
Hypoxia, Acidification and Inflammation: Partners in Crime in Parkinson’s Disease Pathogenesis?


1
Department of Biomedical Sciences, University of Lausanne, CH-1015 Lausanne, Switzerland
2
Institute of Sport Sciences, University of Lausanne, CH-1015 Lausanne, Switzerland
*
Author to whom correspondence should be addressed.
Immuno 2021, 1(2), 78-90; https://0-doi-org.brum.beds.ac.uk/10.3390/immuno1020006
Submission received: 31 March 2021
/
Revised: 30 April 2021
/
Accepted: 10 May 2021
/
Published: 12 May 2021
(This article belongs to the Section Neuroimmunology)
{kind=link}
Abstract
:Like in other neurodegenerative diseases, protein aggregation, mitochondrial dysfunction, oxidative stress and neuroinflammation are hallmarks of Parkinson’s disease (PD). Differentiating characteristics of PD include the central role of α-synuclein in the aggregation pathology, a distinct vulnerability of the striato-nigral system with the related motor symptoms, as well as specific mitochondrial deficits. Which molecular alterations cause neurodegeneration and drive PD pathogenesis is poorly understood. Here, we summarize evidence of the involvement of three interdependent factors in PD and suggest that their interplay is likely a trigger and/or aggravator of PD-related neurodegeneration: hypoxia, acidification and inflammation. We aim to integrate the existing knowledge on the well-established role of inflammation and immunity, the emerging interest in the contribution of hypoxic insults and the rather neglected effects of brain acidification in PD pathogenesis. Their tight association as an important aspect of the disease merits detailed investigation. Consequences of related injuries are discussed in the context of aging and the interaction of different brain cell types, in particular with regard to potential consequences on the vulnerability of dopaminergic neurons in the substantia nigra. A special focus is put on the identification of current knowledge gaps and we emphasize the importance of related insights from other research fields, such as cancer research and immunometabolism, for neurodegeneration research. The highlighted interplay of hypoxia, acidification and inflammation is likely also of relevance for other neurodegenerative diseases, despite disease-specific biochemical and metabolic alterations.
1. Parkinson’s Disease—A Very Brief Background
The second most common neurodegenerative disease, Parkinson’s Disease (PD), is characterized by the degeneration of dopaminergic (especially neuromelanin-containing [1]) neurons of the substantia nigra pars compacta that elicits the characteristic motor-symptoms, including bradykinesia, tremor and rigidity [2]. There are also other neuronal populations that degenerate in PD, including cholinergic neurons of the pedunculopontine nucleus and dorsal motor nucleus of the vagus, some glutamatergic neuronal populations in the intralaminar nuclei of the thalamus and basolateral amygdala, noradrenergic neurons of the locus coeruleus or serotonergic neurons of the raphe nuclei (summarized in detail in [3]). The particular vulnerability of specific neuronal populations in PD, however, is still enigmatic.
Non-motor symptoms are common as well in PD and may precede motor symptoms by decades [4]. Like in other neurodegenerative diseases, mitochondrial dysfunction [5,6], oxidative stress [7], neuroinflammation [8] and pathological protein aggregation [9] are involved in PD pathogenesis but their causative contributions are poorly understood. PD is classified as an α-synucleinopathy, together with related diseases, such as multiple systems atrophy (MSA) and dementia with Lewy bodies. The defining feature of this group of diseases is the aggregation of the protein α-synuclein. While mutations and multiplications of the α-synuclein-encoding gene, SNCA, can cause PD—as reviewed in [10] —most PD cases (about 90%) cannot be clearly linked to genetic factors. The main risk factor to develop such idiopathic PD is age [11].
The aim of this review is to highlight a possible role of the interplay of a set of specific cell-environmental alterations—that is at least partially modulated by aging—in PD pathogenesis. These alterations are characterized by deficiencies in cellular oxygen supply (hypoxia) and its interplay with acidification of the cellular milieu, as well as with inflammation. While much evidence indicates involvement of these factors, their mechanistic roles in PD etiopathogenesis are poorly understood. We argue that these conditions contribute to the initiation of neurodegenerative processes in vulnerable neurons. Some of the main knowledge gaps are emphasized in order to better understand the metabolic and biochemical alterations of the cellular milieu that render specific neuronal populations vulnerable to neurodegeneration. Such understanding is necessary to promote the development of novel therapeutic strategies able to target and prevent or even reverse these alterations.
2. Regulation of Oxygen Levels and Acidity in the Brain
The regulation of both oxygen levels and pH is critical in the brain and their perturbation may be even more critical in cells vulnerable to neurodegeneration, such as dopaminergic neurons of the substantia nigra in PD. A brief outline on how such regulation is effectuated and how it may be impaired in PD is given below.
2.1. Oxygen-Sensing and Consumption in PD Brain
The brain is one of the major oxygen-consuming organs. This is due to the heavy reliance of neurons in general on oxidative energy metabolism; neurons consume around 80% of the oxygen delivered to the brain [12,13], although the number of non-neuronal cells in the brain is similar to that of neurons [14]. Certain structural and functional features of neurons even increase their dependence on adequate energy levels—and thus on oxygen—as is the case for, e.g., dopaminergic projection neurons. The few (around 300,000–600,000) dopaminergic neurons in the human ventral midbrain (substantia nigra and ventral tegmental area) project and innervate the striatum by means of an estimated 75,000–200,000 presynaptic terminals per dopaminergic neuron [15,16,17,18]. For this purpose, these neurons rely on long, poorly myelinated and highly branched axons [19]. In conjunction with their numerous dendrites, this results in cell body volumes of less than 1% of the whole cell [20] and is associated with a high demand of ATP and oxygen, mitochondrial strain and oxidative stress, as excellently outlined in [3]. Together with pronounced ATP demands for unusually high Na+/K+ ATPase (which maintains the neuronal membrane potential) activities [21] and their general electrophysiological properties [3,20] these features likely contribute to the vulnerability of dopaminergic neurons of the substantia nigra pars compacta in PD.
Conversely, the main source of energy of many non-neuronal brain cells, such as astrocytes and oligodendrocytes, is glycolysis [13]. The energy metabolism of several other cell types in the brain is less understood, but—for example, in the resident immune cells of the brain, microglia—often involves a change in the reliance on metabolic pathways upon activation. Microglia become activated in inflammatory conditions; reduced mitochondrial respiration then is accompanied by increased rates of glycolysis [22]. Increased microglial activation has been shown in PD patients’ brains [23] and the metabolic consequences likely contribute to adverse alterations of the brain environment. Over-activation of microglia can be directly damaging to neurons but also indirectly by influencing other cell types (e.g., by inducing a conversion of normally neuroprotective astrocytes into a neurotoxic phenotype [24]).
Severe oxygen deficiency (hypoxia) is detrimental for the brain, as neurons have to satisfy their high energy demands via oxidative phosphorylation in order to maintain their characteristic energy-intensive functions, such as regulation of action potentials and trans-synaptic signaling. These functions require even more energy in the case of neurons with long projection axons and numerous pre-synaptic terminals, which is the case for the vulnerable neurons in PD. While many other brain cell types are able to dynamically upregulate glycolysis [13] and thus partially compensate for decreased oxygen availability, this does not apply to neurons due to their lack of the positive glycolysis-modulator 6 phosphofructose 2 kinase, fructose 2,6 bisphosphatase 3 (Pfkfb3) [25]. Instead of using glucose for glycolysis, neurons primarily process it through the pentose phosphate pathway, which results in the generation of reduced nicotinamide adenine dinucleotide phosphate (NADPH) that can regenerate glutathione disulfide (HSSG) to glutathione, an important reactive oxygen species (ROS) scavenger [13]. The importance of this anti-oxidant defense mechanism is illustrated by the observation that Pfkfb3 stabilization, and thus the redirection of glucose processing towards glycolysis, results in oxidative stress and cell death [25]. The neuronal vulnerability to oxidative stress is a key aspect of neurodegenerative processes [5]. Therefore, hypoxia and reoxygenation, due to their capacity to induce oxidative stress, are also risk factors from this point of view [26]. With regard to the specific vulnerability of dopaminergic neurons of the substantia nigra, dopamine metabolism is also associated with high ROS and reactive dopamine quinone formation [27,28] that may induce oxidative stress, which is increased in the substantia nigra of the PD patient brain [29]. In line with this observation, the antioxidant glutathione is reduced in the substantia nigra of the PD brain [30]. Altogether, these features may predispose dopaminergic neurons of the substantia nigra to oxidative damage. Indeed, oxidatively modified derivatives of dopamine are formed in the substantia nigra and can react with proteins and lipids, which become constituents of neuromelanin. Notably, this happens to a lower extent in the (less vulnerable) dopaminergic neurons of the ventral tegmental area [20] and is thus maybe one of the distinguishing neuronal vulnerability factors in PD.
The potential key role of hypoxia in PD pathogenesis has been recently outlined [31] and is supported by common respiratory deficits of PD patients [32,33] as well as potentially impaired hypoxia sensing [34]. Interestingly, neuronal loss of potentially chemosensitive respiratory neurons has been demonstrated for another α-synucleinopathy, MSA [35]. Central modulators of adaptations to hypoxia are hypoxia inducible factors (HIF) [36]. An upregulation of HIFs was recently reported in MSA and PD brains [37]. Together with reports on beneficial pharmacological modulation of HIFs in preclinical PD-models (e.g., [38,39,40]) and polymorphisms of HIFs as potential risk factors to develop PD [41], these results further substantiate the role of impaired hypoxia responses in α-synucleinopathy and PD pathogenesis.
Although it is clear that PD is associated with respiratory deficits, reduced tolerance to hypoxia or impaired adaptations to hypoxic stress, and that the vulnerable neurons in PD are particularly sensitive to reduced oxygen levels, it is unclear how these deficiencies are related to PD pathology and symptoms, at which time they appear during pathogenesis or whether they characterize sub-forms of PD. Additional knowledge gaps concern the interaction, metabolic alterations and consequential contributions to PD pathogenesis of different brain cell types that cooperate closely in the response to hypoxia and may promote neuroprotection or neurodegeneration depending on their activation status.
2.2. pH in PD Brain
The acid/base balance is essential for cellular functions and needs to be tightly controlled, in particular in conditions of metabolic stress. In the brain, tissue acidification by an increase in partial pressure of CO2 or of acidic metabolites can cause brain acidosis and severe brain damage [42]. Acidity/basicity, expressed as the potential of hydrogen (pH), in tissue mainly depends on the glycolytic rate and the generation of CO2 [43] and is regulated on the systemic and cellular level by sophisticated buffer systems [44]. Proteins are one example of pH-sensitive cellular components. Based on their H+ affinity (KH), the ambient pH determines protein protonation—and thus the interaction with other molecules and post-translational modifications—and protein structure and function. The systemic, local extracellular and intracellular pH defines cellular programs (e.g., proliferation and cell death) and therefore is decisive for the cellular fate and homeostasis [45,46]. Unsurprisingly, the cellular pH also influences protein aggregation: a lower pH enhances the aggregation of α-synuclein [47]. Furthermore, a tight control of vesicular pH has been demonstrated to be necessary for the regulation of dopamine auto-oxidation [48] and low pH has been shown to increase formation of the toxic 6-hydroxydopamine [49]. The regulation of the pH specifically in the brain furthermore is a requirement for neuronal signaling and is controlled by various transporter proteins and acid-sensing ion channels (ASICs) [44,50]. ASICs are responsive to extracellular acids and exhibit a varying degree of permeability for cations. The ASIC1A subunit provides a sufficient permeability to Ca2+ to confer the threat of neuronal damage upon activation. ASIC1A has also been demonstrated to be the crucial ASIC subunit for acid-sensing in rodent neurons and it is a putative key component of synaptic physiology [51].
Acidification of the extracellular milieu in the context of neurodegenerative disease is associated with an over-activation of ASICs. This at least partially mediates acid-induced toxicity in the brain due to impaired regulation of intracellular Ca2+ levels, which may be aggravated by ASIC-mediated modulation of Ca2+ translocation via the Ca2+ permeable AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) and NMDA (N-methyl-D-aspartate) receptors, as reviewed in [51]. The pace-making function of dopaminergic neurons in the substantia nigra (summarized by [3]) is associated with an unusual reliance on L-type Cav1.3 Ca2+ channels [52] and relatively high Ca2+ fluxes [53] concomitant with low Ca2+ buffering capacity [54]. High levels of Ca2+, which can initiate apoptosis (reviewed in [55]), are obviously dangerous for neurons and Ca2+ also enhances the aggregation and toxicity of α-synuclein (summarized in [3]). pH alterations and the resulting impairments of Ca2+ homeostasis may aggravate these endogenous vulnerabilities of degenerating dopaminergic neurons in PD.
A role of ASICs has indeed been found in several models of neurodegenerative diseases [51], including in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of PD, in which pharmacological ASICs inhibition by amiloride was neuroprotective [56]. ASIC1A deficiency in mice, however, did not confer protection in the same model, indicating that the reported amiloride effects may not be related to its pharmacological action on ASICs [57].
Impaired pH regulation is directly implicated in PD by reducing the cellular capacity of lysosome acidification [58]. Additionally, intracellular acidification may contribute to α-synuclein pathology in PD by favoring α-synuclein fibrillization [59], α-synuclein liquid–liquid phase separation [60] and α-synuclein–mitochondria interactions [61]. This latter process might aggravate toxic α-synuclein pathology formation [31].
A reported increased lactate accumulation in the PD patient brain (if associated with dementia) [62]—indicating a potential role of pH dysregulation in PD—is debated [63] and requires confirmation. The potentially higher PD incidence after use of proton pump inhibitors [64,65] may be a further indication of a role of pH dysregulation in PD progression. However, no causal implications can be derived from these studies. For example, proton pump inhibitors are sometimes prescribed for mood disorders, which are common prodromal symptoms in PD [4]. This suggests that there may be indirect associations between the incidence of PD and the use of proton pump inhibitors.
Notably, acidosis has also been linked to neurodegenerative processes in various other neurodegenerative diseases, such as Alzheimer’s disease [66,67] or amyotrophic lateral sclerosis (ALS) [68].
Taken together, one may assume that a dysregulation of pH is involved in PD pathogenesis, although this notion is not yet well established. More research is needed to understand the development of such deficiencies in PD and whether rescuing them can disrupt adverse pathological cascades. The conflicting results from targeting the pH buffer (e.g., ASICs) systems in the PD model brain also deserve further clarification.
3. Inflammation and pH Alterations in Brain Aging
Age is the main risk factor for the development of idiopathic neurodegenerative diseases [11], including PD [69,70]. Normal metabolic alterations in the aging brain support the assumption of an important interplay between hypoxia, pH alterations and inflammation constituting a vulnerability for age-related neurological diseases. Mitochondrial dysfunction and associated oxidative and inflammatory stress are positively correlated with age [71]. Hypoxia is also an important modulator of aging. However, the directionality of its effects can vary. This mainly depends on the severity of the hypoxic stimulus. Severe intermittent (shown, for example, in human white preadipocytes [72]) and sustained [73] hypoxia can promote cellular senescence and hormonal aging (shown in rodents in [74]). There is also some evidence that the HIF system becomes downregulated during aging [75,76], indicating that the cellular management of hypoxia is impaired in older individuals. In contrast, mild reductions in oxygen supply are associated with increased lifespans in various non-vertebrate [77,78,79,80] and vertebrate [81,82] organisms. Moreover, mild intermittent hypoxia (i.e., hypoxia conditioning, which improves the tolerance to hypoxic insults) may be protective in neurodegenerative diseases, including PD [31,83].
Increasing lysosomal membrane damage with aging has recently been reported to contribute to more acidic cellular environments [84]. Johmura et al. demonstrated that activation of glutaminase 1 protected senescent cells from acidification-induced clearance with detrimental consequences on organ function [84]. Therefore, by implication, cellular acidification may promote neurodegeneration-related pathology but, on the other hand, could also be protective by contributing to senescent cell clearance. This may be particularly relevant in PD, as lysosomal damage can promote diseases with protein aggregation pathology and is a characteristic of the PD brain [85,86]. Indeed, the activity of the lysosomal enzyme glucocerebrosidase decreases not only with aging but is also negatively correlated with PD pathology [87].
Senescence of different cell types in the brain may be an important factor in the development of neurodegenerative diseases in general. One example is astrocyte senescence, which has been prominently linked to neurodegeneration (reviewed recently by [88]). Senescence of these cells is thought to facilitate neurodegeneration both by gain of function (i.e., release of senescence-associated substances and the induction of neuroinflammation) and by loss of function effects. The latter comprises, for example, impairments in the regulation of the blood–brain barrier, the glymphatic system (a waste clearance system of the brain) and metabolic support for other cells (e.g., neurons by the provision of lactate). Astrocyte senescence has been directly linked to glutamate toxicity [89] and has been suggested as a contributor to PD pathogenesis [90].
In summary, cellular hypoxia, pH regulation and the inflammatory status of cells are modulated by increasing age. A reduced cellular capacity to deal with these stressors might facilitate PD pathogenesis. They may be “permissive” factors for the development of neurodegeneration, as discussed by Majdi et al. for acidification [91]. Senescence of different cell types in the brain likely contributes to hypoxic conditions, impaired pH regulation and inflammation and reduces the tolerance of neurons to these conditions.
4. The Interplay between Hypoxia, Acidification and Inflammation
Hypoxia and alterations in the pH of the cellular milieu are known to be strongly associated with inflammatory processes [26]. Brain hypoxia is also clearly linked to tissue pH via resulting lactate accumulation [92,93] and both affect inflammation. Hypoxia has been causally linked to neuroinflammatory diseases [94] and pH alterations are generally intimately associated with inflammatory responses [95]. Thus, as a consequence of an initial brain insult or infection (hypothetic causative trigger), hypoxia, pH alterations and/or inflammation may ensue. Under permissive conditions, these factors then might aggravate each other. Mechanistically, hypoxia [96] and cellular acidification [97] have been demonstrated to activate the nucleotide-binding domain leucine-rich repeat-containing family, pyrin domain-containing 3 (NLRP3) inflammasome. Like hypoxia and reoxygenation, acidification furthermore induces the production of reactive oxygen species [98]. In addition, hypoxic and acidic conditions are associated with the formation of mitochondria-derived damage-associated molecular patterns that can trigger inflammation and neuronal death [96,98]. Conversely, increased levels of reactive oxygen species also trigger intra-cellular acidification by various mechanisms, as summarized by Majdi et al. [91].
While still insufficiently understood, much evidence supports an association of immunity, infection and inflammation with PD and the related α-synuclein pathology. The involvement of immune system dysfunctions in PD has been summarized in an excellent review [24]. Recently, influenza virus infection has been demonstrated to induce α-synuclein aggregation by impairing autophagy [99]. An impairment of lysosome acidification—and thus of α-synuclein clearance—by pro-inflammatory cytokines [100] also is in line with observations of increased infection burden in PD [101].
Aggregated α-synuclein has furthermore been shown to induce an inflammatory response in PD patient blood [102]. This is in line with previous reports in rodent models of PD, in which aggregated—but not monomeric—α-synuclein triggered inflammation, including peripheral immune cell infiltration of the brain [103]. Furthermore, increased levels of pro-inflammatory cytokines have been found in the PD patient brain and cerebrospinal fluid, as reviewed in [104]. It is also possible that the dopaminergic neurons of the substantia nigra are particularly vulnerable to inflammation. This assumption is supported by upregulation of the proinflammatory cyclooxygenase 2 in the substantia nigra of the PD brain (which further might promote the formation of toxic dopamine-quinones) [105] and by high basal levels of other components of the immune response, such as major histocompatibility complex class I heavy chain and β2-microglobulin mRNAs [106]. In addition, the release of neuromelanin from dying neurons may induce microglial activation and neuroinflammation [20].
It is increasingly acknowledged that HIFs are crucial, not only in the adaptation to hypoxia, but that they are induced also under normoxia, for example in response to acidification [107] or inflammation [108]. HIFs also exert complex effects on immune responses [109] and hypoxia and inflammation likely exacerbate each other [110]. Lactate levels also influence inflammation, however, possibly by reducing it [93]. One mechanism for this effect is the capacity of lactate to bind mitochondrial antiviral signaling protein and thereby to inhibit the cellular interferon response [111]. Lactate has also been demonstrated to inhibit glutamate re-uptake by astrocytes [112], possibly contributing to NMDA receptor-mediated glutamate excitotoxicity [113].
While our understanding of the inter-dependence of metabolism and inflammation in response to hypoxia is rapidly expanding, these processes are insufficiently understood in the context of neurodegeneration. Metabolic alterations (including in relation to lactate and other metabolites at the crossroads of hypoxia-related adaptations and inflammation, such as succinate, citrate and NAD+ [114]) of different cell types and the consequences on energy availability, oxidative stress and inflammation are, however, likely of great relevance for PD and other neurodegenerative diseases.
The reported neuroprotective potential of lactate, for example in traumatic brain injury (reviewed by [115]) or in ischemia [116], and their possible anti-inflammatory effects [93], may contradict the assumption of a detrimental role of lactate in PD pathogenesis. However, toxic effects of high levels of lactate have been reported in models in which low levels were neuroprotective (e.g., [116]). This suggests a threshold of lactate levels, at which its actions turn from beneficial to detrimental. It is also conceivable that certain neurons, which are vulnerable in neurodegeneration, are less capable of handling—and profiting from—high lactate levels, in particular at a higher age.5. Conclusions and Implications
We here summarized evidence that hypoxia, brain acidification and inflammation are involved in the pathogenesis of neurodegenerative diseases and specifically of PD. These factors (apart from neuroinflammation) are still rather considered as consequences or epiphenomena and not as potentially causative factors in disease development. The many failures of clinical trials that have often targeted pathological protein aggregation and oxidative stress, and were considered as more important factors in PD and other neurodegenerative diseases, necessitate the exploration of alternative target pathways and novel therapeutic strategies. We hypothesize that the interplay of hypoxia, brain acidification and inflammation represents a central parameter driving PD pathogenesis. Due to their strong interdependence, the occurrence of any of these events in the brain has the potential to aggravate the others. In the sense of the model proposed by Johnson et al. [117], acute insults from hypoxia, pH dysregulation or inflammation may act to “trigger” neurodegenerative diseases, but may also contribute to a sustained impairment of cell metabolism and biochemistry based on ion homeostasis dysregulation, energy deficiency, oxidative stress and inflammation, thus “aggravating” pathogenesis. These conditions are influenced by different cell types and may be particularly detrimental for dopaminergic neurons of the substantia nigra and other vulnerable neurons in PD, based on their characteristically high energy demands and oxidative stress potential (see Figure 1). These neurons rely on mitochondrial efficiency and an adequate supply of oxygen and nutrients but also on efficient antioxidant defense systems. Hypoxia, pH alterations and inflammation may result in mitochondrial failure (and consequential energy deficits). They can also disrupt vesicular storage of dopamine and related cellular antioxidant defense mechanisms, proteostasis (e.g., by inhibition of autophagy, the proteasome and chaperones) and Ca2+ buffering. Consequently hypoxia, pH alterations and inflammation can initiate neurodegenerative processes.
Considerable advancements in the understanding of metabolic alterations and cellular vulnerabilities related to hypoxia and acidification come from cancer research, in which their inter-dependent roles have been recognized as integral to the pathological process [118]. The increasing knowledge on metabolic regulation of the immune system and its effects on cellular/tissue pH and inflammation from the booming research field of immunometabolism is also remarkable [119,120,121]. They provide a solid basis and a well of inspiration to investigate the complex interplay of hypoxia, acidification and inflammation in (models of) neurodegenerative diseases, and the derivation of related neuroprotective strategies.
As one example to achieve an enhanced resistance of the brain to hypoxic insults and inflammation, we recently provided rationales for the strengthening of brain resilience by hypoxia conditioning to counteract hypoxia-related brain insults in dementias [122], PD [31] and Huntington’s disease [123]. While these neurodegenerative diseases certainly are characterized by distinct pathologies and metabolic abnormalities [124], the shared outcomes of dysregulated proteostasis and REDOX homeostasis, as well as mitochondrial deficits and neuroinflammation, suggest an involvement of the interplay of hypoxia, impaired pH and inflammatory processes in many of them. The localization of related insults within the brain and individual tolerance—in combination with other genetic and environmental risk factors—may contribute to an explanation of the great pathological and symptomatic variability also within heterogeneous neurodegenerative disease spectra, such as PD or Alzheimer’s disease.
Author Contributions
J.B.; writing—original draft preparation and visualization, G.P.M.; writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hirsch, E.; Graybiel, A.M.; Agid, Y.A. Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson’s disease. Nature 1988, 334, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Fahn, S. Description of Parkinson’s disease as a clinical syndrome. Ann. N. Y. Acad. Sci. 2003, 991, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 101–113. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Monzio Compagnoni, G.; Di Fonzo, A.; Corti, S.; Comi, G.P.; Bresolin, N.; Masliah, E. The role of mitochondria in neurodegenerative diseases: The lesson from Alzheimer’s disease and Parkinson’s disease. Mol. Neurobiol. 2020, 57, 2959–2980. [Google Scholar] [CrossRef]
- Beal, M.F.; Oakes, D.; Shoulson, I.; Henchcliffe, C. A randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease: No evidence of benefit. JAMA Neurol. 2014, 71, 543–552. [Google Scholar]
- Hirsch, E.C.; Standaert, D.G. Ten Unsolved Questions About Neuroinflammation in Parkinson’s Disease. Mov. Disord. 2021, 36, 16–24. [Google Scholar] [CrossRef]
- Spires-Jones, T.L.; Attems, J.; Thal, D.R. Interactions of pathological proteins in neurodegenerative diseases. Acta Neuropathol. 2017, 134, 187–205. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of α-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2012, 14, 38–48. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Howarth, C.; Gleeson, P.; Attwell, D. Updated Energy Budgets for Neural Computation in the Neocortex and Cerebellum. J. Cereb. Blood Flow Metab. 2012, 32, 1222–1232. [Google Scholar] [CrossRef]
- Magistretti, P.J.; Allaman, I. A cellular perspective on brain energy metabolism and functional imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azevedo, F.A.C.; Carvalho, L.R.B.; Grinberg, L.T.; Farfel, J.M.; Ferretti, R.E.L.; Leite, R.E.P.; Filho, W.J.; Lent, R.; Herculano-Houzel, S. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J. Comp. Neurol. 2009, 513, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Oorschot, D.E. Total number of neurons in the neostriatal, pallidal, subthalamic, and substantia nigral nuclei of the rat basal ganglia: A stereological study using the cavalieri and optical disector methods. J. Comp. Neurol. 1996, 366, 580–599. [Google Scholar] [CrossRef]
- Roberts, R.C.; Force, M.; Kung, L. Dopaminergic synapses in the matrix of the ventrolateral striatum after chronic haloperidol treatment. Synapse 2002, 45, 78–85. [Google Scholar] [CrossRef]
- Matsuda, W.; Furuta, T.; Nakamura, K.C.; Hioki, H.; Fujiyama, F.; Arai, R.; Kaneko, T. Single Nigrostriatal Dopaminergic Neurons Form Widely Spread and Highly Dense Axonal Arborizations in the Neostriatum. J. Neurosci. 2009, 29, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Parent, M.; Parent, A. Relationship between axonal collateralization and neuronal degeneration in basal ganglia. In Parkinson’s Disease and Related Disorders; Springer: Vienna, Austria, 2006; pp. 85–88. [Google Scholar]
- Braak, H.; Bohl, J.R.; Müller, C.M.; Rüb, U.; de Vos, R.A.; Del Tredici, K. Stanley Fahn Lecture 2005: The staging procedure for the inclusion body pathology associated with sporadic Parkinson’s disease reconsidered. Mov. Disord. 2006, 21, 2042–2051. [Google Scholar] [CrossRef]
- Sulzer, D. Multiple hit hypotheses for dopamine neuron loss in Parkinson’s disease. Trends Neurosci. 2007, 30, 244–250. [Google Scholar] [CrossRef]
- Seutin, V.; Shen, K.-Z.; North, R.; Johnson, S. Sulfonylurea-sensitive potassium current evoked by sodium-loading in rat midbrain dopamine neurons. Neuroscience 1996, 71, 709–719. [Google Scholar] [CrossRef]
- Ghosh, S.; Castillo, E.; Frias, E.S.; Swanson, R.A. Bioenergetic regulation of microglia. Glia 2018, 66, 1200–1212. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Tan, E.-K.; Chao, Y.-X.; West, A.; Chan, L.-L.; Poewe, W.; Jankovic, J. Parkinson disease and the immune system—Associations, mechanisms and therapeutics. Nat. Rev. Neurol. 2020, 16, 303–318. [Google Scholar] [CrossRef] [PubMed]
- Herrero-Mendez, A.; Almeida, A.; Fernández, E.; Maestre, C.; Moncada, S.; Bolaños, J.P. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C–Cdh1. Nat. Cell Biol. 2009, 11, 747–752. [Google Scholar] [CrossRef]
- McGarry, T.; Biniecka, M.; Veale, D.J.; Fearon, U. Hypoxia, oxidative stress and inflammation. Free Radic. Biol. Med. 2018, 125, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.G. Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol. Pharmacol. 1978, 14, 633–643. [Google Scholar]
- Sulzer, D.; Zecca, L. Intraneuronal dopamine-quinone synthesis: A review. Neurotox. Res. 1999, 1, 181–195. [Google Scholar] [CrossRef]
- Przedborski, S.; Jackson-Lewis, V.R. ROS and Parkinson’s Disease: A View to Kill. In Free Radicals in Brain Pathophysiology; Taylor & Francis Group: Boca Raton, FL, USA, 2003. [Google Scholar]
- Sian, J.; Dexter, D.T.; Lees, A.J.; Daniel, S.; Agid, Y.; Javoy-Agid, F.; Jenner, P.; Marsden, C.D. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann. Neurol. 1994, 36, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Burtscher, J.; Syed, M.M.K.; Lashuel, H.A.; Millet, G.P. Hypoxia Conditioning as a Promising Therapeutic Target in Parkinson’s Disease? Mov. Disord. 2021, 36, 857–861. [Google Scholar] [CrossRef]
- Pokusa, M.; Hajdúchová, D.; Buday, T.; Kralova Trancikova, A. Respiratory Function and Dysfunction in Parkinson-Type Neurodegeneration. Physiol. Res. 2020, 69, S69–S79. [Google Scholar] [CrossRef]
- Vijayan, S.; Singh, B.; Ghosh, S.; Stell, R.; Mastaglia, F.L. Brainstem ventilatory dysfunction: A plausible mechanism for dyspnea in Parkinson’s Disease? Mov. Disord. 2020, 35, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Onodera, H.; Okabe, S.; Kikuchi, Y.; Tsuda, T.; Itoyama, Y. Impaired chemosensitivity and perception of dyspnoea in Parkinson’s disease. Lancet 2000, 356, 739–740. [Google Scholar] [CrossRef]
- Benarroch, E.E.; Schmeichel, A.M.; Low, P.A.; Parisi, J.E. Depletion of putative chemosensitive respiratory neurons in the ventral medullary surface in multiple system atrophy. Brain 2007, 130, 469–475. [Google Scholar] [CrossRef]
- Semenza, G.L. Regulation of Mammalian O2 Homeostasis by Hypoxia-Inducible Factor 1. Annu. Rev. Cell Dev. Biol. 1999, 15, 551–578. [Google Scholar] [CrossRef] [PubMed]
- Heras-Garvin, A.; Msc, C.D.; Eschlböck, S.; Holton, J.L.; Wenning, G.K.; Stefanova, N. Signs of Chronic Hypoxia Suggest a Novel Pathophysiological Event in α-Synucleinopathies. Mov. Disord. 2020, 35, 2333–2338. [Google Scholar] [CrossRef]
- Zakharova, E.T.; Sokolov, A.V.; Pavlichenko, N.N.; Kostevich, V.A.; Abdurasulova, I.N.; Chechushkov, A.V.; Voynova, I.V.; Elizarova, A.Y.; Kolmakov, N.N.; Bass, M.G.; et al. Erythropoietin and Nrf2: Key factors in the neuroprotection provided by apo-lactoferrin. BioMetals 2018, 31, 425–443. [Google Scholar] [CrossRef]
- Xu, S.-F.; Zhang, Y.-H.; Wang, S.; Pang, Z.-Q.; Fan, Y.-G.; Li, J.-Y.; Wang, Z.-Y.; Guo, C. Lactoferrin ameliorates dopaminergic neurodegeneration and motor deficits in MPTP-treated mice. Redox Biol. 2019, 21, 101090. [Google Scholar] [CrossRef]
- Guo, C.; Hao, L.-J.; Yang, Z.-H.; Chai, R.; Zhang, S.; Gu, Y.; Gao, H.-L.; Zhong, M.-L.; Wang, T.; Li, J.-Y.; et al. Deferoxamine-mediated up-regulation of HIF-1α prevents dopaminergic neuronal death via the activation of MAPK family proteins in MPTP-treated mice. Exp. Neurol. 2016, 280, 13–23. [Google Scholar] [CrossRef]
- Qin, L.; Shu, L.; Zhong, J.; Pan, H.; Guo, J.; Sun, Q.; Yan, X.; Tang, B.; Xu, Q. Association of HIF1A and Parkinson’s disease in a Han Chinese population demonstrated by molecular inversion probe analysis. Neurol. Sci. 2019, 40, 1927–1931. [Google Scholar] [CrossRef]
- Rehncrona, S. Brain acidosis. Ann. Emerg. Med. 1985, 14, 770–776. [Google Scholar] [CrossRef]
- Mookerjee, S.A.; Goncalves, R.L.; Gerencser, A.A.; Nicholls, D.G.; Brand, M.D. The contributions of respiration and glycolysis to extracellular acid production. Biochim. Biophys. Acta Bioenerg. 2015, 1847, 171–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deitmer, J. Acid-base transport and pH regulation. Handb. Neurochem. Mol. Neurobiol. 2007, 5, 469–486. [Google Scholar]
- Swietach, P.; Hulikova, A.; Vaughan-Jones, R.D.; Harris, A.L. New insights into the physiological role of carbonic anhydrase IX in tumour pH regulation. Oncogene 2010, 29, 6509–6521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagadicgossmann, D.; Huc, L.; Lecureur, V. Alterations of intracellular pH homeostasis in apoptosis: Origins and roles. Cell Death Differ. 2004, 11, 953–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, C.L.; Leong, S.L.; Ali, F.E.; Kenche, V.B.; Hill, A.F.; Gras, S.L.; Barnham, K.J.; Cappai, R. Dopamine and the dopamine oxidation product 5, 6-dihydroxylindole promote distinct on-pathway and off-pathway aggregation of α-synuclein in a pH-dependent manner. J. Mol. Biol. 2009, 387, 771–785. [Google Scholar] [CrossRef]
- Umek, N.; Geršak, B.; Vintar, N.; Šoštarič, M.; Mavri, J. Dopamine Autoxidation Is Controlled by Acidic pH. Front. Mol. Neurosci. 2018, 11, 467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Moreno, M.; Rodríguez-López, J.; Martínez-Ortiz, F.; Tudela, J.; Varón, R.; García-Cánovas, F. Effect of pH on the oxidation pathway of dopamine catalyzed by tyrosinase. Arch. Biochem. Biophys. 1991, 288, 427–434. [Google Scholar] [CrossRef]
- Wemmie, J.A.; Price, M.P.; Welsh, M.J. Acid-sensing ion channels: Advances, questions and therapeutic opportunities. Trends Neurosci. 2006, 29, 578–586. [Google Scholar] [CrossRef]
- Wemmie, J.A.; Taugher, R.J.; Kreple, C.J. Acid-sensing ion channels in pain and disease. Nat. Rev. Neurosci. 2013, 14, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.S.; Guzman, J.N.; Ilijic, E.; Mercer, J.N.; Rick, C.; Tkatck, T.; Meredith, G.E.; Surmeier, D.J. ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature 2007, 447, 1081–1086. [Google Scholar] [CrossRef]
- Kuznetsov, A.S.; Kopell, N.J.; Wilson, C.J. Transient High-Frequency Firing in a Coupled-Oscillator Model of the Mesencephalic Dopaminergic Neuron. J. Neurophysiol. 2006, 95, 932–947. [Google Scholar] [CrossRef] [PubMed]
- Foehring, R.C.; Zhang, X.F.; Lee, J.; Callaway, J.C. Endogenous Calcium Buffering Capacity of Substantia Nigral Dopamine Neurons. J. Neurophysiol. 2009, 102, 2326–2333. [Google Scholar] [CrossRef] [Green Version]
- Nagley, P.; Higgins, G.C.; Atkin, J.D.; Beart, P.M. Multifaceted deaths orchestrated by mitochondria in neurones. Biochim. Biophys. Acta Mol. Basis Dis. 2010, 1802, 167–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arias, R.L.; Sung, M.-L.A.; Vasylyev, D.; Zhang, M.-Y.; Albinson, K.; Kubek, K.; Kagan, N.; Beyer, C.; Lin, Q.; Dwyer, J.M.; et al. Amiloride is neuroprotective in an MPTP model of Parkinson’s disease. Neurobiol. Dis. 2008, 31, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Komnig, D.; Imgrund, S.; Reich, A.; Gründer, S.; Falkenburger, B.H. ASIC1a Deficient Mice Show Unaltered Neurodegeneration in the Subacute MPTP Model of Parkinson Disease. PLoS ONE 2016, 11, e0165235. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Martinez-Vicente, M.; Caldwell, G.A.; Caldwell, K.A.; Yue, Z.; Cookson, M.R.; Klein, C.; Vila, M.; Bezard, E. Lysosomal impairment in Parkinson’s disease. Mov. Disord. 2013, 28, 725–732. [Google Scholar] [CrossRef] [PubMed]
- McClendon, S.; Rospigliosi, C.C.; Eliezer, D. Charge neutralization and collapse of the C-terminal tail of alpha-synuclein at low pH. Protein Sci. 2009, 18, 1531–1540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, S.; Singh, N.; Kumar, R.; Patel, K.; Pandey, S.; Datta, D.; Mahato, J.; Panigrahi, R.; Navalkar, A.; Mehra, S.; et al. α-Synuclein aggregation nucleates through liquid–liquid phase separation. Nat. Chem. 2020, 12, 705–716. [Google Scholar] [CrossRef]
- Cole, N.B.; DiEuliis, D.; Leo, P.; Mitchell, D.C.; Nussbaum, R.L. Mitochondrial translocation of α-synuclein is promoted by intracellular acidification. Exp. Cell Res. 2008, 314, 2076–2089. [Google Scholar] [CrossRef] [Green Version]
- Bowen, B.C.; Block, R.E.; Sanchez-Ramos, J.; Pattany, P.M.; Lampman, D.A.; Murdoch, J.B.; Quencer, R.M. Proton MR spectroscopy of the brain in 14 patients with Parkinson disease. Am. J. Neuroradiol. 1995, 16, 61–68. [Google Scholar]
- Firbank, M.J.; Harrison, R.M.; O’Brien, J.T. A comprehensive review of proton magnetic resonance spectroscopy studies in dementia and Parkinson’s disease. Dement. Geriatr. Cogn. Disord. 2002, 14, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.-W.; Liao, K.-F.; Lin, C.-L.; Lin, C.-H. Association between Parkinson’s disease and proton pump inhibitors therapy in older people. Biomedicine 2020, 10, 1–4. [Google Scholar] [PubMed]
- Nielsen, H.H.; Qiu, J.; Friis, S.; Wermuth, L.; Ritz, B. Treatment for Helicobacter pylori infection and risk of parkinson’s disease in Denmark. Eur. J. Neurol. 2012, 19, 864–869. [Google Scholar] [CrossRef] [Green Version]
- Pirchl, M.; Marksteiner, J.; Humpel, C. Effects of acidosis on brain capillary endothelial cells and cholinergic neurons: Relevance to vascular dementia and Alzheimer’s disease. Neurol. Res. 2006, 28, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Fang, B.; Wang, D.; Huang, M.; Yu, G.; Li, H. Hypothesis on the relationship between the change in intracellular pH and incidence of sporadic Alzheimer’s disease or vascular dementia. Int. J. Neurosci. 2010, 120, 591–595. [Google Scholar] [CrossRef]
- Kuo, S.-W.; Jiang, M.; Heckman, C. Potential involvement of intracellular pH in a mouse model of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2013, 15, 151–153. [Google Scholar] [CrossRef] [Green Version]
- Reeve, A.; Simcox, E.; Turnbull, D. Ageing and Parkinson’s disease: Why is advancing age the biggest risk factor? Ageing Res. Rev. 2014, 14, 19–30. [Google Scholar] [CrossRef]
- Collier, T.J.; Kanaan, N.M.; Kordower, J.H. Aging and Parkinson’s disease: Different sides of the same coin? Mov. Disord. 2017, 32, 983–990. [Google Scholar] [CrossRef] [Green Version]
- Campisi, J.; Kapahi, P.; Lithgow, G.J.; Melov, S.; Newman, J.C.; Verdin, E. From discoveries in ageing research to therapeutics for healthy ageing. Nature 2019, 571, 183–192. [Google Scholar] [CrossRef] [Green Version]
- Polonis, K.; Becari, C.; Chahal, C.A.A.; Zhang, Y.; Allen, A.M.; Kellogg, T.A.; Somers, V.K.; Singh, P. Chronic Intermittent Hypoxia Triggers a Senescence-like Phenotype in Human White Preadipocytes. Sci. Rep. 2020, 10, 6846. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Hypoxia-Inducible Histone Lysine Demethylases: Impact on the Aging Process and Age-Related Diseases. Aging Dis. 2016, 7, 180–200. [Google Scholar]
- Wilson, E.N.; Anderson, M.; Snyder, B.; Duong, P.; Trieu, J.; Schreihofer, D.A.; Cunningham, R.L. Chronic intermittent hypoxia induces hormonal and male sexual behavioral changes: Hypoxia as an advancer of aging. Physiol. Behav. 2018, 189, 64–73. [Google Scholar] [CrossRef]
- Kim, H.; Lee, D.-K.; Choi, J.-W.; Kim, J.-S.; Park, S.C.; Youn, H.-D. Analysis of the effect of aging on the response to hypoxia by cDNA microarray. Mech. Ageing Dev. 2003, 124, 941–949. [Google Scholar] [CrossRef]
- Frenkel-Denkberg, G.; Gershon, D.; Levy, A.P. The function of hypoxia-inducible factor 1 (HIF-1) is impaired in senescent mice. FEBS Lett. 1999, 462, 341–344. [Google Scholar] [CrossRef] [Green Version]
- Kayser, E.-B.; Sedensky, M.M.; Morgan, P.G.; Hoppel, C.L. Mitochondrial Oxidative Phosphorylation Is Defective in the Long-lived Mutant clk-1. J. Biol. Chem. 2004, 279, 54479–54486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, J.; Bussière, F.; Hekimi, S. Mitochondrial Electron Transport Is a Key Determinant of Life Span in Caenorhabditis elegans. Dev. Cell 2001, 1, 633–644. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.S.; Lee, R.Y.; Fraser, A.G.; Kamath, R.S.; Ahringer, J.; Ruvkun, G. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat. Genet. 2003, 33, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Copeland, J.M.; Cho, J.; Lo, T., Jr.; Hur, J.H.; Bahadorani, S.; Arabyan, T.; Rabie, J.; Soh, J.; Walker, D.W. Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr. Biol. 2009, 19, 1591–1598. [Google Scholar] [CrossRef] [Green Version]
- Lapointe, J.; Hekimi, S. Early mitochondrial dysfunction in long-lived Mclk1+/-mice. J. Biol. Chem. 2008, 283, 26217–26227. [Google Scholar] [CrossRef] [Green Version]
- Dell’Agnello, C.; Leo, S.; Agostino, A.; Szabadkai, G.; Tiveron, C.; Zulian, A.; Prelle, A.; Roubertoux, P.; Rizzuto, R.; Zeviani, M. Increased longevity and refractoriness to Ca2+-dependent neurodegeneration in Surf1 knockout mice. Hum. Mol. Genet. 2007, 16, 431–444. [Google Scholar] [CrossRef] [Green Version]
- Burtscher, J.; Mallet, R.T.; Burtscher, M.; Millet, G.P. Hypoxia and brain aging: Neurodegeneration or neuroprotection? Ageing Res. Rev. 2021, 68, 101343. [Google Scholar] [CrossRef] [PubMed]
- Johmura, Y.; Yamanaka, T.; Omori, S.; Wang, T.-W.; Sugiura, Y.; Matsumoto, M.; Suzuki, N.; Kumamoto, S.; Yamaguchi, K.; Hatakeyama, S.; et al. Senolysis by glutaminolysis inhibition ameliorates various age-associated disorders. Science 2021, 371, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Minakaki, G.; Krainc, D.; Burbulla, L.F. The Convergence of Alpha-Synuclein, Mitochondrial, and Lysosomal Pathways in Vulnerability of Midbrain Dopaminergic Neurons in Parkinson’s Disease. Front. Cell Dev. Biol. 2020, 8, 580634. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, S.S.; Petersen, D.; Marlet, F.R.; Kücükköse, E.; Galvagnion, C. The interplay between GCase, α-synuclein and lipids in human models of Parkinson’s disease. Biophys. Chem. 2020, 273, 106534. [Google Scholar] [CrossRef] [PubMed]
- Gegg, M.E.; Verona, G.; Schapira, A.H. Glucocerebrosidase deficiency promotes release of α-synuclein fibrils from cultured neurons. Hum. Mol. Genet. 2020, 29, 1716–1728. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.; Torres, C. Faculty Opinions recommendation of Astrocyte senescence: Evidence and significance. Fac. Opin. Post Publ. Peer Rev. Biomed. Lit. 2019, 18, e12937. [Google Scholar]
- Limbad, C.; Oron, T.R.; Alimirah, F.; Davalos, A.R.; Tracy, T.E.; Gan, L.; Desprez, P.-Y.; Campisi, J. Astrocyte senescence promotes glutamate toxicity in cortical neurons. PLoS ONE 2020, 15, e0227887. [Google Scholar] [CrossRef]
- Chinta, S.J.; Woods, G.; DeMaria, M.; Rane, A.; Zou, Y.; McQuade, A.; Rajagopalan, S.; Limbad, C.; Madden, D.T.; Campisi, J.; et al. Cellular Senescence Is Induced by the Environmental Neurotoxin Paraquat and Contributes to Neuropathology Linked to Parkinson’s Disease. Cell Rep. 2018, 22, 930–940. [Google Scholar] [CrossRef] [Green Version]
- Majdi, A.; Mahmoudi, J.; Sadigh-Eteghad, S.; Golzari, S.E.; Sabermarouf, B.; Reyhani-Rad, S. Permissive role of cytosolic pH acidification in neurodegeneration: A closer look at its causes and consequences. J. Neurosci. Res. 2016, 94, 879–887. [Google Scholar] [CrossRef]
- Swenson, E.R. Hypoxia and Its Acid–Base Consequences: From Mountains to Malignancy. In Hypoxia: Translation in Progress; Roach, R.C., Hackett, P.H., Wagner, P.D., Eds.; Springer: Boston, MA, USA, 2016; pp. 301–323. [Google Scholar]
- Ivashkiv, L.B. The hypoxia–lactate axis tempers inflammation. Nat. Rev. Immunol. 2020, 20, 85–86. [Google Scholar] [CrossRef]
- Davies, A.L.; Desai, R.A.; Bloomfield, P.S.; McIntosh, P.R.; Chapple, K.J.; Linington, C.; Fairless, R.; Diem, R.; Kasti, M.; Murphy, M.P.; et al. Neurological deficits caused by tissue hypoxia in neuroinflammatory disease. Ann. Neurol. 2013, 74, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Okajima, F. Regulation of inflammation by extracellular acidification and proton-sensing GPCRs. Cell. Signal. 2013, 25, 2263–2271. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nat. Cell Biol. 2015, 520, 553–557. [Google Scholar]
- Rajamäki, K.; Nordström, T.; Nurmi, K.; Åkerman, K.E.O.; Kovanen, P.T.; Öörni, K.; Eklund, K.K. Extracellular acidosis is a novel danger signal alerting innate immunity via the NLRP3 inflammasome. J. Biol. Chem. 2013, 288, 13410–13419. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, J.; Basit, F.; Swarts, H.G.; Forkink, M.; Oliveira, P.J.; Willems, P.H.G.M.; Koopman, W.J.H. Extracellular acidification induces ROS- and mPTP-mediated death in HEK293 cells. Redox Biol. 2018, 15, 394–404. [Google Scholar] [CrossRef]
- Marreiros, R.; Müller-Schiffmann, A.; Trossbach, S.V.; Prikulis, I.; Hänsch, S.; Weidtkamp-Peters, S.; Moreira, A.R.; Sahu, S.; Soloviev, I.; Selvarajah, S.; et al. Disruption of cellular proteostasis by H1N1 influenza A virus causes α-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2020, 117, 6741–6751. [Google Scholar] [CrossRef]
- Wang, M.-X.; Cheng, X.-Y.; Jin, M.; Cao, Y.-L.; Yang, Y.-P.; Wang, J.-D.; Li, Q.; Wang, F.; Hu, L.-F.; Liu, C.-F. TNF compromises lysosome acidification and reduces α-synuclein degradation via autophagy in dopaminergic cells. Exp. Neurol. 2015, 271, 112–121. [Google Scholar] [CrossRef]
- Bu, X.-L.; Wang, X.; Xiang, Y.; Zeng, F.; Liu, Y.-H.; Xiang, Y.; Liang, C.-R.; Wang, Q.H.; Wang, X.; Cao, H.-Y.; et al. The association between infectious burden and Parkinson’s disease: A case-control study. Parkinsonism Relat. Disord. 2015, 21, 877–881. [Google Scholar] [CrossRef]
- Piancone, F.; Saresella, M.; La Rosa, F.; Marventano, I.; Meloni, M.; Navarro, J.; Clerici, M. Inflammatory Responses to Monomeric and Aggregated α-Synuclein in Peripheral Blood of Parkinson Disease Patients. Front. Neurosci. 2021, 15, 235. [Google Scholar] [CrossRef]
- Harms, A.S.; Delic, V.; Thome, A.D.; Bryant, N.; Liu, Z.; Chandra, S.; Jurkuvenaite, A.; West, A.B. α-Synuclein fibrils recruit peripheral immune cells in the rat brain prior to neurodegeneration. Acta Neuropathol. Commun. 2017, 5, 85. [Google Scholar] [CrossRef] [Green Version]
- Jankovic, J.; Tan, E.K. Parkinson’s disease: Etiopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatry 2020, 91, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Teismann, P.; Tieu, K.; Choi, D.-K.; Wu, D.-C.; Naini, A.; Hunot, S.; Vila, M.; Jackson-Lewis, V.; Przedborski, S. Cyclooxygenase-2 is instrumental in Parkinson’s disease neurodegeneration. Proc. Natl. Acad. Sci. USA 2003, 100, 5473–5478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindå, H.; Hammarberg, H.; Piehl, F.; Khademi, M.; Olsson, T. Expression of MHC class I heavy chain and β2-microglobulin in rat brainstem motoneurons and nigral dopaminergic neurons. J. Neuroimmunol. 1999, 101, 76–86. [Google Scholar] [CrossRef]
- Xu, J.; Peng, Z.; Li, R.; Dou, T.; Xu, W.; Gu, G.; Liu, Y.; Kang, Z.; Tao, H.; Zhang, J.H.; et al. Normoxic induction of cerebral HIF-1α by acetazolamide in rats: Role of acidosis. Neurosci. Lett. 2009, 451, 274–278. [Google Scholar] [CrossRef] [PubMed]
- McGettrick, A.F.; O’Neill, L.A. The Role of HIF in Immunity and Inflammation. Cell Metab. 2020, 32, 524–536. [Google Scholar] [CrossRef]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF Transcription Factors, Inflammation, and Immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef] [Green Version]
- Watts, E.R.; Walmsley, S.R. Inflammation and hypoxia: HIF and PHD isoform selectivity. Trends Mol. Med. 2019, 25, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Wang, G.; Xu, Z.-G.; Tu, H.; Hu, F.; Dai, J.; Chang, Y.; Chen, Y.; Lu, Y.; Zeng, H.; et al. Lactate Is a Natural Suppressor of RLR Signaling by Targeting MAVS. Cell 2019, 178, 176–189. [Google Scholar] [CrossRef]
- Bender, A.S.; Young, L.P.; Norenberg, M.D. Effect of lactic acid on l-glutamate uptake in cultured astrocytes: Mechanistic considerations. Brain Res. 1997, 750, 59–66. [Google Scholar] [CrossRef]
- Xiang, Z.; Yuan, M.; Hassen, G.W.; Gampel, M.; Bergold, P.J. Lactate induced excitotoxicity in hippocampal slice cultures. Exp. Neurol. 2004, 186, 70–77. [Google Scholar] [CrossRef]
- Mills, E.; O’Neill, L.A. Succinate: A metabolic signal in inflammation. Trends Mol. Med. 2014, 24, 313–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason, S. A Novel, Multi-Faceted Perception of Lactate in Neurology. Front. Neurosci. 2020, 14, 460. [Google Scholar] [CrossRef] [PubMed]
- Berthet, C.; Lei, H.; Thevenet, J.; Gruetter, R.; Magistretti, P.J.; Hirt, L. Neuroprotective Role of Lactate after Cerebral Ischemia. Br. J. Pharmacol. 2009, 29, 1780–1789. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.E.; Stecher, B.; Labrie, V.; Brundin, L.; Brundin, P. Triggers, Facilitators, and Aggravators: Redefining Parkinson’s Disease Pathogenesis. Trends Neurosci. 2019, 42, 4–13. [Google Scholar] [CrossRef] [Green Version]
- Harguindey, S.; Stanciu, D.; Devesa, J.; Alfarouk, K.; Cardone, R.A.; Orozco, J.D.P.; Devesa, P.; Rauch, C.; Orive, G.; Anitua, E.; et al. Cellular acidification as a new approach to cancer treatment and to the understanding and therapeutics of neurodegenerative diseases. Semin. Cancer Biol. 2017, 43, 157–179. [Google Scholar] [CrossRef] [PubMed]
- Makowski, L.; Chaib, M.; Rathmell, J.C. Immunometabolism: From basic mechanisms to translation. Immunol. Rev. 2020, 295, 5–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lercher, A.; Baazim, H.; Bergthaler, A. Systemic Immunometabolism: Challenges and Opportunities. Immunity 2020, 53, 496–509. [Google Scholar] [CrossRef]
- Wang, A.; Luan, H.H.; Medzhitov, R. An evolutionary perspective on immunometabolism. Science 2019, 363, eaar3932. [Google Scholar] [CrossRef]
- Mallet, R.T.; Burtscher, J.; Manukhina, E.B.; Downey, H.F.; Glazachev, O.S.; Serebrovskaya, T.V.; Burtscher, M. Hypoxic–hyperoxic conditioning and dementia. In Diagnosis and Management in Dementia; Elsevier: Amsterdam, The Netherlands, 2020; pp. 745–760. [Google Scholar]
- Burtscher, J.; Maglione, V.; Di Pardo, A.; Millet, G.P.; Schwarzer, C.; Zangrandi, L. A Rationale for Hypoxic and Chemical Conditioning in Huntington’s Disease. Int. J. Mol. Sci. 2021, 22, 582. [Google Scholar] [CrossRef]
- Griffith, H.R.; den Hollander, J.A.; Okonkwo, O.C.; O’Brien, T.; Watts, R.L.; Marson, D.C. Brain metabolism differs in Alzheimer’s disease and Parkinson’s disease dementia. Alzheimers Dement. 2008, 4, 421–427. [Google Scholar] [CrossRef] [Green Version]
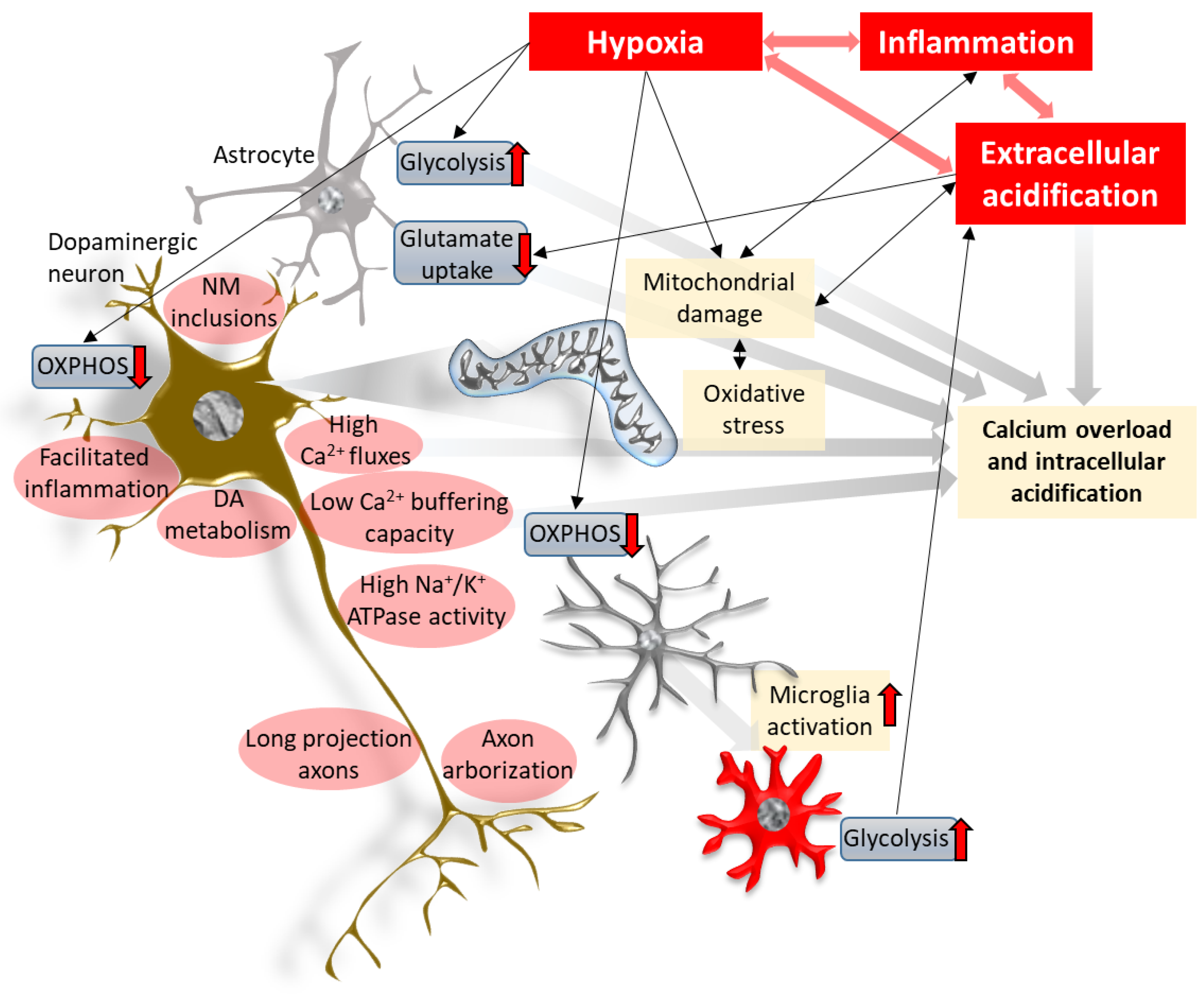
Figure 1.
Hypoxia, acidification and inflammation in the center of the neurodegenerative process. Hypoxic insults in the brain induce shifts in energy metabolism that may lead to oxidative stress and mitochondrial damage, inducing pro-inflammatory signaling. Linked with the activation of glial cells, this results in neuroinflammation. Metabolic changes—strongly mediated by hypoxia inducible factors—of the different cell types lead to acidification of the cellular milieu, in part via the accumulation of lactate and other metabolites. Extracellular acidification increases the activity of cation-permeable acid-sensing ion channels and, in conjunction with impaired mitochondrial proton transport, this results in disturbances of the intracellular ion homeostasis, yielding acidification and Ca2+ toxicity. Together with associated mitochondrial damage, oxidative stress and inflammation, they are prominent mechanisms leading to neurodegeneration. The cell-autonomous features of dopaminergic projection neurons in the substantia nigra pars compacta (red ellipses) likely exacerbate the detrimental effects of such cellular environments. NM: neuromelanin, OXPHOS: oxidative phosphorylation.
Figure 1.
Hypoxia, acidification and inflammation in the center of the neurodegenerative process. Hypoxic insults in the brain induce shifts in energy metabolism that may lead to oxidative stress and mitochondrial damage, inducing pro-inflammatory signaling. Linked with the activation of glial cells, this results in neuroinflammation. Metabolic changes—strongly mediated by hypoxia inducible factors—of the different cell types lead to acidification of the cellular milieu, in part via the accumulation of lactate and other metabolites. Extracellular acidification increases the activity of cation-permeable acid-sensing ion channels and, in conjunction with impaired mitochondrial proton transport, this results in disturbances of the intracellular ion homeostasis, yielding acidification and Ca2+ toxicity. Together with associated mitochondrial damage, oxidative stress and inflammation, they are prominent mechanisms leading to neurodegeneration. The cell-autonomous features of dopaminergic projection neurons in the substantia nigra pars compacta (red ellipses) likely exacerbate the detrimental effects of such cellular environments. NM: neuromelanin, OXPHOS: oxidative phosphorylation.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Burtscher, J.; Millet, G.P. Hypoxia, Acidification and Inflammation: Partners in Crime in Parkinson’s Disease Pathogenesis? Immuno 2021, 1, 78-90. https://0-doi-org.brum.beds.ac.uk/10.3390/immuno1020006
AMA Style
Burtscher J, Millet GP. Hypoxia, Acidification and Inflammation: Partners in Crime in Parkinson’s Disease Pathogenesis? Immuno. 2021; 1(2):78-90. https://0-doi-org.brum.beds.ac.uk/10.3390/immuno1020006
Chicago/Turabian StyleBurtscher, Johannes, and Grégoire P. Millet. 2021. "Hypoxia, Acidification and Inflammation: Partners in Crime in Parkinson’s Disease Pathogenesis?" Immuno 1, no. 2: 78-90. https://0-doi-org.brum.beds.ac.uk/10.3390/immuno1020006