021, 1, 334-343. https://0-doi-org.brum.beds.ac.uk/10.3390/biophysica1030025
AMA Style
Ojeda-May P. Exploring the Mechanism of Shikimate Kinase through Quantum Mechanical and Molecular Mechanical (QM/MM) Methods. Biophysica. 2021; 1(3):334-343. https://0-doi-org.brum.beds.ac.uk/10.3390/biophysica1030025
Chicago/Turabian StyleOjeda-May, Pedro. 2021. "Exploring the Mechanism of Shikimate Kinase through Quantum Mechanical and Molecular Mechanical (QM/MM) Methods" Biophysica 1, no. 3: 334-343. https://0-doi-org.brum.beds.ac.uk/10.3390/biophysica1030025