
PIK3CA Mutation Assessment in HR+/HER2− Metastatic Breast Cancer: Overview for Oncology Clinical Practice


Abstract
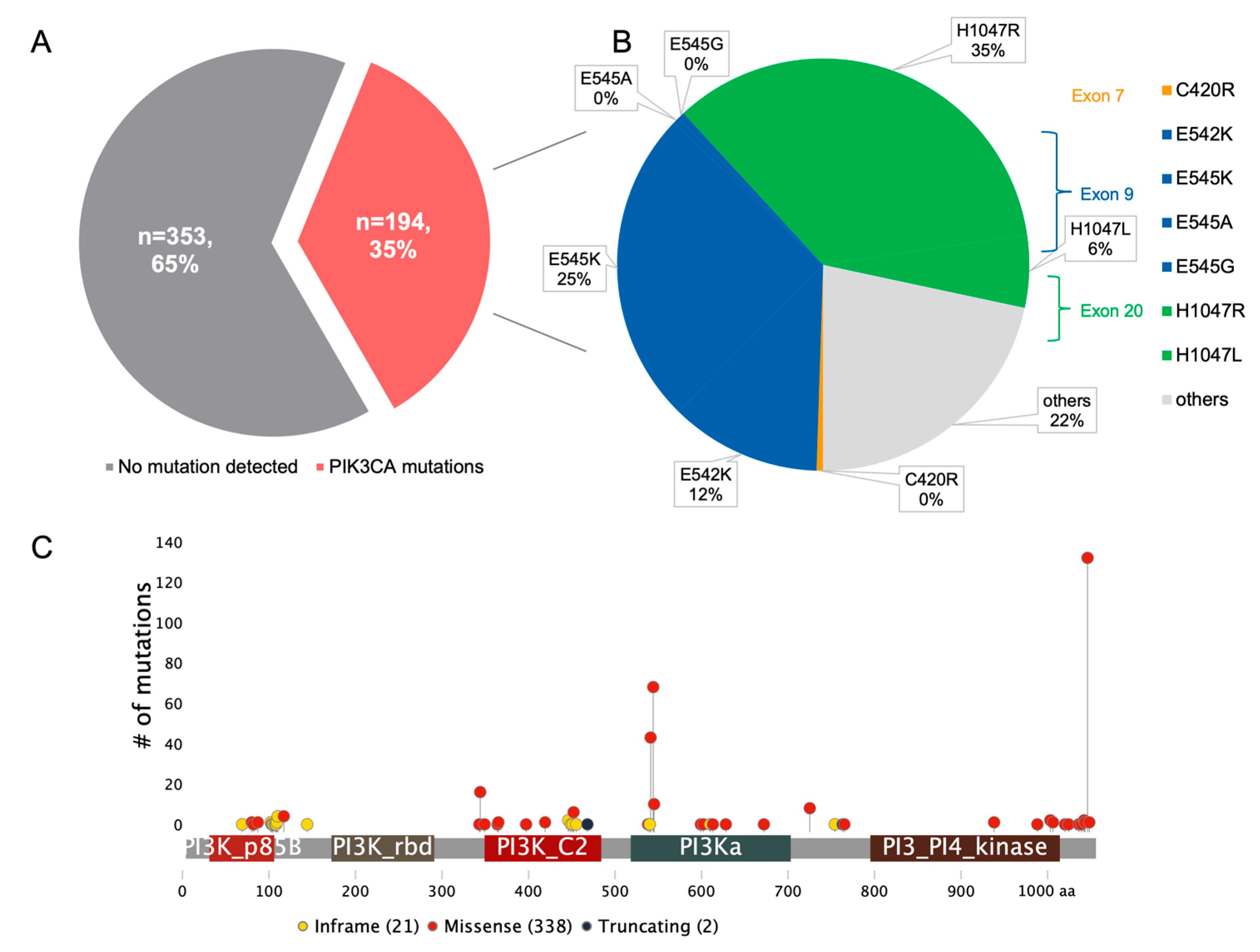
:1. Introduction
2. PI3K Pathway Alterations in ER-Positive BC
3. Prognostic and Predictive Value of PIK3CA Mutational Status in ER-Positive BC
4. Diagnostic Methods for the Detection of PIK3CA Mutations in Breast Cancer
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [Green Version]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Engelman, J.A. Targeting PI3K signalling in cancer: Opportunities, challenges and limitations. Nat. Rev. Cancer 2009, 9, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.J.; Loh, K.; Yap, Y.S. PI3K/Akt/mTOR inhibitors in breast cancer. Cancer Biol. Med. 2015, 12, 342–354. [Google Scholar] [CrossRef]
- Maira, S.M.; Pecchi, S.; Huang, A.; Burger, M.; Knapp, M.; Sterker, D.; Schnell, C.; Guthy, D.; Nagel, T.; Wiesmann, M.; et al. Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol. Cancer Ther. 2012, 11, 317–328. [Google Scholar] [CrossRef] [Green Version]
- Ndubaku, C.O.; Heffron, T.P.; Staben, S.T.; Baumgardner, M.; Blaquiere, N.; Bradley, E.; Bull, R.; Do, S.; Dotson, J.; Dudley, D.; et al. Discovery of 2-{3-[2-(1-isopropyl-3-methyl-1H-1,2-4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl]-1H-pyrazol-1-yl}-2-methylpropanamide (GDC-0032): A beta-sparing phosphoinositide 3-kinase inhibitor with high unbound exposure and robust in vivo antitumor activity. J. Med. Chem. 2013, 56, 4597–4610. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, C.; Huang, A.; Chatenay-Rivauday, C.; Schnell, C.; Reddy, A.; Liu, M.; Kauffmann, A.; Guthy, D.; Erdmann, D.; De Pover, A.; et al. Characterization of the novel and specific PI3Kalpha inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol. Cancer Ther. 2014, 13, 1117–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, A.; Viale, G.; Curigliano, G. Safety, Tolerability, and Management of Toxic Effects of Phosphatidylinositol 3-Kinase Inhibitor Treatment in Patients With Cancer: A Review. JAMA Oncol. 2019. [Google Scholar] [CrossRef]
- Andre, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef]
- Vasan, N.; Toska, E.; Scaltriti, M. Overview of the relevance of PI3K pathway in HR-positive breast cancer. Ann. Oncol. 2019, 30 (Suppl. 10), x3–x11. [Google Scholar] [CrossRef]
- Stemke-Hale, K.; Gonzalez-Angulo, A.M.; Lluch, A.; Neve, R.M.; Kuo, W.L.; Davies, M.; Carey, M.; Hu, Z.; Guan, Y.; Sahin, A.; et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res 2008, 68, 6084–6091. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018, 34, 427–438.e426. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Xing, M.; Mambo, E.; Huang, X.; Liu, J.; Guo, Z.; Chatterjee, A.; Goldenberg, D.; Gollin, S.M.; Sukumar, S.; et al. Somatic mutation and gain of copy number of PIK3CA in human breast cancer. Breast Cancer Res. 2005, 7, R609–R616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isakoff, S.J.; Engelman, J.A.; Irie, H.Y.; Luo, J.; Brachmann, S.M.; Pearline, R.V.; Cantley, L.C.; Brugge, J.S. Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res. 2005, 65, 10992–11000. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Vogt, P.K. Helical domain and kinase domain mutations in p110alpha of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc. Natl. Acad. Sci. USA 2008, 105, 2652–2657. [Google Scholar] [CrossRef] [Green Version]
- Koren, S.; Bentires-Alj, M. Mouse models of PIK3CA mutations: One mutation initiates heterogeneous mammary tumors. FEBS J. 2013, 280, 2758–2765. [Google Scholar] [CrossRef] [PubMed]
- Kalinsky, K.; Jacks, L.M.; Heguy, A.; Patil, S.; Drobnjak, M.; Bhanot, U.K.; Hedvat, C.V.; Traina, T.A.; Solit, D.; Gerald, W.; et al. PIK3CA mutation associates with improved outcome in breast cancer. Clin. Cancer Res. 2009, 15, 5049–5059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dogruluk, T.; Tsang, Y.H.; Espitia, M.; Chen, F.; Chen, T.; Chong, Z.; Appadurai, V.; Dogruluk, A.; Eterovic, A.K.; Bonnen, P.E.; et al. Identification of Variant-Specific Functions of PIK3CA by Rapid Phenotyping of Rare Mutations. Cancer Res. 2015, 75, 5341–5354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hon, W.C.; Berndt, A.; Williams, R.L. Regulation of lipid binding underlies the activation mechanism of class IA PI3-kinases. Oncogene 2012, 31, 3655–3666. [Google Scholar] [CrossRef] [Green Version]
- Kechagioglou, P.; Papi, R.M.; Provatopoulou, X.; Kalogera, E.; Papadimitriou, E.; Grigoropoulos, P.; Nonni, A.; Zografos, G.; Kyriakidis, D.A.; Gounaris, A. Tumor suppressor PTEN in breast cancer: Heterozygosity, mutations and protein expression. Anticancer Res. 2014, 34, 1387–1400. [Google Scholar]
- Saal, L.H.; Holm, K.; Maurer, M.; Memeo, L.; Su, T.; Wang, X.; Yu, J.S.; Malmstrom, P.O.; Mansukhani, M.; Enoksson, J.; et al. PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res. 2005, 65, 2554–2559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juric, D.; Castel, P.; Griffith, M.; Griffith, O.L.; Won, H.H.; Ellis, H.; Ebbesen, S.H.; Ainscough, B.J.; Ramu, A.; Iyer, G.; et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kalpha inhibitor. Nature 2015, 518, 240–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razavi, P.; Dickler, M.N.; Shah, P.D.; Toy, W.; Brown, D.N.; Won, H.H.; Li, B.T.; Shen, R.; Vasan, N.; Modi, S.; et al. Alterations in PTEN and ESR1 promote clinical resistance to alpelisib plus aromatase inhibitors. Nat. Cancer 2020, 1, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.K.; Schiff, R. Mechanisms of endocrine resistance in breast cancer. Annu. Rev. Med. 2011, 62, 233–247. [Google Scholar] [CrossRef] [Green Version]
- Klinge, C.M. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001, 29, 2905–2919. [Google Scholar] [CrossRef] [Green Version]
- Petz, L.N.; Nardulli, A.M. Sp1 binding sites and an estrogen response element half-site are involved in regulation of the human progesterone receptor A promoter. Mol. Endocrinol. 2000, 14, 972–985. [Google Scholar] [CrossRef]
- Webb, P.; Nguyen, P.; Valentine, C.; Lopez, G.N.; Kwok, G.R.; McInerney, E.; Katzenellenbogen, B.S.; Enmark, E.; Gustafsson, J.A.; Nilsson, S.; et al. The estrogen receptor enhances AP-1 activity by two distinct mechanisms with different requirements for receptor transactivation functions. Mol. Endocrinol. 1999, 13, 1672–1685. [Google Scholar] [CrossRef]
- Schiff, R.; Massarweh, S.A.; Shou, J.; Bharwani, L.; Mohsin, S.K.; Osborne, C.K. Cross-talk between estrogen receptor and growth factor pathways as a molecular target for overcoming endocrine resistance. Clin. Cancer Res. 2004, 10, 331S–336S. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Fu, X.; Hennessy, B.T.; Casa, A.J.; Zhang, Y.; Gonzalez-Angulo, A.M.; Lluch, A.; Gray, J.W.; Brown, P.H.; Hilsenbeck, S.G.; et al. Proteomic and transcriptomic profiling reveals a link between the PI3K pathway and lower estrogen-receptor (ER) levels and activity in ER+ breast cancer. Breast Cancer Res. 2010, 12, R40. [Google Scholar] [CrossRef] [Green Version]
- Bosch, A.; Li, Z.; Bergamaschi, A.; Ellis, H.; Toska, E.; Prat, A.; Tao, J.J.; Spratt, D.E.; Viola-Villegas, N.T.; Castel, P.; et al. PI3K inhibition results in enhanced estrogen receptor function and dependence in hormone receptor-positive breast cancer. Sci. Transl. Med. 2015, 7, 283ra251. [Google Scholar] [CrossRef] [Green Version]
- Toska, E.; Osmanbeyoglu, H.U.; Castel, P.; Chan, C.; Hendrickson, R.C.; Elkabets, M.; Dickler, M.N.; Scaltriti, M.; Leslie, C.S.; Armstrong, S.A.; et al. PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D. Science 2017, 355, 1324–1330. [Google Scholar] [CrossRef] [Green Version]
- Zardavas, D.; Te Marvelde, L.; Milne, R.L.; Fumagalli, D.; Fountzilas, G.; Kotoula, V.; Razis, E.; Papaxoinis, G.; Joensuu, H.; Moynahan, M.E.; et al. Tumor PIK3CA Genotype and Prognosis in Early-Stage Breast Cancer: A Pooled Analysis of Individual Patient Data. J. Clin. Oncol. 2018, 36, 981–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosele, F.; Stefanovska, B.; Lusque, A.; Tran Dien, A.; Garberis, I.; Droin, N.; Le Tourneau, C.; Sablin, M.P.; Lacroix, L.; Enrico, D.; et al. Outcome and molecular landscape of patients with PIK3CA-mutated metastatic breast cancer. Ann. Oncol. 2020, 31, 377–386. [Google Scholar] [CrossRef] [Green Version]
- Saura, C.; Hlauschek, D.; Oliveira, M.; Zardavas, D.; Jallitsch-Halper, A.; de la Pena, L.; Nuciforo, P.; Ballestrero, A.; Dubsky, P.; Lombard, J.M.; et al. Neoadjuvant letrozole plus taselisib versus letrozole plus placebo in postmenopausal women with oestrogen receptor-positive, HER2−negative, early-stage breast cancer (LORELEI): A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2019, 20, 1226–1238. [Google Scholar] [CrossRef]
- Schmid, P.; Pinder, S.E.; Wheatley, D.; Macaskill, J.; Zammit, C.; Hu, J.; Price, R.; Bundred, N.; Hadad, S.; Shia, A.; et al. Phase II Randomized Preoperative Window-of-Opportunity Study of the PI3K Inhibitor Pictilisib Plus Anastrozole Compared With Anastrozole Alone in Patients With Estrogen Receptor-Positive Breast Cancer. J. Clin. Oncol. 2016, 34, 1987–1994. [Google Scholar] [CrossRef] [Green Version]
- Baselga, J.; Im, S.A.; Iwata, H.; Cortes, J.; De Laurentiis, M.; Jiang, Z.; Arteaga, C.L.; Jonat, W.; Clemons, M.; Ito, Y.; et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2−negative, advanced breast cancer (BELLE-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 904–916. [Google Scholar] [CrossRef]
- Di Leo, A.; Johnston, S.; Lee, K.S.; Ciruelos, E.; Lonning, P.E.; Janni, W.; O’Regan, R.; Mouret-Reynier, M.A.; Kalev, D.; Egle, D.; et al. Buparlisib plus fulvestrant in postmenopausal women with hormone-receptor-positive, HER2−negative, advanced breast cancer progressing on or after mTOR inhibition (BELLE-3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2018, 19, 87–100. [Google Scholar] [CrossRef]
- Krop, I.E.; Mayer, I.A.; Ganju, V.; Dickler, M.; Johnston, S.; Morales, S.; Yardley, D.A.; Melichar, B.; Forero-Torres, A.; Lee, S.C.; et al. Pictilisib for oestrogen receptor-positive, aromatase inhibitor-resistant, advanced or metastatic breast cancer (FERGI): A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2016, 17, 811–821. [Google Scholar] [CrossRef] [Green Version]
- Vuylsteke, P.; Huizing, M.; Petrakova, K.; Roylance, R.; Laing, R.; Chan, S.; Abell, F.; Gendreau, S.; Rooney, I.; Apt, D.; et al. Pictilisib PI3Kinase inhibitor (a phosphatidylinositol 3-kinase [PI3K] inhibitor) plus paclitaxel for the treatment of hormone receptor-positive, HER2−negative, locally recurrent, or metastatic breast cancer: Interim analysis of the multicentre, placebo-controlled, phase II randomised PEGGY study. Ann. Oncol. 2016, 27, 2059–2066. [Google Scholar] [CrossRef]
- Juric, D.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Abstract GS3-08: Alpelisib + fulvestrant for advanced breast cancer: Subgroup analyses from the phase III SOLAR-1 trial. Cancer Res. 2019, 79, GS3–GS8. [Google Scholar] [CrossRef]
- Dent, S.; Cortes, J.; Im, Y.H.; Dieras, V.; Harbeck, N.; Krop, I.E.; Wilson, T.R.; Cui, N.; Schimmoller, F.; Hsu, J.Y.; et al. Phase III randomized study of taselisib or placebo with fulvestrant in estrogen receptor-positive, PIK3CA-mutant, HER2−negative, advanced breast cancer: The SANDPIPER trial. Ann. Oncol. 2020. [Google Scholar] [CrossRef]
- Lefebvre, C.; Bachelot, T.; Filleron, T.; Pedrero, M.; Campone, M.; Soria, J.C.; Massard, C.; Levy, C.; Arnedos, M.; Lacroix-Triki, M.; et al. Mutational Profile of Metastatic Breast Cancers: A Retrospective Analysis. PLoS Med. 2016, 13, e1002201. [Google Scholar] [CrossRef] [PubMed]
- Avivar-Valderas, A.; McEwen, R.; Taheri-Ghahfarokhi, A.; Carnevalli, L.S.; Hardaker, E.L.; Maresca, M.; Hudson, K.; Harrington, E.A.; Cruzalegui, F. Functional significance of co-occurring mutations in PIK3CA and MAP3K1 in breast cancer. Oncotarget 2018, 9, 21444–21458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Renuse, S.; Sahasrabuddhe, N.A.; Zahari, M.S.; Chaerkady, R.; Kim, M.S.; Nirujogi, R.S.; Mohseni, M.; Kumar, P.; Raju, R.; et al. Activation of diverse signalling pathways by oncogenic PIK3CA mutations. Nat. Commun. 2014, 5, 4961. [Google Scholar] [CrossRef] [PubMed]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasan, N.; Razavi, P.; Johnson, J.L.; Shao, H.; Shah, H.; Antoine, A.; Ladewig, E.; Gorelick, A.; Lin, T.Y.; Toska, E.; et al. Double PIK3CA mutations in cis increase oncogenicity and sensitivity to PI3Kalpha inhibitors. Science 2019, 366, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; Ciruelos, E.M.; Juric, D.; Loibl, S.; Campone, M.; Mayer, I.A.; Rubovszky, G.; Yamashita, T.; Kaufman, B.; Lu, Y.S.; et al. Alpelisib Plus Fulvestrant for PIK3CA-Mutated, Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor-2-Negative Advanced Breast Cancer: Final Overall Survival Results From SOLAR-1. Ann. Oncol. 2020. [Google Scholar] [CrossRef]
- Vora, S.R.; Juric, D.; Kim, N.; Mino-Kenudson, M.; Huynh, T.; Costa, C.; Lockerman, E.L.; Pollack, S.F.; Liu, M.; Li, X.; et al. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell 2014, 26, 136–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juric, D.; Kalinsky, K.; Oliveira, M.; Cervantes, A.; Bedard, P.; Krop, I.; Hamilton, E.; Schmid, P.; Varga, A.; Turner, N.; et al. Abstract OT1-08-04: A first-in-human phase Ia dose escalation study of GDC-0077, a p110a-selective and mutant-degrading PI3K inhibitor, in patients with PIK3CA-mutant solid tumors. Cancer Res. 2020, 80. [Google Scholar] [CrossRef]
- Scheerens, H.; Malong, A.; Bassett, K.; Boyd, Z.; Gupta, V.; Harris, J.; Mesick, C.; Simnett, S.; Stevens, H.; Gilbert, H.; et al. Current Status of Companion and Complementary Diagnostics: Strategic Considerations for Development and Launch. Clin. Transl. Sci. 2017, 10, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Rugo, H.S.; Mayer, I.; Conte, P.; Loibl, S.; Campone, M.; Juric, D.; Andre, F.; Fritzemeier, M.; He, W.; Babbar, N.; et al. Abstract CT142: Prevalence of PIK3CA mutations in patients with hormone receptor-positive, human epidermal growth factor-2-negative advanced breast cancer from the SOLAR-1 trial. Cancer Res. 2019, 79, CT142. [Google Scholar] [CrossRef]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Eccles, S.A.; Aboagye, E.O.; Ali, S.; Anderson, A.S.; Armes, J.; Berditchevski, F.; Blaydes, J.P.; Brennan, K.; Brown, N.J.; Bryant, H.E.; et al. Critical research gaps and translational priorities for the successful prevention and treatment of breast cancer. Breast Cancer Res. 2013, 15, R92. [Google Scholar] [CrossRef] [Green Version]
- Bertucci, F.; Finetti, P.; Guille, A.; Adelaide, J.; Garnier, S.; Carbuccia, N.; Monneur, A.; Charafe-Jauffret, E.; Goncalves, A.; Viens, P.; et al. Comparative genomic analysis of primary tumors and metastases in breast cancer. Oncotarget 2016, 7, 27208–27219. [Google Scholar] [CrossRef] [Green Version]
- O’Leary, B.; Cutts, R.J.; Liu, Y.; Hrebien, S.; Huang, X.; Fenwick, K.; Andre, F.; Loibl, S.; Loi, S.; Garcia-Murillas, I.; et al. The Genetic Landscape and Clonal Evolution of Breast Cancer Resistance to Palbociclib plus Fulvestrant in the PALOMA-3 Trial. Cancer Discov. 2018, 8, 1390–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chae, Y.K.; Davis, A.A.; Jain, S.; Santa-Maria, C.; Flaum, L.; Beaubier, N.; Platanias, L.C.; Gradishar, W.; Giles, F.J.; Cristofanilli, M. Concordance of Genomic Alterations by Next-Generation Sequencing in Tumor Tissue versus Circulating Tumor DNA in Breast Cancer. Mol. Cancer Ther. 2017, 16, 1412–1420. [Google Scholar] [CrossRef] [Green Version]
- Moynahan, M.E.; Chen, D.; He, W.; Sung, P.; Samoila, A.; You, D.; Bhatt, T.; Patel, P.; Ringeisen, F.; Hortobagyi, G.N.; et al. Correlation between PIK3CA mutations in cell-free DNA and everolimus efficacy in HR(+), HER2(-) advanced breast cancer: Results from BOLERO-2. Br. J. Cancer 2017, 116, 726–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merker, J.D.; Oxnard, G.R.; Compton, C.; Diehn, M.; Hurley, P.; Lazar, A.J.; Lindeman, N.; Lockwood, C.M.; Rai, A.J.; Schilsky, R.L.; et al. Circulating Tumor DNA Analysis in Patients With Cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. J. Clin. Oncol. 2018, 36, 1631–1641. [Google Scholar] [CrossRef]
- Cree, I.A.; Deans, Z.; Ligtenberg, M.J.; Normanno, N.; Edsjo, A.; Rouleau, E.; Sole, F.; Thunnissen, E.; Timens, W.; Schuuring, E.; et al. Guidance for laboratories performing molecular pathology for cancer patients. J. Clin. Pathol. 2014, 67, 923–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn 2017, 19, 4–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Trial | Phase | Population | Treatment | Tested Tissue | HR in PIK3CA Mutated | HR in PIK3CA WT/ITT Pop | Conclusion |
|---|---|---|---|---|---|---|---|
| BELLE-2 [38] | III | ER+/HER2−, after AI | Fulvestrant ± buparlisib | Archived tissue | 0.78 (0.67–0.89) | 0.76 (0.60–0.97) | No predictive value |
| Blood (ctDNA) | 0.58 (0.41–0.82) | 1.02 (0.79–1.30) | Benefit only in PIK3CA mutated | ||||
| BELLE-3 [39] | III | ER+/HER2−, after ET + everolimus | Fulvestrant ± buparlisib | Archived tissue | 0.39 (0.23–0.65) | 0.81 (0.59–1.12) | Benefit only in PIK3CA mutated |
| Blood (ctDNA) | 0.46 (0.29–0.73) | 0.73 (0.53–1.00) | |||||
| FERGI [40] | II | ER+/HER2−, after AI | Fulvestrant ± pictilisib | Tissue (not specified) | 0.73 (0.42–1.28) | 0.74 (0.52–1.06) | No predictive value |
| PEGGY [41] | II | ER+/HER2−, 1st/2nd line CT | Paclitaxel ± pictilisib | Tissue (not specified) | 1.06 (0.52–2.12) | 0.95 (0.62–1.46) | No predictive value |
| SOLAR-1 [10,42] | III | ER+/HER2−, after ET | Fulvestrant ± alpelisib | Archived tissue | 0.65 (0.50–0.85) | 0.85 (0.58-1.25) | Benefit only in PIK3CA mutated |
| Blood (ctDNA) | 0.55 (0.39–0.79) | 0.80 (0.60–1.06) | |||||
| SANDPIPER [43] | III | ER+/HER2−, after AI | Fulvestrant ± taselisib | Archived tissue | 0.70 (0.56–0.89) | 0.69 (0.44–1.08) | Benefit only in PIK3CA mutated |
| Blood (ctDNA) | 0.62 (0.47–0.83) | 0.86 (0.57–1.27 |
| Real-time PCR | ddPCR | BEAMing | Sanger seq | NGS | |
|---|---|---|---|---|---|
| Pros | Sensitive: can detect mutant DNA present at 1–5% | High level of sensitivity and specificity | High level of sensitivity | Unknown mutations can be detected | Sensitive for low-abundance mutations |
| Relatively inexpensive | Relatively inexpensive | Can detect gene fusions using RNA | Can detect a wide range of genetic changes in numerous genes | ||
| Cons | Only detects known targeted mutations | Only detects known targeted mutations | Only detects known targeted mutations | Labor intensive | Expensive and requires different DNA preparation method than other molecular mutation assays |
| Limited in the types of mutations detected | Limited in the types of mutations detected | Not as sensitive: requires mutant DNA to be present at 20%–25% | Requires more tumor tissue and sophisticated bioinformatics | ||
| Limited multiplexing capability | Limited multiplexing capability | Cannot detect changes in exon or gene copy number | |||
| Most common sample type | Tumor tissue and plasma | Plasma | Plasma | Tumor tissue | Tumor tissue and plasma |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Criscitiello, C.; Marra, A.; Curigliano, G. PIK3CA Mutation Assessment in HR+/HER2− Metastatic Breast Cancer: Overview for Oncology Clinical Practice. J. Mol. Pathol. 2021, 2, 42-54. https://0-doi-org.brum.beds.ac.uk/10.3390/jmp2010005
Criscitiello C, Marra A, Curigliano G. PIK3CA Mutation Assessment in HR+/HER2− Metastatic Breast Cancer: Overview for Oncology Clinical Practice. Journal of Molecular Pathology. 2021; 2(1):42-54. https://0-doi-org.brum.beds.ac.uk/10.3390/jmp2010005
Chicago/Turabian StyleCriscitiello, Carmen, Antonio Marra, and Giuseppe Curigliano. 2021. "PIK3CA Mutation Assessment in HR+/HER2− Metastatic Breast Cancer: Overview for Oncology Clinical Practice" Journal of Molecular Pathology 2, no. 1: 42-54. https://0-doi-org.brum.beds.ac.uk/10.3390/jmp2010005