
Identification of Natural Products for the Treatment of Alzheimer’s Disease: 3D Similarity Search †


“Coriolan Dragulescu” Institute of Chemistry Timisoara, 24 Mihai Viteazul Av., 300223 Timisoara, Romania
*
Author to whom correspondence should be addressed.
†
Presented at the 24th International Electronic Conference on Synthetic Organic Chemistry, 15 November–15 December 2020; Available online: https://ecsoc-24.sciforum.net/.
Chem. Proc. 2021, 3(1), 75; https://0-doi-org.brum.beds.ac.uk/10.3390/ecsoc-24-08341
Published: 14 November 2020
(This article belongs to the Proceedings of The 24th International Electronic Conference on Synthetic Organic Chemistry)
Abstract
:Glycogen synthase kinase-3 (GSK-3), one of the main tau kinases involved in a variety of cellular processes, has been evidenced as a promising target for Alzheimer’s disease (AD) treatment. In recent years, great efforts have been made to discover new molecules with an enhanced profile that inhibit GSK-3 and display efficacy in AD treatment. SAR502250, a newly discovered selective GSK-3 inhibitor with AD therapeutic potential, represents a good alternative to design future specific inhibitors against this condition. SAR502250 was used as a query in a 3D similarity search on the SPECS database to select new natural compounds as possible GSK-3 inhibitors. According to ShapeTanimoto, TanimotoCombo, and ComboScore matrics, the first 10 SPECS natural compounds were selected and structurally analyzed. The ADME (Absorption, Distribution, Metabolism, and Excretion), physicochemical parameters, and toxicity-related risk profiles of the selected natural compounds were also investigated. The 3D similarity results in conjunction with pharmaceutical profiles revealed the potential use of natural compounds as GSK-3 inhibitors for Alzheimer’s disease therapy.
1. Introduction
In many studies, glycogen synthase kinase-3 (GSK-3) has arisen as a promising therapeutic target for the treatment of inflammatory disorders, diabetes, neurodegenerative diseases, different types of cancers, etc. [1,2,3,4,5,6,7]. GSK-3 has been proven to be a promising target for the treatment of Alzheimer’s disease (AD); hence, its inhibition has been proposed as a strategy for treating AD patients [6]. The search for GSK-3 inhibitors has led to the discovery of a variety of chemical scaffolds that can be grouped into natural inhibitors, inorganic metal ions, maleimides, indirubins, paullones, purines, peptide inhibitors, etc. [8,9,10,11,12].
Alzheimer’s is considered a chronic disease that necessitates treatment for one’s entire life. Therefore, drugs must be safe and well-tolerated for long periods. In this context, attention has been focused on natural products that are considered true sources of active ingredients for medicine. They also play a key role in the drug discovery process. In the last 20 years, many drugs used in the treatment of various diseases like diabetes, cancer, cardiovascular diseases, etc. have often been natural products or their derivatives [13].
The main goal of this study was to select new natural products (NP) from the SPECS NP database [14] as possible GSK-3 inhibitors. In this light, SAR502250, a selective GSK-3 inhibitor used as a query and 3D molecular similarity search with ROCS (Rapid Overlay of Chemical Structures) [15,16], was employed to identify similar natural products with potential inhibition against GSK-3. The physicochemical parameters, toxicity-related risk profiles, and ADME (Absorption, Distribution, Metabolism, and Excretion) parameters were also explored.
2. Methods
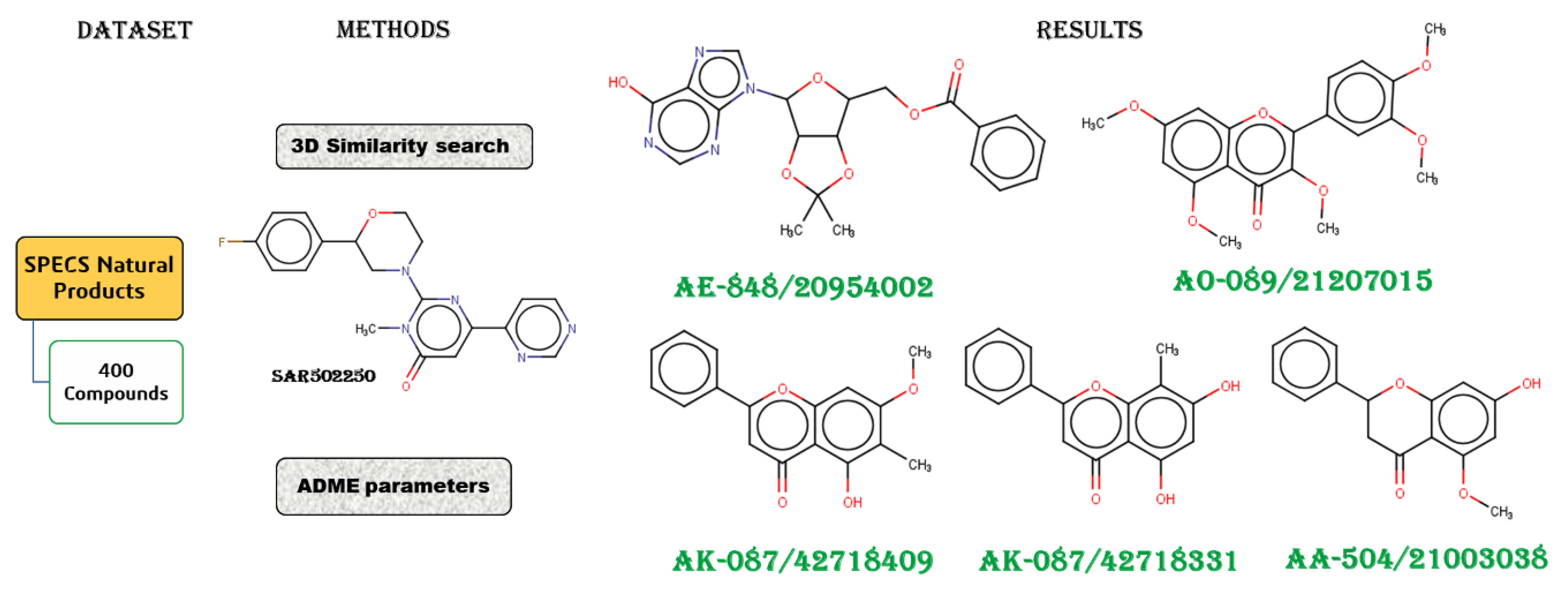
2.1. Workflow
The workflow diagram (Figure 1) followed in this paper comprises (a) the SPECS NP download and preparation, (b) the 3D similarity search, and (c) the ADME and toxicity risk profile predictions of the selected compounds.
2.2. Dataset Preparation
The dataset of 400 natural products [14] was prepared using LigPrep [17] from the Schrödinger suite and Omega (OMEGA v.2.5.1.4, OpenEye Scientific Software, Santa Fe, NM, USA. www.eyesopen.com (accessed on 12 February 2020)) [18,19] from the OpenEye. The ionization states and tautomers were engendered at pH = 7.2 ± 0.2, and the conformational space of each molecule was carried out, generating at most 200 conformations per ligand with an RMSD (root mean square deviation) of 0.8 Å in an energy window of 10 kcal/mol. The resulting dataset was used in the 3D similarity search process.
2.3. 3D Similarity Search
ROCS (ROCS v. 3.2.1.4, OpenEye Scientific Software, Santa Fe, NM, USA) [15,16] uses rigid conformations to evaluate shape similarity and implicit binding site complementarity. The similarity rule is based on the principle that two molecules that are structurally similar have similar biological and physical properties. When overlapping the structures, ROCS utilizes only the heavy atoms of a query, while the hydrogen atoms are ignored. This method is very often used in hit/lead identification to accelerate the speed and efficiency of the drug discovery and development process.
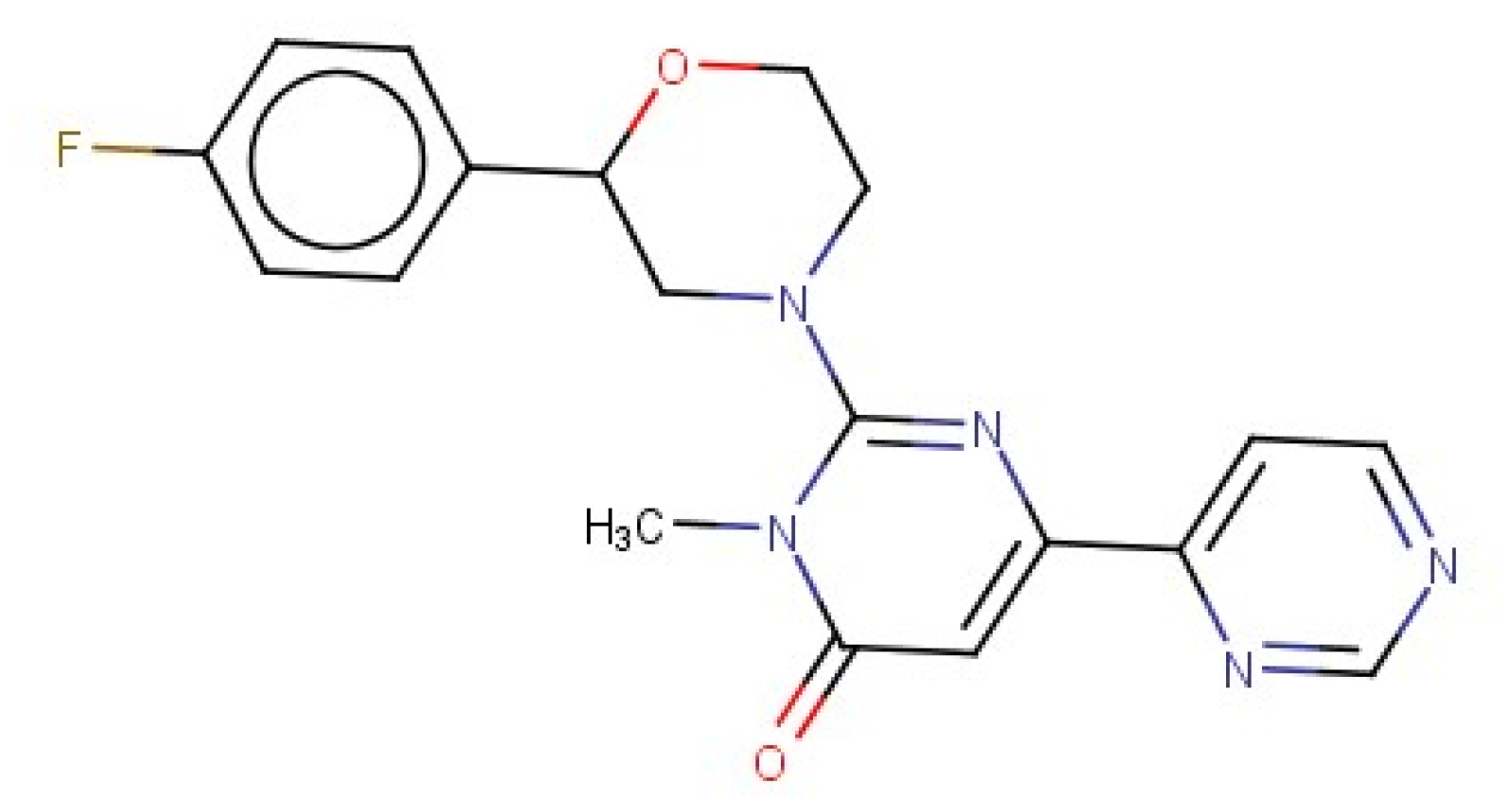
For 3D similarity analysis, the selective SAR502250 inhibitor (2-[2-(4-Fluorophenyl)morpholin-4-yl]-3-methyl-6-pyrimidin-4-ylpyrimidin-4-one; IC50 = 12 nM) [5] was chosen as the query molecule (Figure 2), and the SPECS NP database was engaged.
The ROCS program calculates 13 similarity coefficients as follows: TanimotoCombo, ShapeTanimoto, ColorTanimoto, ScaledColor, ComboScore, ColorScore, FitTverskyCombo, RefTverskyCombo, FitTversky, RefTversky, FitColorTversky, RefColorTversky, and Overlap. The Tanimoto similarity score is one of the most utilized coefficients in the early stage of virtual screening experiments to discover new molecules as potential drugs. ROCS quantifies and ranks the database molecules by Tanimoto coefficients (Equation (1)), based on the best shape overlap between a query molecule and a database molecule. The ROCS maximizes the volume overlap between them.
where the I are the terms for the self-volume overlaps, and the O terms are the overlaps between molecules X and Y.
2.4. ADME and Toxicity-Related Risk Profiles
The ADME (absorption, distribution, metabolism, and excretion) properties of the selected natural products were estimated using the open-source programs, Osiris Property Explorer [20] and SwissADME [21]. Additionally, the risks of side-effects (irritation, mutagenicity, tumorigenicity, and reproduction effectivity) and the passive gastrointestinal absorption (HIA) and brain penetration (BBB) were predicted.
3. Results and Discussions
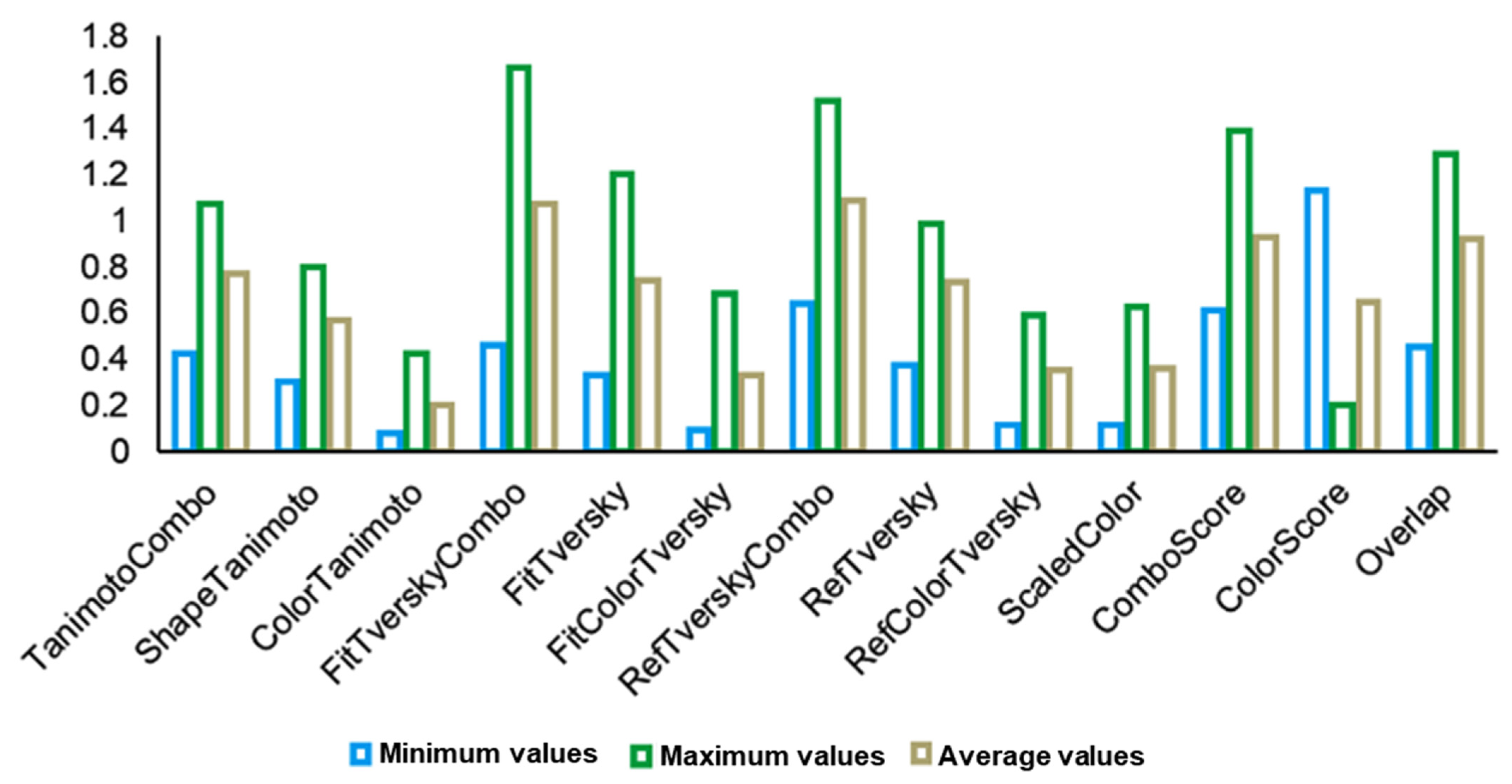
In virtual screening experiments, ROCS has been demonstrated to be robust vis-à-vis to the substitution of bioactive conformation with the lowest energy conformer [18,19]. In this respect, we investigated the SPECS NP database against the lowest energy conformation of SAR502250 (Figure 2). The minimum, maxim, and average values of the 3D similarity coefficients registered are shown in Figure 3.
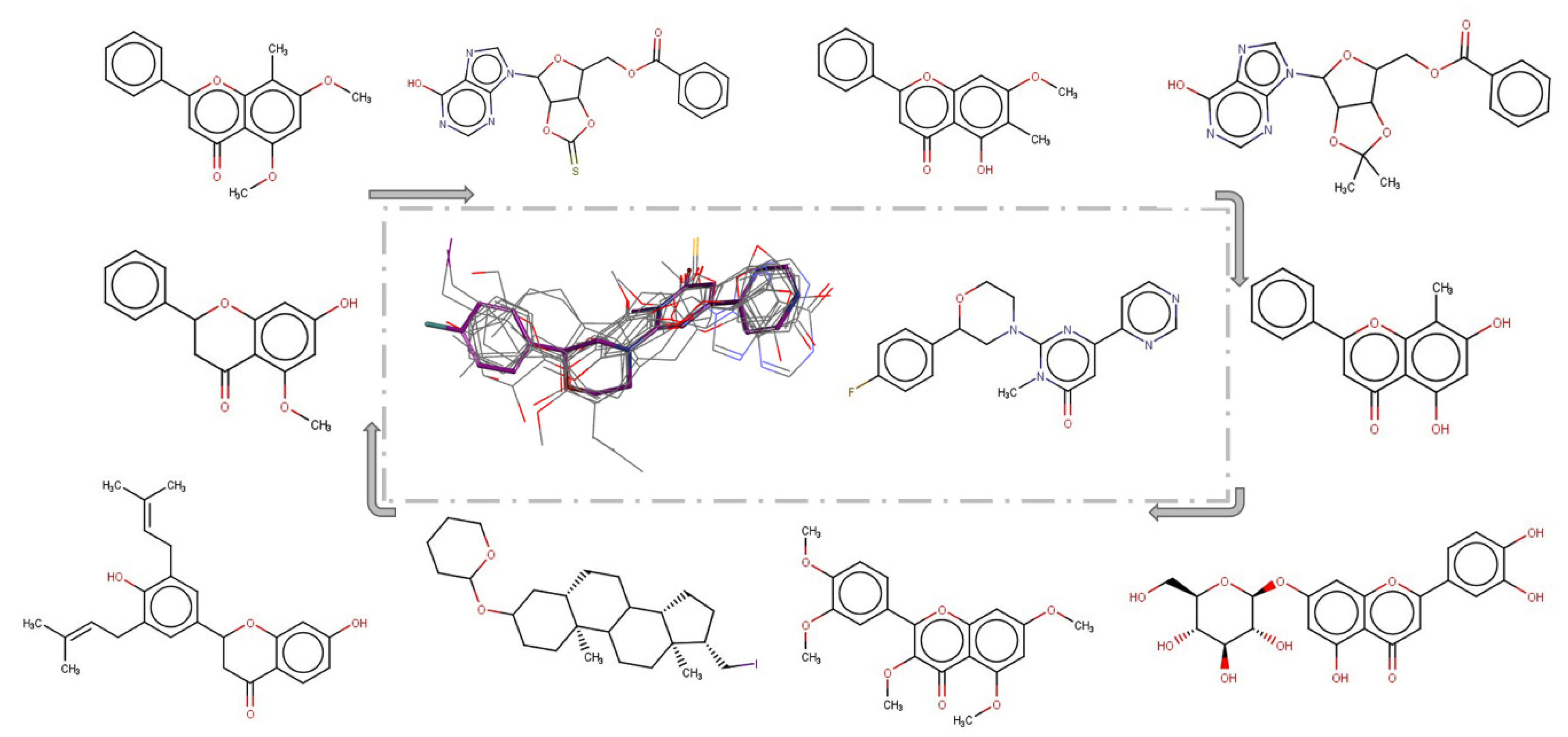
The ROCS overlays between the lowest energy conformer of SAR502250 and SPECS NP generated satisfactory results. To illustrate these observations, BIOVIA Discovery Studio v.4.5.0.15071 software was engaged (Figure 4).
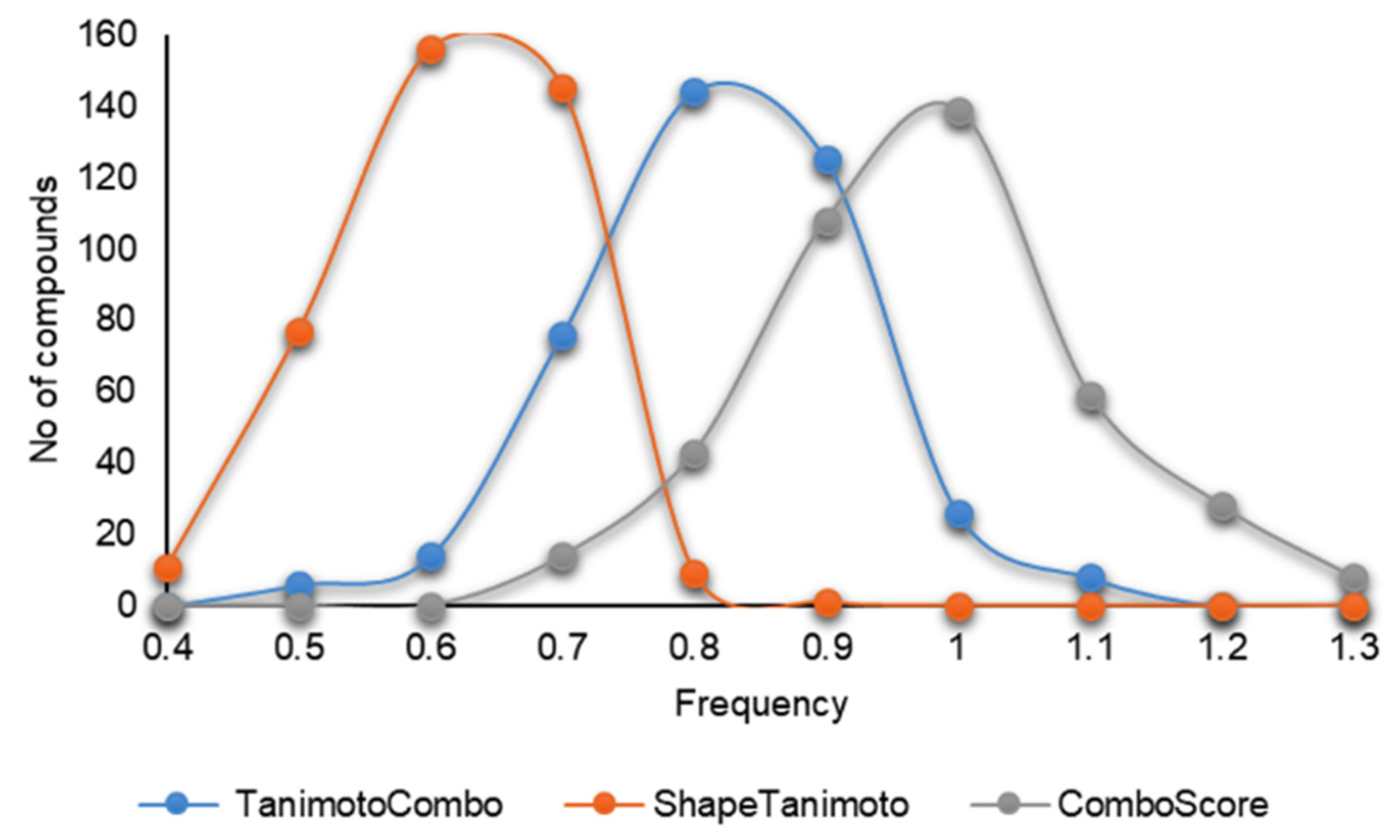
Three (TanimotoCombo, ShapeTanimoto, and ComboScore) out of 13 coefficients were used to assist the prioritization of the natural compounds (Figure 5). Natural compounds with coefficient values greater than 1 for TanimotoCombo, greater than 0.7 for ShapeTanimoto, and greater than 1.2 for ComboScore [19,22,23] were analyzed in detail.
The first 10 (Figure 3 and Figure 4) compounds in the SPECS NP database ordered by TanimotoCombo values were further investigated by applying ADME and toxicity-risk filters to assess their drug-like properties (Table 1).
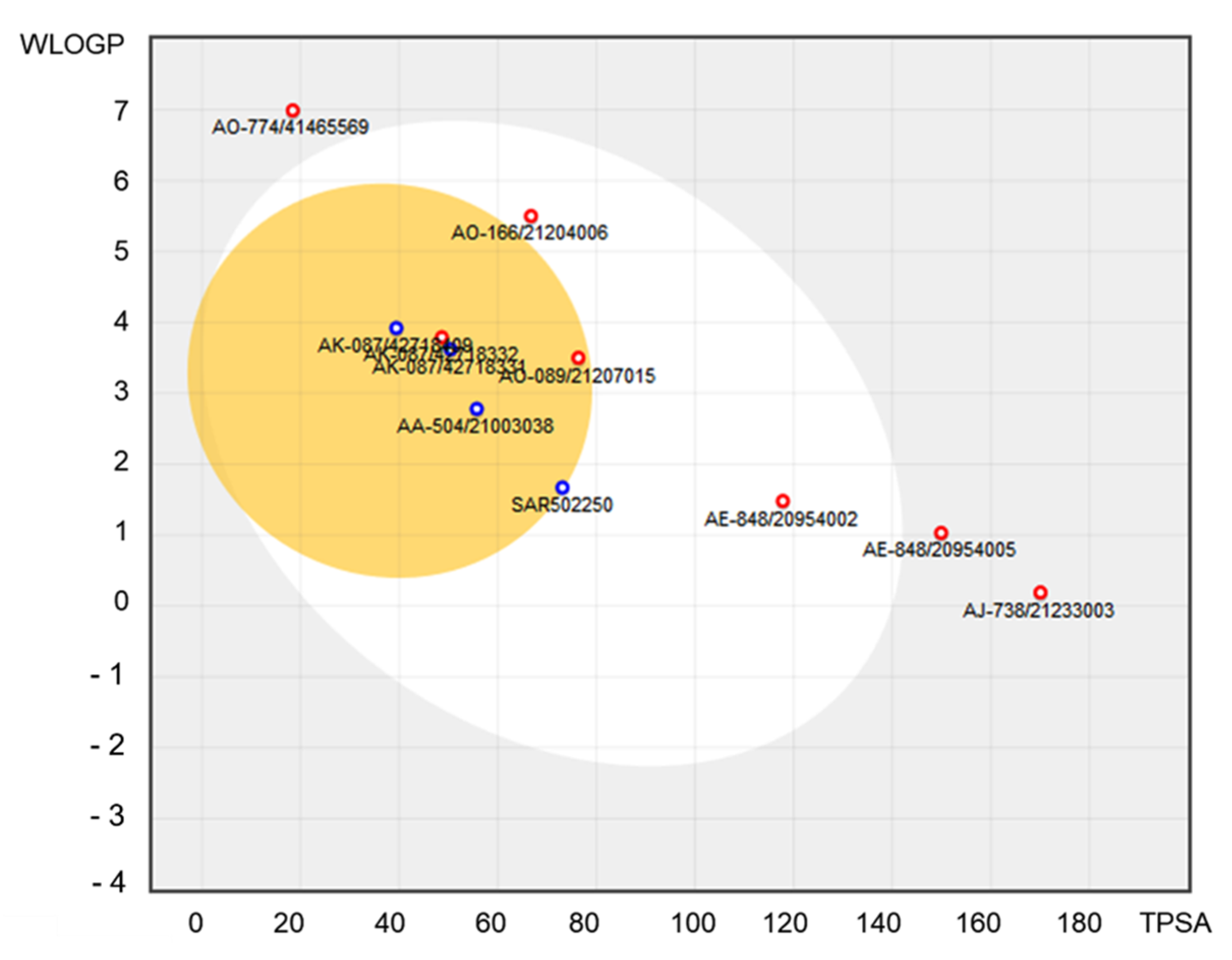
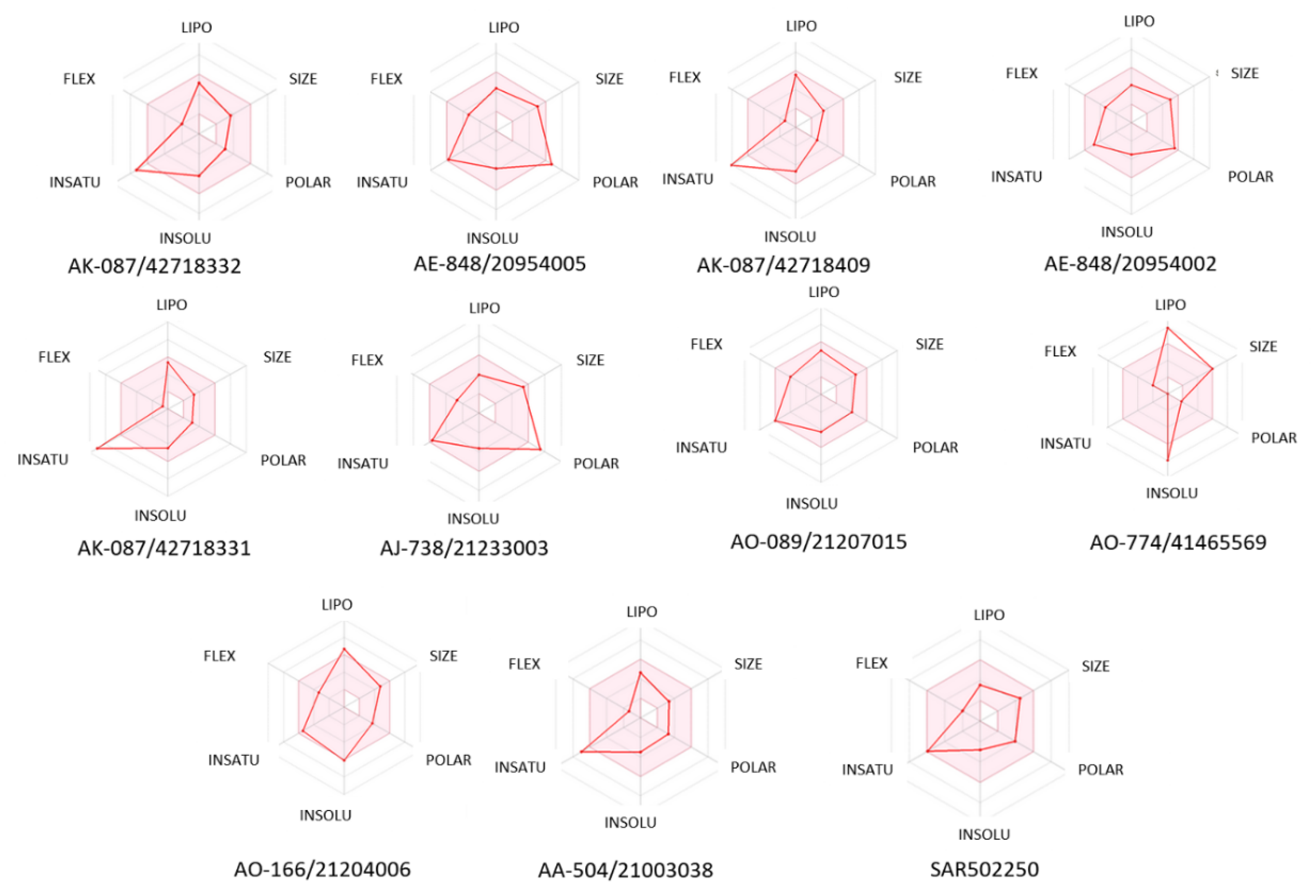
The blood-brain barrier (BBB) permeation and passive gastrointestinal absorption (HIA) were predicted at the same time with SwissADME software for all 10 natural compounds. The so-called BOILED-Egg model was constructed (Figure 6). This is a simple and intuitive graph prediction of HIA and BBB as a function of apparent polarity and lipophilicity (defined by TPSA (Topological Polar Surface Area) and WLOGP (lipophilicity descriptor—the partition coefficient between n-octanol and water), respectively). Also, the bioavailability radar (Figure 7) for each selected natural compound based on size, polarity, lipophilicity, flexibility, solubility, and saturation was built. In this graph, the pink area represents the optimum range for each property: MW between 150 and 500 g/mol, TPSA between 20 and 130 Å2, XLOGP3 between −0.7 and +5.0, no more than nine rotatable bonds, log S not higher than six, and the fraction of carbons in the sp3 hybridization not less than 0.25.
The molecules plotted in the white ellipse had a high probability of good intestinal absorption, while the molecules plotted in the yellow ellipse had a high probability of a good BBB crossing. The molecules which are located in the grey area were predicted as not absorbed by the GI and as BBB non-permeant. The red and blue circles indicate a relation to predict the active efflux by the P-glycoprotein (PGP+ code for blue circle, and PGP− for red circle).
The ADME and toxicity risk profiles suggest that AK-087/42718409, AK-087/42718331, and AA-504/21003038 exhibit excellent drug-like properties, similar to those of selective inhibitor SAR202250 (Table 1, Figure 6). The AK-087/42718331 compound showed only the mutagenic effect risk. In Figure 7, we observe that AE-848/20954002 and AO-089/21207015 compounds are entirely plotted in the pink area like SAR502250 and may be considered to have drug-like properties.
4. Conclusions
In this study, a 3D similarity search, ADME, and toxicity-related predictions were employed to identify novel natural products from the SPECS NP database, with similar profiles compared to those of the GSK-3 selective inhibitor, SAR202250. Three out of 13 3D similarity coefficients (TanimotoCombo, ShapeTanimoto, and ComboScore), together with ADME and toxicity risk analyzed parameters, indicated five SPECS natural products (AK-087/42718409, AK-087/42718331, AA-504/21003038, AE-848/20954002, and AO-089/21207015) as displaying great predicted drug-like properties and pharmacological profiles. These five natural compounds are recommended to be analyzed in-depth as an option for Alzheimer’s disease treatment. We assume that the 3D similarity search in conjunction with pharmaceutical profiles can be applied as a starting routine in the discovery of potentially selective GSK-3 inhibitors.
Author Contributions
L.C. and A.B. conceived of the presented ideas, performed the computational studies, and edited the manuscript; L.P. analyzed some resulting data. All authors discussed the outcomes, contributed to the writing of the paper, and commented on the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
The authors thank ChemAxon Ltd., OpenEye Ltd., and BIOVIA software Inc. (Discovery Studio Visualizer) for providing the academic license. The authors wish to thank Schrödinger Inc. for providing an academic trial license to complete the calculations for this paper. Project No. 1.2 of the “Coriolan Dragulescu” Institute of Chemistry, Timisoara, financially supported the current work.
Conflicts of Interest
The authors indicate no potential conflict of interest.
References
- Duda, P.; Akula, S.M.; Abrams, S.L.; Steelman, L.S.; Martelli, A.M.; Cocco, L.; Ratti, S.; Candido, S.; Libra, M.; Montalto, G.; et al. Targeting GSK3 and Associated Signaling Pathways Involved in Cancer. Cells 2020, 9, 1110. [Google Scholar] [CrossRef] [PubMed]
- Lal, H.; Ahmad, F.; Woodgett, J.; Force, T. The GSK-3 Family as Therapeutic Target for Myocardial Diseases. Circ. Res. 2015, 116, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Kazi, A.; Xiang, S.; Yang, H.; Delitto, D.; Trevino, J.; Jiang, R.H.J.; Ayaz, M.; Lawrence, H.R.; Kennedy, P.; Sebti, S.M. GSK3 suppression upregulates β-catenin and c-Myc to abrogate KRas-dependent tumors. Nat. Commun. 2018, 9, 5154. [Google Scholar] [CrossRef] [PubMed]
- Nabben, M.; Neumann, D. GSK-3 Inhibitors: Anti-Diabetic Treatment Associatedwith Cardiac Risk? Cardiovasc. Drugs 2016, 30, 233–235. [Google Scholar] [CrossRef] [PubMed]
- Griebel, G.; Stemmelin, J.; Lopez-Grancha, M.; Boulay, D.; Boquet, G.; Slowinski, F.; Pichat, P.; Beeské, S.; Tanaka, S.; Mori, A.; et al. The selective GSK3 inhibitor, SAR502250, displays neuroprotective activity and attenuates behavioral impairments in models of neuropsychiatric symptoms of Alzheimer’s disease in rodents. Sci. Rep. 2019, 9, 18045. [Google Scholar] [CrossRef]
- Maqbool, M.; Mobashir, M.; Hoda, N. Pivotal role of glycogen synthase kinase-3: A therapeutic target for Alzheimer’s disease. Eur. J. Med. Chem. 2016, 107, 63–81. [Google Scholar] [CrossRef]
- Ivan, D.; Crisan, L.; Funar-Timofei, S.; Mracec, M. A quantitative structure-activity relationships study for the anti-HIV-1 activities of 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine derivatives using the multiple linear regression and partial least squares methodologies. J. Serb. Chem. Soc. 2013, 78, 495–506. [Google Scholar] [CrossRef]
- Smith, D.G.; Buffet, M.; Fenwick, A.E.; Haigh, D.; Ife, R.J.; Saunders, M.; Slingsby, B.P.; Stacey, R.; Ward, R.W. 3-Anilino-4-arylmaleimides: Potent and selective inhibitors of glycogen synthase kinase-3 (GSK-3). Bioorg. Med. Chem. Lett. 2001, 11, 635–639. [Google Scholar] [CrossRef]
- Polychronopoulos, P.; Magiatis, P.; Skaltsounis, A.L.; Myrianthopoulos, V.; Mikros, E.; Tarricone, A.; Musacchio, A.; Roe, S.M.; Pearl, L.; Leost, M.; et al. Structural basis for the synthesis of indirubins as potent and selective inhibitors of glycogen synthase kinase-3 and cyclin-dependent kinases. J. Med. Chem. 2004, 47, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Kunick, C.; Lauenroth, K.; Leost, M.; Meijer, L.; Lemcke, T. 1-Azakenpaullone is a selective inhibitor of glycogen synthase kinase-3. Bioorg. Med. Chem. Lett. 2004, 14, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, N.; Mouawad, L.; Legraverend, M. Novel 8-arylated purines as inhibitors of glycogen synthase kinase. Eur. J. Med. Chem. 2010, 45, 3389–3393. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.C.; Gaisina, I.N.; El-Khodor, B.F.; Ramboz, S.; Makhortova, N.R.; Rubin, L.L.; Kozikowski, A.P. Identification of a maleimide-based glycogen synthase kinase-3 (GSK-3) inhibitor, BIP-135, that prolongs the median survival time of D7 SMA KO mouse model of spinal muscular atrophy. ACS Chem. Neurosci. 2012, 3, 5–11. [Google Scholar] [CrossRef]
- Sorokina, M.; Steinbeck, C. Review on natural products databases: Where to find data in 2020. J. Cheminform. 2020, 12, 20. [Google Scholar] [CrossRef]
- Specs. Compound Management Services and Research Compounds for the Life Science Industry. Available online: https://www.specs.net/index.php (accessed on 12 February 2020).
- Hawkins, P.C.D.; Skillman, A.G.; Nicholls, A. Comparison of Shape-Matching and Docking as Virtual Screening Tools. J. Med. Chem. 2007, 50, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Rush, T.S.; Grant, J.A.; Mosyak, L.; Nicholls, A. A shape-based 3-D scaffold hopping method and its application to a bacterial protein−protein interaction. J. Med. Chem. 2005, 48, 1489–1495. [Google Scholar] [CrossRef]
- Schrödinger, LLC. LigPrep; Schrödinger, LLC: New York, NY, USA, 2014. [Google Scholar]
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef]
- Hawkins, P.C.D.; Nicholls, A. Conformer generation with OMEGA: Learning from the data set and the analysis of failures. J. Chem. Inf. Model. 2012, 52, 2919–2936. [Google Scholar] [CrossRef] [PubMed]
- Osiris. Osiris Property Explorer. Available online: http://www.organic-chemistry.org/prog/peo/ (accessed on 3 August 2020).
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, druglikeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Muchmore, S.W.; Debe, D.A.; Metz, J.T.; Brown, S.P.; Martin, Y.C.; Hajduk, P.J. Application of Belief Theory to Similarity Data Fusion for Use in Analog Searching and Lead Hopping. J. Chem. Inf. Model. 2008, 48, 941–948. [Google Scholar] [CrossRef]
- Bortolato, A.; Perruccio, F.; Moro, S. Successful Applications of In Silico Approaches for Lead/Drug Discovery; Miteva, M.A., Ed.; Bentham Science Publishers: Sharjah, United Arab Emirates, 2011. [Google Scholar]
Figure 1.
Workflow diagram.

Figure 2.
The structure of the query compound.

Figure 3.
The minimum, maximum, and average 3D similarity coefficient values for SPECS NP concerning the SAR502250 query; for easier graphical representation, the values of the ColorScore coefficient were divided by −5, while the Overlap coefficients values were divided by −1000.
Figure 3.
The minimum, maximum, and average 3D similarity coefficient values for SPECS NP concerning the SAR502250 query; for easier graphical representation, the values of the ColorScore coefficient were divided by −5, while the Overlap coefficients values were divided by −1000.

Figure 4.
ROCS overlay of the top ten natural products prioritized against SAR502250, ordered by TanimotoCombo (see the gray arrow directions).
Figure 4.
ROCS overlay of the top ten natural products prioritized against SAR502250, ordered by TanimotoCombo (see the gray arrow directions).

Figure 5.
The distribution of 3D similarity coefficients (TanimotoCombo, ShapeTanimoto, ComboScore) values for SPECS NP against SAR502250.
Figure 5.
The distribution of 3D similarity coefficients (TanimotoCombo, ShapeTanimoto, ComboScore) values for SPECS NP against SAR502250.

Figure 6.
The WLOGP–TPSA referential for predicted SPECS NP.

Figure 7.
The bioavailability radar of predicted compounds for SPECS NP and SAR502250.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Physiochemical parameters and toxicity-related risk profiles of the 10 SPECS NP predicted compounds by SwissADME and OSIRIS Property Explorer software *. The 3D similarity coefficients (TanimotoCombo, ShapeTanimoto, ComboScore) were calculated with ROCS.
Table 1.
Physiochemical parameters and toxicity-related risk profiles of the 10 SPECS NP predicted compounds by SwissADME and OSIRIS Property Explorer software *. The 3D similarity coefficients (TanimotoCombo, ShapeTanimoto, ComboScore) were calculated with ROCS.
| Molecule | AK-087/ 42718332 | AE-848/ 20954005 | AK-087/ 42718409 | AE-848/ 20954002 | AK-087/ 42718331 | AJ-738/ 21233003 | AO-089/ 21207015 | AO-774/ 41465569 | AO-166/ 21204006 | AA-504/ 21003038 | SAR502250 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| BBB permeant | Yes | No | Yes | No | Yes | No | Yes | No | No | Yes | Yes |
| MW | 296.32 | 414.39 | 281.28 | 412.4 | 267.26 | 447.37 | 372.37 | 500.5 | 392.49 | 270.28 | 367.38 |
| RBN | 3 | 5 | 2 | 5 | 1 | 4 | 6 | 3 | 5 | 2 | 3 |
| MR | 85.87 | 100.26 | 79.51 | 102.12 | 75.04 | 106.24 | 100.38 | 126.33 | 116.99 | 74.02 | 100.96 |
| TPSA | 48.67 | 149.91 | 39.44 | 117.82 | 50.44 | 170.05 | 76.36 | 18.46 | 66.76 | 55.76 | 73.14 |
| XLOGP3 | 3.49 | 2.14 | 4.21 | 1.66 | 3.88 | 1.46 | 3.25 | 8.3 | 6.15 | 2.65 | 0.68 |
| WLOGP | 3.79 | 1.03 | 3.92 | 1.48 | 3.62 | 0.19 | 3.5 | 6.99 | 5.5 | 2.78 | 1.67 |
| GI absorption | High | Low | High | High | High | Low | High | Low | High | High | High |
| Lipinski #violations | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 2 | 0 | 0 | 0 |
| TanimotoCombo | 1.075 | 1.049 | 1.04 | 1.025 | 1.013 | 1.009 | 1.007 | 1.006 | 0.997 | 0.996 | 2 |
| ShapeTanimoto | 0.651 | 0.769 | 0.65 | 0.739 | 0.63 | 0.802 | 0.681 | 0.696 | 0.695 | 0.611 | 1 |
| ComboScore | 1.18 | 1.304 | 1.18 | 1.332 | 1.153 | 1.392 | 1.2 | 1.143 | 1.211 | 1.136 | 2 |
| Toxicity risk * | • M | • M | • M | • M | • M | • M | • M | • M | • M | • M | • M |
| • T | • T | • T | • T | • T | • T | • T | • T | • T | • T | • T | |
| • I | • I | • I | • I | • I | • I | • I | • I | • I | • I | • I | |
| • RE | • RE | • RE | • RE | • RE | • RE | • RE | • RE | • RE | • RE | • RE |
* M = mutagenic; T = tumorigenic; I = irritant; RE = reproductive effective; the red color circles designate properties with high risks of undesired effects, like reproductive or irritant effect, or mutagenicity, while the green color circles indicate drug-like conforming behavior; BBB (Blood Brain Barrier); MW (Molecular Weight); RBN (Rotatable Bonds); MR (Molar Refractivity); TPSA (Topological Polar Surface Area); XLOGP3 and WLOGP (lipophilicity descriptor - the partition coefficient between n-octanol and water); GI (Gastrointestinal Absorption).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Crisan, L.; Bora, A.; Pacureanu, L. Identification of Natural Products for the Treatment of Alzheimer’s Disease: 3D Similarity Search. Chem. Proc. 2021, 3, 75. https://0-doi-org.brum.beds.ac.uk/10.3390/ecsoc-24-08341
AMA Style
Crisan L, Bora A, Pacureanu L. Identification of Natural Products for the Treatment of Alzheimer’s Disease: 3D Similarity Search. Chemistry Proceedings. 2021; 3(1):75. https://0-doi-org.brum.beds.ac.uk/10.3390/ecsoc-24-08341
Chicago/Turabian StyleCrisan, Luminita, Alina Bora, and Liliana Pacureanu. 2021. "Identification of Natural Products for the Treatment of Alzheimer’s Disease: 3D Similarity Search" Chemistry Proceedings 3, no. 1: 75. https://0-doi-org.brum.beds.ac.uk/10.3390/ecsoc-24-08341