
Pathophysiology of Myelodysplastic Syndromes


1
Institut Cochin INSERM U1016, Université de Paris, CNRS UMR8104, 75014 Paris, France
2
Assistance Publique-Hôpitaux de Paris, Centre-Université de Paris, Hôpital Cochin, 75014 Paris, France
*
Author to whom correspondence should be addressed.
Hemato 2021, 2(3), 477-495; https://0-doi-org.brum.beds.ac.uk/10.3390/hemato2030030
Submission received: 31 May 2021
/
Revised: 19 July 2021
/
Accepted: 21 July 2021
/
Published: 26 July 2021
(This article belongs to the Special Issue Challenges in the Treatment of Myelodysplastic Syndrome)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Ineffective hematopoiesis is the major characteristic of early myelodysplastic syndromes. Its pathophysiology relies on a diversity of mechanisms supported by genetic events that develop in aging hematopoietic stem cells. Deletion and mutations trigger epigenetic modifications, and co-transcriptional and post-transcriptional deregulations of gene expression. Epistatic interactions between mutants may aggravate the phenotype. Amplification of minor subclones containing mutations that promote their growth and suppress the others drives the clonal evolution. Aging also participates in reprogramming the immune microenvironment towards an inflammatory state, which precedes the expansion of immunosuppressive cells such as Tregs and myeloid-derived suppressive cells that alters the anti-tumor response of effector cells. Integrating biomarkers of transcription/translation deregulation and immune contexture will help the design of personalized treatments.
1. Introduction
The myelodysplastic syndromes (MDS) are a heterogeneous group of clonal diseases of the hematopoietic stem cell (HSC) that can evolve into secondary acute myeloid leukaemia (AML). MDS patients with a lower risk of AML evolution (LR-MDS), require the treatment of their cytopenias while patients with high-risk MDS (HR-MDS) are expected for premptive therapies of disease progression and curative treatments of secondary leukemia [1,2]. Recurrent cytogenetic aberrations (5q deletion, deletion or monosomy 7, 20q deletion, trisomy 8), genetic mutations affecting epigenetic regulators (TET2, IDH1/2, DNMT3A, ASXL1, EZH2), splicing factors (SF3B1, SRSF2, U2AF1, ZRSR2), transcription factors (TP53, RUNX1), signaling adapters (NRAS, KRAS, JAK2, CBL, KIT) and cohesins (STAG2, SMC1, SMC3) and altered gene expression patterns have been linked to MDS and variably associated with MDS subtypes.
Although these gene mutations or deletions belong to a common set of genetic alterations in cancer, their contribution to disease initiation remains unclear, as the presence of mutations at low allelic burden without cytopenia or bone marrow dysplasia may precede for several years the onset of myeloid malignancies. Such a clonal hematopoiesis may develop in the context of HSC aging and the inflammatory state of the bone marrow microenvironment. Then, epistatic interactions between two or several genes within a clone and further competition between clones participate in disease initiation and progression.
2. Natural Course of MDS Disease
2.1. Cell of Origin
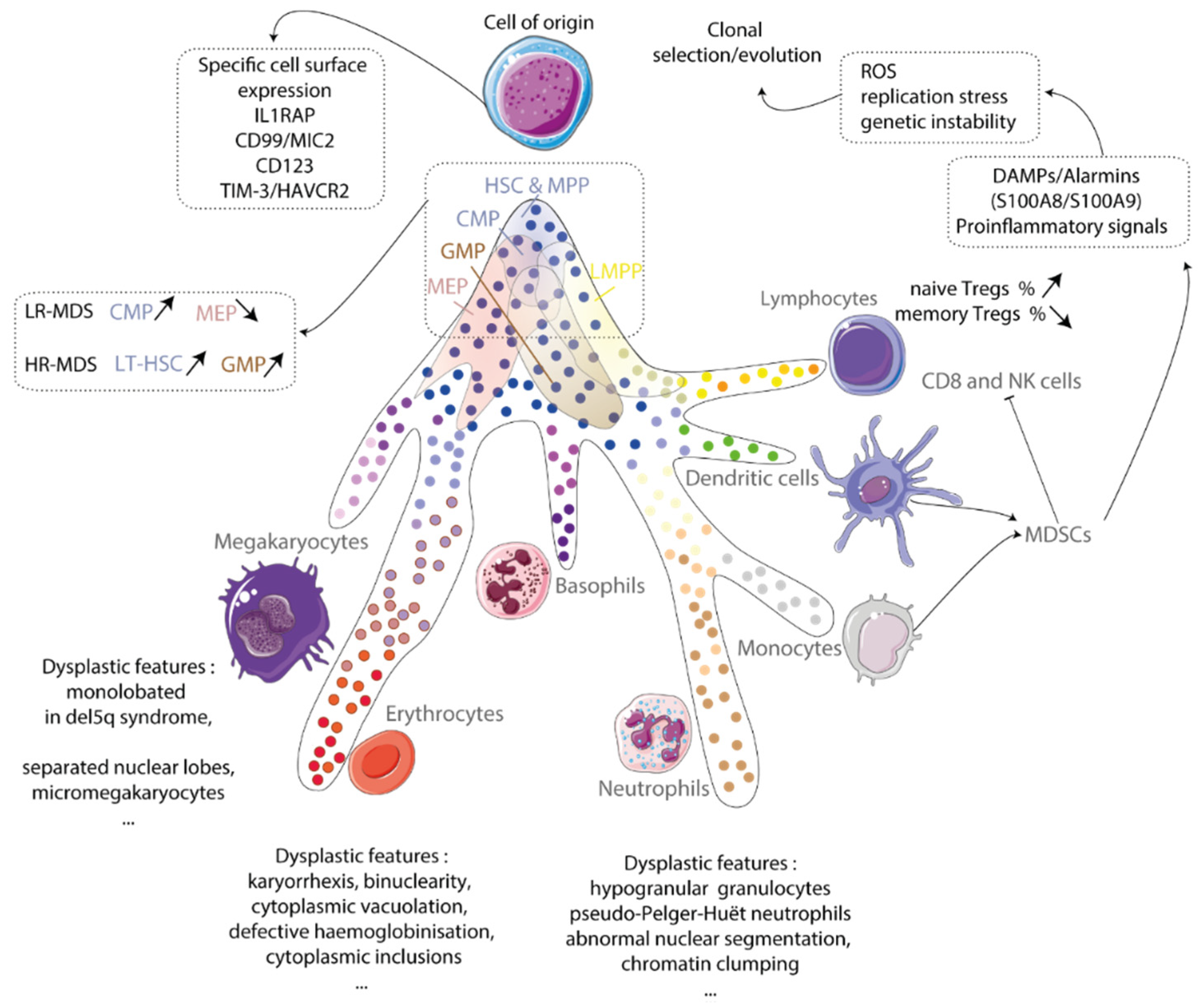
HSCs are immunophenotypically and functionally defined as Lin−/lo CD34+CD38− CD90+CD45RA− cells able to produce myeloid progenitors in vitro and reconstitute hematopoiesis in immunocompromised mice [3,4,5]. Recent works showed that MDS arise from a population of rare MDS cells identified by the presence of recurrent deletions or mutations, sharing these phenotypic features, engrafting into mice bone marrow in which clonal myeloid cells are predominant over lymphoid cells, demonstrating a myeloid-biased HSC [3,4,5,6]. MDS HSCs persist and expand under conventional therapies, therefore contributing to disease progression. Immunophenotyping of long-term (LT)-HSCs (Lin−CD34+CD38−CD90+), short-term HSCs (Lin−CD34+CD38−CD90−), common myeloid progenitor (CMP: Lin−CD34+CD38+CD123+CD45RA−), megakaryocyte erythroid progenitor (MEP: Lin−CD34+CD38+CD123−CD45RA−) and granulocyte monocyte progenitor (GMP: Lin−CD34+CD38+CD123+CD45RA+) compartments demonstrates quantitative anomalies with CMP expansion and MEP reduction in LR-MDS correlated with anemia and thrombocytopenia and LT-HSC and GMP expansion in HR-MDS [3,4,5,6,7]. While the pattern of mutations in the dominant HSC subclone is transmitted to its myeloid progeny, it may differ between CMP, GMP and MEP compartments depending on the functional impact of a given mutation on granulomonocytic or erythroid development [6,7].
The clonal hematopoietic stem and progenitor cells (HSPCs) can be recognized by cell surface expression of specific leukemic markers that may eventually stimulate anti-tumoral immune response. These include interleukin-1 (IL-1) receptor accessory protein (IL1RAP), CD99/MIC2, CD123/IL-3 receptor α chain and the negative regulatory immune checkpoint T-cell immunoglobulin mucin-3/Hepatitis A virus cellular receptor 2 (TIM-3/HAVCR2). IL1RAP is over-expressed on HSCs and GMPs in preleukemic and leukemic stem and progenitor cells [8]. CD99 is a cell surface marker overexpressed on clonal HSCs and CD34+CD38−CD90−CD45RA+ lymphoid-primed multipotent progenitors (LMPPs) known to harbor leukemic stem cell activity in AML [9], and also in high-risk MDS but not in LR-MDS [10,11]. CD123 is strongly expressed on leukemic CD34+/CD38− cells, and MDS CD34+ HSPCs [12]. Finally, TIM-3 usually expressed on innate immune cells and T-cells has been found aberrantly expressed on leukemic blasts in AML and MDS [13]. However, these signals generally block the immune response. Notably, expression of TIM-3 on leukemic cells increases their proliferation through an autocrine mechanism implicating the secretion of one of the TIM-3 ligands galectin-9 (Gal-9), and downstream activation of the β-catenin and NF-κB pathways [14]. Abundant release of Gal-9 can induce T-cell death by binding TIM-3 on T-cells, therefore suppressing T-cell immune response and inducing immune tolerance. Gal-9 is also overexpressed by mesenchymal stromal cells, leading to increased resistance to chemotherapy. Therefore, TIM-3 may be an interesting therapeutic target in AML and MDS (Figure 1).
2.2. Clonal Hematopoiesis
Clonal hematopoiesis and MDS arise with the acquisition of mutations in HSCs throughout life. The C→T transition is the most frequent base-pair change, a consequence of methyl-deamination associated with age [15]. Mutations at low variant allele frequency (VAF) could pre-exist the onset of hematological malignancies and their frequency increases with age in the general population. These mutations mostly affect the DNMT3A and the TET2 gene [16]. DNMT3A mutation has been recognized as a pre-leukemic lesion of HSC in AML because of its persistence at remission [17]. Exome sequencing of peripheral blood samples confirmed that the most frequently mutated genes in the elderly are DNMT3A, TET2, ASXL1, TP53, JAK2 and SF3B1, in 10% of individuals over the age of 65 years and more than 20% over the age of 90 years, suggesting pre-existing clonal hematopoiesis of indeterminate potential or CHIP [18,19,20]. Among aged individuals presenting anemia, DTA (for DNMT3A, TET2, ASXL1) mutations seem to be as frequent as in aged individuals without anemia while TP53 and SF3B1 mutations are significantly more frequent [21]. The presence of CHIP is a strong predictor of the development of subsequent hematologic malignancy as compared to age-matched controls [18,19], although the presence of small clones not growing with time might not be associated with an increased risk of death [21]. Importantly, a CHIP can also associate with an increased propensity to thrombosis, coronary artery disease and stroke, underlying the closed relationship between hematological malignancies and inflammatory diseases, the frequency of which increases in the elderly [22].
2.3. Early MDS
Early MDS emerges thanks to the amplification of a founder clone in HSC compartment followed by the onset of secondary mutations forming a linear or a branched architecture. In the former condition, a single founder clone acquires either a co-dominant mutation with similar VAF or a complex pattern of secondary mutations with lower VAFs. In the later conditions, two or more subclones with the common genetic background of the founder clone co-exist. Mechanistically, some types of mutations such as those targeting signaling adapters (KIT, CBL, JAK2, FLT3) drive the proliferation of clonal cells. In addition, autocrine or paracrine production of cytokines by the leukemic cells or stromal and immune cells further contribute to the clonal selection. The presence of cytopenias and bone marrow dysplasia, since it fulfills the morphologic criteria of the WHO classification together with one or several recurrent mutations, are consistent with the diagnosis of MDS. In the 2016 version of the WHO classification, the isolated deletion of chromosome 5q or the mutation in splicing factor 3B1 SF3B1 gene are included as diagnosis criteria, because they pinpoint distinct subtypes of MDS: the 5q syndrome with bone marrow erythroid hypoplasia and monolobated megakaryocytes and the MDS with ring sideroblasts (MDS-RS) with bone marrow erythroid hyperplasia, respectively [23]. Importantly, isolated del(5q) without TP53 mutation and MDS-RS with SF3B1 mutation, a normal karyotype or any cytogenetic abnormality other than del(5q), monosomy 7, inv(3) or abnormal 3q26, complex (≥3) and any additional somatically mutated gene other than RUNX1 or EZH2 are two distinct MDS entities of good prognosis [24]. Whole genome sequencing has revealed that MDS patients with a low blast count under 10% may have more than 60% of mutated cells embedded in one or two clones in their bone marrow [25]. The genetic lesions that initiate MDS promote self-renewal, leading to a proliferative advantage over normal HSCs, i.e., clonal expansion. Lin−CD34+CD38−CD90+ CD45RA− HSC compartment is enriched of cells with cytogenetic alterations and/or mutations compared with whole bone marrow [3]. Several studies have demonstrated that mutations in the epigenetic modifiers DTA occur early in the evolution of MDS, suggesting initiating events, while mutations in transcription factors or signaling adapters could rather be late events more probably affecting hematopoietic cell proliferation and differentiation [5,6,7]. Splicing factors gene mutations SF3B1, ZRSR2 and U2AF1 are also early events but less common in CHIP, suggesting that they account for a rapid emergence of dysplasia and MDS phenotype. Importantly, the order of mutation onset may drive the disease phenotype.
2.4. Clonal Evolution to AML
The emergence of a secondary AML is associated with the accumulation of multiple new mutations within the leukemic stem cell compartment, but also in myeloid progenitors outside the HSC compartment, the amplification of which is related to increased self-renewal capacities [5,6]. The investigation of clonal dynamics by whole-genome sequencing of paired samples at MDS and secondary AML stages showed that the progression to AML goes along with the persistence of the founding clone and the emergence of at least one additional subclone showing an increased genetic heterogeneity [25]. Whole exome and/or targeted sequencing of sequential samples also showed that during progression, the diversity of mutations and the size of clones increase, with an enrichment in FLT3, PTPN11, WT1, IDH1, NPM1, IDH2 and NRAS mutations associated with rapid progression and short survival, while TP53, GATA2, KRAS, RUNX1, STAG2, ASXL1, ZRSR2 and TET2 mutations may have a lower impact on AML progression and survival [26,27]. After disease-modifying therapy, the clonal architecture may become more complex with the emergence of unwanted new clones [27]. The number of driver mutations correlated with median leukemia-free survival, and an increasing number of driver mutations decreased survival significantly [28]. Among a set of 18 genes most commonly mutated in MDSs, mutations in TP53, EZH2, ETV6, RUNX1 and ASXL1 are independent predictors of poor survival in a large cohort of 439 patients [29]. Combining age, gender, Hb level, platelet count and cytogenetics with the mutational status of a set of 14 genes provides a prognostic model separating patients into four risk groups [30].
Recently, Steidl’s group revealed that stem cells at the MDS stage have a significantly higher complexity of subclonal mutations compared to blast cells using targeted deep sequencing of sorted stem cells premalignant MDS or AML stem cells (Lin−CD34+ CD38−CD45RA−CD123−IL1RAP−) in comparison to MDS or AML blast cells (Lin−CD34+ CD38−CD45RA+CD123+IL1RAP+). They also showed that subclonal mutations dramatically increase in size towards progression to sAML. Therefore, subclonal mutations at diagnosis must be considered for prediction models and for treatment strategy to eliminate MDS stem cells and disrupt disease maintenance [31].
The tumor suppressor gene TP53 encodes a transcription regulator involved in the maintenance of the balance between cell cycle, differentiation and apoptosis. Mutations in the TP53 gene occur in approximately 8–13% of MDS cases and are found at an early disease stage in 20% of MDS patients with del(5q) [32]. TP53 mutations frequently drive the poor prognosis of patients with complex karyotype defined as more than two cytogenetic abnormalities [33]. TP53 allelic state clearly identifies two groups of patients with distinct phenotypes and outcomes: one group is characterized by multiple hits in TP53 gene, a complex karyotype, few co-occurring mutations and an increased risk of death and leukemic transformation independently of other variables of the Revised International Prognostic Scoring System (IPSS-R); the other group of MDS patients has monoallelic mutations and do not differ from TP53 wild-type patients in terms of outcome and response to therapy [34]. Taken together, these studies establish that the recurrence and the link to disease stage indicate that genetic events influence the phenotype and outcome of the disease.
2.5. Epistatic Interactions of Genes Involved in Co-/Post-Transcriptional Regulation of Transcription
Studies of clonal architecture in MDS and sAML demonstrated the bone marrow oligoclonality, each clone containing one to several mutations [24]. Some mutations recurrently occur simultaneously, such as DNMT3A and SF3B1, TET2 and SRSF2 or IDH2 and SRSF2, and STAG2/RUNX1/SRSF2 and ASXL1, suggesting cooperating oncogenic mechanisms [5,6,25,26,30]. On the other hand, certain mutations co-occur much less often than expected by chance, which implies functional redundancy, for example, between TET2 and IDH1/2, or splicing factors or cohesins [27,29] (Figure 2). Studies in human and mice models of gene invalidation or mutation editing have demonstrated quantitative and qualitative alterations in stem and progenitor cell populations. However, the clone interactions are much more difficult to address. Patient-derived xenografts of HSPCs and the generation of induced pluripotent stem cells (iPSc) from patients with MDS will help in understanding how one given clone may overcome the others and how subclones raise during the course of the disease may drive the transformation. Interestingly, important studies addressed the mechanisms of oncogenic cooperation and revealed how biological pathways interconnect in pathological settings.
The first example is the cooperation between IDH2 and SRSF2 mutants. Abdel-Wahab’s group established that IDH2/SRSF2 double mutant cells, specifically, exhibit an aberrant splicing of a member of the Integrator complex, INTS3, that leads to its downregulated expression [35]. The mechanism implicates an “extra-spliceosome” function of SRSF2 in the co-transcriptional regulation of gene expression. During the transition from transcription initiation to elongation in most genes, RNA polymerase II (RNAPII) pauses stably with the 25–60 nt long nascent RNA while awaiting signals for release into the gene body. Conversion of promoter paused RNAPII into elongating polymerase depends on the transcription elongation factor b (P-TEFb), the activity of which is controlled mainly by the 7SK small nuclear ribonucleoprotein. 7SK sequesters active P-TEFb into inactive 7SK/P-TEFb snRNP [36]. SRSF2 associates with 7SK at gene promoters, mediates the release of P-TEFb from the 7SK complex and facilitates the recruitment of P-TEFb and other key transcription elongation factors to activate transcription [37]. However, in some genes, paused RNAPII actively destabilized by the Integrator complex dissociates from template DNA and releases a short, non-productive RNA [38]. Integrator utilizes its RNA endonuclease activity to cleave nascent RNA and drive premature transcription termination, potently attenuating the activity of target genes [38]. Mutant SRSF2 inhibits RNAPII pausing release [39] and attenuates INTS3 expression, causing a blockade of myeloid differentiation further enhanced in double IDH2/SRSF2 mutant cells [35].
A second example comes from the Ogawa’s group who demonstrated a functional interplay between cohesin STAG2 and transcription factor RUNX1 mutants. Both co-localize at enhancer-rich sites across the genome and synergistically attenuate enhancer-promoter loops, particularly at sites enriched for RNAPII, leading to deregulated gene expression, myeloid skewing of HSPC and MDS development in mice. Interestingly, genes with high transcriptional pausing were the most downregulated genes, including genes involved in interferon and inflammatory response pathways [40]. This highlights the pressure applied on immune microenvironment by malignant clones.
Altogether, these studies reveal that deregulation of co-transcriptional processes drives cell reprogramming and may influence interactions with tumor-associated immune cells.
3. Mechanisms of Cytopenias in Early MDS
3.1. Hematopoietic Stem Cell Aging
Hematopoietic stem cells numerically expand with age, but their functional activity declines over time, resulting in degraded blood production. Age-related defects in HSC function are characterized by altered survival, dormancy and regenerative capacity, and the age-related inflammatory microenvironment triggers the genetic insult and the onset of mutations that will further drive the development of the disease.
3.1.1. Aged HSC Functional Defects
Aged murine HSC have altered reconstitution and homing ability with an increased self-renewal under transplant conditions that impairs their lineage potential and their ability to differentiate [41,42,43]. The composition of the HSC compartment changes with age, with an increased frequency of myeloid-biased HSCs, a major increased of megakaryocytic-biased HSCs and a decrease in lymphoid-biased HSCs in which myeloid genes are more frequently expressed [44]. This means that HSCs divide more frequently and lose their multipotency [45,46]. In humans, donor age affects the recipient survival [47], suggesting that HSCs from aged donors may have reduced transplantation capacities. However, although several groups have shown that HSC frequency increases with age in humans [48,49], bone marrow reconstitution of immunocompromised mice by xenotransplant of young versus aged human HSCs demonstrates no decrease in engraftment, and a variable myeloid bias depending on whether when Lin−CD34+CD38−CD90+CD45RA− cells or CD34+CD38− cells were transplanted [48,49], highlighting the important role of the microenvironment.
3.1.2. Aged HSC Changes in Gene Expression Profiles
Changes in HSC pools are associated with modifications in gene expression patterns showing altered metabolism, decreased activation of autophagy, impaired proteostasis, accumulation of DNA damage and epigenetic remodeling. RNA sequencing data comparing aged versus young murine HSCs also identify changes in epigenetic regulator expression and alternative splicing [50]. In humans, RNA sequencing of young versus aged Lin−CD34+CD38− HSCs or Lin−CD34+CD38+ hematopoietic progenitor cells (HPC) also demonstrates metabolic changes such as decreased oxidative phosphorylation, proteostasis and DNA replication with deregulation of DNA repair. Inflammation pathways are activated in aged versus young HPCs [51]. Mechanistically, changes in gene expression profiles may be supported by changes in splicing gene isoforms. Notably, alternative splicing leading to a decrease in BCL2 pro-survival isoform is implicated in HPC aging while leukemic stem cell generation could be supported by an increased expression of a pro-survival isoform BCL-XL [51]. In addition, changes in myeloid transcription factor ETV6 and CEBPB expression suggest their contribution to myeloid bias of aging HSCs.
3.1.3. Epigenetic Changes with Aging in HSC
Studies of DNA methylation in aged human hematopoietic cells identified methylation changes in mononuclear cells and hypomethylation of differentiation-associated genes in CD34+ cells [52,53]. Histone modifications such as increases in H3K4me3, H3K36me3 and H3K27me3 chromatin marks and decreases in H3K27ac and H3K9ac marks associate with human HSC aging [54]. A recently published work of Figueroa’s group has compared, by chromatin immunoprecipitation with sequencing, activating histone modifications H3K4me1, H3K4me3 and H3K27ac and repressive mark H3K27me3 of young and aged human Lin−CD34+CD38−. This study identifies thousands of focal histone modification alterations related to aging [55]. Notably, in Lin−CD34+CD38− cells, promoters of the WNT signaling and developmental genes and promoters and enhancers associated with immune pathways are repressed, suggesting that increased risk of leukemias and immune impairment associated with age are epigenetically controlled. Consistently, transcription factors KLF6, BCL6 and RUNX3 and epigenetic modifiers KAT7, SETD1A and KDM2A are deregulated [55].
3.1.4. Replication Stress in Aged HSC
The accumulation of DNA damage eventually, through defective DNA repair, is a driver of HSC aging, contributing to malignant transformation. The physiological source of DNA damage in HSCs from both normal and diseased individuals is directly related to the exit of HSCs from their quiescent state into an activation state in response to physiological stress, such as infection or chronic blood loss. Repeated activation of HSCs results in the attrition of HSCs and a trend towards a reduction in hematopoiesis [56]. DNA replication stress has been recognized as a driver of HSC function alteration with aging: in old mice, HSCs entering the cell cycle exhibit features of replication stress eventually associated with chromosome break, and replication fork speed is reduced in correlation with a low expression of the helicase components of the replisome [57]. Interestingly, when old HSCs return to quiescence state, replication stress remains in the nucleolus, with γH2AX signals marking the transcriptional silencing of ribosomal DNA [57]. In MDS, the decline function of CD34+ cells could rely on focal replication stress, particularly in the context of splicing factor mutations, while a low transcription level of rRNA by increased methylation of the rDNA promoter is a common feature of all MDS subtypes [58,59].
3.2. Ineffective Erythropoiesis
Anemia is the most frequent symptom of MDS patients related to ineffective erythropoiesis (IE) in the bone marrow. IE, which is also commonly observed in other inherited erythroid disorders, results from a reduced capacity to produce mature differentiated erythroblasts hardly compensated by an increased proliferation of the more immature erythroblasts. Normal erythroid differentiation is predominantly marked by ribosome biogenesis, GATA1-regulated erythroid gene transcription controlling heme-globin balance and metabolic reprogramming, and negatively regulated by programmed cell death; each of these cellular processes are altered in MDS.
3.2.1. Excessive Cell Death
The Fas/Fas ligand pathway, which is normally activated when erythropoietin levels are low in an erythroblastic bone marrow, is over-activated in MDS, thus diminishing the expansion of erythroblastic islands even when the amounts of erythropoietin are elevated. Intrinsic mitochondrial pathway of apoptosis can be prevented by overexpression of anti-apoptotic Bcl-2 [60,61]. Other forms of inflammatory cell death such as necroptosis and pyroptosis, resulting in plasma membrane permeabilization and release of damage-associated molecular patterns (DAMPs) such as S100A8/S100A9 alarmins, which are found in excess in MDS plasma, are also reported [62]. Necroptosis requires the serine/threonine kinase activity of Ripk1 downstream of death receptors, mainly tumor necrosis factor (TNF) receptor, Fas, TNF-related apoptosis-inducing ligand (TRAIL) receptor and pattern recognition receptors (PRRs) Toll-like receptor (TLR)3 and TLR4. Mice with pro-apoptotic Bcl-2 family member deletion, in which apoptosis cascade is disrupted, develop an MDS-like disease related to the activation of TNF-α dependent necroptosis through the release of DAMPS that triggers the production of inflammatory cytokines in the bone marrow [63,64].
Pyroptosis, a caspase-1 dependent pro-inflammatory cell death induced by DAMPs, is a critical driver of HSPC cell death. In MDS bone marrow, alarmins are produced and secreted by myeloid-derived suppressive cells (MDSCs). Alarmins signal through TLR4 and CD33 and trigger activation of NADPH oxidase, leading to ROS production. Then, pyroptosis is executed through the production of pro-IL-1β by NF-κB and the formation of cytosolic inflammasome NLRP3 (NLR family pyrin domain-containing 3). When activated, inflammasome mediates the conversion of pro-caspase-1 into active caspase-1 which catalyzes the maturation of IL-1β and IL-18. The release of pro-inflammatory cytokines by the cells contribute to the inflammatory state of the microenvironment and a vicious cycle of cell death [65,66,67,68,69]. These exogenous signals trigger a genotoxic stress, leading to the clonal evolution of hematopoietic cells. In mice models mimicking defective ribosome biogenesis, pro-inflammatory alarmins S100a8/S100a9 are produced either by stromal osteoblastic cells or hematopoietic cells themselves, such as macrophages or erythroblasts. ROS accumulation participates in the genetic instability of HSC through induction of replication stress and DNA double strand breaks [70,71] (Figure 1). Mesenchymal stem cells (MSCs) interact with HSPCs through adhesion molecules and secrete factors contributing to HSPC maintenance, self-renewal and differentiation [72]. The inflammatory signaling from the niche, extensively reviewed elsewhere [73,74] may favor the occurrence and selection of clonal cells making MDS, a disease that affects the BM microenvironment.
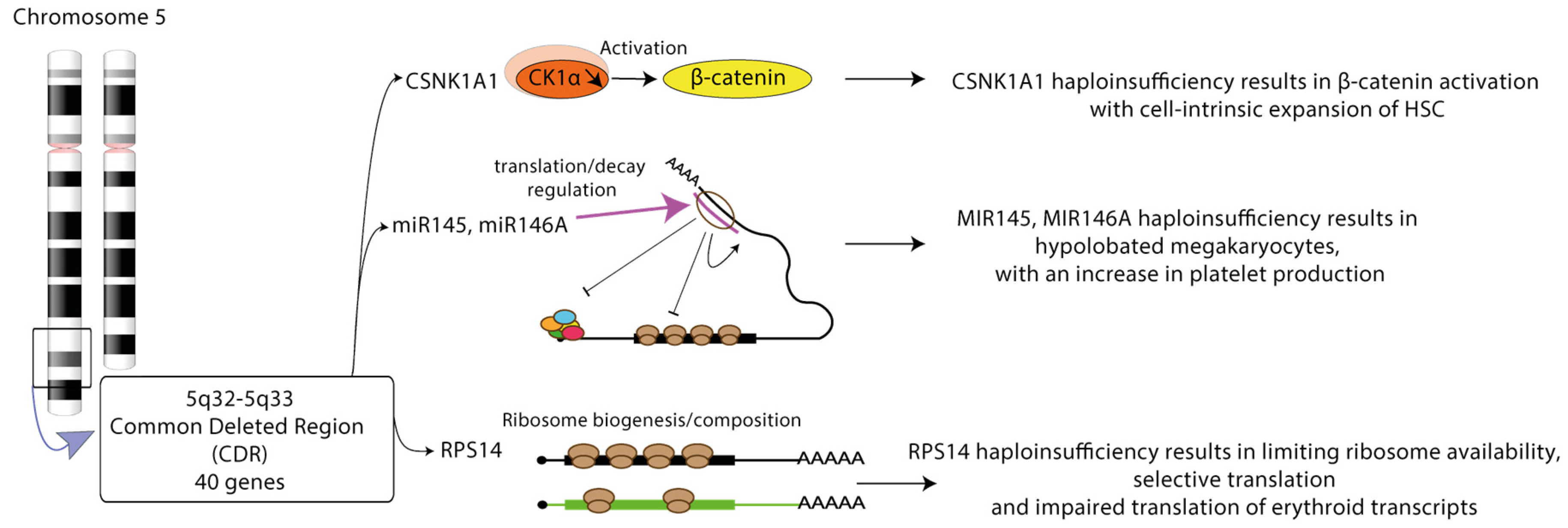
3.2.2. Post-Transcriptional Deregulation of Gene Expression in Del(5q) MDS
Isolated del(5q) is associated with a favorable prognosis, but when the deletion is located in the centromeric side of the common deleted region (CDR) or associated to other cytogenetic abnormalities or mutations, del(5q) MDS becomes a more aggressive disease, with an increased risk of transformation to AML. Phenotypically, del(5q) MDS patients present with a bone marrow hypoplasia, leading to anemia associated in some cases with thrombocytosis. The CDR contains 40 genes, most of them expressed in HSCs. The dyserythropoiesis likely occurs as result of haploinsufficiency of ribosomal protein 14 (RPS14) gene and casein kinase 1α (CSNK1A1) gene encoding a serine/threonine kinase. Haploinsufficiency of CSNK1A1 in murine models results in β-catenin activation, and cell-intrinsic expansion of HSCs [75]. Inactivation of CSNK1A1 s allele by mutation is linked to a poor prognosis. Casein kinase 1 inhibition or its degradation by lenalidomide, the standard treatment of del(5q) MDS, leads to cell apoptosis [76]. Two miRNAs, miR-145 and miR-146a can be lost in humans and their knockdown in mouse HSPCs results in the megakaryocytic phenotype of 5q-syndrome. Their respective targets Toll-interleukin-1 receptor domain-containing adaptor protein (TIRAP) and tumor necrosis factor receptor-associated factor-6 (TRAF6) are involved in innate immune signaling. Their loss in mouse macrophages together with Rps14 and Csnk1a1 invalidation induces S100a8 production in the niche [77,78] (Figure 3).
Given that RPS14 is a component of the 40S ribosomal subunit, the haploinsufficiency of its gene impairs ribosome biogenesis (RiBi), which is crucial for support of normal erythroid cell growth and proliferation and globin synthesis [79]. In normal erythropoiesis, RiBi progressively interrupted from the stage of late basophilic erythroblasts with a balanced diminution of ribosomal proteins renewal, global decrease in protein synthesis and translation selectivity toward erythroid transcripts [80]. A p53 activation may orchestrate the transition from proliferation to differentiation. p53 chromatin immunoprecipitation and sequencing in immature erythroblasts demonstrates the establishment of p53-dependent gene expression profile involving cell cycle arrest, DNA repair and myeloid differentiation, but not apoptosis [80]. By contrast, RiBi is unbalanced, with twice the elevated production of one subunit compared to the other in del(5q) MDS or Diamond-Blackfan anemia (DBA) with heterozygous mutation of ribosomal protein gene. RiBi collapses precociously, leading to the stabilization and over-activation of p53 and the induction of cell cycle arrest and apoptosis [81,82]. Crossing syntenic del(5q) mice with TP53 knock-out mice rescues the apoptotic phenotype [83].
GATA1 is the master erythroid transcription factor controlling erythroid maturation through the transcription of EPO-R and BCL2L1 (BCL-XL), genes involved in heme biosynthesis such as ALAS2 (5’-aminolevulinate synthase 2), HRI (heme-regulated inhibitor or heme-regulated eIF2α kinase (HRI) and globins. GATA1 is post-transcriptionally impaired in MDS with dyserythropoiesis by an inappropriate cleavage by caspases [84]. Its chaperone, HSP70, either not expressed in DBA [85] or absent from the nucleus in MDS [84], does not protect GATA1 against cleavage. Furthermore, in conditions of low ribosome availability, protein translation becomes selective at the expense of GATA-1 and other erythroid transcripts, which are critical for the completion of erythroid differentiation [86,87]. This results in lower globin synthesis and excess of free heme, which contributes to ROS production and oxidative stress [88]. Although the common characteristics of the transcripts less translated in conditions of low ribosome availability have been established, whether the reduction in the translation efficiency of these transcripts depends on their cytosolic sequestration or impaired ribosome recycling remains to be determined (Figure 2).
3.2.3. Co-Transcriptional and Post-Transcriptional Deregulation of Gene Expression by Mutant Splicing Factors
DNA Replication Stress in Splicing Factor Mutated MDS
Splicing factors are involved in the co-transcriptional regulation of gene expression. SRSF2 is involved in RNA polymerase II pausing release [36]. Mutation in this factor induces a defect of pausing release favoring the formation of unscheduled R-loops [38]. These triple-strand structures are formed of a DNA:RNA hybrid due to the hybridization of neosynthesized RNA with the template DNA and a displaced DNA strand. They form obstacles to DNA replication, leading to replication fork stalling, single-strand DNA exposure and eventually double-strand breaks. Additional works suggested that splicing factor-mutated CD34+ progenitors may exhibit enhanced R-loops and DNA damage although technical issues may exist [58,89].
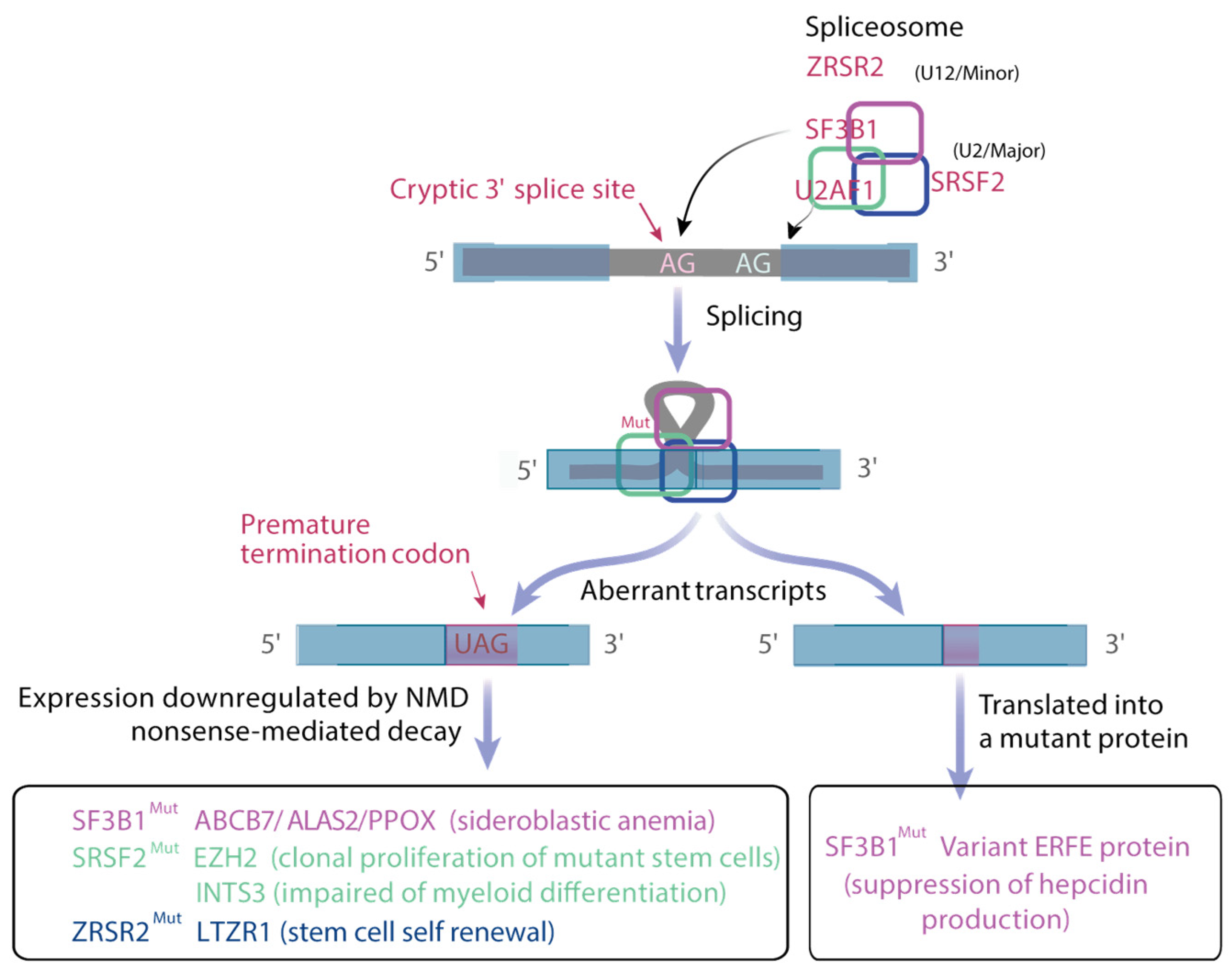
Deregulation of the Splicing Machinery
RNA splicing is a nuclear process accomplished by more than 200 RNA binding proteins (RBP), splicing proteins and small nuclear ribonucleoproteins (snRNP). In snRNP complexes, small nuclear RNAs hybridize with dinucleotides GU and AG at the 5′ and 3′ splice sites (ss), respectively, and with the branchpoint (BP) sequence (usually an A) in the intron. The polypyrimidine tract closed to the BP promotes its recognition by the spliceosome. RBPs bind splicing enhancers and silencers to modulate spliceosome assembly and promote or inhibit splicing. The U2 snRNP complex associates SF3B1, p14, SF3B3 and U2 snRNA and participates in the recognition of the BP. Mutations in the SF3B1 gene affecting its HEAT repeat domains that align as a platform to interact with pre-mRNA cause the recognition of an ectopic BP, leading to the use of an alternative 3′ ss [90,91]. Mutations in U2AF1, SRSF2 and ZRSR2 genes have distinct consequences. U2AF1, in complex with U2AF2, recruits the U2 snRNP to the BP. U2AF1 binds the AG dinucleotide at the 3′ ss, whereas U2AF2 binds the polypyrimidine tract. U2AF1 is affected by hotspot mutations at the S34 and Q157 residues, each of which occurs in one of its two zinc fingers. Aberrant splicing driven by mutant U2AF1 occurs in sequences surrounding the AG dinucleotide at the 3′ ss [92]. For instance, U2AF1 S34 mutants promote inclusion of exon cassette at the 3′ ss containing a C-nucleotide at −3 position. SRSF2 is a member of the serine/arginine-rich (SR) family of RBPs that binds splicing enhancers in exons to recruit the core spliceosome. SRSF2 binds CCNG and GGNG sequences equally well to promote splicing. P95 SRSF2 mutants have a reduced affinity for G-rich sequences promoting preferential splicing of C-rich sequences [93,94]. Interestingly, mutations in the SRSF2 gene promote the expression of an unannotated isoform of EZH2 due to the presence of C-rich exon splicing enhancers within a weak exon that is usually skipped. This retained exon contains a premature termination codon, suggesting that this EZH2 mRNA undergoes NMD [93,94]. EZH2 loss of functionally promotes myeloid malignancy development in vivo whereas restoration of EZH2 expression rescues the impaired hematopoiesis of mutant SRSF2 [93]. Less than 1% of introns with distinct 5′ and 3′ ss are recognized by the minor U12 spliceosome containing ZRSR2. Loss-of-function mutations in ZRSR2 globally increase minor intron retention in U12-type transcripts such as leucine zipper-like transcription regulator 1 LTZR1 and results in enhancement of HSC renewal [95] (Figure 4).
Defective Metabolic Reprogramming
Mutations in splicing factors strongly affect the phenotype of MDS patients. In historical cohorts, patients with SRSF2 mutation exhibit neutropenia and eventual pronounced thrombocytopenia, and patients with ZRSR2 mutation had high percentages of bone marrow blasts and displayed neutropenia [96,97]. The relationship between unusual splicing events and disease phenotype often remains unclear.
Patients with SF3B1 mutation present with low hemoglobin levels as expected in regard to major dyserythropoiesis related to the presence of ringed-sideroblasts defined by a mitochondrial iron overload. During normal erythropoiesis, immature erythroid precursors, which are characterized by high mitochondrial oxidative phosphorylation (OXPHOS), undergo metabolic reprogramming toward a reduction in OXPHOS, ROS production and mitochondrial mass, which are necessary for terminal maturation and enucleation. In SF3B1-mutated MDS, gene expression profiling indicates that iron metabolism and heme biosynthesis are impaired. Multiple changes in splicing are responsible for the down-regulation of numerous transcripts involved in mitochondrial metabolism regulation, heme biosynthesis (ALAS2), iron/sulfur cluster export (ABCB7), sirtuin signaling pathway and OXPHOS through NMD [90,98]. Therefore, sideroblastic anemia is an emblematic example of aberrant metabolic reprogramming.
In SF3B1-mutated MDS, systemic hyperferritinemia is known to precede red blood cell transfusion and to expose the risk of parenchymal iron overload. Alternative 3′ss use may create neojunction if the sequence is kept in frame. In these conditions, RNAs could be translated into neo proteins. Our data identified a variant erythroferrone (ERFE) encoding a variant protein, which contributes to overproduction of ERFE, the transcriptional repressor of hyposideremic hormone hepcidin, in SF3B1-mutated MDS. Excessive ERFE is an independent predictor of systemic hyperferritinemia in low transfusion burden patients [99].
Distinct mechanisms account for ineffective erythropoiesis in SF3B1-mutated MDS or RPS14-deficient MDS, arguing for a personalized management of anemia.
4. Immune Dysregulation in MDS
Immunity in Aging
Aging is defined by an impaired immune response affecting the adaptive response more than the innate immune response. This translates into an increased susceptibility to bacterial and viral infections. Both T and B cell lineages are altered. The T cell repertoire is restricted, and naïve and memory T CD8 cells expand oligoclonally. B cells show reduced class-switch recombination and antibodies produced by B cells have a reduced specificity and affinity for antigens. Immunosenescence also relies on impaired macrophage function, impaired cytokines and chemokines production.
Low-risk MDS development is accompanied by expansion of immune-suppressive cells such as Tregs and MDSCs of granulocytic origin (CD33 and CD15/CD66b with low or no HLA-DR levels) or monocytic origin (CD11b, CD33 and CD14 and lacking or low expression levels of HLA-DR) that result in a defective immune response and compromised immune surveillance and favor the expansion of the malignant clone [62,100,101,102]. This is consistent with a reduced number and altered function of peripheral T CD8 cells and natural killer (NK) cells and also of bone marrow dendritic cells (DC) subsets, including slan+ monocytes, of which capacities to induce T cell proliferation are impaired [103].
The proliferative capacities of Tregs are reduced during the early steps of the disease and restored during disease progression. Tregs activation and/or expansion meaning the phenotypic features of Treg subsets including naïve, central memory and effector memory cells change during the progression of MDS to leukemia. In MDS, naïve CD4+CD25hiFOXP3+CD27+CD45RO− Tregs significantly increase in higher-risk MDS with a trend towards a decrease in memory CD4+CD25hiFOXP3+CD27+CD45RO+ [100]. In another study, MDS patients when acquiring a higher percentage of bone marrow myeloblasts exhibited more Tregs with an activated suppressive effector memory phenotype (CD3+CD4+FOXP3+CD25+CD127dimCD27−CD45RA−), while the majority of peripheral Tregs in healthy individuals display a central (long-term) memory inactive phenotype (CD3+CD4+FOXP3+CD25+ CD127dimCD27+CD45RA−) [104]. Altogether, these data reflect a transition from central memory to effector memory phenotype, which are thought to exhibit higher immunosuppressive functions of the T cell anti-tumor response.
NK cell function is determined by a balance of stimulatory (NKp30, NKp46, NKG2D) and inhibitory (KIR and NKG2A) receptors. Stimulatory receptors recognized stress-induced and tumor-induced ligand. Inhibitory receptors sense the MHC class I molecules expression to eliminate tumor variants. Most studies found a decrease in cytotoxicity, proliferation and increased apoptosis of peripheral NK cells without changes in inhibitory or stimulatory receptors or with a lower frequency of NKG2D+ NK cells [105]. Overall, either NK cells are lacking in high-risk MDS, or NK cells are predominantly composed of immature phenotypes such as NKG2AKIR CD56 NK cells, with poor granules and immature NK cell receptor repertoire [106,107]. MDSC expansion and activation have been associated with cancer and impaired immune effector cell function, including NK cells.
No fortuitous association between MDS and systemic inflammatory and auto-immune diseases (SIAD) is largely reported [108,109], suggesting that chronic inflammation driven by a dysregulated myeloid-driven innate immune response of autoimmunity, which associates autoreactive T cells and possibly autoantibodies, is related to MDS pathophysiology. In a recent study, TET2/IDH mutations associated or not with SRSF2 mutation are the most frequent genetic events in MDS with SIAD. Furthermore, MDS patients with SIAD have a reduced CD4+CD25hiCD127lo Treg population, and a reduction in the stem cell T CD8+ memory cells and T central memory (TCM) subsets. TCM is significantly more decreased in TET2/IDH-mutated patients. Immune-checkpoint receptors (ICR) play a major role in suppressing autoimmunity and maintaining self-tolerance. Assessing ICR expression, the study showed that TCM CD8+CD96+ cells are reduced in TET2/IDH-mutated patients. TET2/IDH mutation deeply modifies Treg and CD8+ T-cell subsets distribution with a disruption of naive/memory balance [110]. Other studies showed elevated PD1+ and Tim-3+ T-cell frequencies in MDS compared to patients in remission, which increase further in MDS at relapse [111]. An increased expression of PD-1 on HSPCs and PD-L1 on MDSCs is reported in MDS in comparison to healthy donors [112]. Tim-3 expression levels increase on MDS CD45/SS or CD34/CD45-gated blasts as MDS progress to advanced stages [13]. Finally, PD-L1+ CD34+ CD38− and PD-L1+ CD34+ CD38+ stem cell frequencies increase in MDS patients compared to stem cell recipients in remission [112]. Interestingly, MDSCs in MDS patients express CD155 that ligates the T-cell immunoreceptor with immunoglobulin and ITIM domain (TIGIT) and delivers an inhibitory signal to natural killer (NK) cells, suggesting that overcoming the TIGIT negative regulatory checkpoint could be a strategy to rescue a productive immune response [113]. Taken together, these studies establish the role of immune dysregulation in MDS pathogenesis.
5. Conclusions
Studies conducted during the past few decades have highlighted the complexity of MDS pathophysiology involving the genetic diversity of clones, inflammatory conditions related to aging and the impaired response of the immune environment. Although a unifying mechanism of disease initiation has not yet been identified, epistatic interactions between mutated genes usually result in alterations in transcription regulatory pathways. Combining the specific targeting of transcriptional regulators with immune checkpoint inhibitors may successfully implement the treatment of fitted MDS patients. This implies to identify and prospectively validate composite biomarkers as a way of personalizing patient management.
Funding
This research is funded by HORIZON 2020 research and innovation program MDS-RIGHT under grant agreement No. 634789, Institut National du Cancer Site de Recherche Intégrée sur le Cancer CARPEM under grant agreement No. 6039.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors acknowledge Valeria Santini, Azienda Ospedaliero Universitaria Careggi, University of Florence, Italy; Eric Solary, INSERM U1287, Villejuif, France; and Lionel Adès, Service d’hématologie senior, Hôpital Saint-Louis, Paris, France, for very helpful discussions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fenaux, P.; Platzbecker, U.; Ades, L. How we manage adults with myelodysplastic syndrome. Br. J. Haematol. 2020, 189, 1016–1027. [Google Scholar] [CrossRef]
- Santini, V. How I treat MDS after hypomethylating agent failure. Blood 2019, 133, 521–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Will, B.; Zhou, L.; Vogler, T.O.; Ben-Neriah, S.; Schinke, C.; Tamari, R.; Yu, Y.; Bhagat, T.D.; Bhattacharyya, S.; Barreyro, L.; et al. Stem and progenitor cells in myelodysplastic syndromes show aberrant stage-specific expansion and harbor genetic and epigenetic alterations. Blood 2012, 120, 2076–2086. [Google Scholar] [CrossRef]
- Pang, W.W.; Pluvinage, J.V.; Price, E.A.; Sridhar, K.; Arber, D.A.; Greenberg, P.L.; Schrier, S.L.; Park, C.Y.; Weissman, I.L. Hematopoietic stem cell and progenitor cell mechanisms in myelodysplastic syndromes. Proc. Natl. Acad. Sci. USA 2013, 110, 3011–3016. [Google Scholar] [CrossRef] [Green Version]
- Woll, P.S.; Kjällquist, U.; Chowdhury, O.; Doolittle, H.; Wedge, D.; Thongjuea, S.; Erlandsson, R.; Ngara, M.; Anderson, K.; Deng, Q.; et al. Myelodysplastic Syndromes Are Propagated by Rare and Distinct Human Cancer Stem Cells In Vivo. Cancer Cell 2014, 25, 794–808. [Google Scholar] [CrossRef] [Green Version]
- Chesnais, V.; Arcangeli, M.-L.; Delette, C.; Rousseau, A.; Guermouche, H.; Lefevre, C.; Bondu, S.; Diop, M.; Cheok, M.; Chapuis, N.; et al. Architectural and functional heterogeneity of hematopoietic stem/progenitor cells in non-del(5q) myelodysplastic syndromes. Blood 2017, 129, 484–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mian, S.A.; Rouault-Pierre, K.; Smith, A.E.; Seidl, T.; Pizzitola, I.; Kizilors, A.; Kulasekararaj, A.G.; Bonnet, D.; Mufti, G.J. SF3B1 mutant MDS-initiating cells may arise from the haematopoietic stem cell compartment. Nat. Commun. 2015, 6, 10004. [Google Scholar] [CrossRef] [PubMed]
- Barreyro, L.; Will, B.; Bartholdy, B.; Zhou, L.; Todorova, T.I.; Stanley, R.F.; Ben-Neriah, S.; Montagna, C.; Parekh, S.; Pellagatti, A.; et al. Overexpression of IL-1 receptor accessory protein in stem and progenitor cells and outcome correlation in AML and MDS. Blood 2012, 120, 1290–1298. [Google Scholar] [CrossRef] [Green Version]
- Goardon, N.; Marchi, E.; Atzberger, A.; Quek, L.; Schuh, A.; Soneji, S.; Woll, P.S.; Mead, A.; Alford, K.A.; Rout, R.; et al. Coexistence of LMPP-like and GMP-like Leukemia Stem Cells in Acute Myeloid Leukemia. Cancer Cell 2011, 19, 138–152. [Google Scholar] [CrossRef] [Green Version]
- Shastri, A.; Will, B.; Steidl, U.; Verma, A. Stem and progenitor cell alterations in myelodysplastic syndromes. Blood 2017, 129, 1586–1594. [Google Scholar] [CrossRef]
- Chung, S.S.; Eng, W.S.; Hu, W.; Khalaj, M.; Garrett-Bakelman, F.E.; Tavakkoli, M.; Levine, R.L.; Carroll, M.; Klimek, V.M.; Melnick, A.M.; et al. CD99 is a therapeutic target on disease stem cells in myeloid malignancies. Sci. Transl. Med. 2017, 9, eaaj2025. [Google Scholar] [CrossRef] [Green Version]
- Li, L.J.; Tao, J.L.; Fu, R.; Wang, H.Q.; Jiang, H.J.; Yue, L.; Zhang, W.; Liu, H.; Shao, Z.H. Increased CD34+CD38−CD123+ cells in myelodysplastic syndrome displaying malignant features similar to those in AML. Int. J. Hematol. 2014, 100, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Asayama, T.; Tamura, H.; Ishibashi, M.; Kuribayashi-Hamada, Y.; Onodera-Kondo, A.; Okuyama, N.; Yamada, A.; Shimizu, M.; Moriya, K.; Takahashi, H.; et al. Functional expression of Tim-3 on blasts and clinical impact of its ligand galectin-9 in myelodysplastic syndromes. Oncotarget 2017, 8, 88904–88917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikushige, Y.; Shima, T.; Takayanagi, S.I.; Urata, S.; Miyamoto, T.; Iwasaki, H.; Takenaka, K.; Teshima, T.; Tanaka, T.; Inagaki, Y.; et al. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell 2010, 7, 708–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busque, L.; Patel, J.P.; Figueroa, M.E.; Vasanthakumar, A.; Provost, S.; Hamilou, Z.; Mollica, L.; Li, J.; Viale, A.; Heguy, A.; et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat. Genet. 2012, 44, 1179–1181. [Google Scholar] [CrossRef]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haemato-poietic stem cells in acute leukaemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef]
- Genovese, G.; Kähler, A.K.; Handsaker, R.; Lindberg, J.; Rose, S.; Bakhoum, S.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef]
- Van Zeventer, I.A.; de Graaf, A.O.; Wouters, H.J.C.M.; van der Reijden, B.A.; van der Klauw, M.M.; de Witte, T.; Jonker, M.A.; Malcovati, L.; Jansen, J.H.; Huls, G. Mutational spectrum and dynamics of clonal hematopoiesis in anemia of older individuals. Blood 2020, 135, 1161–1170. [Google Scholar] [CrossRef]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Athero-sclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Malcovati, L.; Stevenson, K.; Papaemmanuil, E.; Neuberg, D.; Bejar, R.; Boultwood, J.; Bowen, D.T.; Campbell, P.J.; Ebert, B.L.; Fenaux, P.; et al. SF3B1-mutant MDS as a distinct disease subtype: A proposal from the International Working Group for the Prognosis of MDS. Blood 2020, 136, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.J.; Shen, D.; Ding, L.; Shao, J.; Koboldt, D.C.; Chen, K.; Larson, D.E.; McLellan, M.D.; Dooling, D.; Abbott, R.; et al. Clonal architecture of secondary acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1090–1098. [Google Scholar] [CrossRef] [Green Version]
- Makishima, H.; Yoshizato, T.; Yoshida, K.; Sekeres, M.A.; Radivoyevitch, T.; Suzuki, H.; Przychodzen, B.; Nagata, Y.; Meggendorfer, M.; Sanada, M.; et al. Dynamics of clonal evolution in myelodysplastic syndromes. Nat. Genet. 2016, 49, 204–212. [Google Scholar] [CrossRef]
- Da Silva-Coelho, P.; Kroeze, L.I.; Yoshida, K.; Koorenhof-Scheele, T.N.; Knops, R.; van de Locht, L.T.; de Graaf, A.O.; Massop, M.; Sandmann, S.; Dugas, M.; et al. Clonal evolution in myel-odysplastic syndromes. Nat. Commun. 2017, 8, 15099. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical effect of point mutations in myelodysplastic syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2013, 28, 241–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Kao, Y.-R.; Sun, D.; Todorova, T.I.; Reynolds, D.; Narayanagari, S.-R.; Montagna, C.; Will, B.; Verma, A.; Steidl, A.U. Myelodysplastic syndrome progression to acute myeloid leukemia at the stem cell level. Nat. Med. 2018, 25, 103–110. [Google Scholar] [CrossRef]
- Jaedersten, M.; Saft, L.; Smith, A.; Kulasekararaj, A.; Pomplun, S.; Goehring, G.; Hedlund, A.; Hast, R.; Schlegelberger, B.; Porwit, A.; et al. TP53 Mutations in Low-Risk Myelodysplastic Syndromes With del(5q) Predict Disease Progression. J. Clin. Oncol. 2011, 29, 1971–1979. [Google Scholar] [CrossRef] [PubMed]
- Haase, D.; Stevenson, K.E.; Neuberg, D.; Maciejewski, J.P.; Nazha, A.; Sekeres, M.; Ebert, B.L.; Garcia-Manero, G.; Haferlach, C.; Haferlach, T.; et al. TP53 mutation status divides myelodysplastic syndromes with complex karyotypes into distinct prognostic subgroups. Leukemia 2019, 33, 1747–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.; Malcovati, L.; et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat. Med. 2020, 26, 1549–1556. [Google Scholar] [CrossRef] [PubMed]
- Yoshimi, A.; Lin, K.-T.; Wiseman, D.; Rahman, M.A.; Pastore, A.; Wang, B.; Lee, S.; Micol, J.-B.; Zhang, X.J.; De Botton, S.; et al. Coordinated alterations in RNA splicing and epigenetic regulation drive leukaemogenesis. Nat. Cell Biol. 2019, 574, 273–277. [Google Scholar] [CrossRef]
- Peterlin, B.M.; Price, D.H. Controlling the Elongation Phase of Transcription with P-TEFb. Mol. Cell 2006, 23, 297–305. [Google Scholar] [CrossRef]
- Ji, X.; Zhou, Y.; Pandit, S.; Huang, J.; Li, H.; Lin, C.Y.; Xiao, R.; Burge, C.B.; Fu, X.D. SR proteins collaborate with 7SK and promoter-associated nascent RNA to release paused polymerase. Cell 2013, 153, 855–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elrod, N.D.; Henriques, T.; Huang, K.-L.; Tatomer, D.C.; Wilusz, J.; Wagner, E.J.; Adelman, K. The Integrator Complex Attenuates Promoter-Proximal Transcription at Protein-Coding Genes. Mol. Cell 2019, 76, 738–752.e7. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, J.Y.; Huang, Y.J.; Gu, Y.; Qiu, J.; Qian, H.; Shao, C.; Zhang, X.; Hu, J.; Li, H.; et al. The augmented R-loop is a unifying mechanism for myelodys-plastic syndromes induced by high-risk splicing factor mutations. Mol. Cell 2018, 69, 412–425. [Google Scholar] [CrossRef] [Green Version]
- Ochi, Y.; Kon, A.; Sakata, T.; Nakagawa, M.M.; Nakazawa, N.; Kakuta, M.; Kataoka, K.; Koseki, H.; Nakayama, M.; Morishita, D.; et al. Combined Cohesin-RUNX1 Deficiency Syner-gistically Perturbs Chromatin Looping and Causes Myelodysplastic Syndromes. Cancer Discov. 2020, 10, 836–853. [Google Scholar] [CrossRef] [Green Version]
- Morrison, S.; Wandycz, A.M.; Akashi, K.; Globerson, A.; Weissman, I.L. The aging of hematopoietic stem cells. Nat. Med. 1996, 2, 1011–1016. [Google Scholar] [CrossRef]
- Nakamura-Ishizu, A.; Suda, T. Aging of the hematopoietic stem cells niche. Int. J. Hematol. 2014, 100, 317–325. [Google Scholar] [CrossRef] [Green Version]
- Chambers, S.M.; Shaw, C.; Gatza, C.; Fisk, C.J.; Donehower, L.A.; A Goodell, M. Aging Hematopoietic Stem Cells Decline in Function and Exhibit Epigenetic Dysregulation. PLoS Biol. 2007, 5, e201. [Google Scholar] [CrossRef]
- Beerman, I.; Bhattacharya, D.; Zandi, S.; Sigvardsson, M.; Weissman, I.L.; Bryder, D.; Rossi, D.J. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc. Natl. Acad. Sci. USA 2010, 107, 5465–5470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernitz, J.; Kim, H.S.; MacArthur, B.; Sieburg, H.; Moore, K. Hematopoietic Stem Cells Count and Remember Self-Renewal Divisions. Cell 2016, 167, 1296–1309.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Säwen, P.; Eldeeb, M.; Erlandsson, E.; Kristiansen, T.A.; Laterza, C.; Kokaia, Z.; Karlsson, G.; Yuan, J.; Soneji, S.; Mandal, P.K.; et al. Murine HSCs contribute actively to native hematopoiesis but with reduced differentiation capacity upon aging. Elife 2018, 7, e41258. [Google Scholar] [CrossRef]
- Kollman, C.; Howe, C.W.S.; Anasetti, C.; Antin, J.H.; Davies, S.M.; Filipovich, A.H.; Hegland, J.; Kamani, N.; Kernan, N.; King, R.; et al. Donor characteristics as risk factors in recipients after transplantation of bone marrow from unrelated donors: The effect of donor age. Blood 2001, 98, 2043–2051. [Google Scholar] [CrossRef] [PubMed]
- Pang, W.W.; Price, E.A.; Sahoo, D.; Beerman, I.; Maloney, W.J.; Rossi, D.J.; Schrier, S.L.; Weissman, I.L. Human bone marrow hematopoietic stem cells are increased in frequency and mye-loid-biased with age. Proc. Natl. Acad. Sci. USA 2011, 108, 20012–20017. [Google Scholar] [CrossRef] [Green Version]
- Kuranda, K.; Vargaftig, J.; De La Rochère, P.; Dosquet, C.; Charron, D.; Bardin, F.; Tonnelle, C.; Bonnet, D.; Goodhardt, M. Age-related changes in human hematopoietic stem/progenitor cells. Aging Cell 2011, 10, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Luo, M.; Jeong, M.; Rodriguez, B.; Xia, Z.; Hannah, R.; Wang, H.; Le, T.; Faull, K.F.; Chen, R.; et al. Epigenomic Profiling of Young and Aged HSCs Reveals Concerted Changes during Aging that Reinforce Self-Renewal. Cell Stem Cell 2014, 14, 673–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crews, L.A.; Balaian, L.; Santos, N.D.; Leu, H.S.; Court, A.C.; Lazzari, E.; Sadarangani, A.; Zipeto, M.A.; La Clair, J.J.; Villa, R.; et al. RNA Splicing Modulation Selectively Impairs Leukemia Stem Cell Maintenance in Secondary Human AML. Cell Stem Cell 2016, 19, 599–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bocker, M.T.; Hellwig, I.; Breiling, A.; Eckstein, V.; Ho, A.D.; Lyko, F. Genome-wide promoter DNA methylation dynamics of human hematopoietic pro-genitor cells during differentiation and aging. Blood 2011, 117, e182–e189. [Google Scholar] [CrossRef] [Green Version]
- Bacalini, M.G.; Boattini, A.; Gentilini, D.; Giampieri, E.; Pirazzini, C.; Giuliani, C.; Fontanesi, E.; Remondini, D.; Capri, M.; Del Rio, A.; et al. A meta-analysis on age-associated changes in blood DNA methylation: Results from an original analysis pipeline for Infinium 450k data. Aging 2015, 7, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, P.; Vallania, F.; Warsinske, H.C.; Donato, M.; Schaffert, S.; Chang, S.E.; Dvorak, M.; Dekker, C.L.; Davis, M.M.; Utz, P.J.; et al. Single-Cell Chromatin Modification Profiling Reveals Increased Epigenetic Variations with Aging. Cell 2018, 173, 1385–1397.e14. [Google Scholar] [CrossRef] [Green Version]
- Adelman, E.R.; Huang, H.T.; Roisman, A.; Olsson, A.; Colaprico, A.; Qin, T.; Lindsley, R.C.; Bejar, R.; Salomonis, N.; Grimes, H.L.; et al. Aging human hematopoietic stem cells manifest profound epigenetic repro-gramming of enhancers that may predispose to leukemia. Cancer Discov. 2019, 9, 1080–1101. [Google Scholar] [CrossRef]
- Walter, D.; Lier, A.; Geiselhart, A.; Thalheimer, F.B.; Huntscha, S.; Sobotta, M.C.; Moehrle, B.; Brocks, D.; Bayindir, I.; Kaschutnig, P.; et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nat. Cell Biol. 2015, 520, 549–552. [Google Scholar] [CrossRef] [PubMed]
- Flach, J.; Bakker, S.T.; Mohrin, M.; Conroy, P.C.; Pietras, E.M.; Reynaud, D.; Alvarez, S.; Diolaiti, M.E.; Ugarte, F.; Forsberg, E.C.; et al. Replication stress is a potent driver of func-tional decline in ageing haematopoietic stem cells. Nature 2014, 512, 198–202. [Google Scholar] [CrossRef] [Green Version]
- Flach, J.; Jann, J.-C.; Knaflic, A.; Riabov, V.; Streuer, A.; Altrock, E.; Xu, Q.; Schmitt, N.; Obländer, J.; Nowak, V.; et al. Replication stress signaling is a therapeutic target in myelodysplastic syndromes with splicing factor mutations. Haematology 2020. [Google Scholar] [CrossRef]
- Raval, A.; Sridhar, K.J.; Patel, S.; Turnbull, B.B.; Greenberg, P.L.; Mitchell, B.S. Reduced rRNA expression and increased rDNA promoter methylation in CD34+ cells of patients with myelodysplastic syndromes. Blood 2012, 120, 4812–4818. [Google Scholar] [CrossRef] [Green Version]
- Tehranchi, R.; Invernizzi, R.; Grandien, A.; Zhivotovsky, B.; Fadeel, B.; Forsblom, A.-M.; Travaglino, E.; Samuelsson, J.; Hast, R.; Nilsson, L.; et al. Aberrant mitochondrial iron distribution and maturation arrest characterize early erythroid precursors in low-risk myelodysplastic syndromes. Blood 2005, 106, 247–253. [Google Scholar] [CrossRef]
- Gyan, E.; Frisan, E.; Beyne-Rauzy, O.; Deschemin, J.-C.; Pierre-Eugene, C.; Randriamampita, C.; Dubart-Kupperschmitt, A.; Garrido, C.; Dreyfus, F.; Mayeux, P.; et al. Spontaneous and Fas-induced apoptosis of low-grade MDS erythroid precursors involves the endoplasmic reticulum. Leukemia 2008, 22, 1864–1873. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Eksioglu, E.A.; Zhou, J.; Zhang, L.; Djeu, J.; Fortenbery, N.; Epling-Burnette, P.; Van Bijnen, S.; Dolstra, H.; Cannon, J.; et al. Induction of myelodysplasia by myeloid-derived suppressor cells. J. Clin. Investig. 2013, 123, 4595–4611. [Google Scholar] [CrossRef] [PubMed]
- Montalban-Bravo, G.; Class, C.A.; Ganan-Gomez, I.; Kanagal-Shamanna, R.; Sasaki, K.; Richard-Carpentier, G.; Naqvi, K.; Wei, Y.; Yang, H.; Soltysiak, K.A.; et al. Tran-scriptomic analysis implicates necroptosis in disease progression and prognosis in myelodysplastic syndromes. Leukemia 2020, 34, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Wagner, P.N.; Shi, Q.; Salisbury-Ruf, C.T.; Zou, J.; Savona, M.R.; Fedoriw, Y.; Zinkel, S.S. Increased Ripk1-mediated bone marrow necroptosis leads to myelodysplasia and bone marrow failure in mice. Blood 2019, 133, 107–120. [Google Scholar] [CrossRef]
- Sallman, D.A.; Cluzeau, T.; Basiorka, A.A.; List, A. Unraveling the Pathogenesis of MDS: The NLRP3 Inflammasome and Pyroptosis Drive the MDS Phenotype. Front. Oncol. 2016, 6, 151. [Google Scholar] [CrossRef]
- Basiorka, A.A.; McGraw, K.L.; Eksioglu, E.A.; Chen, X.; Johnson, J.; Zhang, L.; Zhang, Q.; Irvine, B.A.; Cluzeau, T.; Sallman, D.A.; et al. The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood 2016, 128, 2960–2975. [Google Scholar] [CrossRef]
- A Basiorka, A.; McGraw, K.L.; Abbas-Aghababazadeh, F.; McLemore, A.; Vincelette, N.D.; A Ward, G.; Eksioglu, E.A.; Sallman, D.A.; Al Ali, N.; Padron, E.; et al. Assessment of ASC specks as a putative biomarker of pyroptosis in myelodysplastic syndromes: An observational cohort study. Lancet Haematol. 2018, 5, e393–e402. [Google Scholar] [CrossRef]
- Mei, Y.; Zhao, B.; Basiorka, A.A.; Yang, J.; Cao, L.; Zhang, J.; List, A.; Ji, P. Age-related inflammatory bone marrow microen-vironment induces ineffective erythropoiesis mimicking del(5q) MDS. Leukemia 2018, 32, 1023–1033. [Google Scholar] [CrossRef] [Green Version]
- Cluzeau, T.; McGraw, K.L.; Irvine, B.; Masala, E.; Ades, L.; Basiorka, A.A.; Maciejewski, J.; Auberger, P.; Wei, S.; Fenaux, P.; et al. Pro-inflammatory proteins S100A9 and tumor necrosis factor-α suppress erythropoietin elaboration in myelodysplastic syndromes. Haematology 2017, 102, 2015–2020. [Google Scholar] [CrossRef] [Green Version]
- Schneider, R.K.; Schenone, M.; Ferreira, M.S.V.; Kramann, R.; Joyce, C.E.; Hartigan, C.; Beier, F.; Brümmendorf, T.H.; Gehrming, U.; Platzbecker, U.; et al. Rps14 haploinsufficiency causes a block in erythroid differentiation mediated by S100A8 and S100A. Nat. Med. 2016, 22, 288–297. [Google Scholar] [CrossRef]
- Zambetti, N.A.; Ping, Z.; Chen, S.; Kenswil, K.J.; Mylona, M.A.; Sanders, M.A.; Hoogenboezem, R.M.; Bindels, E.M.; Adisty, M.N.; Van Strien, P.M.; et al. Mesenchymal Inflammation Drives Genotoxic Stress in Hematopoietic Stem Cells and Predicts Disease Evolution in Human Pre-leukemia. Cell Stem Cell 2016, 19, 613–627. [Google Scholar] [CrossRef] [Green Version]
- Medyouf, H.; Mossner, M.; Jann, J.C.; Nolte, F.; Raffel, S.; Herrmann, C.; Lier, A.; Eisen, C.; Nowak, V.; Zens, B.; et al. Myelodysplastic cells in patients repro-gram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell 2014, 14, 824–837. [Google Scholar] [CrossRef] [Green Version]
- Bulycheva, E.; Rauner, M.; Medyouf, H.; Theurl, I.; Bornhauser, M.; Hofbauer, L.C.; Platzbecker, U. Myelodysplasia is in the niche: Novel concepts and emerging therapies. Leukemia 2014, 29, 259–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pronk, E.; Raaijmakers, M.H.G.P. The mesenchymal niche in MDS. Blood 2019, 133, 1031–1038. [Google Scholar] [CrossRef]
- Schneider, R.K.; Ademà, V.; Heckl, D.; Järås, M.; Mallo, M.; Lord, A.M.; Chu, L.P.; McConkey, M.E.; Kramann, R.; Mullally, A.; et al. Role of Casein Kinase 1A1 in the Biology and Targeted Therapy of del(5q) MDS. Cancer Cell 2014, 26, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Krönke, J.; Fink, E.C.; Hollenbach, P.W.; MacBeth, K.J.; Hurst, S.N.; Udeshi, N.D.; Chamberlain, P.P.; Mani, D.R.; Man, H.W.; Gandhi, A.K.; et al. Lenalidomide induces ubiquitination and deg-radation of CK1α in del(5q) MDS. Nature 2015, 523, 183–188. [Google Scholar] [CrossRef]
- Starczynowski, D.T.; Kuchenbauer, F.; Argiropoulos, B.; Sung, S.; Morin, R.; Muranyi, A.; Hirst, M.; Hogge, D.; Marra, M.; Wells, R.A.; et al. Identification of miR-145 and miR-146a as mediators of the 5q–syndrome phenotype. Nat. Med. 2009, 16, 49–58. [Google Scholar] [CrossRef]
- Ribezzo, F.; Snoeren, I.A.; Ziegler, S.; Stoelben, J.; Olofsen, P.A.; Henic, A.; Ferreira, M.V.; Chen, S.; Stalmann, U.S.; Buesche, G.; et al. Rps14, Csnk1a1 and miRNA145/miRNA146a defi-ciency cooperate in the clinical phenotype and activation of the innate immune system in the 5q- syndrome. Leukemia 2019, 33, 1759–1772. [Google Scholar] [CrossRef] [PubMed]
- Ebert, B.L.; Pretz, J.; Bosco, J.; Chang, C.Y.; Tamayo, P.; Galili, N.; Raza, A.; Root, D.E.; Attar, E.; Ellis, S.R.; et al. Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nat. Cell Biol. 2008, 451, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Le Goff, S.; Boussaid, I.; Floquet, C.; Raimbault, A.; Hatin, I.; Andrieu-Soler, C.; Salma, M.; LeDuc, M.; Gautier, E.-F.; Guyot, B.; et al. p53 activation during ribosome biogenesis regulates normal erythroid differentiation. Blood 2021, 137, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Dutt, S.; Narla, A.; Lin, K.; Mullally, A.; Abayasekara, N.; Megerdichian, C.; Wilson, F.H.; Currie, T.; Khanna-Gupta, A.; Berliner, N.; et al. Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in human erythroid progenitor cells. Blood 2011, 117, 2567–2576. [Google Scholar] [CrossRef] [Green Version]
- Moniz, H.; Gastou, M.; Leblanc, T.; Hurtaud, C.; Cretien, A.; Lecluse, Y.; Raslova, H.; Larghero, J.; Croisille, L.; Faubladier, M.; et al. Group of Société d’Hématologie et d’Immunologie Pédiatrique-SHIP. Primary hematopoietic cells from DBA patients with mutations in RPL11 and RPS19 genes exhibit distinct erythroid phenotype in vitro. Cell Death Dis. 2012, 3, e356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barlow, J.L.; Drynan, L.F.; Hewett, D.R.; Holmes, L.R.; Lorenzo-Abalde, S.; Lane, A.L.; Jolin, H.E.; Pannell, R.; Middleton, A.J.; Wong, S.H.; et al. A p53-dependent mechanism underlies macrocytic anemia in a mouse model of human 5q- syndrome. Nat. Med. 2010, 16, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Frisan, E.; Vandekerckhove, J.; de Thonel, A.; Pierre-Eugène, C.; Sternberg, A.; Arlet, J.B.; Floquet, C.; Gyan, E.; Kosmider, O.; Dreyfus, F.; et al. Defective nuclear localization of Hsp70 is associated with dyserythropoiesis and GATA-1 cleavage in myelodysplastic syndromes. Blood 2012, 119, 1532–1542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gastou, M.; Rio, S.; Dussiot, M.; Karboul, N.; Moniz, H.; Leblanc, T.; Sevin, M.; Gonin, P.; Larghéro, J.; Garrido, C.; et al. The severe phenotype of Diamond-Blackfan anemia is modulated by heat shock protein 70. Blood Adv. 2017, 1, 1959–1976. [Google Scholar] [CrossRef] [Green Version]
- Khajuria, R.K.; Munschauer, M.; Ulirsch, J.C.; Fiorini, C.; Ludwig, L.S.; McFarland, S.K.; Abdulhay, N.J.; Specht, H.; Keshishian, H.; Mani, D.R.; et al. Ribosome Levels Selectively Regulate Translation and Lineage Commitment in Human Hematopoiesis. Cell 2018, 173, 90–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boussaid, I.; Le Goff, S.; Floquet, C.; Gautier, E.F.; Raimbault, A.; Viailly, P.J.; Al Dulaimi, D.; Burroni, B.; Dusanter-Fourt, I.; Hatin, I.; et al. Integrated analyses of translatome and proteome identify the rules of translation selectivity in RPS14-deficient cells. Haematologica 2021, 106, 746–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Keel, S.B.; Shimamura, A.; Liu, L.; Gerds, A.T.; Li, H.Y.; Wood, B.L.; Scott, B.L.; Abkowitz, J.L. Delayed globin synthesis leads to excess heme and the macrocytic anemia of Diamond Blackfan anemia and del(5q) myelodysplastic syndrome. Sci. Transl. Med. 2016, 8, 338ra67. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Ahmed, D.; Dolatshad, H.; Tatwavedi, D.; Schulze, U.; Sanchi, A.; Ryley, S.; Dhir, A.; Carpenter, L.; Watt, S.M.; et al. SF3B1 mutations induce R-loop accumulation and DNA damage in MDS and leukemia cells with therapeutic implications. Leukemia 2020, 34, 2525–2530. [Google Scholar] [CrossRef]
- Darman, R.B.; Seiler, M.; Agrawal, A.A.; Lim, K.H.; Peng, S.; Aird, D.; Bailey, S.L.; Bhavsar, E.B.; Chan, B.; Colla, S.; et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3′ Splice Site Selection through Use of a Different Branch Point. Cell Rep. 2015, 13, 1033–1045. [Google Scholar] [CrossRef] [Green Version]
- Shiozawa, Y.; Malcovati, L.; Galli, A.; Sato-Otsubo, A.; Kataoka, K.; Sato, Y.; Watatani, Y.; Suzuki, H.; Yoshizato, T.; Yoshida, K.; et al. Aberrant splicing and defective mRNA production induced by somatic spliceosome mutations in myelodysplasia. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef]
- Ilagan, J.O.; Ramakrishnan, A.; Hayes, B.; Murphy, M.E.; Zebari, A.S.; Bradley, P.; Bradley, R.K. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res. 2015, 25, 14–26. [Google Scholar] [CrossRef]
- Kim, E.; Ilagan, J.O.; Liang, Y.; Daubner, G.M.; Lee, S.C.W.; Ramakrishnan, A.; Li, Y.; Chung, Y.R.; Micol, J.B.; Murphy, M.E.; et al. Faculty Opinions recommendation of SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 2018, 27, 617–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.A.; Lin, K.T.; Bradley, R.K.; Abdel-Wahab, O.; Krainer, A.R. Recurrent SRSF2 mutations in MDS affect both splicing and NMD. Genes Dev. 2020, 34, 413–427. [Google Scholar] [CrossRef] [PubMed]
- Inoue, D.; Polaski, J.T.; Taylor, J.; Castel, P.; Chen, S.; Kobayashi, S.; Hogg, S.J.; Hayashi, Y.; Pineda, J.M.B.; El Marabti, E.; et al. Faculty Opinions recommendation of Minor intron retention drives clonal hematopoietic disorders and diverse cancer predisposition. Nat. Genet. 2021, 53, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Damm, F.; Kosmider, O.; Gelsi-Boyer, V.; Renneville, A.; Carbuccia, N.; Hidalgo-Curtis, C.; Della Valle, V.; Couronné, L.; Scourzic, L.; Chesnais, V.; et al. Mutations affecting mRNA splicing define distinct clinical phenotypes and correlate with patient outcome in myelodysplastic syndromes. Blood 2012, 119, 3211–3218. [Google Scholar] [CrossRef]
- Pellagatti, A.; Armstrong, R.N.; Steeples, V.; Sharma, E.; Repapi, E.; Singh, S.; Sanchi, A.; Radujkovic, A.; Horn, P.; Dolatshad, H.; et al. Impact of spliceosome mutations on RNA splicing in myelodysplasia: Dysregulated genes/pathways and clinical associations. Blood 2018, 132, 1225–1240. [Google Scholar] [CrossRef] [Green Version]
- Dolatshad, H.; Pellagatti, A.; Liberante, F.G.; Llorian, M.; Repapi, E.; Steeples, V.; Roy, S.; Scifo, L.; Armstrong, R.N.; Shaw, J.; et al. Cryptic splicing events in the iron trans-porter ABCB7 and other key target genes in SF3B1-mutant myelodysplastic syndromes. Leukemia 2016, 30, 2322–2331. [Google Scholar] [CrossRef] [Green Version]
- Bondu, S.; Alary, A.-S.; Lefèvre, C.; Houy, A.; Jung, G.; Lefebvre, T.; Rombaut, D.; Boussaid, I.; Bousta, A.; Guillonneau, F.; et al. A variant erythroferrone disrupts iron homeostasis inSF3B1-mutated myelodysplastic syndrome. Sci. Transl. Med. 2019, 11, eaav5467. [Google Scholar] [CrossRef]
- Kordasti, S.Y.; Ingram, W.; Hayden, J.; Darling, D.; Barber, L.; Afzali, B.; Lombardi, G.; Wlodarski, M.; Maciejewski, J.P.; Farzaneh, F.; et al. CD4+CD25high Foxp3+ regulatory T cells in myelodysplastic syndrome (MDS). Blood 2007, 110, 847–850. [Google Scholar] [CrossRef] [Green Version]
- Kotsianidis, I.; Bouchliou, I.; Nakou, E.; Spanoudakis, E.; Margaritis, D.; Christophoridou, A.V.; Anastasiades, A.; Tsigalou, C.; Bourikas, G.; Karadimitris, A.; et al. Kinetics, function and bone marrow trafficking of CD4+CD25+FOXP3+ regulatory T cells in myelodysplastic syndromes (MDS). Leukemia 2008, 23, 510–518. [Google Scholar] [CrossRef] [Green Version]
- Kittang, A.O.; Kordasti, S.; Sand, K.E.; Costantini, B.; Kramer, A.M.; Perezabellan, P.; Seidl, T.; Rye, K.P.; Hagen, K.M.; Kulasekararaj, A.; et al. Expansion of myeloid derived suppressor cells correlates with number of T regulatory cells and disease progression in myelodysplastic syndrome. OncoImmunology 2015, 5, e1062208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Leeuwen-Kerkhoff, N.; Westers, T.M.; Poddighe, P.J.; Povoleri, G.A.; Timms, J.A.; Kordasti, S.; De Gruijl, T.D.; Van de Loosdrecht, A.A. Reduced frequencies and functional impairment of dendritic cell subsets and non-classical monocytes in myelodysplastic syndromes. Haematologica 2021. [Google Scholar] [CrossRef]
- Mailloux, A.W.; Sugimori, C.; Komrokji, R.S.; Yang, L.; Maciejewski, J.P.; Sekeres, M.A.; Paquette, R.; Loughran, T.P.; List, A.F.; Epling-Burnette, P.K. Expansion of effector memory regulatory T cells represents a novel prognostic factor in lower risk myelodysplastic syndrome. J. Immunol. 2012, 189, 3198–3208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiladjian, J.J.; Bourgeois, E.; Lobe, I. Cytolytic function and survival of natural killer cells are severely altered in myelodysplastic syndromes. Leukemia 2006, 20, 463–470. [Google Scholar] [CrossRef] [Green Version]
- Epling-Burnette, P.K.; Bai, F.; Painter, J.S.; Rollison, D.E.; Salih, H.R.; Krusch, M.; Zou, J.; Ku, E.; Zhong, B.; Boulware, D.; et al. Reduced natural killer (NK) function associated with high-risk myelodysplastic syndrome (MDS) and reduced expression of activating NK receptors. Blood 2007, 109, 4816–4824. [Google Scholar] [CrossRef]
- Hejazi, M.; Manser, A.R.; Fröbel, J.; Kündgen, A.; Zhao, X.; Schönberg, K.; Germing, U.; Haas, R.; Gattermann, N.; Uhrberg, M. Impaired cytotoxicity associated with defective natural killer cell differentiation in myelodysplastic syndromes. Haematologica 2015, 100, 643–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mekinian, A.; Dervin, G.; Lapidus, N.; Kahn, J.E.; Terriou, L.; Liozon, E.; Grignano, E.; Piette, J.C.; Rauzy, O.B.; Grobost, V.; et al. Biologics in myelodysplastic syndrome-related systemic inflammatory and autoimmune diseases: French multicenter retrospective study of 29 patients. Autoimmun. Rev. 2017, 16, 903–910. [Google Scholar] [CrossRef] [Green Version]
- Roupie, A.L.; Guedon, A.; Terrier, B.; Lahuna, C.; Jachiet, V.; Regent, A.; de Boysson, H.; Carrat, F.; Seguier, J.; Terriou, L.; et al. Vasculitis associated with myelodysplastic syndrome and chronic mye-lomonocytic leukemia: French multicenter case-control study. Semin. Arthritis Rheum. 2020, 50, 879–884. [Google Scholar] [CrossRef]
- Zhao, L.-P.; Boy, M.; Azoulay, C.; Clappier, E.; Sébert, M.; Amable, L.; Klibi, J.; Benlagha, K.; Espéli, M.; Balabanian, K.; et al. Genomic landscape of MDS/CMML associated with systemic inflammatory and autoimmune disease. Leukemia 2021, 1–5. [Google Scholar] [CrossRef]
- Moskorz, W.; Cosmovici, C.; Jäger, P.S.; Cadeddu, R.P.; Timm, J.; Haas, R. Myelodysplastic syndrome patients display alterations in their immune status reflected by increased PD-L1-expressing stem cells and highly dynamic exhausted T-cell frequencies. Br. J. Haematol. 2021, 193, 941–945. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Eksioglu, E.A.; Chen, X.; Kandell, W.; Le Trinh, T.; Cen, L.; Qi, J.; Sallman, D.A.; Zhang, Y.; Tu, N.; et al. S100A9-induced overexpression of PD-1/PD-L1 contributes to ineffective hematopoiesis in myelodysplastic syndromes. Leukemia 2019, 33, 2034–2046. [Google Scholar] [CrossRef] [PubMed]
- Sarhan, D.; Brandt, L.; Felices, M.; Guldevall, K.; Lenvik, T.; Hinderlie, P.; Curtsinger, J.; Warlick, E.; Spellman, S.R.; Blazar, B.R.; et al. 161533 TriKE stimulates NK-cell function to overcome myeloid-derived suppressor cells in MDS. Blood Adv. 2018, 2, 1459–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Cell of origin. Myelodysplastic syndrome originates from a clonal hematopoietic stem cell. This cell expresses specific surface markers and drives a myeloid-biased hematopoiesis at the expense of the lymphoid lineage. Hematopoietic stem and progenitor compartments are disturbed and produce dysplastic megakaryocytes, erythroblasts and granulocytes. Inflammatory microenvironment mainly myeloid-derived suppressive cells (MDSCs) producing alarms contribute to ROS generation, replication stress and genetic instability that forces the clonal evolution. IL1RAP: IL-1 receptor accessory protein; TIM-3/HAVCR2: T-cell immunoglobulin mucin-3/Hepatitis A virus cellular receptor 2; LR-MDS: low-risk MDS; HR-MDS: high-risk MDS; HSC: hematopoietic stem cell; LT-HSC: long-term hematopoietic stem cell; MPP: multipotent progenitors; CMP: common myeloid progenitor; GMP: granulocytic myeloid progenitor; MEP: megakaryocytic-erythroid progenitor; Treg: regulator T cells; NK: natural killer; ROS: reactive oxygen species; DAMPs: damage-associated molecular patterns.
Figure 1.
Cell of origin. Myelodysplastic syndrome originates from a clonal hematopoietic stem cell. This cell expresses specific surface markers and drives a myeloid-biased hematopoiesis at the expense of the lymphoid lineage. Hematopoietic stem and progenitor compartments are disturbed and produce dysplastic megakaryocytes, erythroblasts and granulocytes. Inflammatory microenvironment mainly myeloid-derived suppressive cells (MDSCs) producing alarms contribute to ROS generation, replication stress and genetic instability that forces the clonal evolution. IL1RAP: IL-1 receptor accessory protein; TIM-3/HAVCR2: T-cell immunoglobulin mucin-3/Hepatitis A virus cellular receptor 2; LR-MDS: low-risk MDS; HR-MDS: high-risk MDS; HSC: hematopoietic stem cell; LT-HSC: long-term hematopoietic stem cell; MPP: multipotent progenitors; CMP: common myeloid progenitor; GMP: granulocytic myeloid progenitor; MEP: megakaryocytic-erythroid progenitor; Treg: regulator T cells; NK: natural killer; ROS: reactive oxygen species; DAMPs: damage-associated molecular patterns.

Figure 2.
Schematic representation of mutations in MDS. Co-occurring mutations are shown in brown and mutually exclusive mutations are shown in blue. The power of linkage or exclusion is indicated by the color gradient.
Figure 2.
Schematic representation of mutations in MDS. Co-occurring mutations are shown in brown and mutually exclusive mutations are shown in blue. The power of linkage or exclusion is indicated by the color gradient.

Figure 3.
Multiple phenotypic alterations in MDS with 5q deletion. Cell-intrinsic expansion of hematopoietic stem cell (HSC) is a consequence of CSNK1A1 haploinsufficiency. Dysplastic megakaryopoiesis relies on miR145/miR146a haploinsufficiency. Selective translation is a feature of limiting ribosome availability in RPS14 haploinsufficient conditions. CSKN1A1: casein kinase 1A1 gene; CK1a: casein kinase 1a; miR: microRNA; RPS14: ribosomal protein 14.
Figure 3.
Multiple phenotypic alterations in MDS with 5q deletion. Cell-intrinsic expansion of hematopoietic stem cell (HSC) is a consequence of CSNK1A1 haploinsufficiency. Dysplastic megakaryopoiesis relies on miR145/miR146a haploinsufficiency. Selective translation is a feature of limiting ribosome availability in RPS14 haploinsufficient conditions. CSKN1A1: casein kinase 1A1 gene; CK1a: casein kinase 1a; miR: microRNA; RPS14: ribosomal protein 14.

Figure 4.
Deregulation of splicing. The U2 major spliceosome involves SF3B1, which recognizes the branch point and U2AF1 that binds to AG 3′ junction to drive intron excision. SRSF2 enhances splicing through the binding of specific exonic regions. Mutations in these genes either produce premature stop codon, leading to the degradation of the transcript by the non-sense mediated decay, or a variant in-frame transcript that can be translated giving variant protein. ZRSR2 is a component of U12 minor spliceosome and its loss increases intron retention.
Figure 4.
Deregulation of splicing. The U2 major spliceosome involves SF3B1, which recognizes the branch point and U2AF1 that binds to AG 3′ junction to drive intron excision. SRSF2 enhances splicing through the binding of specific exonic regions. Mutations in these genes either produce premature stop codon, leading to the degradation of the transcript by the non-sense mediated decay, or a variant in-frame transcript that can be translated giving variant protein. ZRSR2 is a component of U12 minor spliceosome and its loss increases intron retention.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Fontenay, M.; Farhat, B.; Boussaid, I. Pathophysiology of Myelodysplastic Syndromes. Hemato 2021, 2, 477-495. https://0-doi-org.brum.beds.ac.uk/10.3390/hemato2030030
AMA Style
Fontenay M, Farhat B, Boussaid I. Pathophysiology of Myelodysplastic Syndromes. Hemato. 2021; 2(3):477-495. https://0-doi-org.brum.beds.ac.uk/10.3390/hemato2030030
Chicago/Turabian StyleFontenay, Michaela, Batoul Farhat, and Ismael Boussaid. 2021. "Pathophysiology of Myelodysplastic Syndromes" Hemato 2, no. 3: 477-495. https://0-doi-org.brum.beds.ac.uk/10.3390/hemato2030030