
Current State and Challenges in Development of Targeted Therapies in Myelodysplastic Syndromes (MDS)


1
Department of Hematology/Oncology, Jackson Memorial Hospital, Miami, FL 33136, USA
2
Sylvester Comprehensive Cancer Center, Miller School of Medicine, University of Miami, Miami, FL 33136, USA
3
Department of Internal Medicine, Icahn School of Medicine Mt. Sinai West/Morningside, New York, NY 10019, USA
4
Division of Hematology, Miller School of Medicine, University of Miami, Miami, FL 33136, USA
*
Author to whom correspondence should be addressed.
Hemato 2021, 2(2), 217-236; https://0-doi-org.brum.beds.ac.uk/10.3390/hemato2020013
Submission received: 26 March 2021
/
Revised: 16 April 2021
/
Accepted: 28 April 2021
/
Published: 30 April 2021
(This article belongs to the Section Chronic Myeloid Disease)
Abstract
:Myelodysplastic syndromes (MDS) encompass a variety of myeloid neoplasms characterized by ineffective hematopoiesis. The interaction of abnormal clonal hematopoiesis and changes in the bone marrow microenvironment propagate abnormal clones. Advances in next generation sequencing has identified over 100 somatic mutations, but despite deepened understanding of the genetics of MDS, therapeutic discoveries have remained limited. To date, only five drugs have been approved for MDS: Azacitidine, Decitabine, Lenalidomide, Luspatercept, and oral Decitabine with Cedazuridine. Current strategies for low-risk MDS continue to focus on symptomatic management and correction of cytopenias, while treatment for high-risk MDS focuses on delaying progression of disease and improving survival. In this review we discuss some of the challenges in developing pre-clinical models of MDS in which to test therapeutics, the advances that have been made, and promising novel therapeutics in the pipeline.
1. Introduction
Myelodysplastic syndromes (MDS) represent a spectrum of myeloid neoplasms involving clonal disorders of hematopoietic stems cells and progenitor cells (HSPC), varying degrees of cytopenias, dysplasia, and clinical course, with the feared risk of transformation into acute myeloid leukemia (AML). The clinical heterogeneity of MDS led to the World Health Organization (WHO) 2016 Classification of Myeloid Neoplasms, which classifies subtypes of MDS according to karyotype, morphologic and clinical features, as well as overlap syndromes that include both myelodysplastic and myeloproliferative characteristics [1]. The incidence of MDS per year is 4.9 per 100,000 people according to the SEER [Surveillance, Epidemiology and End Results Program] Registry data, and is more common in males, Caucasians and the elderly [2,3].
MDS is primarily a disease of older individuals, usually diagnosed over age 65 with a median age of 76, complicating both diagnosis and treatment [4]. Age-related clonal hematopoiesis (ARCH) can lead to alterations in B and T cells, and the gradual clonal expansion of HSPCs carrying genetic variants, without progression to a hematologic malignancy [4,5]. Better understanding of the role of the bone marrow microenvironment and its synergistic effects with genetic mutations has helped elucidate when abnormal clonal hematopoiesis (CH) will foster propagation and proliferation of somatically mutated HSPC leading to MDS versus ARCH [2,4]. The presence of myeloid mutations, or CH, at a variant allele fraction ≥2%, is known as clonal hematopoiesis of indeterminate potential (CHIP), with a prevalence of ~20% in people over 70, and increases the risk of MDS by 10-fold [4].
The development of MDS is likely a complex interaction between intrinsic properties of the CH gene mutations, and changes in the bone marrow microenvironment favoring the growth of mutated clones. HSPC failure in MDS can be de novo or secondary to a prior bone marrow insult, such as therapy related MDS from prior chemotherapies including alkylating agents or topoisomerase inhibitors [2]. Cytogenetic studies have shown that the majority of abnormalities in MDS are unbalanced chromosomal alterations, leading to either the addition or loss of genetic material, including −7/del(7q), −5/del (5q), trisomy 8, −17/del(17p)/iso (17q), del(20q), del(11q) del(12p) and +21 g, as opposed to the balanced translocations of AML [4]. Cytogenetic abnormalities are seen in 50–80% of patients, and are often associated with complex karyotype [2,4]. Cytogenetic abnormalities play a large role in prognostication and are included in the revised International Prognostic Scoring System (IPSS-R).
Advancements in next generation sequencing (NGS) have identified over 100 somatic point mutations recurrent in MDS, including driver mutations such as SF3B1, TET2, SRSF2, ASXL1, DNMT3A, RUNX1, U2AF1, TP53, and EZH2 [2,4]. The types and number of mutations, along with the size of the clone provide additional prognostic information. For example, the most commonly mutated genes involve spliceosome function such as SF3B1, SRSF2 and U2AF1, which are all heterozygous genes mutually exclusive of each other [6]. SF3B1 (Splicing factor 3b, subunit 1) is a specific mutation that portends good prognosis and is associated with MDS with ringed sideroblasts (MDS-RS) while SRSF2 has been linked to adverse outcomes. Additionally, mutations in TP53, a tumor suppressor, RUNX1, a myeloid transcription factor, ASXL1 and EZH2 chromatin modifiers, have also been linked to poor prognosis [4]. TP53 mutations are often associated with complex karyotype and along with RUNX1, worsening thrombocytopenia. Epigenetic mutations, such as TET2 and DNMT3A involved in DNA methylation, and ASXL1 involved in histone modification, serve as molecular mediators allowing for propagation of abnormal clones.
Increased knowledge and improved understanding of genetic mutations in MDS has led to increased therapeutic options. In the past two decades, a total of five drugs have been approved by the Food and Drug Administration (FDA): Azacitidine, Decitabine, Lenalidomide, Luspatercept, and oral Decitabine with Cedazuridine [4]. Although these agents can be effective in the treatment and overall survival of MDS, none are curative and new therapies for MDS remains a major unmet need. The only curative option available is hematopoietic stem cell transplantation (HSCT) however, many patients remain ineligible due to advanced age and comorbidities. In this review, we discuss currently approved therapies, therapies under investigation and challenges in creating novel targeted therapeutic drug development (Figure 1).
2. Currently Available Therapies
Given the heterogeneous nature of MDS, there have been several prognostic scoring systems developed to facilitate treatment plans based on the level of risk. Upon diagnosis, patients undergo risk stratification using IPSS-R. This scoring system classifies patients based on cytogenetic risk group, blast count, and degree of cytopenias into five prognostic categories: Very low ≤ 1.5, Low > 1.5–3, Intermediate > 3–4.5, High > 4.5–6, and very high >6. As a whole, low risk disease is considered ≤3.5 points and high risk >3.5 points. The original classification system delineated ≤1 point as low risk and > 1 point as high risk [7,8,9]. A summary of the currently approved drugs in MDS can be found in Table 1.
2.1. Low-Risk MDS
For patients with low-risk MDS (LR-MDS), therapeutic goals focus on quality of life (QoL) as early interventions have not been shown to improve mortality [4]. In general, patients with LR-MDS with Hb > 10 g/dL, absolute neutrophil count (ANC) > 0.5 G/L and platelets > 100 G/L will be asymptomatic and can undergo observation, with close attention to patients with excessive blasts or high-risk molecular features [4]. Standard of care (SOC) therapy for symptomatic patients with LR-MDS includes erythropoiesis-stimulating agents (ESAs), such as recombinant erythropoietin (EPO) or darbepoetin (DAR), as well as transfusion support which has been reviewed elsewhere [8,10,11,12].
Although the use of ESAs has decreased transfusion requirements and increased QoL, some patients fail ESAs or have a loss of response in which case immunomodulating drugs such as lenalidomide may be an option. Patients with del(5q) can often have refractory anemia secondary to increased p53 levels and p53-mediated destruction of erythroid precursors [4,13,14,15]. Lenalidomide is a thalidomide analogue, and displays anti-angiogenic, antineoplastic, anti-inflammatory and pro-erythropoietic properties. Patients with del (5q) given lenalidomide have rapid and sustained responses, with reduced need for transfusions regardless of karyotype, with high rates of transfusion independence ≥26 weeks [13,16]. TP53 mutations can be present in 20% of del(5q) MDS either as early sub-clones or acquired mutations over the course of the disease, and may be related to lower OS with lenalidomide and greater risk of progression to AML [17,18,19]. Alternatively, patients without del(5q) mutations have also shown response to lenalidomide which appears to restore some sensitivity to ESA in refractory patients, albeit responses are strongest with favorable karyotype and low transfusion burden.
Anemia in MDS can be linked to ineffective erythropoiesis with increased proliferation of erythroid progenitors but impaired erythroid maturation. Increased concentrations of transforming growth factor beta (TGF β) superfamily ligands can lead to ineffective erythroblast maturation. Luspatercept is a recombinant fusion protein that acts as a TGF β ligand trap, restoring late-stage erythropoiesis [20]. Luspatercept showed erythroid response rate of 63%, with 38% of patients achieving transfusion independence in the PACE-MDS study [20]. Luspatercept has shown several clinical advantages including response in patients with high EPO levels >500 U/L, a traditionally challenging population to treat, and robust responses in patients with SF3B1 mutations. The MEDALIST trial, as discussed below, led to the approval of Luspatercept for LR-MDS-RS and/or SF3B1 mutations [21]. Lastly, patients who fail first line therapy, or patients with high EPO levels >500 U/L who are unlikely to respond to ESAs, immunosuppressive therapy (IST) such as anti-thymocyte globulin (ATG) with or without cyclosporine or a hypomethylating agents (HMA) can be considered [22,23].
2.2. High Risk MDS
High intensity induction chemotherapy can halt progression of high risk MDS (HR-MDS) and improve survival, but the only potentially curative option at this time is allogenic HSCT. HSCT as a treatment option has been reviewed elsewhere and is outside the scope of this review but is an important option that should be discussed with all eligible patients [24,25,26,27]. Unfortunately, many barriers exist to HSCT including the advanced age of patients at presentation and high rates of co-morbidities, inability to find a donor match or lack of ability to cover the associated financial costs in some countries. Although transplant eligibility has increased over the last few decades due to less toxic conditioning regimens and alternative donor sources, more accessible and less toxic regimens remain highly sought after to slow disease progression.
The approval of HMAs such as AZA and decitabine (DAC) in 2004 and 2006, respectively, have changed the landscape of MDS treatment. Epigenetic modifications such as DNA methylation play a large role in the propagation of abnormal clones. In normal cells, the promotor of active genes is normally unmethylated. Methylation of cytosine residues within CpG islands via DNA methyltransferases (DNMT), an enzyme often increased in malignant cells, alters the configuration of chromatin inhibiting gene transcription causing gene silencing [10]. Cancer cells exhibit complex changes in DNA methylation including simultaneous global demethylation, increased expression of DNMT and de novo methylation at previously unmethylated CpG islands [28]. HMA are DNMT inhibitors allowing for reversal of epigenetically silenced genes, restoration of normal growth patterns and differentiation of immature cells [10].
AZA approval was based in part on studies by the Cancer and Leukemia Group B (CALGB) comparing AZA 75 mg/m2 for 7 days every 28 days versus best supportive care (BSC). Patients who received AZA, regardless of their subtype, had a 60% overall response rate (ORR) including 7% complete response (CR), 16% partial response (PR), and 37% hematological improvement (HI) compared with 5% response rate for BSC [29]. Among patients who received BSC, it is important to note that 5% met criteria for HI, however, leukocytosis was actually secondary to AML transformation. AZA improved time to transformation to AML or death from 13 to 21 months over BSC (p = 0.007), and improved QoL including fatigue, dyspnea, and physical functioning [29]. AZA-001, a phase 3 randomized controlled trial (RCT) confirmed trends in the CALGB study, showing higher rates of CR, longer time to disease progression and death, longer duration of hematological response and statistically significant benefit in overall survival (OS) from 15 months to 24.5 months [30]. Alternative dosing, and AZA as maintenance therapy has been reviewed elsewhere [31].
DAC is thought to have a dual mechanism of action, with higher doses associated with cytotoxicity and lower doses associated with demethylation, and has also shown benefit in treating MDS [32]. Early small studies of DAC showed 50% ORR in elderly patients with HR-MDS, with subsequent larger phase 2 studies of 15 mg/m2 DAC infused over 4 h, every 8 h for 3 days, showing ORR of 49%, which improved to 64% for HR-MDS [33]. A phase 3 study using the same dosing regimen exhibited ORR 30% in the DAC arm, including HI, and 7% in the placebo arm. Overall, there was no statistically significant different in time to AML progression or death, although there was a trend. However, when patients who responded to DAC were examined, median time to AML progression or death was 17.5 months versus 9.8 months for non-responders (p = 0.01), indicating that there was a benefit in OS for patients who responded to DAC [34]. This study ultimately led to the approval of DAC by the FDA in 2006.
ASTX727, the combination of oral DAC with cedazuridine, a cytidine deaminase inhibitor, was recently approved 7th July 2020 for previously treated and untreated de novo and secondary MDS with intermediate-1, intermediate-2, and high-risk IPSS [35]. Oral and IV DAC were found to be equivalent in the ASCERTAIN study, and data from phase 2 NCT02103478 had ORR of 60%; 21% of patients had CR with median duration of 7.5 months and more than half of transfusion dependent individuals became transfusion independent (TI) [35,36].
3. Emerging Therapeutics
3.1. Low Risk MDS
The clinical pipeline in LR-MDS is more robust than it has been in a decade, with the recent approval of luspatercept, and three actively recruiting phase 3 studies. Key details regarding these agents are summarized below, with additional information detailed in Table 2.
3.1.1. Imetelstat
Imetelstat, a novel telomerase inhibitor is under investigation as a single agent in LR-MDS. Telomerase is an enzyme involved in cellular replication that maintains telomeres and prevents them from shortening during cell division. Telomerase is repressed in most normal cells, allowing telomere length to gradually decrease over time, preventing uncontrolled proliferation. Telomerase expression can be upregulated in malignant cells leading to uncontrolled proliferation. Phase 2 data in a single-arm, open label study examining the effects of imetelstat 7.5 mg/kg IV every four weeks in 38 non-del(5q) LR-MDS patients has been optimistic, revealing 8-week TI of 42%, as well as HI of 68%. Importantly, many of the responders experienced durable responses, with 29% of patients remaining TI for ≥1 year. The drug was relatively well tolerated, with reversible cytopenias being most common adverse effects (AE) [37].
Currently, there is a phase 3 RCT study targeting patients with high baseline transfusion rates (>4 pRBC in last 8 weeks) and either ESA failure or serum EPO > 500 U/L, a specific subset of MDS that requires improved treatment options [38,39]. Given the success of the phase 2 trial, replicating these results in a larger double-blind RCT would likely lead to regulatory submission to the FDA.
3.1.2. Luspatercept
In 2020, luspatercept was approved for the treatment of anemia in patients with low to intermediate-risk MDS-RS that failed ESA and required 2 or more RBC units over 8 weeks. Approval was based on the phase 3 MEDALIST trial demonstrating TI > 8 weeks in 38% of patients vs. 13% of placebo (p < 0.001). Survival data is still maturing at this juncture, although progression to HR-MDS/AML has been nearly identical for both arms to date (5.2% for luspatercept vs. 5.3% for placebo) [21].
The COMMANDS trial (NCT03682536), another phase 3 trial, is currently underway comparing luspatercept head-to-head against ESA therapy in ESA-naïve, transfusion dependent, non-del(5q), non-RS MDS. The endpoint of this study is the proportion of patients who remain transfusion free the first 24 weeks after randomization [40]. If successful, the COMMANDS trial would represent a significant paradigm shift in the MDS landscape, with luspatercept potentially replacing ESA as first line therapy in non-del(5q), transfusion dependent LR-MDS patients.
3.1.3. Roxadustat
Poor response to ESAs has been associated with inflammation, IL-6 cytokine release and increased levels of hepcidin which causes iron insufficiency which contributes to difficulties with erythroblast maturation. Roxadustat is an oral hypoxia inducible factor (HIF) inhibitor that is already approved in China for anemia of chronic kidney disease, that works to increase EPO production, decrease hepcidin and promote erythroblast maturation [41]. It is currently being investigated in the phase 3 MATTERHORN RCT (NCT03263091) for patients with non-del(5q) LR-MDS patients with low transfusion burden (≤4 RBC units in 8 weeks) and EPO < 400. It is important to highlight that Roxadustat is targeting a different population than luspatercept’s COMMANDS trial, which targets patients with high transfusion burden [42].
MATTERHORN is designed to mirror the current utilization of ESAs in MDS, and the primary endpoint in the study is TI > 56 days. The open-label portion of this study was performed in 24 patients, and 38% (n = 9) achieved TI > 56 days. Of these nine, seven (78%) of them were on the dose selected for the larger, randomized MATTERHORN study [43]. Similar efficacy to ESA’s may form the basis of a regulatory submission to the FDA, however, future studies will likely need to confirm its superiority, or at least non-inferiority, to ESAs.
3.1.4. New Hypomethylating Agents
Since the approval of oral DAC-cedazuridine in 2020 for HR-MDS, ASTX727 is now being examined in phase 1–2 trials for patients with LR-MDS. Oral HMAs are highly preferred by patients as this minimizes required office visits, which has been of particular importance during the current COVID pandemic.
Other novel HMAs are also being investigated in LR-MDS such as CC-486. CC-486, an oral form of AZA, demonstrated clinical activity in a phase 1 study (NCT00528983) with clinical responses reported in 35% of previously treated patients and 73% of treatment-naïve patients with MDS or chronic myelomonocytic leukemia (CMML) [44]. LR-MDS patients with transfusion dependent anemia achieved TI ≥ 56 days in 31% patients who were on a 14 day regimen, and 38% on 21 day regimen with 21 day regimen exhibiting more durable responses at the 84 day mark (31% vs. 13%) [45]. Based off of these studies, the phase 3 RCT trial AZA-MDS-003 (NCT01566695) was designed to evaluate TI in previously transfusion-dependent LR-MDS patients using 300 mg for 21 day dosing regimen [46]. In July 2020, it was announced that the study had achieved its primary endpoint of TI, with 30.8% in treatment arm versus 11.1% in placebo (p = 0.0002), however, the study was halted early due to a higher incidence of death in the treatment arm (n = 16 CC-486 vs. n = 6 placebo). The study did not reveal any difference in OS between the two arms (17.3 months treatment vs. 16.2 months placebo, though it was underpowered to do so. Future development of CC-486 is unclear at this time, though it may be explored at a modified dosing regimen [47].
3.2. High Risk MDS
While the treatment of LR-MDS has focused on improving symptoms and cytopenias, treatment for HR-MDS centers around improvement of clinical response, halting of disease progression and improvement in OS. An overview of recent therapeutic developments is discussed below and detailed in Table 3.
3.2.1. Pevonedistat (NEDD8)
Pevonedistat, a novel NEDD8 inhibitor, is currently under development for HMA naïve, HR-MDS patients. Neddylation is a post-translational modification that adds a ubiquitin-like protein known as NEDD8, that aids in the proliferation of human cancers, allowing malignant cells to evade apoptosis [48]. Pevonedistat, which inhibits this pathway, was granted breakthrough therapy designation by the FDA in July 2020 after the phase 2 Pevonedistat-2001 trial (NCT02610777), which compared pevonedistat/AZA vs. AZA alone in HMA naïve, HR-MDS patients. Data was released at ASCO June 2020, which demonstrated a trend towards improved OS in HR-MDS patients treated with combination therapy (23.9 vs. 19.1 months, p = 0.24). There was a statistically significant improvement in event-free survival in HR-MDS patients (20.2 vs. 14.8 months, p = 0.045), and thus this was the endpoint selected for the phase 3 pivotal study. Combination therapy also had a statistically significant effect in CR rate in HR-MDS patients, with CR rate of 52% vs. 27% AZA alone (p = 0.050) [49]. The phase 3 PANTHER trial (NCT03268954) is currently underway [50].
3.2.2. Immunotherapies
Magrolimab is an anti-CD47 antibody that interferes with the signal regulatory protein alpha (SIRPα) receptors on macrophages, a method cancer cells use to avoid immune detection. CD47 is a cell surface protein in the immunoglobulin family that is found ubiquitously on cells and plays roles in integrin function, intracellular signaling and interactions with other cell surface glycoproteins such as SIRPα. CD47 is often over expressed on malignant cells and the CD47- SIRPα complex can often shield these cells from macrophage mediated phagocytosis [51]. Magrolimab is unique in that it is the only monoclonal antibody in development for MDS, and was granted Breakthrough Therapy status from the FDA for an accelerated development pathway [52]. This designation was awarded off the strength of phase 1B data where patients were treated with magrolimab in combination with AZA, with ORR of 91% and CR of 42%. The drug was overall well tolerated with myelosuppression noted to be the most common AE. The phase 3 ENHANCE trial, a study of 520 treatment naïve HR-MDS patients receiving magrolimab with AZA vs. AZA alone is actively enrolling [53].
Additional immunotherapy is also under investigation. TIM-3 is expressed on immune cells and leukemic stem cells (LSC) but not HSC, and its inhibition may lead to restoration of immune function and targeting of LSCs. Phase 1b combination data in HR-MDS patients showed ORR of 61% with DAC and 65% with AZA [54]. STIMULUS-MDS-1 is a single arm phase 2 study combining MBG-453 with HMAs in treatment naïve HR-MDS with primary endpoints of CR rate and progression free survival (PFS), while STIMULUS-MDS-2 is a placebo-controlled phase 3 study combining MBG-453 with AZA in treatment naïve HR-MDS and CMML. The primary endpoint of the latter study is OS [55].
3.2.3. Venetoclax
Venetoclax is a BH3 mimetic that functions as an inhibitor of BCL2, an anti-apoptotic protein commonly expressed in cancer cells. It is currently approved for AML in the upfront setting in combination with HMAs for patients over 75 with co-morbidities who are unfit for intensive chemotherapy and is being investigated for first line therapy in MDS. An initial phase 1b, open-label study examining AZA and venetoclax demonstrated an ORR of 77%, CR rate of 42%, and median OS had not yet been reached [56]. Given this strong early stage data, the phase 3 VERONA study (NCT04401748) was initiated in 500 treatment naïve HR-MDS patients in September 2020 with CR and OS as primary endpoints [57]. ASTX-727 is also being examined in combination with venetoclax for HR-MDS. If these trials are successful, it would represent a significant paradigm shift in the HR-MDS landscape, with the combination of HMA and venetoclax becoming standard of care (SOC) in the front-line setting.
3.2.4. Isocitrate Dehydrogenase (IDH) Inhibitors
IDH is a key enzyme in the TCA cycle, and when mutated, it can lead to leukemogenesis. Although only approximately 10% of MDS patients have an IDH mutation, it has been associated with increased risk of AML transformation. Ivosidenib, an IDH-1 inhibitor, provided the proof of concept for IDH inhibition in MDS in a phase I trial where ORR was 92% and CR was 42% [58].
Currently there are several studies underway testing both ivosidenib and the IDH-2 inhibitor enasidenib, NCT03503409 and NCT03744390, respectively. The trials are designed similarly, both multi-cohort, open-label studies examining HMA-naïve patients, patients refractory to HMAs, and LR-MDS patients with anemia resistant to ESAs. The primary endpoint is overall hematological response. They began enrolling in early 2019 with estimated primary completion in January 2022 and February 2023, respectively [59,60]. Other studies in the pipeline are examining ORR of enasidenib in combination with AZA for HMA-naïve and refractory HR-MDS patients (NCT03383575), ivosidenib in combination with venetoclax and AZA(NCT03471260), and ivosidenib and nivolumab in HR-MDS (NCT04044209) [61]. Lastly, a new IDH-1 inhibitor, FT-2102, is being tested in combination with HMA for patients who failed first line IDH inhibitors [62]. The phase 1 portion of this study demonstrated an ORR of 73% and a CR of 55% in IDH-1 mutant MDS patients treated with combination therapy [63].
3.2.5. New Hypomethylating Agents
Novel oral HMAs are under investigation for HR-MDS. ASTX-030 is an oral form of AZA with cedazuridine that is being studied in a multi-arm phase 1–3 trial of HR-MDS/CMM and ASTX-727 is being evaluated in combination with venetoclax [64,65].
Guadecitabine is similar to DAC but with increased resistance to cytidine deaminase. Guadecitabine was effective in a phase 2 single arm study (NCT02131597) of treatment-naïve HR-MDS patients with a response rate of 61% (CR 22%) and OS of 15 months [66]. The ASTRAL-3 study (NCT02907359) was a randomized, open-label, parallel group trial in patients who either failed or were refractory to other HMAs and did not have TP53 mutation, but unfortunately failed to meet primary endpoint of OS [67]. The fate of guadecitabine in MDS remains to be determined, as full study data and subgroup analysis are still pending [68].
3.2.6. Other Targeted Therapies
Selinexor is a first in class selective inhibitor of nuclear export (XPO1) which is over-expressed in malignant cells leading to oncogenesis. Selinexor has already gained approval for other hematologic malignancies and now is under investigation in HR-MDS. A single-arm phase 2 study of selinexor as a single agent in HMA relapsed/refractory HR-MDS or oligoblastic AML patients demonstrated an ORR of 26%. Of note, patients with SF3B1 mutations showed increased responses. Further development with a larger sample of these patients may be explored [69].
Given the prevalence of splicing factor mutations in MDS, spliceosome modulators are also being explored as a novel therapeutic strategy. A phase 1 study (NCT02841540) of a splicing modulating agent, H3B-8800, was successful in preclinical models at preferential killing of splicing mutated cells [70]. The phase 1 study population included cohorts of HR-MDS (n = 24), LR-MDS (n = 21), and CMML (n = 4). Unfortunately, no CR or PR were noted, but 18% in the MDS/CMML cohorts did not require RBC transfusions for ≥8 weeks while on study [71].
TP53 mutations are present in about 10–20% of MDS patients and are associated with worse outcomes due to anti-apoptotic mechanisms and uncontrolled cellular proliferation, thus representing an intriguing therapeutic target for this patient population [72]. A TP53 activating prodrug, APR-246, was examined in a phase 3 study (NCT03745716) in 154 patients with TP53 mutant MDS who were HMA naïve, and compared APR-246 + AZA versus AZA alone [73]. Unfortunately, APR-246 did not meet its primary endpoint of CR, though the combination arm did trend towards better response (33.3% vs. 22.4%, p = 0.13). OS remains to be evaluated after longer follow-up [74].
Rigosertib is a Tyrosine Kinase Inhibitors (TKI) that has been investigated in HR-MDS. Despite promising results in combination with AZA in early phase studies in treatment naïve HR-MDS, the ONTIME study failed to meet its primary endpoint of OS, though there was a trend towards improvement in the rigosertib arm (8.2 mos vs. 5.9 mos, p = 0.33). Post hoc analysis showed a more significant trend in patients with primary HMA failure (8.6 mos vs. 5.3 mos, p = 0.06) and a statistically significant improvement in patients with very HR-MDS (7.6 mos vs. 3.2 mos, p = 0.015) [75]. The INSPIRE study was designed based on these subgroups, comparing rigosertib versus physician’s choice in HR-MDS patients R/R to HMA. Unfortunately, the study failed to meet its primary endpoint of OS [76,77].
4. Limitations in Preclinical Models
Part of the challenges facing therapeutic advances in MDS lies in the difficulty recreating clinical characteristics of MDS in preclinical models. Although there are some MDS cell lines, in vitro models remain limited. Direct testing of patient samples ex vivo is limited by potentially insufficient cell numbers, the inability of cells to survive in culture, and difficulty in genetically engineering primary patient material. In animal models, there has been more success with transplantation of AML cells into immunocompromised mice, however, MDS patient-derived xenografts have been met with poor engraftment and difficulty recapitulating disease phenotypes. Described below are some of the existing preclinical models and their limitations.
4.1. Cell Lines
There is currently only one true cell line available representing MDS without leukemic progression. MDS92 was derived in 1991 from a 52-year-old male bone marrow sample that had del (5q) and proliferation in the presence of interleukin-3 [78]. From MDS92, there were five sublines established including blastic subline MDS-L. MDS-L has helped with the molecular study of del(5q) MDS and the understanding of the mechanism of action of lenalidomide [79].
Many existing cell lines were established from a leukemic phase that evolved from MDS including MOLM-13, MOLM-14, SKM-1, and MUTZ-8 [80]. MOLM-13 and MOLM-14 were derived from peripheral blood of a patient during relapse of secondary AML, and SKM-1 and MUTZ-8 were established from a different patient with secondary AML. SKM-1 had point mutations in NRAS and KRAS. Although these cell lines can help with understanding the pathophysiology of MDS blasts, there remains a need for true MDS cell lines.
4.2. Mouse Models
For mouse models, the hematopathology subcommittee of the Mouse Models of Human Cancer Consortium has a set of guidelines used for the identification of MDS in mice (Table 4) [81].
Xenograft Mice
One approach to studying hematological malignancies is using immunodeficient mice with malignant human xenografts, so-called patient-derived xenografts (PDX). This approach has been shown to work in AML and acute lymphoblastic leukemia (ALL). However, there has been limited success in MDS.
One immunocompromised mouse strain often used in PDX is the severe combined immunodeficiency in the nonobese diabetes background (NOD/SCID) because they have reduced natural killer (NK) cell activity and defective B and T cells. These mice were used in a study in which researchers implanted them with del(5q) HSC from seven MDS patients. One of seven mice had poor implantation (12%) and although the CD45+ CD15+ expression showed the 5q deficiency, there was no evidence of clinical disease found in the recipient mice [82]. Another study used bone marrow from MDS patients and healthy controls and injected them into irradiated NOD/SCID mice with and without cytokines. Cells from the MDS patients had reduced proliferation compared to the healthy controls, but no abnormal karyotypes were observed, suggesting that the implanted human cells were derived from normal bone marrow cells and NOD/SCID mice were unable to reliably support the proliferation of the human MDS cells [83].
One explanation for poor engraftment is the lack of a human-specific microenvironment and thus the murine recipient was unable to support the MDS samples. There have been many different immunodeficient mouse strains with various human cytokines expression levels generated over the years. Among them is the NSG-SGM3 (NSG-S) mice that have transgenic expression of human stem cell factor, granulocyte-macrophage colony-stimulating factor (GM-CSF), and interleukin 3 in the NSG (NOD/scid-IL-2Rγcnull) background. Krevvata et al. compared the engraftment of AML and MDS cells in NSG versus NSG-S mice. They found that 50% of AML samples responded more robustly, had useful levels of engraftment, and had a positive response to human cytokines, whereas MDS sample engraftment remained low at <2% and did not increase over time [84].
Co-injecting MDS-derived CD34+ cells with patient-derived mesenchymal stromal cell (MSC) into the bone marrow cavity of NSG-SGM3A is an approach that has been used to manipulate the murine microenvironment [85]. This initially appeared to result in enhanced engraftment of MDS PDX models however, later studies showed no increase in engraftment with the co-injection [84]. A recent study used humanized scaffolds to support cell growth and differentiation and found that implantation of human MSC scaffolds into immunodeficient mice enabled long-term engraftment in MDS stem cells [86].
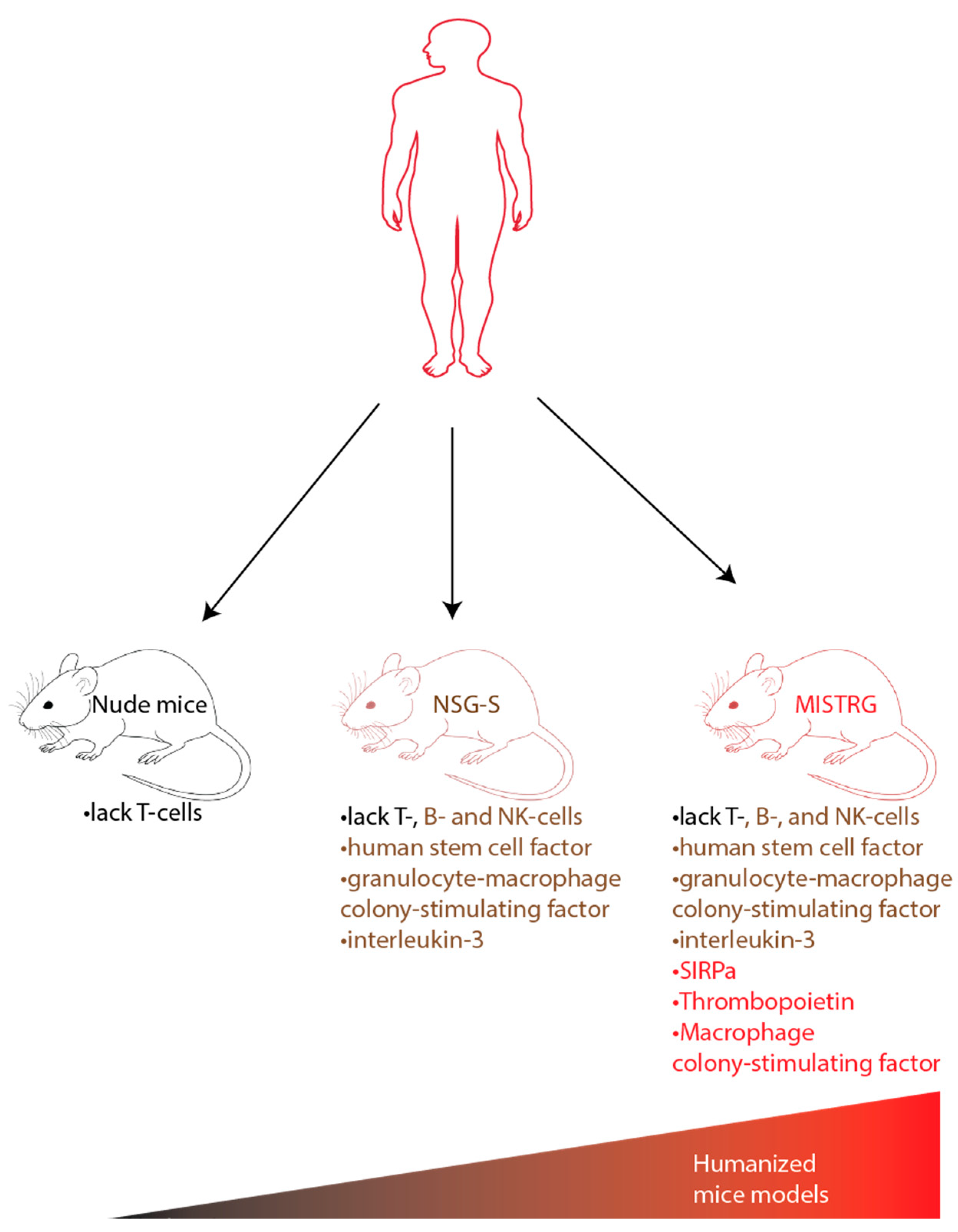
Another humanized mouse model that was developed is MISTRG which stands for the 7 modified genes found in these mice (M-CSFh/h IL-3/GM-CSFh/h SIRPah/h TPOh/h RAG2−/− IL2R γ −/−). These mice are immunodeficient without B/T lymphocytes, or NK cells. They express non-cross reactive human cytokines in place of their murine counterparts including macrophage colony-stimulating factor, interleukin-3, GM-CSF, and thrombopoietin. MISTRG also expresses SIRPα protein which protects human cells from phagocytosis. MISTRG was able to replicate patients’ dysplastic morphology and support all risk MDS PDX with multi-lineage output. The humanized MISTRG can also be propagated via serial transplantation allowing the ability to test therapeutics as described in Figure 2 [87].
4.3. Emerging Technology and Advances
Although human MDS cells can be transplanted into immunodeficient mice, challenges remain in replicating clinical models due to the inability to grow HSPCs in vitro, the inability to xenograft in immunodeficient mice, and the failure to recapitulate MDS in these models. Despite these limitations, new developments have emerged hoping to overcome challenges in developing preclinical models of MDS. Advances include CRISPR/Cas9 and induced pluripotent stem cells (iPSCs) for in vitro models, and genetically engineered mice.
4.3.1. CRISPR/Cas9
Advances in genome engineering have allowed for genomic editing through methods such as CRISPR/Cas9. CRISPR/Cas9 introduces a site-specific double-stranded break in DNA based on a guide RNA-directing Cas9, an endonuclease. This double-stranded break can be used to create gene knockouts from insertions or deletions by non-homologous end joining (NHEJ) or can be used to introduce an engineered template DNA by homology directed repair (HDR).
CRISPR provides the ability to create and test multiple MDS-specific mutations together and insert them into preclinical models to better portray the course of actual clinical disease. Tothova et al. used CRISPR/Cas9 to generate a combination of loss of function mutations commonly mutated in MDS (TET2, ASXL1, DNMT3A, RUNX1, TP53, NF1, EZH2) along with two cohesin genes (STAG2 and SMC3) in human CD34+ cells and then transplanted them into immunodeficient mice. The immunodeficient mice were treated with AZA and TET-2 mutated cells showed a response, whereas ASXL-1 mutated cells displayed resistance. This data mimics findings seen in clinical trials indicating that CRISPR/Cas9 is an incredibly beneficial tool in modeling MDS and testing novel therapeutics [88].
4.3.2. Induced Pluripotent Stem Cells
iPSCs are adult cells that can be reprogrammed to adopt an early cell-like state from which other types of cells can be grown, revolutionizing how MDS cell lines are grown. Since iPSCs can be differentiated into other HSC types, it allows for functional studies on the premalignant clonal intermediates derived from HSC of MDS patients.
The first iPSC-MDS lines came from two patient samples with chromosome 7q deletions. Using iPSCs, Kotini et al. modeled del7q with different ranges of deletions followed by a phenotypic rescue screen. They were able to map the chr7q minimal region and find the location of haplo-insufficient genes [89].
In Hsu et al., MDS samples were used to generate iPSC lines that had premalignant clonal intermediates. Using this reprogramming approach, they showed that SF3B1 mutations can be a second hit mutation preceded by epigenetic mutations, and that SF3B1 mutations are required to cooperate with EZH2 mutations to impair mitochondria and induce apoptosis [90].
iPSCs serve as a good model to study hematological malignancies and transformation of pre-malignant to malignant clones. Patient derived iPSCs provide a novel method to perform controlled mechanistic studies, perform functional studies of mutations, and predict drug responses.
4.3.3. Genetically Engineered Mice
Given that human MDS cells grow poorly in xenografted mice, genetically engineered murine models have been developed, which can be accomplished in two ways. The first approach is the reverse transcription of bone marrow transduction/transplantation mice. In this method, the bone marrow nucleated cells of the mice are harvested and then infected in vitro with the retroviral construct which expresses the gene of interest. These are then transplanted into lethally irradiated homologous host mice. The second approach is to modify the mouse germline to generate mice with altered expression (knock out or knock in) of a gene of interest. This is typically done through homologous recombination of mouse embryonic stem cells which can be injected into blastocysts to create chimeric mice displaying characteristics of MDS. This approach can be refined further using the Cre-Lox recombination system allowing for both temporal and spatial control [91].
The NUP98-HOXD13 mouse model has successfully recreated many of the characteristics of MDS. NUP98-HOXD13 involves the fusion of two genes: nucleoporin protein, NUP98, with homeobox D13, HOXD13. NUP98-HOXD13(NHD13), a conditional transgenic mouse, was developed by utilizing the Vav1 promoter to drive transgenic NHD13 expression in hematopoietic tissue. At four to seven months, these mice developed neutropenia, anemia, and some degree of thrombocytopenia and lymphopenia. At ten to fourteen months, about half of the mice had developed acute leukemia (AML and T-ALL) [92].
The most common cytogenic abnormality found in MDS is del(5q). Barlow et al., were able to generate the first mouse model for human 5q- syndrome using Cre-loxP recombination to delete a region flanked by Cd74-Nid67. Haploinsufficiency of the Cd74-Nid67 region caused macrocytic anemia, prominent dyserythropoiesis, and mono-lobated megakaryocytes in the bone marrow—characteristics similar to that of human 5q-syndrome [93].
Advances in sequencing technologies have allowed for the discovery of chromosomal abnormalities (del(5q), trisomy 8, etc.) and gene mutations (SRSF2, RUNX1, TET2, ASXL1, U2AF1, etc.) found in MDS which has been used to create genetically engineered murine models. The continued development of these animal models will allow for better understanding of the mechanism of action of current therapeutics, and for the analysis of emerging compounds.
5. Conclusions
MDS encompasses a heterogeneous group of myeloid neoplasms where the complexity has made it difficult to find new successful therapeutic modalities. Currently, there are only five approved drugs for MDS including AZA, DAC, lenalidomide for 5q deletions, Luspatercept and oral DAC-cedazuridine.
LR-MDS has seen some development in improving symptomatic management but no significant advances in modifying the disease course. Luspatercept has now been approved for LR to intermediate risk MDS-RS for refractory anemias. Imetelstat and Roxadustat are being investigated for similar indications which may expand treatment options for refractory anemias beyond ESAs. HMA, which have been the backbone for MDS treatment, are also under investigation in oral form.
Targeted therapies in development for HR-MDS have also expanded recently. Pevonedistat, a NEDD8 inhibitor, has shown significant benefit in CR in combination with AZA in HMA naive HR-MDS patients and if data from the phase 3 PANTHER trial are successful, it could alter the treatment paradigm for HR-MDS. Similarly, venetoclax in combination with AZA is being researched in the first line setting and if results of the phase 3 VERONA prove to be significant, there will be a shift in the SOC for HR-MDS patients. IDH-1 and IDH-2 inhibitors, which have proven efficacious in AML, are being investigated in different combinations. Immunotherapies such as magrolimab are in phase 3 trials in combination with AZA and other immunotherapies are in development which can hopefully lead to restoration of normal immune function.
MDS is likely a complex interaction between genetic mutations and the bone marrow microenvironment. Challenges in creating accurate preclinical models on which to test novel therapeutics, such as the need for additional MDS cell lines and better engraftment into mouse models, has hindered previous progress. Improvements in iPSCs have revolutionized how MDS cell lines are grown, CRISPR/Cas9 technology has enhanced the ability to recreate different mutations present in MDS and the creation of genetically engineered mice has resolved some of the issues with xenograft models. The continued development of animal models will improve the ability to recreate MDS clinical models, deepen understanding of MDS genetics and interaction with the bone marrow microenvironment, and advance the testing of future therapeutics that will hopefully revolutionize the landscape of MDS treatments.
Author Contributions
M.S., S.C., M.K. and J.T. wrote and revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
J.T. is supported in part by NIH/NCI (K08CA230319) funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Figure 1 Created with BioRender.com (accessed on 10 March 2021).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Dotson, J.L.; Lebowicz, Y. Myelodysplastic Syndrome; StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Saygin, C.; Carraway, H.E. Current and emerging strategies for management of myelodysplastic syndromes. Blood Rev. 2020, 100791. [Google Scholar] [CrossRef] [PubMed]
- Shlush, L.I. Age-related clonal hematopoiesis. Blood 2018, 131, 496–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, J.; Lee, S.C. Mutations in spliceosome genes and therapeutic opportunities in myeloid malignancies. Genes Chromosom. Cancer 2019, 58, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Sole, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood 2012, 120, 2454–2465. [Google Scholar] [CrossRef]
- Steensma, D.P.; Bennett, J.M. The myelodysplastic syndromes: Diagnosis and treatment. Mayo Clin. Proc. 2006, 81, 104–130. [Google Scholar] [CrossRef]
- Greenberg, P.; Cox, C.; LeBeau, M.M.; Fenaux, P.; Morel, P.; Sanz, G.; Sanz, M.; Vallespi, T.; Hamblin, T.; Oscier, D.; et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997, 89, 2079–2088. [Google Scholar] [CrossRef]
- Gotze, K.; Muller-Thomas, C.; Peschel, C. The role of azacitidine in the management of myelodysplastic syndromes (MDS). Cancer Manag. Res. 2009, 1, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, P.L.; Sun, Z.; Miller, K.B.; Bennett, J.M.; Tallman, M.S.; Dewald, G.; Paietta, E.; van der Jagt, R.; Houston, J.; Thomas, M.L.; et al. Treatment of myelodysplastic syndrome patients with erythropoietin with or without granulocyte colony-stimulating factor: Results of a prospective randomized phase 3 trial by the Eastern Cooperative Oncology Group (E1996). Blood 2009, 114, 2393–2400. [Google Scholar] [CrossRef] [Green Version]
- Hellstrom-Lindberg, E.; Gulbrandsen, N.; Lindberg, G.; Ahlgren, T.; Dahl, I.M.; Dybedal, I.; Grimfors, G.; Hesse-Sundin, E.; Hjorth, M.; Kanter-Lewensohn, L.; et al. A validated decision model for treating the anaemia of myelodysplastic syndromes with erythropoietin + granulocyte colony-stimulating factor: Significant effects on quality of life. Br. J. Haematol. 2003, 120, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- List, A.; Dewald, G.; Bennett, J.; Giagounidis, A.; Raza, A.; Feldman, E.; Powell, B.; Greenberg, P.; Thomas, D.; Stone, R.; et al. Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion. N. Engl. J. Med. 2006, 355, 1456–1465. [Google Scholar] [CrossRef] [Green Version]
- Dutt, S.; Narla, A.; Lin, K.; Mullally, A.; Abayasekara, N.; Megerdichian, C.; Wilson, F.H.; Currie, T.; Khanna-Gupta, A.; Berliner, N.; et al. Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in human erythroid progenitor cells. Blood 2011, 117, 2567–2576. [Google Scholar] [CrossRef] [Green Version]
- Fumagalli, S.; Di Cara, A.; Neb-Gulati, A.; Natt, F.; Schwemberger, S.; Hall, J.; Babcock, G.F.; Bernardi, R.; Pandolfi, P.P.; Thomas, G. Absence of nucleolar disruption after impairment of 40S ribosome biogenesis reveals an rpL11-translation-dependent mechanism of p53 induction. Nat. Cell Biol. 2009, 11, 501–508. [Google Scholar] [CrossRef] [Green Version]
- Fenaux, P.; Giagounidis, A.; Selleslag, D.; Beyne-Rauzy, O.; Mufti, G.; Mittelman, M.; Muus, P.; Te Boekhorst, P.; Sanz, G.; Del Canizo, C.; et al. A randomized phase 3 study of lenalidomide versus placebo in RBC transfusion-dependent patients with Low-/Intermediate-1-risk myelodysplastic syndromes with del5q. Blood 2011, 118, 3765–3776. [Google Scholar] [CrossRef]
- Santini, V.; Fenaux, P.; Giagounidis, A.; Platzbecker, U.; List, A.F.; Haferlach, T.; Zhong, J.; Wu, C.; Mavrommatis, K.; Beach, C.L.; et al. Impact of somatic mutations on response to lenalidomide in lower-risk non-del(5q) myelodysplastic syndromes patients. Leukemia 2021, 35, 897–900. [Google Scholar] [CrossRef]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical effect of point mutations in myelodysplastic syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef] [Green Version]
- Lode, L.; Menard, A.; Flet, L.; Richebourg, S.; Loirat, M.; Eveillard, M.; Le Bris, Y.; Godon, C.; Theisen, O.; Gagez, A.L.; et al. Emergence and evolution of TP53 mutations are key features of disease progression in myelodysplastic patients with lower-risk del(5q) treated with lenalidomide. Haematologica 2018, 103, e143–e146. [Google Scholar] [CrossRef] [Green Version]
- Platzbecker, U.; Germing, U.; Götze, K.S.; Kiewe, P.; Mayer, K.; Chromik, J.; Radsak, M.; Wolff, T.; Zhang, X.; Laadem, A.; et al. Luspatercept for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes (PACE-MDS): A multicentre, open-label phase 2 dose-finding study with long-term extension study. Lancet Oncol. 2017, 18, 1338–1347. [Google Scholar] [CrossRef]
- Fenaux, P.; Platzbecker, U.; Mufti, G.J.; Garcia-Manero, G.; Buckstein, R.; Santini, V.; Diez-Campelo, M.; Finelli, C.; Cazzola, M.; Ilhan, O.; et al. Luspatercept in Patients with Lower-Risk Myelodysplastic Syndromes. N. Engl. J. Med. 2020, 382, 140–151. [Google Scholar] [CrossRef]
- Passweg, J.R.; Giagounidis, A.A.; Simcock, M.; Aul, C.; Dobbelstein, C.; Stadler, M.; Ossenkoppele, G.; Hofmann, W.K.; Schilling, K.; Tichelli, A.; et al. Immunosuppressive therapy for patients with myelodysplastic syndrome: A prospective randomized multicenter phase III trial comparing antithymocyte globulin plus cyclosporine with best supportive care—SAKK 33/99. J. Clin. Oncol. 2011, 29, 303–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahl, M.; DeVeaux, M.; de Witte, T.; Neukirchen, J.; Sekeres, M.A.; Brunner, A.M.; Roboz, G.J.; Steensma, D.P.; Bhatt, V.R.; Platzbecker, U.; et al. The use of immunosuppressive therapy in MDS: Clinical outcomes and their predictors in a large international patient cohort. Blood Adv. 2018, 2, 1765–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartenstein, M.; Deeg, H.J. Hematopoietic stem cell transplantation for MDS. Hematol. Clin. New Am. 2010, 24, 407–422. [Google Scholar] [CrossRef] [Green Version]
- de Witte, T.; Bowen, D.; Robin, M.; Malcovati, L.; Niederwieser, D.; Yakoub-Agha, I.; Mufti, G.J.; Fenaux, P.; Sanz, G.; Martino, R.; et al. Allogeneic hematopoietic stem cell transplantation for MDS and CMML: Recommendations from an international expert panel. Blood 2017, 129, 1753–1762. [Google Scholar] [CrossRef]
- Parmar, S.; de Lima, M. Hematopoietic stem cell transplantation for myelodysplastic syndrome. Biol. Blood Marrow Transplant. 2010, 16, S37–S44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cutler, C.S.; Lee, S.J.; Greenberg, P.; Deeg, H.J.; Perez, W.S.; Anasetti, C.; Bolwell, B.J.; Cairo, M.S.; Gale, R.P.; Klein, J.P.; et al. A decision analysis of allogeneic bone marrow transplantation for the myelodysplastic syndromes: Delayed transplantation for low-risk myelodysplasia is associated with improved outcome. Blood 2004, 104, 579–585. [Google Scholar] [CrossRef]
- Issa, J.P. Epigenetic changes in the myelodysplastic syndrome. Hematol. Oncol. Clin. N. Am. 2010, 24, 317–330. [Google Scholar] [CrossRef] [Green Version]
- Silverman, L.R.; Demakos, E.P.; Peterson, B.L.; Kornblith, A.B.; Holland, J.C.; Odchimar-Reissig, R.; Stone, R.M.; Nelson, D.; Powell, B.L.; DeCastro, C.M.; et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: A study of the cancer and leukemia group B. J. Clin. Oncol. 2002, 20, 2429–2440. [Google Scholar] [CrossRef]
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; Finelli, C.; Giagounidis, A.; Schoch, R.; Gattermann, N.; Sanz, G.; List, A.; et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: A randomised, open-label, phase III study. Lancet Oncol. 2009, 10, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Khan, C.; Pathe, N.; Fazal, S.; Lister, J.; Rossetti, J.M. Azacitidine in the management of patients with myelodysplastic syndromes. Ther. Adv. Hematol. 2012, 3, 355–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saba, H.I. Decitabine in the treatment of myelodysplastic syndromes. Ther. Clin. Risk. Manag. 2007, 3, 807–817. [Google Scholar] [PubMed]
- Wijermans, P.; Lubbert, M.; Verhoef, G.; Bosly, A.; Ravoet, C.; Andre, M.; Ferrant, A. Low-dose 5-aza-2′-deoxycytidine, a DNA hypomethylating agent, for the treatment of high-risk myelodysplastic syndrome: A multicenter phase II study in elderly patients. J. Clin. Oncol. 2000, 18, 956–962. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Issa, J.P.; Rosenfeld, C.S.; Bennett, J.M.; Albitar, M.; DiPersio, J.; Klimek, V.; Slack, J.; de Castro, C.; Ravandi, F.; et al. Decitabine improves patient outcomes in myelodysplastic syndromes: Results of a phase III randomized study. Cancer 2006, 106, 1794–1803. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Decitabine/Cedazuridine: First Approval. Drugs 2020, 80, 1373–1378. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G.; Griffiths, E.A.; Steensma, D.P.; Roboz, G.J.; Wells, R.; McCloskey, J.; Odenike, O.; DeZern, A.E.; Yee, K.; Busque, L.; et al. Oral cedazuridine/decitabine for MDS and CMML: A phase 2 pharmacokinetic/pharmacodynamic randomized crossover study. Blood 2020, 136, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P.; Fenaux, P.; Van Eygen, K.; Raza, A.; Santini, V.; Germing, U.; Font, P.; Diez-Campelo, M.; Thepot, S.; Vellenga, E.; et al. Imetelstat Achieves Meaningful and Durable Transfusion Independence in High Transfusion-Burden Patients with Lower-Risk Myelodysplastic Syndromes in a Phase II Study. J. Clin. Oncol. 2021, 39, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Geron Announces Fifty Percent Enrollment Milestone in IMerge Phase 3 Clinical Trial in Lower Risk MDS. Available online: https://www.businesswire.com/news/home/20201210005269/en/Geron-Announces-Fifty-Percent-Enrollment-Milestone-in-IMerge-Phase-3-Clinical-Trial-in-Lower-Risk-MDS (accessed on 12 March 2021).
- Study to Evaluate Imetelstat (GRN163L) in Subjects with International Prognostic Scoring System (IPSS) Low or Intermediate-1 Risk Myelodysplastic Syndrome (MDS). Available online: https://clinicaltrials.gov/ct2/show/NCT02598661 (accessed on 10 March 2021).
- Efficacy and Safety Study of Luspatercept (ACE-536) Versus Epoetin Alfa for the Treatment of Anemia Due to IPSS-R Very Low, Low or Intermediate Risk Myelodysplastic Syndromes (MDS) in ESA Naïve Subjects Who Require Red Blood Cell Transfusions (COMMANDS). Available online: https://clinicaltrials.gov/ct2/show/NCT03682536 (accessed on 9 March 2021).
- Yan, Z.; Xu, G. A Novel Choice to Correct Inflammation-Induced Anemia in CKD: Oral Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitor Roxadustat. Front. Med. (Lausanne) 2020, 7, 393. [Google Scholar] [CrossRef]
- Efficacy and Safety of FG-4592 for Treatment of Anemia in Patients with Lower Risk MDS with Low Red Blood Cell Transfusion Burden. Available online: https://clinicaltrials.gov/ct2/show/NCT03263091 (accessed on 9 March 2021).
- Henry, D.H.; Glaspy, J.; Harrup, R.A.; Mittelman, M.; Zhou, A.; Bradley, C.; Saha, G.; Bartels, P.; Robert, L.; Yu, K.-H.P. Roxadustat (FG4592; ASP1517; AZD9941) in the Treatment of Anemia in Patients with Lower Risk Myelodysplastic Syndrome (LR-MDS) and Low Red Blood Cell (RBC) Transfusion Burden (LTB). Blood 2019, 134, 843. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Gore, S.D.; Cogle, C.; Ward, R.; Shi, T.; Macbeth, K.J.; Laille, E.; Giordano, H.; Sakoian, S.; Jabbour, E.; et al. Phase I study of oral azacitidine in myelodysplastic syndromes, chronic myelomonocytic leukemia, and acute myeloid leukemia. J. Clin. Oncol. 2011, 29, 2521–2527. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Gore, S.D.; Kambhampati, S.; Scott, B.; Tefferi, A.; Cogle, C.R.; Edenfield, W.J.; Hetzer, J.; Kumar, K.; Laille, E.; et al. Efficacy and safety of extended dosing schedules of CC-486 (oral azacitidine) in patients with lower-risk myelodysplastic syndromes. Leukemia 2016, 30, 889–896. [Google Scholar] [CrossRef]
- The Efficacy and Safety of Oral Azacitidine Plus Best Supportive Care Versus Placebo and Best Supportive Care in Subjects with Red Blood Cell (RBC) Transfusion-Dependent Anemia and Thrombocytopenia Due to International Prognostic Scoring System (IPSS) Low Risk Myelodysplastic Syndrome (MDS). Available online: https://clinicaltrials.gov/ct2/show/NCT01566695 (accessed on 9 March 2021).
- Garcia-Manero, G.; Santini, V.; Almeida, A.; Platzbecker, U.; Jonasova, A.; Silverman, L.R.; Falantes, J.; Reda, G.; Buccisano, F.; Fenaux, P.; et al. Phase III, Randomized, Placebo-Controlled Trial of CC-486 (Oral Azacitidine) in Patients with Lower-Risk Myelodysplastic Syndromes. EHA Open Access Libr. 2021, 39, 1426–1436. [Google Scholar] [CrossRef]
- Zhou, L.; Jiang, Y.; Luo, Q.; Li, L.; Jia, L. Neddylation: A novel modulator of the tumor microenvironment. Mol. Cancer 2019, 18, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ades, L.; Watts, J.M.; Radinoff, A.; Arnan, M.; Cerrano, M.; Lopez, P.F.; Zeidner, J.F.; Diez-Campelo, M.; Graux, C.; Liesveld, J.; et al. Phase II study of pevonedistat (P) + azacitidine (A) versus A in patients (pts) with higher-risk myelodysplastic syndromes (MDS)/chronic myelomonocytic leukemia (CMML), or low-blast acute myelogenous leukemia (LB AML) (NCT02610777). ). J. Clin. Oncol. 2020, 38, 7506. [Google Scholar] [CrossRef]
- Pevonedistat Plus Azacitidine versus Single-Agent Azacitidine as First-Line Treatment for Participants with Higher-Risk Myelodysplastic Syndromes (HR MDS), Chronic Myelomonocytic Leukemia (CMML), or Low-Blast Acute Myelogenous Leukemia (AML) (PANTHER). Available online: https://clinicaltrials.gov/ct2/show/NCT03268954 (accessed on 11 March 2021).
- Oldenborg, P.-A. CD47: A Cell Surface Glycoprotein Which Regulates Multiple Functions of Hematopoietic Cells in Health and Disease. ISRN Hematol. 2013, 2013, 614619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilead’s Magrolimab, an Investigational Anti-CD47 Monoclonal Antibody, Receives FDA Breakthrough Therapy Designation for Treatment of Myelodysplastic Syndrome. Available online: https://www.gilead.com/news-and-press/press-room/press-releases/2020/9/gileads-magrolimab-an-investigational-anticd47-monoclonal-antibody-receives-fda-breakthrough-therapy-designation-for-treatment-of-myelodysplastic (accessed on 11 March 2011).
- Magrolimab + Azacitidine Versus Azacitidine + Placebo in Untreated Participants with Myelodysplastic Syndrome (MDS) (ENHANCE). Available online: https://clinicaltrials.gov/ct2/show/NCT04313881 (accessed on 11 March 2021).
- Brunner, A.M.; Esteve, J.; Porkka, K.; Knapper, S.; Vey, N.; Scholl, S.; Garcia-Manero, G.; Wermke, M.; Janssen, J.; Traer, E.; et al. Efficacy and Safety of Sabatolimab (MBG453) in Combination with Hypomethylating Agents (HMAs) in Patients with Acute Myeloid Leukemia (AML) and High-Risk Myelodysplastic Syndrome (HR-MDS): Updated Results from a Phase 1b Study. In Proceedings of the 2020 ASH Annual Meeting & Exposition, San Diego, CA, USA, 5 December 2020. [Google Scholar]
- Zeidan, A.M.; Esteve, J.; Giagounidis, A.; Kim, H.-J.; Miyazaki, Y.; Platzbecker, U.; Schuh, A.C.; Sekeres, M.A.; Westermann, J.; Xiao, Z.; et al. The STIMULUS Program: Clinical Trials Evaluating Sabatolimab (MBG453) Combination Therapy in Patients (Pts) with Higher-Risk Myelodysplastic Syndromes (HR-MDS) or Acute Myeloid Leukemia (AML). Blood 2020, 136, 45–46. [Google Scholar] [CrossRef]
- Garcia, J.S. Safety, efficacy, and patient-reported outcomes of venetoclax in combination with azacitidine for the treatment of patients with higher-risk myelodysplastic syndrome: A phase 1b study. In Proceedings of the ASH Annual Meeting, San Diego, CA, USA, 5 December 2020. [Google Scholar]
- Study of Venetoclax Tablet with Intravenous or Subcutaneous Azacitidine to Assess Change in Disease Activity in Adult Participants with Newly Diagnosed Higher-Risk Myelodysplastic Syndrome (Verona). Available online: https://clinicaltrials.gov/ct2/show/NCT04401748 (accessed on 11 March 2021).
- DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef] [PubMed]
- IDH1 (AG 120) Inhibitor in Patients with IDH1 Mutated Myelodysplastic Syndrome. Available online: https://clinicaltrials.gov/ct2/show/NCT03503409 (accessed on 11 March 2021).
- IDH2 (AG 221) Inhibitor in Patients with IDH2 Mutated Myelodysplastic Syndrome. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03744390 (accessed on 11 March 2021).
- Azacitidine and Enasidenib in Treating Patients with IDH2-Mutant Myelodysplastic Syndrome. Available online: https://clinicaltrials.gov/ct2/show/NCT03383575 (accessed on 11 March 2021).
- Open-label Study of FT-2102 with or without Azacitidine or Cytarabine in Patients with AML or MDS with an IDH1 Mutation. Available online: https://clinicaltrials.gov/ct2/show/NCT02719574 (accessed on 11 March 2021).
- Cortes, J.E.; Wang, E.S.; Watts, J.M.; Lee, S.; Baer, M.R.; Dao, K.-H.; Dinner, S.; Yang, J.; Donnellan, W.B.; Schwarer, A.P.; et al. Olutasidenib (FT-2102) Induces Rapid Remissions in Patients with IDH1-Mutant Myelodysplastic Syndrome: Results of Phase 1/2 Single Agent Treatment and Combination with Azacitidine. Blood 2019, 134, 674. [Google Scholar] [CrossRef]
- A Study of ASTX030 (Cedazuridine in Combination with Azacitidine) in MDS, CMML, or AML. Available online: https://clinicaltrials.gov/ct2/show/NCT04256317 (accessed on 9 March 2021).
- Venetoclax in Combination with ASTX727 for the Treatment of Treatment-Naive High-Risk Myelodysplastic Syndrome or Chronic Myelomonocytic Leukemia. Available online: https://clinicaltrials.gov/ct2/show/NCT04655755 (accessed on 11 March 2011).
- Garcia-Manero, G.; Sasaki, K.; Montalban-Bravo, G.; Bodden, K.R.; Bose, P.; Alvarado, Y.; Daver, N.G.; Borthakur, G.; Ravandi, F.; Takahashi, K.; et al. Final Report of a Phase II Study of Guadecitabine (SGI-110) in Patients (pts) with Previously Untreated Myelodysplastic Syndrome (MDS). Blood 2018, 132, 232. [Google Scholar] [CrossRef]
- Guadecitabine (SGI-110) vs Treatment Choice in Adults with MDS or CMML Previously Treated with HMAs. Available online: https://clinicaltrials.gov/ct2/show/NCT02907359 (accessed on 11 March 2021).
- Astex and Otsuka Announce Results of Phase 3 ASTRAL-2 and ASTRAL-3 Studies of Guadecitabine (SGI-110) in Patients with Previously Treated Acute Myeloid Leukemia (AML) and Myelodysplastic Syndromes or Chronic Myelomonocytic Leukemia (MDS/CMML). Available online: https://astx.com/astex-and-otsuka-announce-results-of-phase-3-astral-2-and-astral-3-studies-of-guadecitabine-sgi-110-in-patients-with-previously-treated-acute-myeloid-leukemia-aml-and-myelodysplastic-syndromes-or/ (accessed on 10 March 2021).
- Taylor, J.; Mi, X.; Penson, A.V.; Paffenholz, S.V.; Alvarez, K.; Sigler, A.; Chung, S.S.; Rampal, R.K.; Park, J.H.; Stein, E.M.; et al. Safety and activity of selinexor in patients with myelodysplastic syndromes or oligoblastic acute myeloid leukaemia refractory to hypomethylating agents: A single-centre, single-arm, phase 2 trial. Lancet Haematol. 2020, 7, e566–e574. [Google Scholar] [CrossRef]
- A Phase 1 Study to Evaluate H3B-8800 in Participants with Myelodysplastic Syndromes, Acute Myeloid Leukemia, and Chronic Myelomonocytic Leukemia. Available online: https://clinicaltrials.gov/ct2/show/NCT02841540 (accessed on 11 March 2021).
- Steensma, D.P.; Wermke, M.; Klimek, V.M.; Greenberg, P.L.; Font, P.; Komrokji, R.S.; Yang, J.; Brunner, A.M.; Carraway, H.E.; Ades, L.; et al. Results of a Clinical Trial of H3B-8800, a Splicing Modulator, in Patients with Myelodysplastic Syndromes (MDS), Acute Myeloid Leukemia (AML) or Chronic Myelomonocytic Leukemia (CMML). Blood 2019, 134, 673. [Google Scholar] [CrossRef]
- Sallman, D.A. To target the untargetable: Elucidation of synergy of APR-246 and azacitidine in TP53 mutant myelodysplastic syndromes and acute myeloid leukemia. Haematologica 2020, 105, 1470–1472. [Google Scholar] [CrossRef]
- APR-246 & Azacitidine for the Treatment of TP53 Mutant Myelodysplastic Syndromes (MDS). Available online: https://clinicaltrials.gov/ct2/show/NCT03745716 (accessed on 15 March 2021).
- Aprea Therapeutics Announces Results of Primary Endpoint from Phase 3 Trial of Eprenetapopt in TP53 Mutant Myelodysplastic Syndromes (MDS). Available online: https://www.globenewswire.com/news-release/2020/12/28/2150874/0/en/Aprea-Therapeutics-Announces-Results-of-Primary-Endpoint-from-Phase-3-Trial-of-Eprenetapopt-in-TP53-Mutant-Myelodysplastic-Syndromes-MDS.html (accessed on 3 March 2021).
- Garcia-Manero, G.; Fenaux, P.; Al-Kali, A.; Baer, M.R.; Sekeres, M.A.; Roboz, G.J.; Gaidano, G.; Scott, B.L.; Greenberg, P.; Platzbecker, U.; et al. Rigosertib versus best supportive care for patients with high-risk myelodysplastic syndromes after failure of hypomethylating drugs (ONTIME): A randomised, controlled, phase 3 trial. Lancet Oncol. 2016, 17, 496–508. [Google Scholar] [CrossRef]
- Onconova Therapeutics Announces Topline Results from the Pivotal Phase 3 INSPIRE Trial. Available online: https://www.globenewswire.com/news-release/2020/08/24/2082504/0/en/Onconova-Therapeutics-Announces-Topline-Results-from-the-Pivotal-Phase-3-INSPIRE-Trial.html (accessed on 12 March 2021).
- Controlled Study of Rigosertib Versus Physician’s Choice of Treatment in MDS Patients after Failure of an HMA (INSPIRE). Available online: https://clinicaltrials.gov/ct2/show/NCT02562443 (accessed on 11 March 2021).
- Tohyama, K.; Tsutani, H.; Ueda, T.; Nakamura, T.; Yoshida, Y. Establishment and characterization of a novel myeloid cell line from the bone marrow of a patient with the myelodysplastic syndrome. Br. J. Haematol. 1994, 87, 235–242. [Google Scholar] [CrossRef]
- Kida, J.-I.; Tsujioka, T.; Suemori, S.-I.; Okamoto, S.; Sakakibara, K.; Takahata, T.; Yamauchi, T.; Kitanaka, A.; Tohyama, Y.; Tohyama, K. An MDS-derived cell line and a series of its sublines serve as an in vitro model for the leukemic evolution of MDS. Leukemia 2018, 32, 1846–1850. [Google Scholar] [CrossRef] [PubMed]
- Drexler, H.G.; Dirks, W.G.; Macleod, M.R. Many are called MDS cell lines: One is chosen. Leuk Res. 2009, 33, 1011–1016. [Google Scholar] [CrossRef] [PubMed]
- Kogan, S.C.; Ward, J.M.; Anver, M.R.; Berman, J.J.; Brayton, C.; Cardiff, R.D.; Carter, J.S.; de Coronado, S.; Downing, J.R.; Fredrickson, T.N.; et al. Bethesda proposals for classification of nonlymphoid hematopoietic neoplasms in mice. Blood 2002, 100, 238–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, L.; Åstrand-Grundström, I.; Arvidsson, I.; Jacobsson, B.; Hellström-Lindberg, E.; Hast, R.; Jacobsen, S.E.W. Isolation and characterization of hematopoietic progenitor/stem cells in 5q-deleted myelodysplastic syndromes: Evidence for involvement at the hematopoietic stem cell level. Blood 2000, 96, 2012–2021. [Google Scholar] [CrossRef]
- Benito, A.I.; Bryant, E.; Loken, M.R.; Sale, G.E.; Nash, R.A.; John Gass, M.; Deeg, H.J. NOD/SCID mice transplanted with marrow from patients with myelodysplastic syndrome (MDS) show long-term propagation of normal but not clonal human precursors. Leuk Res. 2003, 27, 425–436. [Google Scholar] [CrossRef]
- Krevvata, M.; Shan, X.; Zhou, C.; Dos Santos, C.; Habineza Ndikuyeze, G.; Secreto, A.; Glover, J.; Trotman, W.; Brake-Silla, G.; Nunez-Cruz, S.; et al. Cytokines increase engraftment of human acute myeloid leukemia cells in immunocompromised mice but not engraftment of human myelodysplastic syndrome cells. Haematologica 2018, 103, 959–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medyouf, H.; Mossner, M.; Jann, J.-C.; Nolte, F.; Raffel, S.; Herrmann, C.; Lier, A.; Eisen, C.; Nowak, V.; Zens, B.; et al. Myelodysplastic Cells in Patients Reprogram Mesenchymal Stromal Cells to Establish a Transplantable Stem Cell Niche Disease Unit. Cell Stem Cell 2014, 14, 824–837. [Google Scholar] [CrossRef] [Green Version]
- Mian, S.A.; Abarrategi, A.; Kong, K.L.; Rouault-Pierre, K.; Wood, H.; Oedekoven, C.A.; Smith, A.E.; Batsivari, A.; Ariza-McNaughton, L.; Johnson, P.; et al. Ectopic Humanized Mesenchymal Niche in Mice Enables Robust Engraftment of Myelodysplastic Stem Cells. Blood Cancer Discov. 2021, 2, 135. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Rongvaux, A.; Taylor, A.; Jiang, T.; Tebaldi, T.; Balasubramanian, K.; Bagale, A.; Terzi, Y.K.; Gbyli, R.; Wang, X.; et al. A highly efficient and faithful MDS patient-derived xenotransplantation model for pre-clinical studies. Nat. Commun. 2019, 10, 366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tothova, Z.; Krill-Burger, J.M.; Popova, K.D.; Landers, C.C.; Sievers, Q.L.; Yudovich, D.; Belizaire, R.; Aster, J.C.; Morgan, E.A.; Tsherniak, A.; et al. Multiplex CRISPR/Cas9-Based Genome Editing in Human Hematopoietic Stem Cells Models Clonal Hematopoiesis and Myeloid Neoplasia. Cell Stem Cell 2017, 21, 547–555.e548. [Google Scholar] [CrossRef] [PubMed]
- Kotini, A.G.; Chang, C.-J.; Boussaad, I.; Delrow, J.J.; Dolezal, E.K.; Nagulapally, A.B.; Perna, F.; Fishbein, G.A.; Klimek, V.M.; Hawkins, R.D.; et al. Functional analysis of a chromosomal deletion associated with myelodysplastic syndromes using isogenic human induced pluripotent stem cells. Nat. Biotechnol. 2015, 33, 646–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, J.; Reilly, A.; Hayes, B.J.; Clough, C.A.; Konnick, E.Q.; Torok-Storb, B.; Gulsuner, S.; Wu, D.; Becker, P.S.; Keel, S.B.; et al. Reprogramming identifies functionally distinct stages of clonal evolution in myelodysplastic syndromes. Blood 2019, 134, 186–198. [Google Scholar] [CrossRef] [Green Version]
- Beachy, S.H.; Aplan, P.D. Mouse models of myelodysplastic syndromes. Hematol. Clin. New Am. 2010, 24, 361–375. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-W.; Slape, C.; Zhang, Z.; Aplan, P.D. NUP98-HOXD13 transgenic mice develop a highly penetrant, severe myelodysplastic syndrome that progresses to acute leukemia. Blood 2005, 106, 287–295. [Google Scholar] [CrossRef] [Green Version]
- Barlow, J.L.; Drynan, L.F.; Hewett, D.R.; Holmes, L.R.; Lorenzo-Abalde, S.; Lane, A.L.; Jolin, H.E.; Pannell, R.; Middleton, A.J.; Wong, S.H.; et al. A p53-dependent mechanism underlies macrocytic anemia in a mouse model of human 5q– syndrome. Nat. Med. 2010, 16, 59–66. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Currently approved and emerging therapies for myelodysplastic syndromes (MDS). New targeted therapies and their mechanism of action on MDS cells and the bone marrow microenvironment.
Figure 1.
Currently approved and emerging therapies for myelodysplastic syndromes (MDS). New targeted therapies and their mechanism of action on MDS cells and the bone marrow microenvironment.

Figure 2.
Advances to humanized MDS mice models; MDS xenotransplantation has been challenging due to low engraftment and lack of maintenance of HSCs in mice models. Advances on existing mice models have been made to humanize mice models to allow for the mice models to allow for improved engraftment and myeloid differentiation. Some of these humanized mice models include NSG-S and MISTRG. Legend: NK = natural killer cells.
Figure 2.
Advances to humanized MDS mice models; MDS xenotransplantation has been challenging due to low engraftment and lack of maintenance of HSCs in mice models. Advances on existing mice models have been made to humanize mice models to allow for the mice models to allow for improved engraftment and myeloid differentiation. Some of these humanized mice models include NSG-S and MISTRG. Legend: NK = natural killer cells.


{kind=link}
{kind=link}
Table 1.
Overview of pivotal trials in approved drugs for MDS.
| Drug Candidate | Design | MOA | FDA Indication | NCT # | Primary Endpoint | Approval Date | Notes |
|---|---|---|---|---|---|---|---|
| Lenalidomide (MDS-003) | Single-arm, open-label (phase 2) | Immuno-modulator | TDA in LR-IR Del (5q) MDS | NCT00065156 | 67% TI > 8 weeks | 28 December 2005 | Long term f/u shows TI > 8 weeks has OS benefit (4.3 vs. 2.0 years) |
| Lenalidomide (MDS-004) | RCT | Immuno-modulator | TDA of LR- and IR- MDS with Del(5q) Chromosomal Abnormality | NCT00179621 | 56.1% transfusion independence > 26 weeks vs. 5.9% placebo (p < 0.001) | 28 December 2005 | Unable to assess; early crossover to lenalidomide in non-responders |
| Luspatercept (MEDALIST) | RCT | TGF-ß family ligand trap | TDA in LR-MDS-RS and failed/unlikely to respond to ESA | NCT02631070 | 38% TI > 8 weeks vs. 13% placebo (p < 0.001) | 3 April 2020 | * Excluded del(5q), prior HMA, IMID * Progression to HR-MDS or AML: 5.2% in luspatercept vs. 5.3% in placebo |
| Azacitidine | Randomized, parallel group,-open-label | HMA | LR/HR-MDS patients ineligible for BMT | NCT00071799 | OS 24.5 months in AZA vs. 15 months SOC (HR = 0.58, p < 0.0001) | 19 May 2004 | * Time to AML transformation: 17.8 mos AZA vs. 11.5 mos SOC * ORR: 77% AZA vs. 41% SOC |
| Decitabine | Randomized, open-label | HMA | LR/HR-MDS patients ineligible for BMT | N/A | * ORR: 17% DAC vs. 0% BSC (p < 0.001) * Time to AML or death: 12.1 months DAC vs. 7.8 months BSC (p < 0.16) | 1 May 2006 | * HI: 13% DAC vs. 7% BSC (p < 0.001) * Subgroup of IPS-2/HR-MDS has time to AML/death benefit (12.0 vs. 6.8 months, p < 0.03) |
| ASTX-727 (ASCERTAIN) | Randomized, open-label, crossover study | Oral DAC + Cytadine deaminase inhibitor | IR/HR-MDS | NCT03306264 | AUC of oral DAC-cedazuridine vs. IV DAC; AUC ratio of 98.9% | 7 July 2020 | * Excludes prior treatment with HMA |
TDA = transfusion dependent anemia, TI—transfusion independent, SOC = standard of care, BSC = best supportive care, ORR = overall response rate, OS = overall survival, HI = hematological improvement, HMA = hypomethylating agent, AZA = azacitidine, DAC = decitabine, BMT = bone marrow transplant, LR = low risk, IR = intermediate risk, HR = high risk, AUC = area under the curve, # = number, * = outcomes of special interest.
Table 2.
Overview of LR-MDS Potential Therapeutics.
| Drug Candidate | Phase/Design | MOA | NCT # | Primary Endpoint | Date Complete | Status | Notes |
|---|---|---|---|---|---|---|---|
| Imetelstat | Phase 3 RCT | Telomerase Inhibitor | NCT02598661 | Percentage of TI during any consecutive 8-week period over 2 years | 22 August | Recruiting | Excludes ESA within 4 weeks of study entry |
| Luspatercept (COMMANDS) | Phase 3 RCT | TGF-β family ligand trap | NCT03682536 | TI for 24 weeks | 22 November | Recruiting | Excludes prior ESA use, del(5q), SF3B1 subtype |
| Roxadustat (MATTERHORN) | Phase 3 RCT | HIF Stabilizer | NCT03263091 | TI ≥ 56 consecutive days | 22 January | Recruiting | No restrictions on prior ESA use, excludes del(5q) |
| ASTX727 | Phase 1/2 Randomizedopen-label, dose-finding | Oral HMA + Cytadine deaminase inhibitor | NCT03502668 | Normalization of baseline cytopenia | 21 December | Recruiting |
RCT = randomized control trial, TI = transfusion independence, HMA = hypomethylating agent, ESA = erythropoietin stimulating agent.
Table 3.
Overview of HR-MDS Potential Therapeutics.
| Drug Candidate | Phase + Design | MOA | Single or Combo | NCT # | Primary Endpoint | Completion Date | Status | Notes |
|---|---|---|---|---|---|---|---|---|
| Pevonedistat (PANTHER) | Phase 3 RCT open-label | NEDD8-i | Combo w/HMA | NCT03268954 | EFS | 22 July | Active, not recruiting | Treatment naïve HR-MDS/CMML, and low-blast AML |
| Magrolimab (ENHANCE) | Phase 3 RCT | Anti-CD47 MAB | Combo w/HMA | NCT04313881 | *CR *OS | 25 February | Recruiting | Treatment naïve HR-MDS |
| MBG453 (STIMULUS-MDS2) | Phase 3 RandomizedDouble-blind, active comparator | TIM-3-i | Combo w/HMA | NCT04266301 | OS | 21 August | Recruiting | HMA naïve HR-MDS/CMML |
| Venetoclax (VERONA) | Phase 3 RCT active comparator | BCL2-i | Combo w/HMA | NCT04401748 | *OS *CR | 25 February | Recruiting | Treatment naïve HR-MDS |
| Ivosidenib | Phase 2 Multi-cohort, open-label | IDH1-i | Single | NCT03503409 | Overall HI | 22 January | Recruiting | *Patients with IDH1-mutated MDS *Arm 1: R/R HMA *Arm 2: HMA-naïve *Arm 3: LR-MDS |
| Enasidenib | Phase 2 Multi-cohort, open label | IDH2-i | Single | NCT03744390 | Overall HI | 23 February | Recruiting | *Patients with IDH2-mutated MDS *Arm 1: R/R HMA *Arm 2: HMA-naïve *Arm 3: LR-MDS |
| Enasidenib | Phase 2 Non-randomized, parallel assignment | IDH2-i | Single and combo w/HMA | NCT03383575 | *Incidence of AE *ORR | 22 February | Recruiting | *Patients with IDH2-mutated MDS *Arm 1: HMA-naïve *Arm 2: HMA R/R |
| ASTX030 | Multi-phase, dose-escalation followed by an open-label, randomized, crossover study | Oral AZA + Cytadine deaminase inhibitor | Single | NCT04256317 | AUC oral ASTX030 vs. SC AZA | 23 April | Recruiting | HMA naïve HR-MDS/CMML/AML |
| ASTX727 | Phase 1/2a: Single arm, open label | Oral DAC + Cytadine deaminase inhibitor | Combo w/venetoclax | NCT04655755 | *Phase 1: Safety/tolerability *Phase 2: ORR | 22 July | Not yet recruiting | Treatment naïve HR-MDS/CMML |
| Selinexor | Phase 2 Single arm, open label | XPO1-i | Single | NCT02228525 | ORR | 21 August | Complete | HR-MDS R/R to HMA |
| APR-246 | Phase 3 Randomizedopen label, active comparator | TP53 modulator | Combo w/HMA | NCT03745716 | CR rate of APR-246 + AZA vs. AZA alone | 20 November | Active, not recruiting | HMA naïve, TP53 mutated MDS |
| Rigosertib (INSPIRE) | Phase 3 Randomizedopen label, controlled study | TKI | Single | NCT02562443 | *OS *OS of IPSS-R very high-risk group | 20 December | Complete | *R/R to HMA *OS failed [64] |
OS = overall survival, R/R = relapsed/refractory, EFS = event free survival, CR = clinical response, HI = hematologic response, ORR= overall response rate, HMA = hypomethylating agent, AZA = azacitidine, DAC = decitabine, HR= high risk, AE = adverse event, AUC = area under the curve, SC = subcutaneous, * = outcome of special interest.
Table 4.
Criteria for myeloid dysplasia in mice.
| Criteria Number | Guidelines for MDS in Mice |
|---|---|
| 1 | Neutropenia, Thrombocytopenia, and Anemia found in peripheral blood without leukocytosis and erythrocytosis |
| 2 | Non-lymphoid hematopoietic cells showed dysgranulopoiesis, dyserythropoiesis, or dysplastic megakaryocytes with or without increased non-lymphoid immature forms or blasts |
| 3 | Non-lymphoid leukemia had been excluded out |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Stanchina, M.; Chaudhry, S.; Karr, M.; Taylor, J. Current State and Challenges in Development of Targeted Therapies in Myelodysplastic Syndromes (MDS). Hemato 2021, 2, 217-236. https://0-doi-org.brum.beds.ac.uk/10.3390/hemato2020013
AMA Style
Stanchina M, Chaudhry S, Karr M, Taylor J. Current State and Challenges in Development of Targeted Therapies in Myelodysplastic Syndromes (MDS). Hemato. 2021; 2(2):217-236. https://0-doi-org.brum.beds.ac.uk/10.3390/hemato2020013
Chicago/Turabian StyleStanchina, Michele, Sana Chaudhry, Matthew Karr, and Justin Taylor. 2021. "Current State and Challenges in Development of Targeted Therapies in Myelodysplastic Syndromes (MDS)" Hemato 2, no. 2: 217-236. https://0-doi-org.brum.beds.ac.uk/10.3390/hemato2020013