
Synthesis of (2S,3S)-3-Aroyl Pyroglutamic Acid Amides †


Department of Organic Chemistry, Slovak Technical University, Radlinského 9, SK-812 37 Bratislava, Slovakia
*
Authors to whom correspondence should be addressed.
†
Presented at the 24th International Electronic Conference on Synthetic Organic Chemistry,
15 November–15 December 2020; Available online: https://ecsoc-24.sciforum.net/.
Chem. Proc. 2021, 3(1), 86; https://0-doi-org.brum.beds.ac.uk/10.3390/ecsoc-24-08377
Published: 14 November 2020
(This article belongs to the Proceedings of The 24th International Electronic Conference on Synthetic Organic Chemistry)
Abstract
:A new methodology for the asymmetric synthesis of enantiomerically enriched 3-aroyl pyroglutamic acid derivatives has been developed through effective 5-exo-tet cyclization of N-chloroacetyl aroylalanines. The three-step sequence starts with the synthesis of N-substituted (S,S)-2-amino-4-aryl-4-oxobutanoic acids via highly diastereoselective tandem aza-Michael addition and crystallization-induced diastereomer transformation (CIDT). Their N-chloroacetylation followed by base-catalyzed cyclization and ultimate acid-catalyzed removal of chiral auxiliary without loss of stereochemical integrity furnishes the target substituted pyroglutamic acids. Finally, several series of their benzyl amides were prepared as 3-aroyl analogs of known P2X7 antagonists.
1. Introduction
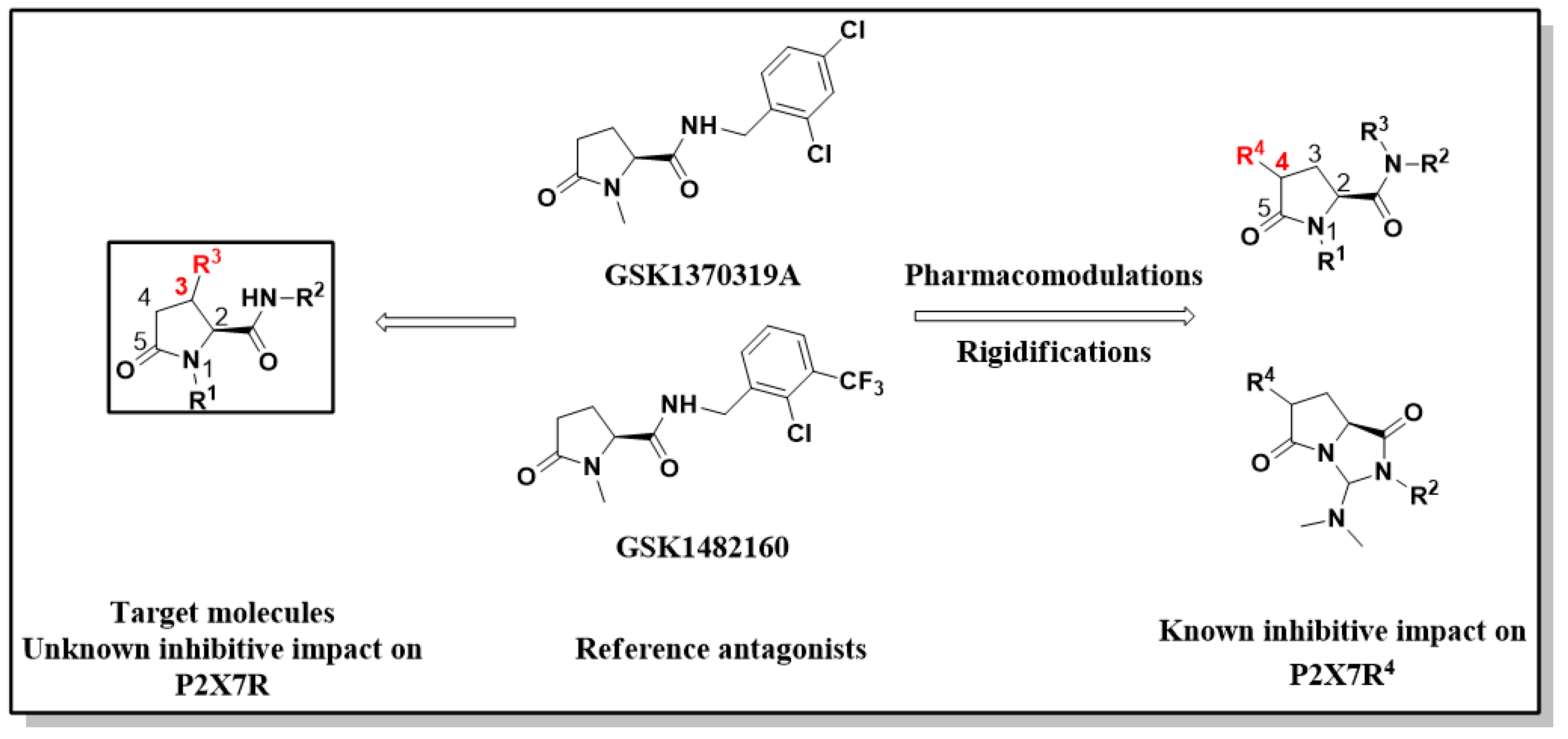
Pyroglutamic acid and its derivatives are a valuable class of compounds found in various natural products and pharmaceuticals, representing either an important chiral auxiliary or building block for the asymmetric synthesis of many biologically and pharmaceutically valuable compounds [1]. Moreover, pyroglutamic acid derivatives have recently appeared to be efficient antagonists of specific types of purinergic receptors. Their inhibition has a promising impact on treating neurodegenerative diseases such as Alzheimer, Huntington, and Parkinson’s disease [2]. This knowledge has led to various studies of the mentioned receptors based on the pyroglutamic derivatives’ inhibitive activity.
Among these, the inhibitive activity of two pyroglutamic compounds, GSK1370319A and GSK1482160, was confirmed by clinical tests [3]. Accordingly, various potential antagonists were designed. The affinity with receptors has been modulated by changing substituents in the first, second, and fourth position of the lactam ring (Scheme 1) [4]. Stereoselective preparation of 3-substituted analogs has not attracted significant attention. The preparation of enantiomerically pure 3-substituted pyroglutamic acid derivatives enables exploring the importance of substitution in the first and third positions of the lactam ring and their impact on biological activity.
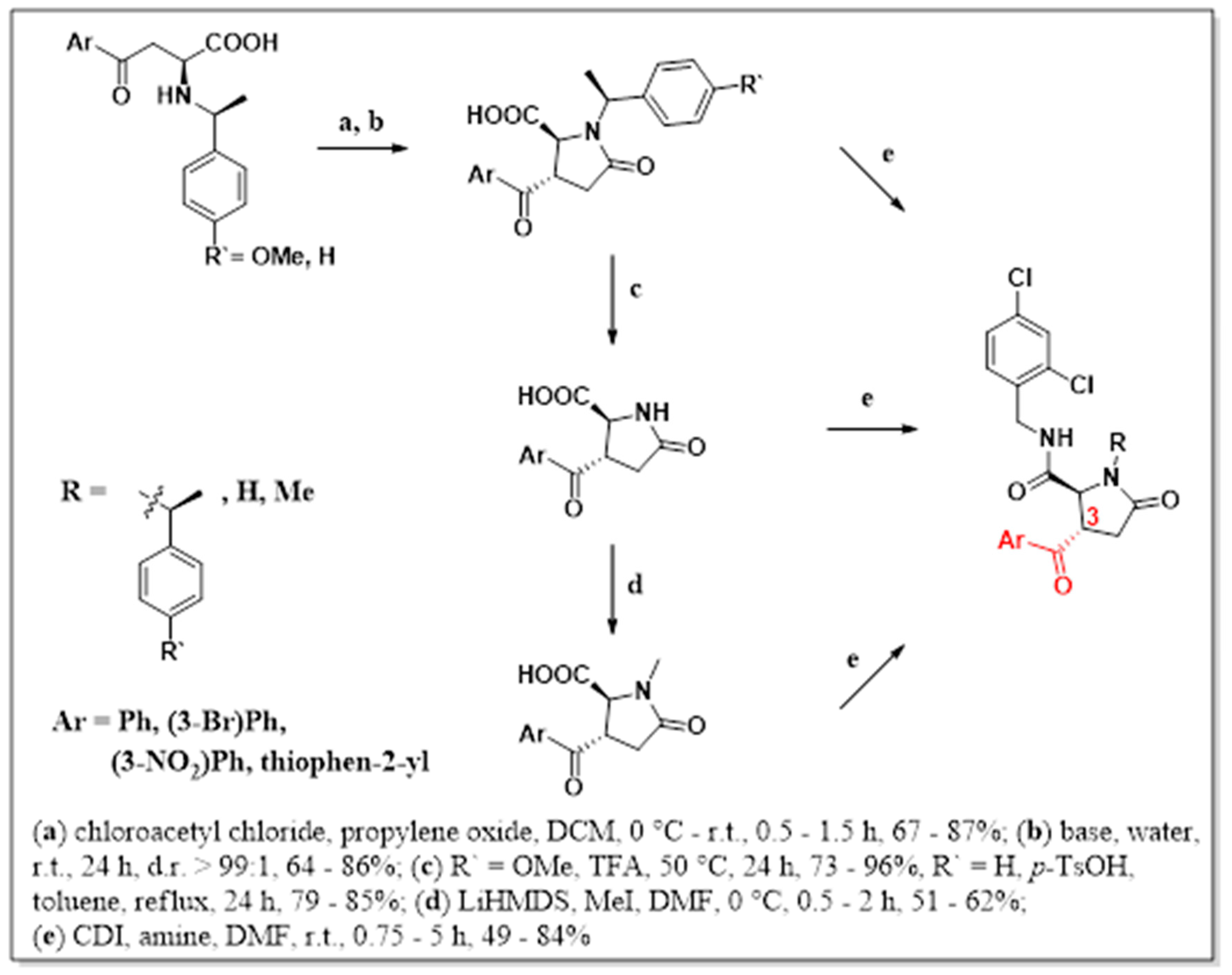
To our surprise, despite the numerous synthetic approaches towards substituted pyroglutamic acid derivatives and the widely recognized importance and utility of these compounds, the synthesis of 3-acyl substituted analogs is still relatively unexplored [5]. This led us to develop a novel approach toward synthesizing 3-aroyl pyroglutamic acid amides in a few steps (Scheme 2).
2. Results and Discussion
The initial step represents the aza-Michael addition of chiral mediators (2a,b) to appropriate aroyl acrylic acids (1a–d) in tandem with CIDT (crystallization-induced diastereomer transformation) (Scheme 3). This efficient methodology spread among our research group allows us to prepare aroyl alanines with high diastereoselectivity. The methodology is based on equilibration of stereoisomers in a solution, and continuous crystallization of a single isomer acts as a driving force for its gradual accumulation in the reaction suspension [6,7,8,9]. This enables isolation of products (3a–g) by simple filtration (Table 1).
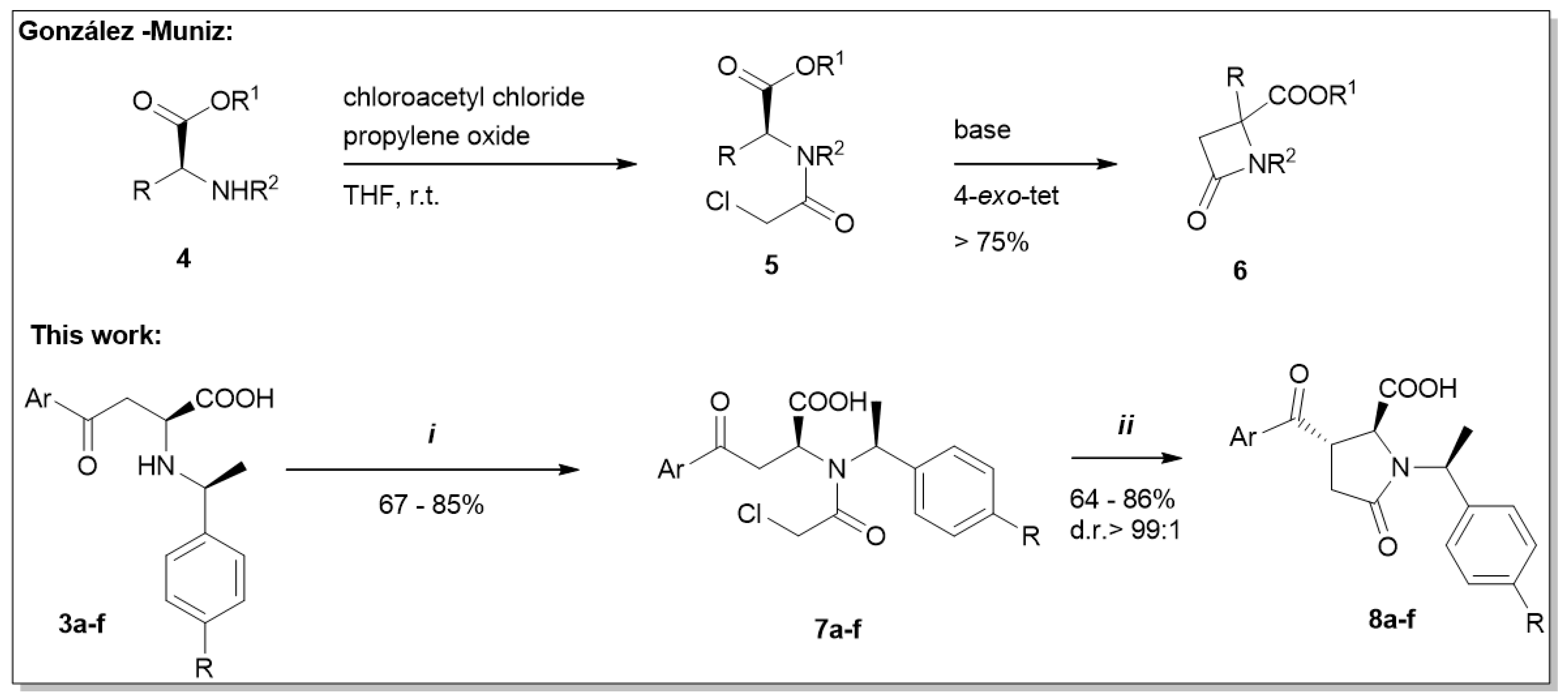
Recently, we published a strategy of N-acylation of amino acids in the presence of chloroacetyl chloride, which yielded the acetylated derivatives [10]. These intermediates represent suitable starting materials for cyclization in the presence of a base. It has been reported previously that N-chloroacetylated amino acids undergo 4-exo-tet cyclization to form β-lactams (6) (Scheme 4) [11]. Due to the occurrence of the enolizable ketone, we expected that the in situ formed enolate undergoes fast 5-exo-tet cyclization instead of the 4-exo-cyclization observed by González-Muniz [11] (Table 2).
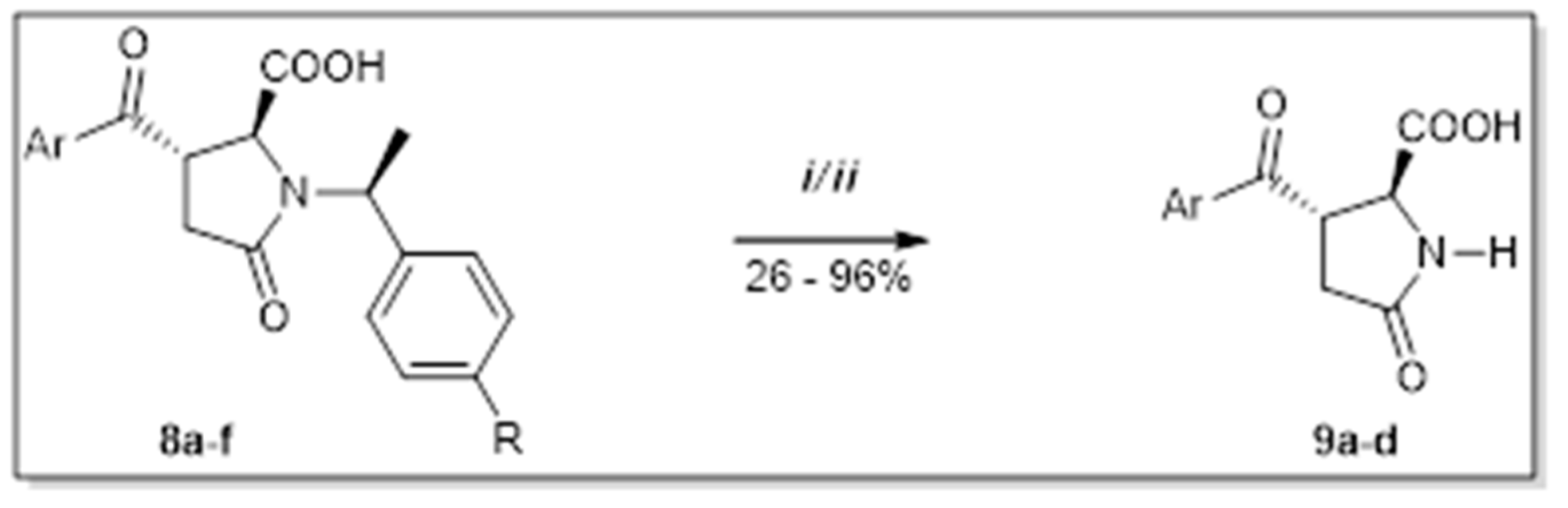
According to Abdi et al. [3], the occurrence of bulky benzylic group on the nitrogen of lactam ring causes inhibitive activity decline towards target receptors. Because of that, we decided to find the suitable conditions for debenzylation of the functional groups descended from the former chiral mediator used in the initial step (Scheme 5). In the case of removing the 1-ethyl-4-methoxybenzylic group, the reaction was successful under conditions i. On the contrary, the removal of the 1-ethylbenzylic group under the same conditions did not proceed, and according to HPLC analysis, there was no conversion to product observed. However, reactions took place in the presence of p-TsOH in refluxing toluene to yield deprotected derivatives 9b,d,f without loss of stereochemical integrity (Table 3).
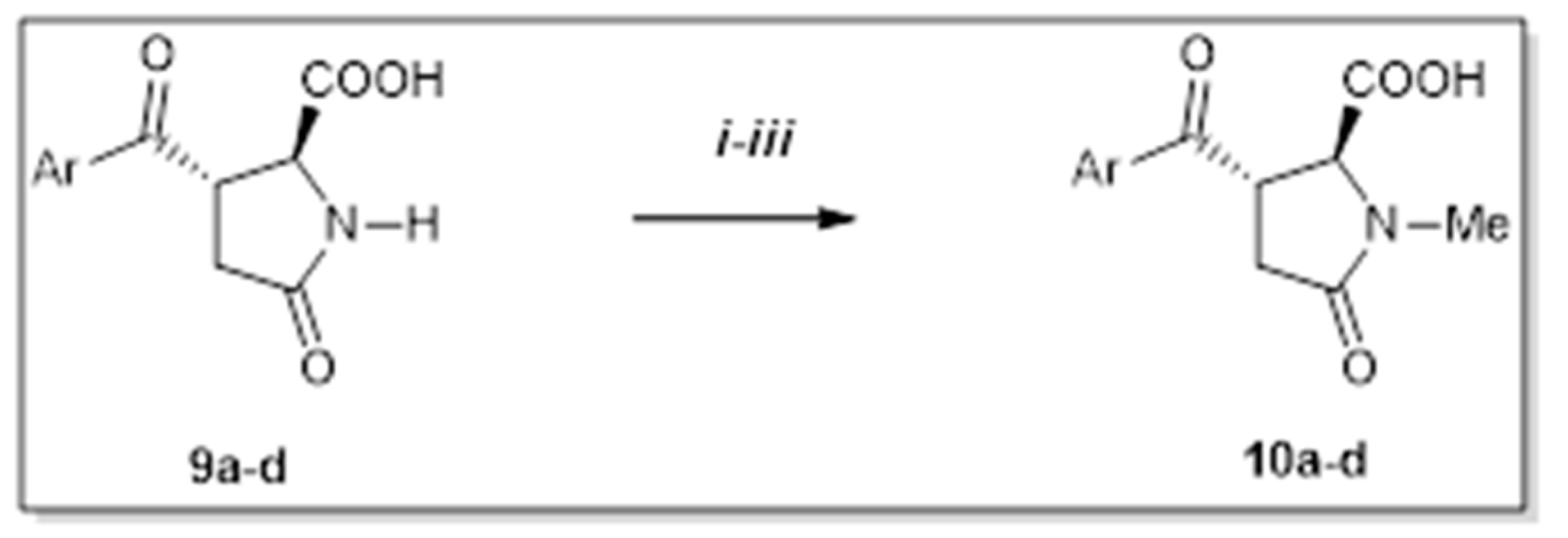
The second part of the synthetic approach represents the preparation of 3-aroyl-N-methyl pyroglutamic acids (10a–d), which covered up a sequence of three reactions (Scheme 6). The first step demands the preparation of methyl esters (Ma–d) due to the low solubility of starting deprotected derivatives (9a–d). The establishment of the methyl group on lactam nitrogen requires a basic environment. Due to the enolizable functional group occurrence, the importance of a sufficient amount of base and low temperature was essential. The 3-aroyl-N-methylated pyroglutamic acids (10a–d) were obtained after three steps with satisfactory overall yields (Table 4).
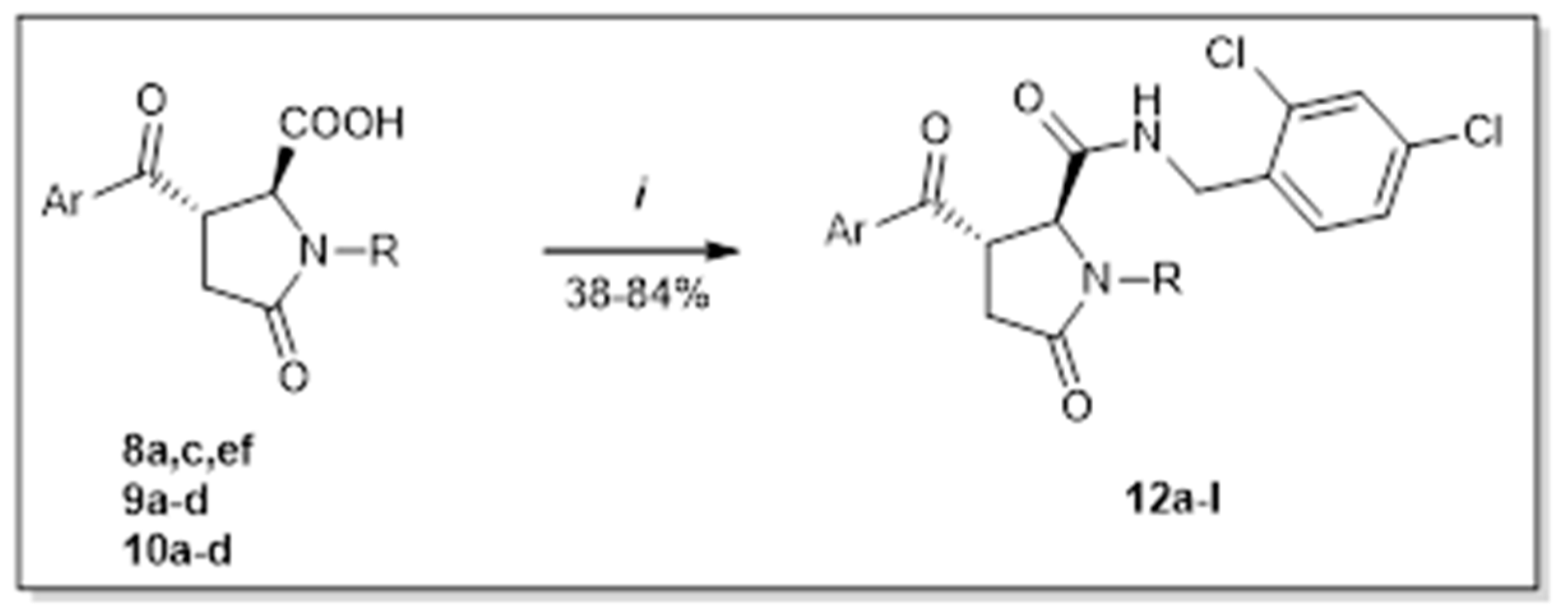
Another valuable source about the antagonism of P2X7 receptors delivered knowledge that disubstituted benzylamides of corresponding pyroglutamic acids are the most effective. We prepared four series of pyroglutamic acid amides that vary in substituents in the first and the third position of the lactam ring. These compounds were obtained as products of well-known acid amine coupling with corresponding activation agent—CDI (Scheme 7) [13]. The desired final products (12) were isolated in yields within the range of 34–84% (Table 5).
3. General Methods
Unless otherwise noted, all chemicals were purchased from commercial sources and used without further purification. Bulk solutions were evaporated under reduced pressure using a Büchi rotary evaporator. HPLC analyses were performed on Varian system using Phenomenex Phenyl-Hexyl 5 µm column. The used mobile phase is specified for each experiment. Due to the long-term use of columns, retention time values are only approximate.
mobile phase A: MeCN/H2O = 1:1 (500/500 mL) + Et3N/H3PO4 = 1:1 (10/10 mL)
mobile phase B: MeCN/H2O = 1:2 (333/666 mL) + Et3N/H3PO4 = 1:1 (10/10 mL)
mobile phase C: MeCN/H2O = 1:3 (250/75 + Et3N/H3PO4 = 2:1 (10/5 mL)
mobile phase D: MeCN/H2O = 2:3 (400/600 mL) + Et3N/H3PO4 = 1:1 (10/10 mL)
Column chromatography was carried out using Silica 60A, particle size 20–45 micron, Davisil, purchased from Fisher Chemical. All reactions were followed by thin-layer chromatography (TLC) where practical, using Macherey-Nagel’s precoated TLC sheets POLYGRAM SIL G/UV254 visualized under UV light (254 nm) or by staining with aqueous basic potassium permanganate or cerium molybdate solutions as appropriate.
All 1H and 13C NMR spectra were recorded using a Varian INOVA 300 MHz and Varian VNMRS 600 MHz spectrometers. Chemical shifts (δ) are given in parts per million (ppm). The 1H NMR chemical shift scale is referenced to TMS internal standard or solvent residual peak. The 13C NMR chemical shift scale is referenced to the solvent peak. Coupling constants (J) are given in hertz (Hz). The multiplicity of 1H NMR signals is reported as follows: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, bs = broad singlet. Optical rotations were recorded using a JASCO P-2000 polarimeter; [α]D values are reported in deg cm3g−1dm−1; concentration (c) is given in g/100 mL at 589 nm. HRMS were measured using Thermo Scientific mass spectrometer with Orbitrap analyzer and HESI and APPI ionization.
4. Experimental
4.1. Procedure A
Michael addition
Starting aroyl acrylic acids 1a–d were prepared according to general procedures described in the literature [14,15,16]. The synthetic strategy and the general procedures are shown on phenyl derivatives (Ar = Ph).
(S)-2-(((S)-1-(4-Methoxyphenyl)ethyl)amino)-4-oxo-4-phenylbutanoic acid—3a
((E)-4-oxo-4-Phenylbut-2-enoic acid (1a, 25.0 g, 0.142 mol) was dissolved in methanol (570 mL) followed by the addition of (S)-1-(4-methoxyphenyl)ethanamine (2b, 1.2 equiv, 0.170 mol, 25 mL). Mixture was stirred for 48 h at 40 °C and monitored by HPLC. The suspension was filtered, washed with Et2O, yielding 3a as a colorless solid (40.0 g, 84 %, d.r. 98:2); m.p. = 174–177 °C, [α]D25 = +58.5 (c 1.0, MeOH: (5 % aq. HCl) 9:1);
HPLC: (mobile phase B): tR = 7.0 min, flow rate 0.7 mL/min;
1H NMR (300 MHz, acetone-d6 + DCl): δ = 8.03–7.91 (m, 2H), 7.69 (d, J = 8.8 Hz, 2H), 7.66–7.59 (m, 1H), 7.55–7.47 (m, 2H), 7.01 (d, J = 8.9 Hz, 2H), 4.87 (q, J = 6.8 Hz, 1H), 4.17 (t, J = 5.2 Hz, 1H), 4.02 (m, 2H), 3.82 (s, 3H), 1.87 (d, J = 6.9 Hz, 3H);
13C NMR (75 MHz, acetone-d6 + DCl): δ = 196.4, 169.7, 161.3, 136.7, 134.4, 131.0, 129.5, 129.1, 128.5, 115.3, 59.3, 55.6, 53.6, 39.7, 21.0.
Accordingly, derivatives 3b–f were prepared.
4.2. Procedure B
N-Chloroacetylation
(S)-2-(2-Chloro-N-((S)-1-(4-methoxyphenyl)ethyl)acetamido)-4-oxo-4-phenylbutanoic acid—7a
(S)-4-oxo-4-Phenyl-2-(((S)-1-phenylethyl)amino)butanoic acid (3a, 18.00 g, 55.0 mmol) was suspended in dichloromethane (275 mL) and cooled to 0 °C. Racemic propylene oxide (10.0 equiv, 0.672 mol, 38.5 mL) was added to the reaction mixture, followed by the solution of chloroacetyl chloride (1.1 equiv, 60.5 mmol, 4.8 mL) in dichloromethane (30 mL). The resulting mixture was stirred for 30 min at r.t. and concentrated under reduced pressure. The obtained crude product was triturated with ethyl acetate/hexane mixture to yield 7a as a pale-yellow solid (16.8 g, 75%); m.p. = 164–165 °C; TLC: Rf = 0.37 (EtOAc/MeOH);
HPLC: (mobile phase A): tR = 17.2 min, flow rate: 0.7 mL/min;
1H NMR (300 MHz, CDCl3): δ = 8.02–7.98 (m, 2H), 7.62–7.53 (m, 1H), 7.49–7.38 (m, 4H), 6.91 (d, J = 8.8 Hz, 2H), 5.19 (q, J = 6.9 Hz, 1H), 4.66 (dd, J = 7.7, 2.5 Hz, 1H), 4.46 (dd, J = 18.3, 7.7 Hz, 1H), 4.28 (d, J = 12.4 Hz, 1H), 4.19 (d, J = 12.4 Hz, 1H), 3.82 (s, 3H), 2.88 (dd, J = 18.3, 2.6 Hz, 1H), 1.60 (d, J = 7.0 Hz, 3H).
Accordingly, derivatives 7b–f were prepared.
4.3. Procedure C
Cyclization
(2S,3S)-3-Benzoyl-1-((S)-1-(4-methoxyphenyl)ethyl)-5-oxopyrrolidine-2-carboxylic acid—8a
(S)-2-(2-Chloro-N-((S)-1-(4-methoxyphenyl)ethyl)acetamido)-4-oxo-4-phenylbutanoic acid (7a, 16.8 g, 41.6 mmol) was dissolved in the solution of sodium hydroxide (2.5 equiv, 140 mmol, 4.14 g) and water (260 mL). The reaction mixture was stirred at r.t. for 24 h. Subsequently, the pH value of reaction mixture was adjusted to 2–3 with 1 M aqueous HCl. The precipitated product was filtered off and washed with water. Crude product was crystallized from MeOH to yield 8a as a colorless solid (11.3 g, 74% yield, d.r. ˃ 99:1); m.p. = 220–222 °C; TLC: Rf = 0.48 (EtOAc/MeOH = 3:2);
HPLC: (mobile phase A): tR = 9.3 min, flow rate: 0.7 mL/min;
1H NMR (300 MHz, DMSO-d6): δ = 8.08–7.90 (m, 2H), 7.75–7.64 (m, 1H), 7.60–7.51 (m, 2H), 7.22 (d, J = 8.7 Hz, 2H), 6.82 (d, J = 8.8 Hz, 2H), 5.01 (q, J = 7.0 Hz, 1H), 4.32 (d, J = 1.6 Hz, 1H), 4.24 (dd, J = 9.3, 1.6 Hz, 1H), 3.72 (s, 3H), 2.81 (dd, J = 16.7, 9.4 Hz, 1H), 2.34 (dd, J = 16.7, 2.0 Hz, 1H), 1.45 (d, J = 7.1 Hz, 3H).
Accordingly, derivatives 8b–f were prepared.
4.4. Procedure D
N-debenzylation—conditions i
(2S,3S)-3-Benzoyl-5-oxopyrrolidine-2-carboxylic acid—9a
Carboxylic acid (8a, 4 g, 10.9 mmol) was dissolved in TFA (24.0 equiv, 0.261 mol, 20.0 mL) and the resulting colorless reaction mixture was stirred at 50 °C for 24 h. The reaction was accompanied with color change from colorless to dark purple. Upon completion, TFA was evaporated under reduced pressure and the dark-purple crude product was triturated with Et2O yielding pale-brown solid (2.4 g, 96%); m.p. = 166–168 °C; TLC: Rf = 0.30 (EtOAc/MeOH = 3:2);
HPLC: (mobile phase B): tR = 8.0 min, flow rate: 0.7 mL/min;
1H NMR (300 MHz, DMSO-d6): δ = 8.12 (bs, 1H), 8.11–8.05 (m, 2H), 7.76–7.55 (m, 3H), 4.48–4.36 (m, 2H), 2.70 (dd, J = 16.8, 10.0 Hz, 1H), 2.27 (dd, J = 16.8, 4.9 Hz, 1H).
Accordingly, derivatives 9c,e were prepared.
N-Bebenzylation—conditions ii
(2S,3S)-3-(3-Nitrobenzoyl)-5-oxopyrrolidine-2-carboxylic acid—9b
Carboxylic acid (8d, 200 mg, 0.582 mmol) was dissolved in toluene (5.8 mL) and p-TsOH (4.0 equiv, 2.33 mmol, 443 mg) was added. The resulting colorless solution was stirred under reflux for 6 h. Reaction was accompanied with color change from colorless to dark brown, indicated styrene polymer formation. Upon completion, the reaction mixture was cooled down to room temperature and poured into water (10 mL). The resulting mixture was extracted with EtOAc (3 × 10 mL). The collected organic layers were washed with 10% solution of K2CO3 (3 × 15 mL). The pH value of water phase was adjusted to 2–3 and extracted with EtOAc (3 × 20 mL). The extract was dried over MgSO4 and concentrated under reduced pressure. Crude product was triturated with a small amount of Et2O (ca. 15 mL), yielding white powder (9b, 118 mg, 85%); m.p. = 221–223 °C; TLC: Rf = 0.33 (EtOAc/MeOH = 3:2);
HPLC: (mobile phase A): tR = 4.5 min, flow rate: 0.7 mL/min;
1H NMR (300 MHz, DMSO-d6): δ = 8.78–8.76 (m, 1H), 8.53–8.44 (m, 2H), 8.17 (bs, 1H), 7.91–7.83 (m, 1H), 4.60–4.51 (m, 1H), 4.40 (dd, J = 3.5, 0.8 Hz, 1H), 2.68 (dd, J = 16.8, 10.0 Hz, 1H), 2.33 (dd, J = 16.8, 4.7 Hz, 1H).
Accordingly, derivatives 9d,f were prepared.
4.5. Procedure E
N-Methylation
(2S,3S)-3-Benzoyl-1-methyl-5-oxopyrrolidine-2-carboxylic acid—10a
To a solution of carboxylic acid (9a, 1.5 g, 6.43 mmol) in dry DMF (21 mL) NaHCO3 (3.0 equiv, 19.3 mmol, 1.61 g) and MeI (5 equiv, 32.2 mmol, 2.0 mL) was added. The reaction mixture was stirred 24 h at 50 °C. Upon completion, a concentrated solution of NH4Cl (60 mL) was added to the reaction mixture, which was consequently extracted with DCM (3 × 40 mL). Collected organic layers were dried over MgSO4 and concentrated under reduced pressure. The crude product was purified by flash column chromatography (100% EtOAc). The obtained colorless solid, methylester Ma (1.35 g, 85%), was used in the subsequent step.
1H NMR (300 MHz, DMSO-d6): δ = 8.22 (bs, 1H), 8.10–7.97 (m, 2H), 7.75–7.65 (m, 1H), 7.62–7.52 (m, 2H), 4.55–4.44 (m, 2H), 3.69 (s, 3H), 2.72 (dd, J = 16.9, 10.0 Hz, 1H), 2.25 (dd, J = 16.9, 4.7 Hz, 1H).
Ma (265 mg, 1.07 mmol) was dissolved in dried DMF (10 mL), cooled down to 0 °C, and LiHMDS (1.3 equiv, 1.39 mmol, 1.4 mL) was added. After 10 min of stirring, MeI (1.3 equiv, 1.39 mmol, 87 μL) was added, and the reaction mixture stirred under an argon atmosphere at 0 °C. After 50 min, a concentrated solution of NH4Cl (50 mL) was added, and resulting solution was extracted with DCM (3 × 60 mL). Collected organic layers were dried over MgSO4 and concentrated under reduced pressure. Crude product was purified by flash column chromatography (60% EtOAc) resulting in a pale-yellow solid substance—N-methylated intermediate (Na,174 mg, 62%); TLC: Rf = 0.4 (EtOAc);
HPLC: (mobile phase B): tR: 22.0 min, flow rate 0.7 mL/min;
1H NMR (300 MHz, CDCl3): δ = 7.98–7.93 (m, 2H), 7.67–7.59 (m, 1H), 7.55–7.48 (m, 2H), 4.70 (d, J = 3.2 Hz, 1H), 4.16 (ddd, J = 10.4, 4.2, 3.3 Hz, 1H), 3.80 (s, 3H), 2.93 (s, 3H), 2.90 (dd, J = 17.0, 11.0 Hz, 1H), 2.57 (dd, J = 17.2, 3.7 Hz, 1H);
13C NMR (151 MHz, CDCl3): δ = 196.22, 172.19, 171.61, 134.36, 134.13, 129.10, 128.87, 62.53, 52.87, 42.55, 33.77, 29.33.
N-Methylated derivative (Na, 300 mg, 1.15 mmol) was dissolved in a mixture of THF/H2O (1:1, 12 mL) and LiOH (2.0 equiv, 2.3 mmol, 55 mg) was added. The reaction mixture was stirred at r.t. for 30 min. Upon completion, water (10 mL) was added to a reaction mixture and pH was adjusted to 2–3 with 1 M HCl. The resulting solution was extracted with EtOAc (3 × 15 mL). The collected organic layers were dried over MgSO4 and concentrated under reduced pressure resulting desired colorless solid substance 10a (237 mg, 84%).
1H NMR (300 MHz, DMSO-d6) δ = 13.30 (s, 1H), 8.12–8.00 (m, 2H), 7.74–7.65 (m, 1H), 7.62–7.51 (m, 3H), 4.44 (d, J = 3.0 Hz, 1H), 4.40–4.33 (m, 1H), 2.87–2.69 (m, 5H), 2.34 (dd, J = 16.9, 3.3 Hz, 1H).
Accordingly, derivatives 10b–d were prepared.
4.6. Procedure F
Amide formation
(2S,3S)-3-Benzoyl-N-(2,4-dichlorobenzyl)-1-((S)-1-(4-methoxyphenyl)ethyl)-5-oxopyrrolidine-2-carboxamide—12a
Carboxylic acid (8a, 300 mg, 0.817 mol) was dissolved in dry DMF (5.4 mL) and CDI (1.2 equiv, 0.980 mol, 159 mg) was added. After 10 min, amine (11, 1.2 equiv, 0.980 mol, 0.132 mL) was added and the reaction mixture was stirred at r.t. for 45 min. Upon completion, the reaction was quenched with a 1 M solution of KHSO4 (10 mL) and extracted with EtOAc (3 × 30 mL). The collected organic layers were washed with water (30 mL) and with brine (30 mL) and concentrated under reduced pressure. Crude product was purified on flash column chromatography (80% EtOAc, 20% Hex). Product 12a was isolated as colorless solid substance (298 mg, 70%); m.p. = 136–137 °C; TLC: Rf = 0.49 (EtOAc);
HPLC: (mobile phase A): tr = 39.6 min, flow rate: 0.7 mL/min;
1H NMR (300 MHz, CDCl3): δ = 8.16–7.87 (m, 2H), 7.65–7.56 (m, 1H), 7.54–7.46 (m, 2H, H), 7.33 (d, J = 2.1 Hz, 1H), 7.26 (d, J = 8.6 Hz, 2H), 7.16 (dd, J = 8.2, 2.1 Hz, 1H), 6.97 (d, J = 8.2 Hz, 1H), 6.77 (d, J = 8.8 Hz, 2H), 5.60 (t, J = 6.1 Hz, 1H), 5.43 (q, J = 7.0 Hz, 1H), 4.51 (d, J = 1.6 Hz, 1H), 4.14 (dd, J = 14.7, 6.3 Hz, 1H), 4.09–4.04 (m, 1H), 3.85 (dd, J = 14.7, 5.6 Hz, 1H), 3.77 (s, 3H), 2.87 (dd, J = 16.9, 9.3 Hz, 1H), 2.61 (dd, J = 16.9, 2.2 Hz, 1H), 1.50 (d, J = 7.1 Hz, 3H).
(2S,3S)-3-Benzoyl-N-(2,4-dichlorobenzyl)-5-oxopyrrolidine-2-carboxamide—12b
Amide 12b was prepared according to procedure B from corresponding acid 9a. Reaction time—30 min. Colorless solid substance, (325 mg, 65%); m.p. = 134–136 °C; TLC: Rf = 0.33 (EtOAc);
HPLC: (mobile phase A): tr = 11.1 min, flow rate: 0.7 mL/min;
1H NMR (300 MHz, CDCl3): δ = 8.02–7.97 (m, 2H), 7.67–7.59 (m, 1H), 7.54–7.46 (m, 2H), 7.32 (d, J = 2.1 Hz, 1H), 7.27 (d, J = 8.3 Hz, 1H), 7.17 (dd, J = 8.2, 2.1 Hz, 1H), 7.13 (bs, 1H), 4.64 (dd, J = 4.6, 1.1 Hz, 1H), 4.48 (d, J = 6.0 Hz, 2H), 4.32 (ddd, J = 9.7, 6.0, 4.6 Hz, 1H), 2.70 (dd, J = 17.0, 9.6 Hz, 1H), 2.50 (dd, J = 17.0, 6.0 Hz, 1H).
(2S,3S)-3-Benzoyl-N-(2,4-dichlorobenzyl)-1-methyl-5-oxopyrrolidine-2-carboxamide—12c
Amide 12c was prepared according to procedure B from corresponding acid 10a. Reaction time—30 min. Pale-yellow foam, (327 mg, 84%); TLC: Rf = 0.32 (EtOAc);
HPLC: (mobile phase A): tr = 13.4 min, flow rate: 0.7 mL/min;
1H NMR (300 MHz, CDCl3): δ = 7.99–7.83 (m, 2H), 7.68–7.59 (m, 1H), 7.54–7.45 (m, 2H), 7.34 (d, J = 2.0 Hz, 1H), 7.28 (d, J = 8.8 Hz, 1H), 7.19 (dd, J = 8.2, 2.1 Hz, 1H), 6.67 (t, J = 5.8 Hz, 2H), 4.64 (d, J = 5.9 Hz, 1H), 4.51 (d, J = 5.0 Hz, 1H), 4.50 (d, J = 5.9 Hz, 1H), 4.08 (ddd, J = 10.3, 6.4, 5.0 Hz, 1H), 2.91 (dd, J = 16.9, 10.3 Hz, 1H), 2.84 (s, 3H), 2.51 (dd, J = 16.7, 6.2 Hz, 1H).
Accordingly, derivatives 12d–l were prepared.
Author Contributions
Synthetic investigation, analytical work, writing—original draft preparation L.P.; Conceptualization, supervision, synthetic investigation, D.B. All authors have read and agreed to the published version of the manuscript.
Funding
This project was supported by the Slovak Research and Development Agency under contract No. APVV-16-0258 and VEGA 1/0489/19.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Khopade, T.M.; Warghude, P.K.; Sonawane, A.D.; Bhat, R.G. Multicomponent synthesis of pyroglutamic acid derivatives via Knoevenagel–Michael-hydrolysis-lactamization-decarboxylation (KMHL-D) sequence. Org. Biomol. Chem. 2019, 17, 561. [Google Scholar] [CrossRef] [PubMed]
- Sperlagh, B.; Illes, P. P2X7 receptor: An emerging target in central nervous system diseases. Trends. Pharmacol. Sci. 2014, 35, 537. [Google Scholar] [CrossRef] [PubMed]
- Abdi, M.H.; Beswick, P.J.; Billinton, A.; Chambers, L.J.; Charlton, A.; Collins, S.D.; Collis, K.L.; Dean, D.K.; Fonfria, E.; Gleave, R.J.; et al. Discovery and structure-activity relationships of a series of pyroglutamic acid amide antagonists of the P2X7 receptor. Bioorg. Med. Chem. Lett 2010, 20, 5080. [Google Scholar] [CrossRef] [PubMed]
- Homerin, G.; Jawhara, S.; Dezitter, X.; Baudelet, D.; Dufrénoy, P.; Rigo, B.; Millet, R.; Furman, C.; Ragé, G.; Lipka, E.; et al. Pyroglutamide-Based P2X7 Receptor Antagonists Targeting Inflammatory Bowel Disease. J. Med. Chem. 2019, 63, 2074–2094. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Yang, X.; Li, S.; Zhu, Y.; Li, Y.; Zhang, Y. Visible light-induced aerobic oxidative cross-coupling of glycine esters with α-angelicalactone: A facile pathway to γ-lactams. Org. Biomol. Chem. 2018, 16, 6728. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.G. Developing processes for crystallization-induced asymmetric transformation. Org. Process. Res. Dev. 2005, 9, 800. [Google Scholar] [CrossRef]
- Berkeš, D.; Jakubec, P.; Winklerová, D.; Považanec, F.; Daich, A. CIAT with simultaneous epimerization at two stereocenters. Synthesis of substituted β-methyl-α-homophenylalanines. Org. Biomol. Chem. 2007, 5, 121. [Google Scholar] [CrossRef] [PubMed]
- Jakubec, P.; Berkeš, D. Crystallisation-induced asymmetric transformation (CIAT) for the synthesis of dipeptides containing homophenylalanine. Tetrahedron Asymmetry 2010, 21, 2807. [Google Scholar] [CrossRef]
- Brands, K.M.J.; Davies, A.J. Crystallization-induced diastereomer transformations. Chem. Rev. 2006, 106, 2711–2733. [Google Scholar] [CrossRef] [PubMed]
- Markus, J.; Puchľová, E.; Pinčeková, L.; Moncol, J.; Doháňošová, J.; Berkeš, D.; Caletková, O. Synthesis and Derivatization of 3-Aroyl Pyroglutamic Acids. ChemistrySelect 2020, 5, 2115. [Google Scholar] [CrossRef]
- Gerona-Navarro, G.; Bonache, M.A.; Herranz, R.; García-López, M.T.; González-Muniz, R. Entry to new conformationally constrained amino acids. First synthesis of 3-unsubstituted 4-alkyl-4-carboxy-2-azetidinone derivatives via an intramolecular N α-C α-cyclization strategy. J. Org. Chem. 2001, 66, 3538. [Google Scholar] [CrossRef]
- Chern, C.-Y.; Huang, Y.-P.; Kan, W.M. Selective N-debenzylation of amides with p-TsOH. Tetrahedron Lett. 2003, 44, 1039. [Google Scholar] [CrossRef]
- Vaidyanathan, R.; Kalthod, V.G.; Ngo, D.P.; Manley, J.M.; Lapekas, S.P. Amidations using N, N′-carbonyldiimidazole: Remarkable rate enhancement by carbon dioxide. J. Org. Chem. 2004, 69, 2565. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.-W.; Liu, Y.-K. Enantio-and diastereoselective synthesis of tetrahydrofuro [2, 3-b] furan-2 (3 H)-one derivatives and related oxygen heterocycles via an asymmetric organocatalytic cascade process. Org. Chem. Front. 2017, 4, 2358. [Google Scholar] [CrossRef]
- Zhao, S.; Lin, J.-B.; Zhao, Y.-Y.; Liang, Y.-M.; Xu, P.-F. Hydrogen-bond-directed formal [5+ 1] annulations of oxindoles with ester-linked bisenones: Facile access to chiral spirooxindole δ-lactones. Org. Lett. 2014, 16, 1802. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Xu, J.; Zhang, G.; Xu, C.; Zhao, L.; You, X.; Wang, Y.; Lu, Y.; Yu, L.; Wang, J. Design, synthesis, and bioevaluation of a novel class of (E)-4-oxo-crotonamide derivatives as potent antituberculosis agents. Bioorg. Med. Chem. Lett. 2019, 29, 539. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Structural analogs of P2X7 antagonists GSK 1370319A, GSK 1482160.

Scheme 2.
Proposed synthetic strategy towards 3-aroyl pyroglutamic acid amides.

Scheme 3.
aza-Michael addition of (S)-1-phenylethanamine (2a) and (S)-1-(4-methoxyphenyl)ethanamine (2b) to aroyl acrylic acids (1a–d) in tandem with CIDT. (i) 1.2 equiv, amine, solvent, r.t.—40 °C, 6–48 h.
Scheme 3.
aza-Michael addition of (S)-1-phenylethanamine (2a) and (S)-1-(4-methoxyphenyl)ethanamine (2b) to aroyl acrylic acids (1a–d) in tandem with CIDT. (i) 1.2 equiv, amine, solvent, r.t.—40 °C, 6–48 h.

Scheme 4.
N-chloroacetylation followed by base-catalyzed cyclization of (S,S)-aroylalanines (3a–f). (i) chloroacetyl chloride, propylene oxide, DCM, 0 °C—r.t.; (ii) NaOH, H2O, r.t.
Scheme 4.
N-chloroacetylation followed by base-catalyzed cyclization of (S,S)-aroylalanines (3a–f). (i) chloroacetyl chloride, propylene oxide, DCM, 0 °C—r.t.; (ii) NaOH, H2O, r.t.

Scheme 5.
Acid-catalyzed debenzylation of lactam nitrogen. (i) TFA, 50 °C, 24 h; (ii) p-TsOH, toluene, reflux, 2–8 h [12].
Scheme 5.
Acid-catalyzed debenzylation of lactam nitrogen. (i) TFA, 50 °C, 24 h; (ii) p-TsOH, toluene, reflux, 2–8 h [12].

Scheme 6.
N-Alkylation of lactam nitrogen. (i) MeI, NaHCO3, DMF, 50 °C, 24 h; (ii) MeI, LiHMDS, DMF, 0 °C—r.t., 0.5–2 h; (iii) LiOH, THF/H2O, r.t., 0.5–3 h.
Scheme 6.
N-Alkylation of lactam nitrogen. (i) MeI, NaHCO3, DMF, 50 °C, 24 h; (ii) MeI, LiHMDS, DMF, 0 °C—r.t., 0.5–2 h; (iii) LiOH, THF/H2O, r.t., 0.5–3 h.

Scheme 7.
Amide formation (i) CDI, amine 11, solvent, r.t., 3–24 h.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Results of aza-Michael addition of (S)-1-phenylethanamine (2a) and (S)-1-(4-methoxyphenyl)ethanamine (2b) to aroyl acrylic acids (1a–d) in tandem with CIDT.
Table 1.
Results of aza-Michael addition of (S)-1-phenylethanamine (2a) and (S)-1-(4-methoxyphenyl)ethanamine (2b) to aroyl acrylic acids (1a–d) in tandem with CIDT.
| Product 3a–g | Ar | R | Yield 3 (%) | d.r. | m.p. (°C) |
|---|---|---|---|---|---|
| 3a | Ph | OMe | 84 | 98:2 | 174–177 |
| 3b | (3-NO2)Ph | H | 89 | 99:1 | 172–173 |
| 3c | OMe | 86 | 99:1 | 179–181 | |
| 3d | (3-Br)Ph | H | 70 | 98:2 | 172–175 |
| 3e | OMe | 73 | 97:3 | 173–178 | |
| 3f | Thiophen-2-yl | H | 80 | 94:6 | 196–198 |
| 3g | OMe | - | - | - |
Table 2.
Results of N-chloroacetylation followed by base-catalyzed cyclization of (S,S)-aroylalanines (3a–f).
Table 2.
Results of N-chloroacetylation followed by base-catalyzed cyclization of (S,S)-aroylalanines (3a–f).
| Substrate 3a–f | Ar | R | Yield 7 (%) | m.p. (°C) | Yield 8 (%) | m.p. (°C) |
|---|---|---|---|---|---|---|
| 3a | Ph | OMe | 75 | 164–165 | 74 | 220–222 |
| 3b | (3-NO2)Ph | H | 85 | 144–146 | 64 | 232–234 |
| 3c | OMe | 67 | 173–175 | 86 | 182–183 | |
| 3d | (3-Br)Ph | H | 85 | 129–132 | 73 | 216–217 |
| 3e | OMe | 78 | 158–160 | 73 | 162–167 | |
| 3f | Thiophen-2-yl | H | 79 | 204–205 | 72 | 227–229 |
Table 3.
Result of acid-catalyzed debenzylation of lactam nitrogen.
| Substrate 8a–g | Ar | R | Conditions | Yield 9 (%) | m.p. (°C) |
|---|---|---|---|---|---|
| 8a | Ph | OMe | i | 96 | 166–167 |
| 8b | (3-NO2)Ph | H | ii | 85 | 221–223 |
| 8c | OMe | i | 94 | 221–222 | |
| 8d | (3-Br)Ph | H | ii | 79 | 212–215 |
| 8e | OMe | i | 73 | 215–216 | |
| 8f | Thiophen-2-yl | H | ii | 26 | - |
Table 4.
Results of N-alkylation of lactam nitrogen.
| Substrate 9a–d | Ar | Overall Yield 10 (3 Steps) (%) |
|---|---|---|
| 9a | Ph | 44 |
| 9b | (3-NO2)Ph | 36 |
| 9c | (3-Br)Ph | 42 |
| 9d | Thiophen-2-yl | - |
Table 5.
Results of amide formation by CDI.
| Substrate | Ar | R | Yield 12 (%) | m.p. (°C) |
|---|---|---|---|---|
| 8a | Ph | (S)-1-((4-OMe)Ph)Et | 70 | 136–137 |
| 9a | H | 65 | 134–136 | |
| 10a | Me | 84 | - | |
| 8c | (3-NO2)Ph | (S)-1-((4-OMe)Ph)Et | 49 | 191–192 |
| 9b | H | 36 | 223–225 | |
| 10b | Me | 56 | - | |
| 8e | (3-Br)Ph | (S)-1-((4-OMe)Ph)Et | 69 | - |
| 9c | H | 49 | 116–118 | |
| 10c | Me | 57 | - | |
| 8f | Thiophen-2-yl | (S)-1-(Ph)Et – | 77 | 154–156 |
| 9d | H | 82 | 157–159 | |
| 10d | Me | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pincekova, L.; Berkes, D. Synthesis of (2S,3S)-3-Aroyl Pyroglutamic Acid Amides. Chem. Proc. 2021, 3, 86. https://0-doi-org.brum.beds.ac.uk/10.3390/ecsoc-24-08377
AMA Style
Pincekova L, Berkes D. Synthesis of (2S,3S)-3-Aroyl Pyroglutamic Acid Amides. Chemistry Proceedings. 2021; 3(1):86. https://0-doi-org.brum.beds.ac.uk/10.3390/ecsoc-24-08377
Chicago/Turabian StylePincekova, Lucia, and Dusan Berkes. 2021. "Synthesis of (2S,3S)-3-Aroyl Pyroglutamic Acid Amides" Chemistry Proceedings 3, no. 1: 86. https://0-doi-org.brum.beds.ac.uk/10.3390/ecsoc-24-08377