
Modern Challenges for Early-Phase Clinical Trial Design and Biomarker Discovery in Metastatic Non-Small-Cell Lung Cancer

 , , , and
, , , and
Abstract
:1. Introduction
2. Modern Versus Old Early-Phase Clinical Trial Design: Lights and Shadows
3. Examples of Drug Development Programs in NSCLC: The Case of Gefitinib, Crizotinib and Osimertinib for Oncogene-Addicted NSCLC
4. Examples of Drug Development Programs in NSCLC: The Case of Nivolumab, Pembrolizumab and Avelumab for Non-Oncogene Addicted NSCLC
5. Conclusions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arrowsmith, J.; Miller, P. Trial watch: Phase II and phase III attrition rates 2011–2012. Nat. Rev. Drug Discov. 2013, 12, 569. [Google Scholar] [CrossRef] [PubMed]
- Amiri-Kordestani, L.; Fojo, T. Why do phase III clinical trials in oncology fail so often? J. Natl. Cancer Inst. 2012, 104, 568–569. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; You, B.; Pond, G.R.; Chen, E.X. Assumptions of expected benefits in randomized phase III trials evaluating systemic treatments for cancer. J. Natl. Cancer Inst. 2012, 104, 590–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz, J.; Swanton, C.; Kurzrock, R. Molecular profiling and the reclassification of cancer: Divide and conquer. Am. Soc. Clin. Oncol. Educ. Book 2013, 33, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, A. Immunotherapy and NSCLC: The long and winding road. Cancers 2020, 12, 2512. [Google Scholar] [CrossRef]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic Non-Small Cell Lung Cancer: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-up. Available online: https://www.esmo.org/guidelines/lung-and-chest-tumours/clinical-practice-living-guidelines-metastatic-non-small-cell-lung-cancer (accessed on 2 April 2021).
- Drilon, A. TRK inhibitors in TRK fusion-positive cancers. Ann. Oncol. 2019, 30 (Suppl. S8), viii23–viii30. [Google Scholar] [CrossRef] [Green Version]
- Jardim, D.L.; Schwaederle, M.; Hong, D.S. An appraisal of drug development timelines in the era of precision oncology. Oncotarget 2016, 7, 53037–53046. [Google Scholar] [CrossRef] [Green Version]
- Mandrekar, S.J.; Dahlberg, S.E.; Simon, R. Improving clinical trial efficiency: Thinking outside the box. Am. Soc. Clin. Oncol. Educ. Book 2015, 35, e141–e147. [Google Scholar] [CrossRef]
- Tsimberidou, A.M.; Iskander, N.G.; Hong, D.S. Personalized medicine in a phase I clinical trials program: The MD Anderson Cancer Center initiative. Clin. Cancer Res. 2012, 18, 6373–6383. [Google Scholar] [CrossRef] [Green Version]
- Schwaederle, M.; Zhao, M.; Lee, J.J. Association of biomarker-based treatment strategies with response rates and progression-free survival in refractory malignant neoplasms: A meta-analysis. JAMA Oncol. 2016, 2, 1452–1459. [Google Scholar] [CrossRef] [Green Version]
- Adashek, J.J.; LoRusso, P.M.; Hong, D.S. Phase I trials as valid therapeutic options for patients with cancer. Nat. Rev. Clin. Oncol. 2019, 16, 773–778. [Google Scholar] [CrossRef] [Green Version]
- Manji, A.; Brana, I.; Amir, E. Evolution of clinical trial design in early drug development: Systematic review of expansion cohort use in single-agent phase I cancer trials. J. Clin. Oncol. 2013, 31, 4260–4267. [Google Scholar] [CrossRef]
- Bugano, D.D.; Hess, K.; Jardim, D.L. Use of expansion cohorts in phase I trials and probability of success in phase II for 381 anticancer drugs. Clin. Cancer Res. 2017, 23, 4020–4026. [Google Scholar] [CrossRef] [Green Version]
- Dahlberg, S.E.; Shapiro, G.I.; Clark, J.W. Evaluation of statistical designs in phase I expansion cohorts: The Dana-Farber/Harvard Cancer Center experience. J. Natl. Cancer Inst. 2014, 106, dju163. [Google Scholar] [CrossRef] [Green Version]
- Chabner, B.A. Early accelerated approval for highly targeted cancer drugs. N. Engl. J. Med. 2011, 364, 1087–1089. [Google Scholar] [CrossRef]
- Ferrara, M.G.; Di Noia, V.; D’Argento, E.; Vita, E.; Damiano, P.; Cannella, A.; Ribelli, M.; Pilotto, S.; Milella, M.; Tortora, G.; et al. Oncogene-addicted non-small-cell lung cancer: Treatment opportunities and future perspectives. Cancers 2020, 12, 1196. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; O’Dwyer, P.J.; Christian, M. Phase I clinical trial design in cancer drug development. J. Clin. Oncol. 2000, 18, 684–692. [Google Scholar] [CrossRef]
- Booth, C.M.; Calvert, A.H.; Giaccone, G.; Lobbezoo, M.W.; Seymour, L.K.; Eisenhauer, E.A. End points and other considerations in Phase I studies of targeted anticancer therapy: Recommendations from the task force on Methodology for the Development of Innovative Cancer Therapies (MDICT). Eur. J. Cancer 2008, 44, 19–24. [Google Scholar] [CrossRef]
- Mansinho, A.; Boni, V.; Miguel, M.; Calvo, E. New designs in early clinical drug development. Ann. Oncol. 2019, 30, 1460–1465. [Google Scholar] [CrossRef] [Green Version]
- Tan, D.S.; Thomas, G.V.; Garret, M.D.; Banerji, U.; de Bono, J.S.; Kaye, S.B.; Workman, P. Biomarker-driven early clinical trials in oncology: A paradigm shift in drug development. Cancer J. 2009, 15, 406–420. [Google Scholar] [CrossRef]
- Banerji, U.; Workman, P. Critical parameters in targeted drug development: The pharmacological audit trail. Semin. Oncol. 2016, 43, 436–445. [Google Scholar] [CrossRef] [Green Version]
- Zou, H.Y.; Li, Q.; Lee, J.H.; McDonnell, S.R.; Yamazaki, S.; Koudriakova, T.B.; Alton, G.; Cui, J.J.; Kung, P.P.; Nambu, M.D.; et al. An orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res. 2007, 67, 4408–4417. [Google Scholar] [CrossRef] [Green Version]
- Clark, J.W.; Camidge, D.R.; Kwak, E.L.; Maki, R.G.; Shapiro, G.I.; Chen, I.; Tan, W.; Randolph, S.; Christensen, J.G.; Ozeck, M.; et al. Dose-escalation trial of the ALK, MET & ROS1 inhibitor, crizotinib, in patients with advanced cancer. Future Oncol. 2020, 16, 4289–4301. [Google Scholar] [PubMed]
- Kwak, E.L.; Bang, Y.J.; Camidge, D.R.; Shaw, A.T.; Solomon, B.; Maki, R.G.; Ou, S.H.; Dezube, B.J.; Jänne, P.A.; Costa, D.B.; et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N. Engl. J. Med. 2010, 363, 1693–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camidge, D.R.; Bang, Y.J.; Kwak, E.L.; Iafrate, A.J.; Varella-Garcia, M.; Fox, S.B.; Riely, G.J.; Solomon, B.; Ou, S.H.; Kim, D.W.; et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: Updated results from a phase 1 study. Lancet Oncol. 2012, 13, 1011–1019. [Google Scholar] [CrossRef] [Green Version]
- Blackhall, F.; Camidge, D.R.; Shaw, A.T.; Soria, J.C.; Solomon, B.J.; Mok, T.; Hirsh, V.; Jänne, P.A.; Shi, Y.; Yang, P.C.; et al. Final results of the large-scale multinational trial PROFILE 1005: Efficacy and safety of crizotinib in previously treated patients with advanced/metastatic ALK-positive non-small-cell lung cancer. ESMO Open 2017, 2, e000219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le-Rademacher, J.; Dahlberg, S.; Lee, J.; Adjei, A.A.; Mandrekar, S.J. Biomarker clinical trials in lung cancer: Design, logistics, challenges, and practical considerations. J. Thorac. Oncol. 2018, 13, 1625–1637. [Google Scholar] [CrossRef] [Green Version]
- Blackhall, F.; Cappuzzo, F. Crizotinib: From discovery to accelerated development to front-line treatment. Ann. Oncol. 2018, 29, 1073. [Google Scholar] [CrossRef] [Green Version]
- Ranson, M.; Hammond, L.A.; Ferry, D.; Kris, M.; Tullo, A.; Murray, P.I.; Miller, V.; Averbuch, S.; Ochs, J.; Morris, C.; et al. ZD1839, a selective oral epidermal growth factor receptor-tyrosine kinase inhibitor, is well tolerated and active in patients with solid, malignant tumors: Results of a phase I trial. J. Clin. Oncol. 2002, 20, 2240–2250. [Google Scholar] [CrossRef]
- Baselga, J.; Rischin, D.; Ranson, M.; Calvert, H.; Raymond, E.; Kieback, D.G.; Kaye, S.B.; Gianni, L.; Harris, A.; Bjork, T.; et al. Phase I safety, pharmacokinetic, and pharmacodynamic trial of ZD1839, a selective oral epidermal growth factor receptor tyrosine kinase inhibitor, in patients with five selected solid tumor types. J. Clin. Oncol. 2002, 20, 4292–4302. [Google Scholar] [CrossRef]
- Herbst, R.S.; Maddox, A.M.; Rothenberg, M.L.; Small, E.J.; Rubin, E.H.; Baselga, J.; Rojo, F.; Hong, W.K.; Swaisland, H.; Averbuch, S.D.; et al. Selective oral epidermal growth factor receptor tyrosine kinase inhibitor ZD1839 is generally well-tolerated and has activity in non-small-cell lung cancer and other solid tumors: Results of a phase I trial. J. Clin. Oncol. 2002, 20, 3815–3825. [Google Scholar] [CrossRef]
- Nakagawa, K.; Tamura, T.; Negoro, S.; Kudoh, S.; Yamamoto, N.; Yamamoto, N.; Takeda, K.; Swaisland, H.; Nakatani, I.; Hirose, M.; et al. Phase I pharmacokinetic trial of the selective oral epidermal growth factor receptor tyrosine kinase inhibitor gefitinib (‘Iressa’, ZD1839) in Japanese patients with solid malignant tumors. Ann. Oncol. 2003, 14, 922–930. [Google Scholar] [CrossRef]
- Fukuoka, M.; Yano, S.; Giaccone, G.; Tamura, T.; Nakagawa, K.; Douillard, J.Y.; Nishiwaki, Y.; Vansteenkiste, J.; Kudoh, S.; Rischin, D.; et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (The IDEAL 1 Trial). J. Clin. Oncol. 2003, 21, 2237–2246. [Google Scholar] [CrossRef]
- Kris, M.G.; Natale, R.B.; Herbst, R.S.; Lynch, T.J., Jr.; Prager, D.; Belani, C.P.; Schiller, J.H.; Kelly, K.; Spiridonidis, H.; Sandler, A.; et al. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: A randomized trial. JAMA 2003, 290, 2149–2158. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.H.; Yu, C.J.; Shih, J.Y.; Chang, Y.C.; Hu, F.C.; Tsai, M.C.; Chen, K.Y.; Lin, Z.Z.; Huang, C.J.; Shun, C.T.; et al. Specific EGFR mutations predict treatment outcome of stage IIIB/IV patients with chemotherapy-naive non-small-cell lung cancer receiving first-line gefitinib monotherapy. J. Clin. Oncol. 2008, 26, 2745–2753. [Google Scholar] [CrossRef]
- Sequist, L.V.; Martins, R.G.; Spigel, D.; Grunberg, S.M.; Spira, A.; Jänne, P.A.; Joshi, V.A.; McCollum, D.; Evans, T.L.; Muzikansky, A.; et al. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J. Clin. Oncol. 2008, 26, 2442–2449. [Google Scholar] [CrossRef]
- Costanzo, R.; Piccirillo, M.C.; Sandomenico, C.; Carillio, G.; Montanino, A.; Daniele, G.; Giordano, P.; Bryce, J.; De Feo, G.; Di Maio, M.; et al. Gefitinib in non small cell lung cancer. J. Biomed. Biotechnol. 2011, 2011, 815269. [Google Scholar] [CrossRef]
- Armour, A.A.; Watkins, C.L. The challenge of targeting EGFR: Experience with gefitinib in nonsmall cell lung cancer. Eur. Respir. Rev. 2010, 117, 186–196. [Google Scholar] [CrossRef]
- Cross, D.A.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef] [Green Version]
- Janne, P.A.; Yang, J.C.-H.; Kim, D.W.; Planchard, D.; Ohe, Y.; Ramalingam, S.S.; Ahn, M.J.; Kim, S.W.; Su, W.C.; Horn, L.; et al. AZD9291 in EGFR inhibitor-resistant non-small cell lung cancer. N. Engl. J. Med. 2015, 372, 1689–1699. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Yang, J.C.-H.; Lee, C.K.; Kurata, T.; Kim, D.W.; John, T.; Nogami, N.; Ohe, Y.; Mann, H.; Rukazenkov, Y.; et al. Osimertinib as first-line treatment of EGFR mutation-positive advanced non-small-cell lung cancer. J. Clin. Oncol. 2018, 36, 841–849. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.C.; Ahn, M.J.; Kim, D.W.; Ramalingam, S.S.; Sequist, L.V.; Su, W.C.; Kim, S.W.; Kim, J.H.; Planchard, D.; Felip, E.; et al. Osimertinib in pretreated T790M-positive advanced non-small-cell lung cancer: AURA study phase II extension component. J. Clin. Oncol. 2017, 35, 1288–1296. [Google Scholar] [CrossRef]
- Goss, G.; Tsai, C.M.; Shepherd, F.A.; Bazhenova, L.; Lee, J.S.; Chang, G.C.; Crino, L.; Satouchi, M.; Chu, Q.; Hida, T.; et al. Osimertinib for pretreated EGFR Thr790Met positive advanced non-small-cell lung cancer (AURA2): A multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2016, 17, 1643–1652. [Google Scholar] [CrossRef]
- Yver, A. Osimertinib (AZD9291)—A science-driven, collaborative approach to rapid drug design and development. Ann. Oncol. 2016, 27, 1165–1170. [Google Scholar] [CrossRef]
- Fox, E.; Curt, G.A.; Balis, F.M. Clinical trial design for target-based therapy. Oncologist 2002, 7, 401–409. [Google Scholar] [CrossRef] [Green Version]
- Russo, A.; McCusker, M.G.; Scilla, K.A.; Arensmeyer, K.E.; Mehra, R.; Adamo, V.; Rolfo, C. Immunotherapy in lung cancer: From a minor god to the olympus. Adv. Exp. Med. Biol. 2020, 1244, 69–92. [Google Scholar]
- Menis, J.; Hasan, B.; Besse, B. New clinical research strategies in thoracic oncology: Clinical trial design, adaptive, basket and umbrella trials, new end-points and new evaluations of response. Eur. Respir. Rev. 2014, 23, 367–378. [Google Scholar] [CrossRef] [Green Version]
- Haanen, J.B.A.G.; Carbonnel, F.; Robert, C.; Kerr, K.M.; Peters, S.; Larkin, J.; Jordan, K. Management of toxicities from immunotherapy: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2017, 28 (Suppl. S4), iv119–iv142. [Google Scholar] [CrossRef]
- Snyder, A.; Wolchok, J.D.; Chan, T.A. Genetic basis for clinical response to CTLA-4 blockade. N. Engl. J. Med. 2015, 372, 783. [Google Scholar] [CrossRef]
- Wang, Z.; Duan, J.; Cai, S.; Han, M.; Dong, H.; Zhao, J.; Zhu, B.; Wang, S.; Zhuo, M.; Sun, J.; et al. Assessment of blood tumor mutational burden as a potential biomarker for immunotherapy in patients with non-small cell lung cancer with use of a next generation sequencing cancer gene panel. JAMA Oncol. 2019, 5, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Duchemann, B.; Remon, J.; Naigeon, M.; Mezquita, L.; Ferrara, R.; Cassard, L.; Jouniaux, J.M.; Boselli, L.; Grivel, J.; Auclin, E.; et al. Integrating circulating biomarkers in the immune checkpoint inhibitor treatment in lung cancer. Cancers 2020, 12, 3625. [Google Scholar] [CrossRef] [PubMed]
- Nishino, M.; Ramaiya, N.H.; Hatabu, H.; Hodi, F.S. Monitoring immune-checkpoint blockade: Response evaluation and biomarker development. Nat. Rev. Clin. Oncol. 2017, 14, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Carbognin, L.; Pilotto, S.; Milella, M.; Vaccaro, V.; Brunelli, M.; Caliò, A.; Cuppone, F.; Sperduti, I.; Giannarelli, D.; Chilosi, M.; et al. Differential activity of nivolumab, pembrolizumab and MPDL3280A according to the tumor expression of programmed death-ligand-1 (PD L1): Sensitivity analysis of trials in melanoma, lung and genitourinary cancers. PLoS ONE 2015, 10, e0130142. [Google Scholar] [CrossRef] [Green Version]
- Aguiar, P.N.; Santoro, I.L.; Tadokoro, H.; de Lima Lopes, G.; Filardi, B.A.; Oliveira, P.; Mountzios, G.; de Mello, R.A. The role of PD-L1 expression as a predictive biomarker in advanced non-small-cell lung cancer: A network meta-analysis. Immunotherapy 2016, 8, 479–488. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Gettinger, S.N.; Horn, L.; Gandhi, L.; Spigel, D.R.; Antonia, S.J.; Rizvi, N.A.; Powderly, J.D.; Heist, R.S.; Carvajal, R.D.; Jackman, D.M.; et al. Overall survival and long-term safety of nivolumab (anti-programmed death 1 antibody, BMS-936558, ONO-4538) in patients with previously treated advanced non-small-cell lung cancer. J. Clin. Oncol. 2015, 33, 2004–2012. [Google Scholar] [CrossRef]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [Green Version]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Gettinger, S.; Horn, L.; Jackman, D.; Spigel, D.; Antonia, S.; Hellmann, M.; Powderly, J.; Heist, R.; Sequist, L.V.; Smith, D.C.; et al. Five-year follow-up of nivolumab in previously treated advanced non-small-cell lung cancer: Results from the CA209-003 study. J. Clin. Oncol. 2018, 36, 1765–1784. [Google Scholar] [CrossRef]
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti–PD-1 therapy. Clin. Cancer Res. 2014, 20, 5064–5074. [Google Scholar] [CrossRef] [Green Version]
- Gettinger, S.; Rizvi, N.A.; Chow, L.Q.; Borghaei, H.; Brahmer, J.; Ready, N.; Gerber, D.E.; Shepherd, F.A.; Antonia, S.; Goldman, J.W.; et al. Nivolumab monotherapy for first-line treatment of advanced non-small-cell lung cancer. J. Clin. Oncol. 2016, 34, 2980–2987. [Google Scholar] [CrossRef]
- Carbone, D.P.; Reck, M.; Paz-Ares, L.; Creelan, B.; Horn, L.; Steins, M.; Felip, E.; van den Heuvel, M.M.; Ciuleanu, T.E.; Badin, F.; et al. First-line nivolumab in stage IV or recurrent non-small-cell lung cancer. N. Engl. J. Med. 2017, 376, 2415–2426. [Google Scholar] [CrossRef]
- Reck, M.; Rodriguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [Green Version]
- Remon, J.; Besse, B.; Soria, J.C. Successes and failures: What did we learn from recent first-line treatment immunotherapy trials in non-small cell lung cancer? BMC Med. 2017, 15, 55. [Google Scholar]
- Leroi, N.; Lallemand, F.; Coucke, P.; Noel, A.; Martinive, P. Impacts of ionizing radiation on the different compartments of the tumor microenvironment. Front. Pharmacol. 2016, 7, 78. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Dolled-Filhart, M.; Roach, C.; Toland, G.; Stanforth, D.; Jansson, M.; Lubiniecki, G.M.; Ponto, G.; Emancipator, K. Development of a companion diagnostic for pembrolizumab in non-small cell lung cancer using immunohistochemistry for programmed death ligand-1. Arch. Pathol. Lab. Med. 2016, 140, 1243–1249. [Google Scholar] [CrossRef] [Green Version]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- European Medicines Agency. Concept Paper on Predictive Biomarker-Based Assay Development in the Context of Drug Development and Lifecycle; European Medicines Agency: London, UK, 2017. [Google Scholar]
- Kang, S.P.; Gergich, K.; Lubiniecki, G.M.; de Alwis, D.P.; Chen, C.; Tice, M.A.B.; Rubin, E.H. Pembrolizumab KEYNOTE-001: An adaptive study leading to accelerated approval for two indications and a companion diagnostic. Ann. Oncol. 2017, 28, 1388–1398. [Google Scholar] [CrossRef]
- Herbst, R.S.; Baas, P.; Kim, D.W.; Felip, E.; Pérez-Gracia, J.L.; Han, J.Y.; Molina, J.; Kim, J.H.; Arvis, C.D.; Ahn, M.J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef]
- Herbst, R.S.; Baas, P.; Perez-Gracia, J.L.; Felip, E.; Kim, D.W.; Han, J.Y.; Molina, J.R.; Kim, J.H.; Dubos Arvis, C.; Ahn, M.J.; et al. Use of archival versus newly collected tumor samples for assessing PD-L1 expression and overall survival: An updated analysis of KEYNOTE-010 trial. Ann. Oncol. 2019, 30, 281–289. [Google Scholar] [CrossRef]
- Hirsch, F.R.; McElhinny, A.; Stanforth, D.; Ranger-Moore, J.; Jansson, M.; Kulangara, K.; Richardson, W.; Towne, P.; Hanks, D.; Vennapusa, B.; et al. PD-L1 immunohistochemistry assays for lung cancer: Results from phase 1 of the Blueprint PDL1 IHC Assay Comparison Project. J. Thorac. Oncol. 2017, 12, 208–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsao, M.S.; Kerr, K.M.; Kockx, M.; Beasley, M.B.; Borczuk, A.C.; Botling, J.; Bubendorf, L.; Chirieac, L.; Chen, G.; Chou, T.Y.; et al. PD-L1 immunohistochemistry comparability study in real-life clinical samples: Results of Blueprint Phase 2 Project. J. Thorac. Oncol. 2018, 13, 1302–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blueprint Working Groups. A Blueprint Proposal for Companion Diagnostic Comparability; Blueprint Working Groups: Washington, DC, USA, 2015. [Google Scholar]
- Heery, C.R.; O’Sullivan-Coyne, G.; Madan, R.A.; Cordes, L.; Rajan, A.; Rauckhorst, M.; Lamping, E.; Oyelakin, I.; Marté, J.L.; Lepone, L.M.; et al. Avelumab for metastatic or locally advanced previously treated solid tumours (JAVELIN Solid Tumor): A phase 1a, multicohort, dose-escalation trial. Lancet Oncol. 2017, 18, 587–598. [Google Scholar] [CrossRef]
- Gulley, J.L.; Rajan, A.; Spigel, D.R.; Iannotti, N.; Chandler, J.; Wong, D.J.L.; Leach, J.; Edenfield, W.J.; Wang, D.; Grote, H.J.; et al. Avelumab for patients with previously treated metastatic or recurrent non-small-cell lung cancer (JAVELIN Solid Tumor): Dose-expansion cohort of a multicentre, open-label, phase 1b trial. Lancet Oncol. 2017, 18, 599–610. [Google Scholar] [CrossRef] [Green Version]
- Barlesi, F.; Vansteenkiste, J.; Spigel, D.; Ishii, H.; Garassino, M.; de Marinis, F.; Özgüroğlu, M.; Szczesna, A.; Polychronis, A.; Uslu, R.; et al. Avelumab versus docetaxel in patients with platinum-treated advanced non-small-cell lung cancer (JAVELIN Lung 200): An open-label, randomised, phase 3 study. Lancet Oncol. 2018, 19, 1468–1479. [Google Scholar] [CrossRef]
- Feng, Z.; Schlichting, M.; Helwig, C.; Chand, V.K.; Gelb, A.; Jin, H.; Grote, H.J. Comparative study of two PD-L1 expression assays in patients with non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2017, 35 (Suppl. 15), e20581. [Google Scholar] [CrossRef]
- Verschraegen, C.F.; Jerusalem, G.; McClay, E.F.; Iannotti, N.; Redfern, C.H.; Bennouna, J.; Chen, F.L.; Kelly, K.; Mehnert, J.; Morris, J.C.; et al. Efficacy and safety of first-line avelumab in patients with advanced non-small cell lung cancer: Results from a phase Ib cohort of the JAVELIN Solid Tumor study. J. Immunother. Cancer 2020, 8, e001064. [Google Scholar] [CrossRef]
- Updated Information on Sotorasib Dose-Comparison Study. Available online: https://ascopost.com/news/april-2021/updated-information-on-sotorasib-dose-comparison-study/ (accessed on 10 June 2021).
- Redig, A.J.; Jänne, P.A. Basket trials and the evolution of clinical trial design in an era of genomic medicine. J. Clin. Oncol. 2015, 33, 975–977. [Google Scholar] [CrossRef] [Green Version]


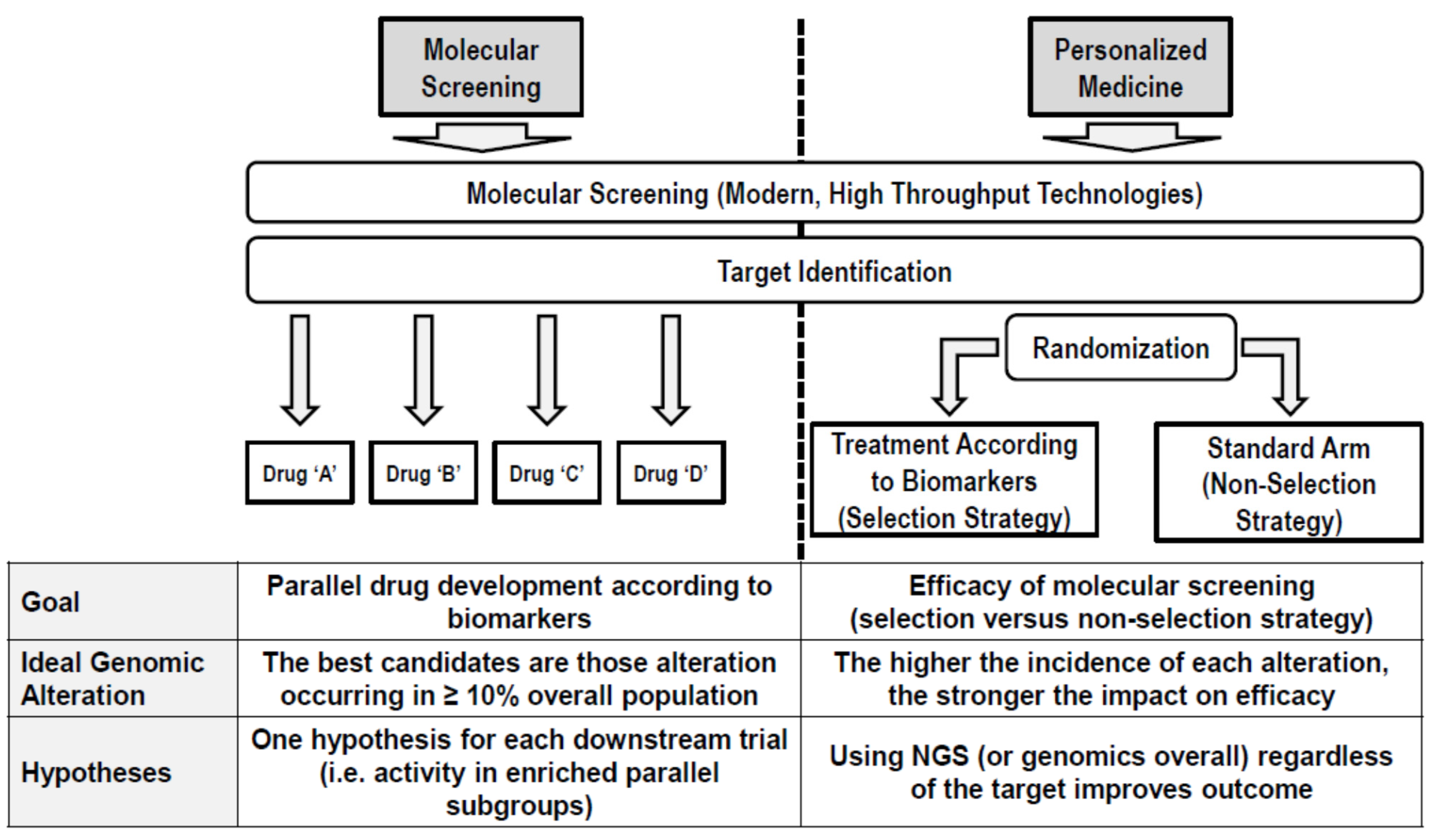{kind=link}
| Characteristics | Gefitinib | Crizotinib | Osimertinib |
|---|---|---|---|
| Year of phase 1 trials starting | 2002 | 2006 | 2012 |
| Time to biomarker identification | Phase 2 trials | Expansion cohort phase 1 trial | Pre-clinical phase |
| Year of drug approval for targeted population | 2014 | 2011 | 2015 |
| Time required for drug approval | 12 years | 5 years | 2.5 years |
| Type of drug development program | Classic drug development from phase 1 to phase 3 trials (with late biomarker identification) | Phase 1 expansion cohort with targeted population | Classic drug development from phase 1 to phase 3 trials in the selected population |
| Characteristics | Nivolumab | Pembrolizumab | Avelumab |
|---|---|---|---|
| Year of phase 1 trials starting | 2009 | 2012 | 2013 |
| Co-development of companion diagnostic from early phases | NO | YES | NO |
| Time required for FDA drug approval | 5 years (approval in second line) | 3 years (approval in second line) * | Not approved |
| Type of drug development program | Classic drug development from phase 1 to phase 3 trials | Classic drug development from phase 1 to phase 3 trials (including adaptive design trials) | Classic drug development from phase 1 to phase 3 trials |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossi, A.; Pilotto, S.; Carbognin, L.; Ferrara, M.G.; Belluomini, L.; Daniele, G.; Bria, E. Modern Challenges for Early-Phase Clinical Trial Design and Biomarker Discovery in Metastatic Non-Small-Cell Lung Cancer. J. Mol. Pathol. 2021, 2, 207-222. https://0-doi-org.brum.beds.ac.uk/10.3390/jmp2030018
Rossi A, Pilotto S, Carbognin L, Ferrara MG, Belluomini L, Daniele G, Bria E. Modern Challenges for Early-Phase Clinical Trial Design and Biomarker Discovery in Metastatic Non-Small-Cell Lung Cancer. Journal of Molecular Pathology. 2021; 2(3):207-222. https://0-doi-org.brum.beds.ac.uk/10.3390/jmp2030018
Chicago/Turabian StyleRossi, Antonio, Sara Pilotto, Luisa Carbognin, Miriam Grazia Ferrara, Lorenzo Belluomini, Gennaro Daniele, and Emilio Bria. 2021. "Modern Challenges for Early-Phase Clinical Trial Design and Biomarker Discovery in Metastatic Non-Small-Cell Lung Cancer" Journal of Molecular Pathology 2, no. 3: 207-222. https://0-doi-org.brum.beds.ac.uk/10.3390/jmp2030018