Is It Possible to Measure Monobromamine Using Colorimetric Methods Based on the Berthelot Reaction, Like for Monochloramine?

Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Solvents
2.2. Artificial Seawater Preparation
2.3. Analytical Devices
3. Results and Discussion
3.1. MCA and MBA Preparation and Standardization
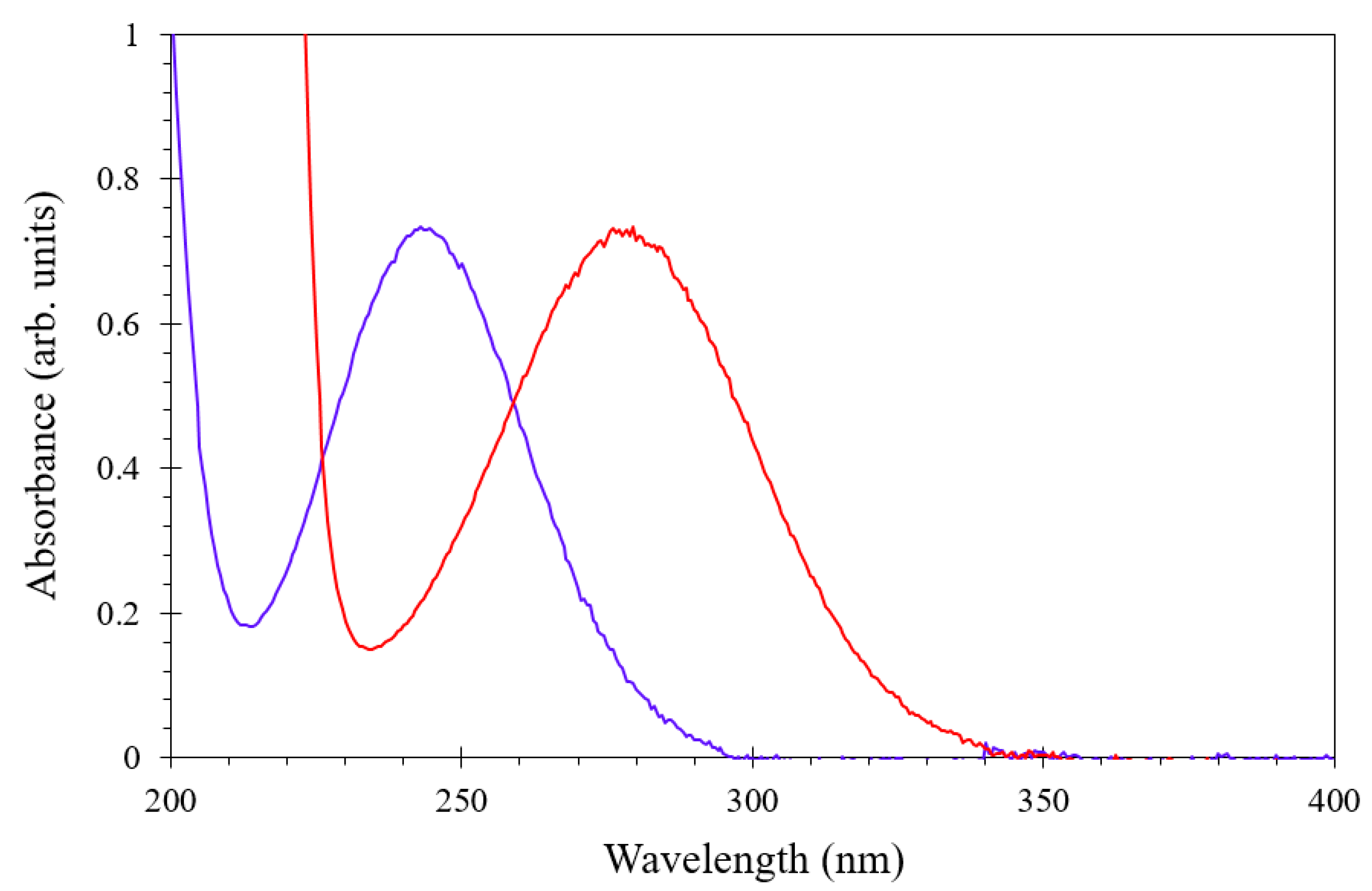
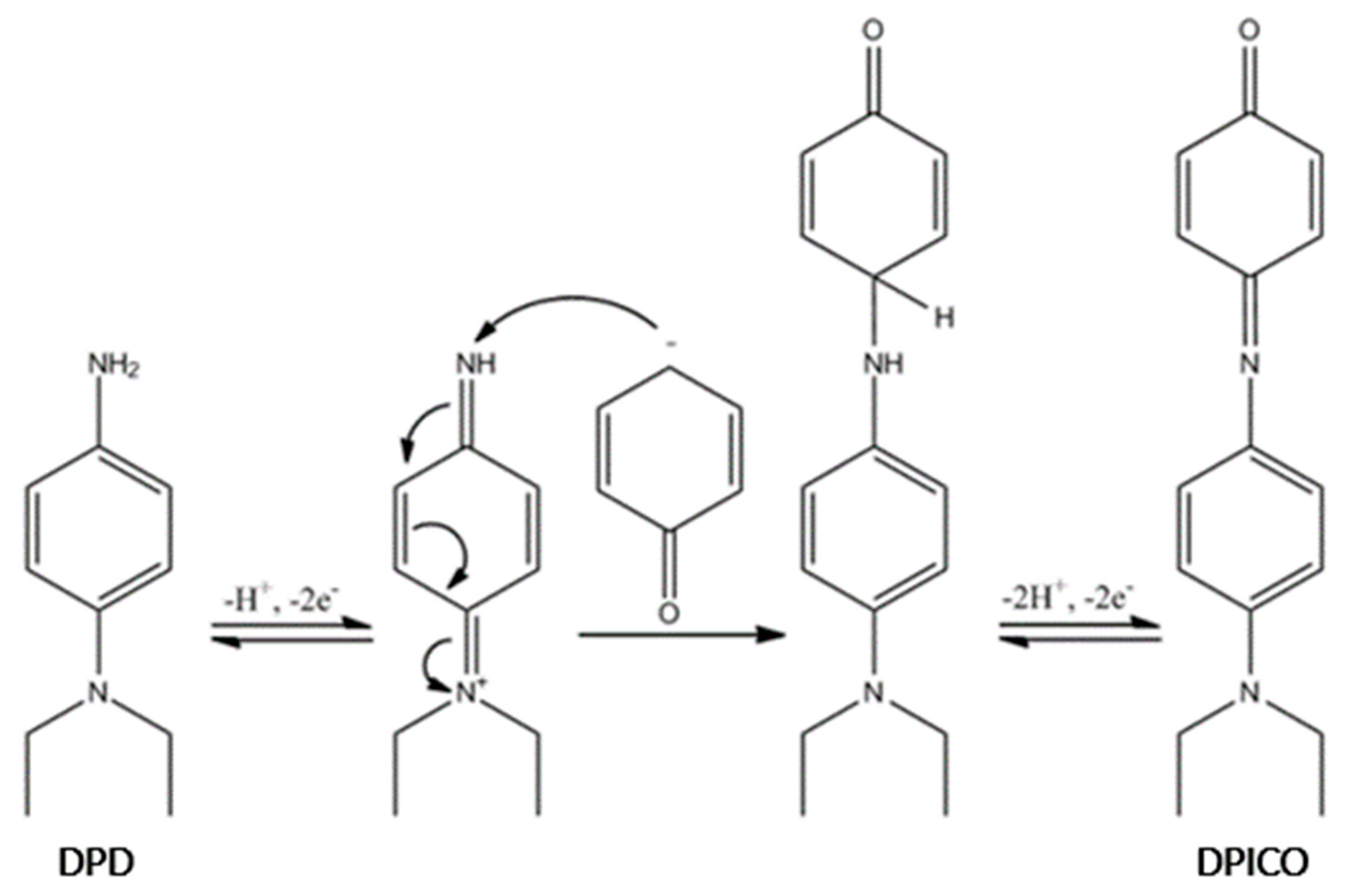
3.2. Spectrophotometric Investigation of the Berthelot Reaction to Determine MBA Level
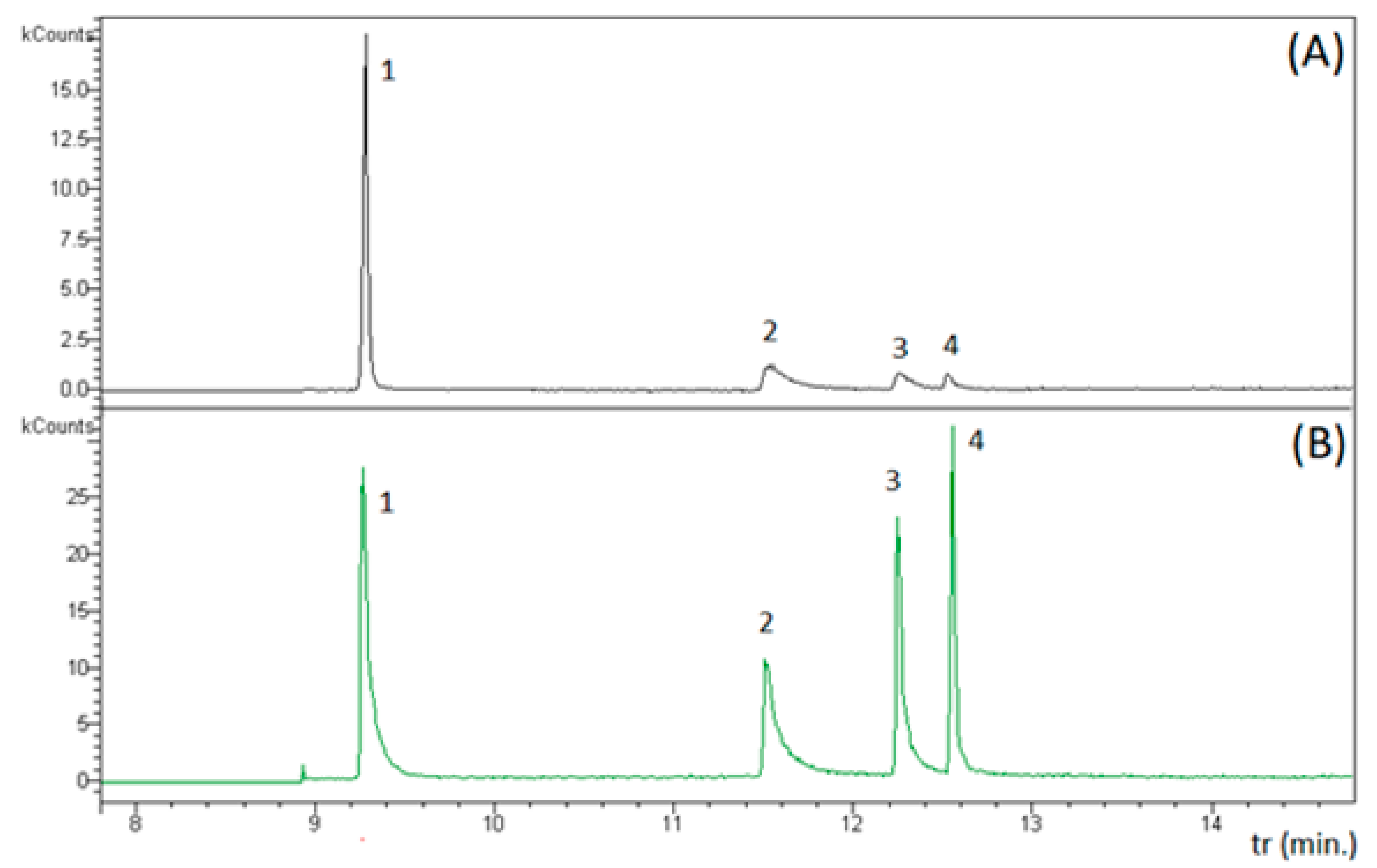
3.3. GC–MS Characterization of the Products of the Reaction between Monobromamine and Berthelot Reagents in Ultrapure Water
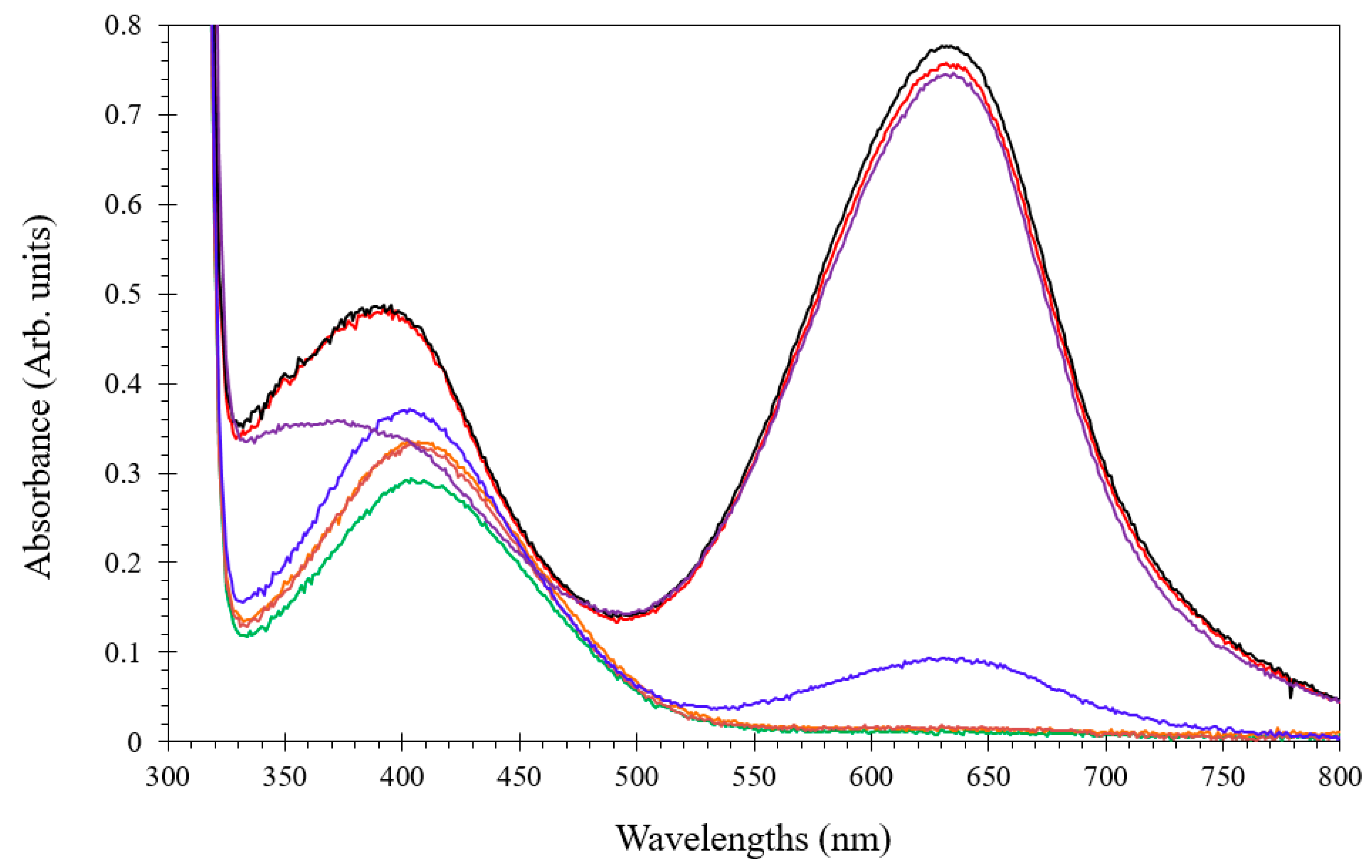
3.4. Investigation of the Berthelot Reaction in Chlorinated Seawater
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kinani, A.; Kinani, S.; Richard, B.; Lorthioy, M.; Bouchonnet, S. Formation and determination of organohalogen by-products in water—Part I. Discussing the parameters influencing the formation of organohalogen by-products and the relevance of estimating their concentration using the AOX (adsorbable organic halide) method. Trends Anal. Chem. 2016, 85, 273–280. [Google Scholar] [CrossRef]
- Timerbaev, A.E.; Fukushi, K. Analysis of seawater and different highly saline natural waters by capillary zone electrophoresis. Mar. Chem. 2003, 82, 221–238. [Google Scholar] [CrossRef]
- Deborde, M.; von Gunten, U. Reactions of chlorine with inorganic and organic compounds during water treatment - Kinetics and mechanisms. Water Res. 2008, 42, 13–51. [Google Scholar] [CrossRef] [PubMed]
- Heeb, M.B.; Kristiana, I.; Trogolo, D.; Arey, J.S.; von Gunten, U. Formation and reactivity of inorganic and organic chloramines and bromamines during oxidative water treatment. Water Res. 2017, 110, 91–101. [Google Scholar] [CrossRef] [PubMed]
- How, Z.T.; Kristiana, I.; Busetti, F.; Linge, K.L.; Joll, C.A. Organic chloramines in chlorine-based disinfected water systems. J. Environ. Sci. 2017, 58, 2–18. [Google Scholar] [CrossRef]
- Na, C.; Olson, T.M. Relative reactivity of amino acids with chlorine in mixtures. Environ. Sci. Technol. 2007, 41, 3220–3225. [Google Scholar] [CrossRef]
- Haag, W.R.; Lietzke, M.H. A Kinetic Model for Predicting the Concentrations of Active Halogens Species in Chlorinated Saline Cooling Waters; ORNL/TM-7942; Oak Ridge National Laboratory: Oak Ridge, TN, USA, 1981. [Google Scholar]
- Sugam, R.; Helz, G.R. Chlorine speciation in seawater; a metastable equilibrium model for ClI and BrI species. Chemosphere 1981, 10, 41–57. [Google Scholar] [CrossRef]
- Heeb, B.M.; Criquet, J.; Zimmermann-Steffens, S.G.; von Gunten, U. Oxidative treatment of bromide-containing waters: Formation of bromine and its reactions with inorganic and organic compounds. Water Res. 2014, 48, 15–42. [Google Scholar] [CrossRef]
- Arnitz, R.; Nagl, M.; Gottardi, W. Microbicidal activity of monochloramine and chloramine T compared. J. Hosp. Infect. 2009, 73, 164–170. [Google Scholar] [CrossRef]
- Kim, B.R.; Anderson, J.E.; Mueller, S.A.; Gaines, W.A.; Kendall, A.M. Literature review-efficacy of various disinfectants against Legionella in water systems. Water Res. 2002, 36, 4433–4444. [Google Scholar] [CrossRef]
- Lee, W.H.; Pressman, J.G.; Wahman, D.G. Three-dimensional free chlorine and monochloramine biofilm penetration: Correlating penetration with biofilm activity and viability. Environ. Sci. Technol. 2018, 52, 1889–1898. [Google Scholar] [CrossRef] [PubMed]
- Donnermair, M.M.; Blatchley, E.R. Disinfection efficacy of organic chloramines. Water Res. 2003, 37, 1557–1770. [Google Scholar] [CrossRef]
- Lee, W.; Westerhoff, P.; Yang, X.; Shang, C. Comparison of colorimetric and membrane introduction mass spectrometry techniques for chloramine analysis. Water Res. 2007, 41, 3097–3102. [Google Scholar] [CrossRef] [PubMed]
- Cooper, W.J.; Jones, A.C.; Whitehead, R.F.; Zika, R.G. Sunlight-induced photochemical decay of oxidants in natural waters: Implications in ballast water treatment. Environ. Sci. Technol. 2007, 41, 3728–3733. [Google Scholar] [CrossRef] [PubMed]
- Jafvert, C.T.; Valentine, R.L. Reaction scheme for the chlorination of ammoniacal water. Environ. Sci. Technol. 1992, 26, 577–586. [Google Scholar] [CrossRef]
- Isaac, R.A.; Morris, J.C. Transfer of active chlorine from chloramine to nitrogenous organic compounds. 2. Mechanism. Environ. Sci. Technol. 1985, 19, 810–814. [Google Scholar] [CrossRef]
- Antelo, J.M.; Arce, F.; Parajó, M. Kinetic study of the decomposition of N-chloramines. J. Phys. Org. Chem. 1996, 9, 447–454. [Google Scholar] [CrossRef]
- Criquet, J.; Rodriguez, E.M.; Allard, S.; Wellauer, S.; Salhi, E.; Joll, C.A.; von Gunten, U. Reaction of bromine and chlorine with phenolic compounds and natural organic matter extracts—Electrophilic aromatic substitution and oxidation. Water Res. 2015, 85, 476–486. [Google Scholar] [CrossRef]
- Westerhoff, P.; Chao, P.; Mash, H. Reactivity of natural organic matter with aqueous chlorine and bromine. Water Res. 2004, 38, 1502–1513. [Google Scholar] [CrossRef]
- Pope, P.G.; Speitel, G.E., Jr. Reactivity of Bromine-Substituted Haloamines in Forming Haloacetic Acids. Disinfection By-Products in Drinking Water; ACS Publications: Washington, DC, USA, 2008. [Google Scholar]
- Kinani, S.; Richard, B.; Souissi, Y.; Bouchonnet, S. Analysis of inorganic chloramines in water. Trends Anal. Chem. 2012, 33, 55–67. [Google Scholar] [CrossRef]
- Roumiguières, A.; Kinani, S.; Bouchonnet, S. Tracking monochloramine decomposition in MIMS analysis. Sensors 2020, 20, 247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sollo, F.W.; Larson, T.E.; McGurk, F.F. Colorimetric methods for bromine. Environ. Sci. Technol. 1971, 5, 240–246. [Google Scholar] [CrossRef]
- Searle, P.L. The Berthelot or indophenol reaction and its use in the analytical chemistry of nitrogen. Analyst 1984, 109, 549–568. [Google Scholar] [CrossRef]
- Tao, H.Z.; Chen, L.; Li, X.; Yang, Y.L.; Li, G.B. Salicylate-spectrophotometric determination of inorganic monochloramine. Anal. Chim. Acta 2008, 615, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Kinani, S.; Layousse, S.; Richard, B.; Kinani, A.; Bouchonnet, S.; Thoma, A.; Sacher, F. Selective and trace determination of monochloramine in river water by chemical derivatization and liquid chromatography/tandem mass spectrometry analysis. Talanta 2015, 140, 189–197. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Kelley, L.P.; Haigis, M.C. An LC-MS Approach to quantitative measurement of ammonia isotopologues. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef]
- Chayata, H.; Lassalle, Y.; Nicol, É.; Manolikakes, S.; Souissi, Y.; Bourcier, S.; Gosmini, C.; Bouchonnet, S. Characterization of the ultraviolet–visible photoproducts of thiophanate-methyl using high performance liquid chromatography coupled with high-resolution tandem mass spectrometry-Detection in grapes and tomatoes. J. Chromatogr. A 2016, 1441, 75–82. [Google Scholar] [CrossRef]
- Sacher, F.; Gerstner, P.; Merklinger, M.; Thoma, A.; Kinani, A.; Roumiguières, A.; Bouchonnet, S.; Richard-Tanaka, B.; Layousse, S.; Ata, R.; et al. Determination of monochloramine dissipation kinetics in various surface water qualities under relevant environmental conditions—Consequences regarding environmental risk assessment. Sci. Total. Environ. 2019, 685, 542–554. [Google Scholar] [CrossRef]
- Heasley, V.L.; Lingner, D.W.; Boerneke, J.L.; Boerneke, M.A.; Hsu, H.; Minnema, R.A.; Moulton, C.A.; Sweeney, A.R. Studies on the syntheses of monobromamine NH2Br and dibromamine NHBr2 in various solvents. Res. J. Chem. Environ. 2013, 17, 45–48. [Google Scholar]
- Lei, H.; Mariñas, B.J.; Minear, R.A. Bromamine decomposition kinetics in aqueous solutions. Environ. Sci. Technol. 2004, 38, 2111–2119. [Google Scholar] [CrossRef]
- NF EN ISO 7393-2, Water Quality-Determination of Free Chlorine and Total Chlorine—Part 2: Colorimetric Method Using N,N-diethyl-1,4-phenylenediamine, for Routine Control Purposes; ISO: Geneva, Switzerland, 2018.
- Adachi, M.; Murata, Y.; Nakamura, S. The relationship between the structures and absorption spectra of cyan color indoaniline dyes. J. Org. Chem. 1993, 58, 5238–5244. [Google Scholar] [CrossRef]
- Beiginejad, H.; Nematollahi, D.; Khazalpour, S. Thermodynamic and electrochemical oxidation of some diamine derivatives: Experimental and theoretical investigation. J. Electrochem. Soc. 2015, 162, 877–883. [Google Scholar] [CrossRef]
- Han, J.; Tao, F.-M. Correlations and predictions of pKa values of fluorophenols and bromophenols using hydrogen-bonded complexes with ammonia. J. Phys. Chem. A 2006, 110, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Sugam, R.; Helz, G.R. Speciation of chlorine produced oxidants in marine waters: Theoretical aspects. Chesapeake Sci. 1977, 18, 113–115. [Google Scholar] [CrossRef]
- Guo, F.L. The bromination kinetics of phenolic compounds in aqueous solution. J. Hazard. Mater. 2009, 170, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Luh, J.; Benito, J.M. Kinetics of Bromochloramine Formation and Decomposition. Environ. Sci. Technol. 2014, 48, 2843–2852. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment | Br− (812 µM) | NH4Cl (7.37 µM) | Chlorine Dose (7.00 µM; Cl/N = 0.95) | Main Oxidizing Agent(s) Formed (a) |
|---|---|---|---|---|
| A | 1 | n.a. (b) | n.a. | No product |
| B (c) | n.a. | 1 | 2 | NH2Cl |
| C | 1 | n.a. | 2 | HOBr/BrO− |
| D | 1 | 3 | 2 | NH2Br |
| E | 3 | 1 | 2 | NH2Cl |
| F | n.a. | 1 | 2 | NH2Cl |
| G | 1 | 2 | 3 | NH2Cl, NH2Br |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roumiguières, A.; Bouchonnet, S.; Kinani, S. Is It Possible to Measure Monobromamine Using Colorimetric Methods Based on the Berthelot Reaction, Like for Monochloramine? Analytica 2020, 1, 1-11. https://0-doi-org.brum.beds.ac.uk/10.3390/analytica1010001
Roumiguières A, Bouchonnet S, Kinani S. Is It Possible to Measure Monobromamine Using Colorimetric Methods Based on the Berthelot Reaction, Like for Monochloramine? Analytica. 2020; 1(1):1-11. https://0-doi-org.brum.beds.ac.uk/10.3390/analytica1010001
Chicago/Turabian StyleRoumiguières, Adrien, Stéphane Bouchonnet, and Said Kinani. 2020. "Is It Possible to Measure Monobromamine Using Colorimetric Methods Based on the Berthelot Reaction, Like for Monochloramine?" Analytica 1, no. 1: 1-11. https://0-doi-org.brum.beds.ac.uk/10.3390/analytica1010001