
Synthesis of Alkenyl Sulfides Catalyzed by CuNPs/TiO2. A Theoretical-Computational Analysis †


Instituto de Química del Sur (INQUISUR-CONICET), Departamento de Química, Universidad Nacional del Sur, Av. Alem 1253, B8000CPB Bahia Blanca, Argentina
*
Authors to whom correspondence should be addressed.
†
Presented at the 24th International Electronic Conference on Synthetic Organic Chemistry, 15 November–15 December 2020; Available online: https://ecsoc-24.sciforum.net/ .
Chem. Proc. 2021, 3(1), 120; https://0-doi-org.brum.beds.ac.uk/10.3390/ecsoc-24-08323
Published: 14 November 2020
(This article belongs to the Proceedings of The 24th International Electronic Conference on Synthetic Organic Chemistry)
Abstract
:Copper nanoparticles (CuNPs) supported on TiO2 have shown to effectively catalyze the synthesis of Z-alkenyl sulfides from activated alkynes and thiol derivatives, through an anti-Markovnikov process. Activated alkynes bearing an adjacent electron-withdrawing group gave good to excellent products conversions, while the unactivated alkynes did not react. In order to give an explanation to the reactivity observed from the experimental results and to know the most simple and representative structure of the catalyst, density functional theory (DFT) computational studies have been applied.
1. Introduction
Alkenyl sulfides are recognized constituents of the sulfur containing organic compounds and they are of great interest because they can be used as versatile building blocks in organic synthesis [1,2]. The alkyne hydrothiolation, is a simple approach to produce alkenyl sulfides from thiols and alkynes [3,4], which in principle can lead to one of the regio- and stereoisomeric alkenyl sulfides through a Markovnikov orientation, E linear and Z linear, or give mixtures of them through an anti-Markovnikov orientation (Scheme 1) [5,6].
Regarding thiols, our research group has been reported a systematic and straightforward procedure for the synthesis of thiols with potential applications in materials chemistry [7,8]. On the other hand, in the last years, we have been working in the development of methodologies based on the use of supported copper nanocatalysts [9,10,11,12,13], and the copper-based catalytic systems could activate both the alkyne and the thiol in this reaction [4]. On this basis, we report our results about the theoretical study of hydrothiolation reaction between different alkynes and thiols derivatives catalyzed by copper nanoparticles (CuNPs) supported on TiO2 (CuNPs/TiO2) in dichloromethane (DCM). Activated alkynes bearing an adjacent electron-withdrawing group gave conversions from good to excellent of anti-Markovnikov Z-alkenyl sulfides, while the unactivated alkynes did not react. To explain this, a theoretical study was carried out with different density functional theory (DFT) methods for deriving partial atomic charges for all the alkynes. Furthermore, since we assumed that the reaction starts when the alkyne is activated by the copper, we computationally modelled the active copper nanocatalyst structure.
2. Methods
2.1. General
All moisture sensitive reactions were carried out under a nitrogen atmosphere. Anhydrous tetrahydrofuran was freshly distilled from sodium/benzophenone ketyl. All starting materials were of the best available grade (Aldrich, Merck, Alfa Aesar) and were used without further purification. Commercially available copper(II) chloride dihydrate was dehydrated upon heating in oven (150 °C, 45 min) prior to use for the preparation of CuNPs. Column chromatography was performed with Merck silica gel 60 (0.040–0.063 μm, 240–400 mesh) and hexane/EtOAc as eluent. Reactions were monitored by thin-layer chromatography on silica gel plates (60F-254) visualized under UV light and/or using FeCl3 in water as stain. Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker ARX-300 spectrometer using CDCl3 or CD3OD as solvents.
The CuNPs/TiO2 catalyst was prepared following the procedure previously reported in our group [13].
2.2. Computational Procedure
Calculations were performed applying DFT [14] methods with the ORCA [15] program and Gaussian09 [16,17] (for the NBO analysis). We employed three members of the Minnesota family functionals (M06, M06-L, M06-2X) [18,19] and the hybrid functional B3LYP [20,21,22], together with def2-TZVP(-f) and 6-311+G* basis sets, respectively. The D3BJ Grimme’s dispersion correction [20,23,24,25] was applied as implemented in ORCA. The initial analysis for some structures was performed with the BP [26,27] and/or PBE [27,28] functionals with the def2-SVP basis set. Characterization of all stationary points was done by Hessian matrix calculations of geometries obtained with full optimization for the minimums. The energies in solution were obtained with full geometry optimization within the conductor-like polarizable continuum model (CPCM) [29]. Partial atomic charges were derived from Mulliken, CHELPG, and NBO analysis.
3. Results and Discussion
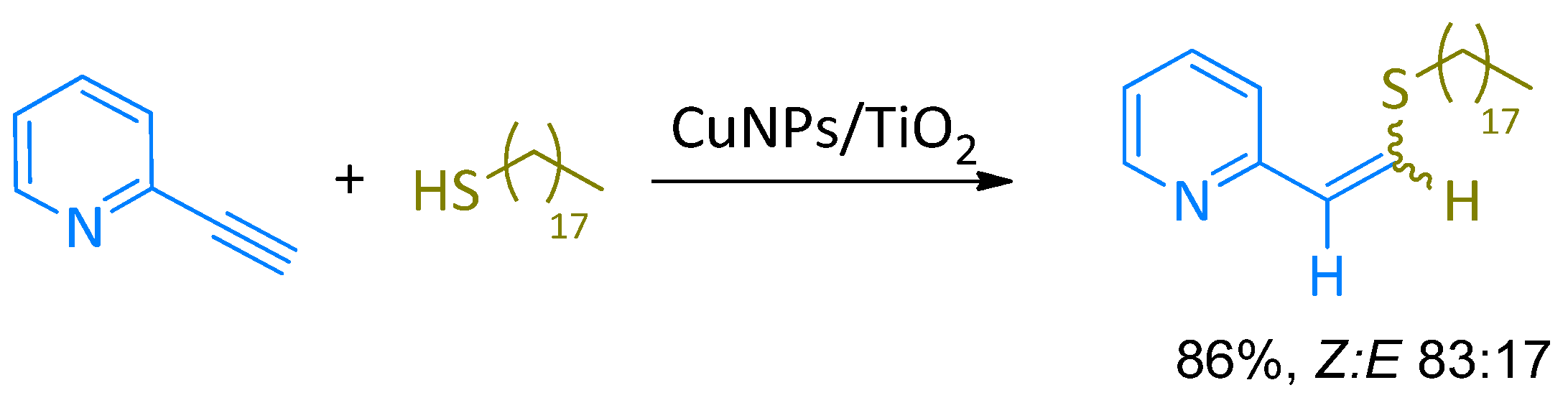
The reaction between 2-ethynylpyridine and 1-octadecanethiol, as model substrates, was tested for the hydrothiolation reaction in the presence of CuNPs supported on a variety of inorganic materials using different solvents and reaction temperatures. In all the tested cases, the regioselectivity was excellent, providing only the anti-Markovnikov adduct but the stereoselectivity was moderate giving an average Z:E isomers. The best results were obtained using TiO2 as support, obtaining a conversion of 86% and a selectivity of 83:17 Z/E (Scheme 2). This result could suggest the requirement for the presence of Lewis acid (Ti4+) and/or Lewis basic (O2−) sites on the support for the reaction to take place. Control experiments carried out in the absence of the CuNPs/TiO2 catalyst gave no conversion of the starting alkyne to the desired product.
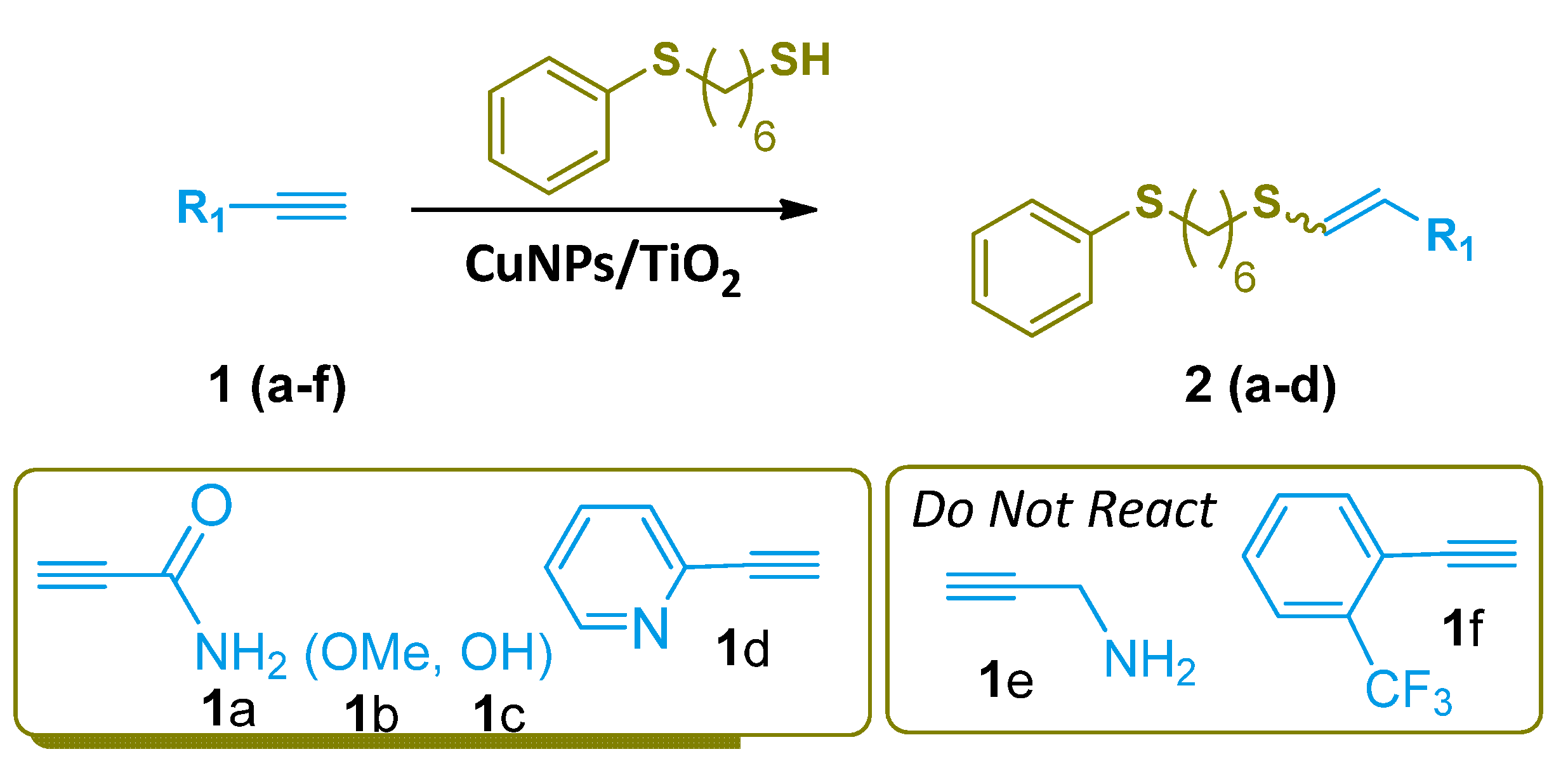
The scope of the hydrothiolation reaction was studied for a series of terminal alkynes, with 6-(phenylthio)hexane-1-thiol. The starting catechol-thiol was synthesized according to the procedure reported by Mancebo-Aracil et al.
As shown in Scheme 3, the alkyne and the thiol reacted in the presence of the CuNPs/TiO2 in DCM as solvent. Activated alkynes bearing an adjacent electron-withdrawing group (1a–d) gave the anti-Markovnikov alkenyl sulfides 2a–d as the main reaction products with good to excellent yield. In all cases, the Z-alkenyl sulfides were the majority stereoisomer obtained. On the other hand, electron-rich alkynes (1e,f) also were tested, but they did not react.
With the aim to understand the experimental results and begin a study of the reaction mechanism, we made a computational analysis with the ORCA and Gaussian09 software packages, the last one only for NBO analysis. Depending on the system under study, the density functional theory (DFT) calculations were performed with different functionals and basis sets. The energies in solution were obtained with the CPCM model.
It is known that partial atomic charges are values that allow quantizing the distribution of electron density in a molecule in order to understand its reactivity. Since there is no way to define the limits of an atom it is not possible to define an atomic charge exactly, however, through computational modeling, using combinations of functionals and basis sets, atomic contributions or atomic partial charges can be derived according to different population analyses. The methods employed to analyze electron density can be grouped into three groups: a—Methods based on wave function analysis (Mulliken, Natural Population Analysis); b—Methods based on a least-squares fit to the electrostatic potential of the molecule (such as CHELPG and Merz–Singh–Kollman (MK)) and c—Methods based on electron density (Atoms In Molecules). Mulliken [30] population analysis is the simplest way to determine partial charges. But this method is highly dependent on the basis set employed, and becomes particularly problematic when diffuse functions are used, besides, it overestimates the covalent character of the bonds. In NBO [31] analysis, the base orbitals are transformed by natural atomic orbitals (NAOs) and hybrid natural orbitals (NHOs) into natural bonding orbitals (NBOs). These orbitals correspond to Lewis structures where two-center bonds and nonbonding electrons are located. The key difference between them is that Mulliken charges are calculated through original basis functions, while NBO charges are derived based on natural atomic orbitals (NAOs). In the CHELPG [32] (CHarges from ELectrostatic Potentials using a Grid based method), charges are adjusted to reproduce the molecular electrostatic potential (MEP) at a set of points around the molecule.
Based on that, we initially made an analysis of these three methods to obtain the partial charge distribution on the alkyne 1a. As can be seen from Table 1, Mulliken charges are shown to be highly dependent of the functional utilized. For example, the charge on C4 was 0.73 (M06), 0.46 (M06-2X), and 0.79 (M06-L), all of them in gas phase. Unlike the Mulliken method, the CHELPG charges shown almost no dependence with the theory level, the charge on C4 was 0.86 (M06), 0.84 (M06-2X), and 0.83 (M06-L), in gas phase. NBO was calculated with Gaussian09, using the B3LYP/6-311+G** method, this basis is very similar to TZVP. In principle, the obtained charges distribution was satisfactory, although the net values are all lower (C4 = 0.59) than those obtained with CHELPG. We also included implicit solvent in the calculations (CPCM=DCM) and observed a redistribution of the charge, particularly on the centers most affected by differences in electronegativity, but this did not imply a noticeable change. For example, using M06-L/TZVP(-f), the CHELPG charges on C4 and O5 were 0.83 and −0.53 in gas phase, and 0.88 and −0.65 with solvent, respectively. The same was observed with the NBO method, although the differences between the charges were minor than with CHELPG method.
As shown Table 2, Table 3, Table 4, Table 5 and Table 6, to continue with the study of the charges of the other alkynes (1b–f), we used as computational calculation methods M06-L and B3LYP with the bases sets already mentioned, since they are the functional ones with which the subsequent mechanistic research will continue. Only the CHELPG and NBO charges were derived, since the former are the best ones that fit with the proposed system and the latter will be used as a control during the computational study.
All alkynes (1a–d) shown to be reactive in the hydrothiolation, are directly attached to a sp2 carbon, and it is known that this makes them more reactive towards nucleophiles. It is interesting to note that CHELPG calculated charges (M06-L/TZVP(-f), CPCM=DCM) on all these carbons are values very positive: 0.88 (C4) for alkyne 1a, 1.00 (C4) for 1b, 0.86 (C4) for 1c, and 0.98 (C5) for 1d (Table 1, Table 2, Table 3 and Table 4).
In the case of the unreacted alkynes (1e,f); in 1e, the triple bond is attached to a sp3 carbon, this could clearly affect its reactivity. However, in the alkyne 1f the triple bond is directly attached to a sp2 carbon. If we observed the CHELPG charge distribution on the carbon directly attached to the triple bond, is 0.68 (C3) for 1e and 0.24 (C4) for 1f, respectively (Table 5 and Table 6), values much less positive than in the case of alkynes 1a–d, and even more in the case of 1f, making the triple bond less reactive to a nucleophilic attack.
This shows that being able to adequately estimate partial charge distribution, can be a molecular reactivity parameter to be considered, along with others, when we computationally study a system.
As we assume that the reaction could start when the alkyne and/or the thiol are activated by the copper catalyst, it was necessary to establish how many support molecules were attached to the metal surface in order to act as stabilizers.
For this purpose, we modeled different structures of the active copper nanocatalyst with the aim to find the most simple and representative structure. Initially, we included monomeric and dimeric species of the support (TiO2) and the initial conformational analysis for the structures was performed with the PBE functional applying the D3BJ dispersion and the def2-SVP basis set. This led us to four stable structures: As can be seen from the figures in Table 7 we obtained Ia with a monomeric TiO2 and IIa–c that include dimeric species of TiO2. Then we optimized them to find the most energetically favorable structure, with two functionals M06-L and B3LYP, and the def2-TZVP(-f) and 6-311+G** basis set respectively, which are known to be appropriate methodologies for the mechanistic studies on Cu-catalyzed reactions [33,34,35,36,37]. The energies in solution (DCM) were obtained with the conductor-like polarizable continuum model (CPCM) as implemented in ORCA.
As can be seen from Table 7, all these proposed structures shown to be stable and the formation of all of them could take place exothermically both in gas phase and in solvent. It is necessary to note that structure IIc could not be obtained as a minimum when it was calculated with solvent. The monomeric structure Ia was the less exothermic of all. With respect to the dimeric ones, flat structures (IIa,c) were less favorable than the basket type (IIb) structure, that showed to be the thermochemically more favored, since it occurs with an exothermicity 236 kcal/mol−1 and 224 kcal/mol with M06-L/TZVP(-f) and B3LYP/6-311+G** respectively in dichloromethane as solvent. From these analyses of nanocatalyst, we could conclude that the copper would be attached to the TiO2 dimer through the oxygen atoms making a basket type structure.
Then, we considered that the first stage of the reaction mechanism would involve the formation of a catalyst-alkynyl complex, through a π-coordination between the alkyne and the copper. For this purpose, we used the catalyst IIb and the alkyne 1a as model structures, and we carried out the computational calculations with the two methodologies already mentioned. As shown in the figure below and Table 8, we obtained this complex as a minimum with M06-L and B3LYP as functionals, and that occurs exothermically both in gas phase and in dichloromethane. It is important to mention that by adding implicit solvent to B3LYP simulations, a much more remarkable stabilization of the complex structure was achieved compared to by modeling the system with M06-L, which led to a much greater difference in exothermicity to pass of the formation energy of the complex in gas phase (−10.9 kcal/mol) to the formation energy in condensed phase (−30.7 kcal/mol).
At present, we are still working on the next steps of this reaction to establish the reaction mechanism and finally justify the selectivity observed experimentally.
4. Conclusions
In conclusion, based on a new simple and economical methodology for the synthesis of vinyl sulfides, catalyzed by CuNPs/TiO2, we performed a computational analysis for the alkynes 1a–f, and were able to show that CHELPG partial atomic charges are values that, together with other parameters, could allow us to explain reactivity towards a nucleophilic attack. Besides, we modelled different structures for the nanocatalyst and we could conclude that the copper would be attached to the TiO2 dimer through the oxygen atoms making a basket type structure. Finally, we studied the first stage of the reaction, and observed that the formation energy of the catalyst-alkynyl complex was exothermic for both of the employee calculation methodologies. Further mechanistic details are now under study.
Funding
This work was generously supported by the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET, PIP-2011-11220100100268), Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT, Prest. BID, PICT-2014-2171, PICT-2016-0385) and Universidad Nacional del Sur (UNS, PGI 24/Q072) from Argentina.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Doroszuk, J.; Musiejuk, M.; Ponikiewski, Ł.; Witt, D. Convenient and Efficient Diastereoselective Preparation of Functionalized Z-Alkenyl Sulfides. Eur. J. Org. Chem. 2018, 2018, 6333–6337. [Google Scholar] [CrossRef]
- Riesco-Domínguez, A.; van de Wiel, J.; Hamlin, T.A.; van Beek, B.; Lindell, S.D.; Blanco-Ania, D.; Bickelhaupt, F.M.; Rutjes, F.P.J.T. Trifluoromethyl Vinyl Sulfide: A Building Block for the Synthesis of CF3S-Containing Isoxazolidines. J. Org. Chem. 2018, 83, 1779–1789. [Google Scholar] [CrossRef] [PubMed]
- Choudhuri, K.; Pramanik, M.; Mandal, A.; Mal, P. S−H···π Driven Anti-Markovnikov Thiol-Yne Click Reaction. Asian J. Org. Chem. 2018, 7, 1849–1855. [Google Scholar] [CrossRef]
- Castarlenas, R.; di Giuseppe, A.; Pérez-Torrente, J.J.; Oro, L.A. The Emergence of Transition-Metal-Mediated Hydrothiolation of Unsaturated Carbon–Carbon Bonds: A Mechanistic Outlook. Angew. Chem. Int. Ed. 2013, 52, 211–222. [Google Scholar] [CrossRef]
- Yang, Y.; Rioux, R.M. Highly regio- and stereoselective hydrothiolation of acetylenes with thiols catalyzed by a well-defined supported Rh complex. Chem. Commun. 2011, 47, 6557–6559. [Google Scholar] [CrossRef] [PubMed]
- Dondoni, A.; Marra, A. Metal-Catalyzed and Metal-Free Alkyne Hydrothiolation: Synthetic Aspects and Application Trends. Eur. J. Org. Chem. 2014, 2014, 3955–3969. [Google Scholar] [CrossRef]
- Mancebo-Aracil, J.; Casagualda, C.; Moreno-Villaécija, M.A.; Nador, F.; García-Pardo, J.; Franconetti-García, A.; Busqué, F.; Alibés, R.; Esplandiu, M.J.; Ruiz-Molina, D.; et al. Bioinspired Functional Catechol Derivatives through Simple Thiol Conjugate Addition. Chem. Eur. J. 2019, 25, 12367–12379. [Google Scholar] [CrossRef] [PubMed]
- Daniel, R.M.; Josep, S.V.; Juan, M.A. Catechol-Derivative Compounds And Their Use. WO2019025498, 7 February 2019. [Google Scholar]
- Nador, F.; Volpe, M.A.; Alonso, F.; Feldhoff, A.; Kirschning, A.; Radivoy, G. Copper nanoparticles supported on silica coated maghemite as versatile, magnetically recoverable and reusable catalyst for alkyne coupling and cycloaddition reactions. Appl. Catal. A Gen. 2013, 455, 39–45. [Google Scholar] [CrossRef]
- Moglie, Y.; Buxaderas, E.; Mancini, A.; Alonso, F.; Radivoy, G. Amide Bond Formation Catalyzed by Recyclable Copper Nanoparticles Supported on Zeolite Y under Mild Conditions. ChemCatChem 2019, 11, 1487–1494. [Google Scholar] [CrossRef]
- Buxaderas, E.; Mayer, M.G.; Volpe, M.A.; Radivoy, G. Bimetallic Cu-Pd Nanoparticles Supported on Bio-silica as an Efficient Catalyst for Selective Aerobic Oxidation of Benzylic Alcohols. Synthesis 2017, 49, 1387–1393. [Google Scholar]
- Gutierrez, V.; Mascaró, E.; Alonso, F.; Moglie, Y.; Radivoy, G. Direct synthesis of β-ketophosphonates and vinyl phosphonates from alkenes or alkynes catalyzed by CuNPs/ZnO. RSC Adv. 2015, 5, 65739–65744. [Google Scholar] [CrossRef]
- Menéndez, C.; Nador, F.; Radivoy, G.; Gerbino, D. One-step synthesis of xanthones catalyzed by a highly efficient copper-based magnetically recoverable nanocatalyst. Org. Lett. 2014, 16, 2846–2849. [Google Scholar] [CrossRef] [PubMed]
- Kohn, W.; Sham, I.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101–194118. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Miehlich, E.; Savin, A.; Stoll, H.; Preuss, H. Results obtained with the correlation energy density functionals of becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Grimme, S.J. Accurate description of van der Waals complexes by density functional theory including empirical corrections. Comput. Chem. 2004, 25, 1463–1473. [Google Scholar] [CrossRef]
- Grimme, S.J. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H.J. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. Chem. Phys. 2010, 132, 154104–154119. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L.J. Effect of the damping function in dispersion corrected density functional theory. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868, Erratum in 1997, 78, 1396. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M.J. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic Population Analysis on LCAO–MO Molecular Wave Functions. II. Overlap Populations, Bond Orders, and Covalent Bond Energies. J. Chem. Phys. 1955, 23, 1841–1846. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinholt, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Valency and Bonding: A Natural Bond Orbital Donor-Acceptor Perspective; Cambridge University Press: Cambridge, UK, 2005.
- Breneman, C.M.; Wiberg, K.B. Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis. J. Comput. Chem. 1990, 11, 361–373. [Google Scholar] [CrossRef]
- Li, H.; Luo, M.; Tao, G.; Qin, S. Theoretical Calculations on the Mechanism of Enantioselective Copper(I)-Catalyzed Addition of Enynes to Ketones. Catalysts 2018, 8, 359. [Google Scholar] [CrossRef]
- Sirijaraensre, J.; Khongpracha, P.; Limtrakul, J. Mechanistic insights into CO2 cycloaddition to propylene oxide over a single copper atom incorporated graphene-based materials: A theoretical study. Appl. Surf. Sci. 2019, 470, 755–763. [Google Scholar] [CrossRef]
- Ananikov, V.P.; Beletskaya, I.P. Alkyne Insertion into the M-P and M-H Bonds (M=Pd, Ni, Pt, and Rh): A Theoretical Mechanistic Study of the C-P and C-H Bond-Formation Steps. Chem. Asian J. 2011, 6, 1423–1430. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wu, Y.; Wang, Z.; Zhu, J.; Zhao, Y. Mechanistic Insight into the Copper-Catalyzed Phosphorylation of Terminal Alkynes: A Combined Theoretical and Experimental Study. J. Org. Chem. 2014, 79, 6816–6822. [Google Scholar] [CrossRef] [PubMed]
- Fortunato, L.; Moglie, Y.; Dorn, V.; Radivoy, G. Hydrophosphorylation of aliphatic alkynes catalyzed by CuNPs/ZnO for the synthesis of vinyl phosphonates. A DFT study on the reaction mechanism. RSC Adv. 2017, 7, 18707–18713. [Google Scholar] [CrossRef]
Scheme 1.
Schematic representation of alkyne hydrothiolation.

Scheme 2.
Reaction between 2-ethynylpyridine and 1-octadecanethiol.

Scheme 3.
Alkyne hydrothiolation.


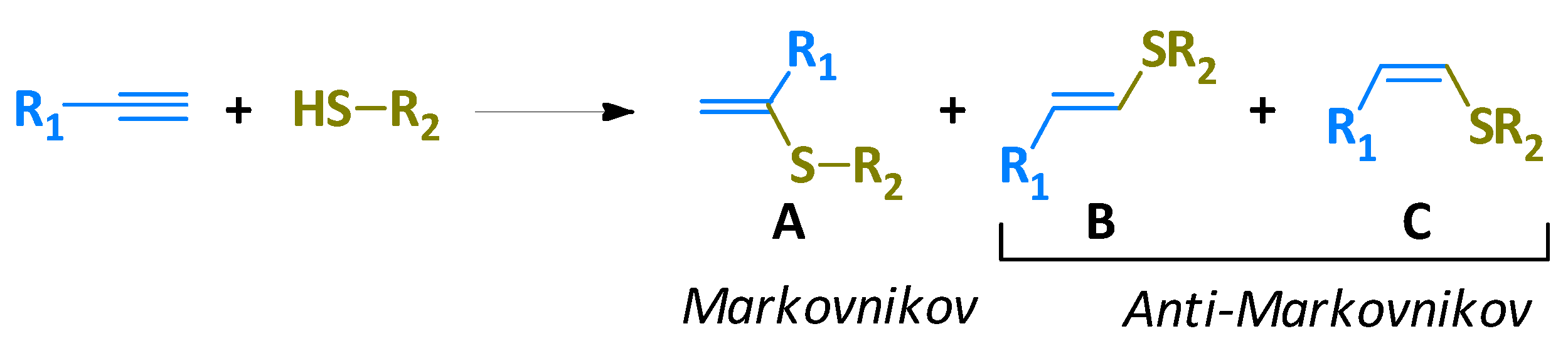{kind=link}
{kind=link}
{kind=link}
Table 1.
Geometry and partial atomic charges for alkyne 1a.
 1a | M06/TZVP(-f) Gas Phase | M06-2X/TZVP(-f) Gas Phase | ||||
| Atom | MULLIKEN | CHELPG | MULLIKEN | CHELPG | ||
| 1 C | −0.098 | −0.303 | −0.135 | −0.324 | ||
| 2 C | −0.299 | −0.165 | −0.075 | −0.153 | ||
| 3 H | 0.223 | 0.286 | 0.192 | 0.303 | ||
| 4 C | 0.726 | 0.863 | 0.464 | 0.842 | ||
| 5 O | −0.524 | −0.551 | −0.465 | −0.544 | ||
| 6 N | −0.470 | −0.980 | −0.436 | −0.997 | ||
| 7 H | 0.219 | 0.423 | 0.229 | 0.432 | ||
| 8 H | 0.223 | 0.428 | 0.225 | 0.442 | ||
| M06-L/TZVP(-f) Gas Phase | M06-L/TZVP(-f) CPCM=DCM | B3LYP/6-311+G** | ||||
| Atom | MULLIKEN | CHELPG | MULLIKEN | CHELPG | NBO Gas Phase | NBO CPCM=DCM |
| 1 C | 0.043 | −0.305 | 0.033 | −0.283 | −0.158 | −0.145 |
| 2 C | −0.329 | −0.157 | −0.339 | −0.219 | −0.105 | −0.124 |
| 3 H | 0.199 | 0.28 | 0.243 | 0.303 | 0.231 | 0.248 |
| 4 C | 0.793 | 0.829 | 0.795 | 0.879 | 0.592 | 0.596 |
| 5 O | −0.630 | −0.533 | −0.739 | −0.647 | −0.574 | −0.643 |
| 6 N | −0.466 | −0.941 | −0.456 | −0.917 | −0.783 | −0.758 |
| 7 H | 0.193 | 0.409 | 0.235 | 0.444 | 0.397 | 0.414 |
| 8 H | 0.197 | 0.418 | 0.228 | 0.44 | 0.401 | 0.414 |
Table 2.
Geometry and partial atomic charges for alkyne 1b.
 1b | M06-L/TZVP(-f) | B3LYP/6-311+G** | |||
| Atom | CHELPG Gas Phase | CHELPG CPCM=DCM | NBO Gas Phase | NBO CPCM=DCM | |
| 1 C | −0.173 | −0.157 | −0.129 | −0.114 | |
| 2 C | −0.329 | −0.387 | −0.106 | −0.131 | |
| 3 H | 0.253 | 0.28 | 0.231 | 0.248 | |
| 4 C | 0.92 | 0.997 | 0.728 | 0.741 | |
| 5 O | −0.523 | −0.602 | −0.552 | −0.583 | |
| 6 O | −0.321 | −0.339 | −0.519 | −0.519 | |
| 7 C | −0.140 | −0.149 | −0.222 | −0.224 | |
| 8 H | 0.127 | 0.148 | 0.189 | 0.198 | |
| 9 H | 0.093 | 0.105 | 0.19 | 0.192 | |
| 10 H | 0.093 | 0.105 | 0.19 | 0.192 | |
Table 3.
Geometry and partial atomic charges for alkyne 1c.
 1c | M06-L/TZVP(-f) | B3LYP/6-311+G** | |||
| Atom | CHELPG Gas Phase | CHELPG CPCM=DCM | NBO Gas Phase | NBO CPCM=DCM | |
| 1 C | −0.238 | −0.216 | −0.124 | −0.098 | |
| 2 C | −0.239 | −0.275 | −0.160 | −0.175 | |
| 3 H | 0.276 | 0.3 | 0.236 | 0.253 | |
| 4 C | 0.78 | 0.857 | 0.724 | 0.739 | |
| 5 O | −0.442 | −0.548 | −0.510 | −0.571 | |
| 6 O | −0.540 | −0.589 | −0.640 | −0.653 | |
| 7 H | 0.403 | 0.471 | 0.481 | 0.505 | |
Table 4.
Geometry and partial atomic charges for alkyne 1d.
 1d | M06-L/TZVP(-f) | B3LYP/6-311+G** | |||
| Atom | CHELPG Gas Phase | CHELPG CPCM=DCM | NBO Gas Phase | NBO CPCM=DCM | |
| 1 C | 0.356 | 0.41 | 0.061 | 0.059 | |
| 2 C | −0.440 | −0.466 | −0.237 | −0.232 | |
| 3 C | 0.22 | 0.244 | −0.167 | −0.160 | |
| 4 C | −0.592 | −0.628 | −0.199 | −0.197 | |
| 5 C | 0.899 | 0.979 | 0.113 | 0.104 | |
| 6 N | −0.638 | −0.765 | −0.430 | −0.465 | |
| 7 H | 0.047 | 0.052 | 0.191 | 0.198 | |
| 8 H | 0.168 | 0.191 | 0.214 | 0.226 | |
| 9 H | 0.078 | 0.095 | 0.212 | 0.224 | |
| 10 H | 0.205 | 0.23 | 0.22 | 0.229 | |
| 11 C | −0.306 | −0.363 | −0.037 | −0.052 | |
| 12 C | −0.253 | −0.239 | −0.167 | −0.176 | |
| 13 H | 0.256 | 0.259 | 0.226 | 0.241 | |
Table 5.
Geometry and partial atomic charges for alkyne 1e.
 1e | M06-L/TZVP(-f) | B3LYP/6-311+G** | |||
| Atom | CHELPG Gas Phase | CHELPG CPCM=DCM | NBO Gas Phase | NBO CPCM=DCM | |
| 1 C | −0.219 | −0.242 | −0.240 | −0.258 | |
| 2 C | −0.385 | −0.416 | −0.036 | −0.036 | |
| 3 C | 0.615 | 0.682 | −0.268 | −0.273 | |
| 4 H | 0.281 | 0.305 | 0.226 | 0.239 | |
| 5 H | −0.018 | −0.022 | 0.214 | 0.221 | |
| 6 N | −1.003 | −1.121 | −0.814 | −0.836 | |
| 7 H | −0.019 | −0.023 | 0.214 | 0.221 | |
| 8 H | 0.374 | 0.418 | 0.352 | 0.361 | |
| 9 H | 0.375 | 0.419 | 0.352 | 0.361 | |
Table 6.
Geometry and partial atomic charges for alkyne 1f.
 1f | M06-L/TZVP(-f) | B3LYP/6-311+G** | |||
| Atom | CHELPG Gas Phase | CHELPG CPCM=DCM | NBO Gas Phase | NBO CPCM=DCM | |
| 1 C | −0.325 | −0.356 | −0.157 | −0.170 | |
| 2 C | −0.104 | −0.116 | −0.042 | −0.053 | |
| 3 H | 0.272 | 0.3 | 0.229 | 0.243 | |
| 4 C | 0.26 | 0.241 | −0.107 | −0.115 | |
| 5 C | −0.090 | −0.079 | −0.108 | −0.118 | |
| 6 C | 0.506 | 0.514 | 1.069 | 1.071 | |
| 7 C | −0.110 | −0.122 | −0.176 | −0.175 | |
| 8 H | 0.133 | 0.147 | 0.228 | 0.235 | |
| 9 C | −0.133 | −0.129 | −0.191 | −0.188 | |
| 10 H | 0.127 | 0.144 | 0.213 | 0.224 | |
| 11 C | −0.090 | −0.101 | −0.184 | −0.181 | |
| 12 H | 0.125 | 0.145 | 0.214 | 0.225 | |
| 13 C | −0.188 | −0.187 | −0.152 | −0.153 | |
| 14 H | 0.141 | 0.157 | 0.22 | 0.227 | |
| 15 F | −0.168 | −0.186 | 0.35 | 0.357 | |
| 16 F | −0.184 | −0.185 | −0.358 | 0.358 | |
| 17 F | −0.171 | −0.188 | −0.35 | 0.358 | |
Table 7.
Geometries and formation energy (kcal/mol) for the different catalyst modeled.
 |  |  |  | |
|---|---|---|---|---|
| Ia | IIa | IIb | IIc | |
| M06-L/TZVP(-f) gas phase | −80 | −228 | −261 | −151 |
| M06-L/TZVP(-f) CPCM=DCM | −74 | −208 | −236 | |
| B3LYP/6-311+G** gas phase | −73 | −208 | −227 | −139 |
| B3LYP/6-311+G** CPCM=DCM | −73 | −207 | −224 |
Table 8.
Geometry and formation energy (kcal/mol) for the complex nanocatalyst-alkyne 1a.
 | Method | Formation Energy (kcal/mol) |
| M06-L/TZVP(-f), gas phase | −30.4 | |
| M06-L/TZVP(-f), CPCM=DCM | −26.0 | |
| B3LYP/6-311+G**, gas phase | −10.9 | |
| B3LYP/6-311+G**, CPCM=DCM | −30.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Capurso, M.; Radivoy, G.; Nador, F.; Dorn, V. Synthesis of Alkenyl Sulfides Catalyzed by CuNPs/TiO2. A Theoretical-Computational Analysis. Chem. Proc. 2021, 3, 120. https://0-doi-org.brum.beds.ac.uk/10.3390/ecsoc-24-08323
AMA Style
Capurso M, Radivoy G, Nador F, Dorn V. Synthesis of Alkenyl Sulfides Catalyzed by CuNPs/TiO2. A Theoretical-Computational Analysis. Chemistry Proceedings. 2021; 3(1):120. https://0-doi-org.brum.beds.ac.uk/10.3390/ecsoc-24-08323
Chicago/Turabian StyleCapurso, Matías, Gabriel Radivoy, Fabiana Nador, and Viviana Dorn. 2021. "Synthesis of Alkenyl Sulfides Catalyzed by CuNPs/TiO2. A Theoretical-Computational Analysis" Chemistry Proceedings 3, no. 1: 120. https://0-doi-org.brum.beds.ac.uk/10.3390/ecsoc-24-08323