
A First Attempt to Identify Repurposable Drugs for Type 2 Diabetes: 3D-Similarity Search and Molecular Docking †


“Coriolan Dragulescu” Institute of Chemistry Timisoara, 24 Mihai Viteazul Av., 300223 Timișoara, Romania
*
Author to whom correspondence should be addressed.
†
Presented at the 24th International Electronic Conference on Synthetic Organic Chemistry, 15 November–15 December 2020; Available online: https://ecsoc-24.sciforum.net/.
Chem. Proc. 2021, 3(1), 7; https://0-doi-org.brum.beds.ac.uk/10.3390/ecsoc-24-08368
Published: 14 November 2020
(This article belongs to the Proceedings of The 24th International Electronic Conference on Synthetic Organic Chemistry)
Abstract
:Drug repositioning involves the investigation of existing drugs for new therapeutic purposes, such as type 2 diabetes. This disease affects the health and quality of life for individuals around the world. Sitagliptin, a highly selective dipeptidyl peptidase-4 (DPP-4) inhibitor, is used to treat type 2 diabetes mellitus by effective fasting and improved glycemic control. Despite this advantage, serious hypersensitivity reactions have been acknowledged for patients receiving sitagliptin. In this context, it is necessary to develop new drugs with enhanced profiles and targeting DPP-4. Sitagliptin, ((2R)-4-oxo-4-[3-(trifluoromethyl)-5,6-dihidro[1,2,4]triazolo[4,3-A]pirazin-7(8H)-yl]-1-(2,4,5-trifluorophenyl)butan-2-amine), was used as a query in a 3D-similarity search on the approved DrugBank. Based on the TanimotoCombo parameter, the first 10 approved DrugBank drugs were docked in the 4FFW active site to identify effective antidiabetic effects for possible repurposable drugs marketed with other indications.
1. Introduction
Diabetes mellitus type 2 (T2DM) is a progressive chronic disease that affects more than 425 million people worldwide [1]. Of the two types of diabetes, T2DM accounts for around 90% of cases. T2DM is characterized by persistent hyperglycemia derived from insulin deficiency and insulin resistance [2]. Chronic hyperglycemia is associated with harmful microvascular and macrovascular difficulties, such as retinopathy, nephropathy, and coronary artery illness [3].
The dipeptidyl peptidase 4 (DPP-4) proteolytic enzyme related to the pathophysiology of T2DM is expressed on the surface of most cell types, and is connected with signal transduction, immune regulation, and apoptosis. Moreover, it is responsible for the degradation of two incretins hormones—glucagon-like peptide 1 (GLP-1) and glucose-dependent-insulinotropic polypeptide (GIP) [4,5]. These two hormones are culpable for improving insulin production and release. DPP-4 catalyzes the N-terminal dipeptides of glucagon-like peptide 1 (GLP-1), which plays a pivotal role in blood glucose level regulation. In this context, the inhibition of DPP-4 and the design of appropriate DPP-4 inhibitors have been considered an essential strategy for T2DM treatment [6,7]. The newly launched DPP-4 inhibitors, in addition to the recognized antihyperglycemic and pancreatic effects, showed new cardiovascular protections and anti-inflammatory effects.
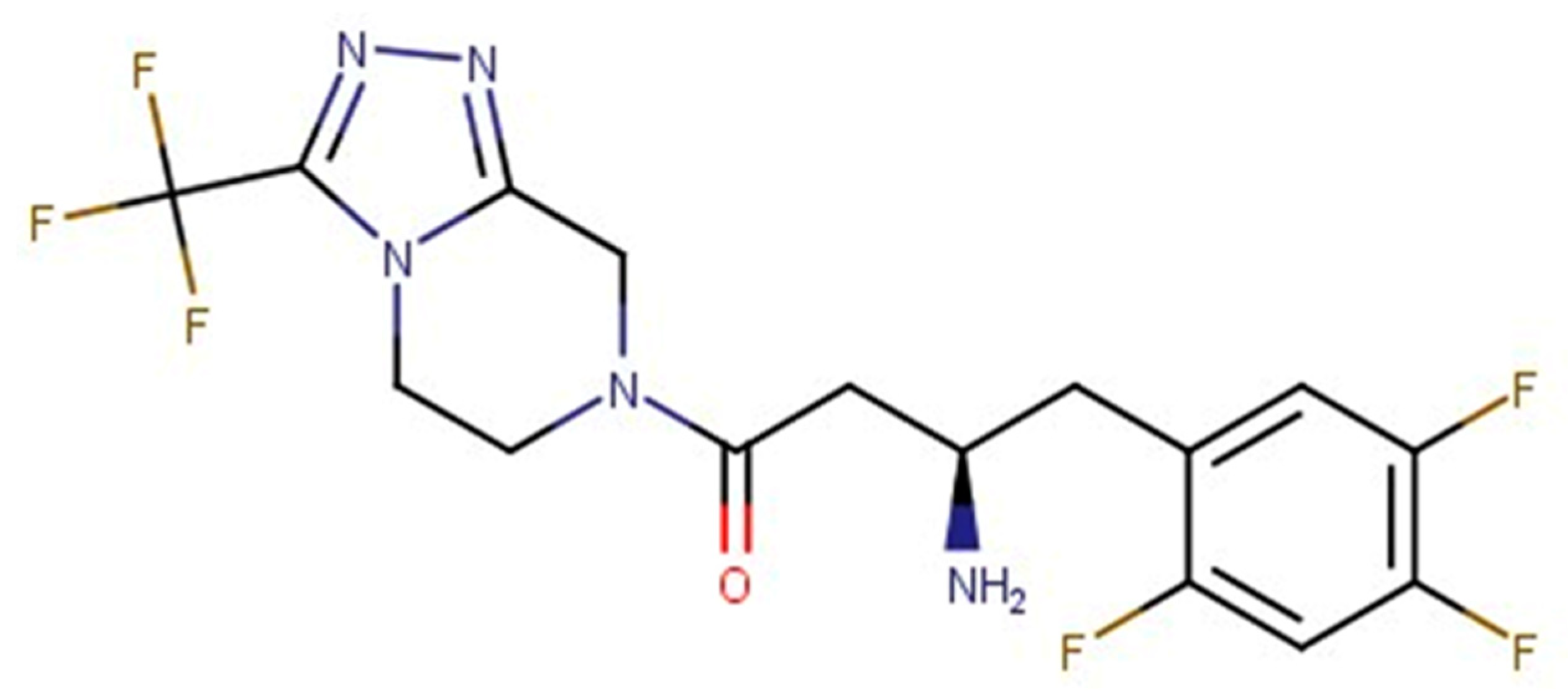
Sitagliptin ((2R)-4-oxo-4-[3-(trifluoromethyl)-5,6-dihidro[1,2,4]triazolo[4,3-A]pirazin-7(8H)-yl]-1-(2,4,5-trifluorophenyl)butan-2-amine)), a highly selective DPP-4 inhibitor, is used primarily for T2DM treatment to improve glycemic control, in addition to the benefits of reducing body weight, blood pressure, and albuminuria [8,9]. Despite these advantages, various side effects during treatment with Sitagliptin have been observed, such as hypersensitivity, pancreatitis, high triglyceride levels, some kidney problems, etc. Based on this, new DPP-4 inhibitors with improved chemical and pharmacokinetic profiles are required. In this light, the drug reposition approach may be a helpful, efficient, and low-cost strategy to find new indications for existing drugs [10].
Our work aims to identify new repurposable drugs, from the approved DrugBank database, as DPP-4 inhibitors with potential antidiabetic activity. To reach this goal, Rapid Overlay of Chemical Structures (ROCS), together with molecular docking, has been applied. Sitagliptin was used as a query in a 3D-similarity search on the approved DrugBank, resulting in the prioritization of ten compounds with a TanimotoCombo score between 0.995–1.2. Further, the relationship between binding interactions of the top ten prioritized ROCS compounds with key amino acids of DPP-4 active site and their inhibitory capacities were investigated.
2. Methodology
2.1. Dataset
The 2454 compounds downloaded from the approved DrugBank database [11] were prepared for ROCS analysis and docking simulation with the aid of LigPrep [12] by generating their ionization states and tautomers at pH = 7.2 ± 0.2. The conformers of each ligand and Sitagliptin were generated with Omega, OpenEye, Sanata Fe, NM, USA [13,14,15] using default settings. The generated Omega conformers were used as input for ROCS analysis and molecular docking. Drug Sitagliptin (Figure 1) was employed as a reference molecule for both processes.
2.2. Protein Preparation
The X-ray structure of the DPP-4 receptor, co-crystallized with Sitagliptin (PDB ID: 4FFW) [16] and downloaded from PDB, was prepared for docking by generating the active site box of 5956 Å3 and two inner/outer contours of 81 Å3/557 Å3, respectively, using the MakeReceptor facility, OpenEye [17]. The water molecules that exceeded 5 Å from the co-crystallized ligand were deleted and only the significant ones were kept. In the absence of any constraints, the default parameters were engaged.
2.3. D-Similarity
The shape similarity search, performed with ROCS, is recognized as an important ligand shape-based virtual screening method. ROCS employs 3D Gaussian functions to picture the shape of molecules [18,19,20]. The use of a chemical force field improves the outcomes provided by shape-based superposition. It is acknowledged that the performance of ROCS strongly depends on the selection of interrogation molecules. In this context, we chose the bioactive conformation of ((2R)-4-oxo-4-[3-(trifluoromethyl)-5,6-dihidro[1,2,4]triazolo[4,3-A]pirazin-7(8H)-yl]-1-(2,4,5-trifluorophenyl)butan-2-amine) co-crystallized ligand (PDB ID: 4FFW) [16] as a template. Out of thirteen similarity parameters [18,19,20], TanimotoCombo was used as a reference to rank the chemicals from the DrugBank database.
2.4. Molecular Docking
Docking analysis of ten prioritized compounds into the active site of the DPP-4 receptor was achieved by Fast Rigid Exhaustive Docking (FRED), Openeye [21]. During the docking process, the ligands and the receptor were treated as rigid structures. Furthermore, ten docking poses were retained for each ligand. The Chemgauss 4 (CG4) score [22,23,24] was used to score ligand poses biding into the 4FFW active site. The best ligand binding conformations were picked based on binding orientation, interactions with key binding site residues, and CG4 score values. To evaluate docking performance, the co-crystallized ligand Sitagliptin was redocked into the DPP-4 active site and the RMSD (root-mean-square deviation) was measured between docked conformations and its pose in X-ray crystal.
2.5. Pharmacokinetic Profile
The pharmacokinetics (absorption, distribution, metabolism, and excretion) properties of the prioritized DrugBank drugs were estimated using a free web-tool, SwissAdme (http://www.swissadme.ch) (accesed on 2 September 2020) [25]. Also, the BOILED-Egg, iLOGP, and Bioavailability Radar are available to be estimated with SwissAdme. The BOILED-Egg model predicts two key parameters of a molecule—passive human gastrointestinal absorption (HIA) and blood-brain barrier (BBB) permeation.
3. Results and Discussion
Molecular similarity is a fundamental concept widely used in the early stages of drug development to design new molecules. 3D-shape similarity is a method of choice in virtual screening campaigns, and a crucial factor for small molecules activity. In this regard, the 3D-shape similarity between query Sitagliptin and DrugBank database molecules was realized. The similarity between these shape representations was estimated by computing various similarity metrics. Analysis in Figure 2 indicates that the top ten prioritized approved drugs arrange more systematically on the query molecule, showing slight structural variability concentrated on the triazolopyrazine unit of Sitagliptin.
3D-similarity coefficients based on shape, color, and a combination of these two are listed in Table 1. One (TanimotoCombo) out of six coefficients were used to assist the prioritization of the approved DrugBank drugs (Figure 3). Approved drugs with coefficient values greater than 1 for TanimotoCombo were discussed in detail (Table 1, Figure 3).
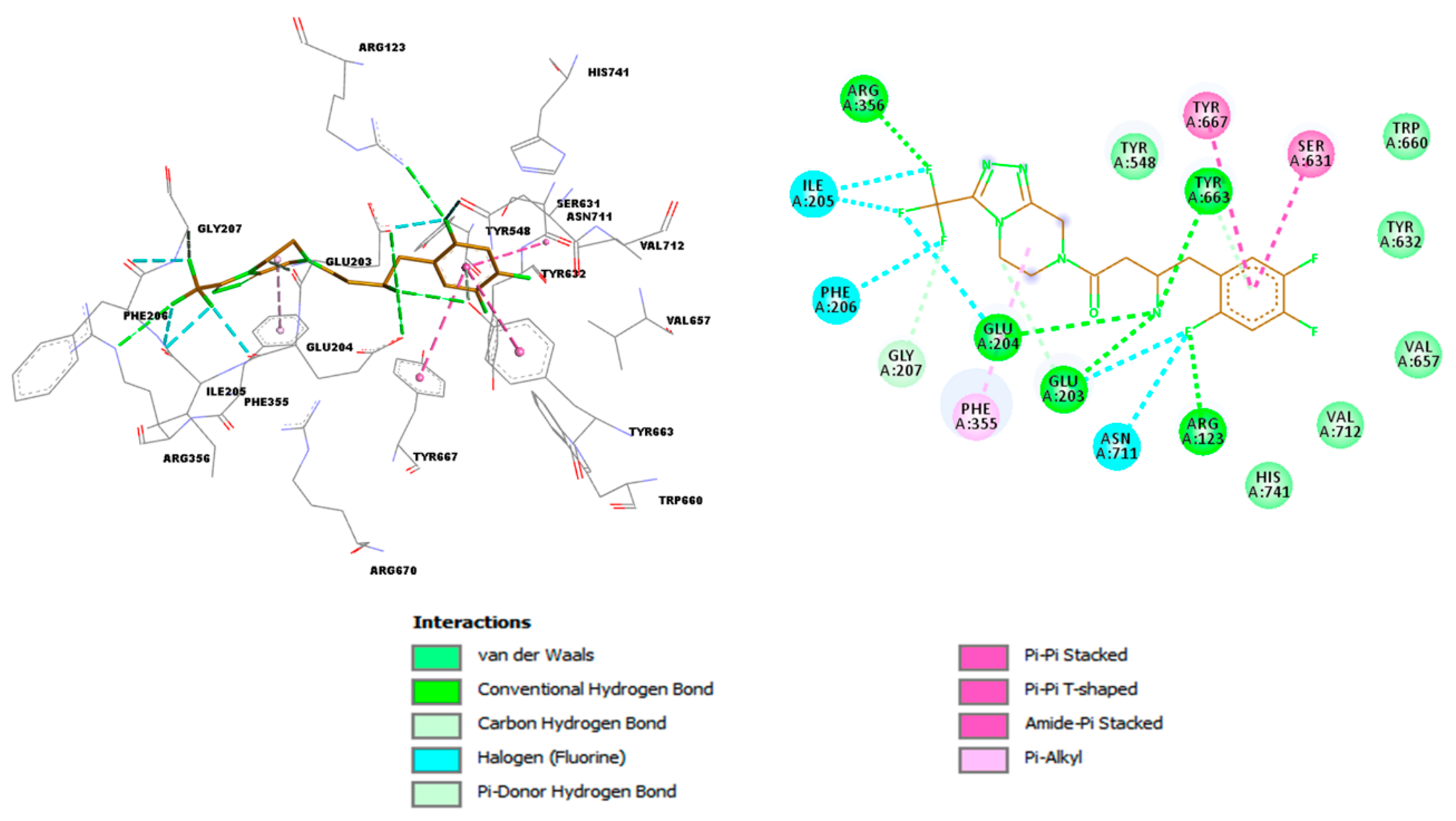
The first ten (Figure 2 and Table 1) compounds in the DrugBank database ranked by TanimotoCombo values were further subjected to the docking process. The performance of the docking experiment was checked by redocking co-crystallized ligand, Sitagliptin, into active sites of DPP4 (4FFW). The RMSD value between the coordinates of the atoms of co-crystallized ligand and the docked pose was 1.706 (Figure 4). The appropriate reproduction of the important interactions of Sitagliptin with the 4FFW active site (Figure 5) confirms that the docking approach can be further applied to the compounds prioritized by a 3D-similarity search.
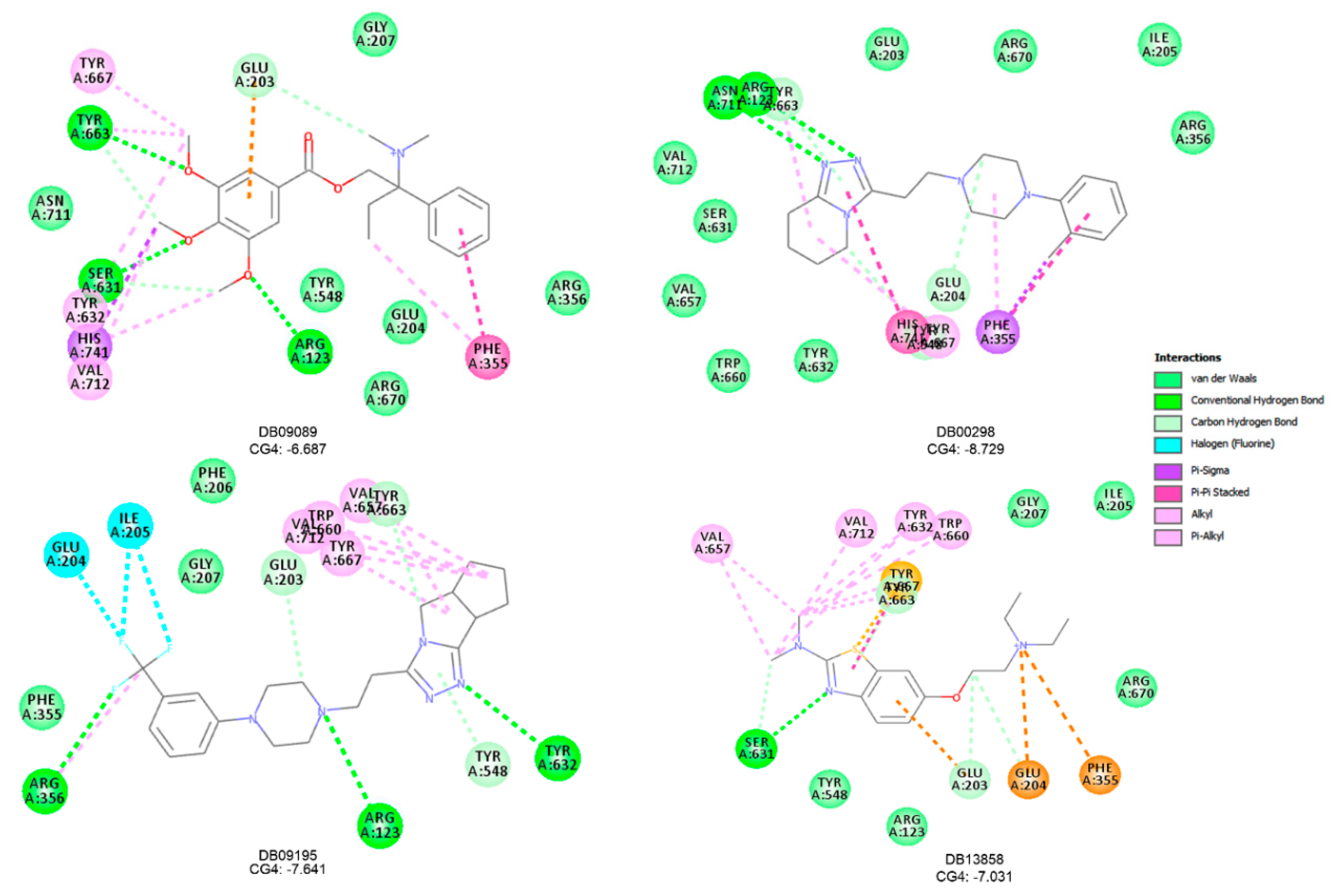
According to the CG4 score values and binding interaction realized by the prioritized compounds in the 4FFW active site, four out of ten prioritized DrugBank compounds were suggested to manifest possible antidiabetic effects, namely: DB09195 (Lorpiprazole), DB09089 (Trimebutine), DB00298 (Dapiprazole), and DB13858 (Dimazole) (Figure 6). The visual analysis of the docking results showed that the orientation of these four approved drugs closely mimics the orientations of the native ligands, Sitagliptin, in the active site of the DPP-4 receptor (Figure 6). Two out of four compounds, DB09089 and DB09195, simulate the most similar interactions with Sitagliptin.
DB09089 (Trimebutine) connects to DPP-4 by forming interactions as follows: (i) hydrogen bonds (3) with Ser631, Arg123, and Tyr663, (ii) π-alkyl (6) with His741, Tyr632, Tyr63, Tyr667, Val712, and Phe355, (iii) π-sigma (1) with His741, (iv) carbon hydrogen bonds (1)—Glu203, (v) π-π-stacked (1)—Phe355, and (vi) π-anion (1)—Glu203.
DB09195 (Lorpiprazole) binds to DPP-4 by establishing interactions as follows: (i) hydrogen bonds (3) with Arg356, Arg123, and Tyr632, (ii) π-alkyl (5) with Arg356, Tyr667, Tyr660, Val712, and Val657, (iii) π-halogen (3) with Ile205 and Glu204, (iv) carbon hydrogen bonds (3)—Tyr548, Tyr663, and Glu203.
The interactions performed by these four inhibitors are consistent with the features required by a DPP-4 inhibitor.
These four DrugBank compounds selected after the docking process include drugs used to treat several health issues, which are as follows: (i) DB09195 (Lorpiprazole) is used for major depressive disorder treatment; (ii) DB09089 (Trimebutine) is marketed for irritable bowel syndrome (IBS) and lower gastrointestinal tract motility disorders treatments; (iii) DB00298 (Dapiprazole) is used to decrease the size of pupils after an eye examination; and iv) DB13858 (Dimazole) is an antifungal used to treat tinea infections.
Analyzing the SwissADME properties [25], passive gastrointestinal absorption (HIA) and brain permeability (BBB) indicate that all four approved drugs passively permeate the BBB (yellow region), while only one compound, DB13858, cannot be effluated from the CNS (red dot) (Figure 7).
The four molecules (DB09195, DB09089, DB00298, and DB13858) plotted in the yellow ellipse have a high probability of a good BBB crossing, while the other four molecules (DB00447, DB00567, DB01333, and DB00833) situated in the white ellipse have a high probability of good HIA. The blue dots suggest molecules predicted as a substrate of P-glycoprotein (PGP+), while the red dots show molecules predicted as a non-substrate of P-glycoprotein (PGP−). In the yellow ellipse, DB09195 and DB09089 drugs are placed closely to reference molecule Sitagliptin. Two compounds, DB01150 and DB01060, are located in the grey area, and are predicted to be unabsorbed by GI and BBB non-permeable.
4. Conclusions
The present work successfully applied 3D-similarity search, molecular docking, and pharmacokinetics estimation approaches to identify and optimize old drugs for new uses, such as T2DM therapy. The ten approved drugs prioritized by similarity search were docked in the active site of the DPP4 receptor. The SwissADME parameters, passive gastrointestinal absorption (HIA), and brain permeability (BBB) indicate that approved drugs passively permeate the BBB (yellow region—DB09195, DB09089, DB00298, DB13858), while only DB13858 cannot be effluated from the CNS (red dot). Four out of ten approved DrugBank compounds, DB09195, DB09089, DB00298, and DB13858, showed excellent pharmacokinetic profiles and specific interactions with the DPP-4 binding site, which recommends them as possible DPP-4 inhibitors and candidates in the treatment of diabetes.
Author Contributions
D.I. and A.B. designed the computational framework of the study, analyzed resulted data, and edited the manuscript; L.C. performed the computational studies and edited the manuscript. All authors discussed the outcomes, contributed to the writing of the paper, and commented on the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
The authors thank ChemAxon Ltd., OpenEye Ltd., and BIOVIA software Inc. (Discovery Studio Visualizer) for providing academic license. The authors wish to thank Schrödinger Inc for providing an academic trial license to complete the calculations for this paper. Project No. 1.2 of the “Coriolan Dragulescu” Institute of Chemistry, Timisoara, Romanian Academy, financially supported the current work.
Conflicts of Interest
The authors indicate no potential conflict of interest.
References
- Goyal, R.; Jialal, I. Diabetes Mellitus Type 2. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Chen, L.; Magliano, D.J.; Zimmet, P.Z. The worldwide epidemiology of type 2 diabetes mellitus-present and future perspectives. Nat. Rev. Endocrinol. 2012, 8, 228–236. [Google Scholar] [CrossRef]
- Kahn, S.E.; Cooper, M.E.; Del Prato, S. Pathophysiology and treatment of type 2 diabetes: Perspectives on the past, present, and future. Lancet 2014, 383, 1068–1083. [Google Scholar] [CrossRef]
- Zhu, Y.; Meng, X.; Cai, Z.; Hao, Q.; Zhou, W. Synthesis of phenylpyridine derivatives and their biological evaluation to-ward dipeptidyl peptidase-4. Chem. Heterocycl. Compd. 2017, 53, 350–356. [Google Scholar] [CrossRef]
- Lai, Z.-W.; Li, C.; Liu, J.; Kong, L.; Wen, X.; Sun, H. Discovery of highly potent DPP-4 inhibitors by hybrid compound design based on linagliptin and alogliptin. Eur. J. Med. Chem. 2014, 83, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Mulakayala, N.; Reddy, U.C.H.; Iqbal, J.; Pal, M. Synthesis of dipeptidylpeptidase-4-inhibitors: A brief overview. Tetrahedron 2010, 66, 4919–4938. [Google Scholar] [CrossRef]
- Augustyns, K.; Van der Veken, P.; Senten, K.; Haerners, A. Dipeptidyl peptidase IV inhibitors as new therapeutic agents for the treatment of Type 2 diabetes. Expert Opin. Ther. Patents 2003, 13, 499–510. [Google Scholar] [CrossRef]
- Rosenstock, J.; Brazg, R.; Andryuk, P.J.; Lu, K.; Stein, P. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing pioglitazone therapy in patients with type 2 diabetes: A 24-week, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Clin. Ther. 2006, 28, 1556–1568. [Google Scholar] [CrossRef] [PubMed]
- Ahrén, B.; Simonsson, E.; Larsson, H.; Landin-Olsson, M.; Torgeirsson, H.; Jansson, P.-A.; Sandqvist, M.; Båvenholm, P.; Efendic, S.; Eriksson, J.W.; et al. Inhibition of Dipeptidyl Peptidase IV Improves Metabolic Control Over a 4-Week Study Period in Type 2 Diabetes. Diabetes Care 2002, 25, 869–875. [Google Scholar] [CrossRef] [PubMed]
- Padhy, B.M.; Gupta, Y.K. Drug repositioning: Re-investigating existing drugs for new therapeutic indications. J. Postgrad. Med. 2011, 57, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Cheng, D.; Shrivastava, S.; Tzur, D.; Gautam, B.; Hassanali, M. DrugBank: A knowledge-base for drugs, drug actions and drug targets. Nucleic Acids Res. 2008, 36, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger. Schrödinger Release 2016-1: LigPrep; v.3.1; Schrödinger, LLC: New York, NY, USA, 2016. [Google Scholar]
- OpenEye Scientific Software. OMEGA; v.2.5.1.4; OpenEye Scientific Software Inc.: Santa Fe, NM, USA, 2013. [Google Scholar]
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.C.D.; Nicholls, A.N. Conformer generation with OMEGA: Learning from the data set and the analysis of failures. J. Chem. Inf. Model. 2012, 52, 2919–2936. [Google Scholar] [CrossRef] [PubMed]
- RCS Protein Data Bank. Available online: https://www.rcsb.org/structure/4FFW (accessed on 16 March 2021).
- OpenEye Scientific Software. MakeReceptor; v.3.2.0.2; OpenEye Scientific Software Inc.: Santa Fe, NM, USA, 2015. [Google Scholar]
- OpenEye Scientific Software. ROCS; v. 3.2.1.4; OpenEye Scientific Software: Santa Fe, NM, USA, 2013. [Google Scholar]
- Hawkins, P.C.D.; Skillman, A.A.G.; Nicholls, A. Comparison of Shape-Matching and Docking as Virtual Screening Tools. J. Med. Chem. 2007, 50, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Venhorst, J.; Núñez, S.; Terpstra, J.W.; Kruse, C.G. Assessment of Scaffold Hopping Efficiency by Use of Molecular Interaction Fingerprints. J. Med. Chem. 2008, 51, 3222–3229. [Google Scholar] [CrossRef] [PubMed]
- OpenEye Scientific Software. FRED; v.3.2.0.2; OpenEye Scientific Software Inc.: Santa Fe, NM, USA, 2015. [Google Scholar]
- McGann, M. FRED Pose Prediction and Virtual Screening Accuracy. J. Chem. Inf. Model. 2011, 51, 578–596. [Google Scholar] [CrossRef] [PubMed]
- McGann, M. FRED and HYBRID Docking Performance on Standardized Datasets. J. Comput.-Aid Mol. Des. 2012, 26, 897–906. [Google Scholar] [CrossRef] [PubMed]
- McGaughey, G.B.; Sheridan, R.P.; Bayly, C.I.; Culberson, J.C.; Kreatsoulas, C.; Lindsley, S.; Maiorov, V.; Truchon, J.-F.; Cornell, W.D. Comparison of Topological, Shape, and Docking Methods in Virtual Screening. J. Chem. Inf. Model. 2007, 47, 1504–1519. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Dassault Systèmes BIOVIA. Discovery Studio Visualizer; v20.1.0, vol19295; Dassault Systèmes: San Diego, CA, USA, 2019. [Google Scholar]
Figure 1.
The structure of Sitagliptin DPP-4 inhibitor, used as a query.

Figure 2.
Rapid Overlay of Chemical Structures (ROCS) overlay of the top ten prioritized drugs against Sitagliptin (thickened stick), ranked by TanimotoCombo (BIOVIA Discovery Studio [26]).
Figure 2.
Rapid Overlay of Chemical Structures (ROCS) overlay of the top ten prioritized drugs against Sitagliptin (thickened stick), ranked by TanimotoCombo (BIOVIA Discovery Studio [26]).

Figure 3.
The best overlap after ROCS (BIOVIA Discovery Studio [26]).
Figure 3.
The best overlap after ROCS (BIOVIA Discovery Studio [26]).

Figure 4.
The best superposition of RX structure (query molecule) in orange, with docked pose in gray (BIOVIA Discovery Studio, [26]).
Figure 4.
The best superposition of RX structure (query molecule) in orange, with docked pose in gray (BIOVIA Discovery Studio, [26]).

Figure 5.
3D and 2D representations of interactions established by Sitagliptin in 4FFW binding site (BIOVIA Discovery Studio [26]).
Figure 5.
3D and 2D representations of interactions established by Sitagliptin in 4FFW binding site (BIOVIA Discovery Studio [26]).

Figure 6.
2D representations of interactions of DB09195, DB09089, DB00298, and DB13858 with 4FFW binding [26]; the CG4 docking score values are also specified (BIOVIA Discovery Studio [26]).

Figure 7.
The WLOGP–versus–TPSA referential for predicted compounds of DrugBank.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The top ten approved drugs prioritized against Sitagliptin, ordered by TanimotoCombo *.
| Name | TC | ShT | CoT | ScCo | CS | CoS |
|---|---|---|---|---|---|---|
| DB09089 | 1.203 | 0.690 | 0.513 | 0.721 | 1.411 | −5.765 |
| DB00298 | 1.098 | 0.770 | 0.328 | 0.463 | 1.233 | −3.705 |
| DB09195 | 1.068 | 0.765 | 0.303 | 0.466 | 1.231 | −3.725 |
| DB01333 | 1.020 | 0.707 | 0.312 | 0.565 | 1.273 | −4.523 |
| DB00447 | 1.017 | 0.704 | 0.313 | 0.566 | 1.270 | −4.528 |
| DB00567 | 1.008 | 0.670 | 0.339 | 0.601 | 1.270 | −4.805 |
| DB13858 | 1.004 | 0.450 | 0.554 | 0.624 | 1.074 | −4.993 |
| DB00833 | 1.000 | 0.677 | 0.322 | 0.579 | 1.257 | −4.633 |
| DB01150 | 1.000 | 0.748 | 0.252 | 0.553 | 1.301 | −4.427 |
| DB01060 | 0.995 | 0.718 | 0.277 | 0.623 | 1.342 | −4.988 |
* TanimotoCombo (TC), ShapeTanimoto (ST), ColorTanimoto (CoT), ScaledColor (ScCo), ComboScore (CS), ColorScore (CoS).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Istrate, D.; Bora, A.; Crisan, L. A First Attempt to Identify Repurposable Drugs for Type 2 Diabetes: 3D-Similarity Search and Molecular Docking. Chem. Proc. 2021, 3, 7. https://0-doi-org.brum.beds.ac.uk/10.3390/ecsoc-24-08368
AMA Style
Istrate D, Bora A, Crisan L. A First Attempt to Identify Repurposable Drugs for Type 2 Diabetes: 3D-Similarity Search and Molecular Docking. Chemistry Proceedings. 2021; 3(1):7. https://0-doi-org.brum.beds.ac.uk/10.3390/ecsoc-24-08368
Chicago/Turabian StyleIstrate, Daniela, Alina Bora, and Luminita Crisan. 2021. "A First Attempt to Identify Repurposable Drugs for Type 2 Diabetes: 3D-Similarity Search and Molecular Docking" Chemistry Proceedings 3, no. 1: 7. https://0-doi-org.brum.beds.ac.uk/10.3390/ecsoc-24-08368