Composition and Potential Function of Fecal Bacterial Microbiota from Six Bird Species

 ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Fecal Sampling
2.2. DNA Extraction and Sequencing
2.3. Bioinformatics
2.4. Differences among Bird Species
2.5. Diversity Analysis
2.6. Prediction of Metabolic Profiles and Organism-Level Microbiome Phenotypes
3. Results
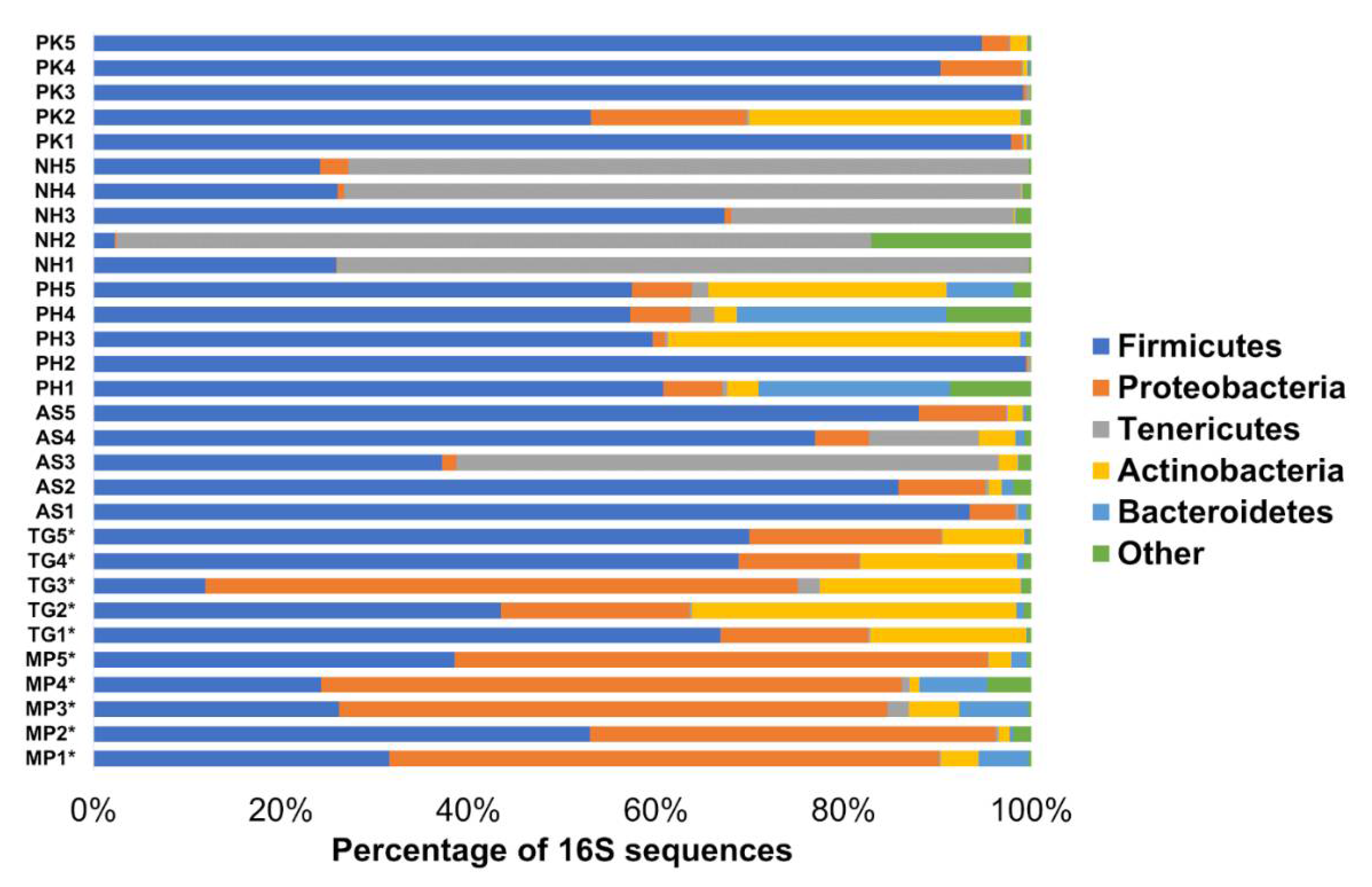
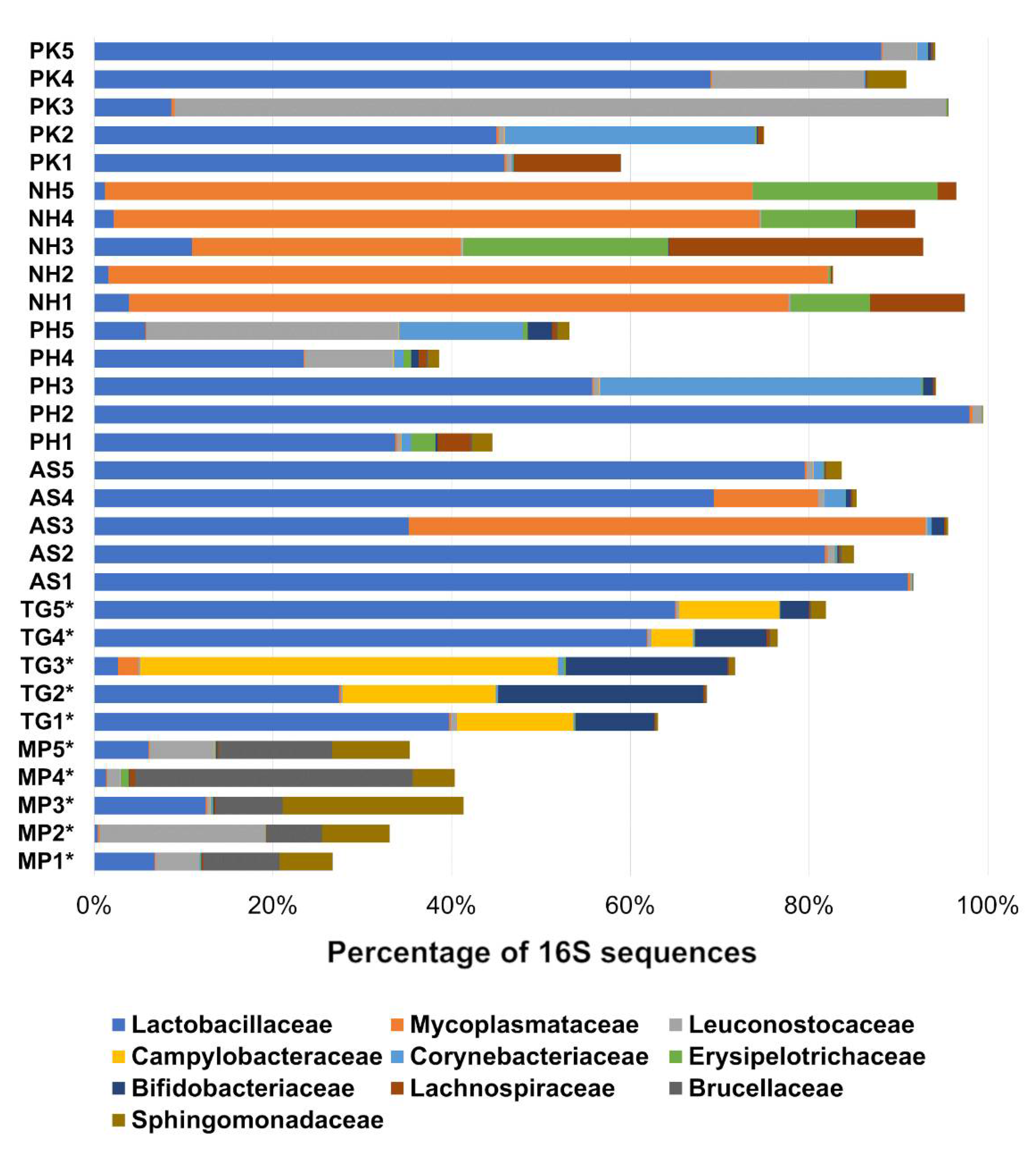
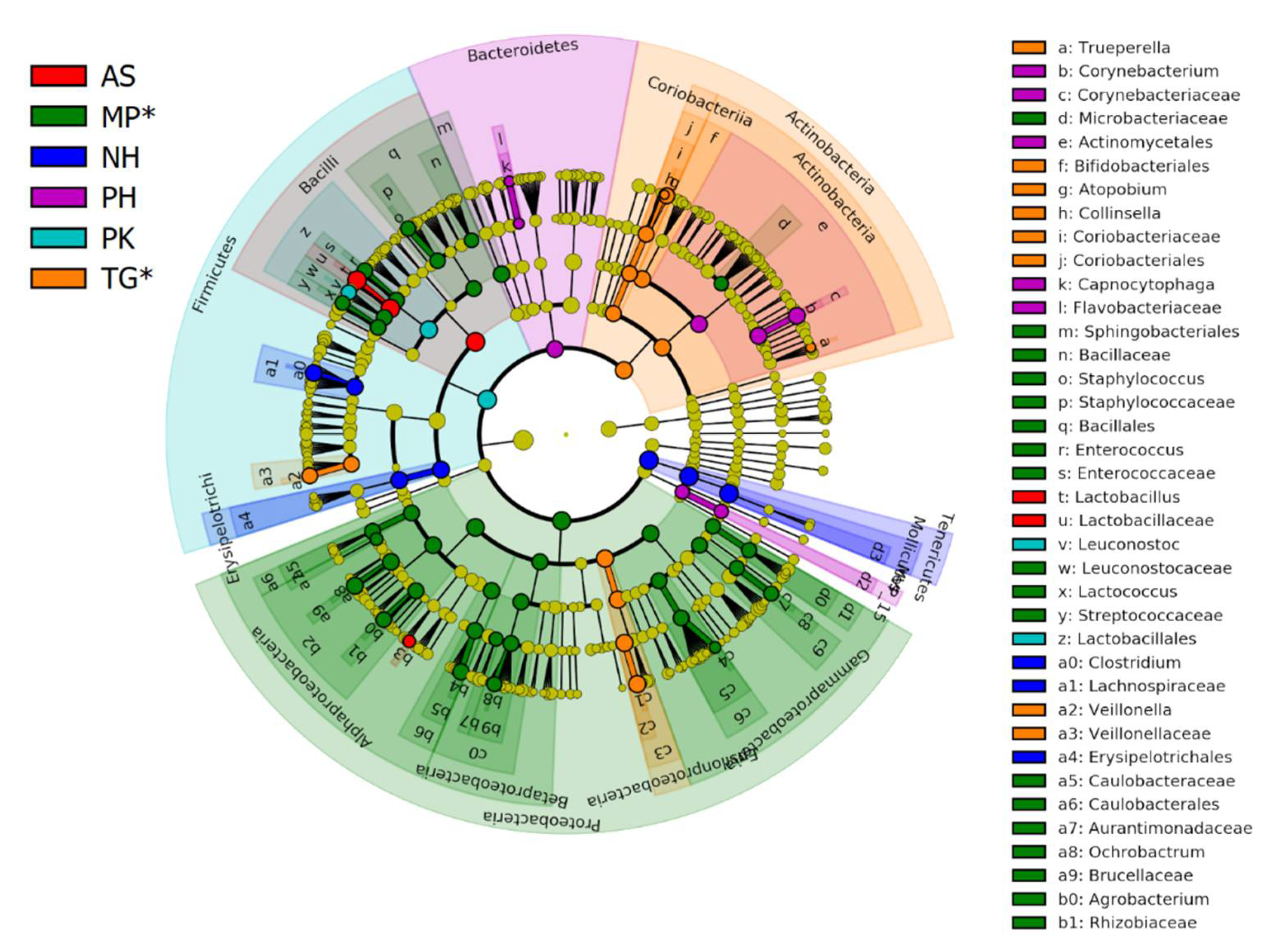
3.1. Fecal Bacterial Composition
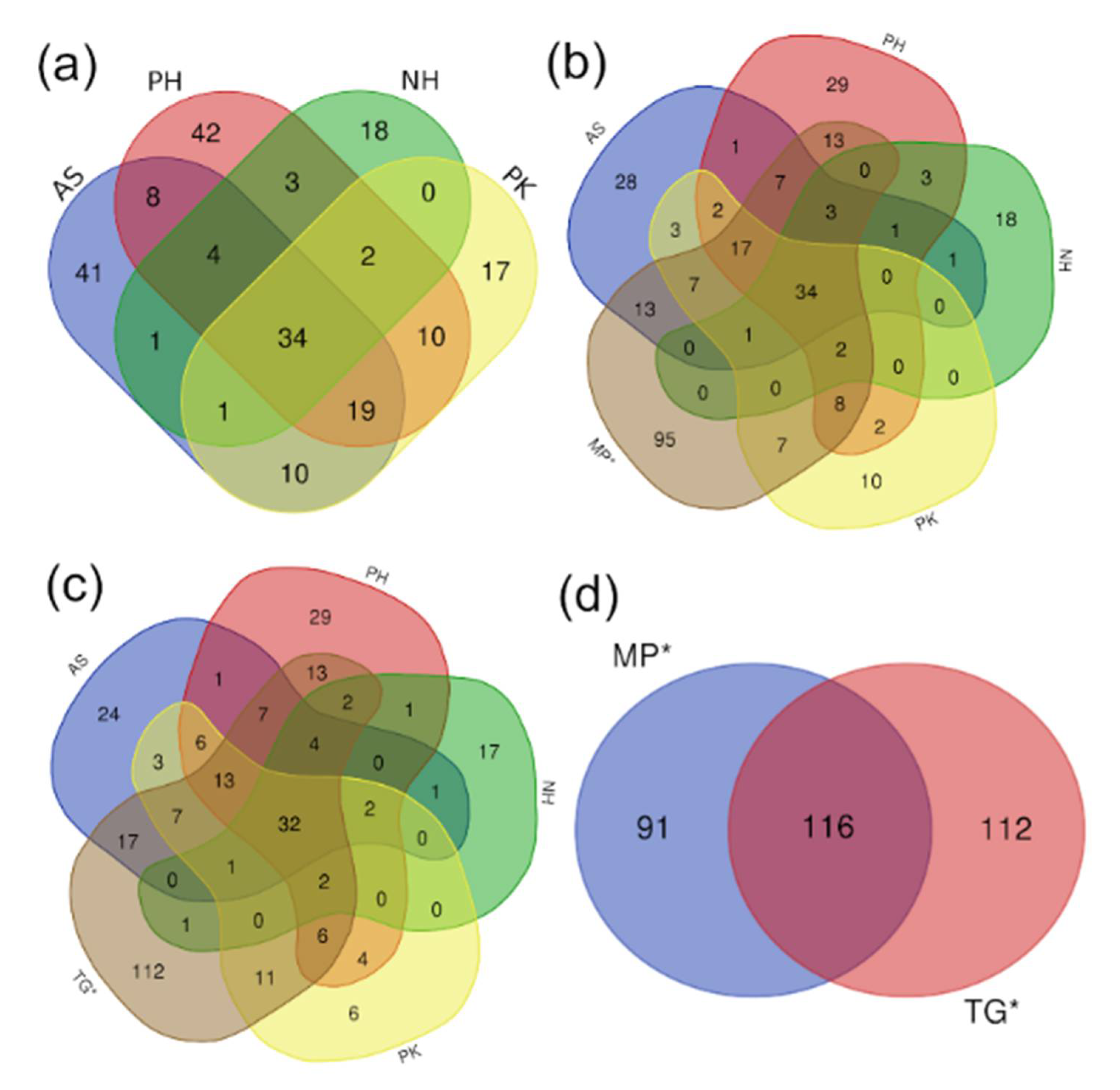
3.2. Core Microbiome
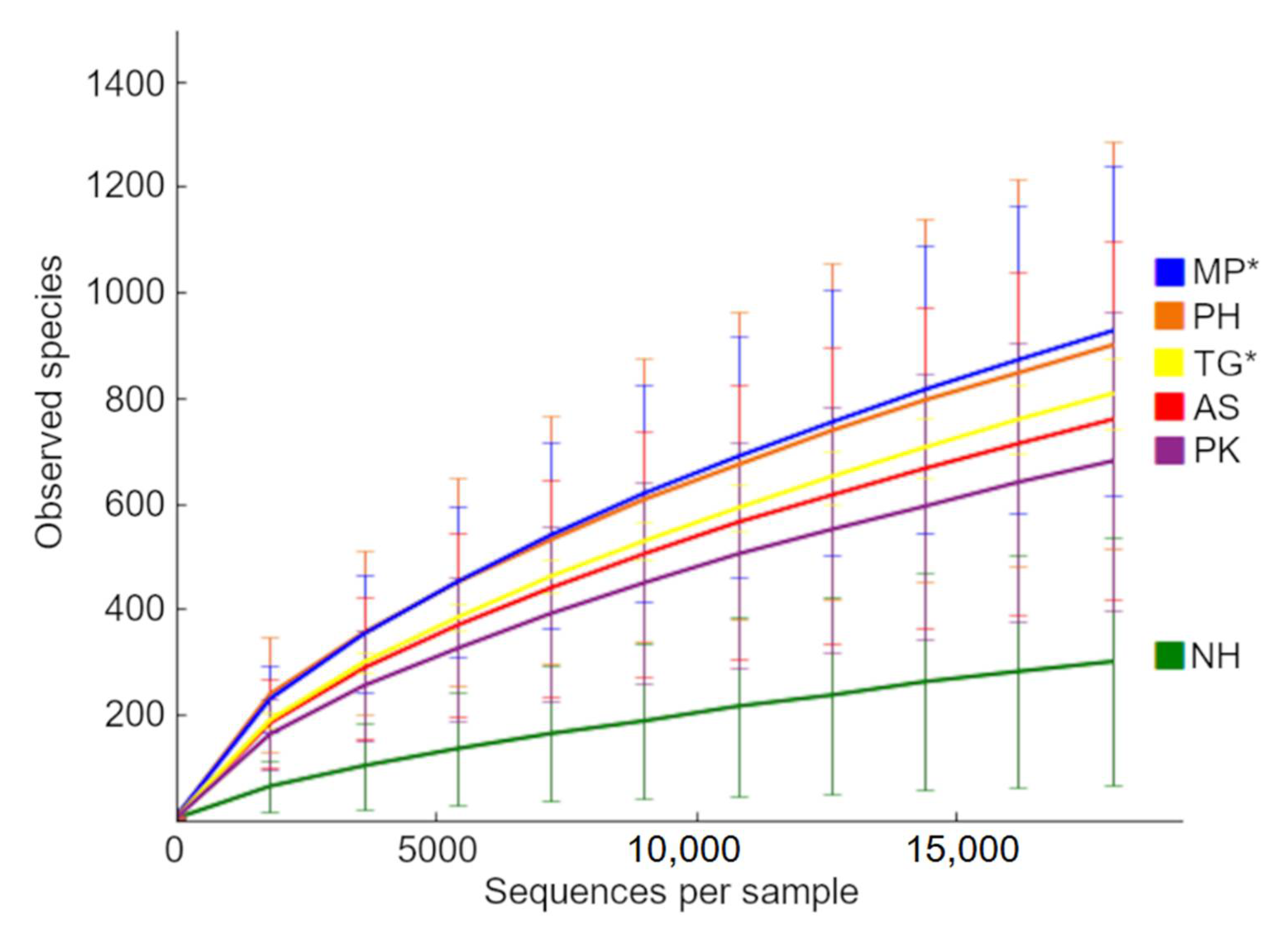
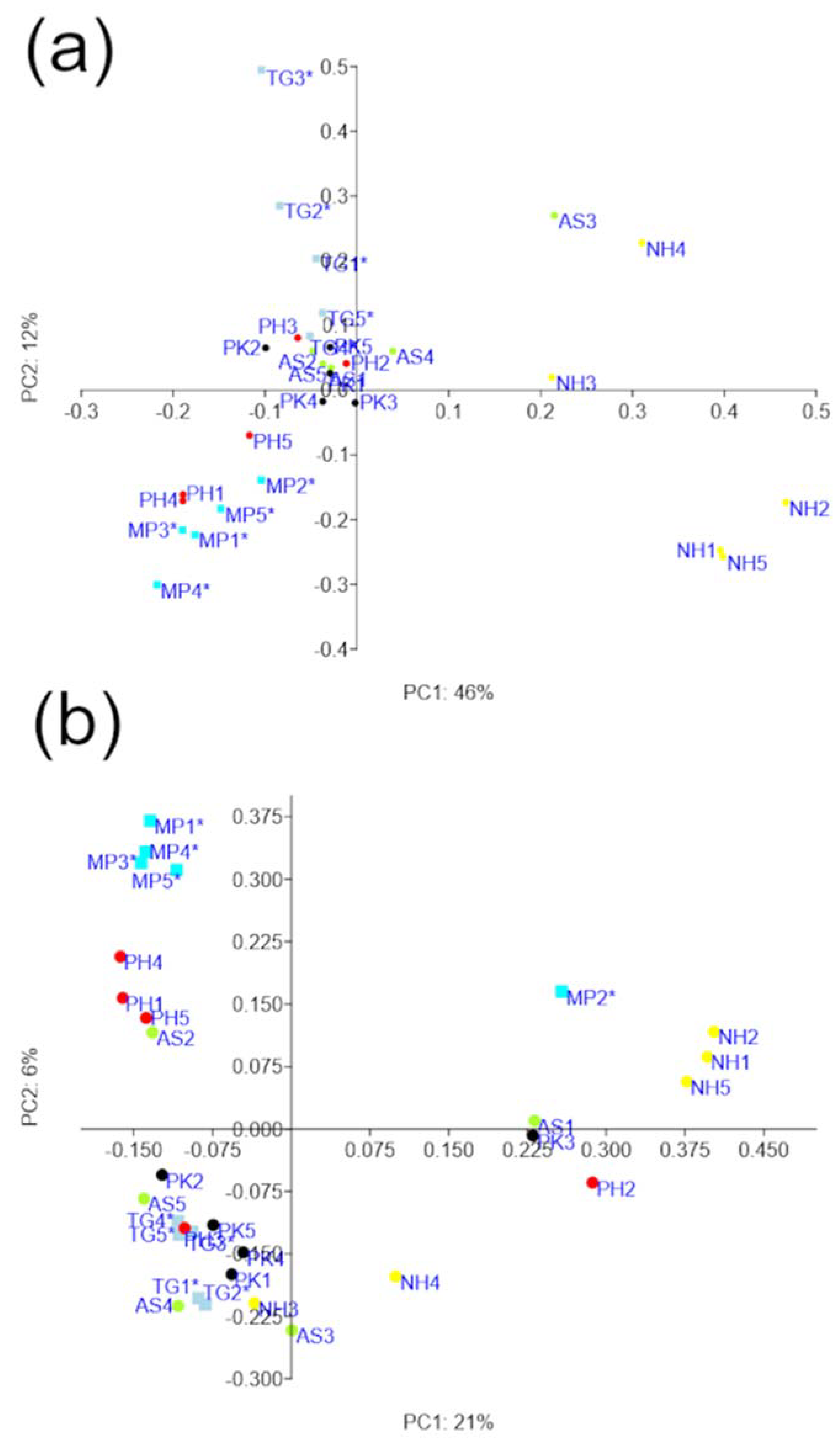
3.3. Diversity Analyses
3.4. Predicted Metabolic Profile
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McFall-Ngai, M.; Hadfield, M.G.; Bosch, T.C.G.; Carey, H.V.; Domazet-Lošo, T.; Douglas, A.E.; Dubilier, N.; Eberl, G.; Fukami, T.; Gilbert, S.F.; et al. Animals in a bacterial world, a new imperative for the life sciences. Proc. Natl. Acad. Sci. USA 2013, 110, 3229–3236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waite, D.W.; Taylor, M.W. Characterizing the avian gut microbiota: Membership, driving influences, and potential function. Front. Microbiol. 2014, 5, 223. [Google Scholar] [CrossRef] [Green Version]
- Waite, D.W.; Taylor, M.W. Exploring the avian gut microbiota: Current trends and future directions. Front. Microbiol. 2015, 6, 673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hird, S.M.; Sanchez, C.; Carstens, B.C.; Brumfield, R.T. Comparative gut microbiota of 59 neotropical bird species. Front. Microbiol. 2015, 6, 1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godon, J.J.; Arulazhagan, P.; Steyer, J.P.; Hamelin, J. Vertebrate bacterial gut diversity: Size also matters. BMC Ecol. 2016, 16, 12. [Google Scholar] [CrossRef] [Green Version]
- Hammons, S.; Oh, P.L.; Martínez, I.; Clark, K.; Schlegel, V.L.; Sitorius, E.; Scheideler, S.E.; Walter, J. A small variation in diet influences the Lactobacillus strain composition in the crop of broiler chickens. Syst. Appl. Microbiol. 2010, 33, 275–281. [Google Scholar] [CrossRef]
- Lewis, W.B.; Moore, F.R.; Wang, S.; Lewis, W.B.; Moore, F.R.; Wang, S. Changes in gut microbiota of migratory passerines during stopover after crossing an ecological barrier. Auk 2017, 134, 137–145. [Google Scholar] [CrossRef]
- Ritz, K. The plate debate: Cultivable communities have no utility in contemporary environmental microbial ecology. FEMS Microbiol. Ecol. 2007, 60, 358–362. [Google Scholar] [CrossRef] [Green Version]
- Oakley, B.B.; Lillehoj, H.S.; Kogut, M.H.; Kim, W.K.; Maurer, J.J.; Pedroso, A.; Lee, M.D.; Collett, S.R.; Johnson, T.J.; Cox, N.A. The chicken gastrointestinal microbiome. FEMS Microbiol. Lett. 2014, 360, 100–112. [Google Scholar] [CrossRef]
- Richards, P.; Fothergill, J.; Bernardeau, M.; Wigley, P. Development of the caecal microbiota in three broiler breeds. Front. Vet. Sci. 2019, 6, 201. [Google Scholar] [CrossRef]
- Brilhante, R.S.N.; Castelo-Branco, D.S.C.M.; Soares, G.D.P.; Astete-Medrano, D.J.; Monteiro, A.J.; Cordeiro, R.A.; Sidrim, J.J.C.; Rocha, M.F.G. Characterization of the gastrointestinal yeast microbiota of cockatiels (Nymphicus hollandicus): A potential hazard to human health. J. Med. Microbiol. 2010, 59, 718–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godoy-Vitorino, F.; Goldfarb, K.C.; Brodie, E.L.; Garcia-Amado, M.A.; Michelangeli, F.; Domínguez-Bello, M.G. Developmental microbial ecology of the crop of the folivorous hoatzin. ISME J. 2010, 4, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Mazcorro, J.F.; Castillo-Carranza, S.A.; Guard, B.; Gomez-Vazquez, J.P.; Dowd, S.E.; Brightsmith, D.J. Comprehensive molecular characterization of bacterial communities in feces of pet birds using 16S marker sequencing. Microb. Ecol. 2017, 73, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Kreisinger, J.; Kropáčková, L.; Petrželková, A.; Adámková, M.; Tomášek, O.; Martin, J.F.; Michálková, R.; Albrecht, T. Transgenerational transfer on fecal microbiota structure in a long distance migratory bird. Front. Microbiol. 2017, 8, 50. [Google Scholar] [CrossRef] [PubMed]
- Grond, K.; Sandercock, B.K.; Jumpponen, A.; Zeglin, L.H. The avian gut microbiota: Community, physiology and function in wild birds. J. Avian Biol. 2018, 49, e01788. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-Y.; Chen, C.-K.; Chen, Y.-Y.; Fang, A.; Shaw, G.T.-W.; Hung, C.-M.; Wang, D. Maternal gut microbes shape the early-life assembly of gut microbiota in passerine chicks via nests. Microbiome 2020, 8, 129. [Google Scholar] [CrossRef]
- Van Veelen, H.P.J.; Salles, J.F.; Matson, K.D.; ven der Velde, M.; Tieleman, B.I. Microbial environment shapes immune function and cloacal microbiota dynamics in zebra finches Taeniopygia Guttata. Anim. Microbiome 2020, 2, 21. [Google Scholar] [CrossRef]
- Videvall, E.; Strandh, M.; Engelbrecht, A.; Cloete, S.; Cornwallis, C.K. Measuring the gut microbiome in birds: Comparison of faecal and cloacal sampling. Mol. Ecol. Resour. 2018, 18, 424–434. [Google Scholar] [CrossRef]
- Berlow, M.; Kohl, K.D.; Derryberry, E.P. Evaluation of non-lethal gut microbiome sampling methods in a passerine bird. Int. J. Avian Sci. 2019, 162, 911–923. [Google Scholar] [CrossRef]
- Lee, S.-J.; Cho, S.; La, T.-M.; Lee, H.-J.; Lee, J.-B.; Park, S.-Y.; Song, C.-S.; Choi, I.-S.; Lee, S.-W. Comparison of microbiota in the cloaca, colon, and magnum of layer chicken. PLoS ONE 2020, 15, e0237108. [Google Scholar] [CrossRef]
- Burton, E.N.; Cohn, L.A.; Reinero, C.N.; Rindt, H.; Moore, S.G.; Ericsson, A.C. Characterization of the urinary microbiome in healthy dogs. PLoS ONE 2017, 12, e0177783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frimodt-Møller, N. The urine microbiome—Contamination or a novel paradigm? EBioMedicine 2019, 44, 20–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boseret, G.; Losson, B.; Mainil, J.G.; Thiry, E.; Saegerman, C. Zoonoses in pet birds: Review and perspectives. Vet. Res. 2013, 44, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcaraz, L.D.; Hernández, A.M.; Peimbert, M. Exploring the cockatiel (Nymphicus hollandicus) fecal microbiome, bacterial inhabitants of a worldwide pet. PeerJ 2016, 4, e2837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, E.E. Zoonotic diseases of common pet birds: Psittacine, passerine, and columbiform species. Vet. Clin. North Am. Exot. Anim. Pract. 2011, 14, 457–476. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Rideout, J.R.; He, Y.; Navas-Molina, J.A.; Walters, W.A.; Ursell, L.K.; Gibbons, S.M.; Chase, J.; McDonald, D.; Gonzalez, A.; Robbins-Pianka, A.; et al. Subsampled open-reference clustering creates consistent, comprehensive OTU definitions and scales to billions of sequences. PeerJ 2014, 2, e545. [Google Scholar] [CrossRef] [Green Version]
- Jousset, A.; Bienhold, C.; Chatzinotas, A.; Gallien, L.; Gobet, A.; Kurm, V.; Küsel, K.; Rillig, M.C.; Rivett, D.W.; Salles, J.F.; et al. Where less may be more: How the rare biosphere pulls ecosystems strings. ISME J. 2007, 11, 853–862. [Google Scholar] [CrossRef]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozupone, C.A.; Hamady, M.; Kelley, S.T.; Knight, R. Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 2007, 73, 1576–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammer, O.; Harper, D.A.T.; Ryan, P.D. PAST: Paleontological statistics software package for education and data analysis. Paleontol. Electron. 2001, 4, 1–9. [Google Scholar]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Parks, D.H.; Beiko, R.G. Identifying biologically relevant differences between metagenomic communities. Bioinformatics 2010, 26, 715–721. [Google Scholar] [CrossRef] [Green Version]
- Ward, T.; Larson, J.; Meulemans, J.; Hillmann, B.; Lynch, J.; Sidiropoulos, D.; Spear, J.R.; Caporaso, G.; Blekhman, R.; Knight, R.; et al. BugBase predicts organism-level microbiome phenotypes. bioRxiv 2017. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [Green Version]
- Thompson, L.R.; Sanders, J.G.; McDonald, D.; Amir, A.; Ladau, J.; Locey, K.J.; Prill, R.J.; Tripathi, A.; Gibbons, S.M.; Ackerman, G.; et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 2017, 551, 457–463. [Google Scholar] [CrossRef] [Green Version]
- Frank, D.N.; St. Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [Green Version]
- Pasolli, E.; Asnicar, F.; Manara, S.; Zolfo, M.; Karcher, N.; Armanini, F.; Beghini, F.; Manghi, P.; Tett, A.; Ghensi, P.; et al. Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell 2019, 176, 649–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Mazcorro, J.F.; Kawas, J.R.; Licona Cassani, C.; Mertens-Talcott, S.; Noratto, G. Different analysis strategies of 16S rRNA gene data from rodent studies generate contrasting views of gut bacterial communities associated with diet, health and obesity. PeerJ 2020, 8, e10372. [Google Scholar] [CrossRef] [PubMed]
- Xenoulis, P.G.; Gray, P.L.; Brightsmith, D.; Palculict, B.; Hoppes, S.; Steiner, J.M.; Tizard, I.; Suchodolski, J.S. Molecular characterization of the cloacal microbiota of wild and captive parrots. Vet. Microbiol. 2010, 146, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, B.C.M.; Murray, M.; Tseng, F.; Widmer, G. The fecal microbiota of wild and captive raptors. Anim. Microbiome 2020, 2, 15. [Google Scholar] [CrossRef]
- Zou, A.; Sharif, S.; Parkinson, J. Lactobacillus elicits a ‘Marmite effect’ on the chicken cecal microbiome. NPJ Biofilms Microbiomes 2018, 4, 27. [Google Scholar] [CrossRef] [Green Version]
- Hauffe, H.C.; Barelli, C. Conserve the germs: The gut microbiota and adaptive potential. Conserv. Genet. 2019, 20, 19–27. [Google Scholar] [CrossRef]
- Ghosh, T.S.; Arnoux, J.; O’Toole, P.W. Metagenomic analysis reveals distinct patterns of gut lactobacillus prevalence, abundance, and geographical variation in health and disease. Gut Microbes 2020, 12, 1–19. [Google Scholar] [CrossRef]
- Walter, J. Ecological role of Lactobacilli in the gastrointestinal tract: Implications for fundamental and biomedical research. Appl. Environ. Microbiol. 2008, 74, 4985–4996. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.; Si, W.; Forster, R.J.; Huang, R.; Yu, H.; Yin, Y.; Yang, C.; Han, Y. 16S rRNA gene-based analysis of mucosa-associated bacterial community and phylogeny in the chicken gastrointestinal tracts: From crops to ceca. FEMS Microbiol. Ecol. 2007, 59, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Cisek, A.A.; Binek, M. Chicken intestinal microbiota function with a special emphasis on the role of probiotic bacteria. Pol. J. Vet. Sci. 2014, 17, 385–394. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Mazcorro, J.F.; Lanerie, D.J.; Dowd, S.E.; Paddock, C.G.; Grützner, N.; Steiner, J.M.; Ivanek, R.; Suchodolski, J.S. Effect of a multi-species synbiotic formulation on fecal bacterial microbiota of healthy cats and dogs as evaluated by pyrosequencing. FEMS Microbiol. Ecol. 2011, 78, 542–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldassarre, M.E.; Di Mauro, A.; Tafuri, S.; Rizzo, V.; Gallone, M.S.; Mastromarino, P.; Capobianco, D.; Laghi, L.; Zhu, C.; Capozza, M.; et al. Effectiveness and safety of a probiotic-mixture for the treatment of infantile colic: A double-blind, randomized, placebo-controlled clinical trial with fecal real-time PCR and NMR-based metabolomics analysis. Nutrients 2018, 10, 195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, B.J.; Rather, I.A.; Kumar, V.J.R.; Choi, U.H.; Moon, M.R.; Lim, J.H.; Park, Y.H. Evaluation of Leuconostoc mesenteroides YML003 as a probiotic against low-pathogenic avian influenza (H9N2) virus in chickens. J. Appl. Microbiol. 2012, 113, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Staley, M.; Bonneaud, C.; McGraw, K.J.; Vleck, C.M.; Hill, G.E. Detection of Mycoplasma gallisepticum in house finches (Haemorhous mexicanus) from Arizona. Avian Dis. 2018, 62, 14–17. [Google Scholar] [CrossRef] [PubMed]
- Sawicka, A.; Durkalec, M.; Tomczyk, G.; Kursa, O. Occurrence of Mycoplasma gallisepticum in wild birds: A systematic review and meta-analysis. PLoS ONE 2020, 15, e0231545. [Google Scholar] [CrossRef] [Green Version]
- Song, S.J.; Lauber, C.; Costello, E.K.; Lozupone, C.A.; Humphrey, G.; Berg-Lyons, D.; Caporaso, J.G.; Knights, D.; Clemente, J.C.; Nakielny, S.; et al. Cohabiting family members share microbiota with one another and with their dogs. eLife 2013, 2, e00458. [Google Scholar] [CrossRef]
- Jaspers, E.; Overmann, J. Ecological significance of microdiversity: Identical 16S rRNA gene sequences can be found in Bacteria with highly divergent genomes and ecophysiologies. Appl. Environ. Microbiol. 2004, 70, 4831–4839. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Jones, R.B.; Fodor, A.A. Inference-based accuracy of metagenome prediction tools varies across sample types and functional categories. Microbiome 2020, 8, 46. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Mazcorro, J.F.; Mills, D.A.; Murphy, K.; Noratto, G. Effect of barley supplementation on the fecal microbiota, caecal biochemistry, and key biomarkers of obesity and inflammation in obese db/db mice. Eur. J. Nutr. 2018, 57, 2513–2528. [Google Scholar] [CrossRef]
- Falony, G.; Joossens, M.; Vieira-Silva, S.; Wang, J.; Darzi, Y.; Faust, K.; Kurilshikov, A.; Bonder, M.J.; Valles-Colomer, M.; Vandeputte, D.; et al. Population-level analysis of gut microbiome variation. Science 2016, 352, 560–564. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Diet | Number of Birds (Female:Male) | Estimated Age for Females and Males |
|---|---|---|---|
| MP1 | Commercial feed * | 4 (undetermined) | 1 year |
| MP2 | Commercial feed | 5 (undetermined) | 6 months–1 year |
| MP3 | Commercial feed | 6 (undetermined) | 2–8 months |
| MP4 | Commercial feed | 4 (undetermined) | 2 years |
| MP5 | Commercial feed | 5 (undetermined) | 6 months–1 year |
| TG1 | Canary grass (Phalaris canariensis) | 6 (undetermined) | 8 months |
| TG2 | Mixture of seeds † | 6 (undetermined) | Undetermined |
| TG3 | Mixture of seeds | 5 (undetermined) | 3–9 months |
| TG4 | Mixture of seeds | 7 (undetermined) | 6 months–1 year |
| TG5 | Mixture of seeds | 6 (undetermined) | 6 months–1 year |
| AS1 | Mixture of seeds | 5 (2:3) | Undetermined |
| AS2 | Mixture of seeds | 5 (undetermined) | 6 months–1 year |
| AS3 | Mixture of sun flower and other seeds | 6 (undetermined) | 6 months–1 year |
| AS4 | Mixture of seeds | 4 (undetermined) | Undetermined |
| AS5 | Mixture of sun flower and other seeds | 5 (undetermined) | 4–5 months |
| PH1 | Breeding paste, croquette, foxtail millet (Setaria italica), fruit, mixture of seeds, wholemeal bread | 4 (2:2) | 2–4 years |
| PH2 | Breeding paste, croquette, foxtail millet (Setaria italica), fruit, mixture of seeds, wholemeal bread | 4 (2:2) | 3–5 years |
| PH3 | Breeding paste, croquette, foxtail millet (Setaria italica), fruit, mixture of seeds, wholemeal bread | 4 (2:2) | 3–5 years |
| PH4 | Fruit, legumes, mixture of sun flower and other seeds | 4 (undetermined) | 1 year |
| PH5 | Fruit, legumes, mixture of sun flower and other seeds | 4 (undetermined) | 5 months–1 year |
| NH1 | Breeding paste, croquette, foxtail millet (Setaria italica), fruit, mixture of seeds, wholemeal bread | 6 (3:3) | 3–4 years (females), 1–2 years (males) |
| NH2 | Breeding paste, croquette, foxtail millet (Setaria italica), fruit, mixture of seeds, wholemeal bread | 5 (2:5) | 2–5 years |
| NH3 | Mixture of sun flower and other seeds | 6 (undetermined) | 1 year |
| NH4 | Sun flower seeds | 6 (3:3) | 5–6 months |
| NH5 | Mixture of sun flower and other seeds | 6 (4:2) | 3 months–1 year |
| PK1 | Fruits, legumes, mixture of sun flower and other seeds | 4 (2:2) | 2–4 years |
| PK2 | Fruits, legumes, mixture of sun flower and other seeds | 4 (2:2) | 2–4 years (females), 1–5 years (males) |
| PK3 | Soft fruit, KAYTEE exact hand-feeding formula | 4 (undetermined) | 4–5 months |
| PK4 | Fruits, legumes, mixture of sun flower and other seeds | 4 (undetermined) | 1 year |
| PK5 | Fruits, legumes, mixture of sun flower and other seeds | 5 (undetermined) | 1–2 years |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia-Mazcorro, J.F.; Alanis-Lopez, C.; Marroquin-Cardona, A.G.; Kawas, J.R. Composition and Potential Function of Fecal Bacterial Microbiota from Six Bird Species. Birds 2021, 2, 42-59. https://0-doi-org.brum.beds.ac.uk/10.3390/birds2010003
Garcia-Mazcorro JF, Alanis-Lopez C, Marroquin-Cardona AG, Kawas JR. Composition and Potential Function of Fecal Bacterial Microbiota from Six Bird Species. Birds. 2021; 2(1):42-59. https://0-doi-org.brum.beds.ac.uk/10.3390/birds2010003
Chicago/Turabian StyleGarcia-Mazcorro, Jose F., Cecilia Alanis-Lopez, Alicia G. Marroquin-Cardona, and Jorge R. Kawas. 2021. "Composition and Potential Function of Fecal Bacterial Microbiota from Six Bird Species" Birds 2, no. 1: 42-59. https://0-doi-org.brum.beds.ac.uk/10.3390/birds2010003