1. Introduction
Diffuse large B-cell lymphoma (DLBCL) is one of the most frequent histological subtypes of non-Hodgkin lymphoma (NHL) in the Western countries, representing approximately 25% of the cases. DLBCL not-otherwise specified (NOS) is characterized for being a heterogeneous disease because of the morphological characteristics, the biological background, and the genetic alterations [
1]. In the current classification of the World Health Organization (WHO) [
2], DLBCL has some separate diagnostic categories including the T-cell/histiocyte rich large B-cell lymphoma, the primary DLBCL of the mediastinum, and the intravascular lymphoma, etc.
DLBCL can be cured in around 50% of the cases with current therapy, mainly based on the R-CHOP (Rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone). Due to the clinical heterogeneity, it is important to identify the patients with a worse outcome. The International Prognostic Index (IPI) and its derivatives are the main tools being used to stratify the prognosis of the patients with DLBCL. The IPI includes the following variables: age, serum lactate dehydrogenase, Eastern Cooperative Oncology Group (ECOG) performance status, clinical stage, and the number of extranodal disease sites. The variants of the original IPI include the age-adjusted, the stage-adjusted, and the National Comprehensive Cancer Network International Prognostic Index (NCCN IPI) [
3]. The gene expression analysis (GEP) classified the DLBCL patients according to the cell-of-origin as germinal centre B-cell-like (GCB) associated with a good prognosis, and as activated B-cell-like (ABC) associated with a poor prognosis [
4,
5,
6]. Importantly, the role of the immune microenvironment was also highlighted [
7].
The microenvironment is comprised of several immune cells including CD8 + cytotoxic T-lymphocytes, CD4 + helper T-lymphocytes, natural killer (NK) cells, FOXP3 + regulatory T-lymphocytes (Treg), and macrophages, among others [
8]. The tumor-associated macrophages (TAMs) are of special interest because the ones with an M2-like phenotype have tumor-promoting capabilities [
9], which involve tumor proliferation, invasion, angiogenesis, metastasis, and suppression of anti-tumor immunity [
10,
11]. In DLBCL, it has been reported that high numbers are associated with a poor prognosis of DLBCL [
9,
12].
Macrophage colony-stimulating factor 1 receptor (CSF1R) is a tyrosine-protein kinase that functions as a cell-surface receptor for CSF1 and IL34 and regulates the survival, proliferation, and differentiation of macrophages [
13]. Due to the association of TAMs with tumorigenesis and the suppression of the anti-tumor immunity, CSF1R is of great interest as a target for cancer treatment using small molecules CSF1R inhibitors [
14,
15]. In case of Hodgkin Lymphoma, an abstract report by Moskowithz CH et al. described the use of a CSF1R inhibitor (PLX3397) in patients with relapsed or refractory disease, a phase 2 single agent clinical trial, and concluded that the efficacy of single agent PLX3397 in that study population was modest, and that the manageable safety profile and evidence of target inhibition might warrant further testing in combination therapy trials. To the best of our knowledge, the use of CSF1R inhibitors in DLBCL has not been performed [
16].
The purpose of this work was to analyze the expression of CSF1R in DLBCL. First, we analyzed the immunohistochemical protein expression of CSF1R in a series of 198 cases of DLBCL from Tokai University Hospital and performed several clinicopathological correlations. Then, we analyzed the gene expression of CSF1R in DLBCL using a robust series from western countries of 414 from the Lymphoma/Leukemia Molecular Profiling Project (LLMPP), and we focused on the identification of genes associated with the CSF1R as a dichotomic variable (high vs. low levels) and then with other relevant cancer-related genes.
3. Results
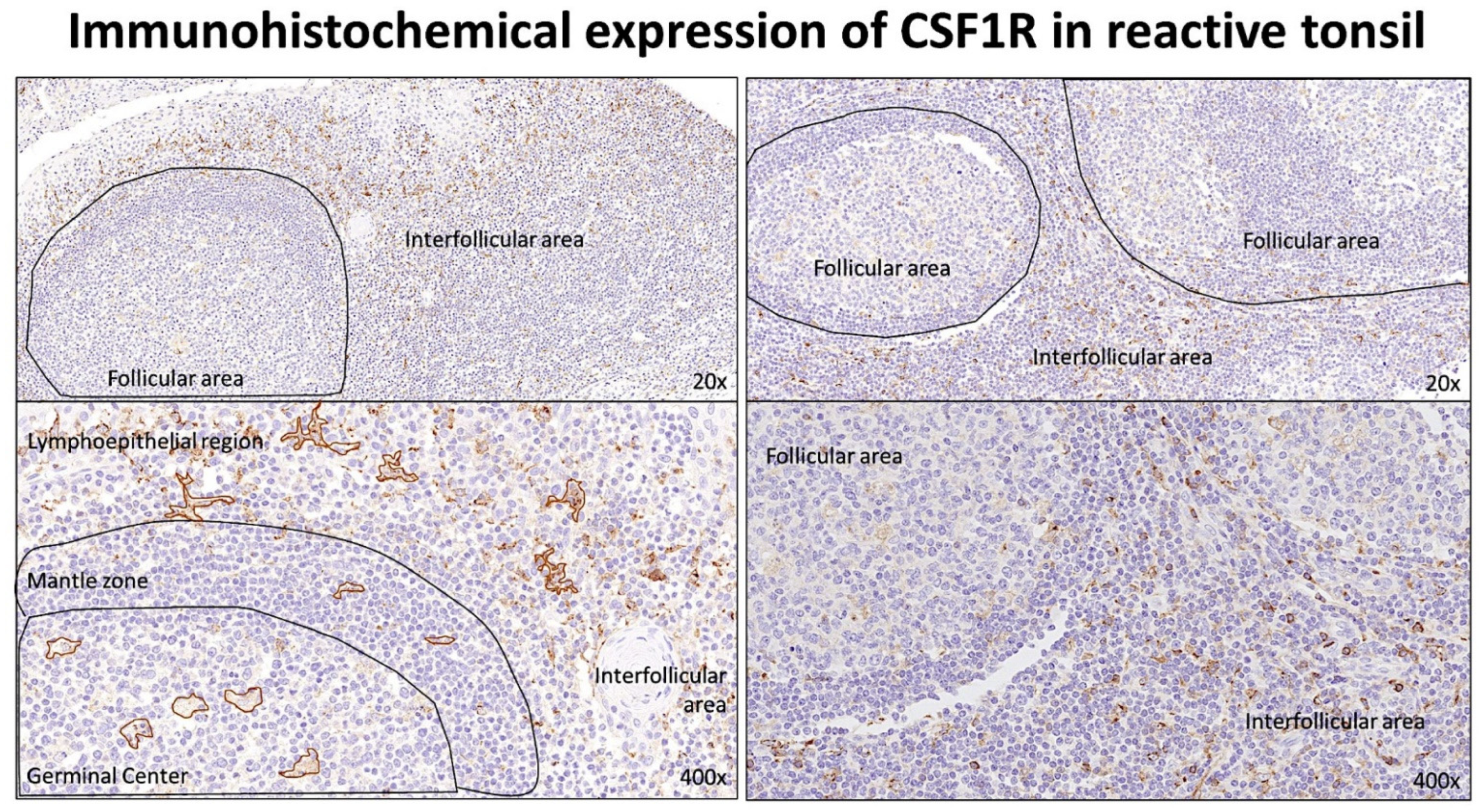
3.1. Immunohistochemical Expression of CSF1R in Reactive Tonsils
The staining for CSF1R was performed in 10 reactive tonsils using an autostainer and, under the microscope, the slides showed that the CSF1R-positive cells had a morphology macrophage. CSF1R-positive cells were distributed both in the follicular and in the interfollicular compartments. In the germinal centers, the CSF1R-positive cells had a morphology compatible with tangible body macrophages. In the interfollicular area, the CSF1R-positive cells had a morphology compatible with macrophages/dendritic cells (
Figure 1).
3.2. Correlation between the Immunohistochemical Expression of CSF1R and Prognosis of the Patients in the Tokai DLBCL Series
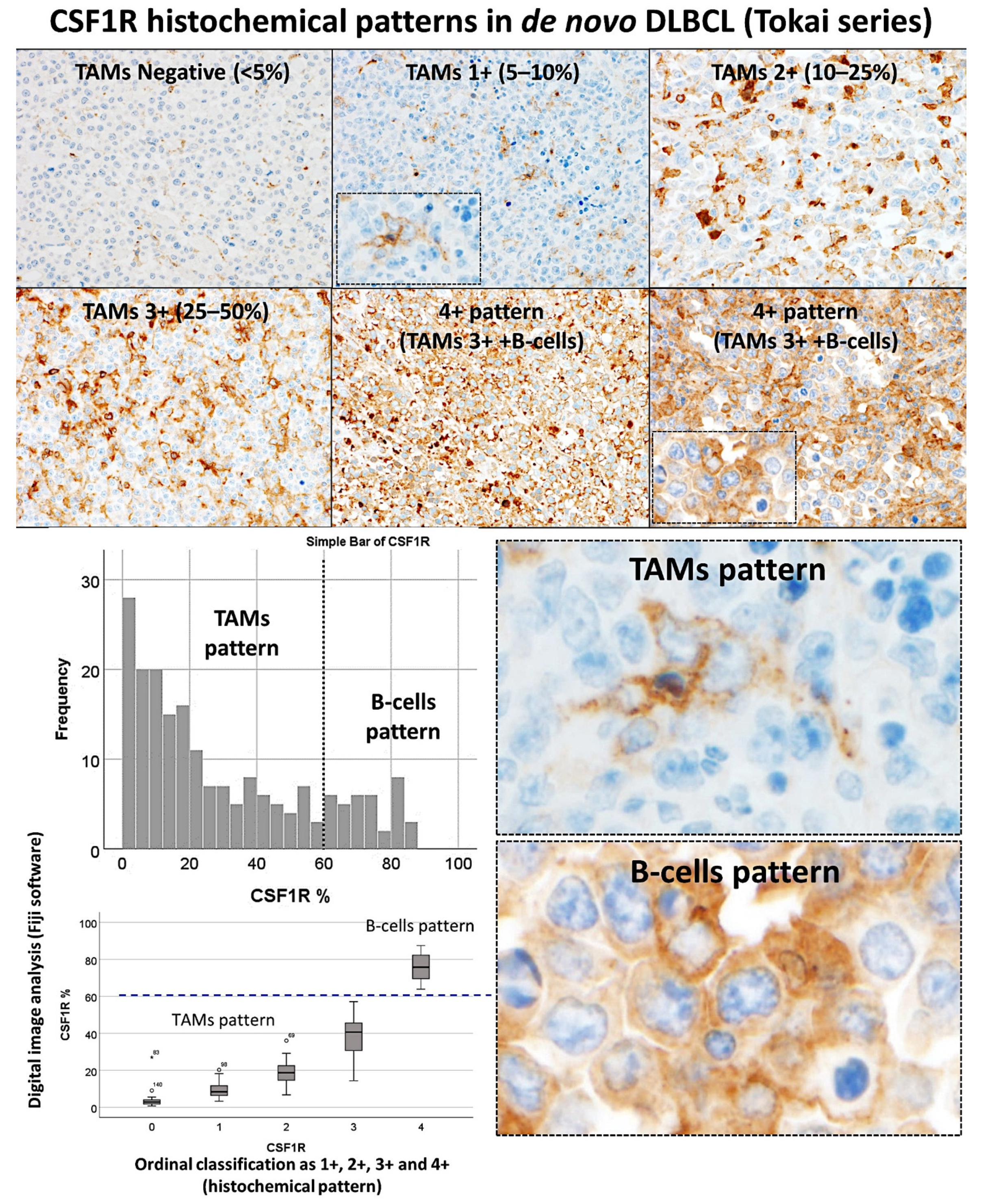
The CSF1R staining was performed in a series of 198 cases of DLBCL. The CSF1R-positive cells had a morphology of tumor-associated macrophages (TAMs). In addition, in some cases, the staining was diffuse (B-cell pattern).
The cases were initially evaluated as an ordinal variable as 0 (no staining, <5%), 1+ (an estimated 5–10% of CSF1R + TAMs), 2+ (10–15%), 3+ (25–50%), and 4+ (diffuse pattern/B-cell pattern). The CSF1R staining in TAMs was of macrophages with dendritiform-like elongations. Conversely, the B-cell pattern showed a diffuse staining of the B-lymphocytes of the DLBCL. After that, the slides were digitalized and a representative area for each case was kept for digital image quantification using Fiji software. The CSF1R expression ranged from 0.37% to 87.45%, with a median of 19.9% and a mean of 29.4% ± 25.6%. The relationship between the ordinal evaluation and the digital image quantification is shown in
Figure 2. At 60% cut-off, the TAMs vs. the B-cell pattern could be differentiated [a receiver operating characteristic (ROC) analysis was not performed in this case].
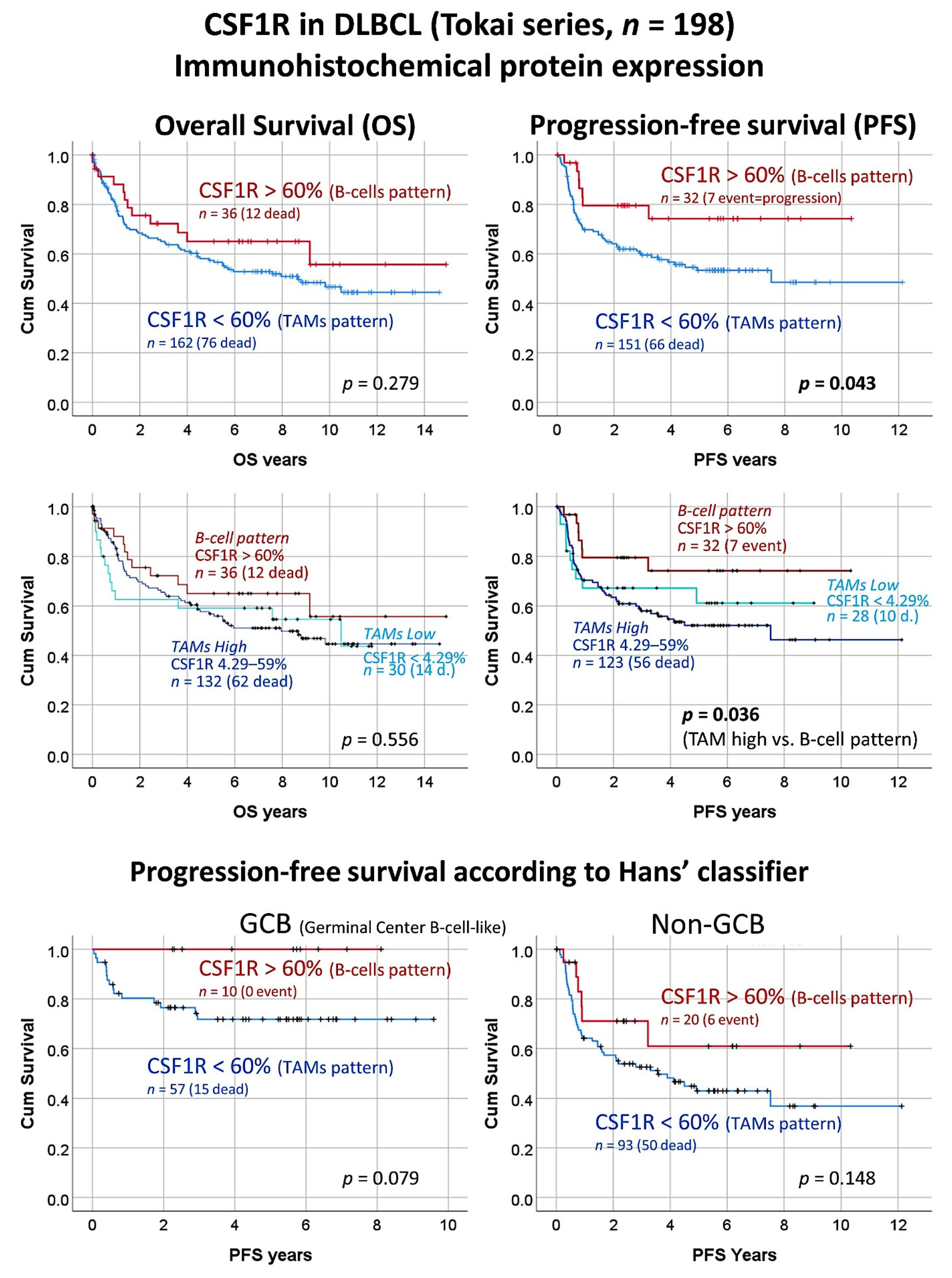
The expression of CSF1R was correlated with several clinicopathological characteristics of the patients from the Tokai series of DLBCL. Using the cut-off of 60% that differentiate the TAMs with the B-cells patterns, two groups of patients with different progression-free survival could be identified. The patients with CSF1R B-cells pattern were characterized with a more favorable progression-free survival (Cox regression, Hazard Risk (HR) = 0.5, 95% confidence interval (CI) for HR 0.2–0.9,
p = 0.049). Conversely, a CSF1R TAMs pattern was associated with an unfavorable progression-free survival (HR = 2.2, 95% CI for HR 1.0–4.8,
p = 0.049) (
Figure 3). Of note, when the group of TAMs pattern was divided into two subgroups, high vs. low, the low CSF1R + TAMs subgroup had a trend of more favorable progression-free survival than the group of high CSF1R + TAMs. When a multivariate Cox regression analysis was performed including the histological pattern of CSF1R (TAMs vs. B-cells patterns) and the IPI (low + low-intermediate vs. high-intermediate + high), only the IPI kept the prognostic relevance for the progression-free survival.
The survival analysis was repeated by stratifying the cases according to the cell-of-origin molecular subtypes based on the Hans’ classifier both for the overall survival and progression-free survival. In case of the overall survival, the CSF1R expression patterns did not correlated with the outcome. Conversely, the progression-free survival tended to keep the prognostic relevance for both the GCB and non-GCB, but this difference was not statistically significant (
p = 0.079 and 0.148, respectively). Of note, our interpretation is that, in a larger series, if the proportion is kept, the difference would be significant because the groups are well separated in the graphs (
Figure 3).
The CSF1R with the 60% cut-off was also correlated with the rest of clinicopathological characteristics of the patients and the samples (
Table 3,
Table 4,
Table 5,
Table 6 and
Table 7). High CSF1R expression (i.e., >60%, B-cells pattern) was associated with a lower MYC proto-oncogene immunohistochemical expression (
p = 0.038), higher MDM2 immunohistochemical expression (
p = 0.051), lower DNA-binding protein IKAROS immunohistochemical expression (
p = 0.042), an absence of
BCL2 translocation (
p = 0.026), an absence of mutation of
MYD88 L265P (
p = 0.028), and lower disease progression (
p = 0.028). No other correlations were found with the other variables, including the cell-of-origin classification (Hans’ classifier).
The expression of CSF1R was correlated with other markers of the CSF1R-pathway including macrophage markers in each of the two histological patterns: The TAMs and the B-cell patterns. In the TAMs pattern group (
n = 162), CSF1R positively correlated with STAT3, TNFAIP8, CD163, and CD68. In the B-cell pattern group (
n = 36), CSF1R inversely correlated with TNFAIP8 and CD163 (
Table 6).
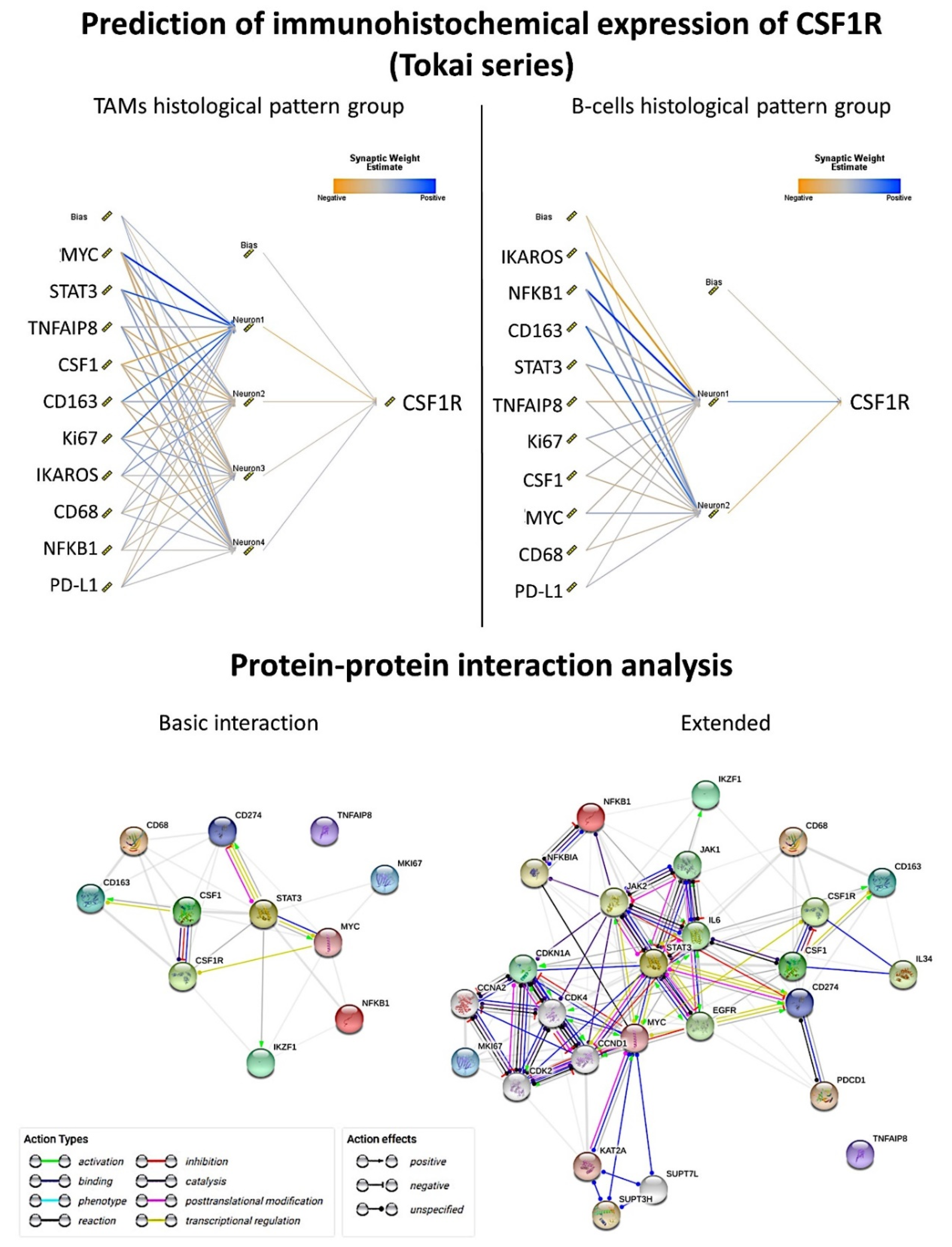
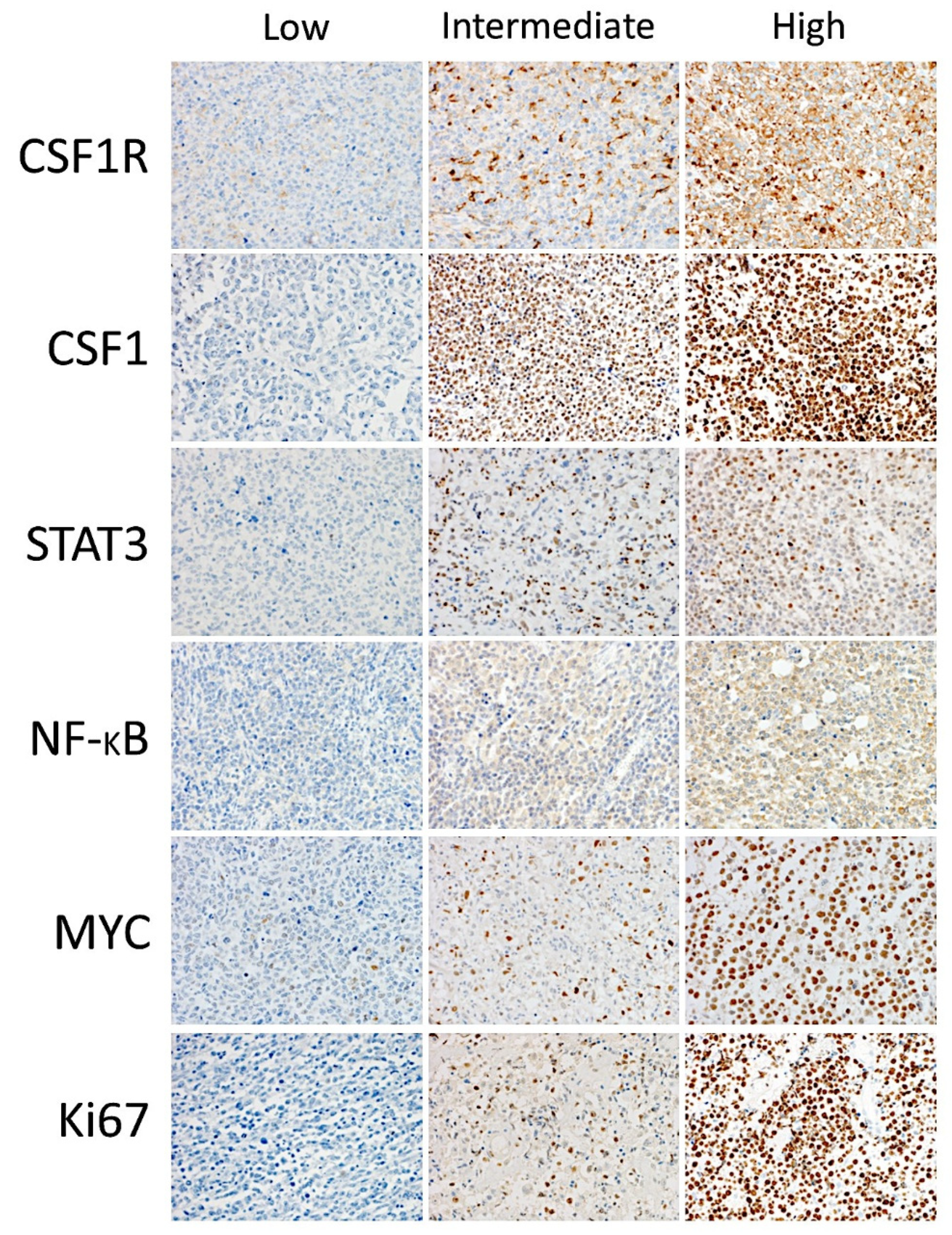
The same type of analysis was performed for each histological pattern using predictive analytics with 12 models including regression, generalized linear, KNN algorithm (nearest neighbor analysis, the number of nearest neighbors to examine is called
k), linear-AS (namely, linear analytic server), LSVM (linear support vector machine), random trees, SVM (support vector machine), tree-AS, linear, CHAID (Chi-squared automatic interaction detection), C&R tree (classification and regression tree), and a neural network (
Figure 4 and
Figure 5). We aimed to predict the CSF1R expression as a quantitative variable by the previous 10 markers (CSF1, STAT3, NFKB1, Ki67, MYC, PD-L1, TNFAIP8, IKAROS, CD163, and CD68).
In the TAMs pattern group (n = 162), good correlation was found using the neural network (multilayer perceptron), regression, and generalized linear model (correlation accuracy >0.70). In the multilayer perceptron analysis, the target CSF1R was predicted by the 10 markers. From most important to least important, the markers were MYC, STAT3, TNFAIP8, CSF1, CD163, Ki67, IKAROS, CD68, NFKB1, and PDL1. The regression analysis showed that CSF1R expression could be calculated as TNFAIP8*0.3 + IKAROS*-3.7 + PD-L1*0.05 + CD163*-0.2 + Ki67*-0.1 + MYC*-0.4 + CSF1*0.04 + NFKB1*-0.1 + CD68*0.4 + STAT3*-0.3 + 35.9.
In the B-cells pattern group (n = 36), a good correlation was found using regression (accuracy 100), generalized linear (100), neural network (0.92), and SVM (0.73). The regression analysis showed that the CSF1R expression could be calculated as TNFAIP8*0.05 + IKAROS*-14.2 + PD-L1*-0.07 + Ki67*-0.3 + CSF1*0.5 + NFKB1*0.1 + CD68*-1.2 + STAT3*0.5 + 125.1. In the multilayer perceptron analysis, the target CSF1R was predicted by the 10 markers. From most important to least important, the markers were IKAROS, NFKB1, CD163, STAT3, TNFAIP8, Ki67, CSF1, MYC, CD68, and PD-L1.
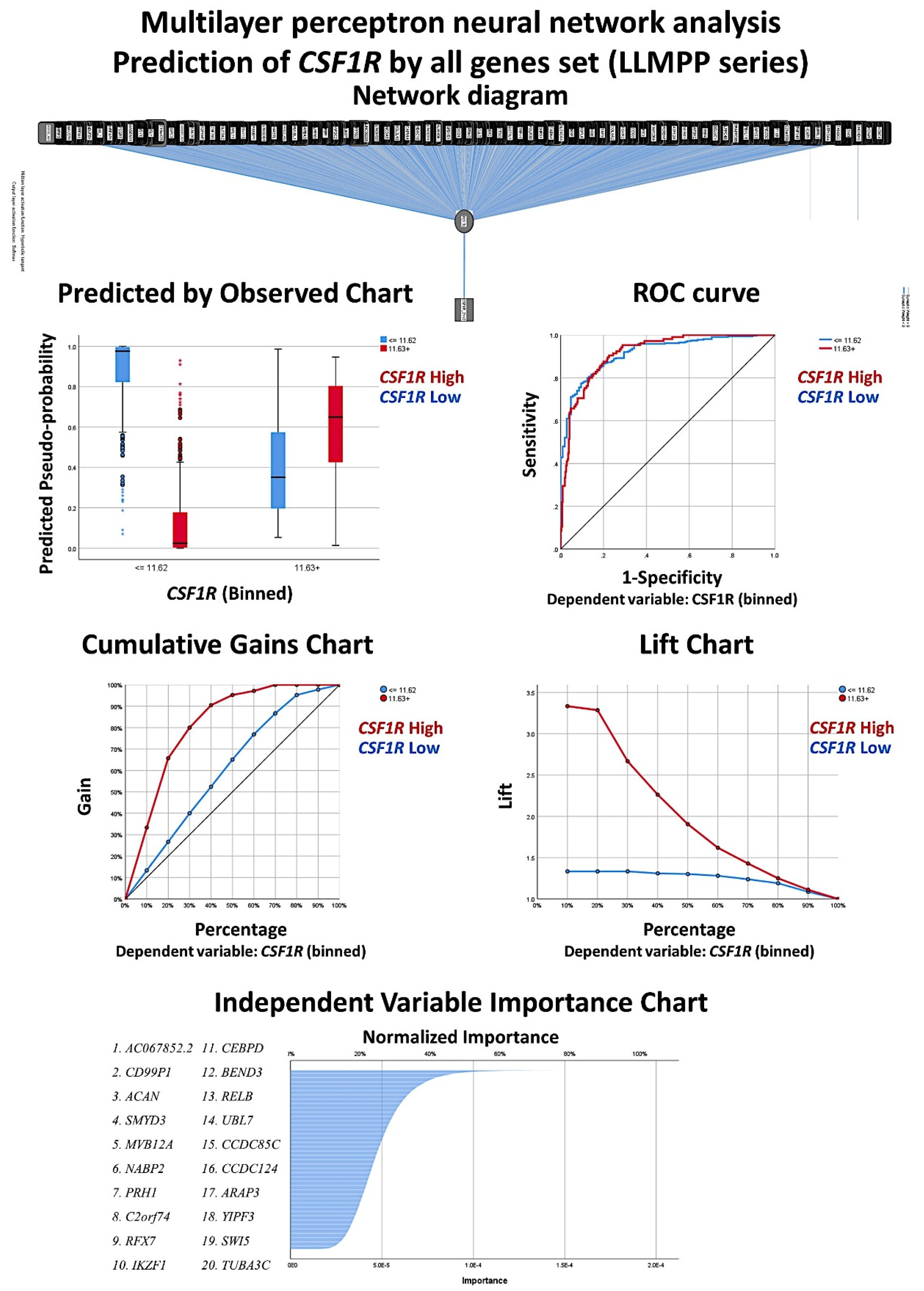
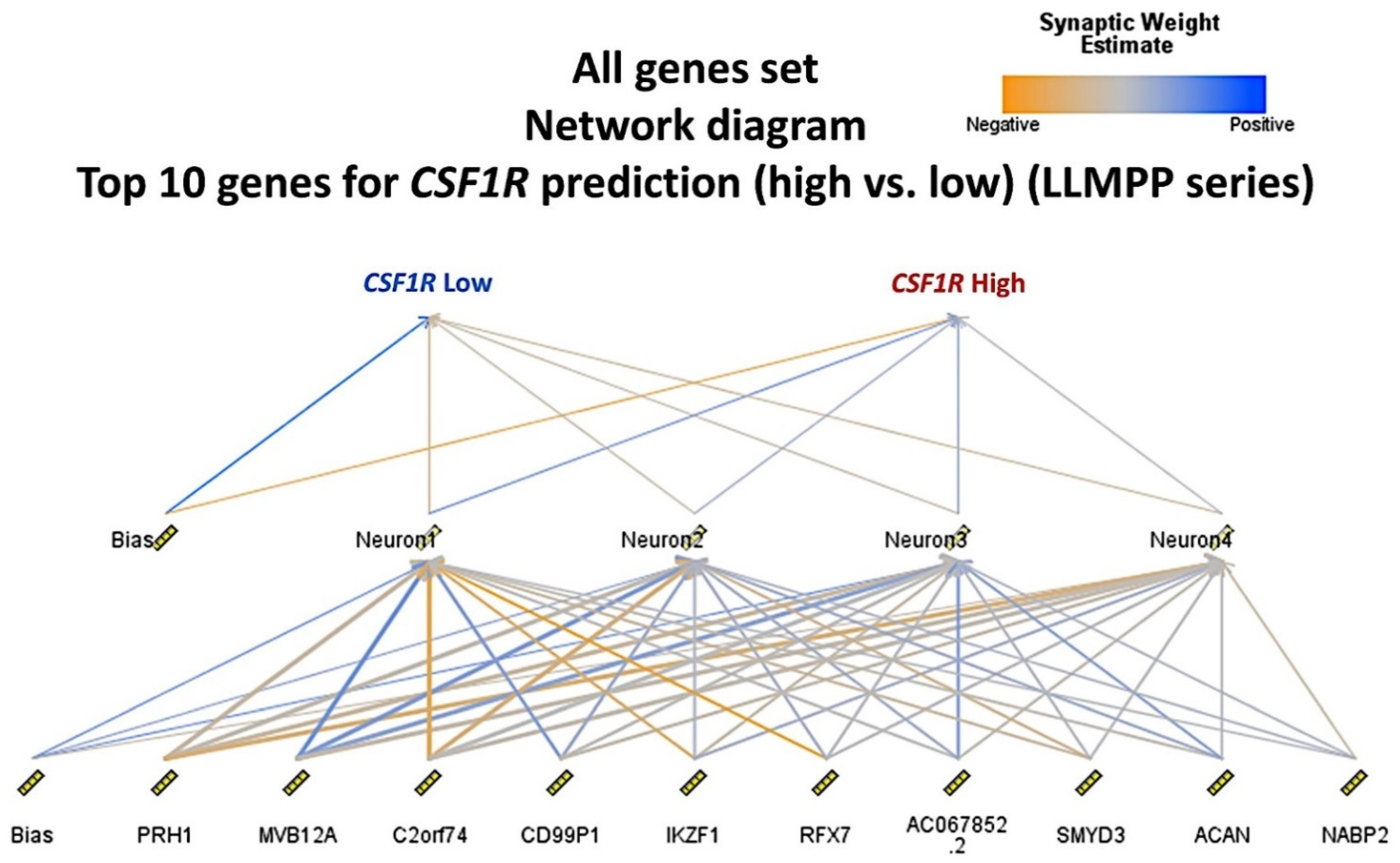
3.3. Identification of the Genes Associated with CSF1R Expression Levels in the LLMPP DLBCL Series
The series of 414 cases was divided into two groups, according to the
CSF1R expression: ≤11.62 (
n = 309, 64.7%), and ≥11.63 (
n = 105, 46.7%). The cut-off was found using the “transform variable“ and the “visual binding“ function of the SPSS software (version 26). When making the cutpoints, equal percentiles were used based on the scanned cases, and the intervals corresponded to the number of desired cutpoints. As a start, 3 cutpoints were set (25% for each interval). Then, the binned variable was subjected to overall survival analysis and a compromise between the statistical significance and a balanced distribution of the samples was found. A multilayer perceptron neural network analysis was performed to identify the most relevant genes associated with the
CSF1R expression (
Figure 6 and
Figure 7). Using this technique, all the genes of the array (
n = 20,683) were ranked according to their normalized importance for predicting the
CSF1R expression as a dichotomic variable (high vs. low, using the cut-off value of 11.63). The neural network performance was good, with an area under the curve of 0.92 and a model with only a 12.2% of incorrect predictions in the training and a 14.9% in the testing set. In this model, the most relevant genes (with a normalized importance >70%) were as follows:
AC067852.2, CD99P1, ACAN, SMYD3, MVB12A, NABP2, PRH1, C2orf74, RFX7, IKZF1, and
CEBPD.
A logistic regression was performed to ascertain the effects of the most relevant genes, that were previously identified in the multilayer perceptron neural network analyses, on the likelihood that the patients have a high CSF1R expression. The genes with a normalized importance >70% were selected and the analysis included univariate and multivariate (backward conditional) tests.
In the multivariate analysis, increasing expression of
CD99P1, MVB12A, IKZF1, and
CEBPD was associated with an increased likelihood of exhibiting high
CSF1R expression, but increasing
PRH1 and
C2orf74 was associated with a reduction in the likelihood of exhibiting high
CSF1R expression (
Table 7).
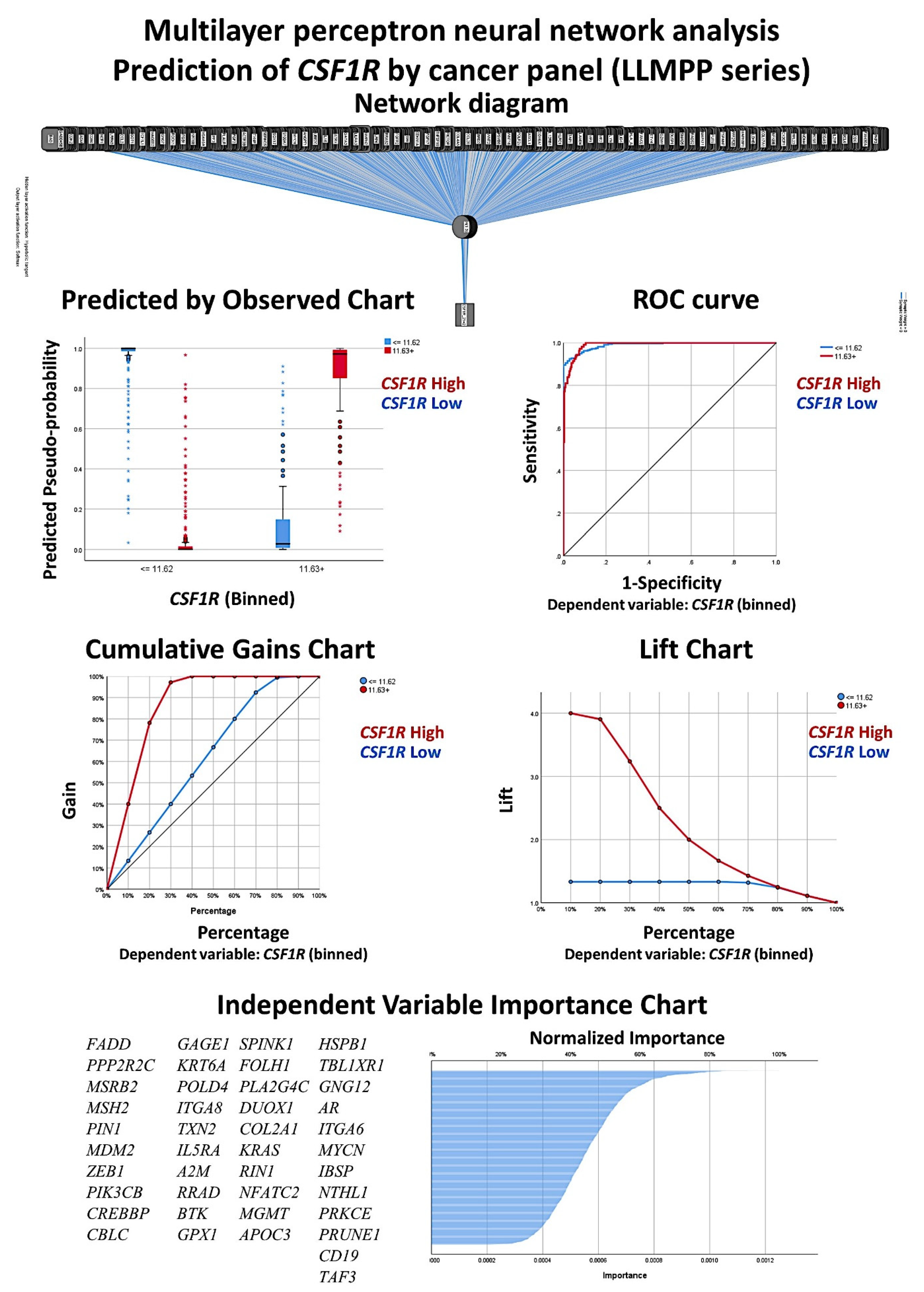
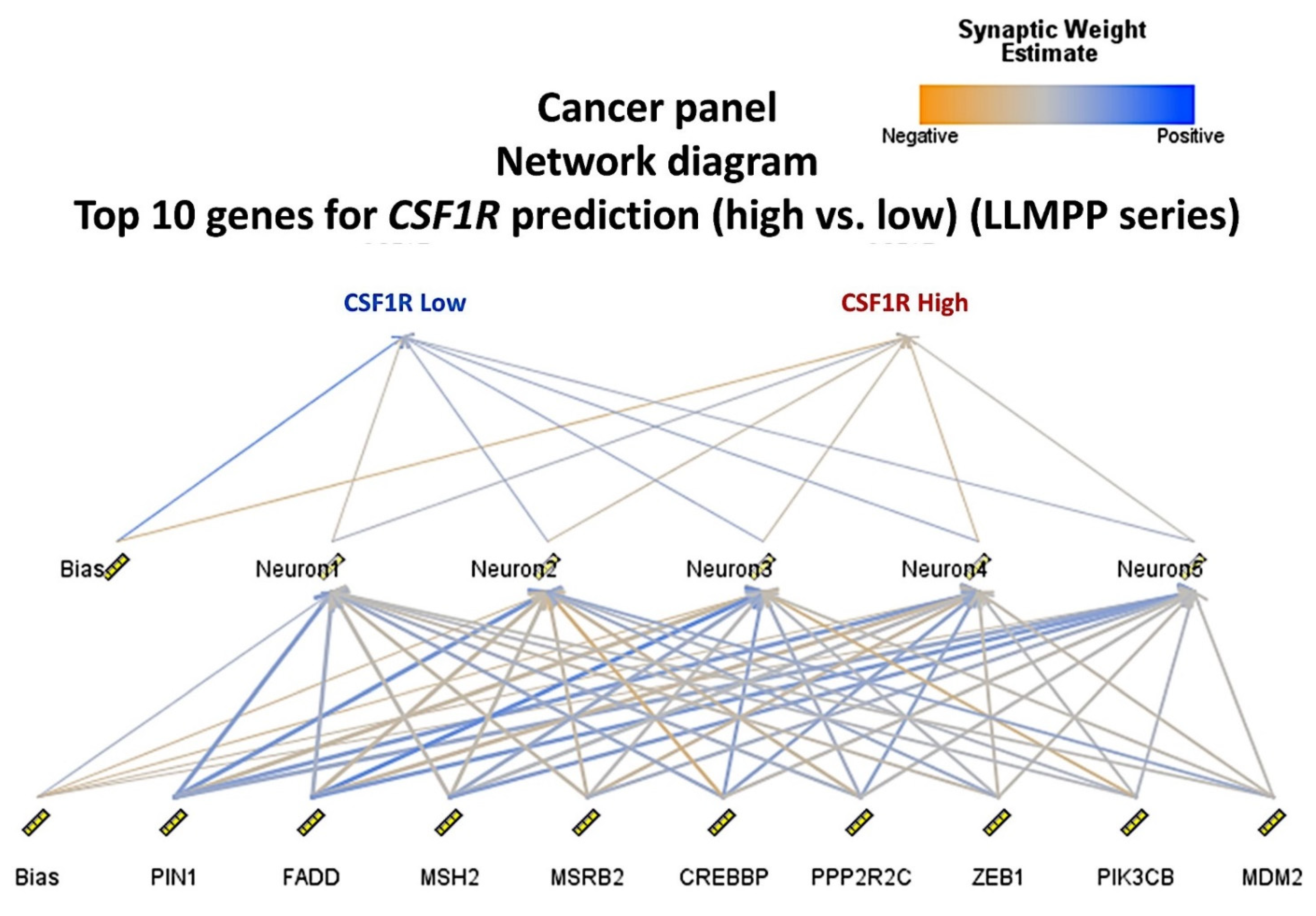
3.4. Identification of the Genes of the Cancer Transcriptome Atlas Panel Associated with CSF1R Levels of the LLMPP DLBCL Series
A multilayer perceptron neural network analysis was performed to identify the most relevant genes of the transcriptome atlas panel associated with the
CSF1R expression (
Figure 8 and
Figure 9). Using this technique, all the genes of the array (
n = 1790) were ranked according to their normalized importance for predicting the
CSF1R expression as a dichotomic variable (high vs. low, using the cut-off value of 11.63). The neural network performance was good, with an area under the curve of 0.99 and a model with only a 3.3% of incorrect predictions in the training and an 11.2% in the testing set. In this model, the most relevant genes (normalized importance >70%) were 42. In order from most to least important were as follows:
FADD (normalized importance 100%)
, PPP2R2C (91%)
, MSRB2 (85%)
, MSH2, PIN1, MDM2, ZEB1, PIK3CB, CREBBP, CBLC, GAGE1, KRT6A, POLD4, ITGA8, TXN2, IL5RA, A2M, RRAD, BTK, GPX1, SPINK1, FOLH1, PLA2G4C, DUOX1, COL2A1, KRAS, RIN1, NFATC2, MGMT, APOC3, HSPB1, TBL1XR1, GNG12, AR, ITGA6, MYCN, IBSP, NTHL1, PRKCE, PRUNE1, CD19, and
TAF3 (70%).
A logistic regression was performed to ascertain the effects of the most relevant genes of the cancer panel, which were previously highlighted in the multilayer perceptron neural network analyses, on the likelihood that the patients have a high CSF1R expression. The genes with a normalized importance >70% were selected and the analysis included univariate and multivariate (backward conditional) tests.
In the multivariate analysis, increasing expression of
PLA2G4C,
RIN1,
NFATC2, and
HSPB1 was associated with an increased likelihood of exhibiting high
CSF1R expression, but increasing
PIN1, TXN2, IL5RA, SPINK1, FOLH1, KRAS, ITGA6, PRKCE, and
TAF3 was associated with a reduction in the likelihood of exhibiting high
CSF1R expression (
Table 8).
3.5. Correlation between Expression Levels of CSF1R and CD47 in the LLMPP DLBCL Series
The
CD47 was one of the genes present in the transcriptome atlas panel set that belongs to the immune checkpoint pathway. It is related to the macrophages’ pathway and it is associated with the prognosis of DLBCL [
31,
32,
33,
34]. An immunohistochemical study showed that CD47 was expressed by the B-lymphocytes of DLBCL, while its receptor SIRPA (namely Tyrosine-protein phosphatase non-receptor type substrate 1) was expressed by the tumor-associated macrophages (TAMs) [
34]. SIRPA (is a relevant immune checkpoint marker because it mediates negative regulation of phagocytosis [
13].
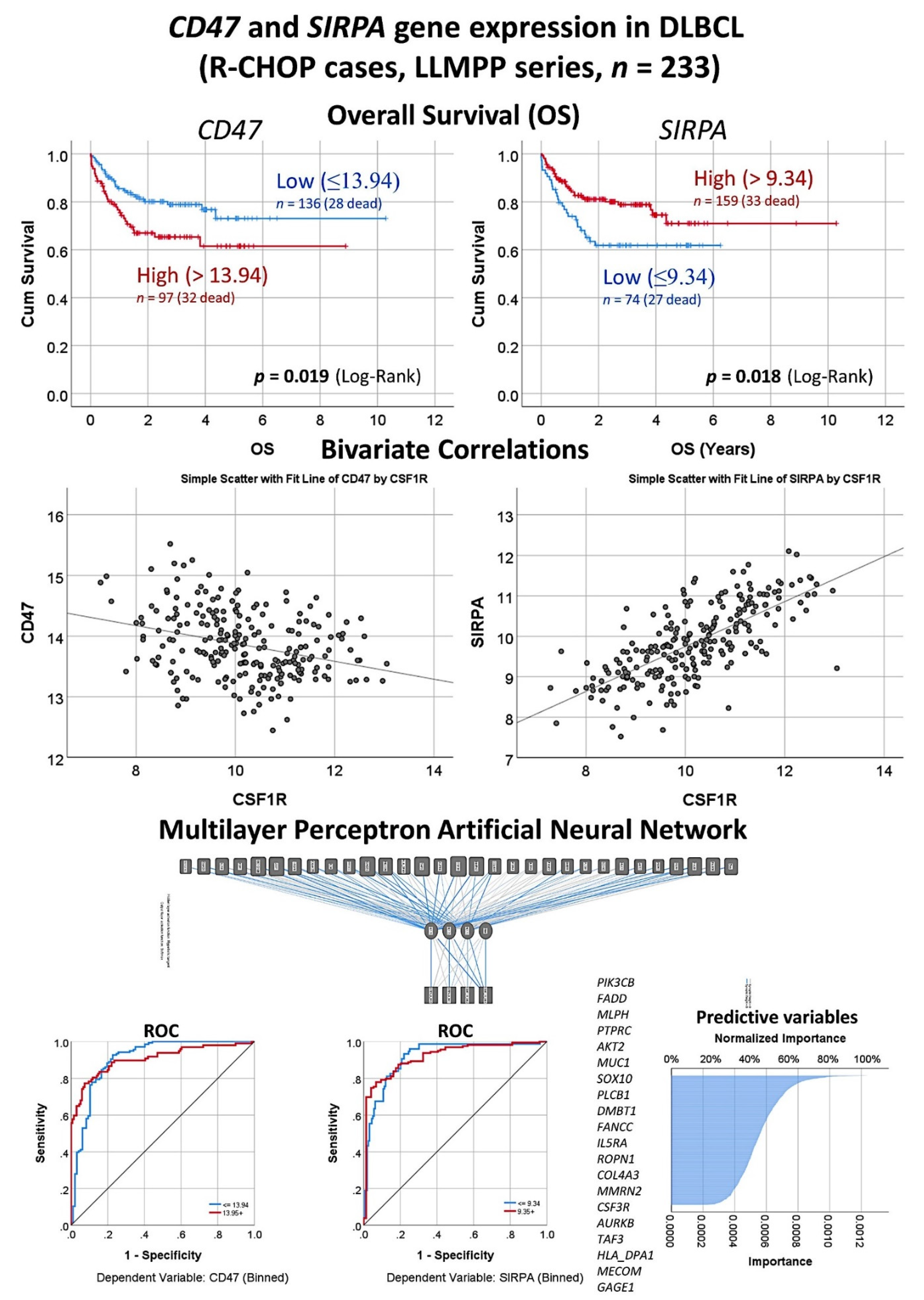
In the 233 DLBCL cases of the LLMPP series with R-CHOP treatment, high gene expression of CD47 correlated with an unfavorable overall survival of the patients (cut-off value = 13.94, Hazard Risk = 1.82,
p = 0.021) (
Figure 10). Conversely, high expression of SIRPA correlated with a favorable overall survival (cut-off value = 9.34, Hazard Risk = 0.55,
p = 0.02). Of note, CD47 and SIRPA gene expression levels inversely correlated between them (Pearson Correlation = −0.3,
p < 0.001). Both markers were correlated with the CSF1R expression (
Figure 10). CSF1R inversely correlated with CD47 (Pearson Correlation = −0.31,
p < 0.001). Conversely, CSF1R strongly correlated positively with SIRPA (Pearson Correlation = 0.71,
p < 0.001). Finally, in order to identify which genes of the transcriptome atlas panel were more associated with the expression of both CD47 and SIRPA, a multilayer perceptron artificial neural network analysis was performed (
Figure 10). The most relevant markers were the following: PIK3CB, FADD, MLPH, PTPRC, AKT2, MUC1, SOX10, PLCB1, DMBT1, and FANCC. Of note, the predictive modeling by the neural network had a high efficiency with an area under the curve (ROC) of 0.91 for both markers.
4. Discussion
Colony stimulating factor 1 receptor (CSF1R), also known as macrophage colony-stimulating factor receptor (M-CSFR) and CD115, is a cell surface protein that functions as a receptor for colony stimulating factor 1 (CSF1) and the Interleukin-34 (IL-34). CSF1R has a role in regulating the homeostatic survival of the tumor-associated macrophages (TAMs). TAMs are relevant because they promote tumorigenesis of many types of cancer, including non-Hodgkin lymphomas [
23,
35,
36,
37,
38]. Therefore, CSF1R is a potentially relevant oncological target.
CSF1R expression was initially thought to be characteristic of myeloid cells, but recent research has shown that non-myeloid cells can also express CSF1R, including malignant B-lymphocytes and classical Hodgkin Lymphoma [
23,
35,
36,
37]. In this research about DLBCL, we have found that the immunohistochemical expression of CSF1R was variable. The most characteristic pattern was of TAMs, present in 82% of the cases. These CSF1R-positive TAMs had a morphology that was like the one seen in M2-like TAMs, with a higher shape and dendritiform elongations, especially when the concentration was high in the tumor immune microenvironment. In addition, a CSF1R-positive B-cells pattern was seen in 18% of the cases. This CSF1R pattern affected the B-lymphocytes of DLBCL and the expression was diffuse. The CSF1R pattern correlated with the prognosis of the patients. The CSF1R-positive TAMs pattern was associated with a poor progression-free survival. Conversely, the B-cells pattern correlated with a favorable progression-free survival. Interestingly, although not statistically significant, the pattern of low CSF1R-positive TAMs had a better survival than the cases with high CSF1R-positive TAMs.
The start point of this research was to check if the gene expression of
CSF1R correlated with the prognosis of the patients with DLBCL. We used the LLMPP series that is comprised of 414 DLBCL cases. This series from western countries is robust and very well annotated. Using a cut-off, two groups with different overall survival could be found. The group with low
CSF1R expression, ≤11.62 (
n = 309, 64.7%), was associated with favorable survival. Conversely, the group with high
CSF1R expression, ≥11.63 (
n = 105, 46.7%), was associated with a poor outcome. We also correlated the
CSF1R with other markers, including
CD163 and
PD-L1 that are markers of M2-like TAMs. The correlation was moderate and positive. Therefore, the hypothesis was that
CSF1R in DLBCL identified only TAMs. In DLBCL, high CD163-positive TAMs have been associated with poor prognosis of the patients [
9,
10,
12], which is the same result seen by gene expression in the LLMPP series. Nevertheless, the presence of a B-cell pattern was not expected. This B-cell pattern was a new finding in the Tokai University Hospital series. Of note, in the reactive tonsils, CSF1R-positive B-cells were not identified. Therefore, this B-cells pattern in DLBCL may be pathological, as seen in Hodgkin Lymphoma.
In the Tokai series, a correlation between the two CSF1R patterns was made with several clinicopathological characteristics of the series. Initially, not many associations were found, but the B-cells pattern was associated with a lower MYC immunohistochemical expression, absence of
BCL2 translocation, absence of mutation of
MYD88 L265P, and higher MDM2 immunohistochemical expression. These characteristics point to a lower pathological background in this group of patients. Correlation with the clinical features of the patients also showed that the CSF1R + TAMs pattern was associated with a poor progression-free survival of the patients, disease progression, higher MYC expression, lower MDM2 expression,
BCL2 translocation, and
MYD88 L265P mutation. In addition, the histological expression of CSF1R was also correlated with 10 CSF1R-related markers including CSF1, STAT3, NF-KB, Ki67, MYC, PD-L1, TNFAIP8, IKAROS, CD163, and CD68, and predictive modeling with high accuracy for CSF1R was found using regression, generalized linear, an artificial intelligence neural network (multilayer perceptron), and SVM. Of note, CSF1R moderately correlated with STAT3, TNFAIP8, CD163, and CD68. Therefore, our results agree with groups that showed that, in DLBCL, high CD163-positive TAMs were associated with poor prognosis of the patients [
9,
10,
12].
Finally, we used artificial intelligence analysis to identify the genes that predicted the
CSF1R expression in the LLMPP series. Many data mining applications use neural networks because of their power, flexibility, and ease of use in situations where the underlying process is complex [
26]. Among them, the multilayer perceptron analysis predicts one or more target variables based on the values of several predictors [
26]. In this research, we performed two types of analysis. First, we used all the genes of the array and the result ranked the genes according to their importance to predict the
CSF1R expression (high vs. low). This analysis was technically successful, as shown by the low percentage of incorrect predictions and the high area under the curve. Second, we used an immune-oncology cancer panel and the multilayer perceptron managed to predict the
CSF1R expression with even better performance. Therefore, it is expected that those genes are not only related to the
CSF1R expression mechanisms but also related to the prognosis of DLBCL.
If the gene
CSF1R is checked in the cBioPortal webpage for cancer genomics and a combined study for DLBCL with 1295 samples is performed, the result shows that there are no alterations in this gene. We think that CSF1R is not relevant in DLBCL for the mutational status or other genomic changes, but it is relevant for their association to macrophage signature. Of note, the relevance of CD163 in DLBCL is well established as a marker for an inferior prognosis [
9,
10].
CSF1R may be relevant in other subtypes of cancer. According to the Human Protein Atlas (
http://www.proteinatlas.org; accessed on 9 April 2021) [
39] that used the TCGA dataset, the RNA expression of
CSF1R shows low cancer specificity. Among the different types of tumors that are being tested, glioma is the subtype that shows more
CSF1R expression. High
CSF1R expression correlated with a poor prognosis of renal and testis cancer. Nevertheless, no information is provided regarding lymphoma and
CSF1R in the Human Protein Atlas. According to the Kaplan-Meier Plotter (
http://kmplot.com/analysis/index.php?p=background; accessed on 9 April 2021) [
40], high expression of CSF1R is associated with favorable overall survival of breast cancer and unfavorable overall survival of ovarian, lung, and gastric cancer. Therefore,
CSF1R seems to be relevant in the pathogenesis of other subtypes of cancer as well. Due to the importance of the CSF1R in cancer, several groups have used CSF1/CSF1R inhibitors as monotherapy in clinical development. For example, small molecules have been used in melanoma, prostate cancer with metastasis, glioblastoma multiforme, solid tumors, relapse or refractory acute myeloid leukemia, and breast cancer. A review manuscript describing the use of CSF1R inhibitors in cancer therapy has recently been written by Cannarile MA et al. [
41]
There are new discriminators in the literature that are worth mentioning. For example, CD47 is a marker of the immune checkpoint that is a potential negative regulator of the DLBCL treatment outcome (“don’t eat me”). In a report by Bouwstra et al., CD47-positive DLBCL is characterized by worse overall survival when treated by R-CHOP [
33]. Therefore, DLBCL patients of the non-GCB cell-of-origin subtype may benefit from CD47-targeted therapy in addition to rituximab and possibly in addition to macrophage-targeted therapy. In the last section of our research, we analyzed the
CD47 and
SIRPA expression in the LLMPP database. We found that high
CD47 correlated with a poor overall survival of the patients, and that high
SIRPA (the receptor for CD47) correlated with good survival and with
CSF1R (
Figure 10). In addition, we also highlighted the genes of the cancer panel associated with the expression of these two markers. Therefore, CD47 is an interesting marker with complex relationships and will require further analysis. TAMs in DLBCL can also be targeted using a legumain inhibitor, which suppressed the tumor progression in an OCI-Ly3 xenograft mouse model of DLBCL [
42]. Wu ZL et al. reported that high nuclear expression of STAT3 associated with an unfavorable prognosis of DLBCL [
43]. In our research, we found that, in the CSF1R histological pattern of TAMs, which was associated with a worse progression-free survival, the CSF1R marker correlated with the STAT3 expression. Finally, high expression of PD-L1 was associated with poor prognosis in DLBCL [
44]. This result was also recently confirmed by our group [
45], but, in this research, the CSF1R did not correlate with the PD-L1 expression. Another marker that we have recently described is the apoptosis inhibitor TNFAIP8, which is associated with a poor prognosis of the patients [
28]. In this research, we found that, in the TAMs histological pattern, CSF1R correlated with TNFAIP8.


 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}