
Molecular Docking Studies on Various Food Grade Dyes as a Potential Inhibitor of COVID-19 †


1
Department of Pharmaceutical Sciences and Technology, Birla Institute of Technology, Mesra, Jharkhand 835215, India
2
Departamento de Química Orgánica, Facultad de Ciencias‐Campus de Lugo, Universidad De Santiago De Compostela, 15705 Santiago de Compostela, Spain
3
Department of Chemical Engineering and Technology, Birla Institute of Technology, Mesra, Jharkhand 835215, India
*
Author to whom correspondence should be addressed.
†
Presented at the 24th International Electronic Conference on Synthetic Organic Chemistry,
15 November–15 December 2020; Available online: https://ecsoc-24.sciforum.net/.
Chem. Proc. 2021, 3(1), 111; https://0-doi-org.brum.beds.ac.uk/10.3390/ecsoc-24-08353
Published: 14 November 2020
(This article belongs to the Proceedings of The 24th International Electronic Conference on Synthetic Organic Chemistry)
Abstract
:In December 2019, the Coronavirus disease-2019 (COVID-19) virus emerged in Wuhan, China. The first resolved COVID-19 crystal structure (main protease) has been developed and various repurposing activities are in process. In this study, a knowledge gap in relation to COVID-19, with the previously known fatal Coronavirus (CoV) epidemics, Severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS) CoVs, is covered by the investigation of sequence statistics, molecular modelling, virtual screening, docking, and sequence comparison statistics of the COVID-19 main protease. COVID-19 main protease Mpro formed a sequence similarity group with SARS CoV that was distant from MERS CoV. The identity % was 96 and 51 for COVID-19/SARS and COVID-19/MERS CoV sequence comparisons, respectively. We used molecular docking and a molecular interaction approach to identify small-molecules that bind to the isolated Viral S-protein at its host receptor region. These molecules have good solubility and pharmacodynamics properties. They also obey Lipinski’s rule, which makes them promising compounds to pursue further biochemical and cell-based assays to explore their potential for use against COVID-19. We hypothesize that the top score identified molecules that may be used to limit viral recognition of host cells and/or disrupt host-virus interactions. A ranked list of selected compounds is given that can be tested experimentally.
1. Introduction
On the penultimate day of 2019, health officials at the Wuhan Municipal Health Commission (Hubei Province, Wuhan, China) reported an occurrence of concentrated pneumonia in the city of Wuhan. Shortly after reporting the outbreak, the Chinese Centre for Disease Control (Chinese CDC) and local Chinese health workers determined that the cause of the outbreak was a novel coronavirus, i.e., nCov-2019 [1,2,3]. On 11 March 2020, WHO declared it as a pandemic. The symptoms of Coronavirus disease-2019 (COVID-19) infection are mild respiratory symptoms and a fever that occurs on an average of 5–6 days after infection (mean incubation period 5–6 days, range 1–14 days) [4,5,6]. The current treatment options are use of antivirals and antimalarials. The first available crystal structure of COVID-19 proteins was Mpro, which was published in February 2020 (Protien data bank (PDB ID) 6lu7). In this study, the first virtual screening study against the first known COVID-19 was performed. The obtained results will help in identifying some potential inhibitors to combat the recent dangerous COVID-19. We propose to use food grade dyes that could acts as a treatment option in case of COVID-19 patients. We have used computational methods, e.g., molecular docking, to evaluate the activity as well as the interactions.
2. Materials and Methods
2.1. Retrieval of Mpro Sequences
The NCBI GenBank or GISAID (Available Online: https://www.gisaid.org/ (accessed on 10 October 2020)) were used to obtain the COVID-19 sequences. SARS Coronavirus (CoV) and MERS CoV sequences were obtained from the GenBank [7,8].
2.2. Sequence Alignment and Multiple Sequence Comparisons
Pairwise and multiple sequence comparisons of Mpro were done using CLC genomics software (Qiagen Inc., USA). The sequence comparison matrix was generated, including the number of gaps, number of different residues, and identity %.
Sequences alignments of Mpro were from SARS CoV, MERS CoV, and COVID-19.
|
| Identities 294/306 (96%) |
| SARS Mpro 2AMD SGFRKMAFPSGKVEGCMVQVTCGTTTLNGLWLDDTVYCPRHVICTAEDMLNPNYEDLLIR 65 |
| COVID-19 Mpro YP_009725301 ...................................................................................................V........................S.................................... 60 |
| SARS Mpro 2AMD KSNHSFLVQAGNVQLRVIGHSMQNCLLRLKVDTSNPKTPKYKFVRIQPGQTFSVLACYNG 125 |
| COVID-19 Mpro YP_009725301........N............................................................................V.K.........A........................................................... 120 |
| SARS Mpro 2AMD SPSGVYQCAMRPNHTIKGSFLNGSCGSVGFNIDYDCVSFCYMHHMELPTGVHAGTDLEGK 185 |
| COVID-19 Mpro YP_009725301..............................................................F.................................................................................................N 180 |
| SARS Mpro 2AMD FYGPFVDRQTAQAAGTDTTITLNVLAWLYAAVINGDRWFLNRFTTTLNDFNLVAMKYNYE 245 |
| COVID-19 Mpro YP_009725301...........................................................................V....................................................................................... 240 |
| SARS Mpro 2AMD PLTQDHVDILGPLSAQTGIAVLDMCAALKELLQNGMNGRTILGSTILEDEFTPFDVVRQC 305 |
| COVID-19 Mpro YP_009725301..............................................................S.................................................AL............................................. 300 |
| SARS Mpro 2AMD SGVTFQ 311 |
| COVID-19 Mpro YP_009725301 ...... 306 |
|
| Identities 157/310 (51%) |
| MERS Mpro 5C3N SGLVKMSHPSGDVEACMVQVTCGSMTLNGLWLDNTVWCPRHVMCPADQLSDPNYDALLIS 60 |
| COVID-19 Mpro YP_009725301.........FR........AF...........K........G...............TT..............DV.......Y............I.......TSEDMLN.........ED..........R 60 |
| MERS Mpro 5C3N MTNHSFSVQKHIGAPANLRVVGHAMQGTLLKLTVDVANPSTPAYTFTTVKPGAAFSVLAC 120 |
| COVID-19 Mpro YP_009725301 KS.......N......L.......---AGNVQ........I.......S.......NCV........K.......T........K.......K......K......VRIQ.......QT..... 117 |
| MERS Mpro 5C3N YNGRPTGTFTVVMRPNYTIKGSFLCGSCGSVGYTKEGSVINFCYMHQMELANGTHTGSAF 180 |
| COVID-19 Mpro YP_009725301...........S.......S........VYQCA...........F............N............FNIDYDCVS..........H........PT......V.......A.......TDL 177 |
| MERS Mpro 5C3N DGTMYGAFMDKQVHQVQLTDKYCSVNVVAWLYAAILNGCAWFVKPNRTSVVSFNEWALAN 240 |
| COVID-19 Mpro YP_009725301 E.......NF........P.......V....R....TA....AAG.....TTIT......L............VI.....DR.....LNRFT....TLND.....LV....MKY 237 |
| MERS Mpro 5C3N QFTEFVGTQSVDM---LAVKTGVAIEQLLYAIQQLY-TGFQGKQILGSTMLEDEFTPEDV 296 |
| COVID-19 Mpro YP_009725301 NY-.....PLTQDH......ILGP.....SAQ......I.....VLDMCASLKE.....LQN.....MN.....RT...........AL..............F.. 296 |
| MERS Mpro 5C3N NMQIMGVVMQ 306 |
| COVID-19 mpro YP_009725301 VR.CS..TF. 306 |
2.3. Docking
The structure of COVID-19 virus Mpro in complex with N3 provides a model for identifying lead inhibitors to target COVID-19 virus Mpro through in silico screening. We used a molecular docking approach to predict the binding energy and inhibition constants of various food grade dyes under study [9,10]. We docked our ligands into the main protease of COVID-19 and screened them for their activity against COVID-19.
2.4. Predictive ADME Studies
Predictive Absorption, Distribution, Metabolism, Excretion (ADME) studies were performed by using SWISS tools*, an online tool that requires the structure or the SMILES for calculating the parameters.
The test compounds were built within the window by using the drawing tools of the online server, otherwise SMILES could be directly copied instead of drawing the structures [11]. To assure a drug-like pharmacokinetic profile in rational drug designing, predictive ADME calculations are done on the basis of Lipinski’s rule of five.
2.5. Toxicity
The toxicity of the molecules were predicted by using Toxtree [12], a free offline tool available for the prediction of toxicity. It requires the SMILES format of structures to calculate the toxicity.
The SMILES format of the compounds were pasted in the chemical identifier bar, and then their toxicity was estimated on the basis of creamer rules. The compounds were categorized into three classes, i.e., Low (Class I), Intermediate (Class II), and High (Class III).
3. Results and Discussions
3.1. Docking
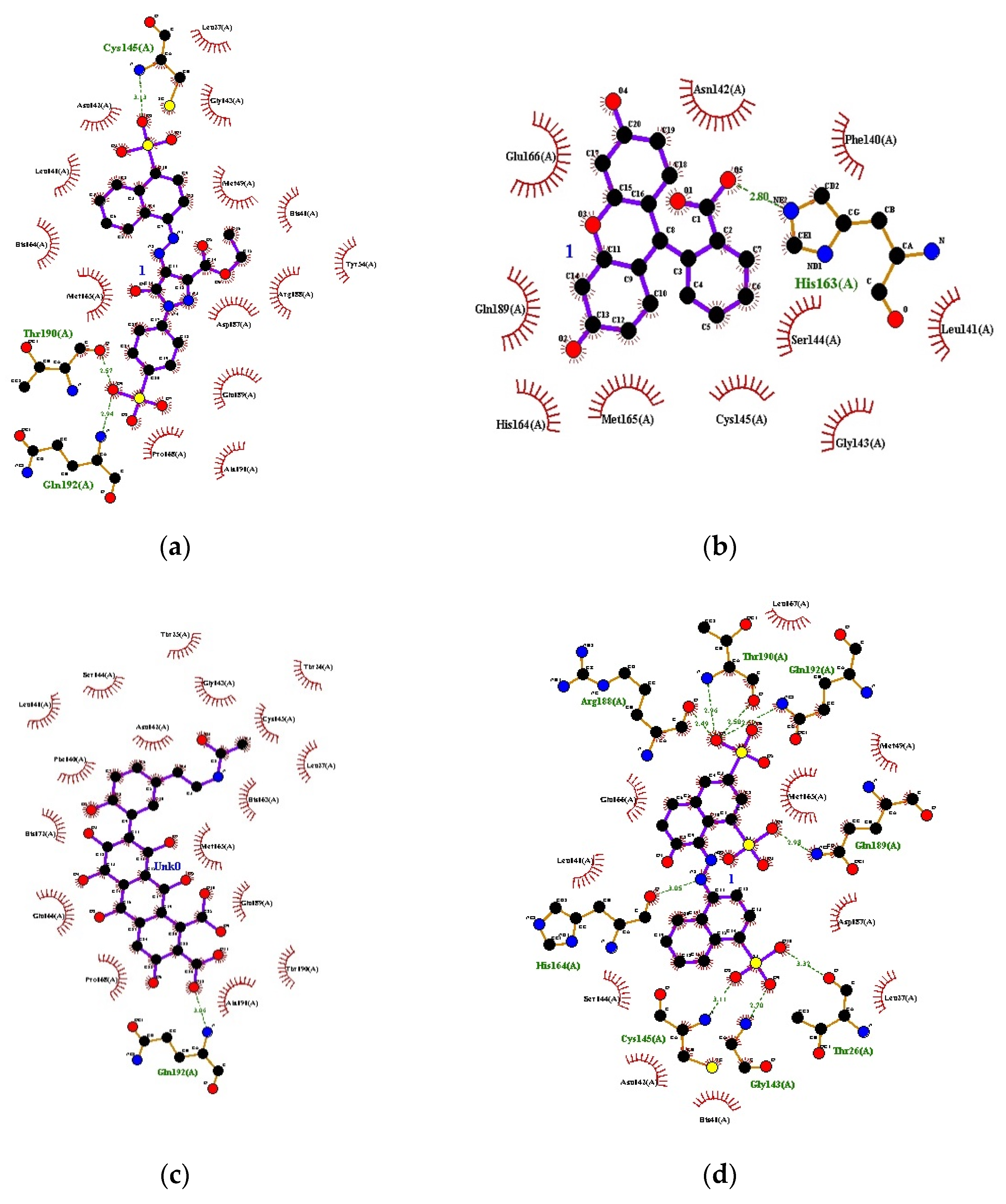
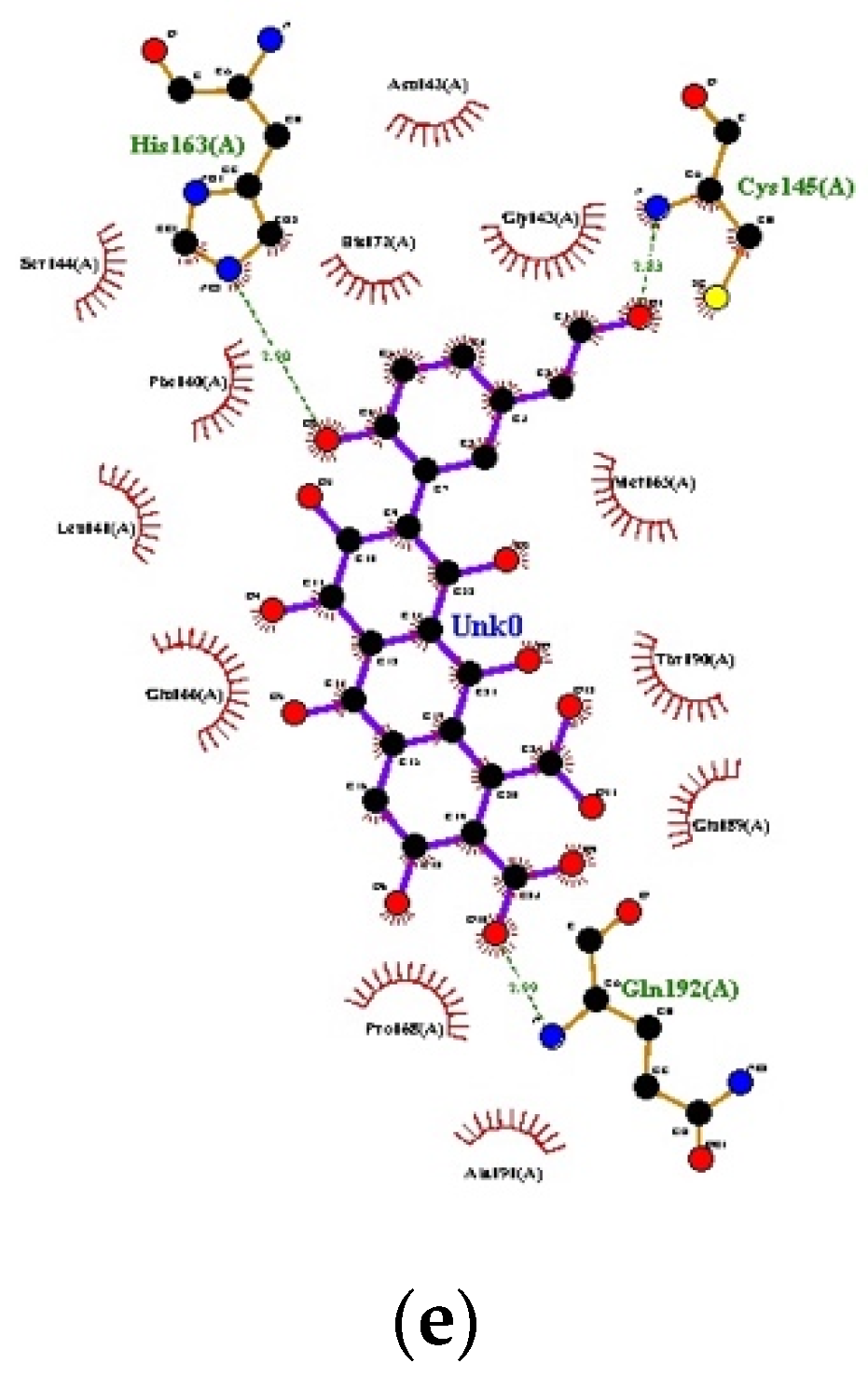
The PDB ID of protein used was 6LU7, which was retrieved from the protein data bank. The validation of the model that was performed redocked the internal ligand/inhibitor into the active site of the macromolecule. The individual ligands were then prepared in Auto Dock 4.2.6 software, as per standard protocols, and docking was carried out. The results are listed below Table 1 and Figure 1.
3.2. Predictive ADME Studies
Analysis of all the compounds was done for the physicochemically and pharmacokinetically important descriptors using SWISS tools. In order to predict the drug-alike properties of molecules, these major descriptors were required:
- ⮚
- Molecular weight (mol MW) (150–650)
- ⮚
- Octanol/water partition coefficient (Log Po/w) (−2–6.5)
- ⮚
- Hydrogen bond donor (≤5)
- ⮚
- Hydrogen bond acceptor (≤10)
- ⮚
- Human oral absorption percentage (≥80% is high, ≤25% is poor)
The entire set of compounds showed appreciable values for the properties analyzed, as well as exhibited drug-like aspects based on Lipinski’s rule of five. The results are summarized in Table 2.
3.3. Toxicity
Toxicity prediction of the compounds is necessary before further development. The toxicity is predicted by using Craemer rules. It categorizes the compounds into the classes, i.e., Low (Class I), Intermediate (Class II), and High (Class III), depending on its toxicity index. The categories are based on different thresholds of toxicological concern, as follows:
- ⮚
- Class I—1800 (30 µg/kg bw/d)
- ⮚
- Class II—540 (9 µg/kg bw/d)
- ⮚
- Class III—90 (1.5 µg/kg bw/d)
The results are summarized in Table 3.
From the ADME studies, it was found that only a few compounds followed all the parameters for being a suitable drug candidate, but all the other compounds violated the parameters by a few factors, which, on further modifications, can be modified to promising drug candidates. The toxicity studies suggest that the therapeutic range of some compounds is very narrow, whereas some have wide therapeutic ranges, and these can be modified as per the purpose. The modifications required can be taken as a future perspective to develop these compounds as promising drug candidates.
4. Conclusions
Researchers are now focusing mainly on synthetic protease inhibitors, but natural compounds have always been found to be better than their synthetic counterparts. As natural chemists, we tried to focus on untouched natural drugs that could provide better drug therapies in the future. As per our study, the sequence identity % was 96 and 51 for COVID-19/SARS and COVID-19/MERS CoV, respectively. Docking studies revealed that Orange B (−10.35 kcal/mol) and Cochineal Red A (−9.52 kcal/mol) had the best binding affinity with the receptor. They had low GI absorption but showed no BLOOD BRAIN BARRIER (BBB) permeation activity. They obeyed the Lipinski rule and bioavailability score was 0.11 and showed drug-like aspects. Cochineal Red A was classified under Low Class I toxicity. Erythrosine, Laccaic Acid A, Laccaic Acid B, Azorubine, and Quinoline yellow also had a comparable binding affinity. These two molecules/compounds proved to be a good inhibitor against the COVID-19 main protease. Further MD simulation studies can be performed to mimic the interaction of the molecules with the receptor. These molecules can further be studied for their in vitro and in vivo activity. This work may be able to pave a new path for the development of potential drugs using food grade dyes and for the selection of compounds, as well as designing new scaffolds or novel combinatorial libraries of analogs/derivatives; however, before coming to any outcome of an in-silico study, proper in-vitro and in-vivo research works should be performed.
Author Contributions
Authors contributed equally: Conceptualization, P.D. and M.G.; Software P.D.; Supervision M.G. and A.M.; Manuscript writing P.D., M.G., A.M. and J.A.S. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors would like to thank the Head, Department of Pharmaceutical Sciences and Technology, BIT, Mesra for providing the research facilities for performing the current study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Xu, X.; Chen, P.; Wang, J.; Feng, J.; Zhou, H.; Li, X.; Zhong, W.; Hao, P. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci. China Life Sci. 2020, 63, 457–460. [Google Scholar] [CrossRef] [PubMed]
- Peiris, J.S.M.; Lai, S.T.; Poon, L.L.M.; Guan, Y.; Yam, L.Y.C.; Lim, W.; Nicholls, J.; Yee, W.K.S.; Yan, W.W.; Cheung, M.T.; et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 2003, 361, 1319–1325. [Google Scholar] [CrossRef]
- Li, W.; Wong, S.-K.; Li, F.; Kuhn, J.H.; Huang, I.-C.; Choe, H.; Farzan, M. Animal Origins of the Severe Acute Respiratory Syndrome Coronavirus: Insight from ACE2-S-Protein Interactions. J. Virol. 2006, 80, 4211–4219. [Google Scholar] [CrossRef] [PubMed]
- Zaki, A.; Van Boheemen, S.; Bestebroer, T.; Osterhaus, A.; Fouchier, R. Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- Desenclos, J.C.; Van der Werf, S.; Bonmarin, I.; Levy-Bruhl, D.; Yazdanpanah, Y.; Hoen, B. Introduction of SARS in France, March–April, 2003. Emerg. Infect. Dis. 2004, 10, 195. [Google Scholar] [CrossRef] [PubMed]
- Guarner, J. Three Emerging Coronaviruses in Two Decades. Am. J. Clin. Pathol. 2020, 153, 420–421. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, e00127-20. [Google Scholar] [CrossRef] [PubMed]
- Hilgenfeld, R. From SARS to MERS: Crystallographic studies on coronaviral pro- teases enable antiviral drug design. FEBS J. 2014, 281, 4085–4096. [Google Scholar] [CrossRef] [PubMed]
- Kandeel, M.; Altaher, A.A.; Alnazawi, M. Molecular Dynamics and Inhibition of MERS CoV Papain-like Protease by Small Molecule Imidazole and Aminopurine Derivatives. Lett. Drug Des. Discov. 2019, 16, 584–591. [Google Scholar] [CrossRef]
- Li, Y.-H.; Hu, C.-Y.; Wu, N.-P.; Yao, H.; Li, L. Molecular Characteristics, Functions, and Related Pathogenicity of MERS-CoV Proteins. Engineering 2019, 5, 940–947. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://www.swissadme.ch (accessed on 15 October 2020).
- Patlewicz, G.; Jeliazkova, N.; Safford, R.J.; Worth, A.P.; Aleksiev, B. An evaluation of the implementation of the Cramer classification scheme in the Toxtree software. SAR QSAR Environ. Res. 2008, 19, 495–524. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Docking interactions: (a) Orange B, (b) Cochineal Red A, (c) Erythrosine, (d) Laccaic acid A, (e) Laccaic acid B.
Figure 1.
Docking interactions: (a) Orange B, (b) Cochineal Red A, (c) Erythrosine, (d) Laccaic acid A, (e) Laccaic acid B.



{kind=link}
{kind=link}
Table 1.
List of ligands with binding energy and inhibition constants.
| S. No. | Ligands | 1st Run | 2nd Run | 3rd Run | |||
|---|---|---|---|---|---|---|---|
| Binding Energy | Inhibition Constant | Binding Energy | Inhibition Constant | Binding Energy | Inhibition Constant | ||
| 1 | DG01 | −10.35 | 26.12 nM | −9.99 | 47.43 nM | −9.91 | 54.73 nM |
| 2 | DG02 | −9.52 | 104.45 nM | −9.07 | 225.6 nM | −8.99 | 259.33 nM |
| 3 | DG03 | −9.43 | 121.71 nM | −9.29 | 154.77 nM | −9.28 | 158.05 nM |
| 4 | DG04 | −9.1 | 214.18 nM | −8.98 | 261.41 nM | −8.66 | 447.14 nM |
| 5 | DG05 | −9.00 | 251.81 nM | −8.89 | 305.47 | −8.87 | 314.38 nM |
| 6 | DG06 | −8.86 | 322.93 nM | −8.63 | 472.32 nM | −8.63 | 475.09 nM |
| 7 | DG07 | −8.53 | 555.76 nM | −8.53 | 561.87 nM | −8.52 | 571.48 nM |
| 8 | DG08 | −7.97 | 1.44 μM | −7.6 | 2.67 uM | −7.11 | 6.1 uM |
| 9 | DG09 | −7.86 | 1.73 μM | −7.72 | 2.2 uM | −7.63 | 2.54 uM |
| 10 | DG10 | −7.81 | 1.87 μM | −7.81 | 1.87 uM | −7.80 | 1.92 uM |
| 11 | DG11 | −7.42 | 3.63 μM | −7.33 | 4.24 uM | −7.28 | 4.6 uM |
| 12 | DG12 | −7.35 | 4.12 μM | −6.33 | 22.87 uM | −6.30 | 24.27 uM |
| 13 | DG13 | −7.34 | 4.14 μM | −7.28 | 4.62 uM | −7.32 | 4.32 uM |
| 14 | DG14 | −6.14 | 31.82 μM | −6.13 | 31.97 uM | −6.12 | 32.46 uM |
| 15 | DG15 | −6.24 | 26.75 μM | −4.79 | 307.68 uM | −5.78 | 58.44 uM |
Table 2.
SWISS ADME for compounds DG01-15.
| Compounds | DG01 | DG02 | DG03 | DG04 | DG05 | DG06 | DG07 | DG08 | DG09 | DG10 | DG11 | DG12 | DG13 | DG14 | DG15 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Properties | |||||||||||||||
| MW | 546.53 | 538.53 | 835.89 | 537.43 | 496.38 | 458.46 | 273.29 | 561.69 | 539.4 | 314.25 | 468.42 | 408.41 | 422.39 | 495.39 | 538.41 |
| HBA | 11 | 11 | 5 | 12 | 12 | 9 | 3 | 7 | 14 | 7 | 12 | 9 | 8 | 12 | 13 |
| HBD | 3 | 4 | 2 | 8 | 8 | 3 | 0 | 3 | 9 | 4 | 3 | 3 | 4 | 8 | 7 |
| MR | 138.93 | 123.99 | 139.61 | 131.61 | 120.15 | 113.81 | 79.7 | 149.36 | 128.28 | 77.74 | 109.69 | 96.31 | 101.04 | 121.7 | 129.89 |
| TPSA | 208.86 | 229.71 | 75.99 | 238.99 | 230.12 | 170.45 | 47.03 | −1.14 | 273.21 | 132.13 | 220.19 | 170.45 | 183.7 | 235.91 | 236.19 |
| LOG Po/w | 1.54 | 1.37 | 5.23 | −1.25 | −1.25 | 2.8 | 2.05 | 2.94 | −4 | 0 | 0.32 | 2.02 | −0.18 | −0.71 | −0.31 |
| Solubility (mg/mL) | 1.13 × 10−2 | 6.97 × 10−2 | 4.44 × 10−7 | 6.15 × 10−3 | 7.05 × 10−3 | 4.58 × 10−3 | 6.63 × 10−3 | 4.22 × 10−6 | 3.51 × 10−1 | 4.22 × 10−2 | 5.74 × 10−1 | 5.59 × 10−2 | 1.63 × 10−1 | 5.74 × 10−4 | 2.90 × 10−3 |
| G.I absorption | Low | Low | High | Low | Low | Low | High | Low | Low | High | Low | Low | Low | Low | Low |
| BBB Permeant | No | No | No | No | No | No | Yes | No | No | No | No | No | No | No | No |
| CYP1A2 | No | No | Yes | No | Yes | No | Yes | No | No | No | No | No | No | Yes | Yes |
| CYP2D6 | No | No | No | No | No | No | No | No | No | No | No | No | No | No | No |
| Veber | No | Yes | Yes | No | No | No | Yes | No | No | Yes | No | No | No | No | No |
| Lipinski | No | No | No | No | No | Yes | Yes | No | No | Yes | No | Yes | Yes | No | No |
| Bioavailability Score | 0.11 | 0.11 | 0.17 | 0.11 | 0.11 | 0.11 | 0.55 | 0.11 | 0.11 | 0.56 | 0.11 | 0.11 | 0.11 | 0.11 | 0.11 |
Molecular weight (MW), hydrogen bond acceptor (HBA), hydrogen bond donor (HBD), total polar surface area (TPSA), octanol/water partition coefficient (Log Po/w), aqueous solubility (Log S), molar refractivity (MR), Cytochrome P450 1A2 (CYP1A2), Cytochrome P450 2D6 (CYP2D6).
Table 3.
Toxicity of the compounds DG01-15.
| Compounds | Toxicity Class |
|---|---|
| DG01 | High Class III |
| DG02 | Low Class I |
| DG03 | High Class III |
| DG04 | High Class III |
| DG05 | High Class III |
| DG06 | Low Class I |
| DG07 | High Class III |
| DG08 | Low Class I |
| DG09 | High Class III |
| DG10 | High Class III |
| DG11 | Low Class I |
| DG12 | Low Class I |
| DG13 | Low Class I |
| DG14 | High Class III |
| DG15 | High Class III |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Dagur, P.; Seijas, J.A.A.; Mishra, A.; Ghosh, M. Molecular Docking Studies on Various Food Grade Dyes as a Potential Inhibitor of COVID-19. Chem. Proc. 2021, 3, 111. https://0-doi-org.brum.beds.ac.uk/10.3390/ecsoc-24-08353
AMA Style
Dagur P, Seijas JAA, Mishra A, Ghosh M. Molecular Docking Studies on Various Food Grade Dyes as a Potential Inhibitor of COVID-19. Chemistry Proceedings. 2021; 3(1):111. https://0-doi-org.brum.beds.ac.uk/10.3390/ecsoc-24-08353
Chicago/Turabian StyleDagur, Pankaj, Julio A. A. Seijas, Amarnath Mishra, and Manik Ghosh. 2021. "Molecular Docking Studies on Various Food Grade Dyes as a Potential Inhibitor of COVID-19" Chemistry Proceedings 3, no. 1: 111. https://0-doi-org.brum.beds.ac.uk/10.3390/ecsoc-24-08353