
A Comparison of Microbial Communities of Mango and Orange Residues for Bioprospecting of Biosurfactant Producers

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. Microbial Enrichments
2.3. DNA Isolation and 16S rRNA Sequencing
2.4. Bioinformatics and Statistical Analysis
3. Results
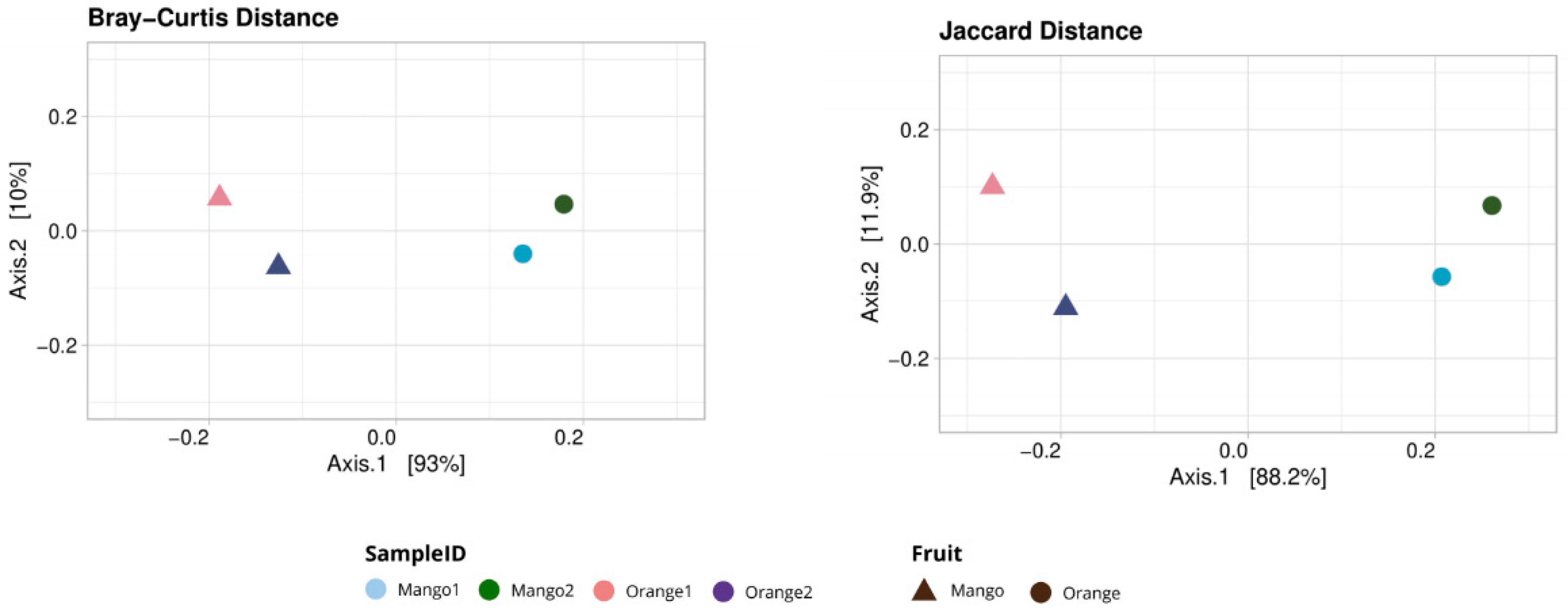
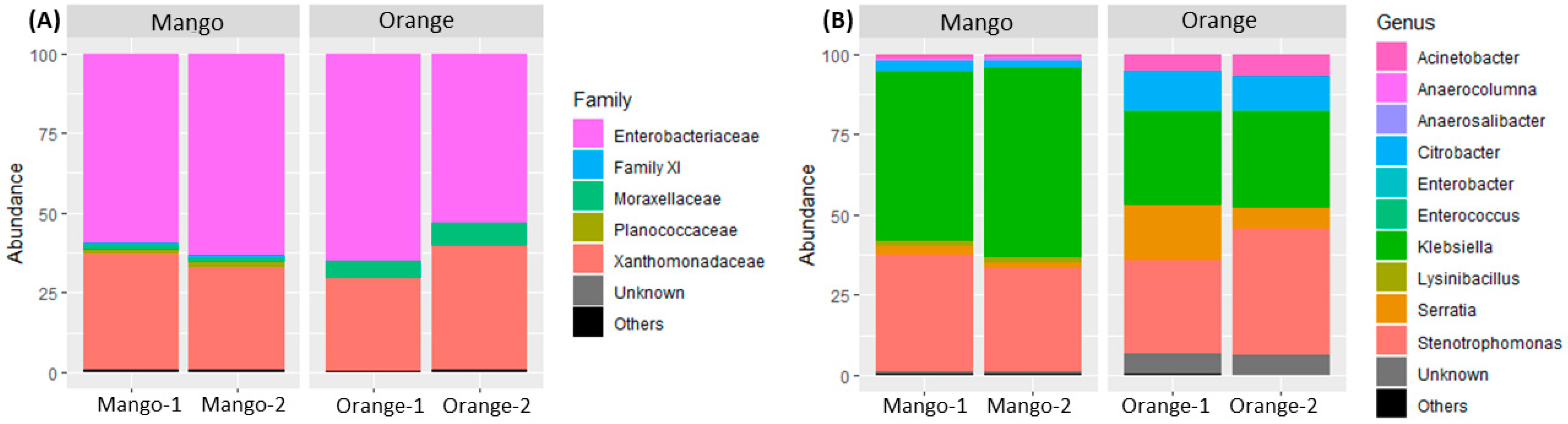
3.1. Taxonomy Analysis
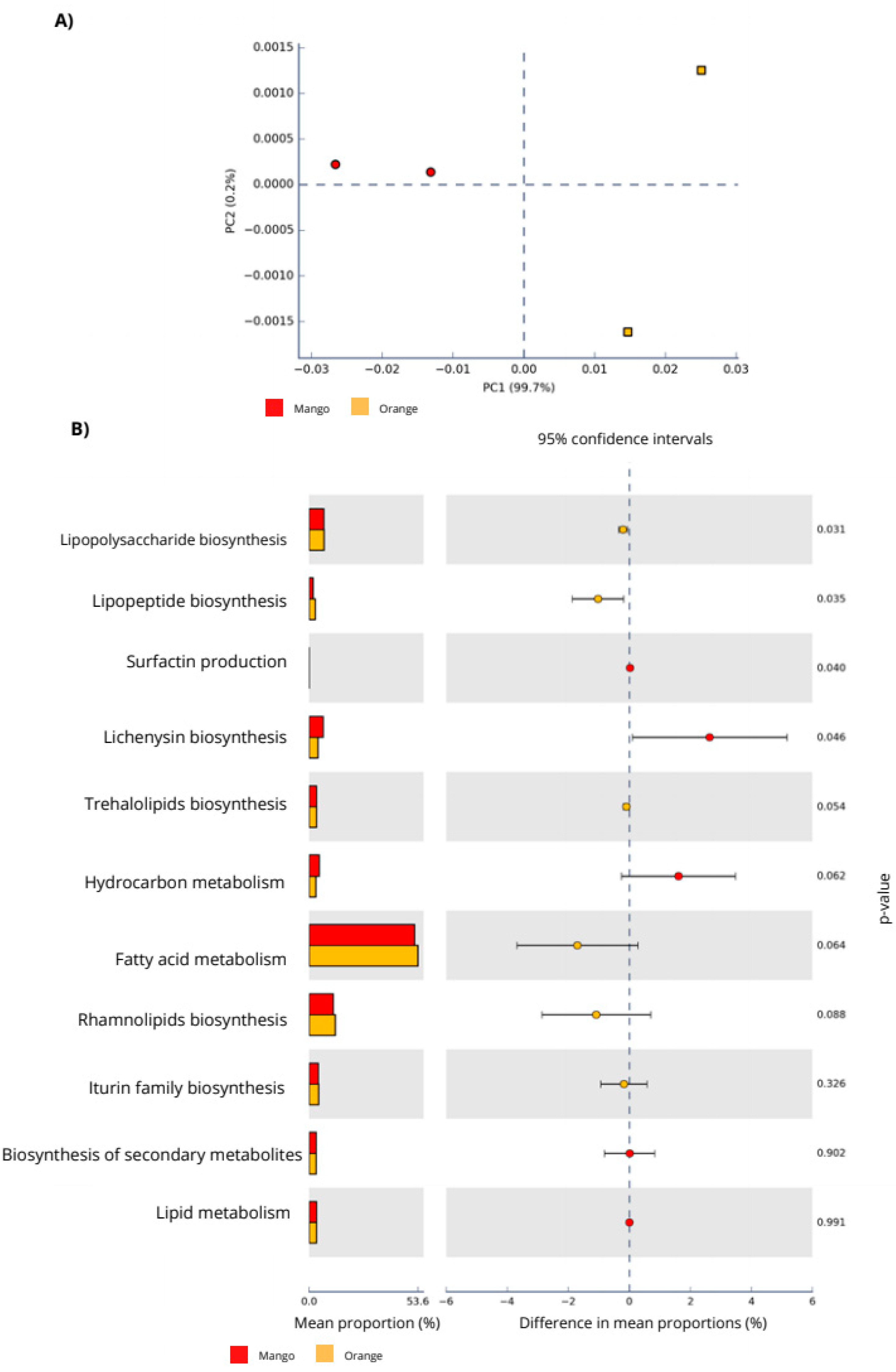
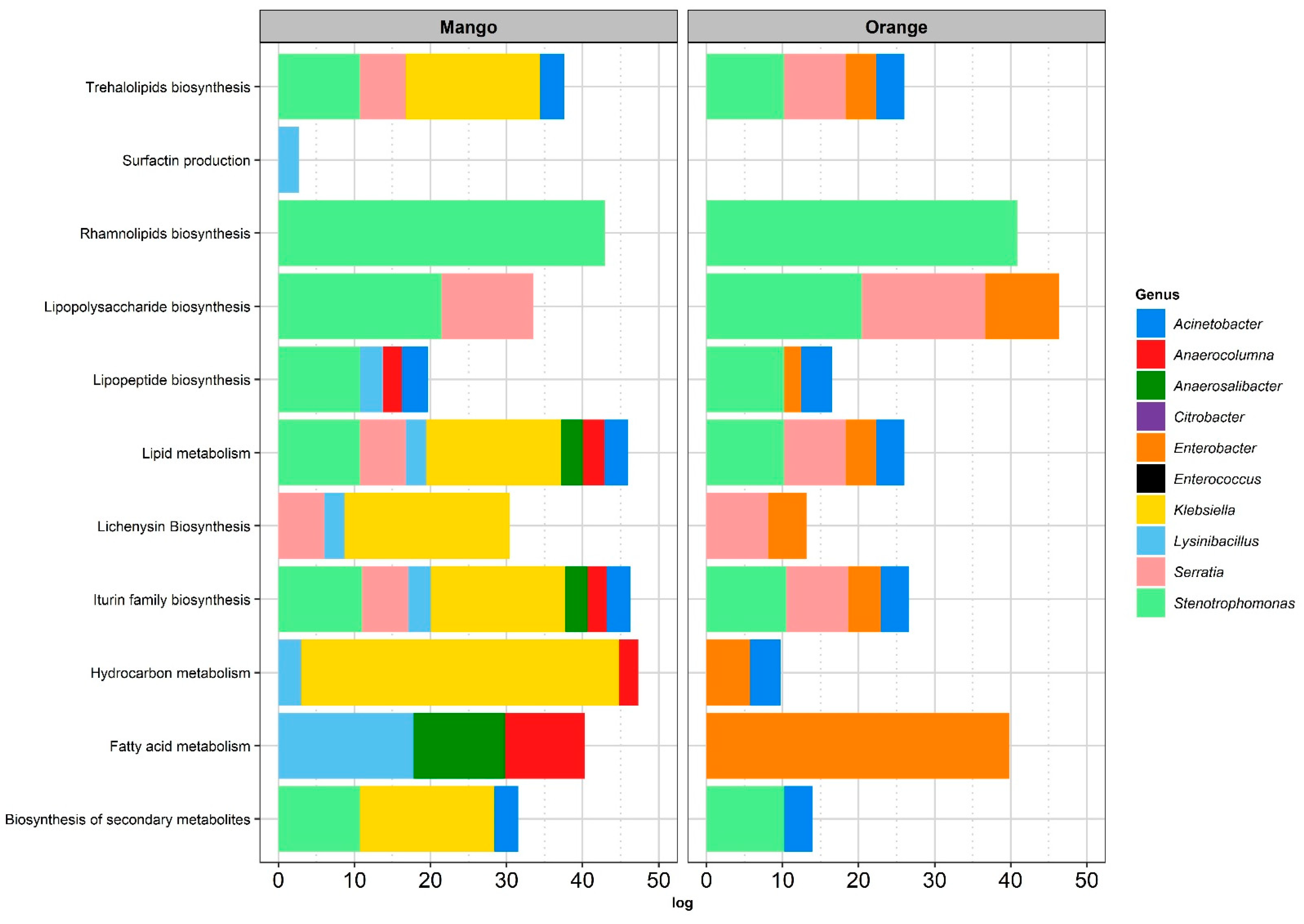
3.2. Functional Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Benincasa, M.; Abalos, A.; Oliveira, I.; Manresa, A. Chemical structure, surface properties and biological activities of the biosurfactant produced by Pseudomonas aeruginosa LBI from soapstock. Antonie Van Leeuwenhoek 2004, 85, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Domingues, R.R.; Trugilho, P.F.; Silva, C.A.; De Melo, I.C.N.A.; Melo, L.C.A.; Magriotis, Z.M.; Sánchez-Monedero, M.A. Properties of biochar derived from wood and high-nutrient biomasses with the aim of agronomic and environmental benefits. PLoS ONE 2017, 12, e0176884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, P.; Patil, Y.; Rale, V. Biosurfactant production: Emerging trends and promising strategies. J. Appl. Microbiol. 2018, 126, 2–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fariq, A.; Yasmin, A. Production, characterization and bioactivities of biosurfactants from newly isolated strictly halophilic bacteria. Process Biochem. 2020, 98, 1–10. [Google Scholar] [CrossRef]
- Ribeiro, B.G.; Guerra, J.M.C.; Sarubbo, L.A. Biosurfactants: Production and application prospects in the food industry. Biotechnol. Prog. 2020, 36, e3030. [Google Scholar] [CrossRef]
- Fracchia, L.; Ceresa, C.; Franzetti, A.; Cavallo, M.; Gandolfi, I.; Van Hamme, J.; Gkorezis, P.; Marchant, R.; Banat, I.M. Industrial applications of biosurfactants. In Biosurfactants: Production and Utilization—Processes, Technologies, and Economics; CRC Press: Boca Raton, FL, USA, 2014; pp. 245–260. [Google Scholar]
- Rizzo, C.; Caldarone, B.; De Luca, M.; De Domenico, E.; Giudice, A.L. Native bilge water bacteria as biosurfactant producers and implications in hydrocarbon-enriched wastewater treatment. J. Water Process Eng. 2021, 43, 102271. [Google Scholar] [CrossRef]
- Ben Belgacem, Z.; Bijttebier, S.; Verreth, C.; Voorspoels, S.; Van De Voorde, I.; Aerts, G.; Willems, K.A.; Jacquemyn, H.; Ruyters, S.; Lievens, B. Biosurfactant production by Pseudomonas strains isolated from floral nectar. J. Appl. Microbiol. 2015, 118, 1370–1384. [Google Scholar] [CrossRef]
- Kumar, A.P.; Janardhan, A.; Viswanath, B.; Monika, K.; Jung, J.-Y.; Narasimha, G. Evaluation of orange peel for biosurfactant production by Bacillus licheniformis and their ability to degrade naphthalene and crude oil. 3 Biotech. 2016, 6, 43. [Google Scholar] [CrossRef] [Green Version]
- FAO. FAOSTAT; Food and Agriculture Organization of the United Nations: Rome, Italy, 2018. [Google Scholar]
- Cypriano, D.Z.; Da Silva, L.L.; Bohórquez, M.A.M.; Tasic, L. Orange Biomass By-products. Rev. Virtual Quím. 2017, 9, 176–191. [Google Scholar] [CrossRef]
- Empresa Brasileiro de Pesquisa Agropecuária. Exportação de manga brasileira bate recorde em 2020, totalizando US$ 246 milhões. Available online: https://www.embrapa.br/busca-de-noticias/-/noticia/60585117/exportacao-de-manga-brasileira-bate-recorde-em-2020-totalizando-us-246-milhoes (accessed on 21 August 2021).
- Mehmood, N.; Iqbal, M.; Asgher, M. Production and Optimization of Biosurfactants from Bacillus subtilis. EC Microbiol. 2018, 5, 211–224. [Google Scholar]
- Napp, A.P.; Pereira, J.E.S.; Oliveira, J.S.; Silva-Portela, R.C.; Agnez-Lima, L.F.; Peralba, M.C.; Bento, F.M.; Passaglia, L.M.; Thompson, C.E.; Vainstein, M.H.; et al. Comparative metagenomics reveals different hydrocarbon degradative abilities from enriched oil-drilling waste. Chemosphere 2018, 209, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Lang, K. Projeto Primer para NGS curtas com amplicon. Imunol. Hum. 2013, 74, 8. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babraham Bioinformatics. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 25 March 2022).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science using QIIME. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [Green Version]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Ferraz, A.C.P.; Gadelha, B.D.Q.; Aguiar-Coelho, V.M. Análise faunística de Calliphoridae (Diptera) da Reserva Biológica do Tinguá, Nova Iguaçu, Rio de Janeiro. Rev. Bras. Èntomol. 2009, 53, 620–628. [Google Scholar] [CrossRef] [Green Version]
- Albuquerque, M.A.; Silva, E.L.; Barros, K.N.N.O.; Junior, S.F.A.X. Comparação entre coeficientes similaridade uma aplicação em ciências florestais. Matemática Estatística Foco 2016, 4, 102–114. [Google Scholar]
- Nwaguma, I.V.; Chikere, C.B.; Okpokwasili, G.C. Isolation, characterization, and application of biosurfactant by Klebsiella pneumoniae strain IVN51 isolated from hydrocarbon-polluted soil in Ogoniland, Nigeria. Bioresour. Bioprocess. 2016, 3, 40. [Google Scholar] [CrossRef] [Green Version]
- Gargouri, B.; Contreras, M.D.M.; Ammar, S.; Segura-Carretero, A.; Bouaziz, M. Biosurfactant production by the crude oil degrading Stenotrophomonas sp. B-2: Chemical characterization, biological activities and environmental applications. Environ. Sci. Pollut. Res. 2017, 24, 3769–3779. [Google Scholar] [CrossRef] [PubMed]
- Adeyemi, J.O.; Onajob, I.B.; Adebajo, L.O.; Orji, F.A.; Ilusany, O.A.F.; Samson, O.J. Biossurfactant production potentials of some bacteria recovered from agro-industrial wastes. Trends Sci. Technol. J. 2019, 4, 870–873. [Google Scholar]
- Clements, T.; Ndlovu, T.; Khan, W. Broad-spectrum antimicrobial activity of secondary metabolites produced by Serratia marcescens strains. Microbiol. Res. 2019, 229, 126329. [Google Scholar] [CrossRef]
- Araújo, H.W.C.; Andrade, R.F.S.; Montero-Rodríguez, D.; Rubio-Ribeaux, D.; Da Silva, C.A.A.; Campos-Takaki, G.M. Sustainable biosurfactant produced by Serratia marcescens UCP 1549 and its suitability for agricultural and marine bioremediation applications. Microb. Cell Fact. 2019, 18, 2. [Google Scholar] [CrossRef]
- Ibrahim, H.M. Characterization of biosurfactants produced by novel strains of Ochrobactrum anthropi HM-1 and Citrobacter freundii HM-2 from used engine oil-contaminated soil. Egypt. J. Pet. 2018, 27, 21–29. [Google Scholar] [CrossRef]
- Anaukwu, C.; Ekwealor, A.; Ezemba, C.; Anakwenze, V.; Okafor, U.; Archibong, E. Pseudomonas monteilii and Citrobacter murliniae, Biosurfactant-Producing Bacteria Isolated from Nigerian Soil. Br. Microbiol. Res. J. 2015, 10, 1–9. [Google Scholar] [CrossRef]
- Teixeira, L.M.; Merquior, V.L.C. The family moraxellaceae. In The Prokaryotes—Gammaproteobacteria, Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; Volume 22, pp. 443–476. [Google Scholar]
- Ohadi, M.; Dehghannoudeh, G.; Shakibaie, M.; Banat, I.M.; Pournamdari, M.; Forootanfar, H. Isolation, characterization, and optimization of biosurfactant production by an oil-degrading Acinetobacter junii B6 isolated from an Iranian oil excavation site. Biocatal. Agric. Biotechnol. 2017, 12, 1–9. [Google Scholar] [CrossRef]
- Fontes, G.C.; Amaral, P.F.F.; Coelho, M.A.Z. Biosurfactants production by yeasts. Química Nova 2008, 31, 2091–2099. [Google Scholar] [CrossRef]
- Jimoh, A.A.; Lin, J. Biosurfactant: A new frontier for greener technology and environmental sustainability. Ecotoxicol. Environ. Saf. 2019, 184, 109607. [Google Scholar] [CrossRef] [PubMed]
- Phetcharat, T.; Dawkrajai, P.; Chitov, T.; Mhuantong, W.; Champreda, V.; Bovonsombut, S. Biosurfactant-Producing Capability and Prediction of Functional Genes Potentially Beneficial to Microbial Enhanced Oil Recovery in Indigenous Bacterial Communities of an Onshore Oil Reservoir. Curr. Microbiol. 2019, 76, 382–391. [Google Scholar] [CrossRef]
- Ryan, R.P.; Monchy, S.; Cardinale, M.; Taghavi, S.; Crossman, L.; Avison, M.B.; Berg, G.; Van Der Lelie, D.; Dow, J.M. The versatility and adaptation of bacteria from the genus Stenotrophomonas. Nat. Rev. Microbiol. 2009, 7, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Coronel, J.R.; Marqués, A.; Manresa, Á.; Aranda, F.J.; Teruel, J.A.; Ortiz, A. Interaction of the Lipopeptide Biosurfactant Lichenysin with Phosphatidylcholine Model Membranes. Langmuir 2017, 33, 9997–10005. [Google Scholar] [CrossRef]
- Parthipan, P.; Preetham, E.; Machuca, L.L.; Rahman, P.; Murugan, K.; Rajasekar, A. Biosurfactant and Degradative Enzymes Mediated Crude Oil Degradation by Bacterium Bacillus subtilis A1. Front. Microbiol. 2017, 8, 193. [Google Scholar] [CrossRef] [Green Version]
- Pacheco, G.J.; Ciapina, E.M.P.; Gomes, E.D.B.; Junior, N.P. Biosurfactant production by Rhodococcus erythropolis and its application to oil removal. Braz. J. Microbiol. 2010, 41, 685–693. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.-W.; Liu, H.-S. Rhodococcus erythropolis strain NTU-1 efficiently degrades and traps diesel and crude oil in batch and fed-batch bioreactors. Process Biochem. 2011, 46, 202–209. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biosurfactant Relation Metabolism | Mango Abundance (%) | Orange Abundance (%) |
|---|---|---|
| Fatty acid metabolism | 51.94 | 53.64 |
| Rhamnolipids biosynthesis | 11.85 | 12.92 |
| Lipopolysaccharide biosynthesis | 7.17 | 7.37 |
| Lichenysin Biosynthesis | 6.92 | 4.29 |
| Hydrocarbon metabolism | 4.96 | 3.34 |
| Iturin family biosynthesis | 4.46 | 4.63 |
| Lipid metabolism | 3.65 | 3.65 |
| Trehalolipids biosynthesis | 3.53 | 3.63 |
| Biosynthesis of secondary metabolites | 3.50 | 3.50 |
| Lipopeptide biosynthesis | 2.00 | 3.00 |
| Surfactin production | 0.02 | 0.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Paula, F.; Vieira, N.V.; da Silva, G.F.; Delforno, T.P.; Duarte, I.C.S. A Comparison of Microbial Communities of Mango and Orange Residues for Bioprospecting of Biosurfactant Producers. Ecologies 2022, 3, 120-130. https://0-doi-org.brum.beds.ac.uk/10.3390/ecologies3020010
de Paula F, Vieira NV, da Silva GF, Delforno TP, Duarte ICS. A Comparison of Microbial Communities of Mango and Orange Residues for Bioprospecting of Biosurfactant Producers. Ecologies. 2022; 3(2):120-130. https://0-doi-org.brum.beds.ac.uk/10.3390/ecologies3020010
Chicago/Turabian Stylede Paula, Fernanda, Natália Vama Vieira, Gabriela Fiori da Silva, Tiago Palladino Delforno, and Iolanda Cristina Silveira Duarte. 2022. "A Comparison of Microbial Communities of Mango and Orange Residues for Bioprospecting of Biosurfactant Producers" Ecologies 3, no. 2: 120-130. https://0-doi-org.brum.beds.ac.uk/10.3390/ecologies3020010