
A Comparative Theoretical and Spectroscopic Study of Aminomethylbenzoic Acid Derivatives as Potential NLO Candidates †


1
Laboratory of Structures, Properties and Interatomic Interactions LASPI2A, Department of Matter Sciences, Faculty of Sciences and Technology, Abbes Laghrour University Khenchela, Khenchela 40000, Algeria
2
Department of Chemistry, Faculty of Science, Cumhuriyet University, 58140 Sivas, Turkey
*
Author to whom correspondence should be addressed.
†
Presented at the 24th International Electronic Conference on Synthetic Organic Chemistry, 15 November–15 December 2020; Available online: https://ecsoc-24.sciforum.net/ .
Chem. Proc. 2021, 3(1), 102; https://0-doi-org.brum.beds.ac.uk/10.3390/ecsoc-24-08099
Published: 13 November 2020
(This article belongs to the Proceedings of The 24th International Electronic Conference on Synthetic Organic Chemistry)
Abstract
:Three aminomethylbenzoic acid derivatives were theoretically studied at M062X/6-311++G(d,p) level in a vacuum, namely 2-ammonio-5-methylcarboxybenzene perchlorate (1), 4-(ammoniomethyl) carboxybenzene nitrate (2) and 4-(ammoniomethyl)carboxybenze perchlorate (3). The compounds’ structures were fully optimized and compared with the single-crystal X-ray diffraction results, showing a very close agreement with the experimental structural parameters. Their IR, 1H- and 13C-NMR spectra were calculated and examined in detail. Furthermore, the molecular electrostatic potential (MEP) maps of the studied compounds were investigated and the strength of the non-covalent interactions evaluated. In addition to these results, the NLO properties of the three compounds were predicted.
1. Introduction
2-carboxy-4-methylaniline (known as 2-amino-5-methylbenzoic acid) and 4-(aminomethyl)benzoic acid (PAMBA) are biologically active molecules serving as pharmaceutical intermediates [1] and their derivatives are considered as potential agents for anti-cancer chemotherapy and evaluated for their cytotoxic activity [2,3,4]. Furthermore, PAMBA, described for its antifibrinolytic action [5,6], is known with its derivatives to inhibit the proliferation of endothelial cells [7,8] and are additionally proven to be antiproteolytic [9].
A structural survey associated with 2-carboxy-4-methylanilinium derivatives and their related amino acids in the structural database (CSD, Version 5.39 [10]) returned only two hits, namely: 2-carboxy-4-methylanilinium perchlorate (1) (CSD refcode: CURKOR [11]) and 2-carboxy-4-methylanilinium chloride monohydrate (CSD refcode: GAZZAK [12]). Furthermore, the zwitterionic form of 4-(aminomethyl)benzoic acid, viz. 4-(ammoniomethyl)benzoate monohydrate (CSD refcode: PONTAP), was reported once in the literature [13], and the database survey showed two hits of (4-carboxyphenyl)methanaminium salts (the nitrate derivative (2), CSD refcode: CURCUX and the perchlorate salt (3), CSD refcode: CURLAE) [11].
Quantum chemical calculations have been considered an attractive research area in the last few years [14,15,16,17]. Therefore, modelling studies of the salts (1)–(3) were carried out by using the M06-2X with a 6-311++G(d,p) basis set in the gas phase and the structures of the three compounds were fully optimized at the same level and compared with the experimental single-crystal X-ray diffraction results previously reported [11]. The IR and the NMR spectra of the mentioned compounds were calculated and examined in detail. Furthermore, the molecular electrostatic potential (MEP) maps of the related compounds were investigated. In addition to these results, the interaction strength between the anions and the cations were calculated and discussed in detail. The NLO behavior of the studied compounds was furthermore theoretically evaluated.
2. Materials and Methods
The computational processes are performed by GaussView 5.0.9 [18] and Gaussian 09 AS64L-G09RevD.01 [19] programs. The M06-2X function of the hybrid density functional theory (DFT) has been derived by Zhao et al. in 2008 [20]. It was adopted in the calculations together with the 6-311++G(d,p) basis set. In the IR computations, no imaginary frequency was observed. As for the NMR spectra of the related compounds, they were calculated with a gauge-independent atomic orbital (GIAO) method. In the calculation of chemical shift values, tetramethylsilane (TMS) was used as a reference substance. Additionally, in the calculations of the MEP maps, the electrostatic potential (ESP) charges were taken into consideration.
3. Results and Discussion
3.1. Comparative Theoretical and Experimental Structural Study
The optimized structures of compounds (1)–(3) are represented in Figure 1. Selected experimental and calculated geometric parameters are given in Table 1. The accuracy of the calculated results towards the experimental ones was investigated by plotting the scatter graphs. Hence, the regression coefficients (R2) were calculated from these graphs for each compound and given in the related table.
According to Table 1, the experimental bond distances vary from 1.203 to 1.507 ♦ for compound (1), from 1.213 to 1.505 ♦ for compound (2), and in the range 1.241–1.516 ♦ for compound (3). These distances are related to the C─CO2H and C─CH3/C─CH2─ bonds. Whereas, the associated calculated values are found to be (1.212–1.505 ♦), (1.200–1.507 ♦) and (1.241–1.516 ♦), respectively. Consequently, the calculated and experimental geometric parameters (bond lengths and angles) are very close to each other and it can be easily seen that there is a good agreement in terms of regression coefficients, with R2 worth 0.9998, 0.9998 and 0.9993 for compounds (1)–(3), respectively.
The cationic and anionic moieties in compounds (1)–(3) were optimized separately at the same level of theory as follow: 2-ammonio-5-methylcarboxybenzene (+1) (A), 4-(ammoniomethyl) carboxybenzene (+1) (B), perchlorate (-1) (C) and nitrate (-1) (D) and the total energy of the whole structures are given in Table 2.
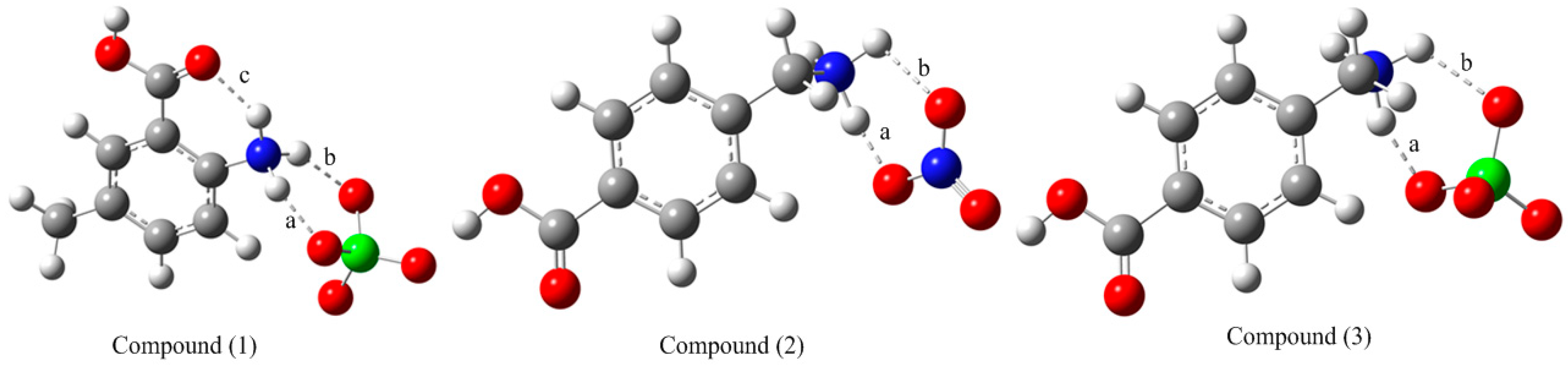
There is an intramolecular hydrogen bond (labeled “c” in Figure 1) in the cationic moiety (A) and it is therefore more stable than the cation (B). Additionally, there are two other intermolecular hydrogen bonds as labeled in Figure 1 as “a” and “b” in compound (1). As for compounds (2) and (3), they exhibit two intermolecular non-covalent interactions (“a” and “b”). The corresponding interaction energies are calculated as −407.916, −477.384 and −434.142 kJ mol−1 for compounds (1)–(3), respectively. These results show that the interaction in compound (2) is stronger than those of compounds (1) and (3). It implies that the associated hydrogen bond is stronger due to the electronegative nitrogen atom. The single-crystal X-ray diffraction results [11] show that compound (2) displays six strong and moderate O–H…O, O–H…N and N–H…O hydrogen bonds. Whereas in (3), it was observed three N–H…O hydrogen bonds only against six O–H…O and N–H…O intermolecular interactions in (1).
3.2. Spectral Analyses
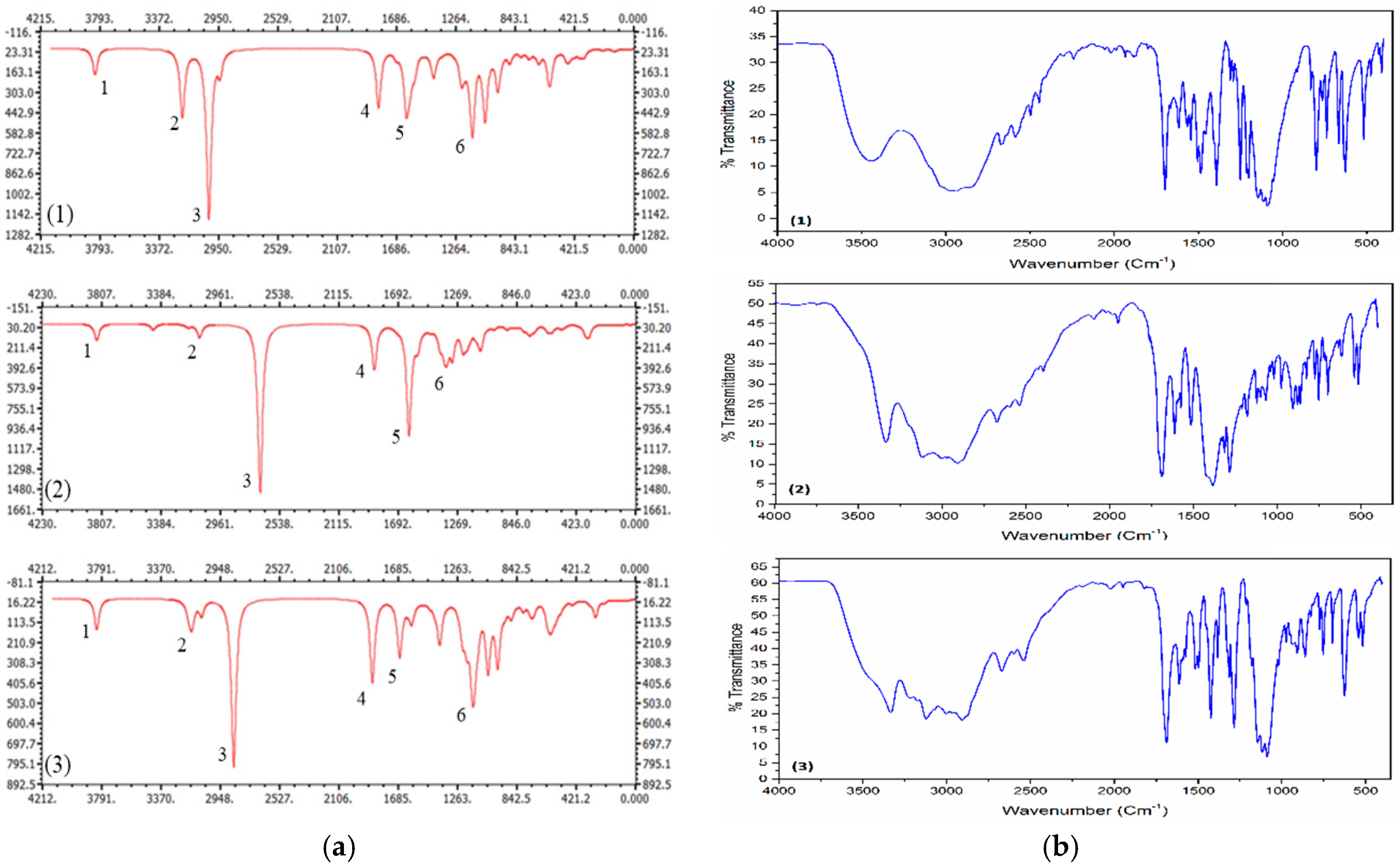
The IR spectrum of the studied compounds are calculated and represented in Figure 2a. The most significant bands are labeled in the same figure and the vibration mode and corresponding frequencies are given in Table 3. In Figure 2b, we illustrated the reported experimental FTIR spectra [11].
Furthermore, the 1H- and 13C-NMR spectra of the studied compounds were calculated at the same level of theory. The chemical shift values of the related atom are calculated by using TMS. The chemical shift values of the hydrogen and carbon atoms are given in Table 4.
According to Table 4, the chemical shift values of the aromatic carbon atoms are in the range of 135–186 ppm. As for the aliphatic carbons, the chemical shift value is calculated to be 24.1 ppm for compound (1) and nearly 50 ppm for compounds (2) and (3), since the amine group is connected to this carbon in both compounds. The chemical shift values of hydrogen atoms in the aromatic ring are in the range 8–10 ppm vs. 2–5 ppm for the hydrogen atoms connected to the aliphatic carbon. The chemical shift values of some hydrogens are high due to the presence of the hydrogen-bonding networks and the electronegative atoms. These results show that there are three hydrogen bonds in compound (1) and two hydrogen bonds in compound (2) and (3).
3.3. Molecular Electrostatic Potential (MEP) Maps and Contour Diagrams
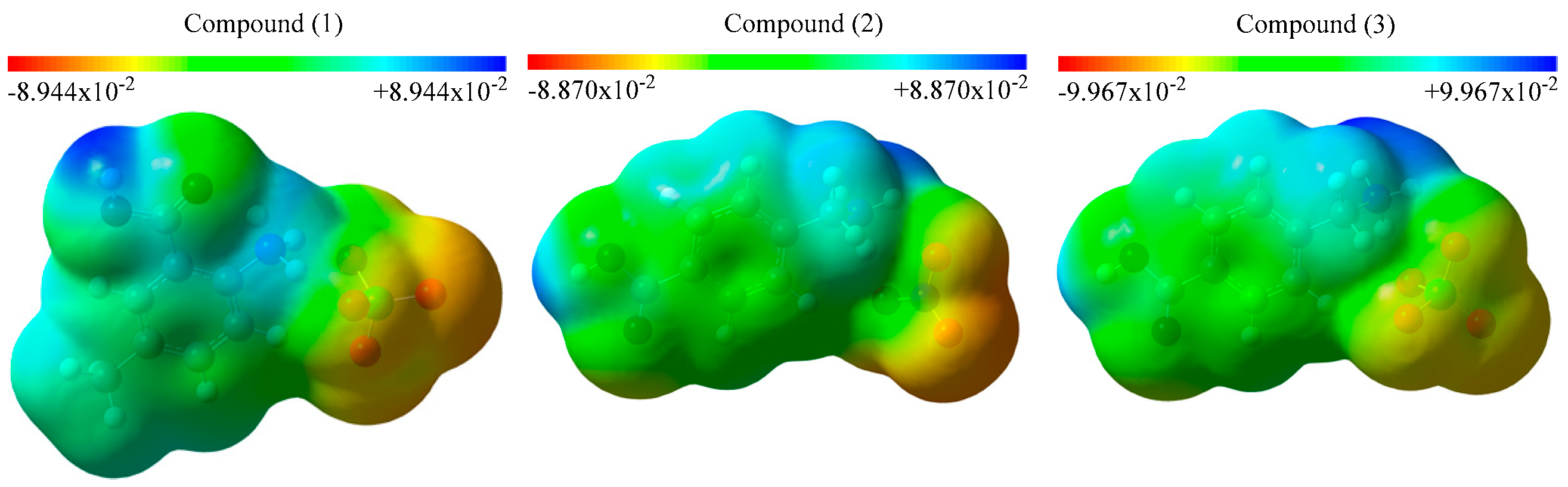
The molecular electrostatic potential (MEP) maps are important because they allow investigating the active regions, the reaction mechanisms or the interaction regions. The MEP maps of the studied structures are calculated and represented in Figure 3. There are red, yellow, green and blue regions in the figure, which are determined in terms of the electron density. The electronically rich regions are mainly represented in red or yellow whereas the electronically poor ones are displayed in blue. The blue regions are dominant around the hydrogen atoms in the studied compounds, while the red ones are dominant nearby the oxygen atoms which are linked to the chlorine or the nitrogen atoms. The red regions are appropriate to nucleophilic attacks. Particularly, the environment of the carboxyl group and the benzene ring are in the green and yellow. The electron densities are decreasing from red to blue and therefore, the red and yellow regions are more active than the green and blue ones.
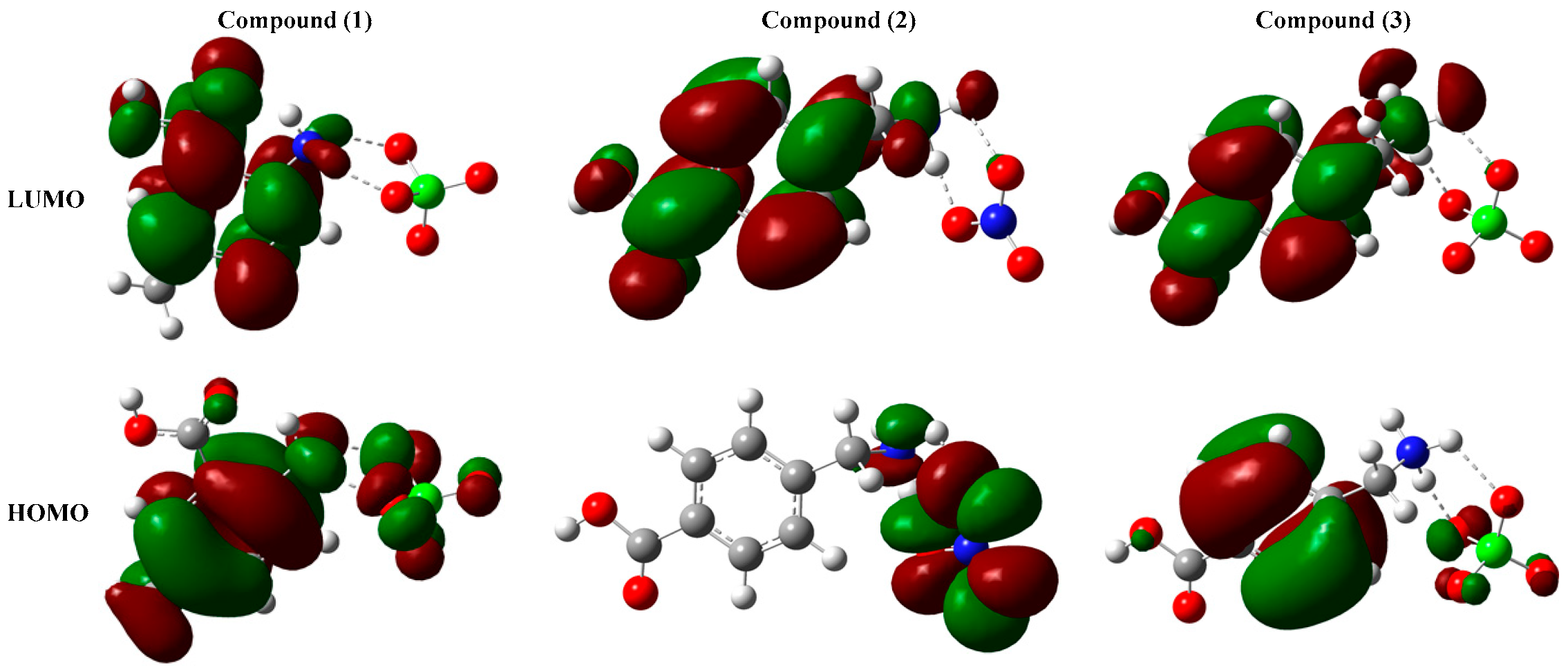
The contour diagrams of the frontier molecular orbitals are calculated at the same level of theory and represented in Figure 4.
3.4. Non-Linear Optical Properties
The non-linear optical (NLO) properties can be affected by the molecular structure, the electromagnetic field, the frequency. It is important in providing the key functions of frequency shifting, the optical modulation, the optical switching, the optical logic and the optical memory for the technologies in areas such as telecommunications, signal processing and optical interactions. The NLO properties of the related compounds are predicted by calculating some quantum chemical parameters, which are the total static dipole moment (μ), the average linear polarizability (α), the anisotropy of polarizability (Δα), and the first hyper polarizability (β). These parameters are calculated by using Equations (1)–(4) and given in Table 5.
The NLO properties increase with the increasing of the total static dipole moment, the average linear polarizability, the anisotropy of polarizability and the first hyper polarizability. The related quantum chemical parameters of compounds (1)–(3) are higher than the urea’s values. Therefore, the three compounds are better than urea and could be good candidates for NLO applications. According to the mentioned descriptors and the following ranking:
Compound (1) > Compound (3) > Compound (2) (for μ)
Compound (1) > Compound (3) > Compound (2) (for α)
Compound (3) > Compound (1) > Compound (2) (for Δα)
Compound (1) > Compound (2) > Compound (3) (for β)
It is worth noted that compound (1) is the best candidate for NLO applications.
4. Conclusions
Computational investigations of three aminomethylbenzoic acid derivatives were performed by using the M062X method with the 6-311++G(d,p) basis sets in the gas phase. The structural and spectral analyses were carried out in detail, presenting a very good agreement between the theoretical results and the experimental values. The interaction energies were furthermore calculated and the hydrogen bond strengths of the mentioned compounds were evaluated, showing the presence of intramolecular and intermolecular O–H…O, O–H…N and N–H…O hydrogen bonds. Additionally, the MEP maps of the related compounds were examined and the NLO properties estimated, concluding that the three compounds are good candidates for NLO applications.
Author Contributions
Conceptualization, A.D. and K.S.; methodology, A.D. and K.S.; software, K.S.; investigation, A.D. and K.S.; resources, A.D.; writing—original draft preparation, A.D. and K.S.; writing—review and editing, A.D. and K.S.; visualization, A.D. and K.S. All authors have read and agreed to the published version of the manuscript.
Data Availability Statement
Full structural details might be found in the CIF files deposited at the Cambridge Crystallographic Data Centre, CCDC No 1055152, 1055153 and 1055154. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html, or from the CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK: fax: (+44) 01223-336-033; e-mail: [email protected].
Acknowledgments
Funding was provided by the General Direction of research and development technologies/Ministry of Higher Education and Research Sciences DGRSDT/MESRS, Algeria. The financial support from Abbes Laghrour University of Khenchela is acknowledged. The numerical calculations reported in this paper are performed at TUBITAK ULAKBIM, High Performance and Grid Computing Center (TRUBA Resources).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Zheng, B.H.; Fang, Z.J.; Jiao, Y.; Jiang, Y.H. Composes a transfert de protons: Synthese, structure et etude des differentes interactions interatomiques. Jiangsu Chem. Ind. 2007, 55, 39–41. [Google Scholar]
- Cao, S.L.; Feng, Y.-P.; Jiang, Y.-Y.; Liu, S.Y.; Ding, G.Y.; Li, R.T. Synthesis and in vitro antitumor activity of 4(3H)-quinazolinone derivatives with dithiocarbamate side chains. Bioorg. Med. Chem. Lett. 2005, 15, 1915–1917. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.L.; Feng, Y.P.; Zheng, X.L.; Jiang, Y.Y.; Zhang, M.; Wang, Y.; Xu, M. Synthesis of substituted benzylamino- and heterocyclylmethylamino carbodithioate derivatives of 4-(3H)-quinazolinone and their cytotoxic activity. Arch. Pharm. 2006, 339, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.L.; Guo, Y.W.; Wang, X.B.; Zhang, M.; Feng, Y.P.; Jiang, Y.Y.; Wang, Y.; Gao, Q.; Ren, J. Synthesis and Cytotoxicity Screening of Piperazine-1-carbodithioate Derivatives of 2-Substituted Quinazolin-4(3H)-ones. Arch. Pharm. 2009, 342, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Steinmetzer, T.; Pilgram, O.; Wenzel, B.M.; Wiedemeyer, S.J.A. Fibrinolysis Inhibitors: Potential Drugs for the Treatment and Prevention of Bleeding. J. Med. Chem. 2020, 63, 1445–1472. [Google Scholar] [CrossRef] [PubMed]
- Markwardt, F.; Neuland, P.; Klöcking, H.P. On the antifibrinolytic activity of the esters of 4-aminomethylbenzoic acid (PAMBA)]. Pharmazie 1966, 21, 345–348. [Google Scholar] [PubMed]
- Pilgrim, H.; Grosse, S. Proliferation of cultured endothelial cells under the effect of aprotinin and 4-aminomethylbenzoic acid. Biomed. Biochim. Acta 1986, 45, 1015–1019. [Google Scholar] [PubMed]
- Kazmirowski, H.G.; Neuland, P.; Landmann, H.; Markwardt, F. Synthesis of antiproteolytic active derivatives of 4-aminomethylbenzoic acid and other structurally similar compounds. Pharmazie 1967, 22, 465–470. [Google Scholar] [PubMed]
- Beyer, S.; Pilgrim, H. The inhibition of endothelial cell proliferation by esters of 4-aminomethylbenzoic acid (pamba). Pharmazie 1991, 46, 597–599. [Google Scholar] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Direm, A.; Altomare, A.; Moliterni, A.; Benali-Cherif, N. Intermolecular interactions of proton transfer compounds: Synthesis, crystal structure and Hirshfeld surface analysis. Acta Cryst. 2015, B71, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Dadda, N.; Nassour, A.; Guillot, B.; Benali-Cherif, N.; Jelsch, C. Charge-density analysis and electrostatic properties of 2-carboxy-4-methylanilinium chloride monohydrate obtained using a multipolar and a spherical-charges model. Acta Cryst. 2014, A68, 452–463. [Google Scholar] [CrossRef]
- Atria, A.M.; Garland, M.T.; Baggio, R. Crystal structure of zwitterionic 4-(ammonio methyl) benzoate: A simple molecule giving rise to a complex supra molecular structure. Acta Cryst. 2014, E70, 385–388. [Google Scholar]
- Sayin, K.; Karakaş, D. Determination of structural, spectral, electronic and biological properties of tosufloxacin boron complexes and investigation of substituent effect. J. Mol. Struct. 2017, 1146, 191–197. [Google Scholar] [CrossRef]
- Üngördü, A.; Tezer, N. The solvent (water) and metal effects on HOMO-LUMO gaps of guanine base pair: A computational study. J. Mol. Graph. Model. 2017, 74, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, R.; Thamaraichelvan, A.; Viswanathan, B. CO2 transformation on the active site of carbonic anhydrase enzyme leading to formation of H2CO3-A biomimetic model through computational study. Turk. Comput. Theor. Chem. 2017, 1, 17–26. [Google Scholar]
- Mirzaei, M. 5–Fluorouracil: Computational Studies of Tautomers and NMR Properties. Turk. Comput. Theor. Chem. 2017, 1, 27–34. [Google Scholar]
- Dennington, R.; Keith, T.; Millam, J. (Eds.) GaussView, version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. (Eds.) Gaussian 09; revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
Figure 1.
Optimized structures of the studied compounds at the M06-2X/6-311++G(d,p) level in the gas phase.
Figure 1.
Optimized structures of the studied compounds at the M06-2X/6-311++G(d,p) level in the gas phase.

Figure 2.
(a) IR spectra of compounds (1)–(3) at the M062X/6-311++G(d,p) level. (b) Experimental FTIR spectra of the corresponding compounds.
Figure 2.
(a) IR spectra of compounds (1)–(3) at the M062X/6-311++G(d,p) level. (b) Experimental FTIR spectra of the corresponding compounds.

Figure 3.
MEP maps of the studied compounds.

Figure 4.
The contour diagram of the studied compounds frontier molecular orbitals.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Selected experimental and calculated structural parameters of compounds (1)–(3).
| Bond Lengths (Å) | Bond Angles (°) | ||||
|---|---|---|---|---|---|
| Experimental | Calculated | Experimental | Calculated | ||
| Compound (1) (R2 = 0.9998) | |||||
| O1-C7 | 1.316 | 1.333 | O1-C7-O2 | 122.5 | 121.6 |
| O2-C7 | 1.203 | 1.212 | O1-C7-C1 | 113.7 | 113.4 |
| N1-C2 | 1.465 | 1.459 | O2-C7-C1 | 123.8 | 124.9 |
| C1-C2 | 1.396 | 1.398 | C2-C1-C6 | 118.7 | 118.7 |
| C1-C7 | 1.499 | 1.486 | C2-C1-C7 | 120.8 | 121.2 |
| C5-C8 | 1.507 | 1.505 | C1-C2-N1 | 121.8 | 121.3 |
| Compound (2) (R2 = 0.9998) | |||||
| O1-C7 | 1.311 | 1.346 | O1-C7-O2 | 123.3 | 122.6 |
| O2-C7 | 1.213 | 1.200 | O1-C7-C1 | 113.9 | 112.7 |
| N1-C8 | 1.486 | 1.494 | O2-C7-C1 | 122.9 | 124.6 |
| C1-C2 | 1.386 | 1.392 | C2-C1-C6 | 119.4 | 120.4 |
| C1-C7 | 1.485 | 1.491 | C2-C1-C7 | 121.2 | 121.7 |
| C4-C8 | 1.505 | 1.507 | C4-C8-N1 | 112.1 | 110.2 |
| Compound (3) (R2 = 0.9993) | |||||
| O1A-C7A | 1.302 | 1.346 | O1A-C7A-O2A | 123.7 | 122.7 |
| O2A-C7A | 1.241 | 1.200 | O1A-C7A-C1A | 115.3 | 112.7 |
| N1A-C8A | 1.499 | 1.503 | O2A-C7A-C1A | 120.9 | 124.6 |
| C1A-C2A | 1.388 | 1.393 | C2A-C1A-C6A | 119.8 | 120.5 |
| C1A-C7A | 1.494 | 1.492 | C2A-C1A-C7A | 119.3 | 121.6 |
| C4A-C8A | 1.516 | 1.504 | C4A-C8A-N1A | 110.6 | 109.5 |
Table 2.
Total energy of the related structures at the M062X/6-311++G(d,p) level in vacuum.
| Moiety | Total Energy (a.u.) | Compound | Total Energy (a.u.) |
|---|---|---|---|
| (A) | −515.608 | Compound (1) | −1276.528 |
| (B) | −515.593 | Compound (2) | −796.100 |
| (C) | −760.764 | Compound (3) | −1276.522 |
| (D) | −280.326 |
Table 3.
Calculated and experimental vibration modes and wavenumbers (cm−1) of the labeled bands in the IR spectra of the mentioned compounds.
Table 3.
Calculated and experimental vibration modes and wavenumbers (cm−1) of the labeled bands in the IR spectra of the mentioned compounds.
| Compound (1) | Compound (2) | Compound (3) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Label | Calculated | Experimental | Mode a | Calculated | Experimental | Mode a | Calculated | Experimental | Mode a |
| 1 | 3832 | 3446 | νOH | 3843 | 3338 | νOH | 3841 | 3337 | νOH |
| 2 | 3212 | 3030 | νNH | 3106 | 3119 | νNH | 3153 | 3118 | νNH |
| 3 | 3024 | νNH | 2696 | νNH | 2873 | νNH | |||
| 4 | 1817 | 1698 | νC = O | 1868 | 1688 | νC = O | 1868 | 1689 | νC = O |
| 5 | 1620 | 1507 | αNH2 | 1618 | 1516 | αNH2, | 1677 | 1514 | αNH2 |
| 1385 | νN = O | ||||||||
| 6 | 1150 | 111512551091 | νCl-O, ωCH, ωNH | 1355 | 1283 | ωCH | 1148 | 111512901086 | νCl-O, ωCH, ωNH |
a Vibration Modes, ν: stretching; α: scissoring; ω: wagging.
Table 4.
Chemical shift values of hydrogen and carbon atoms of the mentioned compounds at the same level of theory.
Table 4.
Chemical shift values of hydrogen and carbon atoms of the mentioned compounds at the same level of theory.
| Assignments | 1H-NMR | Assignments | 13C-NMR | ||||
|---|---|---|---|---|---|---|---|
| (1) | (2) | (3) | (1) | (2) | (3) | ||
| C1 | 135.8 | 151.7 | 152.8 | C2H | - | 9.03 | 9.07 |
| C2 | 151.5 | 153.0 | 153.5 | C3H | 9.52 | 8.07 | 8.10 |
| C3 | 150.0 | 149.1 | 148.3 | C4H | 8.79 | - | - |
| C4 | 163.6 | 160.5 | 158.7 | C5H | - | 9.64 | 9.80 |
| C5 | 164.3 | 154.7 | 157.7 | C6H | 9.15 | 9.47 | 9.50 |
| C6 | 155.5 | 157.4 | 157.3 | C8H′ | 3.04 | 5.03 | 5.51 |
| C7 | 185.6 | 178.3 | 179.0 | C8H″ | 2.67 | 3.67 | 3.66 |
| C8 | 24.1 | 49.4 | 50.7 | C8H′″ | 2.05 | - | - |
| N1H′ | 10.88 | 16.65 | 11.90 | ||||
| N1H″ | 10.79 | 7.02 | 7.30 | ||||
| N1H′″ | 10.72 | 3.19 | 4.29 | ||||
| O1H | 6.48 | 6.64 | 6.34 | ||||
Table 5.
The mentioned quantum chemical parameters for the relevant compounds.
| Compound | µ 1 | α 2 | Δα 2 | β 3 |
|---|---|---|---|---|
| Urea | 1.76 | 2.83 | 10.46 | 5.09 × 10−28 |
| (1) | 4.88 | 16.48 | 32.52 | 1.80 × 10−27 |
| (2) | 4.04 | 15.56 | 30.68 | 1.03 × 10−27 |
| (3) | 4.40 | 16.22 | 33.04 | 8.88 × 10−28 |
1 in Debye, 2 in Å3, 3 in cm5/esu.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Direm, A.; Sayın, K. A Comparative Theoretical and Spectroscopic Study of Aminomethylbenzoic Acid Derivatives as Potential NLO Candidates. Chem. Proc. 2021, 3, 102. https://0-doi-org.brum.beds.ac.uk/10.3390/ecsoc-24-08099
AMA Style
Direm A, Sayın K. A Comparative Theoretical and Spectroscopic Study of Aminomethylbenzoic Acid Derivatives as Potential NLO Candidates. Chemistry Proceedings. 2021; 3(1):102. https://0-doi-org.brum.beds.ac.uk/10.3390/ecsoc-24-08099
Chicago/Turabian StyleDirem, Amani, and Koray Sayın. 2021. "A Comparative Theoretical and Spectroscopic Study of Aminomethylbenzoic Acid Derivatives as Potential NLO Candidates" Chemistry Proceedings 3, no. 1: 102. https://0-doi-org.brum.beds.ac.uk/10.3390/ecsoc-24-08099