Why Immunotherapy Fails in Multiple Myeloma
by
 , and
, and
Luis Gerardo Rodríguez-Lobato
1,2,†, 
Aina Oliver-Caldés
1,2,†,
David F. Moreno
1,2,
Carlos Fernández de Larrea
1,2,‡ and
Joan Bladé
1,2,*,‡ 
1
Amyloidosis and Multiple Myeloma Unit, Department of Hematology, Hospital Clínic of Barcelona, 08036 Barcelona, Spain
2
Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), 08036 Barcelona, Spain
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this manuscript.
‡
These authors share senior authorship.
Hemato 2021, 2(1), 1-42; https://0-doi-org.brum.beds.ac.uk/10.3390/hemato2010001
Submission received: 15 November 2020
/
Revised: 15 December 2020
/
Accepted: 18 December 2020
/
Published: 22 December 2020
(This article belongs to the Special Issue Immunotherapy in Myeloma: A Theme Issue in Honor of Prof. Dr. Gösta Gahrton)
Abstract
:Multiple myeloma remains an incurable disease despite great advances in its therapeutic landscape. Increasing evidence supports the belief that immune dysfunction plays an important role in the disease pathogenesis, progression, and drug resistance. Recent efforts have focused on harnessing the immune system to exert anti-myeloma effects with encouraging outcomes. First-in-class anti-CD38 monoclonal antibody, daratumumab, now forms part of standard treatment regimens in relapsed and refractory settings and is shifting to front-line treatments. However, a non-negligible number of patients will progress and be triple refractory from the first line of treatment. Antibody-drug conjugates, bispecific antibodies, and chimeric antigen receptors (CAR) are being developed in a heavily pretreated setting with outstanding results. Belantamab mafodotin-blmf has already received approval and other anti-B-cell maturation antigen (BCMA) therapies (CARs and bispecific antibodies are expected to be integrated in therapeutic options against myeloma soon. Nonetheless, immunotherapy faces different challenges in terms of efficacy and safety, and manufacturing and economic drawbacks associated with such a line of therapy pose additional obstacles to broadening its use. In this review, we described the most important clinical data on immunotherapeutic agents, delineated the limitations that lie in immunotherapy, and provided potential insights to overcome such issues.
1. Introduction
Multiple myeloma (MM) is a neoplastic plasma cell disease that accounts for 1.8% of all cancers diagnosed annually in the United States (US) and a similar proportion of all cancers diagnosed annually in Western Europe. MM is considered the second most common hematological malignancy after lymphoma or chronic lymphocytic leukemia [1,2,3].
Clonal plasma cells arise on the basis of an initial event—like cytogenetic (CG) abnormalities—that occur in early development of the B-cell maturation process [4]. Once a non-malignant plasma cell acquires a primary CG abnormality, namely trisomies or IgH translocations, the potential clone is able to remain for many years. From a clinical perspective, monoclonal gammopathy of undetermined significance (MGUS) is a well-defined pre-MM stage for detection of CG abnormalities [5,6,7]. However, multiple ways can trigger clonal plasma cells, like the well-recognized “second hits” that include monosomies, 1q aberrations, or del17p. Additionally, with the bone marrow (BM) microenvironment playing a key role, disease progression is characterized by a parallel, altered immune response. Among the most relevant cytokines in MM are interleukin 6 (IL-6) [8,9], B cell activating factor belonging to the TNF family (BAFF), transmembrane activator and calcium-modulator and cytophilin ligand interactor (TACI) [10], and insulin-like growth factor I (IGF-1) [11]. In advanced stages involving extramedullary disease, there appears to be an independent IL-6 pathway that facilitates migration outside the BM [12,13]. Other cytokines involved in MM include interleukin 8 (IL-8), interleukin (IL-10), vascular endothelial growth factor (VEGF), and transforming growth factor-beta (TGF-β), all of which induce tumor growth and inhibit T cell activity [14]. T cell exhaustion relies on the basis of T cell activity loss and sustained expression of inhibitory receptors. Moreover, IL-10 can increase expression of immune checkpoints on T cells such as programmed cell-death-protein-1 (PD-1), T cell immunoglobulin and ITIM domain (TIGIT), and cytotoxic T-lymphocyte antigen 4 (CTLA-4) and thereby reduce their effector activity [15,16,17]. Other immune interactions include stimulation of T-helper 17 (Th-17) by TGF-β or IL-6 to produce bone disease [18]. In summary, multiple interactions from the BM microenvironment and MM cells lead to immune escape and suppression of T cell effector capacity. Cyclical recruitment of exhausted T cells helps maintain the pathological immune microenvironment.
Treatment strategies are based on the combination of proteasome inhibitors (PI) and immunomodulatory drugs (IMiDs) [19,20]; however, in relapse and refractory (R/R) MM scenarios, immunotherapy may play an even stronger role in inhibiting immune checkpoints, targeting plasma cell surface antigens, and even developing cancer vaccines [21,22]. Given post-procedure immune restoration with better immune surveillance, another option for patients with high-risk disease and good performance status is allogeneic transplantation [23]. However, toxicity related to this procedure may not be well tolerated in many patients.
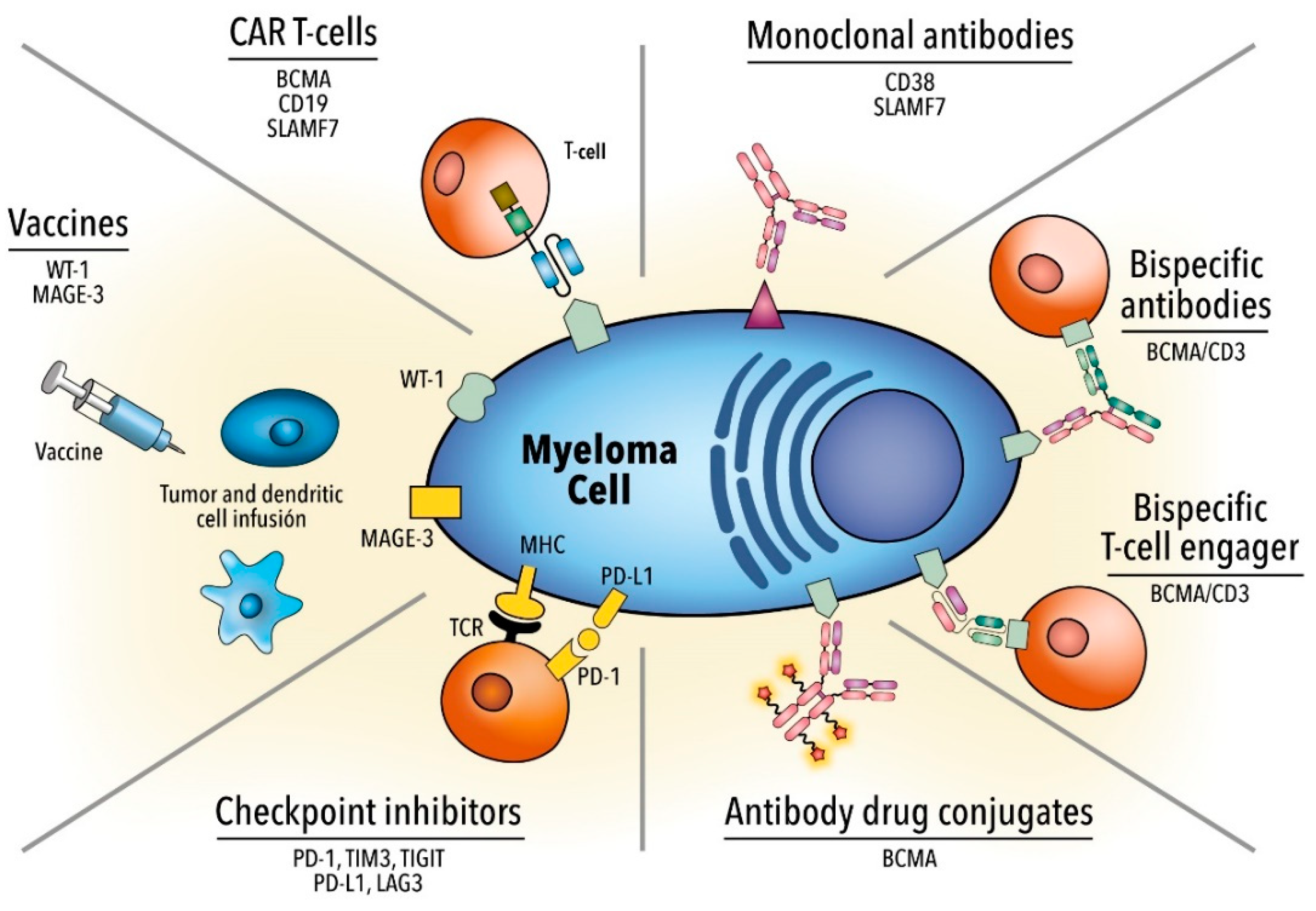
For this reason, designing chimeric antigen receptor (CAR) T cells is an innovative therapeutic option, especially in individuals with R/R MM [24]. While improvements have been made in treatment strategies, MM continues to be an almost incurable disease and novel therapeutic strategies are necessary. In this review, we described the most important clinical data on immunotherapeutic agents (Table 1 and Figure 1), delineated the limitations that lie in immunotherapy, and provided potential insights to overcome such issues.
2. Monoclonal Antibodies
2.1. Anti-CD38
CD38 was first discovered in 1980 when Reinherz and Schlossman were studying the human lymphocyte surface using monoclonal antibodies (MoA) in search of the T cell receptor. A glycoprotein highly expressed in MM cells, CD38 is also found at lower levels in normal lymphoid and myeloid cells, including NK cells, B cells, and activated T cells, and in non-hematological tissues in some cases [61]. The role of CD38 can be observed in several functions. It acts as an adhesion molecule, interacting with the endothelial ligand CD31. It also plays a role in extracellular NAD+ and cytoplasmic NADP metabolism, mobilizing cyclic adenosine diphosphate (ADP) ribose, ADP ribose (ADPR), and nicotinic acid adenine dinucleotide phosphate [62,63].
The high expression of CD38 on MM cells led to the development of several anti-CD38 MoA in the 1990s, with daratumumab (fully human) and isatuximab (chimeric) being the most studied ones. The antitumoral effect of these antibodies correlates with their capacity to induce antibody-dependent cellular toxicity (ADCC), complement-dependent toxicity (CDC), and antibody-dependent cellular phagocytosis (ADCP) of CD38+-opsonized cells. Further, the inhibition of the ectoenzymatic function of CD38 and the induction of direct apoptosis may contribute to the efficacy of these antibodies against MM [64]. Daratumumab interacts with CD38 present in monocytes and can inhibit in vitro osteoclastogenesis and bone resorption, which may improve bone-related alterations in these patients.
Developed in 2008 and approved as a single agent in 2015 and 2016 by the US Food and Drug Administration (FDA) and European Medicines Agency (EMA), respectively, daratumumab administered as monotherapy to heavily pretreated patients with MM showed an overall response rate (ORR) of 31.1%, with 4.7% having a complete response (CR). The median duration of the response was 4 months and median overall survival (mOS) was 20.1 months. This study reported responses in all subgroups, including patients with extramedullary disease and high-risk cytogenetics [25,26].
An ex vivo assay and in vivo xenograft mouse model demonstrated the efficacy of daratumumab when combined with IMiDs such as lenalidomide, proving its capacity to increase daratumumab-mediated lysis and thereby activate the effector function of autologous immune cells. Such improvement in efficacy was also observed when daratumumab was administered in combination with bortezomib and lenalidomide even in bortezomib- and lenalidomide-resistant MM cells. Similarly, the use of lenalidomide in this study proved capable of increasing daratumumab-mediated lysis through activation of NK cells [65].
The number of regimens incorporating daratumumab together with other backbone combinations is increasing. Daratumumab was further tested in a randomized phase II study with lenalidomide and dexamethasone (n = 152)(GEN503) [27,28], in which 88% of patients achieved at least a partial response (PR) and the CR rate was 25%. In the POLLUX [29] phase III study, investigators compared lenalidomide plus dexamethasone (Rd) against daratumumab plus both drugs (DRd). In both groups, patients with lenalidomide-refractory MM were excluded. In the DRd group, 12-month progression-free survival (PFS) and 12-month OS were 83.2% and 91.2%, respectively, whereas 12-month PFS and 12-month OS were 60.1% and 76.4%, respectively, in the Rd group (p < 0.001). Patients treated with the DRd scheme achieved a CR of 43.1%, of which 22.4% were negative minimal residual disease (MRD); patients treated with the Rd scheme achieved a CR of 19.2%, with the stringent complete response (sCR) being 4.2% (p < 0.001). In the CASTOR study, patients with R/R MM receiving bortezomib and dexamethasone (Vd) with or without daratumumab (DVd) were compared. Findings revealed 18-month PFS of 48% and 7.9% in the DVd and Vd groups, respectively, [30] and a benefit conferred in high-risk cytogenetic patients, with a median PFS of 11.2 and 7.2 months in the DVd and Vd groups, respectively [31]. More recently, daratumumab is approved for first-line treatment for patients with MM, including candidates for autologous stem cell transplantation (ASCT) (with bortezomib, thalidomide, and dexamethasone [66]) and non-candidates (with melphalan, bortezomib, and prednisone [67] or lenalidomide and dexamethasone [68]). More combinations in the relapse setting are now in clinical trials, such as daratumumab plus pomalidomide [69] or carfilzomib [70], and results are encouraging.
Isatuximab (chimeric) has shown strong pro-apoptotic activity, independent of cross-linking agents and antitumor activity related to CDC, ADCC, and ADCP. Activity of this antibody is enhanced by pomalidomide; a phase III trial comparing pomalidomide and dexamethasone with or without isatuximab obtained a PFS of 11.5 vs. 6.5 months (isatuximab vs. control, respectively) [71].
The main mechanisms of action of daratumumab include (Table 2):
- Complement-dependent cytotoxicity (CDC): Binding between the Fc tail of the antibody and C1q activates the complement cascade to end with the formation of the membrane attack complex (MAC) [72];
- Antibody-dependent cell-mediated cytotoxicity (ADCC): Binding between FC-gamma receptors on effector cells (T and NK cells) and the Fc tail of daratumumab releases cytotoxic molecules, leading to MM cell death [65];
- Antibody-dependent cellular phagocytosis (ADCP): Opsonization of the tumor cell occurs when the Fc tail of the CD38 antibody binds to the Fc-gamma receptor of phagocytic cells such as monocytes or macrophages [73];
Thus, several mechanisms of resistance of daratumumab have been described:
- CD38 expression: Tests performed on modified MM cell lines that express different levels of CD38 have shown greater CDC and ADCC in cells expressing high levels of CD38 compared to cells with low expression. In MM plasma cells, expression is heterogenic and daratumumab activity is correlated with such expression levels [78]. Analysis performed on samples of patients who had been enrolled in daratumumab clinical trials showed a quick and marked decrease in CD38 levels after treatment in all patients; a decrease in CDC and ADCC was also observed in ex vivo tests. Downregulation of CD38 of this type also occurs in cell subsets other than MM cells and mechanisms are not fully understood. Some strategies to overcome such resistance have been proposed and are based on combinations with other drugs capable of increasing CD38 levels such as IMiDs, panobinostat, all-trans retinoic acid (ATRA), and ricolinostat [79,80,81]. The ability of ATRA to resynthesize CD38 is being analyzed in a clinical trial (NCT02751255);
- Complement inhibitory proteins: Tumor cells are known to be capable of increasing soluble and membrane-bound complement regulatory proteins such as C4-binding protein, CD55, or CD59 to protect themselves from complement attacks, similar to the way in which immune checkpoint inhibitor receptors function [82]. Ex vivo analysis using MM cell lines with low expression of CD55 and CD59, and MM cell lines treated with phospholipase-C to remove GPI-anchored proteins (CD55 and CD59) showed increased daratumumab CDC. These observations were not confirmed with MM cells obtained from daratumumab-naïve patients. In addition, an increase in CD55 and CD59 expression was detected in MM cells obtained from patients who were progressing under monotherapy treatment. In this case, ATRA combination may also decrease upregulation of complement inhibitors [78]. Panobinostat, which has shown to increase CD38 levels, also increases CD55 and CD59 levels, possibly explaining the lack of benefit in terms of CDC, although ADCC improved [83];
- CD47-SIRPα interaction: CD47 expressed in tumor cells of solid tumors and hematological malignancies interacts with regulatory transmembrane protein SIRPα that is expressed on dendritic cells and macrophages, decreasing their phagocytic function [84]. Upregulation of CD47 has been observed in drug-resistant MM cells and blocking the interaction between SIRPα and CD47 restores phagocytosis [85]. Anti-CD47 therapies are under evaluation in other lymphoid malignancies and low-dose cyclophosphamide may decrease CD47 expression to improve ADCP [86,87,88];
- Polymorphisms on Fc-gamma receptors: Mechanism of action of daratumumab ADCC and ADCP depend on the activation of Fc-gamma receptors on effector cells [89]. Affinity may differ based on allelic variants of these receptors. Fc-gamma receptors were genotyped in samples of patients with MM included in daratumumab clinical trials, demonstrating a positive correlation between polymorphisms 3A and 2B and outcome in terms of PFS, albeit not OS [90];
- The way in which the microenvironment plays a crucial role in MM has been well studied. Bone marrow stromal cells (BMSC) protect MM cells from drugs and effector cells such as cytotoxic T cells [91]. Interaction between BMSC and MM cells may upregulate anti-apoptotic molecules like survivin, which could contribute to resistance against daratumumab;
- Soluble CD38 (sCD38) may have a draining effect on daratumumab function and diminish efficacy; however, the presence of sCD38 has been observed in only a few patients and in such cases, did not correlate with response. There are no published data about other CD38 antibodies and the impact of sCD38 [82];
- NK cells play a crucial role in ADCC. Some studies have shown a correlation between daratumumab-induced ADCC and NK cell-to-MM cell ratio [78]. Due to their capacity to activate NK cells, IMiDs could improve NK function and ADCC, even in patients with IMiD-refractory MM [65,92]. An increase in ADCC was observed in ex vivo experiments when interaction between NK inhibitory receptors KIR (KIR2DL-1, -2, -3) and their respective ligands was blocked. Similarly, ADCC was reported to improve synergistically with the addition of lenalidomide to the experiment. As NK cells express CD38 on their surface, fratricide and a diminished effector function can arise. When studied in patients, the reduction in NK levels was similar in responders and non-responders to daratumumab and no correlation with outcome was observed. Some measures have nonetheless been proposed to diminish this eventual effect, including the administration of ex vivo-expanded autologous NK cells to increase the count, and pretreatment of such cells with F(ab’)2 fragments of daratumumab to avoid fratricide [93,94].

{kind=link}
{kind=link}
Table 2.
Mechanisms of action and resistance to daratumumab.
| Mechanisms of Action | Mechanisms of Resistance |
|---|---|
|
|
2.2. Anti-SLAMF7
Signaling lymphocytic molecule F7 (SLAMF7) or cell-surface glycoprotein CD2 subset 1 (CS1) is a glycoprotein expressed on healthy plasma cells, MM cells, and NK-cells, and absent in other tissue. Expression of such is found in more than 95% of MM plasma cells independent of cytogenetics. This molecule belongs to the immunoglobulin superfamily within the SLAM family subgroup [95]. For this reason, generating a MoA directed at this target has been of great interest, with elotuzumab being the most relevant one. Elotuzumab is a humanized immunoglobulin G1 immunostimulatory MoA that works by activating signals in NK cells via interaction with protein EAT-2, and is capable of directly activating NK cells by ADCC via CD16 [96]. In MM plasma cells, this mechanism is compromised by the lack of EAT-2 expression found in tumor cells. For this reason, elotuzumab does not induce proliferation in MM cells.
In a phase I study (n = 35), the maximum tolerated dose of elotuzumab was not reached and the drug was administered at 20 mg/kg iv once every 2 weeks for 8 weeks total. None of the patients achieved a PR or better; 26.5% achieved stable disease; and the rate of infusion-related reactions before prophylaxis initiation was 52% [32].
Elotuzumab is therefore not active in monotherapy. Yet, its potential activity in combination with PI and IMiDs, such as lenalidomide and pomalidomide, was explored. In a randomized phase II study with Vd with or without elotuzumab (n = 152) [33], 63% of patients achieved at least a PR with median PFS (mPFS) of 6.9 months and 1-year OS of 74%, while 65% of patients in the elotuzumab group achieved a PR or better with a PFS of 9.7% and 1-year OS of 85%. No mechanisms of resistance to elotuzumab were described. Furthermore, in a randomized phase III ELOQUENT-2 study testing the combination of Rd +/− elotuzumab in patients with R/R MM, ORR were 79% vs. 66% in the elotuzumab vs. control groups (p = 0.0002), with 1-year OS of 91% vs. 83%, 2-year OS of 73% vs. 69%, and 3-year OS of 60% vs. 53% (p = 0.026). Adverse events (AE) were comparable between groups [34]. Additionally, 117 subjects were enrolled in a multicenter, randomized, open-label phase II trial comparing pomalidomide and dexamethasone with or without elotuzumab in lenalidomide- and bortezomib-refractory patients with R/R MM. With a minimum follow-up of 9.1 months, mPFS were 10.3 and 4.7 months in the elotuzumab and control groups, respectively, and ORR were 53% and 26% in the elotuzumab and control groups, respectively. Infusion reactions were observed in 5% of patients (n = 3) and classified as grades 1 or 2 [35]. A phase III study performed by the German-speaking Myeloma Multicenter Group randomized patients to receive induction therapy based on bortezomib, lenalidomide, and dexamethasone with or without elotuzumab (Elo-VRD vs. VRD) obtaining an ORR of 82.4% vs. 85.6% (p = 0.35), respectively. AEs of grade 3 or higher occurred in 65.4% patients (Elo-VRD) and 66.5% (VRD) mainly related to nervous system disorders, infections, and blood disorders. There were 9 and 4 treatment-related deaths in the Elo-VRD and VRD groups, respectively [97]. At the last American Society of Clinical Oncology (ASCO) meeting, primary analysis of the phase II trial (SWOG-1211) comparing Elo-VRD vs. VRD for ND, high-risk MM patients were presented. One hundred and three patients were included, and after a median follow-up of 53 months, no difference in mPFS (31 vs. 34 months, 68 vs. not reached, respectively; p = 0.45) nor in OS (68 months vs. not reached; p = 0.48) was observed [98]. Recently, data from a phase III clinical trial evaluating Elo-Rd in transplant-ineligible newly diagnosed multiple myeloma (NDMM) patients (ELOQUENT-1) have not shown a benefit with the addition of elotuzumab as front-line therapy [99]. Thus, elotuzumab has shown limited activity in the treatment of MM in terms of response and survival in both first and further lines of therapy. In the future, it will be necessary to determine the best combination for elotuzumab and the best scenario for its use.
A main limitation of both anti-CD38 and anti-SLAMF7 MoAs are infusion-related reactions (IRR), which happen primarily during initial administration and consists of pyrexia, chills, nausea, vomiting, flushing, cough, and dyspnea. Specifically, with elotuzumab, such IRR were mainly observed prior to the administration of premedication based on corticosteroids, acetaminophen, and antihistamines [32]. The rate of IRR due to elotuzumab was 7–10% with proper prophylaxis. In the case of daratumumab, however, IRR were reported in more than 50% of patients during the first infusion, even with prophylaxis, decreasing to 7% in further infusions [27].
In conclusion, monoclonal antibodies, specially CD38 directed agents, have proved to improve outcomes in MM and have reached a starring role in first line treatments.
3. Antibody-Drug Conjugate
Antibody-drug conjugates (ADCs) are MoAs joined to a cytotoxic compound via a chemical linker. These antibodies selectively target specific antigens located on the cell surface of interest. By internalizing the compound, the cytotoxic part can induce cell death. Several targets in MM and their respective antibodies are under study. The most relevant ones are mentioned below:
BCMA (CD269)-targeted ADCs: B-cell maturation antigen is a transmembrane receptor expressed on malignant plasma cells. Belantamab mafodotin (GSK-2857916) is a humanized anti-BCMA IgG1 MoA conjugated to monomethyl auristatin F (MMAF), capable of inducing ADCC activity against myeloma cells. A multicenter, phase I trial with patients with R/R MM (n = 35) showed an ORR of 60%, with 14% CR and mPFS of 12 months. The most frequent AE was thrombocytopenia (35%); similarly, several eye-related events were observed, including blurry vision (52%), dry eyes (37%), and photophobia (29%) [36,37]. Phase II clinical trial for RR MM patients (DREAMM-2) showed 30% and 34% of ORR in the 2.5 and 3.4 mg/kg cohorts, respectively. The most common grade ≥3 AE were keratopathy, thrombocytopenia, and anemia [100]. The keratopathy was further studied in DREAMM-2 patients and microcyst-like epithelial changes were found in 72% of cases. The management of eye-related AEs included dose delays (47%), dose reductions (25%), and discontinuation in 1% of patients [101]. Further studies are being performed to elucidate efficacy of this compound in combination with other MM therapies.
CD138 ADCs: CD138 or syndecan-1 is an extracellular protein receptor involved in cell-to-cell adhesion [102]. It is present in malignant plasma cells and some epithelial neoplasms. Indatuximab ravtansine is a MoA targeting CD138, linked with a disulfide bond to maytansinoid cytotoxic compound. In a phase I/II trial, ORR was 5.9% with no CR; however, 61.8% of patients maintained stable disease, with a mPFS of 3 months [38]. The most frequent toxicities reported were fatigue (47%) and diarrhea (43%) [39].
CD56 ADCs: Neural cell adhesion molecule 1 (NCAM1), otherwise known as CD56, is expressed in 75% of malignant plasma cells, yet in less than 15% of normal plasma cells [103]. Lorvotuzumab-mertansine is a MoA targeting CD56, conjugated to a microtubule inhibitor (MD1) by a disulfide bond linker. In a phase I trial for patients with R/R MM, ORR was reported at 5.7%, with no CR; however, 42.9% of patients maintained stable disease for 15.5 months. Peripheral neuropathy was an AE reported in 5.3% of patients [40].
CD74 ADCs: CD74 is a type II transmembrane glycoprotein of the major histocompatibility complex (MHC) class II, with antigen presentation functions [104]. Milatuzumab doxorubicin is an ADC with a MoA, targeting the CD74 linked via a hydrazine linker to doxorubicin. In a phase I study, this drug showed no objective response; it did, however, maintain 5 of 19 patients in stable disease for at least 3 months [41].
To sum up, ADCs have shown limited clinical results in monotherapy, so further combination studies are required to elucidate their efficacy in MM. Keratopathy could be a limiting factor for its widespread use. It will be necessary to establish adequate preventive measures, make timely diagnoses, and administer effective treatments against this complication.
4. Bispecific Monoclonal Antibodies
Bispecific monoclonal antibodies (Bs MoA) are antibodies that have two different targets and an activating or neutralizing function. Diverse bispecific antibody platforms (BiTE®, DuoBody®, and Dual-Affinity Re-Targeting®) are available, all distinguishable by structural differences among constructs. However, the majority of clinical trial data related to MM treatment are limited to the BiTE® platform [105]. BiTEs (bi-specific T cell engagers) are constructs composed of two different single-chain variable fragments (scFv) obtained from MoA and joined by a flexible peptide linker. One of the scFv acts as a binding domain for tumor cells via recognition of surface antigens and can be modified to specifically bind the malignant cell of interest and the other MoA bound to CD3, the invariable site of the TCR [106]. The junction between tumor cell and T cell leads to proliferation and growth of effector cells. These cells release cytotoxic molecules such as perforin, which creates transmembrane pores in tumor cells, and granzyme B, which acts as an initiator of apoptosis with the consequent tumor cell lysis [107]. This therapy is cytotoxic even without requiring the function of antigen-presenting cells, costimulatory molecules, or MHC-1/peptide complex. In contrast to CAR T cell therapy, Bs MoA have come to be considered as an “off-the-shelf” treatment: Processing and manufacturing time are not necessary and patients can benefit immediately from therapeutic approach [108]. Blinatumomab—targeting CD19 and CD3 in acute lymphoblastic leukemia (ALL)—was the first worldwide-approved Bs MoA (initial approval was conferred in 2014 by the FDA and full approval in 2017) [109,110].
Currently, several target antigens have been studied to treat MM with Bs MoA, with BCMA and CD38 being the most promising ones [107]. AMG-420, a BCMA/CD3 Bs MoA, was tested in a first-in-human, phase I dose-escalation trial. Patients with R/R MM who had received two or more lines of treatment were recruited to obtain a maximum tolerated dose of 400 µg/daily. Of the 10 patients who received that dose, ORR was 70% and 5 (50%) patients achieved MRD-negative CR. Grade 2–3 cytokine release syndrome (CRS) was observed in three patients and non-treatment-related mortality was reported in two patients (pulmonary aspergillosis and fulminant adenovirus hepatitis). One patient developed a dose-limiting, grade 3 peripheral polyneuropathy at 400 µg/dose [42,43]. Due to the miniscule size of Bs MoA (5kDa), its serum half-life is short and results in the continuous need for intravenous administration. With a more extended half-life, BCMA/CD3 Bs MoA (AMG-701) can be administered once per week, having demonstrated in vitro proliferation of central memory and effector memory cells and in vivo MM cytotoxicity [111,112]. A phase I/II study for patients with MM who relapsed after three or more lines of therapy is in progress to estimate the maximum tolerated dose (MTD) and establish safety and tolerability (NCT03287908).
Similarly, teclistamab (JNJ-64007957) is an investigational bispecific antibody targeting both the BCMA and CD3 receptors on T cells. In preclinical studies, the drug proved capable of recruiting and activating T cells to direct their cytotoxicity against BCMA+ MM cells from an MM cell line (H929) and in BM samples obtained from patients with MM as well [113]. Results obtained from these studies led to the development of a phase I clinical trial in patients with R/R MM, enrolling those adult patients who had received a median of 6 lines of treatment. Patients were treated with teclistamab at doses ranging from 0.3–270 µg/kg. Of the 78 patients who were administered teclistamab, 21 responded to treatment. Responses were found to be deep and persistent. Additionally, the treatment achieved an ORR of 67% among the 12 patients who received the highest dosage; three of the patients achieved a CR [44]. CRS was observed in 56% of patients (CRS events were all grades 1–2 and during initial doses). Neurotoxicity was seen in 8% of cases and 3% were grade 3 or higher. In addition, IRR were reported in 9% of patients. There were 2 dose-limiting toxicities: A case of grade 4 thrombocytopenia which resolved after one day and a grade 4 delirium, which resolved after 16 days. A grade 5 AE was reported, consisting of a respiratory failure in the context of pneumonia [44]. Recent results from the last European Hematology Association (EHA) meeting highlight the efficacy of CC-93269, an asymmetric 2 + 1 bispecific with bivalent BCMA binding and monovalent CD3 binding, with a half-life extended domain. This phase I trial (NCT03486067) included 30 patients (median of 5 prior lines of therapy). ORR was 43%, including 17% with a CR or sCR. Among 9 patients receiving 10 mg, the ORR was 89% (≥CR: 44%). The main AEs included cytopenias and infections. CRS was observed in 77% of patients, but most were grade 1 [114]. This study continues including patients in the dose-escalation phase.
GBR-1342 is a CD38/CD3 Bs MoA in current evaluation in a first-in-human, phase I/II study in PI-, IMiD-, and daratumumab-refractory patients with MM; the question of interest is whether subjects previously treated with daratumumab will respond to CD38-targeted Bs MoA. This study is currently recruiting patients (NCT03309111) [115]. Then, there is also the CD38/CD3 Bs MoA AMG-424, which has also shown potent activity against MM cell lines in spite of lower or higher CD38 expression in these cells. As inhibition of tumor growth in a murine model and acceptable toxicities in monkeys have been demonstrated (depletion of peripheral B-lymphocytes), a first-in-human, multi-center, phase I study for patients with R/R MM was approved (NCT03445663) and has, in recent times, finished (June 2020).
Recently, results on Cevostamab-BFCR4350A, a FcRH5-CD3 bispecific antibody have been presented at the last American Society of Hematology (ASH) meeting. The phase I, dose escalation study (NCT03275103), included 51 R/R MM patients (55% with high-risk cytogenetics). The median number of prior lines of therapy was 6. The ORR was 51.7%, including 3 sCRs and 3 CRs. Responses were observed in patients with high-risk cytogenetics, prior exposure to anti-CD38 MoA, ADC, and CAR T cell therapy. Regarding toxicity, CRS was observed in 75% of patients (grade ≥ 3: one patient). Other grade 3 or 4 AEs observed were lymphopenia (11.8%), anemia (5.9%), and thrombocytopenia (5.9%). No fatal (grade 5) AEs have been reported [116].
In addition, the last updated data of talquetamab-JNJ-64407564, a GPRC5D-CD3 Bs MoA, were presented at the last ASH meeting. One-hundred and thirty-seven patients have been included in the phase I trial (NCT03399799). The median number of prior lines was 6, and 15% of the patients had received prior BCMA-directed therapy. Respecting efficacy, this product showed ORRs of 78% and 67% with the IV and the SC route, respectively. CRS was observed in 47% of patients (mostly grades 1 or 2; grade 3 was seen only with the IV route) and neurotoxicity in 5% of patients. IRR have been reported in 14–15% of patients, all of them grades 1 or 2, and usually in the first cycle [117].
Impressive preclinical results are arousing interest in trispecific antibodies such as the trispecific T cell engager targeting CD38, CD3, and CD28, which has shown high killing capacity of MM tumor cell lines in in vitro tests and also suppressed MM growth in mice, with proliferation of memory and effector T cells and downregulation of regulatory T cells in primates [118]. Trispecific NK cell engagers are also under development, targeting the NK antigen CD16A, and BCMA and CD200 in MM cells [119].
The primary results obtained with Bs MoA show that this strategy is a promising approach in the treatment of patients with MMs, although drug-related toxicities, especially CRS and neurotoxicity, days of hospitalization, and patient surveillance should be taken into account.
5. Immune Checkpoint Inhibitors
The immune system plays a crucial role in cancer development and progression. However, cytolytic activity of immune cells during the initial phase of carcinogenesis is the predominant mechanism used to fight against malignant cells. A balance between cancer progression and cancer eradication is then reached during the intermediate phase, mediated by modulatory proteins denominated as checkpoint molecules. When the immune system grows tolerant to the presence of cancer after this phase, tumor cells escape and can progress and induce metastasis [120,121].
Immune checkpoint molecules are a family of proteins composed of receptors—mostly located in T cells and other immune effector cells—and ligands located in antigen-presenting cells (APCs), monocytes, and tumor cells as well. The function of checkpoint receptors is to promote a balance between activating and inhibitory signals [122]. Several examples of checkpoint receptors have been widely studied, such as PD-1, CTLA-4, LAG-3 [123], TIM-3, and TIGIT. Interaction with their respective ligands triggers an inhibitory signal capable of counteracting T cell-mediated immunity. In a physiological setting, checkpoints have a modulatory function that maintains balance between immune response and immune tolerance. This aspect is crucial, as it protects the organism from autoimmunity. Despite that, tumor cells can take advantage of this mechanism, expressing checkpoint ligands on their surface and inducing inhibitory signals, to promote tumor immune tolerance [124,125]. Blocking checkpoint inhibitors has shown impressive tumor response in heavily treated patients with melanoma, lung cancer, or Hodgkin lymphoma [126].
Several immune dysregulations have been described in MM. BM niche cells contribute to tumor growth and immune escape by creating a permissive microenvironment promoted by factors with immunosuppressive properties such as TGF-β, prostaglandin E2, IL-10, and IL-6 [127]. Additionally, an impaired maturation and differentiation pattern has been described in dendritic cells (DCs) of patients with MM [128,129]. Increased levels of PD-L1 have been found in MM plasma cells as well as an increased expression of PD-1 in circulating effector cells like T and NK cells [130,131]. The immunosuppressive role of other checkpoint receptors such as CD85j or TIGIT has also been shown in MM. A study demonstrated lower expression of CD85j, an inhibitory immune checkpoint for B-cell function, in patients with active MM and MGUS (a premalignant condition), suggesting that such a lower expression in malignant PCs may eliminate the inhibitory signal—causing an increase in PC resistance to NK cytotoxicity—and lead to immune escape [132]. TIGIT, an inhibitory checkpoint receptor expressed on lymphocytes, and its ligands poliovirus receptor (PVR) and Nectin-2, could also play a role in immune escape. In vitro functional assays demonstrated inhibition of CD8+ T cell signaling and proliferation, which could be restored by TIGIT blockade. TIGIT blockade also showed an increased proliferation of IFN-γ-secreting CD4+ [17,133]. Although preclinical data suggest that blockade of the PD-1/PD-L1 axis could be effective in the treatment of MM, clinical data published to date do not support such statement. A phase I study with pembrolizumab monotherapy, a PD-L1 blocker, from patients with R/R MM achieved stable disease as the best response [134]. A separate phase I study exploring the use of nivolumab (PD-1 blocker), which comprised patients with different hematological neoplasms, included 27 patients with R/R MM. Of these patients, 63% achieved stable disease, while only one patient achieved an objective response (4%) [45]. Despite the limited outcomes obtained with PD-1/PD-L1 blockers in monotherapy, some studies reported better efficacy when in combination with IMiDs like lenalidomide or pomalidomide, even in patients treated previously with IMiDs. The reason for such efficacy was the enhancing effect conferred by these agents on the PD-1/PD-L1 axis. In the KEYNOTE-183 study, a phase III randomized trial comparing pomalidomide and dexamethasone with or without pembrolizumab, ORR was 34% vs. 40% in the pembrolizumab-PD group and PD group, respectively. Immune-mediated AE occurred in 18% of patients in the pembrolizumab group, with the most frequent being pneumonitis, hyperthyroidism, and rash. Serious AE were reported in 63% vs. 46% of patients in the pembrolizumab group and control group, respectively, with treatment-related mortality occurring in four patients with the following etiologies: Unknown cause, neutropenic sepsis, myocarditis, and Stevens–Johnson syndrome. The FDA indicated that based on the data presented to the monitoring committee, risks associated with the pembrolizumab combination outweighed the benefits and the study was to be discontinued [46]. The phase III study KEYNOTE-185, which compared Rd administration with or without pembrolizumab (200 mg every 3 weeks) in patients with NDMM who were not eligible for ASCT, showed a high rate of immune-mediated AE and mortality, with an interim unplanned analysis suggesting an unfavorable benefit-risk profile [135].
The efficacy of PD-1/PD-L1 blockers seems to be related to a higher immune cell infiltration of the tumor [121]. Mutational burden and neo-antigen expression have also proven to play a crucial role in PD-L1 expression on solid tumors. Results obtained with checkpoint inhibitor blockers may be explained by the fact that MM is known to have a low burden of mutations when compared to other solid, hematological diseases, as well as low immune cell infiltrate [136]. Toxicities observed in KEYNOTE trials raised concerns in other trials that combined an immune checkpoint inhibitor with IMiDs; most were therefore suspended or terminated [137].
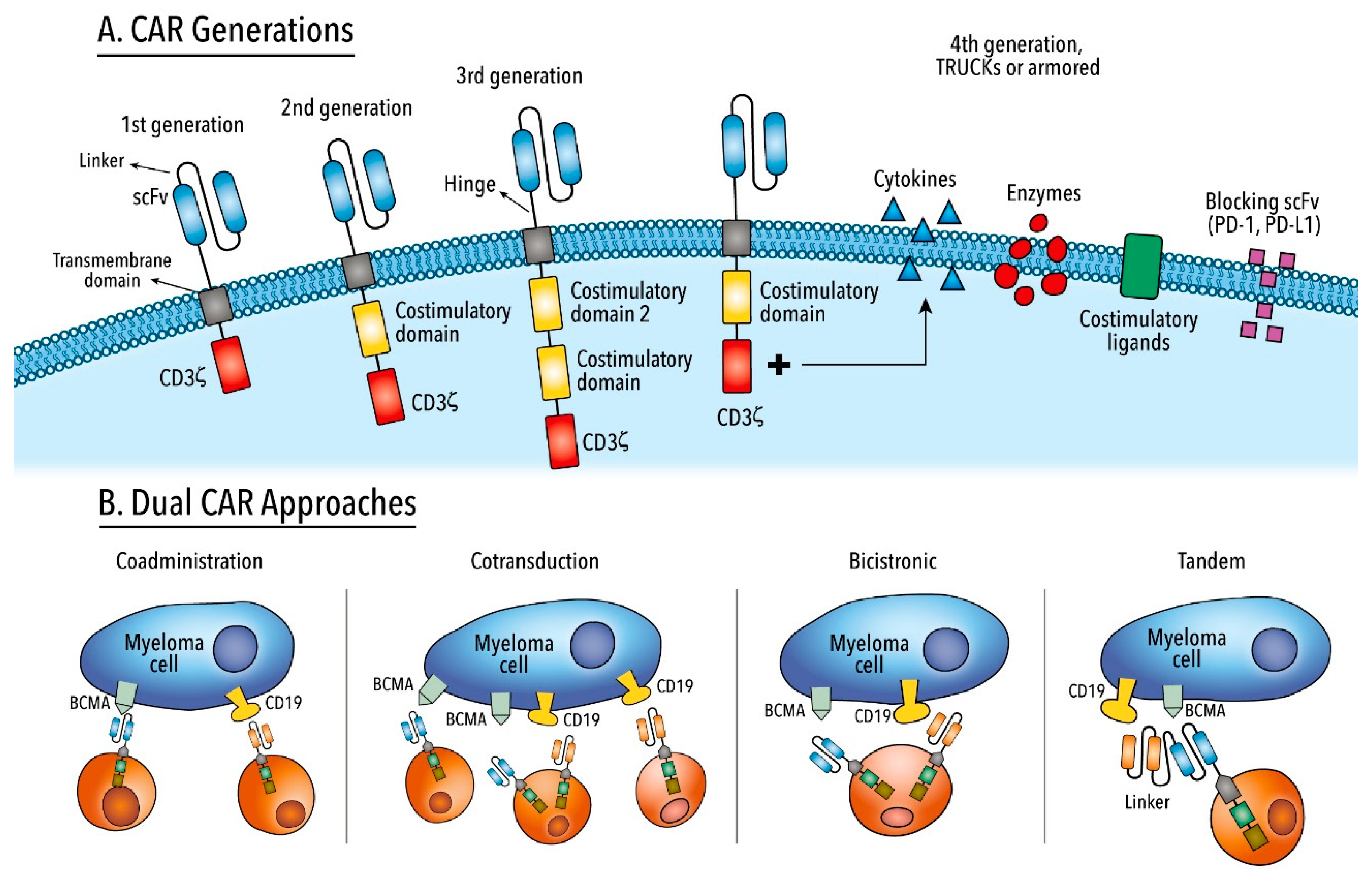
6. Chimeric Antigen Receptor T Cell Therapy
CARs are synthetic fusion proteins designed in a modular fashion that redirect lymphocytes to recognize and eliminate cells that express a target antigen on their surfaces. CARs are endowed with four fundamental components: Either the extracellular antigen-binding domain or scFv derived typically from the light and heavy chains of MoAs to provide antigen-specificity in a non-HLA-restricted manner; either the spacer or hinge based on CD8-, CD28-, IgG1-, or IgG4-derived domains; the transmembrane domain from CD8α or CD28 moieties; and intracellular or activation domains derived from the CD3ζ moiety of the TCR (first generation) and the addition of one (second generation) or two (third generation) costimulatory domains derived from CD28, 4-1BB moieties and others that are necessary for optimal T cell function, proliferation, and persistence. Armored or fourth-generation CARs include immune modulating capacities, suicide genes, controllable on–off protein switches, and molecules to reduce or overcome T cell dysfunction or exhaustion (Figure 2) [138,139,140]. Synthetically engineered T cells expressing CARs against the CD19 antigen have shown outstanding results in B-cell malignancies in clinical trials, and the FDA and EMA have approved the use of tisagenlecleucel, axicabtagene ciloleucel and brexucabtagene autoleucel [141,142,143,144,145,146,147,148]. Indeed, the results led to many more additional clinical trials in diverse hematological and solid cancers, and several encouraging results have been reported with the use of CAR T cell therapy-targeting BCMA in patients with MM. Due to the therapy’s potential as a treatment strategy in patients with R/R MM, the first anti-BCMA CAR is expected to be approved within the coming months. However, an in-depth review of all the clinical trials that are being carried out using CAR T cell therapy in patients with myeloma goes beyond the scope of this manuscript. Exhaustive reviews have been published elsewhere [149,150,151,152,153]. The two most important BCMA CAR T cell products that are currently being evaluated in registration phase clinical trials include idecabtagene vicleucel and ciltacabtagene autoleucel (Table 1). Idecabtagene vicleucel is a second-generation CAR that includes a 4-1BB costimulatory domain and a murine scFv. The latest results from the phase II trial (KarMMa; NCT03361748) were presented at the last ASCO meeting. The trial enrolled 149 patients and the doses of 150 to 450 × 106 CAR T cells were analyzed. The ORR was 73% (CR rate 33%), with a mPFS and OS of 8.8 and 19.4 months, respectively. Patients treated at the highest dose level had an ORR of 82% and a mPFS of 12.1 months. Regarding safety profile, CRS and immune effector cell-associated neurotoxicity syndrome (ICANS) were observed in 84% and 18% of all patients, respectively [53]. Ciltacabtagene autoleucel is also a second-generation CAR that includes a 4-1BB costimulatory domain and two llama-derived variable-heavy chain only fragments against two different BCMA epitopes. The latest results from the phase Ib/II trial (CARTITUDE-1; NCT03548207) were presented at the last ASH meeting. The trial included 97 patients and a single infusion of the product at a target dose of 0.75 × 106 CAR T cell/kg was administered. The ORR was 96.9% (sCR rate 67%), with a one-year PFS and OS of 76.6% and 88.5%, respectively. In terms of toxicity, CRS was observed in 94.8% of all patients (grade ≥ 3 in 4.1%) and ICANS occurred in 20.6% of patients (grade ≥ 3 in 10.3%). Ten deaths occurred during the study due to adverse events (eight patients) and progressive disease (two patients) [52].
BCMA is by far the most predominant antigen used for targeted CAR T cell therapy in MM. Reasons for targeting BCMA include the antigen’s high surface expression in malignant plasma cells, its exclusive expression in some mature B-cell subsets, and its non-expression in normal tissue and hematopoietic stem cells [154,155,156]. BCMA regulates B cell differentiation, survival, and maturation. However, in the malignant plasma cell, BCMA is associated with the cell’s survival and proliferation, and contributes to the immunosuppressive BM microenvironment [157,158]. BCMA expression is higher in patients with MM when compared with non-malignant plasma cells; nevertheless, the levels vary [159,160]. In general, CAR T cells targeting BCMA have shown impressive results in heavily pretreated patients with MM, including achieving deep responses (ORR 60–97% (≥CR in 10–86%)), manageable toxicity (CRS 60–100% (grade ≥ 3 in 0–41%), and ICANS 2–42% (grade ≥ 3 in 0–19%)), and a mPFS of 2–20 months [24,47,48,49,51,52,53,54,161,162,163,164,165,166]. These results are non-homogenous but can be explained by differences in patient inclusion protocols, CAR constructs, conditioning regimens, CAR T cell doses, and toxicity grading scales. Furthermore, despite these impressive remission rates, it should be noted that many patients are resistant and will relapse after CAR T cell therapy. No plateau is observed in PFS curves after CAR T cell infusion, as has been reported in other diseases such as diffuse large B-cell lymphoma or B-cell ALL. The following sections therefore provide a comprehensive analysis of the possible mechanisms of relapse as well as present potential strategies to overcome failure in this type of immunotherapy (Table 3). Table 4 details a summary of differences between BiTEs and CAR T cells. Finally, these sections briefly describe other difficulties, such as toxicity, manufacturing challenges, and economic burden, which could limit the widespread use of CAR T cell therapy.
6.1. Mechanisms or Relapse
Understanding the underlying mechanisms that determine or predict relapses is crucial in order to improve therapeutic approaches. Despite the high CR rates achieved with CD19-targeted CAR T cells (81–90% in B-cell ALL and 50% in B-cell non-Hodgkin lymphoma (NHL)), 10–20% of patients with B-cell ALL and 20–50% of patients with B-cell NHL will be refractory, while 30–60% of those with B-cell ALL who achieve CR and 20–30% of those with B-cell NHL who achieve CR will relapse [141,143,144,167,168]. With respect to BCMA-targeted CAR T cells in patients with MM, 3–40% of such patients will be refractory, while 15–50% of those patients who achieve CR will relapse during the first year of follow-up. Although the data are still very immature, there is evidence that a greater proportion of patients will relapse with longer follow-ups [24,47,48,50,51,52,53,54].
To date, two relapse patterns—antigen-positive and antigen-negative escapes—have been elucidated due to the high number and extensive follow-up period of patients who received CD19-targeted CAR T cells. Knowledge of such patterns may contribute to enhancing BCMA-targeted CAR T cell therapy in patients with MM.
6.1.1. Antigen-Positive Escape
Relapse of this nature most frequently occurs in CAR T cell immunotherapy. It is characterized by the maintenance of antigen expression on the tumor cell surface. Mechanisms found in such relapse underlie poor persistence and the low potency of CAR T cells, as well as mutations in survival or apoptosis pathways in tumor cells and the immunosuppressive tumor microenvironment (TME).
The use of non-human-derived scFv might contribute to CAR T cell inactivation due to the HLA-restricted T cell-mediated immune response and the presence of anti-CAR T cell antibodies [169]. Investigators Xu et al. found anti-CAR T cell antibodies in 6 patients with MM before or after relapse using a llama-derived bi-epitope-targeting BCMA CAR (LCAR-B38M). The presence of these antibodies was also associated with a notable reduction in the number of residual CAR T cells [51]. Different groups are therefore using fully human scFv to reduce antigenicity that would then increase persistence and improve efficacy [48,54,163,164,165,170,171,172]. This strategy may hold potential to re-challenge the targeted antigen and reinfuse the same or different CAR [171].
Intracellular signaling domains also play an important role in the persistence and efficacy of the product. Costimulatory CD28-based CARs enhance activation, proliferation, and cytotoxicity of T cells by promoting effector memory T cell differentiation and increasing aerobic glycolysis, albeit with reduced persistence. Meanwhile, 4-1BB-based CARs promote oxidative metabolism, bolster central memory T cell differentiation and improve T cell persistence [173,174,175,176]. Although the optimal costimulatory molecule to use in myeloma has yet to be elucidated, most products targeting BCMA are based on the 4-1BB moiety. Efforts are currently being undertaken to improve stimulatory signaling. Some examples include incorporating both CD28 and 4-1BB moieties into the CAR to maintain rapid activation kinetics and improved persistence, respectively [177,178]; mutating the activation motif in CD28 or encoding a single CD3ζ ITAM so as to hinder exhaustion and improve the persistence of T cells [179,180,181]; and, either incorporating new costimulatory domains (CD27 or ICOS) to enhance survival or differentiating CD4+ T cells towards a Th1/Th17 phenotype [173,182,183].
T cell fitness and subset composition have been recognized as markers of expansion and response [48,139,184,185]. CAR T cells manufactured from older donor T cells had worst transduction efficiency and impaired effector functions when compared with younger donor T cells, reflected by gene expression, secretory pattern, and transcription factor balance [186]. Quality of harvested T cells might also be compromised due to the disease itself and the type, number, and intensity of prior treatments [187,188,189,190]. Therefore, harvesting T cells during the first line of treatment and not in subsequent relapses may have clinical potential [191], as well as administering CAR T cell therapy as an earlier intervention in patients with MM. There are, in fact, clinical trials underway, evaluating BCMA-targeted CAR T cells in first-line treatment in high-risk patients with MM (KarMMa-4, NCT04196491) and in second-line treatment with high-risk factors (KarMMa-2, NCT03601078). With respect to T cell subpopulations, random compositions of T cell subsets were used in initial BCMA clinical trials; however, growing evidence supports the belief that tailored composition of T cell subsets could increase efficiency and persistency [192]. A higher CD4+/CD8+ T cell ratio in the leukapheresis product was associated with better expansion and response, and less differentiated or more naïve stem cell memory and central memory T cells were predictive biomarkers of expansion and clinical response [48,184]. That stated, orvacabtagene autoleucel (JCARH125, NCT03430011) [154,155] and FCARH143 product (NCT03338972) [164] are manufactured using a 1:1 ratio (CD4+/CD8+) before and after gene transfer, respectively, to homogenize the amount of T cells infused among patients and enhance crosstalk between CD4+ and CD8+ T cells [192]. Another strategy to enrich memory-like T cells includes blocking T cell differentiation signaling derived from constitutive CD3ζ and the phosphoinositide 3-kinase (PI3K), AKT and mTOR activation pathway [193]. bb21217 (NCT03274219) is a next-generation product of idecabtagene vicleucel that adds PI3K inhibitor bb007 during ex vivo culture with the expectation to enhance persistence and potency [161]. The addition of IL-7 and IL-15 to CAR T cell cultures enhances cytolytic and proliferative capacities and enriches naïve central memory T cells [194]. Novel CAR T cell product P-BCMA-101 (NCT03288493), conceived using the non-viral piggyBac® DNA modification system, favors enrichment of T memory stem cells, providing a higher therapeutic index [163].
Ligand-independent chronic activation or tonic signaling leads to detrimental effects on CAR functionality. It is characterized by different growth and phenotype patterns of CAR T cells, and is associated with accelerated differentiation, exhaustion, and impaired anti-tumor effects [195,196]. Adjustments can be made in the configuration of the CAR to avoid it, though. For example, centyrinTM are small monomeric, thermostable, and less immunogenic proteins based on a tenascin fibronectin type III sequence that have binding affinities similar to scFv and no signs of tonic signaling thus far [197]. A clinical trial with such technology is currently underway (P-BCMA-101, NCT03288493) [163]. Additionally, the hinge/spacer is crucial in preventing tonic signaling. Appropriate length of IgG-derived spacers and the replacement of the N-glycosylation site by FcγR-binding might recover CAR T cell functionality [198,199]. However, most CAR myeloma trials use CD8α-derived spacers to reduce the presence of tonic signaling [47,48,200]. Another useful option in third-generation CARs is to place the 4-1BB costimulatory domain distal to the cell membrane [173]. Advancements in genome editing tools using CRISPR/Cas9 have allowed for the targeted genomic knock-in of the CAR sequence to the T cell receptor α constant (TRAC) locus, resulting in a homogeneous expression of the CAR, the prevention of tonic signaling, a delay in effector T cell differentiation and exhaustion, and the enhancement of T cell potency [201].
T cell dysfunction is associated with tumor progression and relapse. T cell exhaustion refers to effector T cells with a reduced capacity to secrete cytokines and is characterized by an increased expression of inhibitory receptors (PD1, TIM3, LAG3, CTLA4, and TIGIT), reduced proliferative capacity, an altered transcriptional factor program (NFAT, IRF4, NR4A, and TOX), and a unique epigenetic landscape. T cell senescence is defined as the terminal differentiation state due to excessive cell replication, and is associated with cell cycle arrest and telomere shortening [202,203,204]. Brudno et al. observed a higher fraction of CAR T cells expressing senescence (KLRG1 and CD57) and exhaustion (PD-1) markers after CAR T cell infusion with CD28-based BCMA CAR in patients with MM [47]. CAR T cell anti-tumor activity may be boosted by the disruption of the immune checkpoint signaling pathway [205], by either combining the cell product with an immune checkpoint inhibitor antibody [205,206,207] or genetic modulations. Several examples of such include deleting PD-1 in CAR T cells with gene silencing techniques such as CRISPR/Cas9 or short hairpins RNAs [208,209]; engineering CARs that secrete PD-L1-targeted IgG antibodies or PD-1-targeted scFv [210,211]; transducing a CAR with a truncated PD-1 receptor that lacks intracellular domains [208]; and transducing a PD-1 switch receptor fused with an intracellular CD28 domain and thus modifying a dominant-negative inhibitory signal via an activating costimulatory signal [212,213]. However, data are conflicting with respect to impairment of anti-tumor function and proliferation activity of CAR T cells due to PD-1 silencing [214,215]. Further investigation is therefore warranted to elucidate the specific role that each immune checkpoint receptor plays and the optimal time for its inhibition in MM [216].
The presence of a soluble target antigen in the bloodstream could be an obstacle in CAR T cell therapy. A reduction in the density of the selected antigen on the tumor cell surface would not make it possible to reach the threshold that triggers the effector functions of CAR T cells and would hamper the scFv domain of the CAR [217]. For example, the soluble fragment of BCMA protein (sBCMA) can be shed into the bloodstream due to cleavage performed by the gamma-secretase (GS) [218]. With respect to MM, sBCMA may have a role in the disease pathophysiology, with increased levels of sBCMA being associated with a worse prognosis [219,220,221]. Preclinical data suggest that high concentrations of sBCMA may interfere with cytokine production and cytolytic capacity of BCMA-targeted CAR T cells [222]. Conversely, though, preclinical [154,160] and clinical [24,47,48,54] evidence highlight that BCMA-targeted CAR T cell activity is not compromised and sBCMA could be an adjunctive biomarker to assess response and progression [47,48]. These differences could be explained by the different epitopes to which the CARs are redirected, as some epitopes could be cryptic or not accessible in the soluble conformation of BCMA [217]. A possible, recently elucidated strategy to reduce the amount of sBCMA is the addition of a GS inhibitor (GSI), which efficiently blocks BCMA shedding [222]. The next section will provide further details about the possible utility of inhibiting the GS in anti-BCMA immunotherapy.
Malignant plasma cells and dysregulated BM TME interactions contribute to the pathogenesis, progression, and therapy resistance in myeloma [223]. However, knowledge concerning the role of immunosuppressive TME in relapse after the use of CAR T cells in patients with myeloma is minimal. Evidence obtained from patients with solid tumors indicates that objective response to CAR T cell therapy is infrequent and ephemeral due to cell stroma and immunosuppressive modulators, aberrant vascularization, hypoxia, and lack of nutrients [224]. In response, a wide set of approaches are being conceived to overcome these challenges. Some examples are as follows: (1) Increasing expression of chemokine receptors (CCR4, CSF-1R, and CCR2b) in CAR T cells to improve migration and anti-tumor activity to boosting T cell trafficking to tumors [225,226,227]; (2) targeting protease activation protein (FAP)-expressing stromal cells or secreting extracellular matrix-modifying enzymes (anti-GD2 CAR) to degrade heparan sulfate proteoglycans to infiltrate physical barriers. However, targeting fibroblast could develop considerable on-target, off-tumor toxicities [228,229,230]; (3) either blocking immune checkpoint pathways as aforementioned; (4) switching inhibitory signals (IL-4) present in the TME to pro-inflammatory signals (IL-2, IL-7 or IL-15) [231,232,233] to overcome T cell inhibitory signals, or disrupting the proapoptotic FAS signal pathway to impair the function as dominant-negative receptors [234]; (5) targeting immunosuppressive immune cells (regulatory T cells, tumor-associated macrophages, MDSC) with CAR T cells [224,235,236]; and (6) engineering bionic CAR T cells (armored CAR T cells or TRUCKs) to secrete stimulatory cytokines (IL-12, IL-15, IL-18) [237,238,239,240] that foster the effector activity of CAR T cells (third stimulatory signal) and propagate the anti-tumor immune response via recruitment of endogenous immune cells [241,242,243]. While the preclinical potential of such advanced engineering is great, clinical utility remains to be defined.
Conditioning regimens or lymphodepletion protocols based on chemotherapy prior to CAR T cell therapy aim to reduce tumor burden, eliminate immunosuppressive cells (Tregs and MDSC), remove homeostatic cytokine sinks (IL-2, IL-7, and IL-15), activate APC, downregulate indoleamine 2,3-dioxygenase in tumor cells, and enhance function, expansion, and persistence of CAR T cells [244,245,246,247]. Beneficial effects such as better clinical response and prognosis have been observed in B-cell malignancies and MM [48,248,249,250,251]. Although no current standard regimen has been established in MM clinical trials, the mainstream combination used is fludarabine plus cyclophosphamide. Further knowledge is needed to determine the most suitable regimen, dosing, and timing of drug administration.
6.1.2. Antigen-Negative Escape
This type of escape is characterized by the loss of or downregulation in the targeted antigen expression. It has been described with different targeted immunotherapies including CAR T cells [252]. Complete antigen loss may not be absolutely necessary to escape CAR T cells; however, decrease in the target antigen expression may suffice. Some evidence suggests that a minimum and individual threshold of antigen expression is needed for CAR T cell activation [252,253]. Apparently, within the MM setting, the antigen-negative or downregulation of the antigen is not the primary mechanism of escape, although some clinical trials using BCMA-targeted CAR T cells have reported it [47,48,164,254]. Investigators Brudno et al. described the case of one patient who lost BCMA expression at the time of progression. BM analysis showed the presence of a mixed cell population, with some maintaining BCMA expression and others losing it [47]. Likewise, Green et al. reported the case of one patient whose tumor biopsy at relapse revealed both a BCMAneg myeloma cell population and 70% reduction in intensity of BCMA expression in remaining myeloma cells [164]. A separate study by Cohen et al. also showed similar findings, reporting a decrease in intensity of BCMA expression in 4 of 9 patients who did not achieve an objective response to the BCMA-targeted CAR [48]. Notably, after CAR T cell infusion, intensity of BCMA expression was minimal in residual myeloma cells; however, in most patients at progression, expression returned to baseline levels [48]. Martin et al. presented three cases of BCMA antigen loss after idecabtagene vicleucel; a biallelic deletion of chromosome 16p encompassing the BCMA locus was confirmed in one case [254,255].
Evidence obtained on CAR T cells targeting the CD19 antigen has elucidated some of the plausible mechanisms related to this subtype of relapse. These mechanisms include: (1) Immune selection pressure: Pre-existing target antigen-negative subclones prior to CAR T cell therapy may transform to dominant clones after selective stress generated by immune-targeted therapy [256,257]. This type of escape is highly probable in myeloma due to intratumor heterogeneity and clonal evolution [258]; (2) gene mutations: Frameshift and missense mutations have been described with the subsequent loss of expression of the targeted antigen. Furthermore, alterations have been identified in the splicing factor that could cause protein isoforms contributing to CAR T cell escape [259,260,261]; (3) lineage switching: Immunotherapy could induce conversion or reprogramming to a different leukemic cell lineage [262,263]; (4) trogocytosis and cooperative killing: Described only in in vitro and xenograft models, this mechanism of escape is characterized by the transfer of the target antigen to CAR T cells during the immune synapse. Such transfer subsequently decreases density of the antigen expressed on the tumor cell surface and triggers fratricide among CAR T cells, resulting in ensuing exhaustion [264,265]; and (5) antigen masking: This mechanism occurs when the CAR gene is unintentionally transduced into a leukemic B-cell during product manufacturing. CAR-transduced blasts will bind to the target antigen expressed on their own cell surface and result in the masking of and resistance to CAR T cells [266]. Overall, though, there may be some strategies to overcome this subtype of escape mechanism.
One of the most relevant and complex factors in determining CAR T cell therapy success is the identification and selection of the most suitable tumor antigens. Although no mainstream definition exists, the ideal tumor antigen should fulfill the following requirements: Have high, homogeneous expression on tumor cell surface; be involved in disease pathophysiology and maintain expression at different stages of the disease; be resistant to therapeutic pressure exerted by immunotherapy to avoid the downregulation or complete loss of the antigen; have no expression in normal tissue to avoid on-target, off-tumor toxicities; and if released into the bloodstream, should be minimal [267,268,269,270]. Potential molecules are currently being evaluated in clinical (e.g., BCMA, SLAMF7, CD19, CD38, CD44v6, GPRC5D) and preclinical (e.g., CD229, integrin β7) settings, and some have shown encouraging outcomes. Providing further details of these evaluations goes beyond the scope of this manuscript; however, comprehensive reviews that address this topic are available [152,153,271]. In all, finding the ideal antigen in myeloma is challenging and grand endeavors are being undertaken to find the balance between safety and effectiveness.
Targeting multiple tumor antigens may counteract antigen escape. Thus far, different approaches have been implemented including sequential treatment or co-administration of different single-target CAR T cell products; co-expression of two different CAR molecules on T cell surfaces using a single bicistronic vector (dual CARs); and expression of two scFvs in extracellular domains in “single-stalk” intracellular module (tandem CARs) (Figure 2) [272,273]. In the preclinical setting, different combinations have been shown to be useful (CD19/BCMA, BCMA/SLAMF7, and BCMA/GPRC5D [215,274,275,276]), while in the clinical setting, preliminary and encouraging results targeting BCMA/CD38, CD19/BCMA, and BCMA/TACI have been presented [277,278,279,280]. However, due to the selection of two antigens, special attention should be given to a potential increase in toxicity. Similarly, at the manufacturing level, bicistronic CARs require codon optimization if DNA recombination is not to occur, and engineering design of tandem CARs must be optimal if adequate antigen recognition and T cell activation are to be achieved [272].
Another plausible method to maintain or upregulate surface density of the target antigen is via in combination with different drugs. CD38 expression can be modulated with ATRA [78] and histone deacetylase inhibitors panobinostat and ricolinostat [80,81] to improve immunotherapy efficacy. BCMA expression could be enhanced with ATRA as an epigenetic modulator and improve CAR T cell efficacy [281]. As mentioned prior, the use of GSI in the preclinical setting reduces the shedding of sBCMA, leading to an increase in BCMA surface expression and improvement in BCMA-targeted CAR T cell therapy [222]. Currently, these findings are being verified in a clinical setting with crenigacestat as the inhibitor of GS. Outcomes of this clinical trial may prove promising, as initial results have been encouraging, especially in patients who relapsed after BCMA-targeted therapies [282].
Myeloma stem cells contribute to the high rates of refractoriness and relapse of this disease. These cells are able to remain in a quiescent state, undergo self-renewal and hold differentiation potential, be resistant to cell death mechanisms, and escape from immunosurveillance. Indeed, due to these characteristics, myeloma stem cells could be a target for CAR T cell therapy [283]. It has been suggested that these less differentiated, myeloma subclones do not express CD138, but they do express other antigens like CD19 and CD229 [284,285,286,287]. Designing CARs targeting these molecules may confer benefits [288,289], even though positive cell fraction is extremely scarce [290]. Nonetheless, while this type of treatment is interesting in theory, further, more comprehensive studies are warranted.
6.2. CAR T Cell-Related Toxicities
This novel therapy revealed new and potentially life-threatening toxicities that could limit its widespread use. The most prominent toxicities are CRS, ICANS and on-target, off-tumor toxicity [291,292]. CRS is characterized by the release of inflammatory cytokines associated with a wide range of symptoms such as fever, hypoxia, hypotension, and organ dysfunction. Treatment may include symptom support, corticosteroids, and IL-6 receptor antagonist tocilizumab [292,293,294,295]. ICANS is associated with the impairment of the blood–brain barrier and a subsequent elevation of cytokines in cerebrospinal fluid. Symptomatology varies from aphasia and confusion to seizures and cerebral edema. Clinical management may include appropriate supportive treatment and corticosteroids; the use of tocilizumab is justified only when CRS is co-existing [292,294,295,296,297]. Great efforts have been made to establish appropriate grading methods and treatment guidelines to improve the diagnosis and management of these complications [298,299,300]. With respect to on-target, off-tumor toxicity, this occurs due to recognition by CAR T cells of a targeted antigen expressed in non-tumor cells. A classic example of such toxicity is the development of B-cell aplasia and hypogammaglobulinemia during the use of CD19-targeted CAR T cells. However, other examples with potentially devastating outcomes have been reported [294,295,296,297,298,299,300,301,302,303].
In an MM setting using BCMA-targeted CAR T cells, CRS and ICANS rates are 60–100% and 2–42%, respectively, with the majority being grade ≤ 2 (59–100% and 81–100%, respectively). Tocilizumab and corticosteroids were required in 28–79% and 15–52% of patients, respectively [24,47,48,53,54,304]. Such data could translate into less severity when compared to CD-19 CARs.
As an in-depth review of different strategies mitigating toxicities has been published elsewhere [272,273,295], only some of the most prominent alternatives will be mentioned. The risk of CRS and ICANS is related to CAR T cell activation kinetics, the dose of CAR T cells infused, and baseline factors or comorbidities. Activation of CAR T cells are modulated by tumor burden, antigen expression, and the construct itself (affinity of scFv and costimulatory domains) [273]. The risk of toxicity may be attenuated by either reducing the number of CAR T cells infused or dividing the doses on different days. Prompt recognition of severe CRS and ICANS with the use of predictive biomarkers may also help. With respect to the construct, tailored modifications can be designed, including (1) optimizing the costimulatory domain (CD28 or 4-1BB), which depends on surface density of target antigen and the degree of expansion or persistence of CAR T cells [294,305]; and (2) engineering CARs with “suicide genes” using an apoptosis-triggering fusion protein comprising caspase 9 (iCasp9) [306] or “OFF-switches” like truncated epidermal growth factor receptor (EGFRt), which can be targeted with cetuximab [307] to dismiss CAR T cells. Dasatinib may also work as an on/off switch for CAR T cells by ablating the lymphocyte-specific protein tyrosine kinase (LCK)-signaling pathway [308]. Strategies designed to limit on-target, off-tumor toxicities include the following: Affinity tuning of the scFv to discern between normal and tumor cells per antigen density level [309,310,311]; “AND” logic-gate CARs that require simultaneous presence of two-cell surface antigens to activate the T cell [312]; “ON-switch” CARs, which need a small, heterodimerizing molecule to bind the dissociated antigen binding domain with the signaling domain for activation [313]; “SPLIT-CARs”, the co-expression of two different CARs that recognize different antigens, in which one encloses the activation domain (CD3ζ) and the other the co-stimulation domain. Both antigens must be present for full T cell activation [314,315,316]; and inhibitory CARs (iCARs) bear an inhibitory signaling domain of immune-checkpoint proteins (CTLA-4 or PD-1) to inhibit T cell activation after recognition of the target antigen expressed in non-tumoral cells [317]. It remains to be determined which of all of these pre-clinical strategies may be useful in the clinical setting. Furthermore, the therapeutic/toxic window of each CAR construct is different; appropriate interventions may therefore differ and should be established for every CAR T cell [252,273].
6.3. Product Manufacturing, Access and Economic Challenges
Manufacturing CAR T cells from autologous T cells has certain limitations, as administration of CAR T cells may not be feasible in some patients. Some primary reasons for these limitations include difficulty in harvesting enough T cells from lymphopenic patients due to the disease itself and previous treatments; disease progression during manufacturing time; and failure in CAR T cell production due to T cell dysfunction [273,318]. To circumvent these hurdles, engineering CAR T cells from healthy allogeneic or “third-party” T cell donors has been proposed. This “off-the-shelf” strategy has many potential advantages, including cryopreserved batches to avoid treatment delays, less T cell quality issues, the possibility to redose and combine different target CAR T cells, possible decrease in manufacturing costs, and a greater number of patients possibly benefiting from such therapy [273,318]. Nevertheless, this approach is associated with two major concerns: (1) Graft versus host disease (GvHD) development and (2) rejection and removal of allogeneic T cells by the host immune system. To reduce the risk of GvHD, different therapeutical approaches are being used such as allogeneic donor-derived T cells in stem cell transplant recipients [319,320], non-αβ T cells (NK cells or umbilical cord blood NK cells) [321,322], and gene-editing tools (zinc-finger, TALEN, and CRISPR/Cas9 technologies) to disrupt the endogenous TCR of CAR T cells [201,323,324]. To avoid allogeneic T cell rejection, it has been suggested to do the following: (1) Creating a T cell bank that matches the majority of HLA-subtypes [325]; (2) elongating the duration of lymphopenia by disrupting TRAC and CD52 locus of CAR T cells to result in alemtuzumab-resistant CAR T cells [324]; (3) and disrupting HLA-A or β2-microglobulin genes on allogeneic CAR T cells [326]. Different promising BCMA (ALLO-715, ALLO-647, and P-BCMA-ALLO1) and SLAMF7 (UCARTCS1)-directed products are being evaluated in clinical trials [327,328,329]. Early data presented in the last ASH meeting showed that ALLO-715 and ALLO-647 have a manageable safety profile and clinical activity [330]. However, more robust data and longer follow-up are needed to determine the true potential and the target population of this strategy.
These “living drugs” are different from other oncological drugs, as various infrastructures, workflows, processes, and regulatory requirements are required to guarantee product quality and manufacturing time (“vein-to-vein time”) [331]. The management of these patients require cooperation among multiple stakeholders and specialized teams with appropriate skill sets, standard operating procedures, and laborious site setup processes (extensive preparation and certifications) [332]. Another critical point in large-scale application is therapy costs. The Institute for Clinical and Economic Research (ICER) performed a cost-effectiveness analysis of previously authorized CAR T cells which are below the threshold to be considered cost effective ($150,000 per quality-adjusted life-year (QALY) gained) [333]. However, this analysis is not yet available for patients with myeloma. Long-term effectiveness will be a key outcome to possibly improve the cost-effectiveness ratio [334]. Other strategies that could reduce costs include non-viral gene delivery with “Sleeping Beauty” or “PiggyBac” transposon/transposase systems—which are more affordable than the use of viral vectors [335,336]—and the creation of community CAR T cell therapy centers and promotion of the outpatient setting to increase the number of patients who benefit from these treatments and improve hospital finances. A shift from centralized to decentralized manufacturing, namely “bedside manufacturing” could increase capacity, reduce costs, and lessen vein-to-vein time [332].
CAR T cell therapy has demonstrated excellent efficacy in clinical trials and some products are expected to be approved during this year. However, this strategy still has only a modest PFS. Therefore, in the near future, new strategies will be necessary to optimize these products.
7. Vaccines
Given the contribution of the immunological profile of MM pathogenesis, a vaccine may be able to stimulate a clinical response achieved with standard therapy. There are many mechanisms involved and clinical trials ongoing. However, this review will only present vaccines with published results. For example, patients treated with vaccines based on dendritic/patient-derived myeloma cells exhibited expansion of CD4+ and CD8+ lymphocytes; 11 of 16 patients achieved stable disease [55]. Furthermore, the same group reported that vaccination after ASCT resulted in expansion of myeloma-specific T cells and deeper minimal residual disease [57]. Other vaccines based on antigens overexpressed in myeloma, such as survivin and human telomerase reverse transcriptase (hTERT), which were transferred after ASCT, have led to higher cellular and humoral reconstitution as well as increased antitumor immunity and improved event-free survival [56]. MAGE-A3 (melanoma-associated antigen 3) is a protein detected in 50% of myeloma cells, becoming more frequent in the advanced stage of the disease. Two studies have therefore used MAGE-A3 as a peptide to conceive a vaccine. Both studies reported high specific T cell immunity after ASCT [58,60]. For smoldering MM, a vaccine targeting three myeloma peptides (XBP1, CD138, and CS1) was safe and well tolerated, achieving acceptable immune response alone and in combination with lenalidomide [59]. In summary, vaccines appear as a safe alternative to stimulate T cell response, possibly increasing or deepening such response after a transplant. A combination of vaccines with other strategies such as anti-PD1 antibodies may improve immune response [337]. The lack of longer follow-up trials to evaluate its real clinical impact and a high number of patients involved makes vaccines an ongoing field of interest, with still so many questions to answer within the coming years.
8. Personal Perspective
Immunotherapy is revolutionizing cancer treatment, uncovering new pathways to harness anti-tumor immune functions. Although novel immunotherapy approaches have proven effective in patients with myeloma, there are still concerns and hurdles that preclude their use. A major challenge is its unpredictable efficacy, namely whether such therapy will prove useful in only a minority of patients [338,339]. Such unpredictability could be due to interpersonal variability, tumor heterogeneity, and clonal evolution [216,258,340]. The reductionist approach that targets single molecular pathways or mutations might be upgraded with the use of combinatorial strategies targeting different pathways and possible synergistic effects [341]. Another way to improve the response would be to implement immunotherapy during the early stages of the disease, as restoration of robust anti-tumor activity would be easier than in advanced or heavily pretreated stages [339]. Additionally, the identification of potential biomarkers remains an unmet need in immunotherapy. Prognostic biomarkers are useful in predicting the likelihood of relapse and survival, irrespective of therapy, while predictive biomarkers foretell outcomes with a specific drug [342]. Molecular profiling technologies have allowed such identification. However, none of these biomarkers have been robustly validated in MM immunotherapy. It may be that, in the future, receptors and ligands of immune checkpoints, sBCMA levels, or the preinfused T cell subset will foresee accurate outcomes. Current medicine is evolving towards precision medicine, mainly in oncology, to personalize and refine health care in all its aspects. This includes medical decisions, diagnostic testing, and treatment selection, as well as elucidating prognosis in a subgroup of patients based on the molecular and cellular features of a tumor, overall environment, and individual lifestyle [343,344]. Several studies have already demonstrated the feasibility of adopting innovative approaches in precision oncology [345,346,347]. However, the percentage of patients who are eligible and will benefit for targeted-driven therapy remains a minority [348]. Different challenges will therefore have to be addressed. For example, as delayed responses could be observed with immunotherapy, traditional endpoints will need to be optimized for chemotherapy-based treatments [349]. Additionally, as progress is being made with biomarkers in precision oncology, next-generation clinical trials or master protocols (basket and umbrella trials) will be needed in order to evaluate specific molecular lesions in small cohorts of patients [350]. Finally, economic sustainability in immunotherapy has become a prominent concern. Immunotherapy agents are expensive, placing a great financial strain on the health system and, in some countries, the patient. Proper cost-effectiveness, cost-benefit, and QALY analyses will be required to resolve this matter of importance [339]. To make it more affordable, outcome-based reimbursement or staged payment models have been developed for gene therapies; however, if immunotherapy is to prove its legitimate value in terms of benefits and grow in accessibility, further real-world evidence and longer follow-ups with more patients will be necessary to leverage the therapeutic approach powerfully [351]. In the following years, close cooperation and coordination among different stakeholders will be necessary to preserve patient access and health care system sustainability.
Immunotherapy has generated a paradigm shift in the treatment of MM. MoA have shown minimal toxicity and outstanding efficacy, so it is expected that anti-CD38 MoA will be used in all standard front-line therapy for younger patients with NDMM and in non-transplant eligible patients. This will generate the need to seek specific therapeutic strategies in R/R patients, since the outcomes of patients with MM R/R to CD38-targeted MoA therapy are poor [352]. There is eagerness that novel immunotherapeutic strategies will provide clinical benefit in this challenging population. However, many BCMA-specific products and approaches are currently being explored in the R/R setting. ADC, Bs MoA, and CAR T cells will be a turning point in the therapeutic arsenal against MM. It will be necessary to analyze the advantages and disadvantages of each product and determine the best scenario to use them. Furthermore, there are currently no direct comparisons to determine which one is superior. Both ADC and Bs MoA are easy to use and available immediately; they do not require specialized centers for their administration, nor apheresis. ADC does not need a predefined number of T cells for their production. CAR T cell requires a few weeks to manufacture and specialized medical centers for administration, narrowing the number of patients who can receive it. Elderly and heavily pre-treated patients with lymphopenia could be a challenge in the manufacture of CAR T cells. T cell fitness could be of great importance for both Bs MoA and CAR T cell function. Regarding toxicities, both Bs MoA and CAR T cell products can develop CRS and ICANS, while ADC can develop keratopathy in the majority of patients. Respecting affordability, ADC and Bs MoA are less expensive when compared to CAR T cell products. So far, it remains unproven which BCMA-targeted strategy provides a better therapeutic index. Real-world data and longer follow-ups will provide new insights to clarify the proper way to integrate these therapies into current treatment algorithms.
9. Conclusions
In recent years, immunotherapy has positioned itself as a cutting-edge therapeutic strategy. It has become a game changer for patients with cancer. In patients with multiple myeloma, immunotherapy remains in a nascent stage; however, its therapeutic potential has already begun to be elucidated. Different products are expected to be approved in the short term. Nonetheless, this type of therapy faces different challenges, from efficacy and safety to manufacturing and economic affordability. To surmount these hurdles, more comprehensive knowledge is necessary to fully understand the mechanisms involved in relapse, boost “bench-to-bedside” research, enhance cooperation among various stakeholders, optimize manufacturing and scalability, and foster sustainability. It is our hope that this novel treatment approach could be used in early stages of the disease and become widely available. Restoring immune balance may just lead to long-lasting remissions long awaited.
Author Contributions
L.G.R.-L., A.O.-C., and D.F.M. wrote and reviewed the manuscript. C.F.d.L. and J.B. reviewed the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
L.G.R.-L. as a BITRECS fellow has received funding from the European Union’s Horizon 2020 research and Innovation Programme under the Marie Sklodowska-Curie grant agreement No 754550 and from “La Caixa” Foundation. This work has been supported in part by grants from the Instituto de Salud Carlos III, Spanish Ministry of Health (FIS PI19/00669 and ICI19/00025), Fondo Europeo de Desarrollo Regional (FEDER), 2017SGR00792 (AGAUR; Generalitat de Catalunya).
Acknowledgments
The authors would like to thank Josep Solanes for his support in the graphic edition. We would like to thank Anthony Armenta for providing medical editing assistance.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Cowan, A.J.; Allen, C.; Barac, A.; Basaleem, H.; Bensenor, I.; Curado, M.P.; Foreman, K.; Gupta, R.; Harvey, J.; Hosgood, H.D.; et al. Global Burden of Multiple Myeloma: A Systematic Analysis for the Global Burden of Disease Study 2016. JAMA Oncol. 2018, 4, 1221–1227. [Google Scholar] [CrossRef] [Green Version]
- Sant, M.; Allemani, C.; Tereanu, C.; De Angelis, R.; Capocaccia, R.; Visser, O.; Marcos-Gragera, R.; Maynadié, M.; Simonetti, A.; Lutz, J.M.; et al. Incidence of hematologic malignancies in Europe by morphologic subtype: Results of the HAEMACARE project. Blood 2010, 116, 3724–3734. [Google Scholar] [CrossRef]
- Kumar, S.K.; Rajkumar, S.V. The multiple myelomas - current concepts in cytogenetic classification and therapy. Nat. Rev. Clin. Oncol. 2018, 15, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Lakshman, A.; Paul, S.; Rajkumar, S.V.; Ketterling, R.P.; Greipp, P.T.; Dispenzieri, A.; Gertz, M.A.; Buadi, F.K.; Lacy, M.Q.; Dingli, D.; et al. Prognostic significance of interphase FISH in monoclonal gammopathy of undetermined significance. Leukemia 2018, 32, 1811–1815. [Google Scholar] [CrossRef] [PubMed]
- Merz, M.; Hielscher, T.; Hoffmann, K.; Seckinger, A.; Hose, D.; Raab, M.S.; Hillengass, J.; Jauch, A.; Goldschmidt, H. Cytogenetic abnormalities in monoclonal gammopathy of undetermined significance. Leukemia 2018, 32, 2717–2719. [Google Scholar] [CrossRef]
- Mikulasova, A.; Wardell, C.P.; Murison, A.; Boyle, E.M.; Jackson, G.H.; Smetana, J.; Kufova, Z.; Pour, L.; Sandecka, V.; Almasi, M.; et al. The spectrum of somatic mutations in monoclonal gammopathy of undetermined significance indicates a less complex genomic landscape than that in multiple myeloma. Haematologica 2017, 102, 1617–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, B.; Zhang, X.G.; Lu, Z.Y.; Bataille, R. Interleukin-6 in human multiple myeloma. Blood 1995, 85, 863–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunn, W.G.; Conley, A.; Deininger, L.; Olson, S.D.; Prockop, D.J.; Gregory, C.A. A crosstalk between myeloma cells and marrow stromal cells stimulates production of DKK1 and interleukin-6: A potential role in the development of lytic bone disease and tumor progression in multiple myeloma. Stem Cells 2006, 24, 986–991. [Google Scholar] [CrossRef] [PubMed]
- Hengeveld, P.J.; Kersten, M.J. B-cell activating factor in the pathophysiology of multiple myeloma: A target for therapy? Blood Cancer J. 2015, 5, e282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprynski, A.C.; Hose, D.; Caillot, L.; Réme, T.; Shaughnessy, J.D.; Barlogie, B.; Seckinger, A.; Moreaux, J.; Hundemer, M.; Jourdan, M.; et al. The role of IGF-1 as a major growth factor for myeloma cell lines and the prognostic relevance of the expression of its receptor. Blood 2009, 113, 4614–4626. [Google Scholar] [CrossRef]
- Mitsiades, C.S.; McMillin, D.W.; Klippel, S.; Hideshima, T.; Chauhan, D.; Richardson, P.G.; Munshi, N.C.; Anderson, K.C. The role of the bone marrow microenvironment in the pathophysiology of myeloma and its significance in the development of more effective therapies. Hematol. Oncol. Clin. N. Am. 2007, 21, 1007–1034. [Google Scholar] [CrossRef] [PubMed]
- Asaoku, H.; Kawano, M.; Iwato, K.; Tanabe, O.; Tanaka, H.; Hirano, T.; Kishimoto, T.; Kuramoto, A. Decrease in BSF-2/IL-6 response in advanced cases of multiple myeloma. Blood 1988, 72, 429–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Görgün, G.T.; Whitehill, G.; Anderson, J.L.; Hideshima, T.; Maguire, C.; Laubach, J.; Raje, N.; Munshi, N.C.; Richardson, P.G.; Anderson, K.C. Tumor-promoting immune-suppressive myeloid-derived suppressor cells in the multiple myeloma microenvironment in humans. Blood 2013, 121, 2975–2987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, D.M.; Bakan, C.E.; Mishra, A.; Hofmeister, C.C.; Efebera, Y.; Becknell, B.; Baiocchi, R.A.; Zhang, J.; Yu, J.; Smith, M.K.; et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: A therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood 2010, 116, 2286–2294. [Google Scholar] [CrossRef]
- Minnie, S.A.; Kuns, R.D.; Gartlan, K.H.; Zhang, P.; Wilkinson, A.N.; Samson, L.; Guillerey, C.; Engwerda, C.; MacDonald, K.; Smyth, M.J.; et al. Myeloma escape after stem cell transplantation is a consequence of T-cell exhaustion and is prevented by TIGIT blockade. Blood 2018, 132, 1675–1688. [Google Scholar] [CrossRef]
- Lozano, E.; Mena, M.P.; Díaz, T.; Martin-Antonio, B.; Leon, S.; Rodríguez-Lobato, L.G.; Oliver-Caldés, A.; Cibeira, M.T.; Bladé, J.; Prat, A.; et al. Nectin-2 Expression on Malignant Plasma Cells Is Associated with Better Response to TIGIT Blockade in Multiple Myeloma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 4688–4698. [Google Scholar] [CrossRef]
- Prabhala, R.H.; Pelluru, D.; Fulciniti, M.; Prabhala, H.K.; Nanjappa, P.; Song, W.; Pai, C.; Amin, S.; Tai, Y.T.; Richardson, P.G.; et al. Elevated IL-17 produced by TH17 cells promotes myeloma cell growth and inhibits immune function in multiple myeloma. Blood 2010, 115, 5385–5392. [Google Scholar] [CrossRef]
- Richardson, P.G.; Weller, E.; Lonial, S.; Jakubowiak, A.J.; Jagannath, S.; Raje, N.S.; Avigan, D.E.; Xie, W.; Ghobrial, I.M.; Schlossman, R.L.; et al. Lenalidomide, bortezomib, and dexamethasone combination therapy in patients with newly diagnosed multiple myeloma. Blood 2010, 116, 679–686. [Google Scholar] [CrossRef] [Green Version]
- Rosiñol, L.; Oriol, A.; Rios, R.; Sureda, A.; Blanchard, M.J.; Hernández, M.T.; Martínez-Martínez, R.; Moraleda, J.M.; Jarque, I.; Bargay, J.; et al. Bortezomib, lenalidomide, and dexamethasone as induction therapy prior to autologous transplant in multiple myeloma. Blood 2019, 134, 1337–1345. [Google Scholar] [CrossRef] [Green Version]
- Leblay, N.; Maity, R.; Hasan, F.; Neri, P. Deregulation of Adaptive T Cell Immunity in Multiple Myeloma: Insights Into Mechanisms and Therapeutic Opportunities. Front. Oncol. 2020, 10, 636. [Google Scholar] [CrossRef] [PubMed]
- Garfall, A.L.; Stadtmauer, E.A. Cellular and vaccine immunotherapy for multiple myeloma. Hematol. Am. Soc. Hematol. Educ. Program. 2016, 2016, 521–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greil, C.; Engelhardt, M.; Ihorst, G.; Schoeller, K.; Bertz, H.; Marks, R.; Zeiser, R.; Duyster, J.; Einsele, H.; Finke, J.; et al. Allogeneic transplantation of multiple myeloma patients may allow long-term survival in carefully selected patients with acceptable toxicity and preserved quality of life. Haematologica 2019, 104, 370–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef]
- Lonial, S.; Weiss, B.M.; Usmani, S.Z.; Singhal, S.; Chari, A.; Bahlis, N.J.; Belch, A.; Krishnan, A.; Vescio, R.A.; Mateos, M.V.; et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): An open-label, randomised, phase 2 trial. Lancet 2016, 387, 1551–1560. [Google Scholar] [CrossRef]
- Usmani, S.Z.; Weiss, B.M.; Plesner, T.; Bahlis, N.J.; Belch, A.; Lonial, S.; Lokhorst, H.M.; Voorhees, P.M.; Richardson, P.G.; Chari, A.; et al. Clinical efficacy of daratumumab monotherapy in patients with heavily pretreated relapsed or refractory multiple myeloma. Blood 2016, 128, 37–44. [Google Scholar] [CrossRef]
- Plesner, T.; Arkenau, H.T.; Lokhorst, H.M.; Gimsing, P.; Krejcik, J.; Lemech, C.; Minnema, M.; Lassen, U.N.; Laubach, J.P.; Ahmadi, T.; et al. Safety and Efficacy of Daratumumab with Lenalidomide and Dexamethasone in Relapsed or Relapsed, Refractory Multiple Myeloma. Blood 2014, 124, 84. [Google Scholar] [CrossRef]
- Plesner, T.; Arkenau, H.T.; Gimsing, P.; Krejcik, J.; Lemech, C.; Minnema, M.C.; Lassen, U.; Laubach, J.P.; Palumbo, A.; Lisby, S.; et al. Daratumumab in Combination with Lenalidomide and Dexamethasone in Patients with Relapsed or Relapsed and Refractory Multiple Myeloma: Updated Results of a Phase 1/2 Study (GEN503). Blood 2015, 126, 507. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Oriol, A.; Nahi, H.; San-Miguel, J.; Bahlis, N.J.; Usmani, S.Z.; Rabin, N.; Orlowski, R.Z.; Komarnicki, M.; Suzuki, K.; et al. Daratumumab, Lenalidomide, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 1319–1331. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, A.; Chanan-Khan, A.; Weisel, K.; Nooka, A.K.; Masszi, T.; Beksac, M.; Spicka, I.; Hungria, V.; Munder, M.; Mateos, M.V.; et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 754–766. [Google Scholar] [CrossRef]
- Weisel, K.C.; San Miguel, J.; Cook, G.; Leiba, M.; Suzuki, K.; Kumar, S.; Cavo, M.; Avet-Loiseau, H.; Quach, H.; Hungria, V.; et al. Efficacy of daratumumab in combination with lenalidomide plus dexamethasone (DRd) or bortezomib plus dexamethasone (DVd) in relapsed or refractory multiple myeloma (RRMM) based on cytogenetic risk status. J. Clin. Oncol. 2017, 35 (Suppl. 15), 8006. [Google Scholar] [CrossRef]
- Zonder, J.A.; Mohrbacher, A.F.; Singhal, S.; Van Rhee, F.; Bensinger, W.I.; Ding, H.; Fry, J.; Afar, D.E.; Singhal, A.K. A phase 1, multicenter, open-label, dose escalation study of elotuzumab in patients with advanced multiple myeloma. Blood 2012, 120, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Jakubowiak, A.; Offidani, M.; Pégourie, B.; De La Rubia, J.; Garderet, L.; Laribi, K.; Bosi, A.; Marasca, R.; Laubach, J.; Mohrbacher, A.; et al. Randomized phase 2 study: Elotuzumab plus bortezomib/dexamethasone vs bortezomib/dexamethasone for relapsed/refractory MM. Blood 2016, 127, 2833–2840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimopoulos, M.A.; Lonial, S.; White, D.; Moreau, P.; Palumbo, A.; San-Miguel, J.; Shpilberg, O.; Anderson, K.; Grosicki, S.; Spicka, I.; et al. Elotuzumab plus lenalidomide/dexamethasone for relapsed or refractory multiple myeloma: ELOQUENT-2 follow-up and post-hoc analyses on progression-free survival and tumour growth. Br. J. Haematol. 2017, 178, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Dytfeld, D.; Grosicki, S.; Moreau, P.; Takezako, N.; Hori, M.; Leleu, X.; LeBlanc, R.; Suzuki, K.; Raab, M.S.; et al. Elotuzumab plus Pomalidomide and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2018, 379, 1811–1822. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.T.; Mayes, P.A.; Acharya, C.; Zhong, M.Y.; Cea, M.; Cagnetta, A.; Craigen, J.; Yates, J.; Gliddon, L.; Fieles, W.; et al. Novel anti-B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood 2014, 123, 3128–3138. [Google Scholar] [CrossRef]
- Trudel, S.; Lendvai, N.; Popat, R.; Voorhees, P.M.; Reeves, B.; Libby, E.N.; Richardson, P.G.; Hoos, A.; Gupta, I.; Bragulat, V.; et al. Antibody-drug conjugate, GSK2857916, in relapsed/refractory multiple myeloma: An update on safety and efficacy from dose expansion phase I study. Blood Cancer J. 2019, 9, 37. [Google Scholar] [CrossRef] [Green Version]
- Schönfeld, K.; Zuber, C.; Pinkas, J.; Häder, T.; Bernöster, K.; Uherek, C. Indatuximab ravtansine (BT062) combination treatment in multiple myeloma: Pre-clinical studies. J. Hematol. Oncol. 2017, 10, 13. [Google Scholar] [CrossRef] [Green Version]
- Jagannath, S.; Heffner Jr, L.T.; Ailawadhi, S.; Munshi, N.C.; Zimmerman, T.M.; Rosenblatt, J.; Lonial, S.; Chanan-Khan, A.; Ruehle, M.; Rharbaoui, F.; et al. Indatuximab Ravtansine (BT062) Monotherapy in Patients With Relapsed and/or Refractory Multiple Myeloma. Clin. Lymphoma. Myeloma. Leuk. 2019, 19, 372–380. [Google Scholar] [CrossRef]
- Ailawadhi, S.; Kelly, K.R.; Vescio, R.A.; Jagannath, S.; Wolf, J.; Gharibo, M.; Sher, T.; Bojanini, L.; Kirby, M.; Chanan-Khan, A. A Phase I Study to Assess the Safety and Pharmacokinetics of Single-agent Lorvotuzumab Mertansine (IMGN901) in Patients with Relapsed and/or Refractory CD-56-positive Multiple Myeloma. Clin. Lymphoma. Myeloma. Leuk. 2019, 19, 29–34. [Google Scholar] [CrossRef]
- Kaufman, J.L.; Niesvizky, R.; Stadtmauer, E.A.; Chanan-Khan, A.; Siegel, D.; Horne, H.; Wegener, W.A.; Goldenberg, D.M. Phase I, multicentre, dose-escalation trial of monotherapy with milatuzumab (humanized anti-CD74 monoclonal antibody) in relapsed or refractory multiple myeloma. Br. J. Haematol. 2013, 163, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Hipp, S.; Tai, Y.T.; Blanset, D.; Deegen, P.; Wahl, J.; Thomas, O.; Rattel, B.; Adam, P.J.; Anderson, K.C.; Friedrich, M. A novel BCMA/CD3 bispecific T-cell engager for the treatment of multiple myeloma induces selective lysis in vitro and in vivo. Leukemia 2017, 31, 1743–1751. [Google Scholar] [CrossRef] [PubMed]
- Topp, M.S.; Duell, J.; Zugmaier, G.; Attal, M.; Moreau, P.; Langer, C.; Krönke, J.; Facon, T.; Salnikov, A.V.; Lesley, R.; et al. Anti-B-Cell Maturation Antigen BiTE Molecule AMG 420 Induces Responses in Multiple Myeloma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Usmani, S.Z.; Mateos, M.V.; Nahi, H.; Krishnan, A.Y.; van de Donk, N.W.; San Miguel, J.; Oriol, A.; Rosiñol, L.; Chari, A.; Adams, H.; et al. Phase I study of teclistamab, a humanized B-cell maturation antigen (BCMA) x CD3 bispecific antibody, in relapsed/refractory multiple myeloma (R/R MM). J. Clin. Oncol. 2020, 38 (Suppl. 15), 100. [Google Scholar] [CrossRef]
- Lesokhin, A.M.; Ansell, S.M.; Armand, P.; Scott, E.C.; Halwani, A.; Gutierrez, M.; Millenson, M.M.; Cohen, A.D.; Schuster, S.J.; Lebovic, D.; et al. Nivolumab in Patients With Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 2698–2704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateos, M.V.; Blacklock, H.; Schjesvold, F.; Oriol, A.; Simpson, D.; George, A.; Goldschmidt, H.; Larocca, A.; Chanan-Khan, A.; Sherbenou, D.; et al. Pembrolizumab plus pomalidomide and dexamethasone for patients with relapsed or refractory multiple myeloma (KEYNOTE-183): A randomised, open-label, phase 3 trial. Lancet Haematol. 2019, 6, e459–e469. [Google Scholar] [CrossRef]
- Brudno, J.N.; Maric, I.; Hartman, S.D.; Rose, J.J.; Wang, M.; Lam, N.; Stetler-Stevenson, M.; Salem, D.; Yuan, C.; Pavletic, S.; et al. T Cells Genetically Modified to Express an Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Poor-Prognosis Relapsed Multiple Myeloma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 2267–2280. [Google Scholar] [CrossRef]
- Cohen, A.D.; Garfall, A.L.; Stadtmauer, E.A.; Melenhorst, J.J.; Lacey, S.F.; Lancaster, E.; Vogl, D.T.; Weiss, B.M.; Dengel, K.; Nelson, A.; et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J. Clin. Investig. 2019, 129, 2210–2221. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.Y.; Zhao, W.H.; Liu, J.; Chen, Y.X.; Cao, X.M.; Yang, Y.; Zhang, Y.L.; Wang, F.X.; Zhang, P.Y.; Lei, B.; et al. Long-Term Follow-up of a Phase 1, First-in-Human Open-Label Study of LCAR-B38M, a Structurally Differentiated Chimeric Antigen Receptor T (CAR-T) Cell Therapy Targeting B-Cell Maturation Antigen (BCMA), in Patients (pts) with Relapsed/Refractory Multiple Myeloma (RRMM). Blood 2019, 134 (Suppl. 1), 579. [Google Scholar]
- Zhao, W.H.; Liu, J.; Wang, B.Y.; Chen, Y.X.; Cao, X.M.; Yang, Y.; Zhang, Y.L.; Wang, F.X.; Zhang, P.Y.; Lei, B.; et al. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J. Hematol. Oncol. 2018, 11, 141. [Google Scholar] [CrossRef]
- Xu, J.; Chen, L.J.; Yang, S.S.; Sun, Y.; Wu, W.; Liu, Y.F.; Xu, J.; Zhuang, Y.; Zhang, W.; Weng, X.Q.; et al. Exploratory trial of a biepitopic CAR T-targeting B cell maturation antigen in relapsed/refractory multiple myeloma. Proc. Natl. Acad. Sci. USA 2019, 116, 9543–9551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madduri, D.; Berdeja, J.G.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D. CARTITUDE-1: Phase 1b/2 Study of Ciltacabtagene Autoleucel, a B-Cell Maturation Antigen-Directed Chimeric Antigen Receptor T Cell Therapy, in Relapsed/Refractory Multiple Myeloma. Blood 2020, 136 (Suppl. 1), 22–25. [Google Scholar] [CrossRef]
- Munshi, N.C.; Anderson, L.D., Jr.; Shah, N.; Jagannath, S.; Berdeja, J.G.; Lonial, S.; Raje, N.S.; Siegel, D.S.; Lin, Y.; Oriol, A.; et al. Idecabtagene vicleucel (ide-cel; bb2121), a BCMA-targeted CAR T-cell therapy, in patients with relapsed and refractory multiple myeloma (RRMM): Initial KarMMa results. J. Clin. Oncol. 2020, 38 (Suppl. 15), 8503. [Google Scholar] [CrossRef]
- Mailankody, S.; Jakubowiak, A.J.; Htut, M.; Costa, L.J.; Lee, K.; Ganguly, S.; Kaufman, J.L.; Siegel, D.S.; Bensinger, W.; Cota, M.; et al. Orvacabtagene autoleucel (orva-cel), a B-cell maturation antigen (BCMA)-directed CAR T cell therapy for patients (pts) with relapsed/refractory multiple myeloma (RRMM): Update of the phase 1/2 EVOLVE study (NCT03430011). J. Clin. Oncol. 2020, 38 (Suppl. 15), 8504. [Google Scholar] [CrossRef]
- Rosenblatt, J.; Vasir, B.; Uhl, L.; Blotta, S.; MacNamara, C.; Somaiya, P.; Wu, Z.; Joyce, R.; Levine, J.D.; Dombagoda, D.; et al. Vaccination with dendritic cell/tumor fusion cells results in cellular and humoral antitumor immune responses in patients with multiple myeloma. Blood 2011, 117, 393–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapoport, A.P.; Aqui, N.A.; Stadtmauer, E.A.; Vogl, D.T.; Fang, H.B.; Cai, L.; Janofsky, S.; Chew, A.; Storek, J.; Akpek, G.; et al. Combination immunotherapy using adoptive T-cell transfer and tumor antigen vaccination on the basis of hTERT and survivin after ASCT for myeloma. Blood 2011, 117, 788–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenblatt, J.; Avivi, I.; Vasir, B.; Uhl, L.; Munshi, N.C.; Katz, T.; Dey, B.R.; Somaiya, P.; Mills, H.; Campigotto, F.; et al. Vaccination with dendritic cell/tumor fusions following autologous stem cell transplant induces immunologic and clinical responses in multiple myeloma patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 3640–3648. [Google Scholar] [CrossRef] [Green Version]
- Rapoport, A.P.; Aqui, N.A.; Stadtmauer, E.A.; Vogl, D.T.; Xu, Y.Y.; Kalos, M.; Cai, L.; Fang, H.B.; Weiss, B.M.; Badros, A.; et al. Combination immunotherapy after ASCT for multiple myeloma using MAGE-A3/Poly-ICLC immunizations followed by adoptive transfer of vaccine-primed and costimulated autologous T cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 1355–1365. [Google Scholar] [CrossRef] [Green Version]
- Nooka, A.K.; Wang, M.L.; Yee, A.J.; Kaufman, J.L.; Bae, J.; Peterkin, D.; Richardson, P.G.; Raje, N.S. Assessment of Safety and Immunogenicity of PVX-410 Vaccine With or Without Lenalidomide in Patients With Smoldering Multiple Myeloma: A Nonrandomized Clinical Trial. JAMA Oncol. 2018, 4, e183267. [Google Scholar] [CrossRef]
- Cohen, A.D.; Lendvai, N.; Nataraj, S.; Imai, N.; Jungbluth, A.A.; Tsakos, I.; Rahman, A.; Mei, A.H.; Singh, H.; Zarychta, K.; et al. Autologous Lymphocyte Infusion Supports Tumor Antigen Vaccine-Induced Immunity in Autologous Stem Cell Transplant for Multiple Myeloma. Cancer Immunol. Res. 2019, 7, 658–669. [Google Scholar] [CrossRef]
- Deaglio, S.; Mehta, K.; Malavasi, F. Human CD38: A (r)evolutionary story of enzymes and receptors. Leuk Res. 2001, 25, 1–12. [Google Scholar] [CrossRef]
- Deaglio, S.; Vaisitti, T.; Billington, R.; Bergui, L.; Omede, P.; Genazzani, A.A.; Malavasi, F. CD38/CD19: A lipid raft-dependent signaling complex in human B cells. Blood 2007, 109, 5390–5398. [Google Scholar] [CrossRef] [PubMed]
- Malavasi, F.; Deaglio, S.; Funaro, A.; Ferrero, E.; Horenstein, A.L.; Ortolan, E.; Vaisitti, T.; Aydin, S. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol. Rev. 2008, 88, 841–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van de Donk, N.W.C.J.; Usmani, S.Z. CD38 Antibodies in Multiple Myeloma: Mechanisms of Action and Modes of Resistance. Front. Immunol. 2018, 9, 2134. [Google Scholar] [CrossRef] [PubMed]
- Nijhof, I.S.; Groen, R.W.; Noort, W.A.; van Kessel, B.; de Jong-Korlaar, R.; Bakker, J.; Van Bueren, J.J.; Parren, P.W.; Lokhorst, H.M.; Van De Donk, N.W.; et al. Preclinical Evidence for the Therapeutic Potential of CD38-Targeted Immuno-Chemotherapy in Multiple Myeloma Patients Refractory to Lenalidomide and Bortezomib. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 2802–2810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreau, P.; Attal, M.; Hulin, C.; Arnulf, B.; Belhadj, K.; Benboubker, L.; Béné, M.C.; Broijl, A.; Caillon, H.; Caillot, D.; et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): A randomised, open-label, phase 3 study. Lancet 2019, 394, 29–38. [Google Scholar] [CrossRef]
- Mateos, M.V.; Cavo, M.; Blade, J.; Dimopoulos, M.A.; Suzuki, K.; Jakubowiak, A.; Knop, S.; Doyen, C.; Lucio, P.; Nagy, Z.; et al. Overall survival with daratumumab, bortezomib, melphalan, and prednisone in newly diagnosed multiple myeloma (ALCYONE): A randomised, open-label, phase 3 trial. Lancet 2020, 395, 132–141. [Google Scholar] [CrossRef]
- Facon, T.; Kumar, S.; Plesner, T.; Orlowski, R.Z.; Moreau, P.; Bahlis, N.; Basu, S.; Nahi, H.; Hulin, C.; Quach, H.; et al. Daratumumab plus Lenalidomide and Dexamethasone for Untreated Myeloma. N. Engl. J. Med. 2019, 380, 2104–2115. [Google Scholar] [CrossRef]
- Chari, A.; Suvannasankha, A.; Fay, J.W.; Arnulf, B.; Kaufman, J.L.; Ifthikharuddin, J.J.; Weiss, B.M.; Krishnan, A.; Lentzsch, S.; Comenzo, R.; et al. Daratumumab plus pomalidomide and dexamethasone in relapsed and/or refractory multiple myeloma. Blood 2017, 130, 974–981. [Google Scholar] [CrossRef]
- Dimopoulos, M.; Quach, H.; Mateos, M.V.; Landgren, O.; Leleu, X.; Siegel, D.; Weisel, K.; Yang, H.; Klippel, Z.; Zahlten-Kumeli, A.; et al. Carfilzomib, dexamethasone, and daratumumab versus carfilzomib and dexamethasone for patients with relapsed or refractory multiple myeloma (CANDOR): Results from a randomised, multicentre, open-label, phase 3 study. Lancet 2020, 396, 186–197. [Google Scholar] [CrossRef]
- Attal, M.; Richardson, P.G.; Rajkumar, S.V.; San-Miguel, J.; Beksac, M.; Spicka, I.; Leleu, X.; Schjesvold, F.; Moreau, P.; Dimopoulos, M.A.; et al. Isatuximab plus pomalidomide and low-dose dexamethasone versus pomalidomide and low-dose dexamethasone in patients with relapsed and refractory multiple myeloma (ICARIA-MM): A randomised, multicentre, open-label, phase 3 study. Lancet 2019, 394, 2096–2107. [Google Scholar] [CrossRef]
- De Weers, M.; Tai, Y.T.; Van Der Veer, M.S.; Bakker, J.M.; Vink, T.; Jacobs, D.C.; Oomen, L.A.; Peipp, M.; Valerius, T.; Slootstra, J.W.; et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J. Immunol. 2011, 186, 1840–1848. [Google Scholar] [CrossRef]
- Overdijk, M.B.; Verploegen, S.; Bögels, M.; van Egmond, M.; van Bueren, J.J.; Mutis, T.; Groen, R.W.; Breij, E.; Martens, A.C.; Bleeker, W.K.; et al. Antibody-mediated phagocytosis contributes to the anti-tumor activity of the therapeutic antibody daratumumab in lymphoma and multiple myeloma. mAbs 2015, 7, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Franssen, L.E.; Stege, C.A.M.; Zweegman, S.; van de Donk, N.W.C.J.; Nijhof, I.S. Resistance Mechanisms Towards CD38-Directed Antibody Therapy in Multiple Myeloma. J. Clin. Med. 2020, 9, 1195. [Google Scholar] [CrossRef] [PubMed]
- Overdijk, M.B.; Jansen, J.M.; Nederend, M.; van Bueren, J.J.; Groen, R.W.; Parren, P.W.; Leusen, J.H.; Boross, P. The Therapeutic CD38 Monoclonal Antibody Daratumumab Induces Programmed Cell Death via Fcγ Receptor-Mediated Cross-Linking. J. Immunol. 2016, 197, 807–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; van de Donk, N.W.; Weiss, B.M.; et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 2016, 128, 384–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitadate, A.; Kobayashi, H.; Abe, Y.; Narita, K.; Miura, D.; Takeuchi, M.; Matsue, K. Pre-treatment CD38-positive regulatory T cells affect the durable response to daratumumab in relapsed/refractory multiple myeloma patients. Haematologica 2020, 105, e37–e40. [Google Scholar] [CrossRef]
- Nijhof, I.S.; Groen, R.W.; Lokhorst, H.M.; Van Kessel, B.; Bloem, A.C.; Van Velzen, J.; de Jong-Korlaar, R.; Yuan, H.; Noort, W.A.; Klein, S.K.; et al. Upregulation of CD38 expression on multiple myeloma cells by all-trans retinoic acid improves the efficacy of daratumumab. Leukemia 2015, 29, 2039–2049. [Google Scholar] [CrossRef]
- Fedele, P.L.; Willis, S.N.; Liao, Y.; Low, M.S.; Rautela, J.; Segal, D.H.; Gong, J.N.; Huntington, N.D.; Shi, W.; Huang, D.; et al. IMiDs prime myeloma cells for daratumumab-mediated cytotoxicity through loss of Ikaros and Aiolos. Blood 2018, 132, 2166–2178. [Google Scholar] [CrossRef] [Green Version]
- García-Guerrero, E.; Gogishvili, T.; Danhof, S.; Schreder, M.; Pallaud, C.; Pérez-Simón, J.A.; Einsele, H.; Hudecek, M. Panobinostat induces CD38 upregulation and augments the antimyeloma efficacy of daratumumab. Blood 2017, 129, 3386–3388. [Google Scholar] [CrossRef]
- García-Guerrero, E.; Götz, R.; Doose, S.; Sauer, M.; Rodríguez-Gil, A.; Nerreter, T.; Kortüm, K.M.; Pérez-Simón, J.A.; Einsele, H.; Hudecek, M.; et al. Upregulation of CD38 expression on multiple myeloma cells by novel HDAC6 inhibitors is a class effect and augments the efficacy of daratumumab. Leukemia 2020, 29, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Zipfel, P.F.; Skerka, C. Complement regulators and inhibitory proteins. Nat. Rev. Immunol. 2009, 9, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Nijhof, I.S.; Casneuf, T.; Van Velzen, J.; van Kessel, B.; Axel, A.E.; Syed, K.; Groen, R.W.; van Duin, M.; Sonneveld, P.; Minnema, M.C.; et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood 2016, 128, 959–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barclay, A.N.; Van den Berg, T.K. The interaction between signal regulatory protein alpha (SIRPα) and CD47: Structure, function, and therapeutic target. Annu. Rev. Immunol. 2014, 32, 25–50. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Iwamura, H.; Kaneko, T.; Ohnishi, H.; Murata, Y.; Okazawa, H.; Kanazawa, Y.; Sato-Hashimoto, M.; Kobayashi, H.; Oldenborg, P.A.; et al. Regulation by SIRPα of dendritic cell homeostasis in lymphoid tissues. Blood 2010, 116, 3517–3525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrisqueta, P.; Sancho, J.M.; Cordoba, R.; Persky, D.O.; Andreadis, C.; Huntington, S.F.; Carpio, C.; Morillo Giles, D.; Wei, X.; Li, Y.F.; et al. Anti-CD47 Antibody, CC-90002, in Combination with Rituximab in Subjects with Relapsed and/or Refractory Non-Hodgkin Lymphoma (R/R NHL). Blood 2019, 134 (Suppl. 1), 4089. [Google Scholar] [CrossRef]
- Naicker, S.; Rigalou, A.; McEllistrim, C.; Natoni, A.; Chiu, C.; Sasser, K.; Ryan, A.; O’Dwyer, M. Patient Data Supports the Rationale of Low Dose Cyclophosphamide to Potentiate the Anti-Myeloma Activity of Daratumumab through Augmentation of Macrophage-Induced ADCP. Blood 2017, 130 (Suppl. 1), 121. [Google Scholar]
- Rigalou, A.; Ryan, A.; Natoni, A.; Chiu, C.; Sasser, K.; O’Dwyer, M.E. Potentiation of Anti-Myeloma Activity of Daratumumab with Combination of Cyclophosphamide, Lenalidomide or Bortezomib Via a Tumor Secretory Response That Greatly Augments Macrophage-Induced ADCP. Blood 2016, 128, 2101. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef]
- van de Donk, N.W.; Casneuf, T.; Di Cara, A.; Parren, P.W.; Zweegman, S.; van Kessel, B.; Lokhorst, H.M.; Usmani, S.Z.; Lonial, S.; Richardson, P.G.; et al. Impact of Fc gamma receptor polymorphisms on efficacy and safety of daratumumab in relapsed/refractory multiple myeloma. Br. J. Haematol. 2019, 184, 475–479. [Google Scholar] [CrossRef] [Green Version]
- Sanne, J.; van de Donk, N.W.; Minnema, M.C.; Huang, J.H.; Aarts-Riemens, T.; Bovenschen, N.; Yuan, H.; Groen, R.W.; McMillin, D.W.; Jakubikova, J.; et al. Accessory cells of the microenvironment protect multiple myeloma from T-cell cytotoxicity through cell adhesion-mediated immune resistance. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 5591–5601. [Google Scholar]
- Van Der Veer, M.S.; de Weers, M.; van Kessel, B.; Bakker, J.M.; Wittebol, S.; Parren, P.W.; Lokhorst, H.M.; Mutis, T. Towards effective immunotherapy of myeloma: Enhanced elimination of myeloma cells by combination of lenalidomide with the human CD38 monoclonal antibody daratumumab. Haematologica 2011, 96, 284–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nijhof, I.S.; van Bueren, J.J.; van Kessel, B.; Andre, P.; Morel, Y.; Lokhorst, H.M.; van de Donk, N.W.; Parren, P.W.; Mutis, T. Daratumumab-mediated lysis of primary multiple myeloma cells is enhanced in combination with the human anti-KIR antibody IPH2102 and lenalidomide. Haematologica 2015, 100, 263–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhang, Y.; Hughes, T.; Zhang, J.; Caligiuri, M.A.; Benson, D.M.; Yu, J. Fratricide of NK Cells in Daratumumab Therapy for Multiple Myeloma Overcome by Ex Vivo-Expanded Autologous NK Cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 4006–4017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannons, J.L.; Tangye, S.G.; Schwartzberg, P.L. SLAM family receptors and SAP adaptors in immunity. Annu. Rev. Immunol. 2011, 29, 665–705. [Google Scholar] [CrossRef]
- Collins, S.M.; Bakan, C.E.; Swartzel, G.D.; Hofmeister, C.C.; Efebera, Y.A.; Kwon, H.; Starling, G.C.; Ciarlariello, D.; Bhaskar, S.; Briercheck, E.L.; et al. Elotuzumab directly enhances NK cell cytotoxicity against myeloma via CS1 ligation: Evidence for augmented NK cell function complementing ADCC. Cancer Immunol. Immunother. 2013, 62, 1841–1849. [Google Scholar] [CrossRef] [Green Version]
- Hartmut, G.; Mai, E.K.; Hans, S.; Uta, B.; Kaya, H.; Christina, K. Bortezomib, Lenalidomide and Dexamethasone with or Without Elotuzumab as Induction Therapy for Newly-Diagnosed, Transplant-Eligible Multiple Myeloma. 2020. Available online: https://library.ehaweb.org/eha/2020/eha25th/295023/hartmut.goldschmidt.bortezomib.lenalidomide.and.dexamethasone.with.or.without (accessed on 14 December 2020).
- Usmani, S.Z.; Ailawadhi, S.; Sexton, R.; Hoering, A.; Lipe, B.; Hita, S.; Durie, B.G.; Zonder, J.A.; Dhodapkar, M.V.; Callander, N.S.; et al. Primary analysis of the randomized phase II trial of bortezomib, lenalidomide, dexamthasone with/without elotuzumab for newly diagnosed, high-risk multiple myeloma (SWOG-1211). J. Clin. Oncol. 2020, 38 (Suppl. 15), 8507. [Google Scholar] [CrossRef]
- Bristol Myers Squibb Reports Primary Results of ELOQUENT-1 Study Evaluating Empliciti (Elotuzumab) Plus Revlimid (lenalidomide) and Dexamethasone in Patients with Newly Diagnosed, Untreated Multiple Myeloma. Available online: https://news.bms.com/news/corporate-financial/2020/Bristol-Myers-Squibb-Reports-Primary-Results-of-ELOQUENT-1-Study-Evaluating-Empliciti-elotuzumab-Plus-Revlimid-lenalidomide-and-Dexamethasone-in-Patients-with-Newly-Diagnosed-Untreated-Multiple-Myeloma/default.aspx (accessed on 14 December 2020).
- Lonial, S.; Lee, H.C.; Badros, A.; Trudel, S.; Nooka, A.K.; Chari, A.; Abdallah, A.O.; Callander, N.; Lendvai, N.; Sborov, D. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): A two-arm, randomised, open-label, phase 2 study. Lancet Oncol. 2020, 21, 207–221. [Google Scholar] [CrossRef]
- Farooq, A.V.; Degli Esposti, S.; Popat, R.; Thulasi, P.; Lonial, S.; Nooka, A.K.; Jakubowiak, A.; Sborov, D.; Zaugg, B.E.; Badros, A.Z.; et al. Corneal Epithelial Findings in Patients with Multiple Myeloma Treated with Antibody-Drug Conjugate Belantamab Mafodotin in the Pivotal, Randomized, DREAMM-2 Study. Ophthalmol. Ther. 2020, 9, 889–911. [Google Scholar] [CrossRef]
- Sanderson, R.D.; Lalor, P.; Bernfield, M. B lymphocytes express and lose syndecan at specific stages of differentiation. Cell Regul. 1989, 1, 27–35. [Google Scholar] [CrossRef]
- Ishitsuka, K.; Jimi, S.; Goldmacher, V.S.; Ab, O.; Tamura, K. Targeting CD56 by the maytansinoid immunoconjugate IMGN901 (huN901-DM1): A potential therapeutic modality implication against natural killer/T cell malignancy. Br. J. Haematol. 2008, 141, 129–131. [Google Scholar] [CrossRef] [PubMed]
- Stein, R.; Mattes, M.J.; Cardillo, T.M.; Hansen, H.J.; Chang, C.H.; Burton, J.; Govindan, S.; Goldenberg, D.M. CD74: A new candidate target for the immunotherapy of B-cell neoplasms. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13 Pt 2, 5556s–5563s. [Google Scholar] [CrossRef] [Green Version]
- Zanwar, S.; Nandakumar, B.; Kumar, S. Immune-based therapies in the management of multiple myeloma. Blood Cancer, J. 2020, 10, 84. [Google Scholar] [CrossRef] [PubMed]
- Baeuerle, P.A.; Kufer, P.; Bargou, R. BiTE: Teaching antibodies to engage T-cells for cancer therapy. Curr. Opin. Mol. Ther. 2009, 11, 22–30. [Google Scholar] [PubMed]
- Velasquez, M.P.; Bonifant, C.L.; Gottschalk SRedirecting, T. cells to hematological malignancies with bispecific antibodies. Blood 2018, 131, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, N. Cancer therapy using bispecific antibodies. Rinsho Ketsueki. 2018, 59, 1942–1947. [Google Scholar]
- Loffler, A.; Kufer, P.; Lutterbuse, R.; Zettl, F.; Daniel, P.T.; Schwenkenbecher, J.M.; Riethmuller, G.; Dorken, B.; Bargou, R.C. A recombinant bispecific single-chain antibody, CD19 x CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood 2000, 95, 2098–2103. [Google Scholar] [CrossRef]
- Gökbuget, N.; Dombret, H.; Bonifacio, M.; Reichle, A.; Graux, C.; Faul, C.; Diedrich, H.; Topp, M.S.; Brüggemann, M.; Horst, H.A.; et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood 2018, 131, 1522–1531. [Google Scholar] [CrossRef] [Green Version]
- Goyos, A.; Li, C.M.; Deegen, P.; Bogner, P.; Thomas, O.; Wahl, J.; Goldstein, R.; Friedrich, M.; Coxon, A.; Balazs, M.; et al. Abstract LB-299: Cynomolgus monkey plasma cell gene signature to quantify the in vivo activity of a half-life extended anti-BCMA BiTE® for the treatment of multiple myeloma. Cancer Res. 2018, 78 (Suppl. 13). [Google Scholar] [CrossRef]
- Cho, S.F.; Lin, L.; Xing, L.; Liu, J.; Yu, T.; Wen, K.; Hsieh, P.; Munshi, N.; Anderson, K.; Tai, Y.T. Anti-BCMA BiTE® AMG 701 Potently Induces Specific T Cell Lysis of Human Multiple Myeloma (MM) Cells and Immunomodulation in the Bone Marrow Microenvironment. Blood 2018, 132 (Suppl. 1), 592. [Google Scholar] [CrossRef]
- Pillarisetti, K.; Powers, G.; Luistro, L.; Babich, A.; Baldwin, E.; Li, Y.; Zhang, X.; Mendonça, M.; Majewski, N.; Nanjunda, R.; et al. Teclistamab is an active T cell-redirecting bispecific antibody against B-cell maturation antigen for multiple myeloma. Blood Adv. 2020, 4, 4538–4549. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.J.; Wong, S.W.; Bermúdez, A.; de la Rubia, J.; Mateos, M.V.; Ocio, E.M.; Rodríguez-Otero, P.; San-Miguel, J.; Li, S.; Sarmiento, R.; et al. Interim Results from the First Phase 1 Clinical Study of the B-cell Maturation Antigen (BCMA) 2+1 T Cell Engager (TCE) CC-93269 in Patients (PTS) with Relapsed/Refractory Multiple Myeloma (RRMM). Available online: https://library.ehaweb.org/eha/2020/eha25th/295025/luciano.j.costa.interim.results.from.the.first.phase.1.clinical.study.of.the (accessed on 14 December 2020).
- Richter, J.R.; Landgren, C.O.; Kauh, J.S.; Back, J.; Salhi, Y.; Reddy, V.; Bayever, E.; Berdej, A. Phase 1, multicenter, open-label study of single-agent bispecific antibody t-cell engager GBR 1342 in relapsed/refractory multiple myeloma. J. Clin. Oncol. 2018, 36 (Suppl. 15), TPS3132. [Google Scholar] [CrossRef]
- Cohen, A.D.; Harrison, S.J.; Krishnan, A.; Fonseca, R.; Forsberg, P.A.; Spencer, A. Initial Clinical Activity and Safety of BFCR4350A, a FcRH5/CD3 T-Cell-Engaging Bispecific Antibody, in Relapsed/Refractory Multiple Myeloma. Blood 2020, 136 (Suppl. 1), 42–43. [Google Scholar] [CrossRef]
- Chari, A.; Berdeja, J.G.; Oriol, A.; van de Donk, N.W.C.J.; Rodriguez, P.; Askari, E. A Phase 1, First-in-Human Study of Talquetamab, a G Protein-Coupled Receptor Family C Group 5 Member D (GPRC5D) x CD3 Bispecific Antibody, in Patients with Relapsed and/or Refractory Multiple Myeloma (RRMM). Blood 2020, 136 (Suppl. 1), 40–41. [Google Scholar] [CrossRef]
- Wu, L.; Seung, E.; Xu, L.; Rao, E.; Lord, D.M.; Wei, R.R.; Cortez-Retamozo, V.; Ospina, B.; Posternak, V.; Ulinski, G.; et al. Trispecific antibodies enhance the therapeutic efficacy of tumor-directed T cells through T cell receptor co-stimulation. Nat. Cancer 2020, 1, 86–98. [Google Scholar] [CrossRef]
- Lancman, G.; Richter, J.; Chari, A. Bispecifics, trispecifics, and other novel immune treatments in myeloma. Hematology 2020, 2020, 264–271. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. 2011, 29, 235–271. [Google Scholar] [CrossRef] [Green Version]
- Salmaninejad, A.; Valilou, S.F.; Shabgah, A.G.; Aslani, S.; Alimardani, M.; Pasdar, A.; Sahebkar, A. PD-1/PD-L1 pathway: Basic biology and role in cancer immunotherapy. J. Cell Physiol. 2019, 234, 16824–16837. [Google Scholar] [CrossRef]
- Costa, F.; Das, R.; Kini Bailur, J.; Dhodapkar, K.; Dhodapkar, M.V. Checkpoint Inhibition in Myeloma: Opportunities and Challenges. Front. Immunol. 2018, 9, 2204. [Google Scholar] [CrossRef] [Green Version]
- Dyck, L.; Mills, K.H.G. Immune checkpoints and their inhibition in cancer and infectious diseases. Eur. J. Immunol. 2017, 47, 765–779. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, J.; Chehrazi-Raffle, A.; Reddi, S.; Salgia, R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: A comprehensive review of registration trials and future considerations. J Immunother. Cancer 2018, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Pratt, G.; Goodyear, O.; Moss, P. Immunodeficiency and immunotherapy in multiple myeloma. Br. J. Haematol. 2007, 138, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Ratta, M.; Fagnoni, F.; Curti, A.; Vescovini, R.; Sansoni, P.; Oliviero, B.; Fogli, M.; Ferri, E.; Della Cuna, G.R.; Tura, S.; et al. cells are functionally defective in multiple myeloma: The role of interleukin-6. Blood 2002, 100, 230–237. [Google Scholar] [CrossRef] [Green Version]
- Bahlis, N.J.; King, A.M.; Kolonias, D.; Carlson, L.M.; Liu, H.Y.; Hussein, M.A.; Terebelo, H.R.; Byrne, G.E.; Levine, B.L.; Boise, L.H.; et al. CD28-mediated regulation of multiple myeloma cell proliferation and survival. Blood 2007, 109, 5002–5010. [Google Scholar] [CrossRef] [Green Version]
- Rosenblatt, J.; Avigan, D. Targeting the PD-1/PD-L1 axis in multiple myeloma: A dream or a reality? Blood 2017, 129, 275–279. [Google Scholar] [CrossRef] [Green Version]
- Paiva, B.; Azpilikueta, A.; Puig, N.; Ocio, E.M.; Sharma, R.; Oyajobi, B.O.; Labiano, S.; San-Segundo, L.; Rodriguez, A.; Aires-Mejia, I.; et al. PD-L1/PD-1 presence in the tumor microenvironment and activity of PD-1 blockade in multiple myeloma. Leukemia 2015, 29, 2110–2113. [Google Scholar] [CrossRef]
- Lozano, E.; Díaz, T.; Mena, M.P.; Suñe, G.; Calvo, X.; Calderón, M.; Pérez-Amill, L.; Rodríguez, V.; Pérez-Galán, P.; Roué, G.; et al. Loss of the Immune Checkpoint CD85j/LILRB1 on Malignant Plasma Cells Contributes to Immune Escape in Multiple Myeloma. J. Immunol. 2018, 200, 2581–2591. [Google Scholar] [CrossRef]
- Guillerey, C.; Harjunpää, H.; Carrié, N.; Kassem, S.; Teo, T.; Miles, K.; Krumeich, S.; Weulersse, M.; Cuisinier, M.; Stannard, K.; et al. TIGIT immune checkpoint blockade restores CD8+ T-cell immunity against multiple myeloma. Blood 2018, 132, 1689–1694. [Google Scholar] [CrossRef] [Green Version]
- Ribrag, V.; Avigan, D.E.; Green, D.J.; Wise-Draper, T.; Posada, J.G.; Vij, R.; Zhu, Y.; Farooqui, M.Z.; Marinello, P.; Siegel, D.S. Phase 1b trial of pembrolizumab monotherapy for relapsed/refractory multiple myeloma: KEYNOTE-013. Br. J. Haematol. 2019, 186, e41–e44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usmani, S.Z.; Schjesvold, F.; Oriol, A.; Karlin, L.; Cavo, M.; Rifkin, R.M.; Yimer, H.A.; LeBlanc, R.; Takezako, N.; McCroskey, R.D.; et al. Pembrolizumab plus lenalidomide and dexamethasone for patients with treatment-naive multiple myeloma (KEYNOTE-185): A randomised, open-label, phase 3 trial. Lancet Haematol. 2019, 6, e448–e458. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jelinek, T.; Paiva, B.; Hajek, R. Update on PD-1/PD-L1 Inhibitors in Multiple Myeloma. Front. Immunol. 2018, 9, 2431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadelain, M.; Brentjens, R.; Rivière, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [Green Version]
- Sadelain, M.; Rivière, I.; Riddell, S. Therapeutic T cell engineering. Nature 2017, 545, 423–431. [Google Scholar] [CrossRef] [Green Version]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Anonymous. Yescarta. European Medicines Agency. 2018. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/yescarta (accessed on 22 July 2020).
- FDA. Research C for BE and YESCARTA (Axicabtagene Ciloleucel). Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/yescarta-axicabtagene-ciloleucel (accessed on 22 July 2020).
- Anonymous. Kymriah. European Medicines Agency. 2018. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/kymriah (accessed on 22 July 2020).
- FDA. Research C for BE and KYMRIAH (Tisagenlecleucel). Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/kymriah-tisagenlecleucel (accessed on 22 July 2020).
- Gagelmann, N.; Riecken, K.; Wolschke, C.; Berger, C.; Ayuk, F.A.; Fehse, B.; Kröger, N. Development of CAR-T cell therapies for multiple myeloma. Leukemia 2020. [Google Scholar] [CrossRef] [PubMed]
- Sellner, L.; Fan, F.; Giesen, N.; Schubert, M.L.; Goldschmidt, H.; Müller-Tidow, C.; Dreger, P.; Raab, M.S.; Schmitt, M. B-cell maturation antigen-specific chimeric antigen receptor T cells for multiple myeloma: Clinical experience and future perspectives. Int. J. Cancer 2020. [Google Scholar] [CrossRef] [PubMed]
- Sidana, S.; Shah, N. CAR T-cell therapy: Is it prime time in myeloma? Hematol. Am. Soc. Hematol. Educ. Program 2019, 2019, 260–265. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, M.; Raje, N. Anti-BCMA CAR T-cell therapy in multiple myeloma: Can we do better? Leukemia 2020, 34, 21–34. [Google Scholar] [CrossRef]
- Rodríguez-Lobato, L.G.; Ganzetti, M.; Fernández de Larrea, C.; Hudecek, M.; Einsele, H.; Danhof, S. CAR T-Cells in Multiple Myeloma: State of the Art and Future Directions. Front. Oncol. Available online: https://www.frontiersin.org/articles/10.3389/fonc.2020.01243/full (accessed on 30 July 2020).
- Carpenter, R.O.; Evbuomwan, M.O.; Pittaluga, S.; Rose, J.J.; Raffeld, M.; Yang, S.; Gress, R.E.; Hakim, F.T.; Kochenderfer, J.N. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 2048–2060. [Google Scholar] [CrossRef] [Green Version]
- Madry, C.; Laabi, Y.; Callebaut, I.; Roussel, J.; Hatzoglou, A.; Le Coniat, M.; Mornon, J.P.; Berger, R.; Tsapis, A. The characterization of murine BCMA gene defines it as a new member of the tumor necrosis factor receptor superfamily. Int. Immunol. 1998, 10, 1693–1702. [Google Scholar] [CrossRef]
- Ng, L.G.; Mackay, C.R.; Mackay, F. The BAFF/APRIL system: Life beyond B lymphocytes. Mol. Immunol. 2005, 42, 763–772. [Google Scholar] [CrossRef]
- Moreaux, J.; Legouffe, E.; Jourdan, E.; Quittet, P.; Rème, T.; Lugagne, C.; Moine, P.; Rossi, J.F.; Klein, B.; Tarte, K. BAFF and APRIL protect myeloma cells from apoptosis induced by interleukin 6 deprivation and dexamethasone. Blood 2004, 103, 3148–3157. [Google Scholar] [CrossRef] [Green Version]
- Tai, Y.T.; Acharya, C.; An, G.; Moschetta, M.; Zhong, M.Y.; Feng, X.; Cea, M.; Cagnetta, A.; Wen, K.; van Eenennaam, H.; et al. APRIL and BCMA promote human multiple myeloma growth and immunosuppression in the bone marrow microenvironment. Blood 2016, 127, 3225–3236. [Google Scholar] [CrossRef] [Green Version]
- Novak, A.J.; Darce, J.R.; Arendt, B.K.; Harder, B.; Henderson, K.; Kindsvogel, W.; Gross, J.A.; Greipp, P.R.; Jelinek, D.F. Expression of BCMA, TACI, and BAFF-R in multiple myeloma: A mechanism for growth and survival. Blood 2004, 103, 689–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, K.M.; Garrett, T.E.; Evans, J.W.; Horton, H.M.; Latimer, H.J.; Seidel, S.L.; Horvath, C.J.; Morgan, R.A. Effective Targeting of Multiple B-Cell Maturation Antigen-Expressing Hematological Malignances by Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor T Cells. Hum. Gene Ther. 2018, 29, 585–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berdeja, J.G.; Alsina, M.; Shah, N.D.; Siegel, D.S.; Jagannath, S.; Madduri, D.; Kaufman, J.L.; Munshi, N.C.; Rosenblatt, J.; Jasielec, J.K.; et al. Updated Results from an Ongoing Phase 1 Clinical Study of bb21217 Anti-Bcma CAR T Cell Therapy. Blood 2019, 134 (Suppl. 1), 927. [Google Scholar] [CrossRef]
- Mailankody, S.; Htut, M.; Lee, K.P.; Bensinger, W.; Devries, T.; Piasecki, J.; Ziyad, S.; Blake, M.; Byon, J.; Jakubowiak, A. JCARH125, Anti-BCMA CAR T-cell Therapy for Relapsed/Refractory Multiple Myeloma: Initial Proof of Concept Results from a Phase 1/2 Multicenter Study (EVOLVE). Blood 2018, 132 (Suppl. 1), 957. [Google Scholar] [CrossRef]
- Costello, C.L.; Gregory, T.K.; Ali, S.A.; Berdeja, J.G.; Patel, K.K.; Shah, N.D.; Ostertag, E.; Martin, C.; Ghoddusi, M.; Shedlock, D.J.; et al. Phase 2 Study of the Response and Safety of P-Bcma-101 CAR-T Cells in Patients with Relapsed/Refractory (r/r) Multiple Myeloma (MM) (PRIME). Blood 2019, 134 (Suppl. 1), 3184. [Google Scholar] [CrossRef]
- Green, D.J.; Pont, M.; Sather, B.D.; Cowan, A.J.; Turtle, C.J.; Till, B.G.; Nagengast, A.M.; Libby, E.N.; Becker, P.S.; Coffey, D.G.; et al. Fully Human Bcma Targeted Chimeric Antigen Receptor T Cells Administered in a Defined Composition Demonstrate Potency at Low Doses in Advanced Stage High Risk Multiple Myeloma. Blood 2018, 132 (Suppl. 1), 1011. [Google Scholar] [CrossRef]
- Mailankody, S.; Ghosh, A.; Staehr, M.; Purdon, T.J.; Roshal, M.; Halton, E.; Diamonte, C.; Pineda, J.; Anant, P.; Bernal, Y.; et al. Clinical Responses and Pharmacokinetics of MCARH171, a Human-Derived Bcma Targeted CAR T Cell Therapy in Relapsed/Refractory Multiple Myeloma: Final Results of a Phase I Clinical Trial. Blood 2018, 132 (Suppl. 1), 959. [Google Scholar] [CrossRef]
- Gagelmann, N.; Ayuk, F.; Atanackovic, D.; Kröger, N. B cell maturation antigen-specific chimeric antigen receptor T cells for relapsed or refractory multiple myeloma: A meta-analysis. Eur. J. Haematol. 2020, 104, 318–327. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1-2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Cao, J.; Wang, G.; Cheng, H.; Wei, C.; Qi, K.; Sang, W.; Zhenyu, L.; Shi, M.; Li, H.; Qiao, J.; et al. Potent anti-leukemia activities of humanized CD19-targeted Chimeric antigen receptor T (CAR-T) cells in patients with relapsed/refractory acute lymphoblastic leukemia. Am. J. Hematol. 2018, 93, 851–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, N.; Trinklein, N.D.; Buelow, B.; Patterson, G.H.; Ojha, N.; Kochenderfer, J.N. Anti-BCMA chimeric antigen receptors with fully human heavy-chain-only antigen recognition domains. Nat. Commun. 2020, 11, 283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Wang, J.; Wang, D.; Hu, G.; Yang, Y.; Zhou, X.; Meng, L.; Hong, Z.; Chen, L.; Mao, X.; et al. Efficacy and Safety of Fully Human Bcma Targeting CAR T Cell Therapy in Relapsed/Refractory Multiple Myeloma. Blood 2019, 134 (Suppl. 1), 929. [Google Scholar] [CrossRef]
- Jie, J.; Hao, S.; Jiang, S.; Li, Z.; Yang, M.; Zhang, W.; Yu, K.; Xiao, J.; Meng, H.; Ma, L.; et al. Phase 1 Trial of the Safety and Efficacy of Fully Human Anti-Bcma CAR T Cells in Relapsed/Refractory Multiple Myeloma. Blood 2019, 134 (Suppl. 1), 4435. [Google Scholar] [CrossRef]
- Guedan, S.; Posey, A.D., Jr.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight. 2018, 11, 3. [Google Scholar] [CrossRef] [Green Version]
- Brocker, T.; Karjalainen, K. Signals through T cell receptor-zeta chain alone are insufficient to prime resting T lymphocytes. J. Exp. Med. 1995, 181, 1653–1659. [Google Scholar] [CrossRef]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Rivière, I.; Sadelain, M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat. Biotechnol. 2002, 20, 70–75. [Google Scholar] [CrossRef]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D., Jr.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 712. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Condomines, M.; van der Stegen, S.J.; Perna, F.; Kloss, C.C.; Gunset, G.; Plotkin, J.; Sadelain, M. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 2015, 28, 415–428. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Palomba, M.L.; Batlevi, C.L.; Riviere, I.; Wang, X.; Senechal, B.; Furman, R.R.; Bernal, Y.; Hall, M.; Pineda, J.; et al. A Phase I First-in-Human Clinical Trial of CD19-Targeted 19-28z/4-1BBL Armored CAR T Cells in Patients with Relapsed or Refractory NHL and CLL Including Richter’s Transformation. Blood 2018, 132 (Suppl.1), 224. [Google Scholar] [CrossRef]
- Boucher, J.C.; Li, G.; Shrestha, B.; Zhang, Y.; Vishwasrao, P.; Cabral, M.L.; Guan, L.; Davila, M.L. Mutation of the CD28 costimulatory domain confers increased CAR T cell persistence and decreased exhaustion. J. Immunol. 2018, 200 (Suppl. 1), 57.28. [Google Scholar]
- Feucht, J.; Sun, J.; Eyquem, J.; Ho, Y.J.; Zhao, Z.; Leibold, J.; Dobrin, A.; Cabriolu, A.; Hamieh, M.; Sadelain, M. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat. Med. 2019, 25, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Madar, A.; Casado-Medrano, V.; Shaw, C.; Wing, A.; Liu, F.; Young, R.M.; June, C.H.; Posey, A.D. Single residue in CD28-costimulated CAR-T cells limits long-term persistence and antitumor durability. J. Clin. Investig. 2020, 130, 3087–3097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, D.-G.; Ye, Q.; Poussin, M.; Harms, G.M.; Figini, M.; Powell, D.J. CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood 2012, 119, 696–706. [Google Scholar] [CrossRef]
- Guedan, S.; Chen, X.; Madar, A.; Carpenito, C.; McGettigan, S.E.; Frigault, M.J.; Lee, J.; Posey, A.D.; Scholler, J.; Scholler, N.; et al. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood 2014, 124, 1070–1080. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Pruteanu, I.; Cohen, A.D.; Garfall, A.L.; Milone, M.C.; Tian, L.; Gonzalez, V.E.; Gill, S.; Frey, N.V.; Barrett, D.M.; et al. Identification and Validation of Predictive Biomarkers to CD19- and BCMA-Specific CAR T-Cell Responses in CAR T-Cell Precursors. Blood 2019, 134 (Suppl. 1), 622. [Google Scholar] [CrossRef]
- Busch, D.H.; Fräßle, S.P.; Sommermeyer, D.; Buchholz, V.R.; Riddell, S.R. Role of memory T cell subsets for adoptive immunotherapy. Semin Immunol. 2016, 28, 28–34. [Google Scholar] [CrossRef] [Green Version]
- Kotani, H.; Li, G.; Yao, J.; Mesa, T.E.; Chen, J.; Boucher, J.C.; Yoder, S.J.; Zhou, J.; Davila, M.L. Aged CAR T Cells Exhibit Enhanced Cytotoxicity and Effector Function but Shorter Persistence and Less Memory-like Phenotypes. Blood 2018, 132 (Suppl. 1), 2047. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Beckwith, K.A.; Patel, P.R.; Ruella, M.; Zheng, Z.; Barrett, D.M.; Lacey, S.F.; Melenhorst, J.J.; McGettigan, S.E.; Cook, D.R.; et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood 2016, 127, 1117–1127. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Perazzelli, J.; Grupp, S.A.; Barrett, D.M. Early memory phenotypes drive T cell proliferation in patients with pediatric malignancies. Sci. Transl. Med. 2016, 8, 320ra3. [Google Scholar] [CrossRef]
- Schubert, M.-L.; Hoffmann, J.-M.; Dreger, P.; Müller-Tidow, C.; Schmitt, M. Chimeric antigen receptor transduced T cells: Tuning up for the next generation. Int. J. Cancer 2018, 142, 1738–1747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suen, H.; Brown, R.; Yang, S.; Weatherburn, C.; Ho, P.J.; Woodland, N.; Nassif, N.; Barbaro, P.; Bryant, C.; Hart, D.; et al. Multiple myeloma causes clonal T-cell immunosenescence: Identification of potential novel targets for promoting tumour immunity and implications for checkpoint blockade. Leukemia 2016, 30, 1716–1724. [Google Scholar] [CrossRef] [PubMed]
- Garfall, A.L.; Dancy, E.K.; Cohen, A.D.; Hwang, W.T.; Fraietta, J.A.; Davis, M.M.; Levine, B.L.; Siegel, D.L.; Stadtmauer, E.A.; Vogl, D.T.; et al. T-cell phenotypes associated with effective CAR T-cell therapy in postinduction vs relapsed multiple myeloma. Blood Adv. 2019, 3, 2812–2815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommermeyer, D.; Hudecek, M.; Kosasih, P.L.; Gogishvili, T.; Maloney, D.G.; Turtle, C.J.; Riddell, S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 2016, 30, 492–500. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; Carol, E.O.; Alli, R.; Basham, J.H.; Abdelsamed, H.A.; Palmer, L.E.; Jones, L.L.; Youngblood, B.; Geiger, T.L. PI3K orchestration of the in vivo persistence of chimeric antigen receptor-modified T cells. Leukemia 2018, 32, 1157–1167. [Google Scholar] [CrossRef]
- Zhou, J.; Jin, L.; Wang, F.; Zhang, Y.; Liu, B.; Zhao, T. Chimeric antigen receptor T (CAR-T) cells expanded with IL-7/IL-15 mediate superior antitumor effects. Protein Cell 2019, 10, 764–769. [Google Scholar] [CrossRef] [Green Version]
- Ajina, A.; Maher, J. Strategies to Address Chimeric Antigen Receptor Tonic Signaling. Mol. Cancer Ther. 2018, 17, 1795–1815. [Google Scholar] [CrossRef] [Green Version]
- Calderon, H.; Mamonkin, M.; Guedan, S. Analysis of CAR-Mediated Tonic Signaling. In Chimeric Antigen Receptor T Cells; Humana: New York, NY, USA, 2020. [Google Scholar]
- Hermanson, D.L.; Barnett, B.E.; Rengarajan, S.; Codde, R.; Wang, X.; Tan, Y.; Martin, C.E.; Smith, J.B.; He, J.; Mathur, R.; et al. A Novel Bcma-Specific, Centyrin-Based CAR-T Product for the Treatment of Multiple Myeloma. Blood 2016, 128, 2127. [Google Scholar] [CrossRef]
- Watanabe, N.; Bajgain, P.; Sukumaran, S.; Ansari, S.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Leen, A.M.; Vera, J.F. Fine-tuning the CAR spacer improves T-cell potency. Oncoimmunology 2016, 5, e1253656. [Google Scholar] [CrossRef] [Green Version]
- Hudecek, M.; Sommermeyer, D.; Kosasih, P.L.; Silva-Benedict, A.; Liu, L.; Rader, C.; Jensen, M.C.; Riddell, S.R. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol. Res. 2015, 3, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.L.; Staehr, M.; Masakayan, R.; Tatake, I.J.; Purdon, T.J.; Wang, X.; Wang, P.; Liu, H.; Xu, Y.; Garrett-Thomson, S.C.; et al. Development and Evaluation of an Optimal Human Single-Chain Variable Fragment-Derived BCMA-Targeted CAR T Cell Vector. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 1447–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining “T cell exhaustion”. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, S.; Zhang, B.; Qiao, L.; Zhang, Y.; Zhang, Y. T Cell Dysfunction and Exhaustion in Cancer. Front. Cell Dev. Biol. 2020, 8, 17. [Google Scholar] [CrossRef] [Green Version]
- Yoon, D.H.; Osborn, M.J.; Tolar, J.; Kim, C.J. Incorporation of Immune Checkpoint Blockade into Chimeric Antigen Receptor T Cells (CAR-Ts): Combination or Built-In CAR-T. Int. J. Mol. Sci. 2018, 19, 340. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Hucks, G.E.; Seif, A.E.; Talekar, M.K.; Teachey, D.T.; Baniewicz, D.; Callahan, C.; Gonzalez, V.; Nazimuddin, F.; Gupta, M.; et al. The effect of pembrolizumab in combination with CD19-targeted chimeric antigen receptor (CAR) T cells in relapsed acute lymphoblastic leukemia (ALL). J. Clin. Oncol. 2017, 35 (Suppl. 15), 103. [Google Scholar] [CrossRef]
- Li, A.M.; Hucks, G.E.; Dinofia, A.M.; Seif, A.E.; Teachey, D.T.; Baniewicz, D.; Callahan, C.; Fasano, C.; McBride, B.; Gonzalez, V.; et al. Checkpoint Inhibitors Augment CD19-Directed Chimeric Antigen Receptor (CAR) T Cell Therapy in Relapsed B-Cell Acute Lymphoblastic. Leuk. Blood 2018, 132 (Suppl. 1), 556. [Google Scholar] [CrossRef]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef] [Green Version]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Chun, J.Y.; Lim, W.A.; Marson, A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci. Rep. 2017, 7, 737. [Google Scholar] [CrossRef]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Siriwon, N.; Zhang, X.; Yang, S.; Jin, T.; He, F.; Kim, Y.J.; Mac, J.; Lu, Z.; Wang, S.; et al. Enhanced Cancer Immunotherapy by Chimeric Antigen Receptor-Modified T Cells Engineered to Secrete Checkpoint Inhibitors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 6982–6992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prosser, M.E.; Brown, C.E.; Shami, A.F.; Forman, S.J.; Jensen, M.C. Tumor PD-L1 co-stimulates primary human CD8(+) cytotoxic T cells modified to express a PD1:CD28 chimeric receptor. Mol. Immunol. 2012, 51, 263–272. [Google Scholar] [CrossRef]
- Liu, X.; Ranganathan, R.; Jiang, S.; Fang, C.; Sun, J.; Kim, S.; Newick, K.; Lo, A.; June, C.H.; Zhao, Y.; et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res. 2016, 76, 1578–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Luo, C.; Wang, Y.; Guo, Y.; Dai, H.; Tong, C.; Ti, D.; Wu, Z.; Han, W. PD-1 silencing impairs the anti-tumor function of chimeric antigen receptor modified T cells by inhibiting proliferation activity. J Immunother. Cancer 2019, 7, 209. [Google Scholar] [CrossRef]
- Zah, E.; Nam, E.; Bhuvan, V.; Tran, U.; Ji, B.Y.; Gosliner, S.B.; Wang, X.; Brown, C.E.; Chen, Y.Y. Systematically optimized BCMA/CS1 bispecific CAR-T cells robustly control heterogeneous multiple myeloma. Nat. Commun. 2020, 11, 2283. [Google Scholar] [CrossRef]
- Minnie, S.A.; Hill, G.R. Immunotherapy of multiple myeloma. J. Clin. Investig. 2020, 130, 1565–1575. [Google Scholar] [CrossRef]
- García-Guerrero, E.; Sierro-Martínez, B.; Pérez-Simón, J.A. Overcoming Chimeric Antigen Receptor (CAR) Modified T-Cell Therapy Limitations in Multiple Myeloma. Front. Immunol. 2020, 11, 1128. [Google Scholar] [CrossRef]
- Laurent, S.A.; Hoffmann, F.S.; Kuhn, P.H.; Cheng, Q.; Chu, Y.; Schmidt-Supprian, M.; Hauck, S.M.; Schuh, E.; Krumbholz, M.; Rübsamen, H.; et al. γ-Secretase directly sheds the survival receptor BCMA from plasma cells. Nat. Commun. 2015, 6, 7333. [Google Scholar] [CrossRef]
- Sanchez, E.; Li, M.; Kitto, A.; Li, J.; Wang, C.S.; Kirk, D.T.; Yellin, O.; Nichols, C.M.; Dreyer, M.P.; Ahles, C.P.; et al. B-cell maturation antigen is elevated in multiple myeloma and correlates with disease status and survival. Br. J. Haematol. 2012, 158, 727–738. [Google Scholar] [CrossRef]
- Ghermezi, M.; Li, M.; Vardanyan, S.; Harutyunyan, N.M.; Gottlieb, J.; Berenson, A.; Spektor, T.M.; Andreu-Vieyra, C.; Petraki, S.; Sanchez, E.; et al. Serum B-cell maturation antigen: A novel biomarker to predict outcomes for multiple myeloma patients. Haematologica 2017, 102, 785–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bujarski, S.; Soof, C.; Chen, H.; Li, M.; Sanchez, E.; Wang, C.S.; Emamy-Sadr, M.; Swift, R.A.; Rahbari, K.J.; Patil, S.; et al. Serum b-cell maturation antigen levels to predict progression free survival and responses among relapsed or refractory multiple myeloma patients treated on the phase I IRUX trial. J. Clin. Oncol. 2018, 36 (Suppl. 15), e24313. [Google Scholar] [CrossRef]
- Pont, M.J.; Hill, T.; Cole, G.O.; Abbott, J.J.; Kelliher, J.; Salter, A.I.; Hudecek, M.; Comstock, M.L.; Rajan, A.; Patel, B.K.; et al. γ-Secretase inhibition increases efficacy of BCMA-specific chimeric antigen receptor T cells in multiple myeloma. Blood 2019, 134, 1585–1597. [Google Scholar] [CrossRef] [PubMed]
- Holthof, L.C.; Mutis, T. Challenges for Immunotherapy in Multiple Myeloma: Bone Marrow Microenvironment-Mediated Immune Suppression and Immune Resistance. Cancers 2020, 12, 988. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Garcia, A.; Palazon, A.; Noguera-Ortega, E.; Powell, D.J.; Guedan, S. CAR-T Cells Hit the Tumor Microenvironment: Strategies to Overcome Tumor Escape. Front. Immunol. 2020, 11, 1109. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.S.Y.; Taylor, J.R.; Farzaneh, F.; Kemeny, D.M.; Dibb, N.J.; Maher, J. Harnessing the tumour-derived cytokine, CSF-1, to co-stimulate T-cell growth and activation. Mol. Immunol. 2008, 45, 1276–1287. [Google Scholar] [CrossRef]
- Di Stasi, A.; De Angelis, B.; Rooney, C.M.; Zhang, L.; Mahendravada, A.; Foster, A.E.; Heslop, H.E.; Brenner, M.K.; Dotti, G.; Savoldo, B. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood 2009, 113, 6392–6402. [Google Scholar] [CrossRef] [Green Version]
- Craddock, J.A.; Lu, A.; Bear, A.; Pule, M.; Brenner, M.K.; Rooney, C.M.; Foster, A.E. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J. Immunother. 2010, 33, 780–788. [Google Scholar] [CrossRef] [Green Version]
- Tran, E.; Chinnasamy, D.; Yu, Z.; Morgan, R.A.; Lee, C.C.; Restifo, N.P.; Rosenberg, S.A. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J. Exp. Med. 2013, 210, 1125–1135. [Google Scholar] [CrossRef]
- Wang, L.C.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.E.; Johnson, L.A.; Durham, A.C.; et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res. 2014, 2, 154–166. [Google Scholar] [CrossRef] [Green Version]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 2015, 21, 524–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leen, A.M.; Sukumaran, S.; Watanabe, N.; Mohammed, S.; Keirnan, J.; Yanagisawa, R.; Anurathapan, U.; Rendon, D.; Heslop, H.E.; Rooney, C.M.; et al. Reversal of tumor immune inhibition using a chimeric cytokine receptor. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 1211–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkie, S.; Burbridge, S.E.; Chiapero-Stanke, L.; Pereira, A.C.; Cleary, S.; van der Stegen, S.J.; Spicer, J.F.; Davies, D.M.; Maher, J. Selective expansion of chimeric antigen receptor-targeted T-cells with potent effector function using interleukin-4. J. Biol. Chem. 2010, 285, 25538–25544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammed, S.; Sukumaran, S.; Bajgain, P.; Watanabe, N.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Fisher, W.E.; Leen, A.M.; Vera, J.F. Improving Chimeric Antigen Receptor-Modified T Cell Function by Reversing the Immunosuppressive Tumor Microenvironment of Pancreatic Cancer. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 249–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, T.N.; Lee, P.H.; Vodnala, S.K.; Gurusamy, D.; Kishton, R.J.; Yu, Z.; Eidizadeh, A.; Eil, R.; Fioravanti, J.; Gattinoni, L.; et al. T cells genetically engineered to overcome death signaling enhance adoptive cancer immunotherapy. J. Clin. Investig. 2019, 129, 1551–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruella, M.; Klichinsky, M.; Kenderian, S.S.; Shestova, O.; Ziober, A.; Kraft, D.O.; Feldman, M.; Wasik, M.A.; June, C.H.; Gill, S. Overcoming the Immunosuppressive Tumor Microenvironment of Hodgkin Lymphoma Using Chimeric Antigen Receptor T Cells. Cancer Discov. 2017, 7, 1154–1167. [Google Scholar] [CrossRef] [Green Version]
- Parihar, R.; Rivas, C.; Huynh, M.; Omer, B.; Lapteva, N.; Metelitsa, L.S.; Gottschalk, S.M.; Rooney, C.M. NK Cells Expressing a Chimeric Activating Receptor Eliminate MDSCs and Rescue Impaired CAR-T Cell Activity against Solid Tumors. Cancer Immunol. Res. 2019, 7, 363–375. [Google Scholar] [CrossRef]
- Hoyos, V.; Savoldo, B.; Quintarelli, C.; Mahendravada, A.; Zhang, M.; Vera, J.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Dotti, G. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia 2010, 24, 1160–1170. [Google Scholar] [CrossRef] [Green Version]
- Yeku, O.O.; Purdon, T.J.; Koneru, M.; Spriggs, D.; Brentjens, R.J. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci. Rep. 2017, 7, 10541. [Google Scholar] [CrossRef] [Green Version]
- Chmielewski, M.; Abken, H. CAR T Cells Releasing IL-18 Convert to T-Bethigh FoxO1low Effectors that Exhibit Augmented Activity against Advanced Solid Tumors. Cell Rep. 2017, 21, 3205–3219. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Ren, J.; Luo, Y.; Keith, B.; Young, R.M.; Scholler, J.; Zhao, Y.; June, C.H. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18. Cell Rep. 2017, 20, 3025–3033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chmielewski, M.; Hombach, A.A.; Abken, H. Of CARs and TRUCKs: Chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol. Rev. 2014, 257, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Wei, Q.; Brzostek, J.; Gascoigne, N.R.J. Signaling from T cell receptors (TCRs) and chimeric antigen receptors (CARs) on T cells. Cell Mol. Immunol. 2020, 17, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Etxeberria, I.; Olivera, I.; Bolaños, E.; Cirella, A.; Teijeira, Á.; Berraondo, P.; Melero, I. Engineering bionic T cells: Signal 1, signal 2, signal 3, reprogramming and the removal of inhibitory mechanisms. Cell Mol. Immunol. 2020, 17, 576–586. [Google Scholar] [CrossRef]
- Gattinoni, L.; Finkelstein, S.E.; Klebanoff, C.A.; Antony, P.A.; Palmer, D.C.; Spiess, P.J.; Hwang, L.N.; Yu, Z.; Wrzesinski, C.; Heimann, D.M.; et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J. Exp. Med. 2005, 202, 907–912. [Google Scholar] [CrossRef]
- Corrigan-Curay, J.; Kiem, H.P.; Baltimore, D.; O’reilly, M.; Brentjens, R.J.; Cooper, L.; Forman, S.; Gottschalk, S.; Greenberg, P.; Junghans, R.; et al. T-cell immunotherapy: Looking forward. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 1564–1574. [Google Scholar] [CrossRef] [Green Version]
- Antony, P.A.; Piccirillo, C.A.; Akpinarli, A.; Finkelstein, S.E.; Speiss, P.J.; Surman, D.R.; Palmer, D.C.; Chan, C.C.; Klebanoff, C.A.; Overwijk, W.W.; et al. CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J. Immunol. 2005, 174, 2591–2601. [Google Scholar] [CrossRef] [Green Version]
- Neelapu, S.S. CAR-T efficacy: Is conditioning the key? Blood 2019, 133, 1799–1800. [Google Scholar] [CrossRef]
- Hirayama, A.V.; Gauthier, J.; Hay, K.A.; Voutsinas, J.M.; Wu, Q.; Gooley, T.; Li, D.; Cherian, S.; Chen, X.; Pender, B.S.; et al. The response to lymphodepletion impacts PFS in patients with aggressive non-Hodgkin lymphoma treated with CD19 CAR T cells. Blood 2019, 133, 1876–1887. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Geyer, M.B.; Brentjens, R.J. CD19-targeted CAR T-cell therapeutics for hematologic malignancies: Interpreting clinical outcomes to date. Blood 2016, 127, 3312–3320. [Google Scholar] [CrossRef]
- Zhang, T.; Cao, L.; Xie, J.; Shi, N.; Zhang, Z.; Luo, Z.; Yue, D.; Zhang, Z.; Wang, L.; Han, W.; et al. Efficiency of CD19 chimeric antigen receptor-modified T cells for treatment of B cell malignancies in phase I clinical trials: A meta-analysis. Oncotarget 2015, 6, 33961–33971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turtle, C.J.; Hanafi, L.A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, N.N.; Fry, T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Fry, T.J.; Shah, N.N.; Orentas, R.J.; Stetler-Stevenson, M.; Yuan, C.M.; Ramakrishna, S.; Wolters, P.; Martin, S.; Delbrook, C.; Yates, B.; et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2018, 24, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Martin, N.; Thompson, E.; Dell’Aringa, J.; Paiva, B.; Munshi, N.; San Miguel, J. Correlation of Tumor BCMA Expression with Response and Acquired Resistance to Idecabtagene Vicleucel in the KarMMa Study in Relapsed and Refractory Multiple Myeloma. 2020. Available online: https://library.ehaweb.org/eha/2020/eha25th/294902/nathan.martin.correlation.of.tumor.bcma.expression.with.response.and.acquired.html?f=listing%3D3%2Abrowseby%3D8%2Asortby%3D1%2Amedia%3D1 (accessed on 8 August 2020).
- Da Via, M.C.; Dietrich, O.; Truger, M.; Arampatzi, P.; Duell, J.; Zhou, X.; Tabares, P.; Danhof, S.; Kraus, S.; Meggendorfer, M. Biallelic Deletion of Chromosome 16p Encompassing the BCMA Locus as a Tumor-Intrinsic Resistance Mechanism to BCMA-Directed CAR T in Multiple Myeloma. 2020. Available online: https://library.ehaweb.org/eha/2020/eha25th/294800/leo.rasche.biallelic.deletion.of.chromosome.16p.encompassing.the.bcma.locus.as.html?f=listing%3D0%2Abrowseby%3D8%2Asortby%3D2%2Asearch%3Dcar-t (accessed on 8 August 2020).
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, J.; Naqvi, A.S.; Luo, M.; Wertheim, G.; Paessler, M.; Thomas-Tikhonenko, A.; Rheingold, S.R.; Pillai, V. Heterogeneity of surface CD19 and CD22 expression in B lymphoblastic leukemia. Am. J. Hematol. 2018, 93, E352–E355. [Google Scholar] [CrossRef] [Green Version]
- Schürch, C.M.; Rasche, L.; Frauenfeld, L.; Weinhold, N.; Fend, F. A review on tumor heterogeneity and evolution in multiple myeloma: Pathological, radiological, molecular genetics, and clinical integration. Virchows Arch. Int. J. Pathol. 2020, 476, 337–351. [Google Scholar] [CrossRef]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; Lanauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef] [Green Version]
- Orlando, E.J.; Han, X.; Tribouley, C.; Wood, P.A.; Leary, R.J.; Riester, M.; Levine, J.E.; Qayed, M.; Grupp, S.A.; Boyer, M.; et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat. Med. 2018, 24, 1504–1506. [Google Scholar] [CrossRef]
- Fischer, J.; Paret, C.; El Malki, K.; Alt, F.; Wingerter, A.; Neu, M.A.; Kron, B.; Russo, A.; Lehmann, N.; Roth, L.; et al. CD19 Isoforms Enabling Resistance to CART-19 Immunotherapy Are Expressed in B-ALL Patients at Initial Diagnosis. J. Immunother. 2017, 40, 187–195. [Google Scholar] [CrossRef] [Green Version]
- Gardner, R.; Wu, D.; Cherian, S.; Fang, M.; Hanafi, L.A.; Finney, O.; Smithers, H.; Jensen, M.C.; Riddell, S.R.; Maloney, D.G.; et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood 2016, 127, 2406–2410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacoby, E.; Nguyen, S.M.; Fountaine, T.J.; Welp, K.; Gryder, B.; Qin, H.; Yang, Y.; Chien, C.D.; Seif, A.E.; Lei, H.; et al. CD19 CAR immune pressure induces B-precursor acute lymphoblastic leukaemia lineage switch exposing inherent leukaemic plasticity. Nat. Commun. 2016, 7, 12320. [Google Scholar] [CrossRef] [PubMed]
- Hamieh, M.; Dobrin, A.; Cabriolu, A.; van der Stegen, S.J.; Giavridis, T.; Mansilla-Soto, J.; Eyquem, J.; Zhao, Z.; Whitlock, B.M.; Miele, M.M.; et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature 2019, 568, 112–116. [Google Scholar] [CrossRef]
- Perez-Amill, L.; Suñe, G.; Antoñana-Vildosola, A.; Castella, M.; Najjar, A.; Bonet, J.; Fernández-Fuentes, N.; Inogés, S.; López, A.; Bueno, C.; et al. Preclinical development of a humanized chimeric antigen receptor against B cell maturation antigen for multiple myeloma. Haematologica 2020. [Google Scholar] [CrossRef] [PubMed]
- Ruella, M.; Xu, J.; Barrett, D.M.; Fraietta, J.A.; Reich, T.J.; Ambrose, D.E.; Klichinsky, M.; Shestova, O.; Patel, P.R.; Kulikovskaya, I.; et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat. Med. 2018, 24, 1499–1503. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.A.; Morgan, R.A.; Dudley, M.E.; Cassard, L.; Yang, J.C.; Hughes, M.S.; Kammula, U.S.; Royal, R.E.; Sherry, R.M.; Wunderlich, J.R.; et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 2009, 114, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.; Barrett, D.M. Current status of chimeric antigen receptor therapy for haematological malignancies. Br. J. Haematol. 2016, 172, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Tamura, H.; Ishibashi, M.; Sunakawa, M.; Inokuchi, K. Immunotherapy for Multiple Myeloma. Cancers 2019, 11, 2009. [Google Scholar] [CrossRef] [Green Version]
- Abbott, R.C.; Cross, R.S.; Jenkins, M.R. Finding the Keys to the CAR: Identifying Novel Target Antigens for T Cell Redirection Immunotherapies. Int. J. Mol. Sci. 2020, 21, 515. [Google Scholar] [CrossRef] [Green Version]
- Hosen, N. Chimeric Antigen Receptor T-Cell Therapy for Multiple Myeloma. Cancers 2019, 11, 2024. [Google Scholar] [CrossRef] [Green Version]
- Guedan, S.; Calderon, H.; Posey, A.D.; Maus, M.V. Engineering and Design of Chimeric Antigen Receptors. Mol. Ther. Methods Clin. Dev. 2019, 12, 145–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Zhang, J.; Li, M.; Xu, N.; Qi, W.; Tan, J.; Lou, X.; Yu, Z.; Sun, J.; Wang, Z.; et al. Characterization of novel dual tandem CD19/BCMA chimeric antigen receptor T cells to potentially treat multiple myeloma. Biomark Res. 2020, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.H.; Wada, M.; Pinz, K.G.; Liu, H.; Shuai, X.; Chen, X.; Yan, L.E.; Petrov, J.C.; Salman, H.; Senzel, L.; et al. A compound chimeric antigen receptor strategy for targeting multiple myeloma. Leukemia 2018, 32, 402–412. [Google Scholar] [CrossRef] [PubMed]
- de Larrea, C.F.; Staehr, M.; Lopez, A.V.; Ng, K.Y.; Chen, Y.; Godfrey, W.D.; Purdon, T.J.; Ponomarev, V.; Wendel, H.G.; Brentjens, R.J.; et al. Defining an Optimal Dual-Targeted CAR T-cell Therapy Approach Simultaneously Targeting BCMA and GPRC5D to Prevent BCMA Escape–Driven Relapse in Multiple Myeloma. Blood Cancer Discov. 2020. Available online: https://bloodcancerdiscov.aacrjournals.org/content/early/2020/06/30/2643-3230.BCD-20-0020 (accessed on 9 August 2020).
- Popat, R.; Zweegman, S.; Cavet, J.; Yong, K.; Lee, L.; Faulkner, J.; Kotsopoulou, E.; Al-Hajj, M.; Thomas, S.; Cordoba, S.P.; et al. Phase 1 First-in-Human Study of AUTO2, the First Chimeric Antigen Receptor (CAR) T Cell Targeting APRIL for Patients with Relapsed/Refractory Multiple Myeloma (RRMM). Blood 2019, 134 (Suppl. 1), 3112. [Google Scholar] [CrossRef]
- Li, C.; Mei, H.; Hu, Y.; Guo, T.; Liu, L.; Jiang, H.; Tang, L.; Wu, Y.; Ai, L.; Deng, J.; et al. A Bispecific CAR-T Cell Therapy Targeting Bcma and CD38 for Relapsed/Refractory Multiple Myeloma: Updated Results from a Phase 1 Dose-Climbing Trial. Blood 2019, 134 (Suppl. 1), 930. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, L.; Liu, L.; Wang, J.; Wang, S.; Gao, L.; Zhang, C.; Liu, Y.; Kong, P.; Liu, J.; et al. A Bcma and CD19 Bispecific CAR-T for Relapsed and Refractory Multiple Myeloma. Blood 2019, 134 (Suppl. 1), 3147. [Google Scholar] [CrossRef]
- Yan, L.; Yan, Z.; Shang, J.; Shi, X.; Jin, S.; Kang, L.; Qu, S.; Zhou, J.; Kang, H.; Wang, R.; et al. Sequential CD19- and Bcma-Specific Chimeric Antigen Receptor T Cell Treatment for RRMM: Report from a Single Center Study. Blood 2019, 134 (Suppl. 1), 578. [Google Scholar] [CrossRef]
- Garcia-Guerrero, E.; Rodríguez-Lobato, L.G.; Danhof, S.; Sierro-Martínez, B.; Goetz, R.; Sauer, M. ATRA Augments BCMA Expression on Myeloma Cells and Enhances Recognition By BCMA-CAR T-Cells. Blood 2020, 136 (Suppl. 1), 13–14. [Google Scholar] [CrossRef]
- Cowan, A.J.; Pont, M.; Sather, B.D.; Turtle, C.J.; Till, B.G.; Nagengast, A.M.; Libby, I.I.I.E.N.; Becker, P.S.; Coffey, D.G.; Tuazon, S.A.; et al. Efficacy and Safety of Fully Human Bcma CAR T Cells in Combination with a Gamma Secretase Inhibitor to Increase Bcma Surface Expression in Patients with Relapsed or Refractory Multiple Myeloma. Blood 2019, 134 (Suppl. 1), 204. [Google Scholar] [CrossRef]
- Pinto, V.; Bergantim, R.; Caires, H.R.; Seca, H.; Guimarães, J.E.; Vasconcelos, M.H. Multiple Myeloma: Available Therapies and Causes of Drug Resistance. Cancers 2020, 12, 407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basak, G.W.; Carrier, E. The Search for Multiple Myeloma Stem Cells: The Long and Winding Road. Biol Blood Marrow Transpl. 2010, 16, 587–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, W.; Wang, Q.; Barber, J.P.; Brennan, S.; Smith, B.D.; Borrello, I.; McNiece, I.; Lin, L.; Ambinder, R.F.; Peacock, C.; et al. Clonogenic multiple myeloma progenitors, stem cell properties, and drug resistance. Cancer Res. 2008, 68, 190–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paiva, B.; Puig, N.; Cedena, M.T.; de Jong, B.G.; Ruiz, Y.; Rapado, I.; Martinez-Lopez, J.; Cordon, L.; Alignani, D.; Delgado, J.A.; et al. Differentiation stage of myeloma plasma cells: Biological and clinical significance. Leukemia 2017, 31, 382–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, M.; Radhakrishnan, S.V.; Luetkens, T.; Atanackovic, D. The role of surface molecule CD229 in Multiple Myeloma. Clin. Immunol. 2019, 204, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Garfall, A.L.; Stadtmauer, E.A.; Hwang, W.T.; Lacey, S.F.; Melenhorst, J.J.; Krevvata, M.; Carroll, M.P.; Matsui, W.H.; Wang, Q.; Dhodapkar, M.V.; et al. Anti-CD19 CAR T cells with high-dose melphalan and autologous stem cell transplantation for refractory multiple myeloma. JCI Insight 2019, 21, 4. [Google Scholar] [CrossRef]
- Atanackovic, D.; Panse, J.; Hildebrandt, Y.; Jadczak, A.; Kobold, S.; Cao, Y.; Templin, J.; Meyer, S.; Reinhard, H.; Bartels, K.; et al. Surface molecule CD229 as a novel target for the diagnosis and treatment of multiple myeloma. Haematologica 2011, 96, 1512–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nerreter, T.; Letschert, S.; Götz, R.; Doose, S.; Danhof, S.; Einsele, H.; Sauer, M.; Hudecek, M. Super-resolution microscopy reveals ultra-low CD19 expression on myeloma cells that triggers elimination by CD19 CAR-T. Nat. Commun. 2019, 10, 3137. [Google Scholar] [CrossRef] [PubMed]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics 2016, 3, 16011. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—Assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef]
- Shimabukuro-Vornhagen, A.; Gödel, P.; Subklewe, M.; Stemmler, H.J.; Schlößer, H.A.; Schlaak, M.; Kochanek, M.; Böll, B.; von Bergwelt-Baildon, M.S. Cytokine release syndrome. J. Immunother. Cancer 2018, 6, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neelapu, S.S. Managing the toxicities of CAR T-cell therapy. Hematol. Oncol. 2019, 37 (Suppl. 1), 48–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yáñez, L.; Sánchez-Escamilla, M.; Perales, M.-A. CAR T Cell Toxicity: Current Management and Future Directions. HemaSphere 2019, 3, e186. [Google Scholar] [CrossRef] [PubMed]
- Gust, J.; Hay, K.A.; Hanafi, L.A.; Li, D.; Myerson, D.; Gonzalez-Cuyar, L.F.; Yeung, C.; Liles, W.C.; Wurfel, M.; Lopez, J.A.; et al. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017, 7, 1404–1419. [Google Scholar] [CrossRef] [Green Version]
- Taraseviciute, A.; Tkachev, V.; Ponce, R.; Turtle, C.J.; Snyder, J.M.; Liggitt, H.D.; Myerson, D.; Gonzalez-Cuyar, L.; Baldessari, A.; English, C.; et al. Chimeric Antigen Receptor T Cell-Mediated Neurotoxicity in Nonhuman Primates. Cancer Discov. 2018, 8, 750–763. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transpl. 2019, 25, 625–638. [Google Scholar] [CrossRef] [Green Version]
- Mahadeo, K.M.; Khazal, S.J.; Abdel-Azim, H.; Fitzgerald, J.C.; Taraseviciute, A.; Bollard, C.M.; Tewari, P.; Duncan, C.; Traube, C.; McCall, D.; et al. Management guidelines for paediatric patients receiving chimeric antigen receptor T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 45–63. [Google Scholar] [CrossRef] [Green Version]
- Yakoub-Agha, I.; Chabannon, C.; Bader, P.; Basak, G.W.; Bonig, H.; Ciceri, F.; Corbacioglu, S.; Duarte, R.F.; Einsele, H.; Hudecek, M.; et al. Management of adults and children undergoing chimeric antigen receptor T-cell therapy: Best practice recommendations of the European Society for Blood and Marrow Transplantation (EBMT) and the Joint Accreditation Committee of ISCT and EBMT (JACIE). Haematologica 2020, 105, 297–316. [Google Scholar] [CrossRef]
- Lamers, C.H.; Sleijfer, S.; Van Steenbergen, S.; Van Elzakker, P.; Van Krimpen, B.; Groot, C.; Vulto, A.; Den Bakker, M.; Oosterwijk, E.; Debets, R.; et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: Clinical evaluation and management of on-target toxicity. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 904–912. [Google Scholar] [CrossRef]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Thistlethwaite, F.C.; Gilham, D.E.; Guest, R.D.; Rothwell, D.G.; Pillai, M.; Burt, D.J.; Byatte, A.J.; Kirillova, N.; Valle, J.W.; Sharma, S.K.; et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol. Immunother. 2017, 66, 1425–1436. [Google Scholar] [CrossRef] [PubMed]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Singh, I.; Zudaire, E.; Yeh, T.M.; Allred, A.J.; Olyslager, Y.; Banerjee, A.; Goldberg, J.D.; et al. Update of CARTITUDE-1: A phase Ib/II study of JNJ-4528, a B-cell maturation antigen (BCMA)-directed CAR-T-cell therapy, in relapsed/refractory multiple myeloma. J. Clin. Oncol. 2020, 38 (Suppl. 15), 8505. [Google Scholar] [CrossRef]
- Salter, A.I.; Ivey, R.G.; Kennedy, J.J.; Voillet, V.; Rajan, A.; Alderman, E.J.; Voytovich, U.J.; Lin, C.; Sommermeyer, D.; Liu, L.; et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci. Signal. 2018, 11, eaat6753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Stasi, A.; Tey, S.K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Chang, W.C.; Wong, C.W.; Colcher, D.; Sherman, M.; Ostberg, J.R.; Forman, S.J.; Riddell, S.R.; Jensen, M.C. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood 2011, 118, 1255–1263. [Google Scholar] [CrossRef]
- Mestermann, K.; Giavridis, T.; Weber, J.; Rydzek, J.; Frenz, S.; Nerreter, T.; Mades, A.; Sadelain, M.; Einsele, H.; Hudecek, M. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci. Transl. Med. 2019, 11, eaau5907. [Google Scholar] [CrossRef]
- Caruso, H.G.; Hurton, L.V.; Najjar, A.; Rushworth, D.; Ang, S.; Olivares, S.; Mi, T.; Switzer, K.; Singh, H.; Huls, H.; et al. Tuning Sensitivity of CAR to EGFR Density Limits Recognition of Normal Tissue While Maintaining Potent Antitumor Activity. Cancer Res. 2015, 75, 3505–3518. [Google Scholar] [CrossRef] [Green Version]
- Arcangeli, S.; Rotiroti, M.C.; Bardelli, M.; Simonelli, L.; Magnani, C.F.; Biondi, A.; Biagi, E.; Tettamanti, S.; Varani, L. Balance of Anti-CD123 Chimeric Antigen Receptor Binding Affinity and Density for the Targeting of Acute Myeloid Leukemia. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 1933–1945. [Google Scholar] [CrossRef]
- Han, C.; Sim, S.J.; Kim, S.H.; Singh, R.; Hwang, S.; Kim, Y.I.; Park, S.H.; Kim, K.H.; Lee, D.G.; Oh, H.S.; et al. Desensitized chimeric antigen receptor T cells selectively recognize target cells with enhanced antigen expression. Nat. Commun. 2018, 9, 468. [Google Scholar] [CrossRef] [Green Version]
- Roybal, K.T.; Rupp, L.J.; Morsut, L.; Walker, W.J.; McNally, K.A.; Park, J.S.; Lim, W.A. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell 2016, 164, 770–779. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-Y.; Roybal, K.T.; Puchner, E.M.; Onuffer, J.; Lim, W.A. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science 2015, 350, aab4077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanitis, E.; Poussin, M.; Klattenhoff, A.W.; Song, D.; Sandaltzopoulos, R.; June, C.H.; Powell, D.J. Chimeric antigen receptor T Cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol. Res. 2013, 1, 43–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloss, C.C.; Condomines, M.; Cartellieri, M.; Bachmann, M.; Sadelain, M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat. Biotechnol. 2013, 31, 71–75. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Feng, Z.; Ma, J.; Ling, S.; Cao, Y.; Gurung, B.; Wu, Y.; Katona, B.W.; O’Dwyer, K.P.; Siegel, D.L.; et al. Bispecific and split CAR T cells targeting CD13 and TIM3 eradicate acute myeloid leukemia. Blood 2020, 135, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, V.D.; Themeli, M.; Sadelain, M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci. Transl. Med. 2013, 5, 215ra172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. “Off-the-shelf” allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Somerville, R.P.; Shi, V.; Rose, J.J.; Halverson, D.C.; Fowler, D.H.; Gea-Banacloche, J.C.; Pavletic, S.Z.; Hickstein, D.D.; Lu, T.L.; et al. Allogeneic T Cells That Express an Anti-CD19 Chimeric Antigen Receptor Induce Remissions of B-Cell Malignancies That Progress After Allogeneic Hematopoietic Stem-Cell Transplantation Without Causing Graft-Versus-Host Disease. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 1112–1121. [Google Scholar] [CrossRef] [Green Version]
- Kochenderfer, J.N.; Dudley, M.E.; Carpenter, R.O.; Kassim, S.H.; Rose, J.J.; Telford, W.G.; Hakim, F.T.; Halverson, D.C.; Fowler, D.H.; Hardy, N.M.; et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood 2013, 122, 4129–4139. [Google Scholar] [CrossRef]
- Chu, J.; Deng, Y.; Benson, D.M.; He, S.; Hughes, T.; Zhang, J.; Peng, Y.; Mao, H.; Yi, L.; Ghoshal, K.; et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia 2014, 28, 917–927. [Google Scholar] [CrossRef] [Green Version]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef]
- Torikai, H.; Reik, A.; Liu, P.Q.; Zhou, Y.; Zhang, L.; Maiti, S.; Huls, H.; Miller, J.C.; Kebriaei, P.; Rabinovitch, B.; et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood 2012, 119, 5697–5705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poirot, L.; Philip, B.; Schiffer-Mannioui, C.; Le Clerre, D.; Chion-Sotinel, I.; Derniame, S.; Potrel, P.; Bas, C.; Lemaire, L.; Galetto, R.; et al. Multiplex Genome-Edited T-cell Manufacturing Platform for “Off-the-Shelf” Adoptive T-cell Immunotherapies. Cancer Res. 2015, 75, 3853–3864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, C.J.; Peacock, S.; Chaudhry, A.N.; Bradley, J.A.; Bolton, E.M. Generating an iPSC bank for HLA-matched tissue transplantation based on known donor and recipient HLA types. Cell Stem Cell 2012, 11, 147–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Quan, Y.; Yan, Q.; Morales, J.E.; Wetsel, R.A. Targeted Disruption of the β2-Microglobulin Gene Minimizes the Immunogenicity of Human Embryonic Stem Cells. Stem Cells Transl. Med. 2015, 4, 1234–1245. [Google Scholar] [CrossRef]
- Sommer, C.; Boldajipour, B.; Valton, J.; Galetto, R.; Bentley, T.; Sutton, J.; Ni, Y.; Leonard, M.; Van Blarcom, T.; Smith, J.; et al. ALLO-715, an Allogeneic BCMA CAR T Therapy Possessing an Off-Switch for the Treatment of Multiple Myeloma. Blood 2018, 132 (Suppl. 1), 591. [Google Scholar] [CrossRef]
- Cranert, S.A.; Richter, M.; Tong, M.; Weiss, L.; Tan, Y.; Ostertag, E.M.; Coronella, J.; Shedlock, D.J. Manufacture of an Allogeneic CAR-T Stem Cell Memory Product Candidate for Multiple Myeloma, P-Bcma-ALLO1, Is Robust, Reproducible and Highly Scalable. Blood 2019, 134 (Suppl. 1), 4445. [Google Scholar] [CrossRef]
- Mathur, R.; Zhang, Z.; He, J.; Galetto, R.; Gouble, A.; Chion-Sotinel, I.; Filipe, S.; Gariboldi, A.; Veeramachaneni, T.; Manasanch, E.E.; et al. Universal SLAMF7-Specific CAR T-Cells As Treatment for Multiple Myeloma. Blood 2017, 130 (Suppl. 1), 502. [Google Scholar]
- Mailankody, S.; Matous, J.V.; Liedtke, M.; Sidana, S.; Malik, S.; Nath, R. Universal: An Allogeneic First-in-Human Study of the Anti-Bcma ALLO-715 and the Anti-CD52 ALLO-647 in Relapsed/Refractory Multiple Myeloma. Blood 2020, 136 (Suppl. 1), 24–25. [Google Scholar] [CrossRef]
- Santomasso, B.; Bachier, C.; Westin, J.; Rezvani, K.; Shpall, E.J. The Other Side of CAR T-Cell Therapy: Cytokine Release Syndrome, Neurologic Toxicity, and Financial Burden. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 433–444. [Google Scholar] [CrossRef]
- Nam, S.; Smith, J.; Yang, G. Driving the next wave of innovation in CAR T-cell Therapies|McKinsey. 2019. Available online: https://www.mckinsey.com/industries/pharmaceuticals-and-medical-products/our-insights/driving-the-next-wave-of-innovation-in-car-t-cell-therapies (accessed on 15 August 2020).
- ICER (Institute for Clinical and Economic Review); CTAF (California Technology Assessment Forum). Chimeric Antigen Receptor T-Cell Therapy for B-Cell Cancers: Effectiveness and Value. In Final Evidence Report; ICER: Boston, MA, USA, 2018. [Google Scholar]
- Lin, J.K.; Lerman, B.J.; Barnes, J.I.; Boursiquot, B.C.; Tan, Y.J.; Robinson, A.Q.; Davis, K.L.; Owens, D.K.; Goldhaber-Fiebert, J.D. Cost Effectiveness of Chimeric Antigen Receptor T-Cell Therapy in Relapsed or Refractory Pediatric B-Cell Acute Lymphoblastic. Leuk. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 13, JCO2018790642. [Google Scholar] [CrossRef] [Green Version]
- Kebriaei, P.; Izsvák, Z.; Narayanavari, S.A.; Singh, H.; Ivics, Z. Gene Therapy with the Sleeping Beauty Transposon System. Trends Genet TIG 2017, 33, 852–870. [Google Scholar] [CrossRef] [PubMed]
- Ramanayake, S.; Bilmon, I.; Bishop, D.; Dubosq, M.C.; Blyth, E.; Clancy, L.; Gottlieb, D.; Micklethwaite, K. Low-cost generation of Good Manufacturing Practice-grade CD19-specific chimeric antigen receptor-expressing T cells using piggyBac gene transfer and patient-derived materials. Cytotherapy 2015, 17, 1251–1267. [Google Scholar] [CrossRef] [PubMed]
- Rosenblatt, J.; Avivi, I.; Binyamini, N.; Uhl, L.; Somaiya, P.; Stroopinsky, D.; Palmer, K.A.; Coll, M.D.; Katz, T.; Bisharat, L.; et al. Blockade of PD-1 in Combination with Dendritic Cell/Myeloma Fusion Cell Vaccination Following Autologous Stem Cell Transplantation Is Well Tolerated, Induces Anti-Tumor Immunity and May Lead to Eradication of Measureable Disease. Blood 2015, 126, 4218. [Google Scholar] [CrossRef]
- Yang, Y. Cancer immunotherapy: Harnessing the immune system to battle cancer. J. Clin. Investig. 2015, 125, 3335–3337. [Google Scholar] [CrossRef] [Green Version]
- Ventola, C.L. Cancer Immunotherapy, Part 3: Challenges and Future Trends. Pharm. Ther. 2017, 42, 514–521. [Google Scholar]
- Pardoll, D. Cancer and the Immune System: Basic Concepts and Targets for Intervention. Semin Oncol. 2015, 42, 523–538. [Google Scholar] [CrossRef] [Green Version]
- Zugazagoitia, J.; Guedes, C.; Ponce, S.; Ferrer, I.; Molina-Pinelo, S.; Paz-Ares, L. Current Challenges in Cancer Treatment. Clin. Ther. 2016, 38, 1551–1566. [Google Scholar] [CrossRef] [Green Version]
- Pawlyn, C.; Davies, F.E. Toward personalized treatment in multiple myeloma based on molecular characteristics. Blood 2019, 133, 660–675. [Google Scholar] [CrossRef] [Green Version]
- Krzyszczyk, P.; Acevedo, A.; Davidoff, E.J.; Timmins, L.M.; Marrero-Berrios, I.; Patel, M.; White, C.; Lowe, C.; Sherba, J.J.; Hartmanshenn, C.; et al. The growing role of precision and personalized medicine for cancer treatment. Technology 2018, 6, 79–100. [Google Scholar] [CrossRef] [Green Version]
- Le Tourneau, C.; Borcoman, E.; Kamal, M. Molecular profiling in precision medicine oncology. Nat. Med. 2019, 25, 711–712. [Google Scholar] [CrossRef]
- Rothwell, D.G.; Ayub, M.; Cook, N.; Thistlethwaite, F.; Carter, L.; Dean, E.; Smith, N.; Villa, S.; Dransfield, J.; Clipson, A.; et al. Utility of ctDNA to support patient selection for early phase clinical trials: The TARGET study. Nat. Med. 2019, 25, 738–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sicklick, J.K.; Kato, S.; Okamura, R.; Schwaederle, M.; Hahn, M.E.; Williams, C.B.; De, P.; Krie, A.; Piccioni, D.E.; Miller, V.A.; et al. Molecular profiling of cancer patients enables personalized combination therapy: The I-PREDICT study. Nat. Med. 2019, 25, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Rodon, J.; Soria, J.C.; Berger, R.; Miller, W.H.; Rubin, E.; Kugel, A.; Tsimberidou, A.; Saintigny, P.; Ackerstein, A.; Braña, I.; et al. Genomic and transcriptomic profiling expands precision cancer medicine: The WINTHER trial. Nat. Med. 2019, 25, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Marquart, J.; Chen, E.Y.; Prasad, V. Estimation of the Percentage of US Patients with Cancer Who Benefit From Genome-Driven Oncology. JAMA Oncol. 2018, 4, 1093–1098. [Google Scholar] [CrossRef]
- Hoos, A.; Eggermont, A.M.; Janetzki, S.; Hodi, F.S.; Ibrahim, R.; Anderson, A.; Humphrey, R.; Blumenstein, B.; Old, L.; Wolchok, J. Improved endpoints for cancer immunotherapy trials. J. Natl. Cancer Inst. 2010, 102, 1388–1397. [Google Scholar] [CrossRef] [Green Version]
- Park, J.J.H.; Hsu, G.; Siden, E.G.; Thorlund, K.; Mills, E.J. An overview of precision oncology basket and umbrella trials for clinicians. CA Cancer J. Clin. 2020, 70, 125–137. [Google Scholar] [CrossRef]
- Jørgensen, J.; Hanna, E.; Kefalas, P. Outcomes-based reimbursement for gene therapies in practice: The experience of recently launched CAR-T cell therapies in major European countries. J. Mark Access Health Policy 2020, 8, 1715536. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, U.H.; Cornell, R.F.; Lakshman, A.; Gahvari, Z.J.; McGehee, E.; Jagosky, M.H.; Gupta, R.; Varnado, W.; Fiala, M.A.; Chhabra, S.; et al. Outcomes of Patients with Multiple Myeloma Refractory to CD38-Targeted Monoclonal Antibody Therapy. Leukemia 2019, 33, 2266–2275. [Google Scholar] [CrossRef]
Figure 1.
Different immunotherapeutic strategies to treat multiple myeloma. BCMA, B-cell maturation antigen; CAR, chimeric antigen receptor; LAG3, lymphocyte activation gene-3; PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1; SLAMF7, signaling lymphocyte activation molecule family 7; TCR, T cell receptor; TIGIT, T cell immunoglobulin and ITIM domain; TIM3, T cell immunoglobulin and mucin domain-containing protein 3; WT-1, Wilms’ tumor 1 protein; MAGE-3, melanoma-associated antigen 3.
Figure 1.
Different immunotherapeutic strategies to treat multiple myeloma. BCMA, B-cell maturation antigen; CAR, chimeric antigen receptor; LAG3, lymphocyte activation gene-3; PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1; SLAMF7, signaling lymphocyte activation molecule family 7; TCR, T cell receptor; TIGIT, T cell immunoglobulin and ITIM domain; TIM3, T cell immunoglobulin and mucin domain-containing protein 3; WT-1, Wilms’ tumor 1 protein; MAGE-3, melanoma-associated antigen 3.
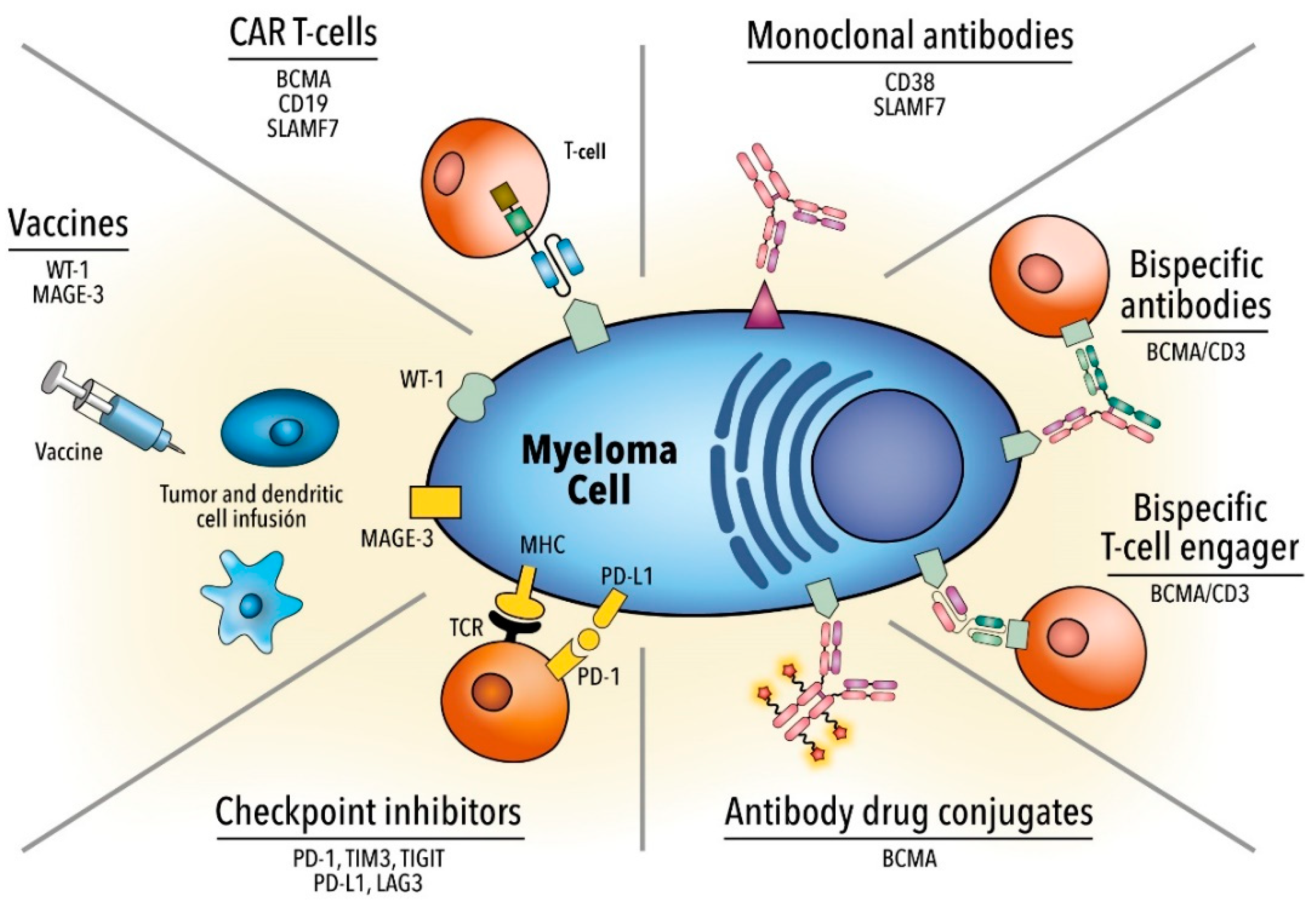
Figure 2.
(A) Structure of a CAR and different generations of chimeric antigen receptors. (B) Schematic representations of different strategies targeting two target antigens simultaneously. TRUCKs, T cells redirected for antigen-unrestricted cytokine-initiated killing; scFv, single-chain variable fragment.
Figure 2.
(A) Structure of a CAR and different generations of chimeric antigen receptors. (B) Schematic representations of different strategies targeting two target antigens simultaneously. TRUCKs, T cells redirected for antigen-unrestricted cytokine-initiated killing; scFv, single-chain variable fragment.

Table 1.
Outcomes of the most important clinical trials using immunotherapy against multiple myeloma.
Table 1.
Outcomes of the most important clinical trials using immunotherapy against multiple myeloma.
| Agent | Target | Specification | Prior Lines | Response | Prognosis | Toxicity |
|---|---|---|---|---|---|---|
| Monoclonal antibodies | CD38 | First-in-human, phase I/II. Monotherapy 16 mg/kg [25,26] | ≥3 | ORR 31.1% sCR 4.7% | PFS 4 mo OS 20.1 mo | IRR 48% (2.7% ≥ grade 3) |
| GEN 503. Part 2: dose expansion with DRd [27,28] | 2 | ORR 81% sCR 25% | PFS 72% OS 90% | IRR 56% (6.3% ≥ grade 3) | ||
| POLLUX phase III DRd vs. Rd. R refractory were excluded [29] | 1 | CR 43.1 vs. 19.2% (p < 0.001) sCR 22.4 vs. 4.6% (p < 0.001) (DRd vs. Rd) | 12 m PFS 83.2 vs. 60.1% OS 91.2 vs. 76.4% (p < 0.001) | IRR 47.7% (6.3% ≥ grade 3); 92% occurring during the first infusion | ||
| CASTOR phase III DVd [30,31] | 2 | ORR 83.8 vs. 63.2% (p < 0.0001) CR or better 28.8 vs. 9.8% (p < 0.0001) sCR 8.8 vs. 2.6% (DVd vs. Vd) | 18 m PFS 48 vs. 7.9% In high-risk cytogenetics PFS 11.2 vs. 7.2% | IRR 45.3% (8.3% ≥ grade 3) | ||
| SLAMF7/CS-1 | E monotherapy. Phase I, dose escalation 0.5–20 mg/kg [32] | ≥2 | No maximum tolerated dose ORR 0% SD 26.5% | NA | IRR 52% before the initiation of prophylaxis | |
| Vd +/− E, randomized phase II [33] | ≥1 | ORR 65 vs. 63% CR 4 vs. 4% (EVd vs. Vd) | PFS 9.7 vs. 6.9 mo OS 85 vs. 74% | IRR 7% (0% ≥ grade 3) | ||
| ELOQUENT-2 Rd +/− E, randomized phase III [34] | 1–3 | ORR 79 vs. 66% (p = 0.0002) (ERd vs. Rd) | 3 y PFS (3y) 26 vs. 18% 3 y-OS 60 vs. 53% (p = 0.026) | Comparable between groups | ||
| Pd +/− E, randomized phase II [35] | ≥2 | ORR 53 vs. 26% (EPd vs. Pd) | PFS 10.3 vs. 4.7 mo | IRR 5% (0% ≥ grade 3) | ||
| ADC | BCMA | GSK-2857916 conjugated to MMAF; phase I [36,37] | ≥3 | ORR 60% CR 9% sCR 6% | PFS 12 mo | Thrombocytopenia 35% Eye-related events: Blurry vision 52%, dry eyes 37%, photophobia 29% |
| CD138 | Indatuximab ravtansine linked to maytansinoid; phases I/II [38,39] | ≥2 | ORR 5.9% CR 0% SD 42.9% | PFS 3 mo | Fatigue 47% Diarrhea 43% | |
| CD56 | Lorvotuzumab-mertansine; phase I [40] | ≥1 | ORR 5.7% CR 0% SD 42.9% | PFS 26.1 weeks in evaluable | Peripheral neuropathy 5.3% | |
| CD74 | Milatuzumab doxorubicin; phase I [41] | ≥2 | No objective responses. SD 5/19 (26%) for 3 mo | NA | n = 1 grade 3 IRR | |
| Bispecific antibodies | BCMA/CD3 | AMG 420: First-in-human, phase I, dose escalation: maximum tolerated 400 µg/day. No extramedullary disease [42,43] | ≥2 | Dose 400 µg/day ORR 70% sCR 50% | Dose 400 µg/day PFS 9 mo | CRS 38.1% (grade ≥ 3 7.1%) Dose-limiting peripheral neuropathy n = 2 |
| Teclistamab; phase I; dose range: 0.3–270 µg/kg [44] | 6 | ORR 78% in patients receiving highest dose | - | CRS 56% (all grade 1/2) Neurotoxicity 8% (3% grade ≥ 3) IRR 9% | ||
| Immune checkpoint inhibitors | PD-1 | Nivolumab monotherapy; phase I including several neoplasms [45] | ≥1 | ORR 4% SD 63% | - | Drug-related AEs 52% any grade, 19% ≥ grade 3 |
| KEYNOTE-183; phase III, randomized Pd +/− Pembrolizumab [46] | ≥2 | ORR 34 vs. 40% (Pembrolizumab + Pd vs. Pd) | PFS 5.6 vs. 8.4 (median time to progression 8.1 vs. 8.7 mo) (Pembrolizumab + Pd vs. Pd) | Serious AE 63 vs. 46% (Pembrolizumab + Pd vs. Pd) TRM n = 4: unknown cause, neutropenic sepsis, myocarditis, Stevens–Johnson syndrome | ||
| KEYNOTE-185; phase III, randomized Rd +/− Pembrolizumab [46] | Newly-diagnosed ASCT ineligible | ORR 64 vs. 62% (Pembrolizumab + Rd vs. Rd) | PFS not reached | Serious AE 54 vs. 39% (Pembrolizumab + Rd vs. Rd) Terminated because of the uneven number of deaths between groups | ||
| CAR T cell | BCMA | NCI scFv murine/CD28 [47] | 9.5 | ORR 81% (≥CR 13%) | mEFS 7.2 mo | CRS 94% (grade ≥ 3 38%) ICANS NA (grade ≥ 3 19%) |
| UPenn/CART-BCMA scFv human/4-1BB [48] | 7 | ORR 64% (≥CR 11%) | mPFS 4.2 mo | CRS 88% (grade ≥ 3 32%) ICANS 32 (grade ≥ 3 12%) | ||
| LCAR-B38M VHH llama/4-1BB [49,50] | 3 | ORR 88% (≥CR 74%) | mPFS 20 mo18 m OS 68% | CRS 89% (grade ≥ 3 7%) ICANS 2 (grade ≥ 3 0%) | ||
| LCAR-B38M VHH llama/4-1BB [51] | 4 | ORR 88% (≥CR 77%) | 1 y PFS 53%1 y OS 82% | CRS 100% (grade ≥ 3 41%) ICANS NA (grade ≥ 3 NA) | ||
| Ciltacabtagene Autolecuel (LCAR-B38M/JNJ68284528) CARTITUDE-1 [52] | 6 | ORR 97% (sCR 67%) | 1 y PFS 76.6% 1 y OS 88.5% | CRS 95% (grade ≥ 3 4%) ICANS 21% (grade ≥ 3 10%) | ||
| Idecabtagene Vicleucel (bb2121)/scFv murine/4-1BB [24] | 7–8 | ORR 85% (≥CR 45%) | mPFS 11.8 mo | CRS 76% (grade ≥ 3 6%) ICANS 42% (grade ≥ 3 3%) | ||
| Idecabtagene Vicleucel (bb2121)/scFv murine/4-1BB KarMMA [53] | 6 | ORR 73% (≥CR 33%) | mPFS 8.8 mo mOS 19.4 mo | CRS 84% (grade ≥ 3 6%) ICANS 18% (grade ≥ 3 3%) | ||
| Orvacabtagene Autoleucel (JCARH125)/scFv human 4-1BB EVOLVE [54] | 6 | ORR 92% (≥CR 36%) | mPFS 9.3 mo | CRS 89% (grade ≥ 3 3%) ICANS 13% (grade ≥ 3 3%) | ||
| Vaccines | Dendritic cells/tumor fusions | Vaccine composed of autologous dendritic cells and patient-derived myeloma cells; 16 patients included [55] | 4 | SD: 11 | - | Site reaction (grade 1) |
| hTERT/Survivin | NCT00499577 [56] | 1 | IR 36% | mEFS 20 mo | Chills 57% Rash > 85% (grades 1–2) | |
| Dendritic cells/tumor fusions | Two cohorts: 24 patients vaccinated post-ASCT 12 patients vaccinated pre- and post- ASCT [57] | - | ORR 78% (CR 47%) | 2 y PFS 57% | Site reaction (grade 1) Myalgia (grade 1) | |
| MAGE-A3 | NCT01245673 [58] | 1–5 | IR 88% | 2 y OS 74% 2 y EFS 56% | Site reaction >90 % | |
| XBP1 CD138 CS1 | NCT01718899: SMM patients; two cohorts: Monotherapy Combination with IMiDs [59] | 1 | IR 95% | mTTP: 36 w mTTP: not reached | Site reaction 58–100% (grades 1–2) | |
| MAGE-A3 | NCT01380145: vaccinated post ASCT [60] | 1–2 | IR 100% | mPFS 27 mo mOS not reached | Site reaction 54% (grade 1)Myalgia 33% (grades 1–2) |
ADC, anti-drug conjugate; AE, adverse events; ASCT, autologous stem cell transplantation; BCMA, B-cell maturation antigen; BiTEs: bi-specific T cell engagers; CAR, chimeric antigen receptor; CR, complete response; CRS, cytokine-release syndrome; D, daratumumab; d, dexamethasone; E, elotuzumab; EFS, event-free survival; ICANS, immune effector cell-associated neurotoxicity syndrome; IMiD, immunomodulatory drug; IR, immune response; IRR, infusion-related reactions; m, median; MMAF, monomethyl auristatin F; mo, months; NA, not available; NCI, National Cancer Institute; ORR, overall response rate; OS, overall survival; P, pomalidomide; PFS, progression-free survival; R, lenalidomide, R/R, relapsed-refractory; SD, stable disease; scFv, single-chain variable fragment; sCR, stringent complete response; TTP, time to progression; UPenn, University of Pennsylvania; V, bortezomib; w, weeks.
Table 3.
Obstacles of CAR T cell therapy and possible strategies to overcome them.
| Limitation | Rationale | Approach |
|---|---|---|
| Antigen-positive escape | Impaired T cell persistence | Optimize CAR design (human scFv, hinge, costimulatory domains) to avoid antigen-independent tonic signaling and reduce antigenicity Younger T cell donors, transduction to stem cell memory T cell and central memory T cells, block T cell differentiation signaling, or use of non-viral transduction systems Genomic knock-in of the CAR sequence to the TRAC locus |
| Impaired T cell potency | Fine-tuning CAR design (human scFv, hinge, costimulatory domains) Avoid antigen-independent tonic signaling Genomic knock-in of the CAR sequence to the TRAC locus Avoid T cell exhaustion (combine with immune check-point inhibitors or disrupt the checkpoint pathway) Reduce the amount of soluble target antigen Optimize lymphodepletion protocol | |
| Tumor microenvironment-induced immunosuppression | Boost T cell trafficking and migration Overcome inhibitory signals by blocking immune check-point pathways or switching inhibitory signals present in the TME into pro-inflammatory signals Targeting immunosuppressive immune cells (regulatory T cells, tumor-associated macrophages, myeloid-derived suppressor cells) Armored CAR T cells or TRUCKs | |
| Antigen-negative escape | Immune selection pressure Gene mutations Lineage switching Trogocytosis Antigen masking | Identification and selection of the most suitable tumor antigen Fine-tuning antigen binding affinity Targeting multiple tumor antigens (sequential or co-administration of single-target CAR products, dual CARs, or tandem CARs) Upregulate surface density of the target antigen Targeting myeloma stem cells |
| Toxicities | CRS and ICANS | Optimizing reduction in the number of CAR T cells infused or dividing doses on different days Prompt recognition with the use of predictive biomarkers Use of tocilizumab or corticosteroids in early stages of the disease Tailored modifications of the construct, optimizing the costimulatory domain CAR T cells with suicide genes or “OFF-switches” |
| On-target, off-tumor | Affinity tuning of the scFv Advanced CAR engineering: “AND” logic-gate, “ON-switch”, “SPLIT”, or inhibitory CARs | |
| Manufacturing | Amount and quality of T cells Vein-to-vein time Production failure | Allogeneic CAR T cells (major concerns: GvHD and CAR T cell rejection) |
| Access and economic | Infrastructure, workflows, processes, regulatory requirements, and economic burden | Cooperation among multiple stakeholders Use of non-viral gene delivery with transposon/transposase systems Creation of community CAR T cell therapy centers Promote the outpatient setting Shift from centralized to decentralized manufacturing, namely “bedside manufacturing” Cost-effectiveness, cost-benefit, and quality-adjusted life-year analyses Outcome-based reimbursement or staged payment models Legitimate value of immunotherapy as shown by real-world evidence and longer follow-ups |
CRS, cytokine-release syndrome; GvHD, graft-versus-host disease; ICANS, immune effector cell-associated neurotoxicity syndrome; scFv, single-chain variable fragment; TME, tumor microenvironment; TRAC, T cell receptor alpha constant; TRUCK, T cells redirected for antigen-unrestricted cytokine-initiated killing.
Table 4.
Some differences between bispecific antibodies and CAR T cell.
| Bispecific Antibodies | CAR T Cell | |
|---|---|---|
| Production | “Off-the-shelf”: No need for manufacturing time, allowing for immediate treatment of the patient | Individual manufacturing for each patient, starting with autologous lymphapheresis Approach: Allogeneic CAR T cells under development |
| Administration | Continuous intravenous infusion Approach: extended half-life bispecific antibodies | Punctual infusion of the product (dose is sometimes split up into several days to reduce AEs) |
| T cell phenotype and effector function | Binding of endogenous CD8+ and CD4+ T cells, which have a superior cytotoxic function than naïve T cells | The product is mostly composed of naïve CD8+ and CD4+ T cells; these cells have higher self-renewal, survival, and penetration in lymphoid tissues |
AE: Adverse events; CAR: Chimeric antigen receptor.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Rodríguez-Lobato, L.G.; Oliver-Caldés, A.; Moreno, D.F.; Fernández de Larrea, C.; Bladé, J. Why Immunotherapy Fails in Multiple Myeloma. Hemato 2021, 2, 1-42. https://0-doi-org.brum.beds.ac.uk/10.3390/hemato2010001
AMA Style
Rodríguez-Lobato LG, Oliver-Caldés A, Moreno DF, Fernández de Larrea C, Bladé J. Why Immunotherapy Fails in Multiple Myeloma. Hemato. 2021; 2(1):1-42. https://0-doi-org.brum.beds.ac.uk/10.3390/hemato2010001
Chicago/Turabian StyleRodríguez-Lobato, Luis Gerardo, Aina Oliver-Caldés, David F. Moreno, Carlos Fernández de Larrea, and Joan Bladé. 2021. "Why Immunotherapy Fails in Multiple Myeloma" Hemato 2, no. 1: 1-42. https://0-doi-org.brum.beds.ac.uk/10.3390/hemato2010001