Detection of Sub-Aroma Threshold Concentrations of Wine Methoxypyrazines by Multidimensional GCMS

 , , and
, , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Reagents and Standards
2.2. Sample Preparation
2.3. Method Optimization
2.3.1. SPME Extraction
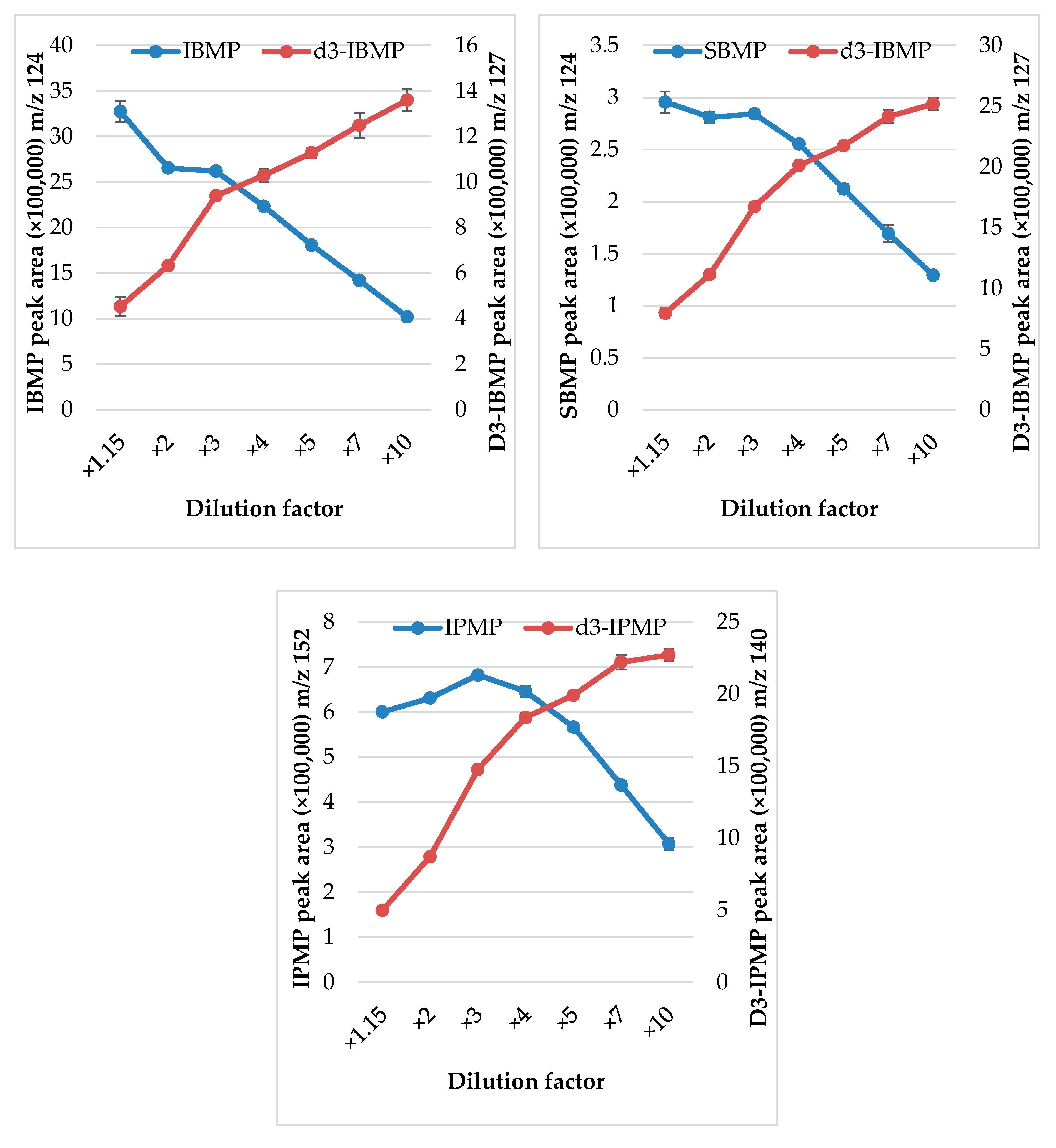
2.3.2. Dilution
2.3.3. Single Dimension GC-FID (Flame Ionization Detector) Analysis
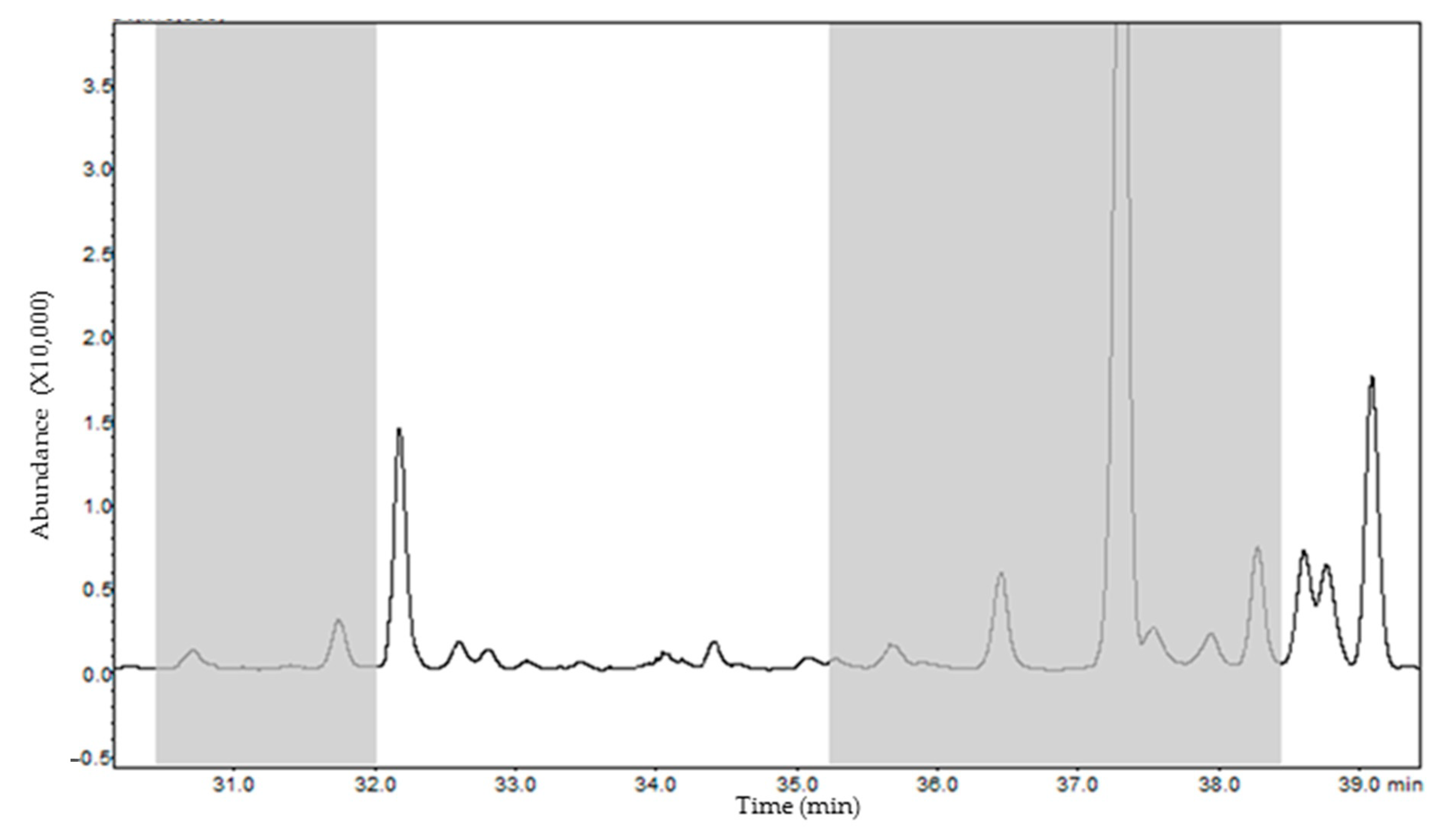
2.3.4. Second Dimension Heart-Cutting MDGC-MS
2.4. Proposed Method
2.5. Method Validation
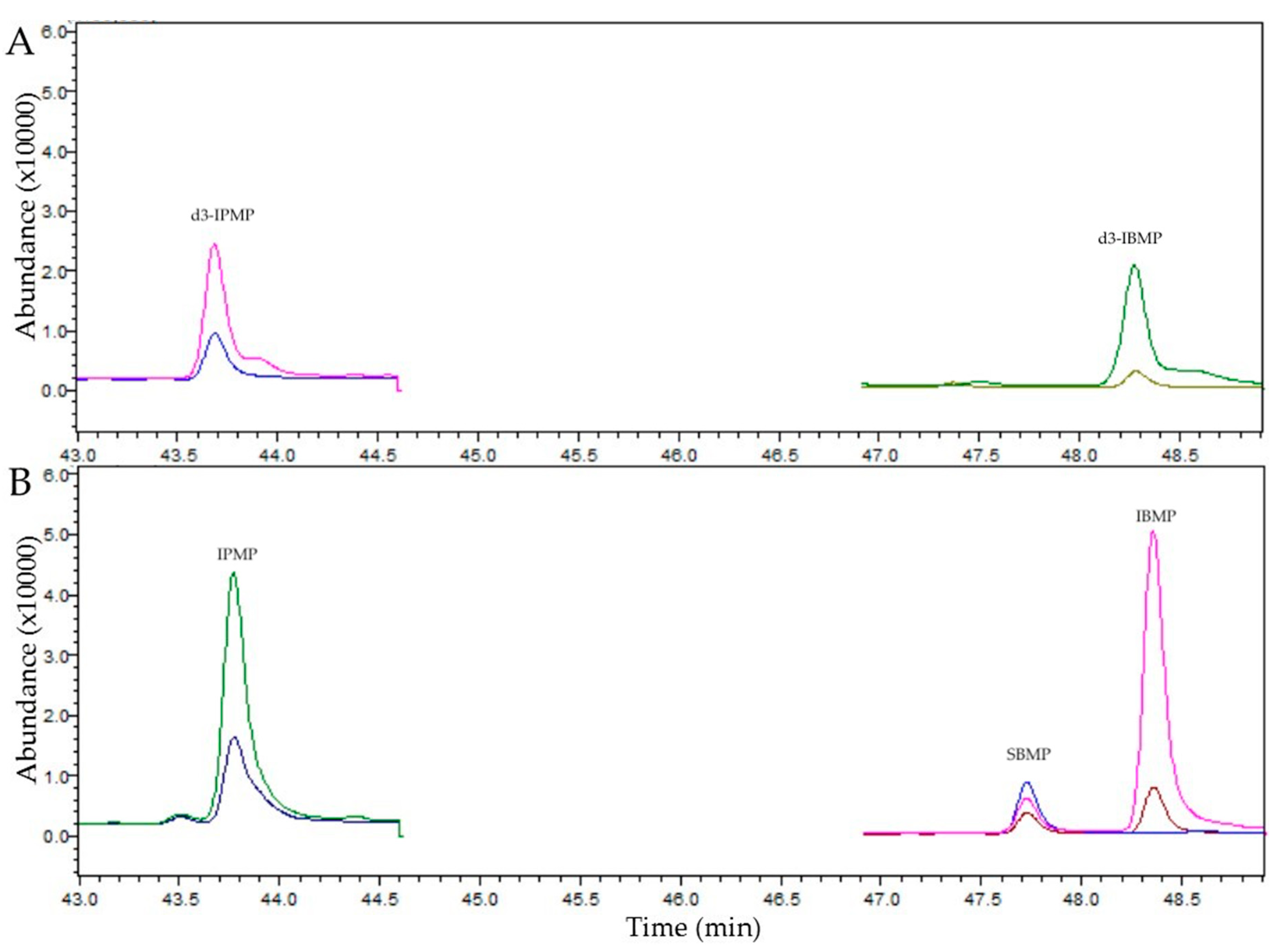
2.5.1. Peak Identification
2.5.2. Linearity
2.5.3. Recovery
2.5.4. Limit of Blank, Limit of Detection, Limit of Quantitation, Accuracy, and Precision
2.6. Data Analysis
3. Results and Discussion
3.1. Extraction Optimization
3.2. Method Validation
3.3. Wine Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lin, J.; Massonnet, M.; Cantu, D. The genetic basis of grape and wine aroma. Hortic. Res. 2019, 6, 81. [Google Scholar] [CrossRef] [Green Version]
- Lei, Y.; Xie, S.; Guan, X.; Song, C.; Zhang, Z.; Meng, J. Methoxypyrazines biosynthesis and metabolism in grape: A review. Food Chem. 2018, 245, 1141–1147. [Google Scholar] [CrossRef]
- Boulton, R.B.; Singleton, V.L.; Bisson, L.F.; Kunkee, R.E. Principle and Practices of Winemaking; Chapman & Hall: New York, NY, USA, 1996; pp. 150–166. [Google Scholar]
- Castro, R.; Natera, R.; Durán, E.; García-Barroso, C. Application of solid phase extraction techniques to analyse volatile compounds in wines and other enological products. Eur. Food Res. Technol. 2008, 228, 1–18. [Google Scholar] [CrossRef]
- Torrens, J.; Riu-Aumatell, M.; López-Tamames, E.; Buxaderas, S. Volatile compounds of red and white wines by headspace-solid-phase microextraction using different fibers. J. Chromatogr. Sci. 2004, 42, 310–316. [Google Scholar] [CrossRef] [Green Version]
- Hjelmeland, A.K.; Wylie, P.L.; Ebeler, S.E. A comparison of sorptive extraction techniques coupled to a new quantitative, sensitive, high throughput GC–MS/MS method for methoxypyrazine analysis in wine. Talanta 2016, 148, 336–345. [Google Scholar] [CrossRef]
- Blumberg, L.M.; Frank, D.; Klee, M.S.; Sandra, P. Comparison of one-dimensional and comprehensive two-dimensional separations by gas chromatography. J. Chromatogr. A 2008, 1188, 2–16. [Google Scholar] [CrossRef]
- Amaral, M.S.S.; Marriott, P.J. The blossoming of technology for the analysis of complex aroma bouquets—A review on flavour and odorant multidimensional and comprehensive gas chromatography applications. Molecules 2019, 24, 2080. [Google Scholar] [CrossRef] [Green Version]
- Wen, Y.; Ontañon, I.; Ferreira, V.; Lopez, R. Determination of ppq-levels of alkylmethoxypyrazines in wine by stirbar sorptive extraction combined with multidimensional gas chromatography-mass spectrometry. Food Chem. 2018, 255, 235–241. [Google Scholar] [CrossRef]
- Parr, W.V.; Green, J.A.; White, K.G.; Sherlock, R.R. The distinctive flavour of New Zealand Sauvignon blanc: Sensory characterisation by wine professionals. Food Qual. Prefer. 2007, 18, 849–861. [Google Scholar] [CrossRef]
- Hartmann, P.J.; McNair, H.M.; Zoecklein, B.W. Measurement of 3-alkyl-2-methoxypyrazine by headspace solid-phase microextraction in spiked model wines. Am. J. Enol. Vitic. 2002, 53, 285. [Google Scholar]
- Shimizu, Y.; Imayoshi, Y.; Kato, M.; Maeda, K.; Iwabuchi, H.; Shimomura, K. Volatiles from leaves of field-grown plants and shoot cultures of Gynura bicolor DC. Flavour Frag. J. 2009, 24, 251–258. [Google Scholar] [CrossRef]
- Jakobsen, H.B.; Hansen, M.; Christensen, M.R.; Brockhoff, P.B.; Olsen, C.E. Aroma volatiles of blanched green peas (Pisum sativum L.). J. Agric. Food Chem. 1998, 46, 3727–3734. [Google Scholar] [CrossRef]
- Lee, K.-G.; Shibamoto, T. Analysis of volatile components isolated from Hawaiian green coffee beans (Coffea arabica L.). Flavour Frag. J. 2002, 17, 349–351. [Google Scholar] [CrossRef]
- Schmarr, H.-G.; Ganß, S.; Koschinski, S.; Fischer, U.; Riehle, C.; Kinnart, J.; Potouridis, T.; Kutyrev, M. Pitfalls encountered during quantitative determination of 3-alkyl-2-methoxypyrazines in grape must and wine using gas chromatography–mass spectrometry with stable isotope dilution analysis. Comprehensive two-dimensional gas chromatography–mass spectrometry and on-line liquid chromatography-multidimensional gas chromatography–mass spectrometry as potential loopholes. J. Chromatogr. A 2010, 1217, 6769–6777. [Google Scholar]
- Machiels, D.; Istasse, L. Evaluation of two commercial solid-phase microextraction fibres for the analysis of target aroma compounds in cooked beef meat. Talanta 2003, 61, 529–537. [Google Scholar] [CrossRef]
- Kalua, C.M.; Boss, P.K. Sample preparation optimization in wine and grapes: Dilution and sample/headspace volume equilibrium theory for headspace solid-phase microextraction. J. Chromatogr. A 2008, 1192, 25–35. [Google Scholar] [CrossRef]
- Onuki, S.; Koziel, J.A.; Jenks, W.S.; Cai, L.; Rice, S.; van Leeuwen, J. Optimization of extraction parameters for quantification of fermentation volatile by-products in industrial ethanol with solid-phase microextraction and gas chromatography. J. Inst. Brew. 2016, 122, 102–109. [Google Scholar] [CrossRef] [Green Version]
- Chin, S.-T.; Eyres, G.T.; Marriott, P.J. System design for integrated comprehensive and multidimensional gas chromatography with mass spectrometry and olfactometry. Anal. Chem. 2012, 84, 9154–9162. [Google Scholar] [CrossRef]
- Schmarr, H.-G.; Slabizki, P.; Legrum, C. Optimization in multidimensional gas chromatography applying quantitative analysis via a stable isotope dilution assay. Anal. Bioanal. Chem. 2013, 405, 6589–6593. [Google Scholar] [CrossRef]
- Stauffer, M. Calibration and Validation of Analytical Methods: A Sampling of Current Approaches; IntechOpen: London, UK, 2018. [Google Scholar] [CrossRef]
- Armbruster, D.A.; Pry, T. Limit of blank, limit of detection and limit of quantitation. Clin. Biochem. Rev. 2008, 29, S49–S52. [Google Scholar]
- Roujou de Boubée, D.; van Leeuwen, C.; Dubourdieu, D. Organoleptic impact of 2-methoxy-3-isobutylpyrazine on red Bordeaux and Loire wines. Effect of environmental conditions on concentrations in grapes during ripening. J. Agric. Food Chem. 2000, 48, 4830–4834. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Ju, Y.; Wei, X.; Dong, S.; Sun, X.; Fang, Y. Significance and transformation of 3-alkyl-2-methoxypyrazines through grapes to wine: Olfactory properties, metabolism, biochemical regulation, and the HP-MP cycle. Molecules 2019, 24, 4598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidhu, D.; Lund, J.; Kotseridis, Y.; Saucier, C. Methoxypyrazine analysis and influence of viticultural and enological procedures on their levels in grapes, musts, and wines. Crit. Rev. Food Sci. 2015, 55, 485–502. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, K.; Samuta, T. Grape maturity and light exposure affect berry methoxypyrazine concentration. Am. J. Enol. Vitic. 1999, 50, 194. [Google Scholar]
- Legrum, C.; Gracia-Moreno, E.; Lopez, R.; Potouridis, T.; Langen, J.; Slabizki, P.; Weiand, J.; Schmarr, H.-G. Quantitative analysis of 3-alkyl-2-methoxypyrazines in German Sauvignon blanc wines by MDGC–MS or MDGC–MS/MS for viticultural and enological studies. Eur. Food Res. Technol. 2014, 239, 549–558. [Google Scholar] [CrossRef]
- Botezatu, A.; Pickering, G.J.; Kotseridis, Y. Development of a rapid method for the quantitative analysis of four methoxypyrazines in white and red wine using multi-dimensional Gas Chromatography—Mass Spectrometry. Food Chem. 2014, 160, 141–147. [Google Scholar] [CrossRef]
- Ishii, A.; Roudnitzky, N.; Béno, N.; Bensafi, M.; Hummel, T.; Rouby, C.; Thomas-Danguin, T. Synergy and masking of odor mixtures: An electrophysiological study of orthonasal vs. retronasal perception. Chem. Senses 2008, 33, 553–561. [Google Scholar] [CrossRef] [Green Version]
- Ilc, T.; Werck-Reichhart, D.; Navrot, N. Meta-analysis of the core aroma components of grape and wine aroma. Front. Plant. Sci. 2016. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Molecular Weight | Retention Time (min) | Retention Indices | Monitored Ions (m/z) * | ||
|---|---|---|---|---|---|---|
| Dimension 1 | Dimension 2 | Calculated | Literature | |||
| IBMP | 166.22 | 37.20 | 48.38 | 1517 | 1516 | 124, 151 |
| IPMP | 152.19 | 31.14 | 43.78 | 1422 | 1423 | 152, 137 |
| SBMP | 166.22 | 35.66 | 47.74 | 1494 | 1494 | 124, 138 |
| d3-IBMP | 169.24 | 37.14 | 48.29 | 1515 | N/A | 127, 154 |
| d3-IPMP | 155.21 | 31.08 | 43.69 | 1421 | N/A | 140, 155 |
| Retention Time (2nd Dimension) | Compound | Retention Indices Literature (1st dimension) | % MS Match |
|---|---|---|---|
| 37.41 | (1H)-Pyrrole | 1499 | 95 |
| 39.01 | 2,3-Butanediol | 1523 | 98 |
| 39.32 | 1-Octen-3-ol | 1423 | 96 |
| 40.56 | cis-Hept-4-enol | 1502 | 88 |
| 40.69 | 6-Hepten-1-ol | n/a | 90 |
| 41.40 | Benzaldehyde | 1525 | 98 |
| 43.48 | p-Cymene | 1426 | 80 |
| 43.78 | IPMP | 1423 | |
| 43.96 | Siloxane (system peak) | n/a | |
| 45.15 | 2-Nonanol | 1521 | 95 |
| 45.67 | β- Linalool | 1537 | 97 |
| 47.74 | SBMP | 1494 | |
| 48.38 | IBMP | 1516 | |
| 49.10 | Decanal | 1512 | 94 |
| 50.50 | 2-Bornene | 1519 | 89 |
| 51.96 | Unknown | ||
| 53.26 | Vitispirane | 1527 | 94 |
| 53.97 | Unknown |
| Compound | Concentration Range (ng/L) | LOB (ng/L) | LOD (ng/L) | %CV b | Equation | R2 c |
|---|---|---|---|---|---|---|
| IPMP | 0.260–9.96 | 0.159 | 0.192 | 1.4 | y = −0.9383x 2 + 5.401x + 0.0334 | 0.9993 |
| SBMP | 0.130–9.99 | 0.016 | 0.061 | 2.8 | y = −0.1479x2 + 13.188x − 0.0216 | 0.9998 |
| IBMP | 0.267–41.16 | 0.110 | 0.175 | 3.5 | y = −0.0615x2 + 3.533x − 0.0238 | 0.9996 |
| Wine | IPMP | SBMP | IBMP | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Spike Concentration (ng/L) | % Recovery | % CV | Spike Concentration (ng/L) | % Recovery | % CV | Spike Concentration (ng/L) | % Recovery | % CV | |
| Sauvignon blanc | 0.66 | 95.1 | 5.47 | 0.66 | 97.1 | 2.95 | 2.73 | 106.3 | 2.30 |
| Sauvignon blanc | 2.26 | 102.0 | 1.41 | 2.21 | 101.2 | 1.15 | 9.14 | 101.8 | 0.57 |
| Model wine (mid-range) | 0.66 | 98.0 | 2.27 | 0.66 | 97.0 | 2.81 | 2.73 | 100.3 | 3.04 |
| Model wine (high-range) | 3.32 | 100.8 | 1.34 | 3.33 | 101.3 | 2.62 | 13.72 | 100.1 | 2.82 |
| Rosé | 0.66 | 97.7 | 3.43 | 0.66 | 94.9 | 5.21 | 2.73 | 99.6 | 6.57 |
| Pinot noir | 0.66 | 100.5 | 0.30 | 0.66 | 94.3 | 4.68 | 2.73 | 95.7 | 3.88 |
| Wine | IPMP (ng/L) | SMBP (ng/L) | IBMP (ng/L) |
|---|---|---|---|
| Sauvignon blanc | 0.259 ± 0.031 | 0.191 ± 0.061 | 0.470 ± 0.164 |
| Rosé | Detected (0.248 ± 0.026) | 0.192 ± 0.058 | 5.80 ± 0.42 |
| Pinot noir | Not Detected | 0.330 ± 0.370 | 0.360 ± 0.070 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olejar, K.J.; Breitmeyer, J.; Wimalasiri, P.M.; Tian, B.; Field, S.K. Detection of Sub-Aroma Threshold Concentrations of Wine Methoxypyrazines by Multidimensional GCMS. Analytica 2021, 2, 1-13. https://0-doi-org.brum.beds.ac.uk/10.3390/analytica2010001
Olejar KJ, Breitmeyer J, Wimalasiri PM, Tian B, Field SK. Detection of Sub-Aroma Threshold Concentrations of Wine Methoxypyrazines by Multidimensional GCMS. Analytica. 2021; 2(1):1-13. https://0-doi-org.brum.beds.ac.uk/10.3390/analytica2010001
Chicago/Turabian StyleOlejar, Kenneth J., Jason Breitmeyer, Pradeep M. Wimalasiri, Bin Tian, and Stewart K. Field. 2021. "Detection of Sub-Aroma Threshold Concentrations of Wine Methoxypyrazines by Multidimensional GCMS" Analytica 2, no. 1: 1-13. https://0-doi-org.brum.beds.ac.uk/10.3390/analytica2010001