Reprogramming of Central Carbon Metabolism in Myeloid Cells upon Innate Immune Receptor Stimulation


Abstract
:1. Introduction
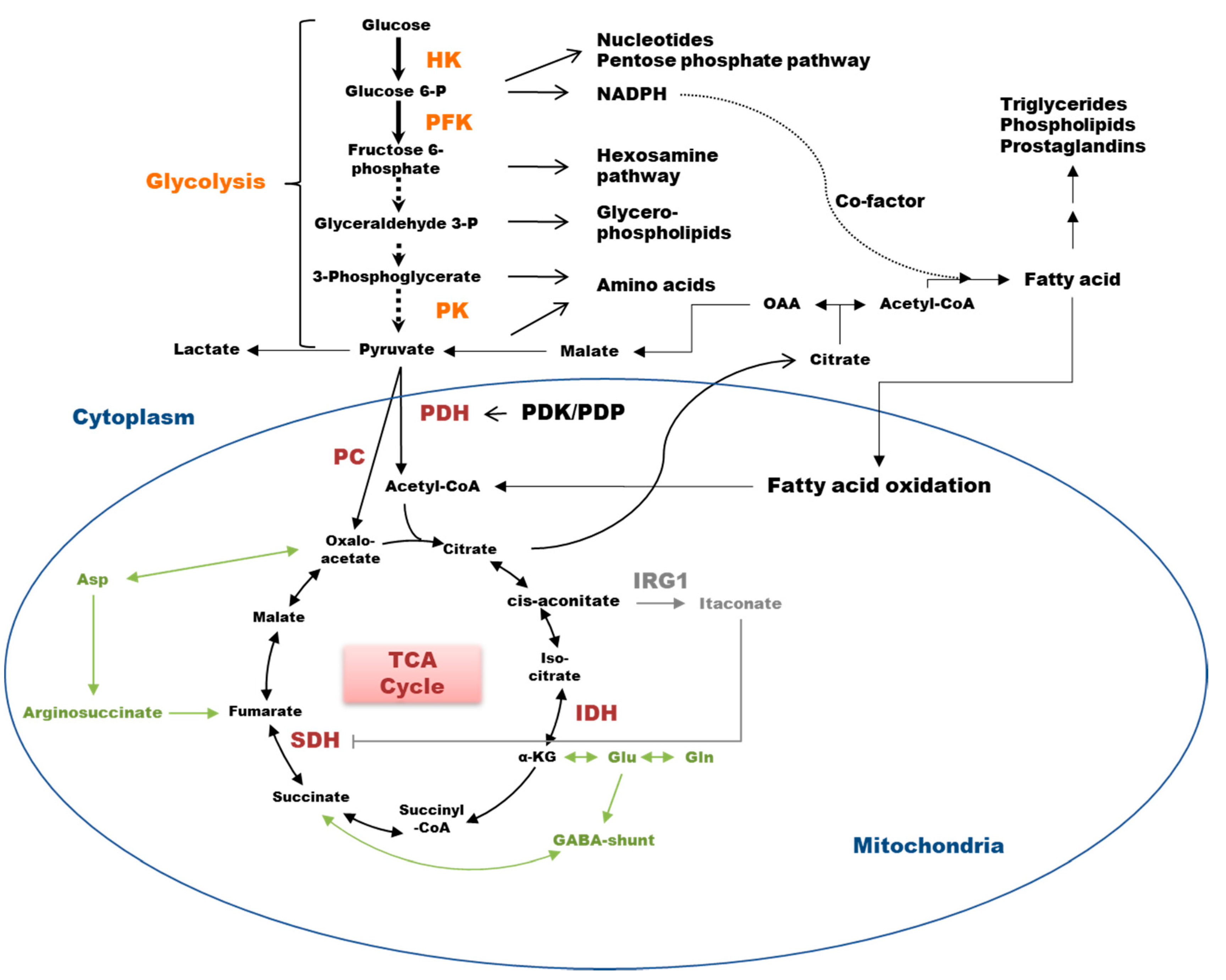
2. Glycolysis in Cell Bioenergetics
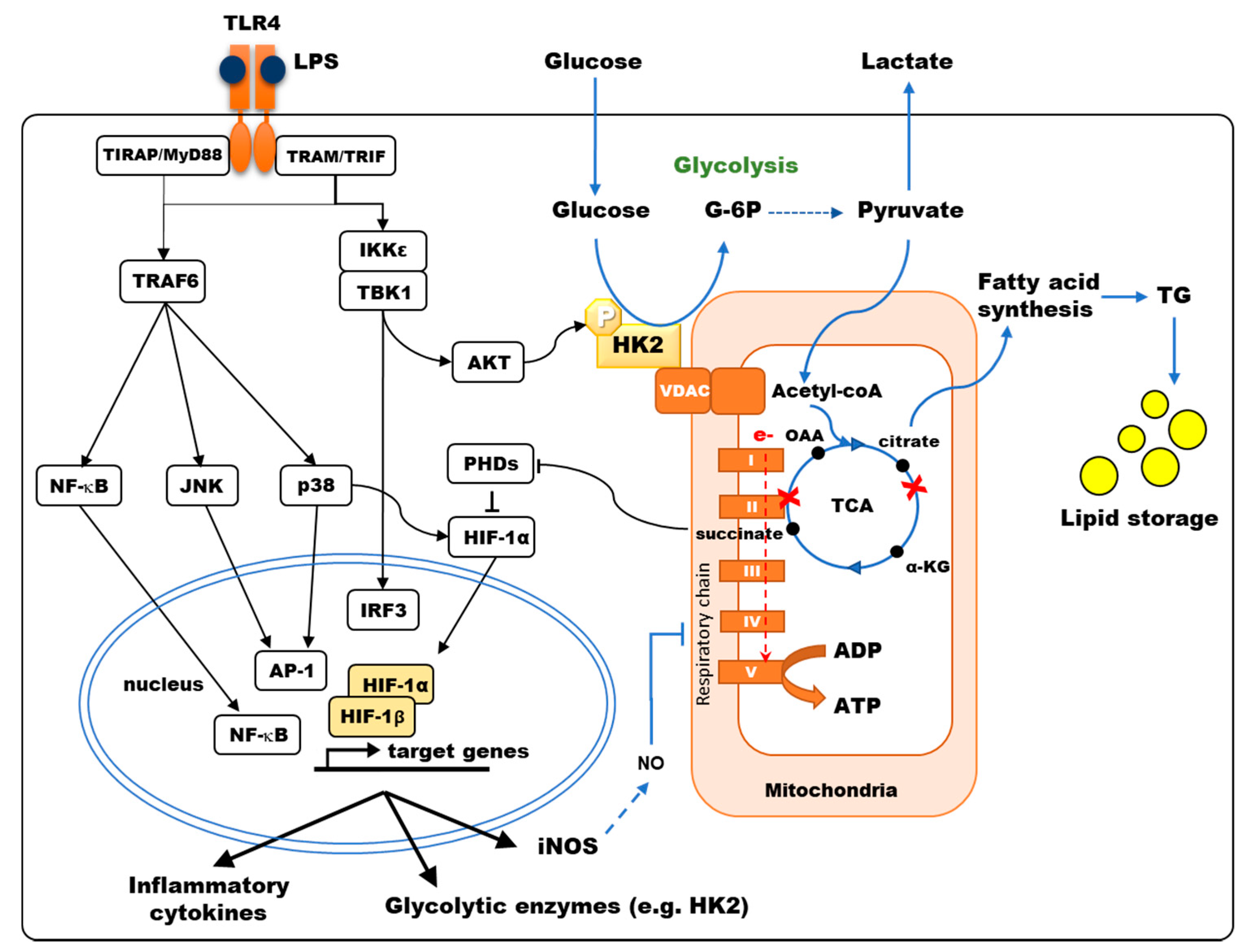
3. PRR Stimulation Triggers a Glycolytic Reprogramming of Immune Cells
4. Different Metabolic Reprogramming According to Cell Type, Species and Origin
5. Mechanisms Controlling Glycolytic Reprogramming in Myeloid Cells
6. TCA Rewiring
7. Lipid Metabolism
8. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Mathis, D.; Shoelson, S.E. Immunometabolism: An emerging frontier. Nat. Rev. Immunol. 2011, 11, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loftus, R.M.; Finlay, D.K. Immunometabolism: Cellular Metabolism Turns Immune Regulator. J. Biol. Chem. 2016, 291, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, L.A.; Pearce, E.J. Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 2016, 213, 15–23. [Google Scholar] [CrossRef]
- Everts, B.; Amiel, E.; van der Windt, G.J.W.; Freitas, T.C.; Chott, R.; Yarasheski, K.E.; Pearce, E.L.; Pearce, E.J. Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood 2012, 120, 1422–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melkonian, E.A.; Schury, M.P. Biochemistry, Anaerobic Glycolysis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Roberts, D.J.; Miyamoto, S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. 2015, 22, 248–257. [Google Scholar] [CrossRef] [Green Version]
- Perrin-Cocon, L.; Aublin-Gex, A.; Diaz, O.; Ramiere, C.; Peri, F.; Andre, P.; Lotteau, V. Toll-like Receptor 4-Induced Glycolytic Burst in Human Monocyte-Derived Dendritic Cells Results from p38-Dependent Stabilization of HIF-1alpha and Increased Hexokinase II Expression. J. Immunol. 2018, 201, 1510–1521. [Google Scholar] [CrossRef] [Green Version]
- Jantsch, J.; Chakravortty, D.; Turza, N.; Prechtel, A.T.; Buchholz, B.; Gerlach, R.G.; Volke, M.; Glasner, J.; Warnecke, C.; Wiesener, M.S.; et al. Hypoxia and hypoxia-inducible factor-1 alpha modulate lipopolysaccharide-induced dendritic cell activation and function. J. Immunol. 2008, 180, 4697–4705. [Google Scholar] [CrossRef]
- Everts, B.; Amiel, E.; Huang, S.C.; Smith, A.M.; Chang, C.H.; Lam, W.Y.; Redmann, V.; Freitas, T.C.; Blagih, J.; van der Windt, G.J.; et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat. Immunol. 2014, 15, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Krawczyk, C.M.; Holowka, T.; Sun, J.; Blagih, J.; Amiel, E.; DeBerardinis, R.J.; Cross, J.R.; Jung, E.; Thompson, C.B.; Jones, R.G.; et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 2010, 115, 4742–4749. [Google Scholar] [CrossRef] [Green Version]
- Perrin-Cocon, L.; Aublin-Gex, A.; Sestito, S.E.; Shirey, K.A.; Patel, M.C.; Andre, P.; Blanco, J.C.; Vogel, S.N.; Peri, F.; Lotteau, V. TLR4 antagonist FP7 inhibits LPS-induced cytokine production and glycolytic reprogramming in dendritic cells, and protects mice from lethal influenza infection. Sci. Rep. 2017, 7, 40791. [Google Scholar] [CrossRef]
- Guak, H.; Al Habyan, S.; Ma, E.H.; Aldossary, H.; Al-Masri, M.; Won, S.Y.; Ying, T.; Fixman, E.D.; Jones, R.G.; McCaffrey, L.M.; et al. Glycolytic metabolism is essential for CCR7 oligomerization and dendritic cell migration. Nat. Commun. 2018, 9, 2463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thwe, P.M.; Pelgrom, L.; Cooper, R.; Beauchamp, S.; Reisz, J.A.; D’Alessandro, A.; Everts, B.; Amiel, E. Cell-Intrinsic Glycogen Metabolism Supports Early Glycolytic Reprogramming Required for Dendritic Cell Immune Responses. Cell Metab. 2017, 26, 558–567.e5. [Google Scholar] [CrossRef] [Green Version]
- Curtis, K.D.; Smith, P.R.; Despres, H.W.; Snyder, J.P.; Hogan, T.C.; Rodriguez, P.D.; Amiel, E. Glycogen Metabolism Supports Early Glycolytic Reprogramming and Activation in Dendritic Cells in Response to Both TLR and Syk-Dependent CLR Agonists. Cells 2020, 9, 715. [Google Scholar] [CrossRef] [Green Version]
- Das Gupta, K.; Shakespear, M.R.; Curson, J.E.B.; Murthy, A.M.V.; Iyer, A.; Hodson, M.P.; Ramnath, D.; Tillu, V.A.; von Pein, J.B.; Reid, R.C.; et al. Class IIa Histone Deacetylases Drive Toll-like Receptor-Inducible Glycolysis and Macrophage Inflammatory Responses via Pyruvate Kinase M2. Cell Rep. 2020, 30, 2712–2728.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amiel, E.; Everts, B.; Fritz, D.; Beauchamp, S.; Ge, B.; Pearce, E.L.; Pearce, E.J. Mechanistic Target of Rapamycin Inhibition Extends Cellular Lifespan in Dendritic Cells by Preserving Mitochondrial Function. J. Immunol. 2014, 193, 2821–2830. [Google Scholar] [CrossRef] [PubMed]
- Bajwa, G.; DeBerardinis, R.J.; Shao, B.; Hall, B.; Farrar, J.D.; Gill, M.A. Cutting Edge: Critical Role of Glycolysis in Human Plasmacytoid Dendritic Cell Antiviral Responses. J. Immunol. 2016, 196, 2004–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fekete, T.; Sütö, M.I.; Bencze, D.; Mázló, A.; Szabo, A.; Biro, T.; Bacsi, A.; Pazmandi, K. Human Plasmacytoid and Monocyte-Derived Dendritic Cells Display Distinct Metabolic Profile Upon RIG-I Activation. Front. Immunol. 2018, 9, 3070. [Google Scholar] [CrossRef]
- Murugina, N.E.; Budikhina, A.S.; Dagil, Y.A.; Maximchik, P.V.; Balyasova, L.S.; Murugin, V.V.; Melnikov, M.V.; Sharova, V.S.; Nikolaeva, A.M.; Chkadua, G.Z.; et al. Glycolytic reprogramming of macrophages activated by NOD1 and TLR4 agonists: No association with proinflammatory cytokine production in normoxia. J. Biol. Chem. 2020, 295, 3099–3114. [Google Scholar] [CrossRef]
- Vijayan, V.; Pradhan, P.; Braud, L.; Fuchs, H.R.; Gueler, F.; Motterlini, R.; Foresti, R.; Immenschuh, S. Human and murine macrophages exhibit differential metabolic responses to lipopolysaccharide—A divergent role for glycolysis. Redox Biol. 2019, 22, 101147. [Google Scholar] [CrossRef]
- Huang, Y.L.; Morales-Rosado, J.; Ray, J.; Myers, T.G.; Kho, T.; Lu, M.; Munford, R.S. Toll-like receptor agonists promote prolonged triglyceride storage in macrophages. J. Biol. Chem. 2014, 289, 3001–3012. [Google Scholar] [CrossRef] [Green Version]
- Chiba, S.; Hisamatsu, T.; Suzuki, H.; Mori, K.; Kitazume, M.T.; Shimamura, K.; Mizuno, S.; Nakamoto, N.; Matsuoka, K.; Naganuma, M.; et al. Glycolysis regulates LPS-induced cytokine production in M2 polarized human macrophages. Immunol. Lett. 2017, 183, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Ménégaut, L.; Thomas, C.; Jalil, A.; Julla, J.B.; Magnani, C.; Ceroi, A.; Basmaciyan, L.; Dumont, A.; Le Goff, W.; Mathew, M.J.; et al. Interplay between Liver X Receptor and Hypoxia Inducible Factor 1α Potentiates Interleukin-1β Production in Human Macrophages. Cell Rep. 2020, 31, 107665. [Google Scholar] [CrossRef] [PubMed]
- Lavrich, K.S.; Speen, A.M.; Ghio, A.J.; Bromberg, P.A.; Samet, J.M.; Alexis, N.E. Macrophages from the upper and lower human respiratory tract are metabolically distinct. Am. J. Physiol. Cell. Mol. Physiol. 2018, 315, L752–L764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basit, F.; de Vries, I.J.M. Dendritic Cells Require PINK1-Mediated Phosphorylation of BCKDE1α to Promote Fatty Acid Oxidation for Immune Function. Front. Immunol. 2019, 10, 2386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dáňová, K.; Klapetková, A.; Kayserová, J.; Šedivá, A.; Špíšek, R.; Jelínková, L.P. NF-κB, p38 MAPK, ERK1/2, mTOR, STAT3 and increased glycolysis regulate stability of paricalcitol/dexamethasone-generated tolerogenic dendritic cells in the inflammatory environment. Oncotarget 2015, 6, 14123–14138. [Google Scholar] [CrossRef]
- Malinarich, F.; Duan, K.; Hamid, R.A.; Bijin, A.; Lin, W.X.; Poidinger, M.; Fairhurst, A.M.; Connolly, J.E. High mitochondrial respiration and glycolytic capacity represent a metabolic phenotype of human tolerogenic dendritic cells. J. Immunol. 2015, 194, 5174–5186. [Google Scholar] [CrossRef] [Green Version]
- Basit, F.; Mathan, T.; Sancho, D.; de Vries, I.J.M. Human Dendritic Cell Subsets Undergo Distinct Metabolic Reprogramming for Immune Response. Front. Immunol. 2018, 9, 2489. [Google Scholar] [CrossRef] [Green Version]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Palsson-McDermott, E.M.; Curtis, A.M.; Goel, G.; Lauterbach, M.A.; Sheedy, F.J.; Gleeson, L.E.; van den Bosch, M.W.; Quinn, S.R.; Domingo-Fernandez, R.; Johnston, D.G.; et al. Pyruvate kinase M2 regulates Hif-1alpha activity and IL-1beta induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 2015, 21, 65–80. [Google Scholar] [CrossRef] [Green Version]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Dabritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457–470. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, E.M.; Gonzalez-Cotto, M.; Baseler, W.A.; Davies, L.C.; Ghesquière, B.; Maio, N.; Rice, C.M.; Rouault, T.A.; Cassel, T.; Higashi, R.M.; et al. Nitric oxide orchestrates metabolic rewiring in M1 macrophages by targeting aconitase 2 and pyruvate dehydrogenase. Nat. Commun. 2020, 11, 698. [Google Scholar] [CrossRef] [PubMed]
- Van den Bossche, J.; Baardman, J.; Otto, N.A.; van der Velden, S.; Neele, A.E.; van den Berg, S.M.; Luque-Martin, R.; Chen, H.-J.; Boshuizen, M.C.S.; Ahmed, M.; et al. Mitochondrial Dysfunction Prevents Repolarization of Inflammatory Macrophages. Cell Rep. 2016, 17, 684–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koenis, D.S.; Medzikovic, L.; van Loenen, P.B.; van Weeghel, M.; Huveneers, S.; Vos, M.; Evers-van Gogh, I.J.; Van den Bossche, J.; Speijer, D.; Kim, Y.; et al. Nuclear Receptor Nur77 Limits the Macrophage Inflammatory Response through Transcriptional Reprogramming of Mitochondrial Metabolism. Cell Rep. 2018, 24, 2127–2140. [Google Scholar] [CrossRef] [Green Version]
- Jha, A.K.; Huang, S.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seim, G.L.; Britt, E.C.; John, S.V.; Yeo, F.J.; Johnson, A.R.; Eisenstein, R.S.; Pagliarini, D.J.; Fan, J. Two-stage metabolic remodelling in macrophages in response to lipopolysaccharide and interferon-γ stimulation. Nat. Metab. 2019, 1, 731–742. [Google Scholar] [CrossRef]
- Cordes, T.; Wallace, M.; Michelucci, A.; Divakaruni, A.S.; Sapcariu, S.C.; Sousa, C.; Koseki, H.; Cabrales, P.; Murphy, A.N.; Hiller, K.; et al. Immunoresponsive Gene 1 and Itaconate Inhibit Succinate Dehydrogenase to Modulate Intracellular Succinate Levels. J. Biol. Chem. 2016, 291, 14274–14284. [Google Scholar] [CrossRef] [Green Version]
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.; Griss, T.; et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 2016, 24, 158–166. [Google Scholar] [CrossRef] [Green Version]
- Moon, J.-S.; Hisata, S.; Park, M.-A.; DeNicola, G.M.; Ryter, S.W.; Nakahira, K.; Choi, A.M.K. mTORC1-Induced HK1-Dependent Glycolysis Regulates NLRP3 Inflammasome Activation. Cell Rep. 2015, 12, 102–115. [Google Scholar] [CrossRef] [Green Version]
- Tan, Z.; Xie, N.; Banerjee, S.; Cui, H.; Fu, M.; Thannickal, V.J.; Liu, G. The monocarboxylate transporter 4 is required for glycolytic reprogramming and inflammatory response in macrophages. J. Biol. Chem. 2015, 290, 46–55. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zhao, Q.; Nie, Y.; Yu, Y.; Misra, B.B.; Zabalawi, M.; Chou, J.W.; Key, C.-C.C.; Molina, A.J.; Quinn, M.A.; et al. Solute Carrier Family 37 Member 2 (SLC37A2) Negatively Regulates Murine Macrophage Inflammation by Controlling Glycolysis. iScience 2020, 23, 101125. [Google Scholar] [CrossRef]
- Lauterbach, M.A.; Hanke, J.E.; Serefidou, M.; Mangan, M.S.J.; Kolbe, C.-C.; Hess, T.; Rothe, M.; Kaiser, R.; Hoss, F.; Gehlen, J.; et al. Toll-like Receptor Signaling Rewires Macrophage Metabolism and Promotes Histone Acetylation via ATP-Citrate Lyase. Immunity 2019, 51, 997–1011. [Google Scholar] [CrossRef]
- Feng, T.-T.; Yang, X.-Y.; Hao, S.-S.; Sun, F.-F.; Huang, Y.; Lin, Q.-S.; Pan, W. TLR-2-mediated metabolic reprogramming participates in polyene phosphatidylcholine-mediated inhibition of M1 macrophage polarization. Immunol. Res. 2020, 68, 28–38. [Google Scholar] [CrossRef]
- Rodriguez-Prados, J.C.; Traves, P.G.; Cuenca, J.; Rico, D.; Aragones, J.; Martin-Sanz, P.; Cascante, M.; Bosca, L. Substrate fate in activated macrophages: A comparison between innate, classic, and alternative activation. J. Immunol. 2010, 185, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Tavakoli, S.; Zamora, D.; Ullevig, S.; Asmis, R. Bioenergetic profiles diverge during macrophage polarization: Implications for the interpretation of 18F-FDG PET imaging of atherosclerosis. J. Nucl. Med. 2013, 54, 1661–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feingold, K.R.; Shigenaga, J.K.; Kazemi, M.R.; McDonald, C.M.; Patzek, S.M.; Cross, A.S.; Moser, A.; Grunfeld, C. Mechanisms of triglyceride accumulation in activated macrophages. J. Leukoc. Biol. 2012, 92, 829–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakhtoura, M.; Chain, R.W.; Sato, P.Y.; Qiu, C.C.; Lee, M.H.; Meissler, J.J.; Eisenstein, T.K.; Koch, W.J.; Caricchio, R.; Gallucci, S. Ethyl Pyruvate Modulates Murine Dendritic Cell Activation and Survival Through Their Immunometabolism. Front. Immunol. 2019, 10, 30. [Google Scholar] [CrossRef] [PubMed]
- Lawless, S.J.; Kedia-Mehta, N.; Walls, J.F.; McGarrigle, R.; Convery, O.; Sinclair, L.V.; Navarro, M.N.; Murray, J.; Finlay, D.K. Glucose represses dendritic cell-induced T cell responses. Nat. Commun. 2017, 8, 15620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loo, Y.-M.; Gale, M. Immune signaling by RIG-I-like receptors. Immunity 2011, 34, 680–692. [Google Scholar] [CrossRef] [Green Version]
- Rezinciuc, S.; Bezavada, L.; Bahadoran, A.; Duan, S.; Wang, R.; Lopez-Ferrer, D.; Finkelstein, D.; McGargill, M.A.; Green, D.R.; Pasa-Tolic, L.; et al. Dynamic metabolic reprogramming in dendritic cells: An early response to influenza infection that is essential for effector function. PLoS Pathog. 2020, 16, e1008957. [Google Scholar] [CrossRef]
- Li, T.; Li, X.; Attri, K.S.; Liu, C.; Li, L.; Herring, L.E.; Asara, J.M.; Lei, Y.L.; Singh, P.K.; Gao, C.; et al. O-GlcNAc Transferase Links Glucose Metabolism to MAVS-Mediated Antiviral Innate Immunity. Cell Host Microbe 2018, 24, 791–803. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Wang, G.; Xu, Z.G.; Tu, H.; Hu, F.; Dai, J.; Chang, Y.; Chen, Y.; Lu, Y.; Zeng, H.; et al. Lactate Is a Natural Suppressor of RLR Signaling by Targeting MAVS. Cell 2019, 178, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Perrin-Cocon, L.; Vidalain, P.O.; Jacquemin, C.; Aublin-Gex, A.; Olmstead, K.; Panthu, B.; Rautureau, G.J.P.; André, P.; Nyczka, P.; Hutt, M.-T.; et al. A hexokinase isoenzyme switch in human liver cancer cells promotes lipogenesis and enhances innate immunity. Commun. Biol. 2020, in press. [Google Scholar]
- Kelly, B.; O’Neill, L.A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Ma, J.; Tang, K.; Huang, B. Beyond energy storage: Roles of glycogen metabolism in health and disease. FEBS J. 2020, in press. [Google Scholar]
- Ma, J.; Wei, K.; Liu, J.; Tang, K.; Zhang, H.; Zhu, L.; Chen, J.; Li, F.; Xu, P.; Chen, J.; et al. Glycogen metabolism regulates macrophage-mediated acute inflammatory responses. Nat. Commun. 2020, 11, 1769. [Google Scholar] [CrossRef] [Green Version]
- Artyomov, M.N.; Sergushichev, A.; Schilling, J.D. Integrating immunometabolism and macrophage diversity. Semin. Immunol. 2016, 28, 417–424. [Google Scholar] [CrossRef] [Green Version]
- Schroder, K.; Irvine, K.M.; Taylor, M.S.; Bokil, N.J.; Le Cao, K.-A.; Masterman, K.-A.; Labzin, L.I.; Semple, C.A.; Kapetanovic, R.; Fairbairn, L.; et al. Conservation and divergence in Toll-like receptor 4-regulated gene expression in primary human versus mouse macrophages. Proc. Natl. Acad. Sci. USA 2012, 109, E944–E953. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.F.; Vachharajani, V.T.; Yoza, B.K.; McCall, C.E. NAD+-dependent sirtuin 1 and 6 proteins coordinate a switch from glucose to fatty acid oxidation during the acute inflammatory response. J. Biol. Chem. 2012, 287, 25758–25769. [Google Scholar] [CrossRef] [Green Version]
- Corcoran, S.E.; O’Neill, L.A. HIF1alpha and metabolic reprogramming in inflammation. J. Clin. Investig. 2016, 126, 3699–3707. [Google Scholar] [CrossRef]
- Haschemi, A.; Kosma, P.; Gille, L.; Evans, C.R.; Burant, C.F.; Starkl, P.; Knapp, B.; Haas, R.; Schmid, J.A.; Jandl, C.; et al. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab. 2012, 15, 813–826. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, J.; Nguyen, A.H.; Rehman, A.; Ochi, A.; Jamal, M.; Graffeo, C.S.; Henning, J.R.; Zambirinis, C.P.; Fallon, N.C.; Barilla, R.; et al. Dendritic cell populations with different concentrations of lipid regulate tolerance and immunity in mouse and human liver. Gastroenterology 2012, 143, 1061–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Infantino, V.; Convertini, P.; Cucci, L.; Panaro, M.A.; Di Noia, M.A.; Calvello, R.; Palmieri, F.; Iacobazzi, V. The mitochondrial citrate carrier: A new player in inflammation. Biochem. J. 2011, 438, 433–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, L.A. A critical role for citrate metabolism in LPS signalling. Biochem. J. 2011, 438, e5–e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weichhart, T.; Hengstschläger, M.; Linke, M. Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 2015, 15, 599–614. [Google Scholar] [CrossRef]
- Roberts, D.J.; Tan-Sah, V.P.; Ding, E.Y.; Smith, J.M.; Miyamoto, S. Hexokinase-II positively regulates glucose starvation-induced autophagy through TORC1 inhibition. Mol. Cell 2014, 53, 521–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, L.A. A broken krebs cycle in macrophages. Immunity 2015, 42, 393–394. [Google Scholar] [CrossRef] [Green Version]
- Bambouskova, M.; Gorvel, L.; Lampropoulou, V.; Sergushichev, A.; Loginicheva, E.; Johnson, K.; Korenfeld, D.; Mathyer, M.E.; Kim, H.; Huang, L.-H.; et al. Electrophilic properties of itaconate and derivatives regulate the IκBζ–ATF3 inflammatory axis. Nature 2018, 556, 501–504. [Google Scholar] [CrossRef]
- Olagnier, D.; Farahani, E.; Thyrsted, J.; Blay-Cadanet, J.; Herengt, A.; Idorn, M.; Hait, A.; Hernaez, B.; Knudsen, A.; Iversen, M.B.; et al. SARS-CoV2-mediated suppression of NRF2-signaling reveals potent antiviral and anti-inflammatory activity of 4-octyl-itaconate and dimethyl fumarate. Nat. Commun. 2020, 11, 4938. [Google Scholar] [CrossRef]
- Rubic, T.; Lametschwandtner, G.; Jost, S.; Hinteregger, S.; Kund, J.; Carballido-Perrig, N.; Schwarzler, C.; Junt, T.; Voshol, H.; Meingassner, J.G.; et al. Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat. Immunol. 2008, 9, 1261–1269. [Google Scholar] [CrossRef]
- Littlewood-Evans, A.; Sarret, S.; Apfel, V.; Loesle, P.; Dawson, J.; Zhang, J.; Muller, A.; Tigani, B.; Kneuer, R.; Patel, S.; et al. GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J. Exp. Med. 2016, 213, 1655–1662. [Google Scholar] [CrossRef]
- van Diepen, J.A.; Robben, J.H.; Hooiveld, G.J.; Carmone, C.; Alsady, M.; Boutens, L.; Bekkenkamp-Grovenstein, M.; Hijmans, A.; Engelke, U.F.H.; Wevers, R.A.; et al. SUCNR1-mediated chemotaxis of macrophages aggravates obesity-induced inflammation and diabetes. Diabetologia 2017, 60, 1304–1313. [Google Scholar] [CrossRef] [Green Version]
- Keiran, N.; Ceperuelo-Mallafre, V.; Calvo, E.; Hernandez-Alvarez, M.I.; Ejarque, M.; Nunez-Roa, C.; Horrillo, D.; Maymo-Masip, E.; Rodriguez, M.M.; Fradera, R.; et al. SUCNR1 controls an anti-inflammatory program in macrophages to regulate the metabolic response to obesity. Nat. Immunol. 2019, 20, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.S.; Lee, S.; Park, M.A.; Siempos, I.; Haslip, M.; Lee, P.J.; Yun, M.; Kim, C.K.; Howrylak, J.; Ryter, S.W.; et al. UCP2-induced fatty acid synthase promotes NLRP3 inflammasome activation during sepsis. J. Clin. Investig. 2015, 125, 665–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raulien, N.; Friedrich, K.; Strobel, S.; Rubner, S.; Baumann, S.; von Bergen, M.; Korner, A.; Krueger, M.; Rossol, M.; Wagner, U. Fatty Acid Oxidation Compensates for Lipopolysaccharide-Induced Warburg Effect in Glucose-Deprived Monocytes. Front. Immunol. 2017, 8, 609. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Carroll, K.C.; Viollet, B.; Suttles, J. AMPKalpha1 deficiency amplifies proinflammatory myeloid APC activity and CD40 signaling. J. Leukoc. Biol. 2013, 94, 1113–1121. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.F.; Vachharajani, V.; Millet, P.; Bharadwaj, M.S.; Molina, A.J.; McCall, C.E. Sequential actions of SIRT1-RELB-SIRT3 coordinate nuclear-mitochondrial communication during immunometabolic adaptation to acute inflammation and sepsis. J. Biol. Chem. 2015, 290, 396–408. [Google Scholar] [CrossRef] [Green Version]
- Panni, R.Z.; Linehan, D.C.; De Nardo, D.G. Targeting tumor-infiltrating macrophages to combat cancer. Immunotherapy 2013, 5, 1075–1087. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Host | Cell Type | Stimulation | Metabolic Consequences | Molecular Mechanism | Ref. |
|---|---|---|---|---|---|
| Human | MDMs | TLR4 | -Glycolysis induction -Intracellular TG accumulation -FAO induction in M2 | AKT-dependent glycolysis induction | [19,20] |
| [21,22] | |||||
| MDMs | TLR2 | -Glycolysis induction | [23] | ||
| Bronchoalveolar macrophages | TLR4 | -No metabolic modification | [24] | ||
| MoDCs | TLR4 | -Glycolysis induction -Glycogen mobilization to support early DC maturation | TLR activation enhanced HK2 expression through a p38-MAPK–dependent HIF-1α accumulation | [7,11] | |
| [13,25] | |||||
| MoDCs | TLR1/2, TLR2/6 | -Glycolysis induction | [7] | ||
| MoDCs | TLR7/8 | -Increased OXPHOS | PINK1-dependent increase of intracellular branched-chain amino acid levels and FAO | [25] | |
| Tolerogenic DCs | TLR4, TLR3 | -Glycolysis induction -Increased OXPHOS and FAO | p38MAPK, ERK1/2, mTOR, STAT3 and mTOR-dependent glycolysis regulates tolerogenic phenotype. | [26] | |
| Shift in redox state | [27] | ||||
| CD1c+ DCs | TLR7/8 | -Glycolysis induction -Reduced OXPHOS | stimulation of BNIP3-dependent mitophagy, which regulates transcriptional activity of AMPKα1. | [28] | |
| Mouse | BMDMs | TLR4 | -Glycolysis induction -Decreased OCR -Rewiring TCA cycle -Decreased ratio of ATP/ADP -Induction of lipid synthesis | ROS production and succinate induced HIF-1α, PKM2 cooperation to induce glycolysis | [15,29,30,31] |
| NO production | [20,32,33,34] | ||||
| Itaconate production | [35,36,37,38] | ||||
| mTORC1/HK1 | [39] | ||||
| Induction of HK2, PFKFB and MCT4 in an NF-κB-dependent manner. | [40] | ||||
| Increased SLC37A2 protein expression | [41] | ||||
| Activation of ATP-citrate lyase (ACLY) promotes histone acetylation. | [42] | ||||
| Transcriptional regulation of lipid synthesis and FAO enzymes | [43] | ||||
| Peritoneal macrophages | TLR2, TLR3, TLR4, TLR9 | -Glycolysis induction -Intracellular TG accumulation | HIF-1α induction of ubiquitous PFK2 isoenzyme in M1 macrophages | [44] | |
| Enhanced HK2 expression | [45] | ||||
| Transcriptional regulation of lipid synthesis, storage and lipolysis enzymes | [21,46] | ||||
| BMDCs | TLR2, TLR3, TLR8, TLR9 | -Induction of glycolysis | [9,10] | ||
| BMDCs | TLR4 | -Induction of glycolysis. -Decreased OCR -Induction of lipid synthesis -Glycogen mobilization | PI3K/Akt pathway; NO production. | [4,10,14,47] | |
| TBK1/IKKε and Akt activation promoting HK2 association to mitochondria. | [9] | ||||
| HIF-1α accumulation | [8,12] | ||||
| Glucose signals via mTORC1, HIF-1α and iNOS induction | [16,48] | ||||
| [13] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perrin-Cocon, L.; Diaz, O.; Aublin-Gex, A.; Vidalain, P.-O.; Lotteau, V. Reprogramming of Central Carbon Metabolism in Myeloid Cells upon Innate Immune Receptor Stimulation. Immuno 2021, 1, 1-14. https://0-doi-org.brum.beds.ac.uk/10.3390/immuno1010001
Perrin-Cocon L, Diaz O, Aublin-Gex A, Vidalain P-O, Lotteau V. Reprogramming of Central Carbon Metabolism in Myeloid Cells upon Innate Immune Receptor Stimulation. Immuno. 2021; 1(1):1-14. https://0-doi-org.brum.beds.ac.uk/10.3390/immuno1010001
Chicago/Turabian StylePerrin-Cocon, Laure, Olivier Diaz, Anne Aublin-Gex, Pierre-Olivier Vidalain, and Vincent Lotteau. 2021. "Reprogramming of Central Carbon Metabolism in Myeloid Cells upon Innate Immune Receptor Stimulation" Immuno 1, no. 1: 1-14. https://0-doi-org.brum.beds.ac.uk/10.3390/immuno1010001