
Can Novel Insights into the Pathogenesis of Myeloproliferative Neoplasm-Related Thrombosis Inform Novel Treatment Approaches?


1
Institute of Hematology, Davidoff Cancer Centre, Beilinson Hospital, Rabin Medical Center, Petah-Tikva 49100, Israel
2
Sackler Faculty of Medicine, Tel Aviv University, Tel Aviv-Yafo 6997801, Israel
*
Author to whom correspondence should be addressed.
Hemato 2021, 2(2), 305-328; https://0-doi-org.brum.beds.ac.uk/10.3390/hemato2020018
Submission received: 10 April 2021
/
Revised: 7 May 2021
/
Accepted: 10 May 2021
/
Published: 16 May 2021
(This article belongs to the Special Issue Advances in the Prevention of Thrombosis in Myeloproliferative Neoplasms)
Abstract
:Despite recent advances in diagnosis and therapy, arterial and venous thrombosis remain a major cause of morbidity and mortality in Philadelphia-negative myeloproliferative neoplasms (MPNs). Preventing and treating arterial and venous thrombosis represent one of the major goals in MPNs. The prothrombotic phenotype of MPNs is the result of a complex interplay between several components. Neutrophils, platelets, red blood cells (RBCs) and endothelial cells assume an activated phenotype in MPNs and undergo morphologic and metabolic changes that render these cells prothrombotic. These changes are in part the result of alterations induced by MPN initiating, driving mutations as well as the effect of extrinsic factors that stem from cell interactions as well as the inflammatory environment and rheological properties that characterize MPNs. In this review, we address current management issues in MPNs and provide an update on recent understanding of the pathogenesis of thrombosis in MPNs. We also address how lessons learned from other thrombo-inflammatory conditions can further inform and improve management of thrombosis in MPNs. Based on the above data and recent discoveries and developments, we discuss potential novel targets and therapeutic approaches to tackle the challenge of thrombosis in MPNs.
1. Introduction
Myeloproliferative neoplasms (MPNs) are stem-cell, clonal hematopoietic disorders characterized by proliferation of one or more myeloid cell lineages in the bone marrow and increased blood cells in the peripheral blood. The classical Philadelphia (Ph) negative MPNs include polycythemia vera (PV), essential thrombocythemia (ET) and primary myelofibrosis (MF). In most cases, clonal proliferation is driven by one of 3 mutually exclusive driver somatic mutations resulting in dysregulation of the Janus kinase-signal transducers and activators of transcription (JAK-STAT) pathway and subsequent activation of downstream signaling pathways [1]. JAK2V617F is the most common driver mutation, found in 96% of patients with PV and in 55% and 65% of patients with ET and MF respectively, while JAK2 exon 12 mutations can be found in about 3% of patients with PV [2]. Calreticulin gene (CALR) and TPO receptor (MPL) gene mutations are found in up to 25% and 4% of patients with ET and up to 35% and 8% of patients with MF, respectively. In about 10–15% of patients with ET and MF no driver mutation can be identified, and these are considered triple negative MPNs [3].
PV, ET and MF are characterized (to different degrees) by a chronic course with a shorter overall survival [4]. They share clinical characteristics such as vasomotor symptoms, extramedullary hematopoiesis with splenomegaly, constitutional symptoms, pruritus, and all carry an increased risk for thrombosis (arterial and venous) and for disease progression/transformation. Although similarities exist in the molecular signatures across the different disease subtypes, disease phenotype, prognosis and therapeutic approaches may significantly differ [4].
Thrombotic and hemorrhagic complications (especially in PV and ET) are central to the clinical phenotype of MPNs and contribute to the inferior quality of life and survival in these patients.
Although arterial and venous thromboses are both increased in MPNs, different pathogenetic pathways are involved [5]. In general, venous thrombosis is associated with hypercoagulability and stasis which leads to endothelial hypoxia and a pro-inflammatory phenotype, characterized by the expression of adhesion molecules, accumulation and activation of leukocytes and platelets. For example, increased formation of neutrophil extracellular traps (NETs) is associated with expression of tissue factor (TF) on platelets, activation of factor XII and subsequently of the intrinsic coagulation pathway [6], and inactivation of anticoagulant molecules such as tissue factor pathway inhibitor (TFPI) [7]. Arterial thrombosis is typically initiated by an atherosclerotic plaque. Expression of adhesion molecules by endothelial cells that cover the atherosclerotic plaque contributes to platelet adhesion, attachment, and migration of monocytes into the vascular wall, increasing plaque growth. Furthermore, increased NET formation is also part of the pathogenesis of arterial thrombosis [7]. Arterial thrombosis is rarely triggered by arterial vasospasm.
Despite the apparent divergent pathways that characterize venous and arterial thrombosis, there is often an overlap in the pathogenic changes induced in MPNs that may affect both pathologies.
In recent years, several genetic and metabolic insights led to better understanding of the pathogenetic processes leading to arterial and venous thrombosis and hemorrhage in MPNs. Some of these insights expose potential targets for prevention and treatment of these complications. In this review, we address current management issues, provide an update on recent understanding of the pathogenesis of thrombosis in MPNs and based on these developments, discuss potential novel targets and therapeutic approaches to tackle the challenge of thrombosis in MPNs.
2. The Scope of the Problem: Thrombosis in MPNs
Thrombosis, and to a lesser extent bleeding are leading causes of morbidity and mortality in MPNs, especially in PV and ET [8,9]. Thrombosis can present in early stages or later during the course of disease [10]. The prevalence of thrombotic events at diagnosis ranges from 12–39% in PV and 11–25% in ET [11] compared to 3–7% in myelofibrosis. The rates of thrombosis in patients with MPNs are increased when compared to the general population, with an estimated 3-fold increased rate of arterial thrombotic events and 10-fold increased rate of venous thrombotic events [10].
Arterial events comprise two-thirds of cases while venous thrombotic events (VTEs) account for a third of total MPN related thrombotic events [10]. VTEs may typically occur in unusual sites including splanchnic (portal, splenic, hepatic and mesenteric) veins and cerebral sinus veins [10,12]. Of note, splanchnic vein thrombosis is exceptionally enriched for in MPNs with high recurrence rates of between 15% and 20% over 10 years [13].
Although primary and secondary prophylaxis reduce the risk for first or recurrent thrombotic events, respectively [14], patients still face high rates of thrombosis in the presence of bleeding complications [15]. Better understanding of the potential mechanisms and risk factors for thrombosis may improve the current treatment approach and encourage the development of new targeted therapies to reduce MPN-associated thrombotic complications, hence improve quality of life and overall survival of these patients.
3. Current Risk Assessment for Thrombosis in MPNs: Established and Suggested Models
Studies of PV and ET have previously used various definitions for the thrombotic risk in PV and ET. In PV, age and history of thrombosis remain the most consistent risk factors [9]. Hence patients are stratified into low risk (age ≤60 years and no history of thrombosis) and high risk (age >60 years old or a history of thrombosis) [16]. In ET, stratification based on the revised international prognostic score for thrombosis in ET (revised IPSET thrombosis) includes age, a history of thrombosis and the status of JAK2V617F mutation. Accordingly, 4 risk groups have been identified: (i) very low risk (age ≤60 years, no history of thrombosis and JAK2 wild type), (ii) low risk (age ≤60 years and no history of thrombosis and JAK2V617F mutated), (iii) intermediate risk (age >60 years old, no history of thrombosis and JAK2 wild type) and (iv) high risk (age >60 years old or a history of thrombosis and JAK2V617F mutated) [17].
Other risk factors which may also contribute to thrombosis propensity such as cardiovascular (CV) risk factors, leukocytosis, JAK2 allele burden and high hematocrit, have not yet been consistently validated. For example, among 891 patients with ET enrolled in an international study, in addition to age >60 years and history of prior thrombosis, other risk factors for arterial thrombosis were identified including CV risk factors (tobacco use, hypertension, or diabetes mellitus), leukocytosis >11 × 109/L, and the presence of JAK2V617F mutation; only male gender predicted venous thrombosis [18]. While CV risk factors were also identified as prothrombotic according to the initial analysis of the International Prognostic Score for Thrombosis in ET (IPSET-thrombosis) [17], a further analysis by Barbui et al. observed a low impact of the CV risk factors on the IPSET thrombosis in patients with ET [19]. Accordingly, CV risk factors are not included in the current risk stratification, nevertheless, they should be managed and be well controlled [16].
The JAK2V617F mutation, and especially a high variant allele burden, increase the risk for thrombotic events compared to the CALR mutations or no mutations (“triple-negative”) in patients with ET [20,21]. Additionally, patients with clonal hematopoiesis of indeterminate significance (CHIP), who harbor the JAK2 mutation without overt MPN have an increased risk for venous thrombosis [22]. Accordingly, a lower risk of thrombosis was reported in MF with CALR mutations compared to JAK2 and MPL [23]. Nonetheless, CALR mutation status did not have a significant impact on the IPSET thrombosis prognostic score [24].
Blood counts were previously studied as possible markers for thrombotic risk. According to the PT 1 trail [25] and the IPSET data set [19,26], thrombocytosis is not associated with increased risk for thrombosis. On the contrary, extreme thrombocytosis (>1000–1500 × 109/L) is associated with a lower risk of thrombosis [18] and increased risk of bleeding complications due to an acquired von Willebrand syndrome [16]. Current guidelines suggest a target reduction of <400 × 109/L platelets although there is conflicting evidence to suggest that specific platelet values are protective against thrombotic events [27,28,29]. It seems that the prothrombotic state is driven by qualitative rather than quantitative changes in platelets [30]. Hyper-viscosity due to erythrocytosis [31] and high hematocrit levels increase the risk for thrombotic events in patients with PV [32]. Reduction of hematocrit levels <45% decreases the risk for thrombotic events in comparison to hematocrit of 45–50% as shown in the phase III CYTOreductive Therapy in Polycythemia Vera (CYTO PV) study [33]. Studies assessing leukocytosis as a risk factor for thrombosis in ET and PV are mostly retrospective in design and demonstrated inconclusive results [34]. In patients with ET, initial reports showed that increased leukocyte counts correlated with both arterial and venous thrombosis [35,36] but these were not consistent in follow-up analyses [37]. Analysis of patients with PV enrolled in the European Collaboration on Low-Dose Aspirin in Polycythemia Vera (ECLAP) study showed an association between leukocytosis (WBC > 17 × 109/L) and an increased risk for thrombosis [38,39]. Similar results were also shown in the CYTO PV study with leukocytosis (WBC > 12 × 109/L) and increased risk for thrombosis [40]. In addition, according to a recent metanalysis of more than 30,000 patients with PV and ET, leukocytosis increased the thrombotic risk, mainly in patients with ET and mainly of arterial events [41]. A more recent multi-center American analysis showed that increased leukocyte counts predict disease progression, but not thrombosis [42]. In fact, it is still debatable whether leukocytosis is a trigger or only a marker of thrombosis [43]. Hence, leukocyte count is not currently incorporated in the current risk stratification for thrombosis.
Additional insights into the pathogenesis of thrombosis in MPNs may identify novel biomarkers and risk factors for thrombosis and refine thrombosis risk models.
4. Pathogenetic Pathways Promoting Thrombosis in MPNs
The prothrombotic phenotype of MPN’s is the result of a complex interplay between several components (Figure 1). Neutrophils, platelets and red blood cells (RBCs) assume an activated phenotype in MPNs and undergo morphologic and metabolic changes that render these cells prothrombotic. These changes are in part the result of changes induced by MPN initiating driving mutations (JAK2, CALR and MPL) as well as the effect of extrinsic factors that stem from cell interactions as well as the inflammatory environment and rheological properties that characterize MPNs. Recent insights also implicate endothelial cells as key players in promoting thrombosis in MPN via cell intrinsic and extrinsic changes.
4.1. RBC’s and Hematocrit
The first and perhaps most intuitive clinical observation was that of the association between elevated hematocrit levels and thrombotic events in patients with PV [32]. This association was validated in several subsequent clinical trials as already indicated and reduction of hematocrit levels is still regarded as an actionable clinical endpoint that reduces thromboses rates in PV [33]. Santisakultarm et al. studied cerebral blood flow in transgenic MPN mouse models by two-photon excited fluorescence microscopy imaging and demonstrated that PV and ET phenotypes are associated with an increase in ‘stalled’ blood flow in an elevated fraction of brain capillaries. In the PV model, a hematocrit over 55% was specifically associated with an increase in capillary flow obstruction [44].
More recently it became clear that the prothrombotic effects of elevated hematocrit result from the distinct biophysical effect of an increased RBC mass as well as from interactions between RBCs with platelets, leukocytes and endothelial cells. Walton et al. showed that elevated hematocrit is associated with an increase in arterial events in an RBC-transfusion model in healthy mice. Additional ex vivo modeling of thrombus propagation highlighted the role of platelets in thrombus growth [45]. The authors propose a model by which elevated hematocrit alters the biophysical dynamics of blood flow in a way that narrows the near-wall RBC depleted zone around a propagating thrombus allowing increased exposure and incorporation of trapped platelets into the growing thrombus [45,46,47]. Additional mechanistic insight into pro-thrombotic RBC platelet interactions was provided by Klatt et al. The activation of platelets in the presence of RBCs prompted FAS ligand/FAS receptor interaction between platelets and RBCs, respectively, that result in the externalization of RBC phosphatidylserine (PS), ultimately promoting thrombin generation and thrombus formation [48].
An increase in RBC adhesion to endothelial cells was suggested by Wautier et al. RBC’s from patients with PV were associated with increased expression and phosphorylation of Lutheran blood group/basal cell adhesion molecule (Lu/BCAM) and adhesion (via Laminin) in an ex-vivo human umbilical vein endothelial cells (HUVECs) model [49]. The increase in Lu/BCAM was later suggested to be a cell-intrinsic property driven by JAK2 mutations [50]. Poisson et al. described the contribution of erythrocyte-derived microvesicles (MV’s) to arterial constriction and thrombosis in a murine model. These effects were mediated via oxidative stress and disruption of the nitric oxide (NO pathway by the myeloperoxidase (MPO)-loaded MV’s [51].
Zhao et al. recently identified Pleckstrin-2 (Plek2) to be involved in the prothrombotic phenotype of MPNs. Plek2 is widely expressed protein that binds to phosphoinositide and was previously shown to play an important role in erythroblast survival and enucleation during erythropoiesis [52]. Pleck2 was shown to be a downstream target of the JAK2/STAT5 pathway in erythroid and myeloid cells and was significantly upregulated in a JAK2V617F MPN mouse model and in patients with MPNs. Loss of Plek2 reverted MPN phenotype in these mice including a reduction in the occurrence of widespread vascular occlusions that was attributed to a decrease in RBC mass in these mice [53].
4.2. Platelets
Platelets are regarded as central contributors to the prothrombotic phenotype in MPN. While thrombocytosis by itself does not directly correlate with vascular events [19,25,26], markers of platelet activation are increased in patients with ET, PV and MF. For example, previous trials demonstrated an increase in plasma levels in patients with MPNs of P-selectin (surface and soluble), CD40L, Platelet factor 4 (PF4), β-thromboglobulin, thrombomodulin and platelet derived growth factor (PDGF) as well as thromboxane B2 in the urine of patients [54,55,56,57,58,59,60,61]. Increased platelet turnover with a higher fraction of immature platelets prone for activation contributes to the activated phenotype of platelets [57,62]. Platelet derived microparticles may have a role in thrombosis in MPNs. In one study, patients with ET had a higher number of circulating microparticles baring platelet and endothelial markers, that were associated with increased endothelial activation and thrombin generation [63]; additional studies implicated platelet derived microparticles in phospholipid-dependent procoagulant activity and acquired “thrombomodulin-resistant” phenotype [12,64,65,66]. Elevated plasma levels of microparticles were shown to be associated with thrombotic events in MPNs in single center observational studies [67,68].
Activated platelets were previously shown to enhance procoagulant effects by an increase in the expression on their membrane of PS and of tissue factor (TF), both initiators of coagulation cascades [62,69]. Platelet-mediated proteolytic down regulation of the activity of protein S may be responsible in part to the activated protein C resistance phenotype in patients with ET and PV [70,71,72].
Despite enhanced platelet activation in MPNs, a paradox of impaired platelet function is evident. Aggregations studies are characterized by an absent second-wave adrenaline aggregation, an increased adenosine diphosphate (ADP) aggregation threshold with normal arachidonic acid or collagen-induced aggregation that fits the phenotype of exhausted defective platelets with secondary storage pool defect [73,74]. These observations were confirmed in a recent systematic review that identified 21 studies assessing platelet aggregation of which 20 demonstrated impaired platelet aggregation in patients with ET [59]. Modeling of platelet-mediated hemostatic defects is challenging, and different studies yield apparent contradicting results. While some studies report defective platelet aggregation and function [75,76], others show no apparent platelet defect [77,78] or a hyper-reactive platelet phenotype [79]. Further complicating the picture is the fact that several of these models result in bleeding-diathesis secondary to an acquired von Willebrand factor (vWF) deficiency-like phenotype [75,77,78].
Moore et al. demonstrated that the PI3 kinase/Rap1 pathway is intrinsically impaired in platelets from JAK2V617F ET patients, resulting in diminished thrombin and thrombopoietin-mediated integrin αIIbβ3 activation that explains the ex-vivo aggregation defect in platelets from patients with ET [80]. On the other hand, Matsuura et al. had shown that Lysyl oxidase (LOX), an enzyme that catalyzes the conversion of specific lysines to aldehydes and that is overexpressed in MPNs [81,82], is associated with platelet activation and thrombosis in murine models, perhaps by affecting platelet adhesion to collagen [83].
This suggests that activation patterns may be related in part to changes induced by somatic driver mutations in MPN as well as increased activation that is the result of cell-cell interaction, inflammation, and microenvironment.
4.3. Leukocytes
The role of leukocytes in thrombosis has recently come under the spotlight.
Leukocytes from patients with MPNs have an activated phenotype. In patients with MPNs, neutrophils express higher levels of CD11b and have increased reactive oxygen species (ROS) production [84] and higher levels of neutrophil activation products such as elastase, catepsin G and myeloperoxidase (MPO) in the plasma [12,85,86,87]. Interestingly, different driver mutations may be associated with differential activation patterns. Torregrosa et al. demonstrated in samples from patients with ET, that CALR-driven disease is associated with reduced leukocyte activation that may account in part for the lower thrombosis rate in this patient subgroup [88].
Recently, neutrophil extracellular traps (NETs) were implicated in the thrombotic tendency in MPNs.
In 2004, Brinkmann and colleagues describe the occurrence of NETs [89]. In response to various stimuli, neutrophils can expel extracellular strands of decondensed DNA in complex with histones and other neutrophil granular proteins. These structures can kill microbes but are also implicated in the pathogenesis of autoimmunity and thrombosis [90,91]. Increased NET formation was also implicated in the pathogenesis of thrombosis in cancer [92]. Neutrophils from patients with MPN were previously shown to be prone for NET formation with increased NET formation following stimulation with ionomycin [76] but not with phorbol 12-myristate 13-acetate (PMA) or inflammatory cytokines [93]. Guy et al. demonstrated that patients with MPNs have increased plasma levels of MPO-DNA complexes, a proposed marker of NET production, with patients with a history of thrombosis demonstrating the highest MPO-DNA levels [94].
In JAK2V617F murine models, neutrophils have also been shown to be primed for NET formation with increased ex-vivo ionomycin-induced NET formation. In-vivo, JAK2 V617F mice had increased rates of venous thrombosis (in a partial inferior vena cava (IVC) ligation model) and thrombus from JAK2 V617F mice had increased expression citrullinated histone 3, a biochemical surrogate for NET formation. Thrombus formation in-vivo and NET formation ex-vivo were both PAD4-dependant in the murine model suggesting that PAD4 is a driver for NETosis and thrombosis in this model [76]. A recent abstract publication validated that JAK V617F neutrophils are characterized by an increase in NET formation in both MPN hematopoietic specific (PF4-iCre) and neutrophil specific (MRP8-iCre) murine models [95].
Activated neutrophils interact with both platelets and the endothelium to promote thrombosis.
Platelet-neutrophil complexes were shown to be abundant in PV, ET and MF [54,55,57]. Adhesion molecules on platelets such as P-selectin and GPIBα or GPIIbIIIa interact with PSGL-1 and CD11b/CD18 (mac-1), respectively and promote activation one upon the other: platelet-derived CCL (RANTES) and PF4 activate leukocytes whereas elastase and cathepsin G that originate in neutrophils promote platelet activation [96].
Monocytes are also primed for activation in MPNs. P-selectin was shown to induce expression of TF on monocytes [97]. Platelets were also shown to induce an increase in cytokine production, specifically in monocytes from patients with MF [98,99].
Interactions between neutrophils and the vascular endothelium are also impacted in MPNs to produce a prothrombotic phenotype. Neutrophils from patients with Jak2V617F MPNs were shown to have hyperactivated β1 (α4β1; VLA-4) integrin-mediated adhesion to vascular cell adhesion molecule 1 (VCAM-1) [100]. Later the same group demonstrated in a vav/cre Jak2V617F MPN murine model that JAK2V617F mutation is associated with increased affinity of β1 and β2 (αLβ2; LFA-1) integrins to their respective receptors, VCAM-1 and intercellular adhesion molecule 1 (ICAM-1). At least for the β1 integrin, a RAP1 mediated conformational change was observed. Venous thrombosis in these mice was reduced by blocking VCAM -1 and ICAM-1 receptors [101].
4.4. Endothelium
Markers of endothelial activation are elevated in the plasma of patients with MPNs and increased levels of vWF, soluble thrombomodulin and soluble P-selectin, E-selectin and L-selectin were previously demonstrated in the plasma of patients with ET and PV [69,102,103]. Markers of angiogenic activity such as vascular endothelial growth factor (VEGF) were found to be increased in patients with MPNs [104,105,106] as were higher levels of circulating endothelial cells (ECs).
The activated endothelium interacts with platelets via vWF-mediated platelet expression of platelet membrane CD40 ligand with endothelial CD40 as well as crosstalk with RBC’s (via expression of Lu/BCAM) and leukocytes (via expression of β1 and β2 integrins), as described above.
Sozer et al. reported a fascinating observation that ECs from terminal hepatic venules obtained from paraffin-embedded sections of liver biopsy specimens in 2 patients with PV and Budd-Chiari syndrome were homozygous for the JAK2V617F [107]. Rosti et al. later validated these findings and demonstrated that ECs carrying the JAK2V617F mutation can be found both in the splenic capillaries and in the splenic vein of patients with MF [108]. ECs bearing the JAK2V617F have a hyperactivated phenotype. In one study, investigators used a pluripotent stem cells strategy to compare JAK2 mutant and wild-type EC’s. JAK2V617F EC’s expressed a pro-inflammatory transcriptome with increased expression of vWF and P-selectin and increased adhesiveness of leukocytes to EC’s [109]. Etheridge et al. elegantly showed in a series of transplant experiments with transgenic Tie2-Cre/FF1 mice, that both hematopoietic and EC expression of JAK2V617F is needed for the full MPN phenotype to be displayed but that mutated EC’s are critical for recapitulating the hemostatic defect in this model (an acquired vWF-like bleeding diathesis) [78]. Similarly, Castiglione et al. demonstrated that the MPN phenotype in Tie2-Cre/FF1 mice, that express JAK2V617F in both hematopoietic and ECs is associated with thrombosis, vasculopathy, and cardiomyopathy but that this phenotype is lost if JAK2V617F expression is restricted only to hematopoietic cells without mutated ECs, suggesting a critical role for mutated ECs in the pro-thrombotic phenotype. Indeed, JAK2V617F expressing ECs displayed a pro-adhesive, pro-inflammatory, and vasculopathic phenotype [110]. Guy et al. showed that EC-specific JAK2V617F expressing mice models (Pdgfb-iCreERT2) are characterized by a pro-thrombotic phenotype due to increased endothelial P-selectin exposure, secondary to degranulation of Weibel–Palade bodies. Blocking P-selectin reverted the increased propensity for thrombosis phenotype [94].
4.5. Thrombo-Inflammation and Clonal Hematopoiesis
MPN’s are accompanied by a systemic inflammatory response that drives disease phenotype and is associated with fibrosis and disease progression [86,111]. Several pro-inflammatory cytokines were shown to be elevated in patients with MPNs including interleukin (IL)-1β, IL-6, IL-8 and tissue necrosis factor (TNF)-α [112,113]. Additionally, increased levels of inflammatory markers such as C-reactive protein (CRP) and pentraxin-3 were described in patients with MPNs [114,115]. Gangaraju et al. conducted unbiased RNA sequencing and found differential expression of several thrombotic, inflammatory, and HIF-regulated genes in granulocytes and platelets of PV patients, especially in patients with a history of thrombosis [116]. Marín Oyarzún et al. recently highlighted an additional inflammation/hypercoagulation cycle by demonstrating that hyperactive toll-like receptor (TLR) responses on platelet are characteristic in patients with ET and promote platelet activation, platelet interaction with leukocytes and the endothelium and secretion of inflammatory mediators [117]. In recent years the concept of ‘thrombo-inflammation’ was conceptualized to express the intimate cross-talk and mutual perpetuating effects of inflammation and thrombosis one upon the other [118]. An increased thrombotic risk is inherent to several pathologies that are characterized by acute or chronic inflammation and this holds true also for MPNs [119]. The term ‘immuno-thrombosis’ was recently coined to characterize the processes by which activated blood cells both trigger and participate in an inflammatory response that results in promotion of intravascular thrombosis [120].
The contribution of the clonal hematopoiesis (CH) to disease phenotype, inflammation, and thrombotic tendency in MPNs is increasingly recognized. Evidence for possible intrinsic prothrombotic effects induced by driver mutations in MPNs and other myeloid neoplasms are emerging and are supported by observations in patients with CHIP. CHIP is an age-dependent process that denotes the presence of pathological clones in the blood of healthy persons without overt hematological disorders or abnormal blood counts [121,122,123,124]. In fact, recent evidence suggests that JAK2V617F mutations may occur in single hematopoietic stem cells decades before the occurrence of overt disease [125]. JAK2-driven CHIP was shown to be associated with increased risk for venous thrombosis [22,76]. In one study, this prothrombotic tendency was attributed to increased NETosis in JAK2 mutated neutrophils (as discussed above) [76]. JAK2-positive CHIP was also shown to be independently associated with coronary heart disease (along with TET2, DNMT3A and ASXL1 mutations) [126]. Wang et al. provided additional mechanistic insight into the atherogenic phenotype induced by JAK2 mutations. They showed that Jak2V617F mice developed neutrophil-rich atheroma that have larger lipid cores with increased iron deposition [127,128].
4.6. COVID-19, MPNs and Thrombosis-(Mechanistic) Lessons Learned from the Pandemic
The Coronavirus 19 disease (COVID-19) pandemic, caused by severe acute respiratory syndrome coronavirus 2 (SARS COV 2) immensely impacted the lives of patients, communities, health systems, economies and basically altered every aspect of our lives [129].
The clinical spectrum of the SARS COV 2 infection is wide [130] and an excessive thrombo-inflammatory response (‘cytokine storm‘) is the hallmark of severe COVID-19, associated with an acute respiratory distress syndrome (ARDS) and in some patients, with widespread thrombotic microangiopathy evolving to a multiorgan failure [131,132].
Venous and arterial thrombosis are commonly seen in patients with COVID-19 disease and are associated with increased morbidity and mortality. A variable incidence of thrombosis, ranging from 7.9% in hospitalized patients in general wards to 22.7% in patients transferred to ICU has been reported [133]. The reason for prothrombotic tendency in patients with COVID-19 infection is not completely understood and seems to be multifactorial, including increased levels of procoagulant factors (e.g., fibrinogen), endothelial damage, and excessive NET formation [99,134,135,136].
Several of these pathological pathways are relevant for thrombosis in MPNs and intriguingly MPN-directed therapies such as JAK inhibitors are currently studied in severe COVID-19. Understanding similarities and disparities between the coagulopathy in MPNs and COVID-19 may inform therapies and therapeutic approaches.
Data regarding COVID-19 infection in patients with MPNs is scarce [137,138]. A recent analysis conducted by Barbui et al. showed a higher risk of mortality in 175 patients with MPNs and COVID-19 infection, enrolled in the MPN-COVID-19 study, in comparison to the general population [139]. Results were more striking in those with MF and in those who discontinued ruxolitinib at the time of COVID-19 diagnosis [140]. Another analysis of 162 patients from the MPN-COVID-19 study reported a high cumulative incidence of thrombosis (8.6%) mostly venous (7.4%) during the acute phase of infection and after 60 days of follow-up [141]. Events were more frequent in patients with ET and in patients transferred to the ICU. Major bleeding events were reported in 7 patients (a cumulative incidence of 5%) [141]. Increased rates of thrombosis and mortality in this population may reflect an impaired cellular immune response (dysregulation of T and natural killer (NK) cells) and augmentation of the already hyperinflammatory and hypercoagulable states associated with MPNs.
Several drugs used to blunt the inflammatory response in COVID-19 are studied, including those frequently used to manage MPNs such as ruxolitinib and interferon alfa (INFa) [142]. Although ruxolitinib may potentially aggravate the course of the viral disease by suppressing T cells and NK cells, it has the potential to decrease the ‘cytokine storm’ effect owing to its extensive anti-inflammatory effect by inhibiting the JAK-STAT pathway. Positive results with ruxolitinib use were reported in patients with COVID-19 infection [143] although its’ role in patients with MPNs and COVID 19 is still unknown.
Recommendations for the management of patients with MPNs and COVID-19 infection are derived from a consensus among experts in the MPN field. Currently, it is not recommended to change MPN specific therapy in patients with MPN and COVID-19 infection, including the use of cytoreduction, JAK inhibitors and anticoagulation. The use of VTE prophylaxis in patients hospitalized with COVID 19 infection and those with MPNs is still undetermined [144].
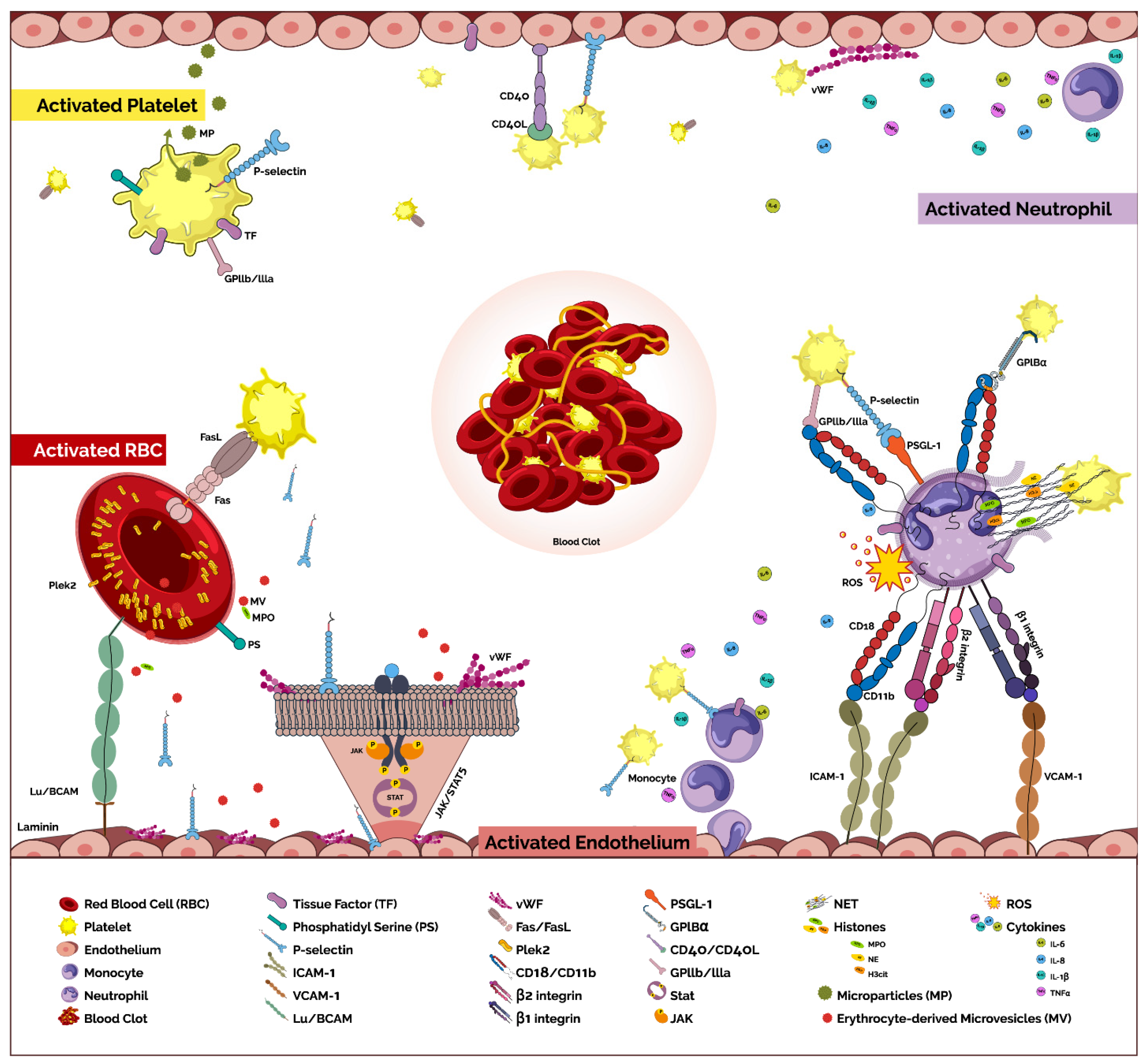
Figure 1.
Pathogenetic pathways promoting thrombosis in MPNs. The prothrombotic phenotype of MPNs is the result of a complex interplay between several components. Despite the apparent divergent pathways that characterize venous and arterial thrombosis, there is often an overlap in the pathogenic process induced in MPNs that may affect both pathologies. RBC’s-The RBC and platelets interact via FAS ligand/FAS receptor that result in the externalization of RBC phosphatidylserine (PS), promoting thrombin generation and thrombus formation [48]. Increased expression and phosphorylation of Lutheran blood group/basal cell adhesion molecule (Lu/BCAM) driven by JAK2 mutations [50] and adhesion (via Laminin) [49], mediate adhesion of RBC to endothelial cells. Erythrocyte-derived microvesicles (MV) contribute to arterial constriction and thrombosis mediated via oxidative stress and disruption of the nitric oxide (NO) pathway by the myeloperoxidase (MPO)-loaded MV’s [51]. In addition, pleckstrin-2 (Plek2) protein, a downstream target of the JAK2/STAT5 pathway in erythroid and myeloid cells, is upregulated in JAK2V617F cells [53] and promotes prothrombotic phenotype. Platelets-Markers of platelet activation are increased in plasma of patients with MPNs [54,55,56,57,58,59,60,61]. Increased platelets derived circulating microparticles (MP) are associated with increased endothelial activation and thrombin generation [63,67,68]. Increased expression of PS and tissue factor (TF) on platelet membrane may initiate coagulation cascades [62,69]. Leukocytes-Markers of neutrophils activation are increased in plasma of patients with MPNs including CD11b [54], reactive oxygen species (ROS) [84] and neutrophil extracellular traps (NETs) [12,85,86,87]. Activated neutrophils interact with both platelets and the endothelium to promote thrombosis. Adhesion molecules on platelets such as P-selectin and GPIBα or GPIIbIIIa interact with PSGL-1 and CD11b/CD18 (mac-1), respectively [61]. Increased affinity of β1 (α4β1; VLA-4) and β2 (αLβ2; LFA-1) integrins to their respective receptors, vascular cell adhesion molecule 1 (VCAM-1) and intercellular adhesion molecule 1 (ICAM-1) contribute to the prothrombotic state [101]. NET formation is increased in MPNs and contributes to both venous and arterial thrombosis [76,129]. P-selectin-mediated expression of TF and platelet-induced cytokine expression are characteristic of monocytes in MPN [97,98,99]. Endothelial-Markers of endothelial activation are increased [69,102,103] as are markers of angiogenic activity [104,105,106]. The activated endothelium interacts with platelets via vWF-mediated platelet expression of CD40 ligand with endothelial CD40 as well as cross-talk with RBC’s (via expression of Lu/BCAM) and leukocytes (via expression of β1 and β2 integrins). Endothelial cells (ECs) carrying the JAK2V617F mutation [108] express a pro-inflammatory transcriptome with increased expression of vWF and P-selectin and increased adhesiveness of leukocytes to ECs [109]. They also display a pro-adhesive, pro-inflammatory, vasculopathic phenotype [110] due to increased endothelial P-selectin exposure, secondary to degranulation of Weibel–Palade bodies. Thrombo-inflammation-Increased levels of pro-inflammatory cytokines such as interleukin (IL)-1β, IL-6, IL-8 and tissue necrosis factor (TNF)-α [112,113], as well as increased inflammatory markers such as C-reactive protein (CRP) and pentraxin-3 [114,115] are found in patients with MPNs.
Figure 1.
Pathogenetic pathways promoting thrombosis in MPNs. The prothrombotic phenotype of MPNs is the result of a complex interplay between several components. Despite the apparent divergent pathways that characterize venous and arterial thrombosis, there is often an overlap in the pathogenic process induced in MPNs that may affect both pathologies. RBC’s-The RBC and platelets interact via FAS ligand/FAS receptor that result in the externalization of RBC phosphatidylserine (PS), promoting thrombin generation and thrombus formation [48]. Increased expression and phosphorylation of Lutheran blood group/basal cell adhesion molecule (Lu/BCAM) driven by JAK2 mutations [50] and adhesion (via Laminin) [49], mediate adhesion of RBC to endothelial cells. Erythrocyte-derived microvesicles (MV) contribute to arterial constriction and thrombosis mediated via oxidative stress and disruption of the nitric oxide (NO) pathway by the myeloperoxidase (MPO)-loaded MV’s [51]. In addition, pleckstrin-2 (Plek2) protein, a downstream target of the JAK2/STAT5 pathway in erythroid and myeloid cells, is upregulated in JAK2V617F cells [53] and promotes prothrombotic phenotype. Platelets-Markers of platelet activation are increased in plasma of patients with MPNs [54,55,56,57,58,59,60,61]. Increased platelets derived circulating microparticles (MP) are associated with increased endothelial activation and thrombin generation [63,67,68]. Increased expression of PS and tissue factor (TF) on platelet membrane may initiate coagulation cascades [62,69]. Leukocytes-Markers of neutrophils activation are increased in plasma of patients with MPNs including CD11b [54], reactive oxygen species (ROS) [84] and neutrophil extracellular traps (NETs) [12,85,86,87]. Activated neutrophils interact with both platelets and the endothelium to promote thrombosis. Adhesion molecules on platelets such as P-selectin and GPIBα or GPIIbIIIa interact with PSGL-1 and CD11b/CD18 (mac-1), respectively [61]. Increased affinity of β1 (α4β1; VLA-4) and β2 (αLβ2; LFA-1) integrins to their respective receptors, vascular cell adhesion molecule 1 (VCAM-1) and intercellular adhesion molecule 1 (ICAM-1) contribute to the prothrombotic state [101]. NET formation is increased in MPNs and contributes to both venous and arterial thrombosis [76,129]. P-selectin-mediated expression of TF and platelet-induced cytokine expression are characteristic of monocytes in MPN [97,98,99]. Endothelial-Markers of endothelial activation are increased [69,102,103] as are markers of angiogenic activity [104,105,106]. The activated endothelium interacts with platelets via vWF-mediated platelet expression of CD40 ligand with endothelial CD40 as well as cross-talk with RBC’s (via expression of Lu/BCAM) and leukocytes (via expression of β1 and β2 integrins). Endothelial cells (ECs) carrying the JAK2V617F mutation [108] express a pro-inflammatory transcriptome with increased expression of vWF and P-selectin and increased adhesiveness of leukocytes to ECs [109]. They also display a pro-adhesive, pro-inflammatory, vasculopathic phenotype [110] due to increased endothelial P-selectin exposure, secondary to degranulation of Weibel–Palade bodies. Thrombo-inflammation-Increased levels of pro-inflammatory cytokines such as interleukin (IL)-1β, IL-6, IL-8 and tissue necrosis factor (TNF)-α [112,113], as well as increased inflammatory markers such as C-reactive protein (CRP) and pentraxin-3 [114,115] are found in patients with MPNs.

5. Current Strategies to Prevent Thrombosis
Treatment approach in PV and ET is mainly guided by the risk for recurrent thrombosis.
Management of low-risk PV includes low dose aspirin and phlebotomy to maintain hematocrit <45% while in high-risk patients, low dose aspirin, phlebotomy and cytoreduction are recommended [16,145].
Management of ET is comprised of observation in very low-risk patients, low dose aspirin in low-risk patients, low dose aspirin +/− cytoreduction in intermediate risk patients and low dose aspirin combined with cytoreduction in high-risk patients.
Assessment and management of CV risk factors (e.g., blood pressure, glucose, hyperlipidemia, smoking, etc.) should be considered in all patients.
5.1. Aspirin
Low dose aspirin is an effective treatment for preventing thrombosis in PV according to the ECLAP study [146] while the data supporting its use in ET is mainly based on the extrapolation from this study. It was also found to be beneficial in a retrospective study, in patients with JAK2V617F-mutated low-risk ET, in preventing venous thrombosis, and in patients with CV risk factors, in preventing arterial thrombosis [147]. Furthermore, Low dose aspirin is effective in relieving vasomotor symptoms in ET [148]. Recent reports suggested that twice-daily aspirin may be more efficient than once-daily aspirin and should be considered in patients with uncontrolled vasomotor symptoms or those with an exceptional high risk for arterial thrombosis (e.g., the presence of CV risk factors, leukocytosis, and a history of arterial thrombosis) [2,140,149]. Nevertheless, it should be used with caution in patients with acquired vWD (von Willebrand disease) and a higher risk of bleeding.
5.2. Hematocrit
The hematocrit goal of treatment is <45% in patients with PV treated with phlebotomy and/or hydroxyurea. A lower hematocrit (e.g., hematocrit <42%) might be appropriate, especially in women and those with uncontrolled vasomotor symptoms.
5.3. Cytoreduction
In high-risk PV/ET cytoreduction is used to reduce the thrombotic risk, although, there is no evidence to imply a risk reduction of VTE recurrence in unusual sites [150]. Cytoreductive agents in PV include hydroxyurea33, [151], recombinant interferon α [152]/peginterferon [153] or busulfan [154]. JAK inhibition is currently utilized as a second-line treatment only in PV. In ET hydroxyurea, recombinant interferon α, busulfan or anagrelide may be used for cytoreduction.
Cytoreduction and specifically hydroxyurea are considered superior to a non-myelosuppressive treatment in ET [27,155] and to phlebotomy in PV [8,156,157]. Cytoreduction is not indicated in low-risk PV/ET in respect to thrombotic risk but might be indicated in certain instances such as uncontrolled PV/ET related symptoms, progressive leukocytosis/thrombocytosis, symptomatic or progressive splenomegaly or poor tolerance to phlebotomy in PV [158].
Recombinant interferon α-2 has been used to treat patients with PV and ET. Peginterferon α-2a, given once weekly, was investigated as 2nd line treatment in patients with PV (n = 50) and ET (n = 65) with resistance or intolerance to hydroxyurea (MPN RC 111 ET and PV trials). Complete hematologic response was documented in 22% of patients and the cumulative incidence of major vascular events at 1 year and 2 years were 2% (95% CI, 1–8%) and 5% (95% CI, 2–15%), respectively [159]. Low and comparable rates of VTEs were also documented in patients with ET (n = 81) who were randomized to either peginterferon α-2a or hydroxyurea (MPN-RC-112 study) [160]. Recently, ropeginterferon α-2b, given every two weeks, was approved as first-line treatment in patients with PV by the European Medicines Agency (EMA). This approval was based on the 5 years results of the PROUD/COUNTIONUATION PV trial, where the rate of complete hematologic response was comparable between ropeginterferon α-2b and the control arm, treated with hydroxyurea, in patients with PV at 12 months. Furthermore, only a few thromboembolic events were recorded in both arms (major cardiovascular events (10% vs. 6%) and major thromboembolic events (3% vs. 3%) [161].
Anagrelide is an oral imidazoquinazoline derivative which is used as cytoreduction in ET. It inhibits proliferation and differentiation of megakaryocytes and reduces platelet counts [162]. It was compared to hydroxyurea in two different studies. In the UK PT1 study, patients with high-risk ET were more prone to develop arterial and venous thrombosis, hemorrhage, or death from any cause while on anagrelide and aspirin in comparison to hydroxyurea and aspirin. In addition, anagrelide was associated with an increased rate of progression to myelofibrosis. Anagrelide advantage over hydroxyurea was a reduced rate of venous thrombosis [155]. The ANAHYDRATE study included 259 patients with ET. This study showed non-inferiority of anagrelide in comparison to hydroxyurea in regard to thrombotic events and in the rates of transformation to MF and leukemia [163].
5.4. JAK Inhibitors
Ruxolitinib (JAK 1/2 inhibitor) has been shown to be effective in respect to hematocrit- and symptom-control in the second-line treatment setting in patients with PV with intolerance or resistance to hydroxyurea (RESPONSE and RESPONSE II trials) [164,165]. Although a meta-analysis of trials utilizing ruxolitinib in PV failed to demonstrate that JAK inhibition reduces thrombosis as compared to best available therapy [166], in the RESPONSE trail 5-year follow-up, the rate of thrombosis was decreased with ruxolitinib therapy [167]. Furthermore, a meta-analysis of trials assessing ruxolitinib in MF was able to demonstrate a significant reduction in thrombotic risk in patients treated with ruxolitinib (risk ratio 0.45, 95% confidence interval (CI) 0.23–0.88) [168].
5.5. Anticoagulation
Anticoagulation therapy is recommended in high-risk patients with PV/ET and venous thrombosis. Although the use of vitamin K antagonists (VKAs) decreases the rate of VTE recurrence, the rate remains high in addition to higher rates of major bleeding events when compared to the general population [169]. There are no convincing data to guide the appropriate duration of anticoagulation treatment. Hence, the duration of anticoagulation therapy is guided by the severity of thrombosis, the risk of recurrence after stopping anticoagulation, and whether disease is controlled. Thus, long-term anticoagulation is recommended in patients with MPN-associated VTE.
Traditionally, VKAs have been used in patients with MPNs and venous thrombosis. It should be noted that there is no compelling evidence to guide the choice of anticoagulation. According to non-inferiority trials, direct oral anticoagulants (DOACS) are effective and safe in preventing and treating VTE in the general population and in patients with solid tumors, in comparison to VKA and low molecular weight heparins (LMWH), respectively [170,171,172,173]. The therapeutic role of DOACS in MPNS is currently being investigated and has been suggested in retrospective studies. In a real-world setting of 71 patients with MPNs affected by VTE (including few cases of VTE in unusual sites) or atrial fibrillation, the use of DOACs was well tolerated, apparently safe, without recurrences of thrombotic events and significant bleeding complications even in high-risk patients [174]. Likewise, a recent large retrospective study of 442 patients with MPNs reported similar rates of VTE with DOACs and VKA for prevention of thrombosis in atrial fibrillation and in secondary prevention of VTE [175]. The use of DOACS was also investigated among those with splanchnic vein thrombosis, where recurrence rates with the use of VKA’s are unsatisfactory [176,177]. Curto-Garcia et al. reported on 102 patients with MPN-associated thrombosis. In this analysis, 32 patients that experienced a total of 38 thrombotic events (15 splanchnic, 12 pulmonary embolisms, eight deep vein thrombosis, two cerebral venous sinus thrombosis, and one superficial thrombophlebitis) received DOACS. DOACS were given for the long term in 25 patients and for a limited course after provoked VTE in 7 patients. With a median follow-up of 2.1 years (range 0.1–7.8), only one case of mesenteric ischemia was recorded, and no major bleedings were recorded apart from 3 patients with non-major significant bleedings under DOACS and aspirin [178].
6. Novel Targets to Prevent Thrombosis
Thrombosis in MPNs represents the clinical endpoint of several concomitant, interdependent disease extrinsic and intrinsic processes (Figure 1). Although this poses many challenges in clinic and in research, it also exposes several potential therapeutic targets and may inform treatment approaches (Table 1).
6.1. Targeting the JAK/STAT Pathway
The hyperactive JAK/STAT pathway is fundamental to the pathogenesis and disease phenotype and drives many of the processes implicated in thrombogenicity in MPNs including inflammation, cell-cell interactions and activation of blood cells and of the endothelium (as exhaustively described above). Thus, inhibiting this pathway may be an attractive approach to prevent thrombosis. In patients with MF, ruxolitinib was shown early on to be associated with significant reductions in the levels of circulating inflammatory cytokines [179]. In a small pilot study, Keohane et al. assessed the impact of JAK inhibition (ruxolitinib, n = 11; fedratinib, n = 4) on biomarkers of thrombosis as compared to patients receiving conventional therapy. No significant difference was observed between the groups in platelet-monocyte aggregates, platelet-neutrophil aggregates, platelet activation markers or neutrophil activation markers. Although monocyte counts were elevated in the JAK inhibition group, a trend towards reduced monocyte TF expression was noted [180]. Another report showed a JAK2-driven increase in ROS production via increased phosphorylation of the p47phox on Ser345 and up-stream of ERK1/2 kinase. JAK inhibition with selective JAK2 inhibitors (AG490 and CEP701) resulted in inhibition of ROS production suggesting ROS as a target for JAK/STAT inhibition in MPNs. A recent report suggests that ruxolitinib is associated with reduced pro-adhesive VWF, VCAM-1, and P-selectin expression in a TNF-α-activated HUVEC endothelial cell model [181]. In a Jak2V617F murine model, ruxolitinib treatment was associated with a significant reduction in venous thrombosis as assessed by partial ligation of the IVC [76]. The large clinical trials assessing ruxolitinib in patients with MF (COMFORT I-II) and PV (RESPONSE) were not designed to detect reduction in thrombotic rates and recent metanalyses yielded inconclusive results [166].
6.2. Targeting Cell-Cell Interaction
Targeting cell-cell interaction may be a practical approach to reduce thrombosis rates in MPNs. P-selectin, a marker of platelet activation, plays a central role in mediating platelet interactions with other blood cells and the endothelium and may be an attractive therapeutic target. In murine models, P-selectin blockade was shown to affect thrombosis. Guy et al. showed that blocking the overexpressed P-selectin (with a blocking antibody) in EC-specific JAK2V617F mice models (Pdgfb-iCreERT2) reverted the thrombotic phenotype [94]. Platelet-induced NET formation was shown in ex-vivo murine models to be P-selectin dependent and blocking PSGL1 resulted in abrogating NET formation in this model [182].
P-selectin inhibitors are available in clinic for other indications. Crizanlizumab is a humanized monoclonal antibody against P-selectin that was shown to significantly lower the rate of sickle cell-related pain crises in a large phase 3 trial [183]. In a recent report, an Italian group assessed a murine p-selectin blocking antibody, RB40.34, that was used in the pre-clinical efforts that supported the clinical trial with crizanlizumab in sickle cell disease. In a p-selectin dependent, Gata1low MF murine model [184], RB40.34 in combination with ruxolitinib reverted MF phenotype with limited toxicity setting the stage for early phase clinical trials with this drug [185].
The leukocyte integrins VLA-4 and LFA-1 were shown to be to have a JAK2-dependent, RAP1 mediated increase in affinity to their respective endothelial receptors that were associated with thrombosis in MPN murine models. Blocking anti–VLA-4 and anti–LFA-1 integrin antibodies suppressed thrombosis in this model [101]. Blocking VCAM-1, the endothelial adhesion molecule that binds VLA-4 inhibited cancer-associated thrombosis and extended survival in a pancreatic ductal adenocarcinoma mouse model [186]. Antibodies to block leukocyte integrin interactions are already in use in clinics to modulate inflammatory responses in various disease states. For example, natalizumab, a monoclonal antibody directed against the alpha chain of the VLA-4 integrin is used to treat multiple sclerosis [187] and Crohn’s disease [188]. A humanized CD11a blocking antibody, Efalizumab, was developed to inhibit LFA-1 and was used to treat psoriasis [189] but was later withdrawn from the market due to reports of JC virus reactivation associated with this drug [190,191].
6.3. Targeting Cell Activation
Targeting prothrombotic components of cell activation may also be a valid therapeutic approach. Several potential inhibitors for NETosis were introduced and some were shown to reduce thrombosis in MPN murine models. Ruxolitinib was shown to inhibit NET formation in neutrophils and significantly reduced venous thrombosis in an MPN IVC patrial ligation model [76]. N-acetylcysteine (NAC), a sulfhydryl-containing compound, that is associated with glutathione replenishment, free radical scavenging, and reducing disulfide bonds was shown to reduce thrombosis in an acute pulmonary thrombosis JAK2VV617F murine model. Complementary analysis showed a reduction in platelet-leukocyte aggregates in mice and inhibition of NET formation in neutrophils derived from MPN patients and controls [192]. Inhibition of PAD4 that is responsible for histone 3 citrullination and the subsequent propagation of NETs was shown to abrogate NET formation and reduce thrombosis in MPN and non-MPN murine models although the utility of such an approach in clinic remains to be studied [76,193]. The use of anti-histone antibodies also demonstrated protective effects against infarcts in a mouse model of transient middle cerebral artery occlusion [194]. DNAse is a deoxyribonuclease (DNA) enzyme produced by recombinant gene technology that is currently used as a nebulizer in clinic to manage patients with Cystic Fibrosis [195]. This drug dismantles NETs and has been shown to be associated with reduced thrombosis in several MPN and non-MPN murine models [76,196,197,198]. Lastly, there is evidence to suggest that standard drugs that are currently used to prevent thrombosis in MPNs have an inhibitory effect on NET production as was shown to be the case with acetylsalicylic acid [199] and hydroxyurea [200].
Plek2 is overexpressed in JAK2V617F positive MPNs and associated with disease phenotype including an increase in RBC mass and vascular occlusions. Han and colleagues recently reported on a Plek2 inhibitor in an MPN murine model. NUP-17d blocks hematopoietic cell proliferation through the disruption of the Plek2 complex and inhibition of the PI3K-Akt pathway. In-vivo, this inhibitor reverted MPN phenotype although its effect on thrombotic tendency was not reported to date [201].
6.4. Targeting Inflammation
Targeting components of the thrombo-inflammatory process have been described in the non-MPN setting. For example, a large phase 3 trial demonstrated that canakinumab, an IL-1β inhibitor led to a significantly lower rate of recurrent CV events as compared to placebo-treated patients, independent of lipid-levels lowering [202].
Poisson et al. described excessive arterial constriction and thrombosis in JAK2V617F murine model that was mediated via oxidative stress and disruption of the NO pathway by MPO-loaded MV’s. These pathologic pathways were blunted by treating mice with NAC. While hydroxyurea improved endothelial dysfunction, ruxolitinib had no effect. Finally, simvastatin was shown to significantly alter endothelial dysfunction and blunt vasoconstriction [51].

{kind=link}
Table 1.
Potential novel approaches to target pro-thrombotic pathways in MPNs.
| Pathway/Target | Target | Drug/Compound | Effect | Context | Ref. |
|---|---|---|---|---|---|
| JAK-STAT | JAK1/2 | Ruxolitinib | Pro-inflammatory cytokines ↓ | MF, Clinical trial | [179] |
| Neutrophil: ROS ↓ NETs ↓ | Ex-vivo studies, Murine models | [76,84] | |||
| RBC: HCT ↓, Lu/BCAM ↓ | Clinical trial Ex-vivo studies | [33] [50] | |||
| Monocyte: TF expression ↓ | Ex-vivo studies | [180] | |||
| Endothelial activation ↓ | Pre-clinical HUVEC model | [181] | |||
| Selectins | P-selectin | P-selectin blocking antibodies | Thrombosis ↓ NETs ↓ PLT-endothelial ↓ PLT-leukocyte ↓ | Murine models Crizanlizumab is a clinically available Ab | [94,182,185] |
| Integrins | VLA4 LFA1 | VLA4 and LFA1 blocking antibodies | Thrombosis ↓ | Murine models | [101] |
| Neutrophil functions* | NETs | NAC | Thrombosis ↓ NETs ↓ PLT-leukocyte ↓ Arterial dysfunction ↓ | Ex-vivo studies, Murine models | [192] [51] |
| PAD4 | Thrombosis ↓ NETs ↓ | Ex-vivo studies, Murine models | [76] [193,203] | ||
| DNAse | Thrombosis ↓ NETs ↓ | Murine models | [196,197,198] | ||
| RBC’s | Plek2 | Plek2 inhibitor | Thrombosis ↓ RBC mass ↓ | Murine models | [53,201] |
| Inflammation | Vasoconstriction | simvastatin | Arterial dysfunction ↓ | Murine models | [51] |
Abbreviations: Ref.: references, MF: myelofibrosis, ROS: reactive oxygen species, NETs: neutrophil extracellular traps, JAK-STAT: janus kinase-signal transducers and activators of transcription, JAK1/2: janus kinase 1/2, RBC: red blood cell, HCT: hematocrit, Lu/BCAM: Lutheran/blood group and basal cell adhesion molecule, TF: tissue factor, HUVEC: human umbilical vein endothelial cells, NAC: N-acetylcysteine, PLT: platelets, Plek2: Pleckstrin-2,. *Ruxolitinib and P-selectin antibodies also target NET formation as detailed in their respective boxes; ruxolitinib also target ROS formation as detailed in the respective box.
7. Summary
Preventing and treating arterial and venous thrombosis represents one of the major challenges in managing MPNs. The expanding understanding of how genetic, epigenetic, metabolic, cellular and biophysical factors contribute to this complication may enable more accurate risk assessment and guide therapy. Identification of biomarkers of cell activation and functional assays to assess prothrombotic tendency as well as bleeding tendency may refine and individualize thrombotic risk stratification.
Several potential therapeutic approaches are informed by recent pathogenetic insights including targeting of hyperactivated pathways downstream to JAK/STAT, interfering with cell-cell interactions and abrogating cell activation and the resultant inflammation. Advancing these therapeutics from the bench to the clinic has the potential to significantly impact how we prevent and treat thrombosis in MPNs. Lessons learned from other thrombo-inflammatory conditions may further inform and improve our understanding and treatment of thrombosis in MPN.
Author Contributions
O.W. and A.S.A. wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rumi, E.; Cazzola, M. Diagnosis, risk stratification, and response evaluation in classical myeloproliferative neoplasms. Blood 2017, 129, 680–692. [Google Scholar] [CrossRef]
- Tefferi, A.; Barbui, T. Polycythemia vera and essential thrombocythemia: 2021 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2020, 95, 1599–1613. [Google Scholar] [CrossRef]
- Grinfeld, J.; Nangalia, J.; Green, A.R. Molecular determinants of pathogenesis and clinical phenotype in myeloproliferative neoplasms. Haematologica 2016, 102, 7–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Guglielmelli, P.; Larson, D.R.; Finke, C.; Wassie, E.A.; Pieri, L.; Gangat, N.; Fjerza, R.; Belachew, A.A.; Lasho, T.L.; et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 2014, 124, 2507–2513. [Google Scholar] [CrossRef] [PubMed]
- Weisel, J.W.; Litvinov, R.I. Red blood cells: The forgotten player in hemostasis and thrombosis. J. Thromb. Haemost. 2019, 17, 271–282. [Google Scholar] [CrossRef] [Green Version]
- Von Brühl, M.L.; Stark, K.; Steinhart, A.; Chandraratne, S.; Konrad, I.; Lorenz, M.; Khandoga, A.; Tirniceriu, A.; Coletti, R.; Köllnberger, M.; et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 2012, 209, 819–835. [Google Scholar] [CrossRef] [PubMed]
- Massberg, S.; Grahl, L.; Von Bruehl, M.-L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010, 16, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Barbui, T. Polycythemia vera and essential thrombocythemia: 2017 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2017, 92, 94–108. [Google Scholar] [CrossRef]
- Marchioli, R.; Finazzi, G.; Landolfi, R.; Kutti, J.; Gisslinger, H.; Patrono, C.; Marilus, R.; Villegas, A.; Tognoni, G.; Barbui, T. Vascular and Neoplastic Risk in a Large Cohort of Patients with Polycythemia Vera. J. Clin. Oncol. 2005, 23, 2224–2232. [Google Scholar] [CrossRef]
- Hultcrantz, M.; Bjorkholm, M.; Dickman, P.W.; Landgren, O.; Derolf, A.R.; Kristinsson, S.Y.; Andersson, T.M.L. Risk for Arterial and Venous Thrombosis in Patients with Myeloproliferative Neoplasms: A Population-Based Cohort Study. Ann. Intern. Med. 2018, 168, 317–325. [Google Scholar] [CrossRef]
- Elliott, M.A.; Tefferi, A. Pathogenesis and management of bleeding in essential thrombocythemia and polycythemia vera. Curr. Hematol. Rep. 2004, 3, 344–351. [Google Scholar]
- Marchetti, M.; Falanga, A. Thrombosis in Myeloproliferative Neoplasms. Semin. Thromb. Hemost. 2014, 40, 348–358. [Google Scholar] [CrossRef]
- Finazzi, G.; De Stefano, V.; Barbui, T. Splanchnic vein thrombosis in myeloproliferative neoplasms: Treatment algorithm 2018. Blood Cancer J. 2018, 8, 64. [Google Scholar] [CrossRef] [PubMed]
- Rungjirajittranon, T.; Owattanapanich, W.; Ungprasert, P.; Siritanaratkul, N.; Ruchutrakool, T. A systematic review and meta-analysis of the prevalence of thrombosis and bleeding at diagnosis of Philadelphia-negative myeloproliferative neoplasms. BMC Cancer 2019, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Barbui, T.; De Stefano, V.; Falanga, A.; Finazzi, G.; Martinelli, I.; Rodeghiero, F.; Vannucchi, A.M.; Barosi, G. Addressing and proposing solutions for unmet clinical needs in the management of myeloproliferative neoplasm-associated thrombosis: A consensus-based position paper. Blood Cancer J. 2019, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Barbui, T.; Barosi, G.; Birgegard, G.; Cervantes, F.; Finazzi, G.; Griesshammer, M.; Harrison, C.; Hasselbalch, H.C.; Hehlmann, R.; Hoffman, R.; et al. Philadelphia-Negative Classical Myeloproliferative Neoplasms: Critical Concepts and Management Recommendations from European LeukemiaNet. J. Clin. Oncol. 2011, 29, 761–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbui, T.; Finazzi, G.; Carobbio, A.; Thiele, J.; Passamonti, F.; Rumi, E.; Ruggeri, M.; Rodeghiero, F.; Randi, M.L.; Bertozzi, I.; et al. Development and validation of an International Prognostic Score of thrombosis in World Health Organization—Essential thrombocythemia (IPSET-thrombosis). Blood 2012, 120, 5128–5133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carobbio, A.; Thiele, J.; Passamonti, F.; Rumi, E.; Ruggeri, M.; Rodeghiero, F.; Randi, M.L.; Bertozzi, I.; Vannucchi, A.M.; Antonioli, E.; et al. Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: An international study of 891 patients. Blood 2011, 117, 5857–5859. [Google Scholar] [CrossRef]
- Barbui, T.; Vannucchi, A.M.; Buxhofer-Ausch, V.; De Stefano, V.; Betti, S.; Rambaldi, A.; Rumi, E.; Ruggeri, M.; Rodeghiero, F.; Randi, M.L.; et al. Practice-relevant revision of IPSET-thrombosis based on 1019 patients with WHO-defined essential thrombocythemia. Blood Cancer J. 2015, 5, e369. [Google Scholar] [CrossRef]
- Falchi, L.; Kantarjian, H.M.; Verstovsek, S. Assessing the thrombotic risk of patients with essential thrombocythemia in the genomic era. Leukemia 2017, 31, 1845–1854. [Google Scholar] [CrossRef]
- Rumi, E.; Pietra, D.; Ferretti, V.; Klampfl, T.; Harutyunyan, A.S.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Elena, C.; Casetti, I.C.; et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood 2014, 123, 1544–1551. [Google Scholar] [CrossRef] [PubMed]
- Cordua, S.; Kjaer, L.; Skov, V.; Pallisgaard, N.; Hasselbalch, H.C.; Ellervik, C. Prevalence and phenotypes of JAK2 V617F and calreticulin mutations in a Danish general population. Blood 2019, 134, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Rumi, E.; Pietra, D.; Pascutto, C.; Guglielmelli, P.; Martinez-Trillos, A.; Casetti, I.; Colomer, D.; Pieri, L.; Pratcorona, M.; Rotunno, G.; et al. Clinical effect of driver mutations of JAK2, CALR, or MPL in primary myelofibrosis. Blood 2014, 124, 1062–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finazzi, G.; Carobbio, A.; Guglielmelli, P.; Cavalloni, C.; Salmoiraghi, S.; Vannucchi, A.M.; Cazzola, M.; Passamonti, F.; Rambaldi, A.; Barbui, T. Calreticulin mutation does not modify the IPSET score for predicting the risk of thrombosis among 1150 patients with essential thrombocythemia. Blood 2014, 124, 2611–2612. [Google Scholar] [CrossRef] [Green Version]
- Campbell, P.J.; MacLean, C.; Beer, P.A.; Buck, G.; Wheatley, K.; Kiladjian, J.-J.; Forsyth, C.; Harrison, C.N.; Green, A.R. Correlation of blood counts with vascular complications in essential thrombocythemia: Analysis of the prospective PT1 cohort. Blood 2012, 120, 1409–1411. [Google Scholar] [CrossRef] [Green Version]
- Haider, M.; Gangat, N.; Lasho, T.; Hussein, A.K.A.; Elala, Y.C.; Hanson, C.; Tefferi, A. Validation of the revised international prognostic score of thrombosis for essential thrombocythemia (IPSET-thrombosis) in 585 Mayo clinic patients. Am. J. Hematol. 2016, 91, 390–394. [Google Scholar] [CrossRef] [Green Version]
- Cortelazzo, S.; Finazzi, G.; Ruggeri, M.; Vestri, O.; Galli, M.; Rodeghiero, F.; Barbui, T. Hydroxyurea for Patients with Essential Thrombocythemia and a High Risk of Thrombosis. N. Engl. J. Med. 1995, 332, 1132–1137. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Larrán, A.; Pereira, A.; Cervantes, F.; Arellano-Rodrigo, E.; Hernández-Boluda, J.-C.; Ferrer-Marín, F.; Angona, A.; Gómez, M.; Muiña, B.; Guillén, H.; et al. Assessment and prognostic value of the European LeukemiaNet criteria for clinicohematologic response, resistance, and intolerance to hydroxyurea in polycythemia vera. Blood 2012, 119, 1363–1369. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Boluda, J.C.; Pereira, A.; Cervantes, F.; Gómez, M.; Arellano-Rodrigo, E.; Alvarez-Larrán, A.; Ferrer-Marín, F.; Kerguelen, A.; Márquez, J.A.; Antelo, M.A.; et al. Clinical evaluation of the European LeukemiaNet response criteria in patients with essential thrombocythemia treated with anagrelide. Ann. Hematol. 2013, 92, 771–775. [Google Scholar] [CrossRef]
- Panova-Noeva, M.; Marchetti, M.; Russo, L.; Tartari, C.J.; Leuzzi, A.; Finazzi, G.; Rambaldi, A.; Cate, H.T.; Falanga, A. ADP-induced platelet aggregation and thrombin generation are increased in Essential Thrombocythemia and Polycythemia Vera. Thromb. Res. 2013, 132, 88–93. [Google Scholar] [CrossRef]
- Gori, T. Viscosity, platelet activation, and hematocrit: Progress in understanding their relationship with clinical and subclinical vascular disease. Clin. Hemorheol. Microcirc. 2011, 49, 37–42. [Google Scholar] [CrossRef]
- Pearson, T.C.; Wetherley-Mein, G. Vascular occlusive episodes and venous haematocrit in primary proliferative polycythaemia. Lancet 1978, 2, 1219–1222. [Google Scholar] [CrossRef]
- McMullin, M.F.; Harrison, C.N.; Kiladjian, J.J.; Leone, G.; Prchal, J.T.; Gordeuk, V.R.; Marchioli, R.; Vannucchi, A.M.; Barbui, T.; Spivak, J.L. Treatment Target in Polycythemia Vera. N. Engl. J. Med. 2013, 368, 1554–1557. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A. Leukocytosis as a risk factor for thrombosis in myeloproliferative neoplasms-biologically plausible but clinically uncertain. Am. J. Hematol. 2010, 85, 93–94. [Google Scholar]
- Tefferi, A.; Gangat, N.; Wolanskyj, A. The interaction between leukocytosis and other risk factors for thrombosis in essential thrombocythemia. Blood 2007, 109, 4105. [Google Scholar] [CrossRef]
- Carobbio, A.; Finazzi, G.; Guerini, V.; Spinelli, O.; Delaini, F.; Marchioli, R.; Borrelli, G.; Rambaldi, A.; Barbui, T. Leukocytosis is a risk factor for thrombosis in essential thrombocythemia: Interaction with treatment, standard risk factors, and Jak2 mutation status. Blood 2006, 109, 2310–2313. [Google Scholar] [CrossRef]
- Gangat, N.; Wolanskyj, A.P.; Schwager, S.M.; Hanson, C.A.; Tefferi, A. Leukocytosis at diagnosis and the risk of subsequent thrombosis in patients with low-risk essential thrombocythemia and polycythemia vera. Cancer 2009, 115, 5740–5745. [Google Scholar] [CrossRef]
- Tefferi, A.; Vannucchi, A.M.; Barbui, T. Polycythemia vera treatment algorithm 2018. Blood Cancer J. 2018, 8, 1–7. [Google Scholar] [CrossRef]
- Landolfi, R.; Di Gennaro, L.; Barbui, T.; De Stefano, V.; Finazzi, G.; Marfisi, R.; Tognoni, G.; Marchioli, R. For the European Collaboration on Low-Dose Aspirin in Polycythemia Vera (ECLAP) Leukocytosis as a major thrombotic risk factor in patients with polycythemia vera. Blood 2006, 109, 2446–2452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbui, T.; Masciulli, A.; Marfisi, M.R.; Tognoni, G.; Finazzi, G.; Rambaldi, A.; Vannucchi, A.M. White blood cell counts and thrombosis in polycythemia vera: A subanalysis of the CYTO-PV study. Blood 2015, 126, 560–561. [Google Scholar] [CrossRef]
- Carobbio, A.; Ferrari, A.; Masciulli, A.; Ghirardi, A.; Barosi, G.; Barbui, T. Leukocytosis and thrombosis in essential thrombocythemia and polycythemia vera: A systematic review and meta-analysis. Blood Adv. 2019, 3, 1729–1737. [Google Scholar] [CrossRef]
- Ronner, L.; Podoltsev, N.; Gotlib, J.; Heaney, M.L.; Kuykendall, A.T.; O’Connell, C.; Shammo, J.; Fleischman, A.G.; Scherber, R.M.; Mesa, R.; et al. Persistent leukocytosis in polycythemia vera is associated with disease evolution but not thrombosis. Blood 2020, 135, 1696–1703. [Google Scholar] [CrossRef] [PubMed]
- Barbui, T.; Carobbio, A.; Rambaldi, A.; Finazzi, G. Perspectives on thrombosis in essential thrombocythemia and polycythemia vera: Is leukocytosis a causative factor? Blood 2009, 114, 759–763. [Google Scholar] [CrossRef] [Green Version]
- Santisakultarm, T.P.; Paduano, C.Q.; Stokol, T.; Southard, T.L.; Nishimura, N.; Skoda, R.C.; Olbricht, W.L.; Schafer, A.I.; Silver, R.T.; Schaffer, C.B. Stalled cerebral capillary blood flow in mouse models of essential thrombocythemia and polycythemia vera revealed by in vivo two-photon imaging. J. Thromb. Haemost. 2014, 12, 2120–2130. [Google Scholar] [CrossRef] [PubMed]
- Walton, B.L.; Lehmann, M.; Skorczewski, T.; Holle, L.A.; Beckman, J.D.; Cribb, J.A.; Mooberry, M.J.; Wufsus, A.R.; Cooley, B.C.; Homeister, J.W.; et al. Elevated hematocrit enhances platelet accumulation following vascular injury. Blood 2017, 129, 2537–2546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machlus, K.R.; Battinelli, E.M. RBCs pin platelets against the (thrombus) wall. Blood 2017, 129, 2460–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogelson, A.L.; Neeves, K.B. Fluid Mechanics of Blood Clot Formation. Annu. Rev. Fluid Mech. 2015, 47, 377–403. [Google Scholar] [CrossRef] [Green Version]
- Klatt, C.; Krüger, I.; Zey, S.; Krott, K.-J.; Spelleken, M.; Gowert, N.S.; Oberhuber, A.; Pfaff, L.; Lückstädt, W.; Jurk, K.; et al. Platelet-RBC interaction mediated by FasL/FasR induces procoagulant activity important for thrombosis. J. Clin. Investig. 2018, 128, 3906–3925. [Google Scholar] [CrossRef]
- Wautier, M.-P.; El Nemer, W.; Gane, P.; Rain, J.-D.; Cartron, J.-P.; Colin, Y.; Le Van Kim, C.; Wautier, J.-L. Increased adhesion to endothelial cells of erythrocytes from patients with polycythemia vera is mediated by laminin α5 chain and Lu/BCAM. Blood 2007, 110, 894–901. [Google Scholar] [CrossRef] [Green Version]
- De Grandis, M.; Cassinat, B.; Kiladjian, J.-J.; Chomienne, C.; El Nemer, W. Lu/BCAM-mediated cell adhesion as biological marker of JAK2V617F activity in erythrocytes of Polycythemia Vera patients. Am. J. Hematol. 2015, 90, 137–138. [Google Scholar] [CrossRef]
- Poisson, J.; Tanguy, M.; Davy, H.; Camara, F.; El Mdawar, M.-B.; Kheloufi, M.; Dagher, T.; Devue, C.; Lasselin, J.; Plessier, A.; et al. Erythrocyte-derived microvesicles induce arterial spasms in JAK2V617F myeloproliferative neoplasm. J. Clin. Investig. 2020, 130, 2630–2643. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Keerthivasan, G.; Mei, Y.; Yang, J.; McElherne, J.; Wong, P.; Doench, J.G.; Feng, G.; Root, D.E.; Ji, P. Targeted shRNA screening identified critical roles of pleckstrin-2 in erythropoiesis. Haematologica 2014, 99, 1157–1167. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Mei, Y.; Cao, L.; Zhang, J.; Sumagin, R.; Yang, J.; Gao, J.; Schipma, M.J.; Wang, Y.; Thorsheim, C.; et al. Loss of pleckstrin-2 reverts lethality and vascular occlusions in JAK2V617F-positive myeloproliferative neoplasms. J. Clin. Investig. 2017, 128, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Arellano-Rodrigo, E.; Alvarez-Larrán, A.; Reverter, J.C.; Villamor, N.; Colomer, D.; Cervantes, F. Increased platelet and leukocyte activation as contributing mechanisms for thrombosis in essential thrombocythemia and correlation with the JAK2 mutational status. Haematologica 2006, 91, 169–175. [Google Scholar]
- Falanga, A.; Marchetti, M.; Vignoli, A.; Balducci, D.; Barbui, T. Leukocyte-platelet interaction in patients with essential thrombocythemia and polycythemia vera. Exp. Hematol. 2005, 33, 523–530. [Google Scholar] [CrossRef]
- Laguna, M.S.; Kornblihtt, L.I.; Marta, R.F.; Molinas, F.C. Thromboxane B2 and platelet derived growth factor in essential thrombocythemia treated with anagrelide. Medicina 2000, 60, 448–452. [Google Scholar]
- Alvarez-Larrán, A.; Arellano-Rodrigo, E.; Reverter, J.C.; Domingo, A.; Villamor, N.; Colomer, D.; Cervantes, F. Increased platelet, leukocyte, and coagulation activation in primary myelofibrosis. Ann. Hematol. 2007, 87, 269–276. [Google Scholar] [CrossRef]
- Lev, P.; Marta, R.; Vassallu, P.; Molinas, F. Variation of PDGF, TGF?, and bFGF levels in essential thrombocythemia patients treated with anagrelide. Am. J. Hematol. 2002, 70, 85–91. [Google Scholar] [CrossRef]
- Kvernberg, J.; Grove, E.L.; Ommen, H.B.; Hvas, A.-M. Platelet Function and Turnover in Essential Thrombocythemia: A Systematic Review. Semin. Thromb. Hemost. 2021, 47, 090–101. [Google Scholar] [CrossRef]
- Van Genderen, P.J.; Lucas, I.S.; Van Strik, R.; Vuzevski, V.D.; Prins, F.J.; Van Vliet, H.H.; Michiels, J.J. Erythromelalgia in essential thrombocythemia is characterized by platelet activation and endothelial cell damage but not by thrombin generation. Thromb. Haemost. 1996, 76, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Oyarzún, C.P.M.; Heller, P.G. Platelets as Mediators of Thromboinflammation in Chronic Myeloproliferative Neoplasms. Front. Immunol. 2019, 10, 1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panova-Noeva, M.; Marchetti, M.; Buoro, S.; Russo, L.; Leuzzi, A.; Finazzi, G.; Rambaldi, A.; Ottomano, C.; Cate, H.T.; Falanga, A. JAK2V617F mutation and hydroxyurea treatment as determinants of immature platelet parameters in essential thrombocythemia and polycythemia vera patients. Blood 2011, 118, 2599–2601. [Google Scholar] [CrossRef]
- Trappenburg, M.C.; Van Schilfgaarde, M.; Marchetti, M.; Spronk, H.M.; Cate, H.T.; Leyte, A.; Terpstra, W.E.; Falanga, A. Elevated procoagulant microparticles expressing endothelial and platelet markers in essential thrombocythemia. Haematologica 2009, 94, 911–918. [Google Scholar] [CrossRef] [Green Version]
- Marchetti, M.; Tartari, C.J.; Russo, L.; Panova-Noeva, M.; Leuzzi, A.; Rambaldi, A.; Finazzi, G.; Woodhams, B.; Falanga, A. Phospholipid-dependent procoagulant activity is highly expressed by circulating microparticles in patients with essential thrombocythemia. Am. J. Hematol. 2013, 89, 68–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mignon, I.; Grand, F.; Boyer, F.; Hunault-Berger, M.; Hamel, J.; Macchi, L. Thrombin generation and procoagulant phospholipids in patients with essential thrombocythemia and reactive thrombocytosis. Am. J. Hematol. 2013, 88, 1007–1011. [Google Scholar] [CrossRef]
- Duchemin, J.; Ugo, V.; Ianotto, J.-C.; Lecucq, L.; Mercier, B.; Abgrall, J.-F. Increased circulating procoagulant activity and thrombin generation in patients with myeloproliferative neoplasms. Thromb. Res. 2010, 126, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, Y.; Tanaka, H.; Luis, E.J.; Sakai, K.; Kumode, T.; Sano, K.; Serizawa, K.; Rai, S.; Morita, Y.; Hanamoto, H.; et al. Elevated plasma levels of procoagulant microparticles are a novel risk factor for thrombosis in patients with myeloproliferative neoplasms. Int. J. Hematol. 2017, 106, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Qi, J.; Zhao, S.; Shen, W.; Dai, L.; Han, W.; Huang, M.; Wang, Z.; Ruan, C.; Wu, D.; et al. Clinical significance of circulating microparticles in Ph− myeloproliferative neoplasms. Oncol. Lett. 2017, 14, 2531–2536. [Google Scholar] [CrossRef] [Green Version]
- Falanga, A.; Marchetti, M.; Vignoli, A.; Balducci, D.; Russo, L.; Guerini, V.; Barbui, T. V617F JAK-2 mutation in patients with essential thrombocythemia: Relation to platelet, granulocyte, and plasma hemostatic and inflammatory molecules. Exp. Hematol. 2007, 35, 702–711. [Google Scholar] [CrossRef]
- Dienava-Verdoold, I.; Marchetti, M.R.; Boome, L.C.J.T.; Russo, L.; Falanga, A.; Koene, H.R.; Mertens, K.; Brinkman, H.J.M. Platelet-mediated proteolytic down regulation of the anticoagulant activity of protein S in individuals with haematological malignancies. Thromb. Haemost. 2012, 107, 468–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchetti, M.; Castoldi, E.; Spronk, H.M.H.; Van Oerle, R.; Balducci, D.; Barbui, T.; Rosing, J.; Cate, H.T.; Falanga, A. Thrombin generation and activated protein C resistance in patients with essential thrombocythemia and polycythemia vera. Blood 2008, 112, 4061–4068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucalossi, A.; Marotta, G.; Bigazzi, C.; Galieni, P.; Dispensa, E. Reduction of antithrombin III, protein C, and protein S levels and activated protein C resistance in polycythemia vera and essential thrombocythemia patients with thrombosis. Am. J. Hematol. 1996, 52, 14–20. [Google Scholar] [CrossRef]
- Avram, S.; Lupu, A.; Angelescu, S.; Olteanu, N.; Mut-Popescu, D. Abnormalities of platelet aggregation in chronic myeloproliferative disorders. J. Cell. Mol. Med. 2001, 5, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Michiels, J.J.; Berneman, Z.; Schroyens, W.; Finazzi, G.; Budde, U.; Van Vliet, H.H. The Paradox of Platelet Activation and Impaired Function: Platelet-von Willebrand Factor Interactions, and the Etiology of Thrombotic and Hemorrhagic Manifestations in Essential Thrombocythemia and Polycythemia Vera. Semin. Thromb. Hemost. 2006, 32, 589–604. [Google Scholar] [CrossRef]
- Lamrani, L.; Lacout, C.; Ollivier, V.; Denis, C.V.; Gardiner, E.; Noe, B.H.T.; Vainchenker, W.; Villeval, J.-L.; Jandrot-Perrus, M. Hemostatic disorders in a JAK2V617F-driven mouse model of myeloproliferative neoplasm. Blood 2014, 124, 1136–1145. [Google Scholar] [CrossRef] [PubMed]
- Wolach, O.; Sellar, R.S.; Martinod, K.; Cherpokova, D.; McConkey, M.; Chappell, R.J.; Silver, A.J.; Adams, D.; Castellano, C.A.; Schneider, R.K.; et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci. Transl. Med. 2018, 10, eaan8292. [Google Scholar] [CrossRef] [Green Version]
- Strassel, C.; Kubovcakova, L.; Mangin, P.H.; Ravanat, C.; Freund, M.; Skoda, R.C.; Denis, C.V.; Dupuis, A.; Herbrecht, R.; Gachet, C.; et al. Haemorrhagic and thrombotic diatheses in mouse models with thrombocytosis. Thromb. Haemost. 2015, 113, 414–425. [Google Scholar] [CrossRef]
- Etheridge, S.L.; Roh, M.E.; Cosgrove, M.E.; Sangkhae, V.; Fox, N.E.; Chen, J.; López, J.A.; Kaushansky, K.; Hitchcock, I.S. JAK2V617F-positive endothelial cells contribute to clotting abnormalities in myeloproliferative neoplasms. Proc. Natl. Acad. Sci. USA 2014, 111, 2295–2300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobbs, C.M.; Manning, H.; Bennett, C.; Vasquez, L.; Severin, S.; Brain, L.; Mazharian, A.; Guerrero, J.A.; Li, J.; Soranzo, N.; et al. JAK2V617F leads to intrinsic changes in platelet formation and reactivity in a knock-in mouse model of essential thrombocythemia. Blood 2013, 122, 3787–3797. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.F.; Hunter, R.W.; Harper, M.T.; Savage, J.S.; Siddiq, S.; Westbury, S.K.; Poole, A.W.; Mumford, A.D.; Hers, I. Dysfunction of the PI3 kinase/Rap1/integrin αIIbβ3 pathway underlies ex vivo platelet hypoactivity in essential thrombocythemia. Blood 2013, 121, 1209–1219. [Google Scholar] [CrossRef] [Green Version]
- Tadmor, T.; Bejar, J.; Attias, D.; Mischenko, E.; Sabo, E.; Neufeld, G.; Vadasz, Z. The expression of lysyl-oxidase gene family members in myeloproliferative neoplasms. Am. J. Hematol. 2013, 88, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Abbonante, V.; Chitalia, V.; Rosti, V.; Leiva, O.; Matsuura, S.; Balduini, A.; Ravid, K. Upregulation of lysyl oxidase and adhesion to collagen of human megakaryocytes and platelets in primary myelofibrosis. Blood 2017, 130, 829–831. [Google Scholar] [CrossRef] [Green Version]
- Matsuura, S.; Mi, R.; Koupenova, M.; Eliades, A.; Patterson, S.; Toselli, P.; Thon, J.; Italiano, J.E.; Trackman, P.C.; Papadantonakis, N.; et al. Lysyl oxidase is associated with increased thrombosis and platelet reactivity. Blood 2016, 127, 1493–1501. [Google Scholar] [CrossRef] [Green Version]
- Hurtado-Nedelec, M.; Csillag-Grange, M.-J.; Boussetta, T.; Belambri, S.A.; Fay, M.; Cassinat, B.; Gougerot-Pocidalo, M.-A.; Dang, P.M.-C.; El-Benna, J. Increased reactive oxygen species production and p47phox phosphorylation in neutrophils from myeloproliferative disorders patients with JAK2 (V617F) mutation. Haematologica 2013, 98, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- Falanga, A.; Marchetti, M.; Evangelista, V.; Vignoli, A.; Licini, M.; Balicco, M.; Manarini, S.; Finazzi, G.; Cerletti, C.; Barbui, T. Polymorphonuclear leukocyte activation and hemostasis in patients with essential thrombocythemia and polycythemia vera. Blood 2000, 96, 4261–4266. [Google Scholar] [CrossRef] [PubMed]
- Barbui, T.; Finazzi, G.; Falanga, A. Myeloproliferative neoplasms and thrombosis. Blood 2013, 122, 2176–2184. [Google Scholar] [CrossRef]
- Schieppati, F.; Russo, D.; Falanga, A. Cancer Tissue Procoagulant Mechanisms and the Hypercoagulable State of Patients with Cancer. Semin. Thromb. Hemost. 2015, 41, 756–764. [Google Scholar] [CrossRef]
- Torregrosa, J.M.; Ferrer-Marin, F.; Lozano, M.L.; Moreno, M.J.; Martínez, C.; Antón, A.I.; Rivera, J.; Vicente, V. Impaired leucocyte activation is underlining the lower thrombotic risk of essential thrombocythaemia patients with CALR mutations as compared with those with the JAK2 mutation. Br. J. Haematol. 2015, 172, 813–815. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Martinod, K.; Wagner, D.D. Thrombosis: Tangled up in NETs. Blood 2014, 123, 2768–2776. [Google Scholar] [CrossRef]
- Granger, V.; Peyneau, M.; Chollet-Martin, S.; De Chaisemartin, L. Neutrophil Extracellular Traps in Autoimmunity and Allergy: Immune Complexes at Work. Front. Immunol. 2019, 10, 2824. [Google Scholar] [CrossRef] [Green Version]
- Demers, M.; Krause, D.S.; Schatzberg, D.; Martinod, K.; Voorhees, J.R.; Fuchs, T.A.; Scadden, D.T.; Wagner, D.D. Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc. Natl. Acad. Sci. USA 2012, 109, 13076–13081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oyarzún, C.P.M.; Carestia, A.; Lev, P.R.; Glembotsky, A.C.; Ríos, M.A.C.; Moiraghi, B.; Molinas, F.C.; Marta, R.F.; Schattner, M.; Heller, P.G. Neutrophil extracellular trap formation and circulating nucleosomes in patients with chronic myeloproliferative neoplasms. Sci. Rep. 2016, 6, 38738. [Google Scholar] [CrossRef] [Green Version]
- Guy, A.; Gourdou-Latyszenok, V.; Le Lay, N.; Peghaire, C.; Kilani, B.; Dias, J.V.; Duplaa, C.; Renault, M.-A.; Denis, C.; Villeval, J.L.; et al. Vascular endothelial cell expression of JAK2V617F is sufficient to promote a pro-thrombotic state due to increased P-selectin expression. Haematologica 2019, 104, 70–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolff, L.; Guy, A.; Gourdou-Latyszenokv, V.; Favre, S.; Colomer, S.; Renault, M.; Couffinhal, T.; James, C. Neutrophils prothrombotic characteristics during myeloproliferative neoplasms. Arch. Cardiovasc. Dis. Suppl. 2020, 12, 205. [Google Scholar] [CrossRef]
- Lisman, T. Platelet-neutrophil interactions as drivers of inflammatory and thrombotic disease. Cell Tissue Res. 2018, 371, 567–576. [Google Scholar] [CrossRef] [Green Version]
- Celi, A.; Pellegrini, G.; Lorenzet, R.; De Blasi, A.; Ready, N.; Furie, B.C. P-selectin induces the expression of tissue factor on monocytes. Proc. Natl. Acad. Sci. USA 1994, 91, 8767–8771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weyrich, A.S.; Elstad, M.R.; McEver, R.P.; McIntyre, T.M.; Moore, K.L.; Morrissey, J.H.; Prescott, S.M.; Zimmerman, G.A. Activated platelets signal chemokine synthesis by human monocytes. J. Clin. Investig. 1996, 97, 1525–1534. [Google Scholar] [CrossRef] [Green Version]
- Fisher, D.A.C.; Miner, C.A.; Engle, E.K.; Hu, H.; Collins, T.B.; Zhou, A.; Allen, M.J.; Malkova, O.N.; Oh, S.T. Cytokine production in myelofibrosis exhibits differential responsiveness to JAK-STAT, MAP kinase, and NFκB signaling. Leukemia 2019, 33, 1978–1995. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Edelmann, B.; Schnoeder, T.M.; Saalfeld, F.C.; Wolleschak, D.; Kliche, S.; Schraven, B.; Heidel, F.H.; Fischer, T. JAK2-V617F activates beta1-integrin-mediated adhesion of granulocytes to vascular cell adhesion molecule. Leukemia 2017, 31, 1223–1226. [Google Scholar] [CrossRef] [Green Version]
- Edelmann, B.; Gupta, N.; Schnoeder, T.M.; Oelschlegel, A.M.; Shahzad, K.; Goldschmidt, J.; Philipsen, L.; Weinert, S.; Ghosh, A.; Saalfeld, F.C.; et al. JAK2-V617F promotes venous thrombosis through β1/β2 integrin activation. J. Clin. Invest. 2018, 128, 4359–4371. [Google Scholar] [CrossRef] [PubMed]
- Cella, G.; Marchetti, M.; Vianello, F.; Panova-Noeva, M.; Vignoli, A.; Russo, L.; Barbui, T.; Falanga, A. Nitric oxide derivatives and soluble plasma selectins in patients with myeloproliferative neoplasms. Thromb. Haemost. 2010, 104, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Arellano-Rodrigo, E.; Alvarez-Larrán, A.; Reverter, J.-C.; Colomer, D.; Villamor, N.; Bellosillo, B.; Cervantes, F. Platelet turnover, coagulation factors, and soluble markers of platelet and endothelial activation in essential thrombocythemia: Relationship with thrombosis occurrence andJAK2 V617F allele burden. Am. J. Hematol. 2009, 84, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Di Raimondo, F.; Palumbo, G.A.; Molica, S.; Giustolisi, R. Angiogenesis in chronic myeloproliferative diseases. Acta Haematol. 2001, 106, 177–183. [Google Scholar] [CrossRef]
- Treliński, J.; Wierzbowska, A.; Krawczyńska, A.; Sakowicz, A.; Pietrucha, T.; Smolewski, P.; Robak, T.; Chojnowski, K. Plasma levels of angiogenic factors and circulating endothelial cells in essential thrombocythemia: Correlation with cytoreductive therapy and JAK2–V617F mutational status. Leuk. Lymphoma 2010, 51, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Subotički, T.; Ajtić, O.M.; Beleslin-Čokić, B.B.; Nienhold, R.; Diklić, M.; Djikić, D.; Leković, D.; Bulat, T.; Marković, D.; Gotić, M.; et al. Angiogenic factors are increased in circulating granulocytes and CD34+cells of myeloproliferative neoplasms. Mol. Carcinog. 2016, 56, 567–579. [Google Scholar] [CrossRef]
- Sozer, S.; Fiel, M.I.; Schiano, T.; Xu, M.; Mascarenhas, J.; Hoffman, R. The presence of JAK2V617F mutation in the liver endothelial cells of patients with Budd-Chiari syndrome. Blood 2009, 113, 5246–5249. [Google Scholar] [CrossRef]
- Rosti, V.; Villani, L.; Riboni, R.; Poletto, V.; Bonetti, E.; Tozzi, L.; Bergamaschi, G.; Catarsi, P.; Dallera, E.; Novara, F.; et al. Spleen endothelial cells from patients with myelofibrosis harbor the JAK2V617F mutation. Blood 2013, 121, 360–368. [Google Scholar] [CrossRef]
- Guadall, A.; Lesteven, E.; Letort, G.; Toor, S.A.; Delord, M.; Pognant, D.; Brusson, M.; Verger, E.; Maslah, N.; Giraudier, S.; et al. Endothelial Cells Harbouring the JAK2V617F Mutation Display Pro-Adherent and Pro-Thrombotic Features. Thromb. Haemost. 2018, 118, 1586–1599. [Google Scholar] [CrossRef] [Green Version]
- Castiglione, M.; Jiang, Y.; Mazzeo, C.; Lee, S.; Chen, J.; Kaushansky, K.; Yin, W.; Lin, R.Z.; Zheng, H.; Zhan, H. Endothelial JAK2V617F mutation leads to thrombosis, vasculopathy, and cardiomyopathy in a murine model of myeloproliferative neoplasm. J. Thromb. Haemost. 2020, 18, 3359–3370. [Google Scholar] [CrossRef]
- Lussana, F.; Rambaldi, A. Inflammation and myeloproliferative neoplasms. J. Autoimmun. 2017, 85, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Vaidya, R.; Caramazza, D.; Finke, C.; Lasho, T.; Pardanani, A. Circulating Interleukin (IL)-8, IL-2R, IL-12, and IL-15 Levels Are Independently Prognostic in Primary Myelofibrosis: A Comprehensive Cytokine Profiling Study. J. Clin. Oncol. 2011, 29, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- Masselli, E.; Pozzi, G.; Gobbi, G.; Merighi, S.; Gessi, S.; Vitale, M.; Carubbi, C. Cytokine Profiling in Myeloproliferative Neoplasms: Overview on Phenotype Correlation, Outcome Prediction, and Role of Genetic Variants. Cells 2020, 9, 2136. [Google Scholar] [CrossRef] [PubMed]
- Lussana, F.; Carobbio, A.; Salmoiraghi, S.; Guglielmelli, P.; Vannucchi, A.M.; Bottazzi, B.; Leone, R.; Mantovani, A.; Barbui, T.; Rambaldi, A. Driver mutations (JAK2V617F, MPLW515L/K or CALR), pentraxin-3 and C-reactive protein in essential thrombocythemia and polycythemia vera. J. Hematol. Oncol. 2017, 10, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbui, T.; Carobbio, A.; Finazzi, G.; Vannucchi, A.M.; Barosi, G.; Antonioli, E.; Guglielmelli, P.; Pancrazzi, A.; Salmoiraghi, S.; Zilio, P.; et al. Inflammation and thrombosis in essential thrombocythemia and polycythemia vera: Different role of C-reactive protein and pentraxin 3. Haematologica 2010, 96, 315–318. [Google Scholar] [CrossRef]
- Gangaraju, R.; Song, J.; Kim, S.J.; Tashi, T.; Reeves, B.N.; Sundar, K.M.; Thiagarajan, P.; Prchal, J.T. Thrombotic, inflammatory, and HIF-regulated genes and thrombosis risk in polycythemia vera and essential thrombocythemia. Blood Adv. 2020, 4, 1115–1130. [Google Scholar] [CrossRef] [Green Version]
- Oyarzún, C.P.M.; Glembotsky, A.C.; Goette, N.P.; Lev, P.R.; De Luca, G.; Pietto, M.C.B.; Moiraghi, B.; Ríos, M.A.C.; Vicente, A.; Marta, R.F.; et al. Platelet Toll-Like Receptors Mediate Thromboinflammatory Responses in Patients with Essential Thrombocythemia. Front. Immunol. 2020, 11, 705. [Google Scholar] [CrossRef]
- Jackson, S.P.; Darbousset, R.; Schoenwaelder, S.M. Thromboinflammation: Challenges of therapeutically targeting coagulation and other host defense mechanisms. Blood 2019, 133, 906–918. [Google Scholar] [CrossRef] [Green Version]
- Schafer, A.I. Thrombotic, Vascular, and Bleeding Complications of the Myeloproliferative Neoplasms. Hematol. Clin. N. Am. 2021, 35, 305–324. [Google Scholar] [CrossRef]
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2012, 13, 34–45. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef]
- Young, A.L.; Challen, G.A.; Birmann, B.M.; Druley, T.E. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat. Commun. 2016, 7, 12484. [Google Scholar] [CrossRef] [PubMed]
- Van Egeren, D.; Escabi, J.; Nguyen, M.; Liu, S.; Reilly, C.R.; Patel, S.; Kamaz, B.; Kalyva, M.; DeAngelo, D.J.; Galinsky, I.; et al. Reconstructing the Lineage Histories and Differentiation Trajectories of Individual Cancer Cells in Myeloproliferative Neoplasms. Cell Stem Cell 2021, 28, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.E.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef]
- Wang, W.; Liu, W.; Fidler, T.; Wang, Y.; Tang, Y.; Woods, B.; Welch, C.; Cai, B.; Silvestre-Roig, C.; Ai, D.; et al. Macrophage Inflammation, Erythrophagocytosis, and Accelerated Atherosclerosis in Jak2 (V617F) Mice. Circ. Res. 2018, 123, e35–e47. [Google Scholar] [CrossRef]
- Libby, P.; Molinaro, R.; Sellar, R.S.; Ebert, B.L. Jak-ing Up the Plaque’s Lipid Core...and Even More. Circ. Res. 2018, 123, 1180–1182. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Wiersinga, W.J.; Rhodes, A.; Cheng, A.C.; Peacock, S.J.; Prescott, H.C. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 324, 782–793. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Wu, D.; Guo, W.; Cao, Y.; Huang, D.; Wang, H.; Wang, T.; Zhang, X.; Chen, H.; Yu, H.; et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Investig. 2020, 130, 2620–2629. [Google Scholar] [CrossRef] [Green Version]
- Nopp, S.; Moik, F.; Jilma, B.; Pabinger, I.; Ay, C. Risk of venous thromboembolism in patients with COVID-19: A systematic review and meta-analysis. Res. Pr. Thromb. Haemost. 2020, 4, 1178–1191. [Google Scholar] [CrossRef]
- Bozkus, C.C.; Roudko, V.; Finnigan, J.P.; Mascarenhas, J.; Hoffman, R.; Iancu-Rubin, C.; Bhardwaj, N. Immune Checkpoint Blockade Enhances Shared Neoantigen-Induced T-cell Immunity Directed against Mutated Calreticulin in Myeloproliferative Neoplasms. Cancer Discov. 2019, 9, 1192–1207. [Google Scholar] [CrossRef]
- Stein, B.L.; Martin, K. From Budd-Chiari syndrome to acquired von Willebrand syndrome: Thrombosis and bleeding complications in the myeloproliferative neoplasms. Hematol. Am. Soc. Hematol. Educ. Program 2019, 2019, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Connors, J.M.; Levy, J.H. COVID-19 and its implications for thrombosis and anticoagulation. Blood 2020, 135, 2033–2040. [Google Scholar] [CrossRef] [PubMed]
- Breccia, M.; Piciocchi, A.; De Stefano, V.; Finazzi, G.; Iurlo, A.; Fazi, P.; Soddu, S.; Martino, B.; Palandri, F.; Siragusa, S.; et al. COVID-19 in Philadelphia-negative myeloproliferative disorders: A GIMEMA survey. Leukemia 2020, 34, 2813–2814. [Google Scholar] [CrossRef]
- Palandri, F.; Piciocchi, A.; De Stefano, V.; Breccia, M.; Finazzi, G.; Iurlo, A.; Fazi, P.; Soddu, S.; Martino, B.; Siragusa, S.; et al. How the coronavirus pandemic has affected the clinical management of Philadelphia-negative chronic myeloproliferative neoplasms in Italy—A GIMEMA MPN WP survey. Leukemia 2020, 34, 2805–2808. [Google Scholar] [CrossRef]
- Barbui, T.; Vannucchi, A.M.; Alvarez-Larran, A.; Iurlo, A.; Masciulli, A.; Carobbio, A.; Ghirardi, A.; Ferrari, A.; Rossi, G.; Elli, E.; et al. High mortality rate in COVID-19 patients with myeloproliferative neoplasms after abrupt withdrawal of ruxolitinib. Leukemia 2021, 35, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Dillinger, J.G.; Sideris, G.; Henry, P.; Bal dit Sollier, C.; Ronez, E.; Drouet, L. Twice daily aspirin to improve biological aspirin efficacy in patients with essential thrombocythemia. Thromb. Res. 2012, 129, 91–94. [Google Scholar] [CrossRef]
- Barbui, T.; De Stefano, V.; Alvarez-Larran, A.; Iurlo, A.; Masciulli, A.; Carobbio, A.; Ghirardi, A.; Ferrari, A.; Cancelli, V.; Elli, E.M.; et al. Among classic myeloproliferative neoplasms, essential thrombocythemia is associated with the greatest risk of venous thromboembolism during COVID-19. Blood Cancer J. 2021, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kamaz, B.; Mullally, A. COVID-19 and myeloproliferative neoplasms: Some considerations. Leukemia 2021, 35, 279–281. [Google Scholar] [CrossRef]
- La Rosee, F.; Bremer, H.C.; Gehrke, I.; Kehr, A.; Hochhaus, A.; Birndt, S.; Fellhauer, M.; Henkes, M.; Kumle, B.; Russo, S.G.; et al. The Janus kinase 1/2 inhibitor ruxolitinib in COVID-19 with severe systemic hyperinflammation. Leukemia 2020, 34, 1805–1815. [Google Scholar] [CrossRef] [PubMed]
- NIH. COVID-19 Treatment Guidelines. 2020. Available online: covid19treatmentguidelines.nih.gov (accessed on 10 March 2021).
- Tefferi, A.; Barbui, T. Polycythemia vera and essential thrombocythemia: 2015 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2015, 90, 162–173. [Google Scholar] [CrossRef]
- Landolfi, R.; Marchioli, R.; Kutti, J.; Gisslinger, H.; Tognoni, G.; Patrono, C.; Barbui, T. Efficacy and Safety of Low-Dose Aspirin in Polycythemia Vera. N. Engl. J. Med. 2004, 350, 114–124. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Larrán, A.; Cervantes, F.; Pereira, A.; Arellano-Rodrigo, E.; Pérez-Andreu, V.; Hernández-Boluda, J.-C.; Ayats, R.; Salvador, C.; Muntañola, A.; Bellosillo, B.; et al. Observation versus antiplatelet therapy as primary prophylaxis for thrombosis in low-risk essential thrombocythemia. Blood 2010, 116, 1205–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Genderen, P.J.J.; Mulder, P.G.H.; Waleboer, M.; Van De Moesdijk, D.; Michiels, J.J. Prevention and treatment of thrombotic complications in essential thrombocythaemia: Efficacy and safety of aspirin. Br. J. Haematol. 1997, 97, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Pascale, S.; Petrucci, G.; Dragani, A.; Habib, A.; Zaccardi, F.; Pagliaccia, F.; Pocaterra, D.; Ragazzoni, E.; Rolandi, G.; Rocca, B.; et al. Aspirin-insensitive thromboxane biosynthesis in essential thrombocythemia is explained by accelerated renewal of the drug target. Blood 2012, 119, 3595–3603. [Google Scholar] [CrossRef] [Green Version]
- Kreher, S.; Ochsenreither, S.; Trappe, R.U.; Pabinger, I.; Bergmann, F.; Petrides, P.E.; Koschmieder, S.; Matzdorff, A.; Tiede, A.; Griesshammer, M.; et al. Prophylaxis and management of venous thromboembolism in patients with myeloproliferative neoplasms: Consensus statement of the Haemostasis Working Party of the German Society of Hematology and Oncology (DGHO), the Austrian Society of Hematology and Oncology (OGHO) and Society of Thrombosis and Haemostasis Research (GTH e.V.). Ann. Hematol. 2014, 93, 1953–1963. [Google Scholar] [PubMed]
- Kiladjian, J.-J.; Chevret, S.; Dosquet, C.; Chomienne, C.; Rain, J.-D. Treatment of Polycythemia Vera With Hydroxyurea and Pipobroman: Final Results of a Randomized Trial Initiated in 1980. J. Clin. Oncol. 2011, 29, 3907–3913. [Google Scholar] [CrossRef]
- Huang, B.-T.; Zeng, Q.-C.; Zhao, W.-H.; Li, B.-S.; Chen, R.-L. Interferon α-2b gains high sustained response therapy for advanced essential thrombocythemia and polycythemia vera with JAK2V617F positive mutation. Leuk. Res. 2014, 38, 1177–1183. [Google Scholar] [CrossRef]
- Masarova, L.; Patel, K.P.; Newberry, K.J.; Cortes, J.; Borthakur, G.; Konopleva, M.; Estrov, Z.; Kantarjian, H.; Verstovsek, S. Pegylated interferon alfa-2a in patients with essential thrombocythaemia or polycythaemia vera: A post-hoc, median 83 month follow-up of an open-label, phase 2 trial. Lancet Haematol. 2017, 4, e165–e175. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Larrán, A.; Martínez-Avilés, L.; Hernández-Boluda, J.C.; Ferrer-Marín, F.; Antelo, M.L.; Burgaleta, C.; Mata, M.I.; Xicoy, B.; Martínez-Trillos, A.; Gómez-Casares, M.T.; et al. Busulfan in patients with polycythemia vera or essential thrombocythemia refractory or intolerant to hydroxyurea. Ann. Hematol. 2014, 93, 2037–2043. [Google Scholar] [CrossRef]
- Harrison, C.N.; Campbell, P.J.; Buck, G.; Wheatley, K.; East, C.L.; Bareford, D.; Wilkins, B.S.; Van Der Walt, J.D.; Reilly, J.T.; Grigg, A.P.; et al. Hydroxyurea Compared with Anagrelide in High-Risk Essential Thrombocythemia. N. Engl. J. Med. 2005, 353, 33–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fruchtman, S.M.; Mack, K.; Kaplan, M.E.; Peterson, P.; Berk, P.D.; Wasserman, L.R. From efficacy to safety: A Polycythemia Vera Study group report on hydroxyurea in patients with polycythemia vera. Semin. Hematol. 1997, 34, 17–23. [Google Scholar] [PubMed]
- De Stefano, V.; Za, T.; Rossi, E.; Vannucchi, A.M.; Ruggeri, M.; Elli, E.; Micò, C.; Tieghi, A.; Cacciola, R.R.; Santoro, C.; et al. Recurrent thrombosis in patients with polycythemia vera and essential thrombocythemia: Incidence, risk factors, and effect of treatments. Haematologica 2008, 93, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Barbui, T.; Tefferi, A.; Vannucchi, A.M.; Passamonti, F.; Silver, R.T.; Hoffman, R.; Verstovsek, S.; Mesa, R.; Kiladjian, J.-J.; Hehlmann, R.; et al. Philadelphia chromosome-negative classical myeloproliferative neoplasms: Revised management recommendations from European LeukemiaNet. Leukemia 2018, 32, 1057–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yacoub, A.; Mascarenhas, J.; Kosiorek, H.; Prchal, J.T.; Berenzon, D.; Baer, M.R.; Ritchie, E.; Silver, R.T.; Kessler, C.; Winton, E.; et al. Pegylated interferon alfa-2a for polycythemia vera or essential thrombocythemia resistant or intolerant to hydroxyurea. Blood 2019, 134, 1498–1509. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas, J.; Kosiorek, M.H.E.; Prchal, J.T.; Rambaldi, A.; Berenzon, D.; Yacoub, A.; Harrison, D.C.N.; McMullin, M.F.; Vannucchi, A.M.; Ewing, B.J.; et al. Results of the Myeloproliferative Neoplasms—Research Consortium (MPN-RC) 112 Randomized Trial of Pegylated Interferon Alfa-2a (PEG) Versus Hydroxyurea (HU) Therapy for the Treatment of High Risk Polycythemia Vera (PV) and High Risk Essential Thrombocythemia (ET). Blood 2018, 132, 577. [Google Scholar] [CrossRef]
- Gisslinger, H.; Klade, C.; Georgiev, P.; Krochmalczyk, D.; Gercheva-Kyuchukova, L.; Egyed, M.; Rossiev, V.; Dulicek, P.; Illes, A.; Pylypenko, H.; et al. Ropeginterferon alfa-2b versus standard therapy for polycythaemia vera (PROUD-PV and CONTINUATION-PV): A randomised, non-inferiority, phase 3 trial and its extension study. Lancet Haematol. 2020, 7, e196–e208. [Google Scholar] [CrossRef]
- Tomer, A. Effects of anagrelide on in vivo megakaryocyte proliferation and maturation in essential thrombocythemia. Blood 2002, 99, 1602–1609. [Google Scholar] [CrossRef] [Green Version]
- Gisslinger, H.; Gotic, M.; Holowiecki, J.; Penka, M.; Thiele, J.; Kvasnicka, H.-M.; Kralovics, R.; Petrides, P.E. Anagrelide compared with hydroxyurea in WHO-classified essential thrombocythemia: The ANAHYDRET Study, a randomized controlled trial. Blood 2013, 121, 1720–1728. [Google Scholar] [CrossRef] [Green Version]
- Vannucchi, A.M.; Kiladjian, J.J.; Griesshammer, M.; Masszi, T.; Durrant, S.; Passamonti, F.; Harrison, C.N.; Pane, F.; Zachee, P.; Mesa, R.; et al. Ruxolitinib versus Standard Therapy for the Treatment of Polycythemia Vera. N. Engl. J. Med. 2015, 372, 426–435. [Google Scholar] [CrossRef] [Green Version]
- Passamonti, F.; Griesshammer, M.; Palandri, F.; Egyed, M.; Benevolo, G.; Devos, T.; Callum, J.; Vannucchi, A.M.; Sivgin, S.; Bensasson, C.; et al. Ruxolitinib for the treatment of inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): A randomised, open-label, phase 3b study. Lancet Oncol. 2017, 18, 88–99. [Google Scholar] [CrossRef]
- Masciulli, A.; Ferrari, A.; Carobbio, A.; Ghirardi, A.; Barbui, T. Ruxolitinib for the prevention of thrombosis in polycythemia vera: A systematic review and meta-analysis. Blood Adv. 2020, 4, 380–386. [Google Scholar] [CrossRef] [Green Version]
- Kiladjian, J.-J.; Zachee, P.; Hino, M.; Pane, F.; Masszi, T.; Harrison, C.N.; Mesa, R.; Miller, C.B.; Passamonti, F.; Durrant, S.; et al. Long-term efficacy and safety of ruxolitinib versus best available therapy in polycythaemia vera (RESPONSE): 5-year follow up of a phase 3 study. Lancet Haematol. 2020, 7, e226–e237. [Google Scholar] [CrossRef]
- Samuelson, B.T.; Vesely, S.K.; Chai-Adisaksopha, C.; Scott, B.L.; Crowther, M.; Garcia, D. The impact of ruxolitinib on thrombosis in patients with polycythemia vera and myelofibrosis: A meta-analysis. Blood Coagul. Fibrinolysis 2016, 27, 648–652. [Google Scholar] [CrossRef]
- De Stefano, V.; Ruggeri, M.; Cervantes, F.; Alvarez-Larrán, A.; Iurlo, A.; Randi, M.; Elli, E.; Finazzi, M.; Finazzi, G.; Zetterberg, E.; et al. High rate of recurrent venous thromboembolism in patients with myeloproliferative neoplasms and effect of prophylaxis with vitamin K antagonists. Leukemia 2016, 30, 2032–2038. [Google Scholar] [CrossRef] [PubMed]
- Kearon, C.; Akl, E.A.; Ornelas, J.; Blaivas, A.; Jimenez, D.; Bounameaux, H.; Huisman, M.; King, C.S.; Morris, T.A.; Sood, N.; et al. Antithrombotic Therapy for VTE Disease: CHEST Guideline and Expert Panel Report. Chest 2016, 149, 315–352. [Google Scholar] [CrossRef]
- Steffel, J.; Verhamme, P.; Potpara, T.S.; Albaladejo, P.; Antz, M.; Desteghe, L.; Haeusler, K.G.; Oldgren, J.; Reinecke, H.; Roldan-Schilling, V.; et al. The 2018 European Heart Rhythm Association Practical Guide on the use of non-vitamin K antagonist oral anticoagulants in patients with atrial fibrillation. Eur. Hearth J. 2018, 39, 1330–1393. [Google Scholar] [CrossRef] [Green Version]
- Raskob, G.E.; Van Es, N.; Verhamme, P.; Carrier, M.; Di Nisio, M.; Garcia, D.; Grosso, M.A.; Kakkar, A.K.; Kovacs, M.J.; Mercuri, M.F.; et al. Edoxaban for the Treatment of Cancer-Associated Venous Thromboembolism. N. Engl. J. Med. 2018, 378, 615–624. [Google Scholar] [CrossRef]
- Young, A.M.; Marshall, A.; Thirlwall, J.; Chapman, O.; Lokare, A.; Hill, C.; Hale, D.; Dunn, J.A.; Lyman, G.H.; Hutchinson, C.; et al. Comparison of an Oral Factor Xa Inhibitor with Low Molecular Weight Heparin in Patients With Cancer With Venous Thromboembolism: Results of a Randomized Trial (SELECT-D). J. Clin. Oncol. 2018, 36, 2017–2023. [Google Scholar] [CrossRef]
- Serrao, A.; Breccia, M.; Napolitano, M.; Fiori, L.; Santoro, M.; Scalzulli, E.; Santopietro, M.; Santoro, C.; Raso, S.; Chistolini, A. A multicenter real-life study on anticoagulant treatment with direct oral anticoagulants in patients with Ph-negative myeloproliferative neoplasms. Am. J. Hematol. 2020, 95, E329–E332. [Google Scholar] [CrossRef] [PubMed]
- Barbui, T.; De Stefano, V.; Carobbio, B.A.; Iurlo, A.; Alvarez-Larran, A.; Vannucchi, A.M.; Palandri, F.; Harrison, D.C.; Sibai, M.H.; Griesshammer, M.; et al. Direct Oral Anticoagulants for Myeloproliferative Neoplasms (MPN-DOACs): Results from an International Study on 442 Patients. Blood 2020, 136, 42–43. [Google Scholar] [CrossRef]
- Sant’Antonio, E.; Guglielmelli, P.; Pieri, L.; Primignani, M.; Randi, M.L.; Santarossa, C.; Rumi, E.; Cervantes, F.; Delaini, F.; Msc, A.C.; et al. Splanchnic vein thromboses associated with myeloproliferative neoplasms: An international, retrospective study on 518 cases. Am. J. Hematol. 2020, 95, 156–166. [Google Scholar] [CrossRef] [Green Version]
- Lavu, S.; Szuber, N.; Mudireddy, M.; Yogarajah, M.; Gangat, N.; Pardanani, A.; Hanson, C.A.; Ketterling, R.P.; Ashrani, A.A.; Kamath, P.S.; et al. Splanchnic vein thrombosis in patients with myeloproliferative neoplasms: The Mayo clinic experience with 84 consecutive cases. Am. J. Hematol. 2018, 93, E61–E64. [Google Scholar] [CrossRef]
- Curto-Garcia, N.; Doyle, A.J.; Breen, K.A.; McLornan, D.P.; Radia, D.H.; Hunt, B.J.; Ling, G.; Harrison, C.N. Outcomes of patients receiving direct oral anticoagulants for myeloproliferative neoplasm-associated venous thromboembolism in a large tertiary centre in the UK. Br. J. Haematol. 2020, 189, e79–e81. [Google Scholar] [CrossRef] [Green Version]
- Verstovsek, S.; Kantarjian, H.; Mesa, R.A.; Pardanani, A.D.; Cortes-Franco, J.; Thomas, D.A.; Estrov, Z.; Fridman, J.S.; Bradley, E.C.; Erickson-Viitanen, S.; et al. Safety and Efficacy of INCB018424, a JAK1 and JAK2 Inhibitor, in Myelofibrosis. N. Engl. J. Med. 2010, 363, 1117–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keohane, C.; McLornan, D.P.; Sanchez, K.; Connor, C.; Radia, D.; Harrison, C.N. The effects of JAK inhibitor therapy upon novel markers of thrombosis in myeloproliferative neoplasms. Haematologica 2015, 100, e348–e350. [Google Scholar] [CrossRef] [Green Version]
- DaSilva, B.A.; Aronovich, E.; Nguyen, A.; Nguyen, J.; Reynolds, D.; Doak, B.G.D.; Vercellotti, G.M.; Wood, D.K.; Beckman, M.J.D. Ruxolitinib Reduces Endothelial Pro-Adhesive Interactions: Implications for JAK2V617+ MPN Thrombosis. Blood 2020, 136, 1. [Google Scholar] [CrossRef]
- Etulain, J.; Martinod, K.; Wong, S.L.; Cifuni, S.M.; Schattner, M.; Wagner, D.D. P-selectin promotes neutrophil extracellular trap formation in mice. Blood 2015, 126, 242–246. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Blankenship, L.; Jaiyesimi, I.; Ataga, K.I.; Kutlar, A.; Kanter, J. Crizanlizumab in Sickle Cell Disease. N. Engl. J. Med. 2017, 376, 1795–1796. [Google Scholar] [CrossRef]
- Spangrude, G.J.; Lewandowski, D.; Martelli, F.; Marra, M.; Zingariello, M.; Sancillo, L.; Rana, R.A.; Migliaccio, A.R. P-Selectin Sustains Extramedullary Hematopoiesis in the Gata1low Model of Myelofibrosis. Stem Cells 2015, 34, 67–82. [Google Scholar] [CrossRef]
- Verachi, P.; Martelli, F.; Zingariello, M.; Chaturvedi, S.; Wilke, C.; Campello-Iddison, V.; Migliaccio, A.R. Preclinical Rationale for the Use of Crizanlizumab (SEG101) in Myelofibrosis. Blood 2020, 136, 26–27. [Google Scholar] [CrossRef]
- Sano, M.; Takahashi, R.; Ijichi, H.; Ishigaki, K.; Yamada, T.; Miyabayashi, K.; Kimura, G.; Mizuno, S.; Kato, H.; Fujiwara, H.; et al. Blocking VCAM-1 inhibits pancreatic tumour progression and cancer-associated thrombosis/thromboembolism. Gut 2020. [Google Scholar] [CrossRef] [PubMed]
- Khoy, K.; Mariotte, D.; Defer, G.; Petit, G.; Toutirais, O.; Le Mauff, B. Natalizumab in Multiple Sclerosis Treatment: From Biological Effects to Immune Monitoring. Front. Immunol. 2020, 11, 549842. [Google Scholar] [CrossRef] [PubMed]
- Ford, A.C.; Sandborn, W.J.; Khan, K.J.; Hanauer, S.B.; Talley, N.J.; Moayyedi, P. Efficacy of Biological Therapies in Inflammatory Bowel Disease: Systematic Review and Meta-Analysis. Am. J. Gastroenterol. 2011, 106, 644–659. [Google Scholar] [CrossRef]
- Krueger, J.G.; Ochs, H.D.; Patel, P.; Gilkerson, E.; Guttman-Yassky, E.; Dummer, W. Effect of Therapeutic Integrin (CD11a) Blockade with Efalizumab on Immune Responses to Model Antigens in Humans: Results of a Randomized, Single Blind Study. J. Investig. Dermatol. 2008, 128, 2615–2624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Major, E.O. Progressive Multifocal Leukoencephalopathy in Patients on Immunomodulatory Therapies. Annu. Rev. Med. 2010, 61, 35–47. [Google Scholar] [CrossRef] [Green Version]
- Bednarczyk, M.; Stege, H.; Grabbe, S.; Bros, M. β2 Integrins-Multi-Functional Leukocyte Receptors in Health and Disease. Int. J. Mol. Sci. 2020, 21, 1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craver, B.M.; Ramanathan, G.; Hoang, S.; Chang, X.; Luque, L.F.M.; Brooks, S.; Lai, H.Y.; Fleischman, A.G. N-acetylcysteine inhibits thrombosis in a murine model of myeloproliferative neoplasm. Blood Adv. 2020, 4, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Martinod, K.; Demers, M.; Fuchs, T.A.; Wong, S.L.; Brill, A.; Gallant, M.; Hu, J.; Wang, Y.; Wagner, D.D. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 8674–8679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Meyer, S.F.; Suidan, G.L.; Fuchs, T.A.; Monestier, M.; Wagner, D.D. Extracellular Chromatin Is an Important Mediator of Ischemic Stroke in Mice. Arter. Thromb. Vasc. Biol. 2012, 32, 1884–1891. [Google Scholar] [CrossRef] [Green Version]
- Hubbard, R.C.; McElvaney, N.G.; Birrer, P.; Shak, S.; Robinson, W.W.; Jolley, C.; Wu, M.; Chernick, M.S.; Crystal, R.G. A Preliminary Study of Aerosolized Recombinant Human Deoxyribonuclease I in the Treatment of Cystic Fibrosis. N. Engl. J. Med. 1992, 326, 812–815. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Hofbauer, T.M.; Ondracek, A.S.; Chausheva, S.; Alimohammadi, A.; Artner, T.; Panzenboeck, A.; Rinderer, J.; Shafran, I.H.; Mangold, A.; et al. Neutrophil extracellular traps promote fibrous vascular occlusions in chronic thrombosis. Blood 2021, 137, 1104–1116. [Google Scholar] [CrossRef] [PubMed]
- Hisada, Y.; Mackman, N. Update from the laboratory: Mechanistic studies of pathways of cancer-associated venous thrombosis using mouse models. Hematologica 2019, 2019, 182–186. [Google Scholar] [CrossRef]
- Jiménez-Alcázar, M.; Rangaswamy, C.; Panda, R.; Bitterling, J.; Simsek, Y.J.; Long, A.T.; Bilyy, R.; Krenn, V.; Renné, C.; Renné, T.; et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science 2017, 358, 1202–1206. [Google Scholar] [CrossRef] [Green Version]
- Cuenca, E.J.; Gisbert, N.G.; Reyes-García, A.M.D.L.; Aroca, C.; Martinez-Montesinos, L.; Andrade, M.; Aguila, S.; Guijarro-Carrillo, P.J.; Cifuentes-Riquelme, R.; González-Conejero, R.; et al. Hydroxyurea Reduces Neutrophil Extracellular Trap Formation in Myeloproliferative Neoplasms. Blood 2020, 136, 20–21. [Google Scholar] [CrossRef]
- Carestia, A.; Davis, R.P.; Grosjean, H.; Lau, M.W.; Jenne, C.N. Acetylsalicylic acid inhibits intravascular coagulation during Staphylococcus aureus–induced sepsis in mice. Blood 2020, 135, 1281–1286. [Google Scholar] [CrossRef]
- Han, X.; Mei, Y.; Schiltz, G.; Mishra, R.; Jain, A.; Ji, M.P. Targeting Pleckstrin-2 for the Treatment of Myeloproliferative Neoplasms. Blood 2020, 136, 10–11. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Lewis, H.D.; Liddle, J.; Coote, J.E.; Atkinson, S.J.; Barker, M.D.; Bax, B.D.; Bicker, K.L.; Bingham, R.P.; Campbell, M.; Chen, Y.H.; et al. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat. Chem. Biol. 2015, 11, 189–191. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wolach, O.; Shacham Abulafia, A. Can Novel Insights into the Pathogenesis of Myeloproliferative Neoplasm-Related Thrombosis Inform Novel Treatment Approaches? Hemato 2021, 2, 305-328. https://0-doi-org.brum.beds.ac.uk/10.3390/hemato2020018
AMA Style
Wolach O, Shacham Abulafia A. Can Novel Insights into the Pathogenesis of Myeloproliferative Neoplasm-Related Thrombosis Inform Novel Treatment Approaches? Hemato. 2021; 2(2):305-328. https://0-doi-org.brum.beds.ac.uk/10.3390/hemato2020018
Chicago/Turabian StyleWolach, Ofir, and Adi Shacham Abulafia. 2021. "Can Novel Insights into the Pathogenesis of Myeloproliferative Neoplasm-Related Thrombosis Inform Novel Treatment Approaches?" Hemato 2, no. 2: 305-328. https://0-doi-org.brum.beds.ac.uk/10.3390/hemato2020018