
The AL Amyloid Fibril: Looking for a Link between Fibril Formation and Structure

Institute of Protein Biochemistry, Ulm University, 89081 Ulm, Germany
Hemato 2021, 2(3), 505-514; https://0-doi-org.brum.beds.ac.uk/10.3390/hemato2030032
Submission received: 15 July 2021
/
Revised: 4 August 2021
/
Accepted: 4 August 2021
/
Published: 6 August 2021
(This article belongs to the Special Issue Advances in Amyloidosis: A Theme Issue in Honor of Prof. Dr. Giampaolo Merlini)
{kind=link}
{kind=link}
{kind=link}
Abstract
:The formation and deposition of fibrils derived from immunglobulin light chains is a hallmark of systemic AL amyloidosis. A particularly remarkable feature of the disease is the diversity and complexity in pathophysiology and clinical manifestations. This is related to the variability of immunoglobulins, as virtually every patient has a variety of mutations resulting in their own unique AL protein and thus a unique fibril deposited in the body. Here, I review recent biochemical and biophysical studies that have expanded our knowledge on how versatile the structure of AL fibrils in patients is and highlight their implications for the molecular mechanism of fibril formation in AL amyloidosis.
1. Introduction
Systemic AL amyloidosis is a patient specific disease caused by an underlying plasma cell dyscrasia in which a specific light chain is overexpressed by a proliferating plasma cell clone, leading to the deposition of amyloid fibrils [1]. The part of the light chain deposited in the fibril is called AL protein. Interestingly, only 10% to 15% of all plasma cell diseases result in AL amyloidosis [1], meaning that only a small fraction of the theoretically possible light chains ever forms amyloid fibrils in the body. Yet, the molecular basis of this circumstance is not clear and the identification of common structural features between the disease-associated light chain protein and AL fibrils of different patients is important to better understand and treat the disease. This review summarizes recent findings on the structure of AL amyloid fibrils and their implications for the molecular mechanism of fibril formation.
2. Structure and Variability of Light Chains
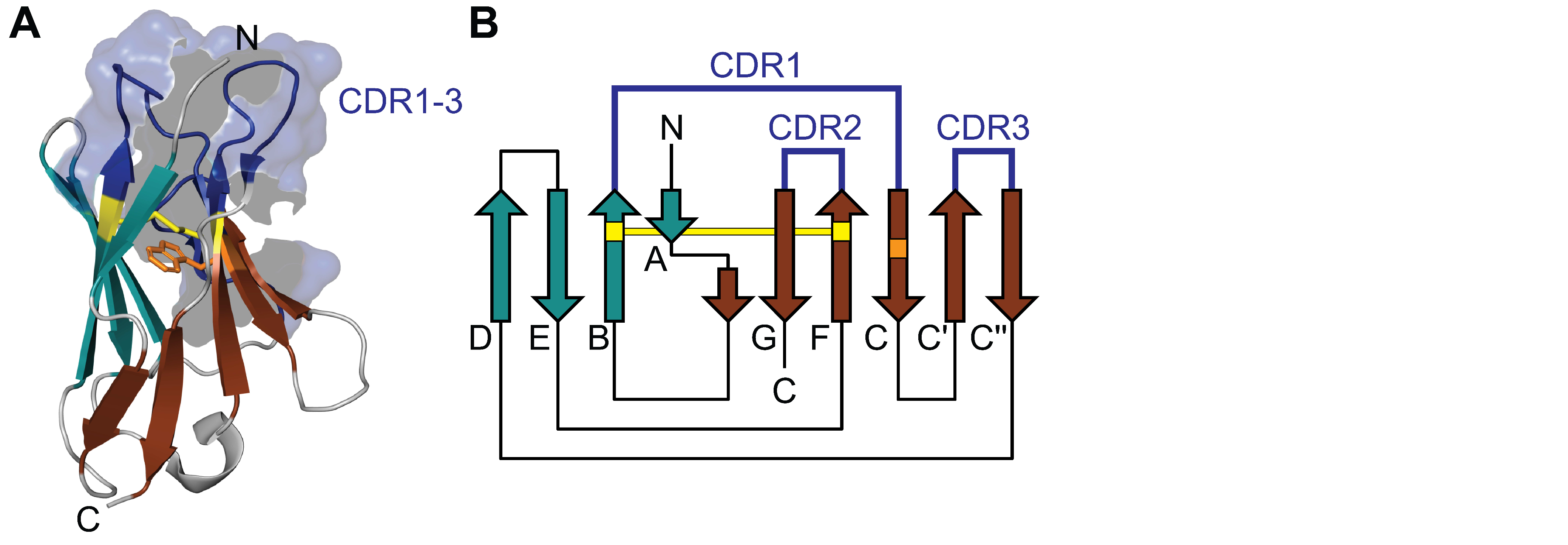
Light chains consist of about 220 amino acids and have a molecular weight of about 25 KDa. Each light chain is composed of two immunoglobulin domains, an N-terminal variable (V) domain and a C-terminal constant (C) domain [2]. Structurally, the C- and V-domains share conserved common features. Both are composed of two β sheets packed tightly against each other forming a Greek-key β-sandwich (Figure 1). The V-domain comprises about 100 amino acids organized in a 4-stranded antiparallel sheet packed against a 5-stranded antiparallel sheet (ABED/CC′C″FG) [3,4]. The C-domain consists of 90 to 100 amino acids arranged in a 4-stranded antiparallel sheet packed against a 3-stranded antiparallel sheet, lacking the C′ and C″ strand present in the V-domain [3,4]. The fold and structure of the domains is stabilized by a highly conserved disulfide bridge between the two β-sheets (connecting strand B and F) and a hydrophobic core located inside the β-sandwich formed mainly by hydrophobic amino acids in strands B, C, E and F as well as a highly conserved tryptophan in strand C (Figure 1) [4]. While the C-domain shows relatively little sequence variation, the V-domain is characterized by considerable sequence variability. This variability is mainly caused by three hypervariable loops (complementary determining regions, CDR) that form the antigen binding site [2] and cluster on the same side of the domain (Figure 1). The rest of the V-domain shows less sequence variability and forms the structurally conserved framework surrounding the CDRs [2].
The diversity of light chains is the reason why virtually every AL amyloidosis patient has a unique person-specific AL protein [5]. It is achieved through a complex maturation process that mainly involves two molecular events: (I) somatic recombination and (II) somatic hypermutation [2,4].
(I) Somatic recombination generates diversity in two ways. (a) Three gene segments (V, J and C) are recombined to generate a full-length light chain gene. The gene segments V and J together encode the V-domain, while the C segment encodes the C-domain. For each gene segment, multiple functional and non-functional copies exist, which are used stochastically in serial rearrangement events [6]. In addition, there are two separate light chain gene loci with their own set of gene segments, subdividing the light chain repertoire into two isotypes, κ (located on Chromosome 2) and λ (located on Chromosome 22). Allelic variations of the gene loci further increase the variability of recombined gene segments [6]. All theoretically possible rearrangements of functional gene segments reach several hundred different light chain sequences, which is called combinatorial diversity. (b) When the V and J gene segment are joined, a random truncation of the 3′ end of the V segment and/or the 5′ end of the J segment by an exonuclease may occur, resulting in P nucleotides [4]. A random addition of nucleotides in the region of the 3′ end of the V segment and/or the 5′ end of the J segment may also occur before the segments are ligated, resulting in N nucleotides [4]. This so-called junctional diversity increases the variability in the CDR3 in particular, however, the occurrence of P and N nucleotides can also lead to frameshifts, which in purely mathematical terms only lead to a functional open reading frame for a light chain in 1/3 of all cases.
(II) After successful somatic recombination, single point mutations are introduced into the rearranged light chain gene [2]. The process takes place mainly in the CDRs with a frequency of approximately 106 times that of a spontaneous gene mutation [7]. Somatic hypermutation potentially alters stability, specificity and binding affinity. However, it can also lead to genes that harbor premature stop codons or light chains that cannot fold correctly.
3. Properties of Amyloidogenic Light Chains
There is evidence that the isotype (κ or λ) or the germline segments used for somatic recombination contribute decisively to the propensity of a light chain to form fibrils. Somatic rearrangement occurs first in the κ locus and only if this is unsuccessful, the λ locus is used [6]. This is the reason why about 2/3 of all B lymphocytes in the polyclonal repertoire of healthy individuals produce a κ light chain [8]. Interestingly, AL amyloidosis is more frequently associated with λ light chains (~75% of cases) than with κ light chains (~25% of cases) [1]. One explanation for such a bias could be genetic abnormalities associated with the AL clonal plasma cell, like an increase or decrease in the number of genes or chromosomes, translocation of genes or dysregulation of transcription factors that increase the accessibility of certain light chain subfamilies or germline segments for somatic recombination [9,10]. Another explanation could be the intrinsic differences in structure and physicochemical properties between λ and κ light chains, which are thought to be encoded in the germline [11,12]. For example, λ light chains show CDR3 regions that are longer, more hydrophobic and more conformationally flexible [11], which potentially make them more prone to aggregation. AL amyloidosis occurs preferentially in individuals with B lymphocytes expressing certain germline subfamilies, namely λ1, λ2, λ3, λ6 or κ1 [8]. Out of these subfamilies, only 5 germline segments, IGLV1-44 (1c), IGLV2-14 (2a2), IGLV3-01 (3r), IGLV6-57 (6a) and IGKV1-33 (O18/O8) encode around 60% of the reported amyloidogenic light chains [13,14,15,16]. In contrast, other germ line segments, such as IGLV1-40 (1e), IGLV3-10 (3p) or IGLV3-25 (3m) are frequently expressed in the polyclonal B cell repertoire, but are rarely or not found in AL amyloidosis [15]. There is also evidence that the isotype or the germline segment may influence the tendency to predominantly deposit in certain organs, so called organ tropism [1,8]. IGLV1-44 and IGLV2-14 are more likely to cause peripheral nerve and cardiac deposition [9,13,16], IGLV6-57 correlates with renal deposition [9,13,14,16] and IGKV1-33 is more likely to cause liver deposition [16]. However, these observations are based on a limited number of patients harboring a specific gene subfamily, which may be difficult to generalize. In summary, the vast majority of light chains in the body are non-amyloidogenic, and the ultimate contribution of the isotype or germline segment to the amyloidogenic properties of light chains is not clear.
Changes in primary structure introduced by somatic hypermutation into the germline sequence or by posttranslational modifications are also discussed as drivers of light chain aggregation [8,17]. The identification of critical amino acid positions and their concentration in specific secondary structure regions showed that the location and nature of the mutation, rather than the number, determines the amyloidogenicity of light chains [18,19,20,21,22]. Some mutations have been attributed to a decrease in thermodynamic stability [18,23,24,25], increase in conformational dynamics [17,20,21] or effects on interdomain interactions [26,27]. However, in most cases it is very difficult to pinpoint specific mutations and the exact molecular mechanism by which certain mutations drive a light chain towards amyloid formation remains unclear. In the context of posttranslational modifications, light chain truncations are often found in the fibril but may vary depending on the primary structure or isotype. Reports identified V-domains with small parts of the C-domain as the main constituent of λ AL proteins [28,29,30,31,32], while κ AL proteins have been found to consist mainly of the C-domain [33,34]. The exact cleavage sites have not been determined in most cases and whether or not truncation occurs before fibril formation and is important in this process or after fibril formation and is just a result of truncation of a part not included in the fibril core is an open question [35,36,37]. In this context, a recent study hypothesized that all light chains have comparable intrinsic amyloidogenicity and that the interplay of protein sequence, environmental conditions and the presence and action of proteases determines whether or not a particular light chain is deposited as an amyloid fibril in a particular patient [38]. Another recent hypothesis is that disease-associated amyloid fibrils are selected in the body by their proteolytic stability, a mechanism termed proteolytic selection [39,40]. That is, protease-stable fibrils have a greater chance of escaping endogenous clearance mechanisms, proliferating and accumulating in a native cellular environment. Regardless of whether truncation occurs before or after fibril formation, both hypotheses propose a central role for proteolysis in AL amyloidosis and suggest that amino acid sequence and proteolytic stability cannot be simply correlated. AL-associated light chains show a higher frequency of acquired glycosylation than non-amyloidogenic light chains, which suggests that glycosylation may also contribute to amyloidogenicity [18,41,42,43,44]. Sugar moieties may also affect interactions with other molecules or cell surfaces and therefore could have an impact on organ tropism [45]. Other posttranslational modifications like disulfide bridge formation [32,46] or pyroglutamate formation [47,48,49] have also been found in AL proteins. Both modifications are common to natural antibodies [2,50] and whether they play a role in fibril formation of light chains is unclear. It is worth noting that most studies on the fibrillogenesis of light chains have mainly focused on V-domains and therefore the involvement of the C-domain in fibril formation and the occurrence of mutations in this domain have been rarely investigated and reported [17,37,51].
4. Structure of AL Fibrils
The structure of patient-derived AL fibrils and the molecular mechanism by which misfolded conformations are formed in the body is not well understood. Most initial knowledge of the structure of light chain-derived fibrils comes from in vitro studies of fibrils formed from recombinant V-domains or short fragments of light chains in the test tube and analyzed by nuclear magnetic resonance spectroscopy or cryo-electron microscopy [52,53,54,55,56,57]. These studies showed that light chain-derived fibrils consist of an in-register parallel β-sheet structure and that the polypeptide chain adopts a conformation with intermolecular contacts that is completely different from its native structure, suggesting that it undergoes dramatic structural rearrangements before fibril formation. However, it is crucial to point out that a growing number of studies show that in vitro fibrils differ in structure and stability from fibrils formed in the body [32,39,40,58,59,60,61,62]. This suggests that it is important to focus on patient-derived fibrils when investigating the structural basis of a disease. In this context, cryo electron microscopy is currently the state of the art for structural analysis of patient-derived amyloid fibrils. Three recent cryo electron microscopy studies shed new light on this topic by determining the structure of AL fibrils and their corresponding AL protein. All fibrils were obtained from patients with cardiac amyloidosis and derived from the λ isotype but different germlines, namely IGLV1-44*01 [63], IGLV3-19*01 [64] and IGLV6-57*02 [65].
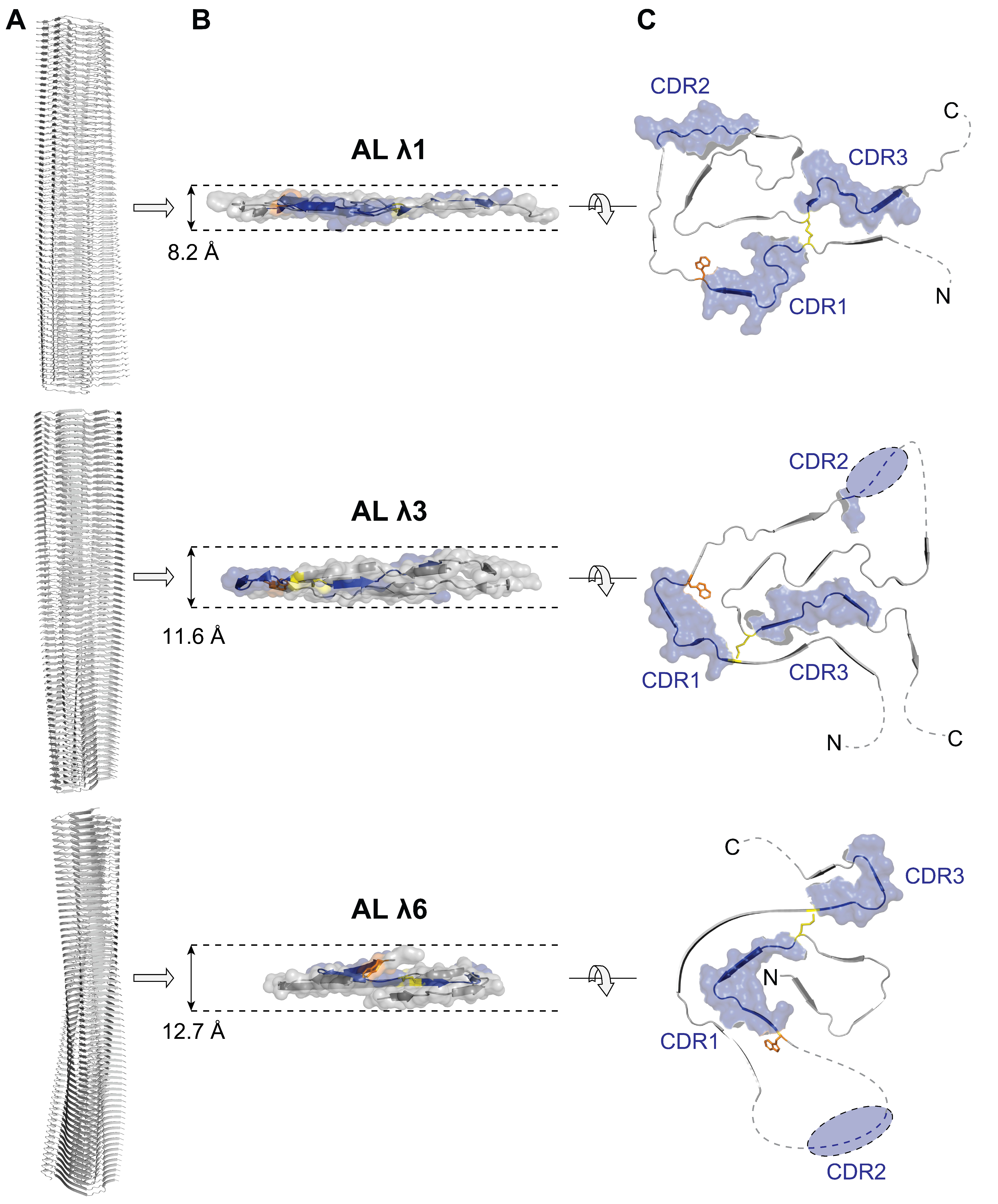
The fibril structures possess some striking similarities. All are asymmetric and show a single protofilament architecture, meaning they consist of only one polypeptide chain per layer (Figure 2). As commonly observed in λ AL proteins [28,29,30,31,32], the polypeptide chain underlying the fibril consists of the V-domain and a short part of the C-domain (Figure 3). The highly ordered fibril core is formed by only a major portion of the V-domain (Figure 3), suggesting that the V-domain is a prerequisite to build the fibril core of λ AL fibrils. The c-terminal remainder of the V-domain and the remaining C-domain form a conformationally flexible structure around the core (Figure 2). The monomers are compactly folded without crossovers of the polypeptide chain and stack on top of each other in a parallel, in-register structure (Figure 2). Superficially, the individual protein layers form a relatively flat structure. But in detail, however, the polypeptide chain along the backbone sometimes exhibits height differences of up to 13 Å (Figure 2). These height differences allow the stacked polypeptide chains to spatially interlock with each other and interact via side chains, thus stabilizing the fibril structure [63,64,65]. Another consequence of these height differences is that the fibril tips do not form a flat surface, but rather grooves and ridges that present hydrophobic and hydrophilic regions that may serve as docking sites for monomers [65]. The fold of the polypeptide chain in the fibril core is substantially different from the native globular fold. Many of the side-chain interactions present in the globular state are not preserved in the fibril and regions that are spatially adjacent in the globular structure, such as the CDRs (Figure 1), are widely separated in the fibril (Figure 2). Another example is the highly conserved tryptophan, which is buried both in the native state and in the fibril, but has different spatial environments (Figure 1 and Figure 2). These differences suggest that dramatic structural rearrangements and unfolding of the native state must occur [63,64,65]. Another remarkable feature is the persistence of the canonical disulfide bridge of the native fold in the fibril structure (Figure 1 and Figure 2). Interestingly, the spatial orientation of the regions connected by the disulfide bridge is rotated by 180° between the native state and fibril, i.e., the backbone runs in the same N to C orientation in the native state (Figure 1) and in the opposite N to C orientation in the fibril (Figure 2). This suggests that the structural rearrangement must occur around the intact disulfide bridge that restricts the conformational freedom of the misfolded polypeptide chain [63].
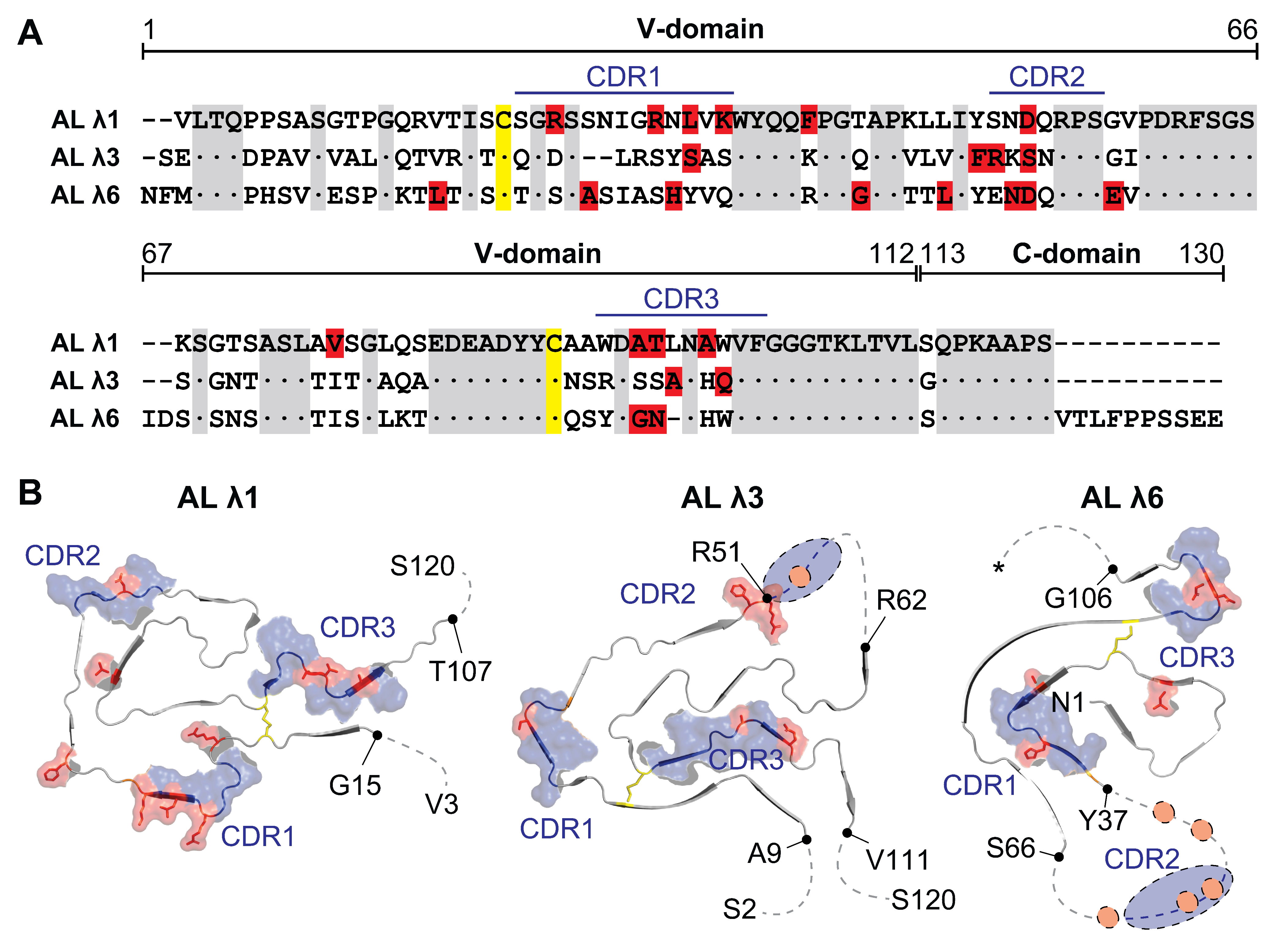
Besides the above-mentioned similarities, the fibril structures also show clear structural differences. The fold of the monomer is highly different and reflects the underlying sequence variability (Figure 2). The N-terminus can be disordered and surface exposed or hidden in the core of the fibril in a β strand conformation (Figure 2). The number of β strands within the polypeptide chain (between 9 and 12) as well as their length and position vary (Figure 2). The location of the CDRs in the fibril fold differs profoundly, but they mainly contribute to the rigid core region (Figure 2). The polypeptide chain may be highly ordered and structurally rigid throughout the entire core of the fibril or may have structurally flexible regions in between the highly ordered parts (Figure 2). The position and number of mutations compared to the germline sequence shows a relatively large variance, although the vast majority is located inside the CDRs (Figure 3). The λ1 AL protein has 10 mutations (2 outside the CDRs), the λ3 AL protein has 6 mutations (1 outside the CDRs) and the λ6 AL protein has 10 mutations (4 outside the CDRs). This variance supports the idea that it is not the number of mutations but rather the position and type of mutation that is important for amyloid formation [19]. However, analysis of the mutations does not provide a conclusive explanation for their role in fibril formation [63,64,65]. Some are exposed on the surface while others are buried, some are in structurally well-ordered regions while others are part of highly flexible regions (Figure 3) and they do not interrupt obvious crucial interactions for protein stability [63,64,65]. In the case of the λ3 fibril, an additional mutation was found outside the AL protein in the C-domain of the light chain [64], raising the question of whether such mutations play a role in fibril formation and whether their effects may be underestimated because they have hardly been studied so far.
One particular position in the germline sequence is consistently mutated in all AL proteins and that is the asparagine at position 53 in CDR2 (Figure 3). Whether this particular position could be critical for fibril formation remains unclear, as it has not been found to be particularly important [27]. None of the AL proteins found in the current fibril structures show a glycosylation or a pyroglutamylation, which thus further leaves open the relevance of these modifications for fibril formation. The AL proteins show different lengths, which is mainly caused by the variability of the c terminal truncation site (Figure 3A). The λ1 and λ3 AL proteins terminate mainly in the region around position 120 [63,64], while for the λ6 AL protein, different truncation sites around position 130 and 150 have been reported [65]. The region around position 120 is in close proximity to the short linker connecting the V- and C-domain and is flexible and exposed in both the natively folded light chain [66] and the fibril (Figure 2 and Figure 3). In addition, the regions around position 130 and 150 are exposed in both states [65]. Whether these regions are common proteolytic sites involved in the generation of AL proteins and whether or not truncation occurs before fibril formation or after fibril formation is a matter for further studies. Based on the current knowledge, it is reasonable to assume that sequence variability leads to molecular interactions that stabilize a particular fibril structure. However, no definite conclusion can be drawn with regard to amino acid mutations and posttranslational modifications and their role in fibrillization, as the amount of structural information is currently limited.
5. Conclusions
Our understanding of how AL fibrils are structured and what the structural determinants of their formation are has improved significantly in the last years. Detailed knowledge of the molecular structure of AL fibrils supports the concept that light chain unfolding is required and showed that dramatic structural rearrangements occur around the canonical disulfide bridge of the immunoglobulin domain. However, as AL amyloidosis is a patient-specific disease, there is great heterogeneity in the clinical phenotypes, the associated polypeptide chains and fibril structures. Therefore, further work is needed to identify common mechanistic and structural patterns to open up possibilities for the development of new diagnostic or therapeutic strategies.
Funding
This article was funded by the Deutsche Forschungsgemeinschaft through the grant HA 7138/3 (Research Unit FOR 2969).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The author thanks Sambhasan Banerjee and Thomas Heerde for software support.
Conflicts of Interest
The author declares no conflict of interest.
References
- Merlini, G.; Dispenzieri, A.; Sanchorawala, V.; Schönland, S.O.; Palladini, G.; Hawkins, P.N.; Gertz, M.A. Systemic immunoglobulin light chain amyloidosis. Nat. Rev. Dis. Primers 2018, 4, 38. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr.; Travers, P.; Walport, M.; Shlomchik, M.J. Immunobiology: The Immune System in Health and Disease, 5th ed.; Garland Science: New York, NY, USA, 2001. [Google Scholar]
- Bork, P.; Holm, L.; Sander, C. The immunoglobulin fold: Structural classification, sequence patterns and common core. J. Mol. Biol. 1994, 242, 309–320. [Google Scholar] [CrossRef]
- Lefranc, M.P.; Lefranc, G. Immunoglobulins or Antibodies: IMGT® Bridging Genes, Structures and Functions. Biomedicines 2020, 8, 319. [Google Scholar] [CrossRef]
- Bellotti, V.; Mangione, P.; Merlini, G. Review: Immunoglobulin light chain amyloidosis—The archetype of structural and pathogenic variability. J. Struct. Biol. 2000, 130, 280–289. [Google Scholar] [CrossRef]
- Collins, A.M.; Watson, C.T. Immunoglobulin Light Chain Gene Rearrangements, Receptor Editing and the Development of a Self-Tolerant Antibody Repertoire. Front. Immunol. 2018, 9, 2249. [Google Scholar] [CrossRef] [Green Version]
- Odegard, V.H.; Schatz, D.G. Targeting of somatic hypermutation. Nat. Rev. Immunol. 2006, 6, 573–583. [Google Scholar] [CrossRef]
- Herrera, G.A.; Teng, J.; Turbat-Herrera, E.A.; Zeng, C.; del Pozo-Yauner, L. Understanding Mesangial Pathobiology in AL-Amyloidosis and Monoclonal Ig Light Chain Deposition Disease. Kidney Int. Rep. 2020, 5, 1870–1893. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.S.; Geyer, S.M.; Price-Troska, T.L.; Allmer, C.; Kyle, R.A.; Gertz, M.A.; Fonseca, R. Immunoglobulin light chain variable (V) region genes influence clinical presentation and outcome in light chain-associated amyloidosis (AL). Blood 2003, 101, 3801–3808. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Huang, X.; Li, J. Light chain amyloidosis: Where are the light chains from and how they play their pathogenic role? Blood Rev. 2017, 31, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Townsend, C.L.; Laffy, J.M.; Wu, Y.B.; Silva O’Hare, J.; Martin, V.; Kipling, D.; Fraternali, F.; Dunn-Walters, D.K. Significant Differences in Physicochemical Properties of Human Immunoglobulin Kappa and Lambda CDR3 Regions. Front. Immunol. 2016, 7, 388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Kant, R.; Bauer, J.; Karow-Zwick, A.R.; Kube, S.; Garidel, P.; Blech, M.; Rousseau, F.; Schymkowitz, J. Adaption of human antibody λ and κ light chain architectures to CDR repertoires. Protein Eng. Des. Sel. 2019, 32, 109–127. [Google Scholar] [CrossRef] [Green Version]
- Comenzo, R.L.; Zhang, Y.; Martinez, C.; Osman, K.; Herrera, G.A. The tropism of organ involvement in primary systemic amyloidosis: Contributions of Ig V(L) germ line gene use and clonal plasma cell burden. Blood 2001, 98, 714–720. [Google Scholar] [CrossRef] [Green Version]
- Perfetti, V.; Casarini, S.; Palladini, G.; Vignarelli, M.C.; Klersy, C.; Diegoli, M.; Ascari, E.; Merlini, G. Analysis of V(lambda)-J(lambda) expression in plasma cells from primary (AL) amyloidosis and normal bone marrow identifies 3r (lambdaIII) as a new amyloid-associated germline gene segment. Blood 2002, 100, 948–953. [Google Scholar] [CrossRef] [PubMed]
- del Pozo-Yauner, L.; Becerril, B.; Ochoa-Leyva, A.; Rodríguez-Ambriz, S.L.; Pérez Carrión, J.I.; Zavala-Padilla, G.; Sánchez-López, R.; Fernández Velasco, D.A. The Structural Determinants of the Immunoglobulin Light Chain Amyloid Aggregation. In Physical Biology of Proteins and Peptides; Olivares-Quiroz, L., Guzmán-López, O., Jardón-Valadez, H., Eds.; Springer: Cham, Switzerland, 2015; pp. 1–28. [Google Scholar]
- Kourelis, T.V.; Dasari, S.; Theis, J.D.; Ramirez-Alvarado, M.; Kurtin, P.J.; Gertz, M.A.; Zeldenrust, S.R.; Zenka, R.M.; Dogan, A.; Dispenzieri, A. Clarifying immunoglobulin gene usage in systemic and localized immunoglobulin light-chain amyloidosis by mass spectrometry. Blood 2017, 129, 299–306. [Google Scholar] [CrossRef]
- Blancas-Mejia, L.M.; Misra, P.; Dick, C.J.; Cooper, S.A.; Redhage, K.R.; Bergman, M.R.; Jordan, T.L.; Maar, K.; Ramirez-Alvarado, M. Immunoglobulin light chain amyloid aggregation. Chem. Commun. 2018, 54, 10664–10674. [Google Scholar] [CrossRef] [PubMed]
- Oberti, L.; Rognoni, P.; Barbiroli, A.; Lavatelli, F.; Russo, R.; Maritan, M.; Palladini, G.; Bolognesi, M.; Merlini, G.; Ricagno, S. Concurrent structural and biophysical traits link with immunoglobulin light chains amyloid propensity. Sci. Rep. 2017, 7, 16809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, F.J. Four structural risk factors identify most fibril-forming kappa light chains. Amyloid 2000, 7, 200–211. [Google Scholar] [CrossRef]
- Poshusta, T.L.; Sikkink, L.A.; Leung, N.; Clark, R.J.; Dispenzieri, A.; Ramirez-Alvarado, M. Mutations in specific structural regions of immunoglobulin light chains are associated with free light chain levels in patients with AL amyloidosis. PLoS ONE 2009, 4, e5169. [Google Scholar] [CrossRef] [PubMed]
- Kazman, P.; Vielberg, M.T.; Pulido Cendales, M.D.; Hunziger, L.; Weber, B.; Hegenbart, U.; Zacharias, M.; Köhler, R.; Schönland, S.; Groll, M.; et al. Fatal amyloid formation in a patient’s antibody light chain is caused by a single point mutation. Elife 2020, 9, e52300. [Google Scholar] [CrossRef]
- Rottenaicher, G.J.; Weber, B.; Rührnößl, F.; Kazman, P.; Absmeier, R.M.; Hitzenberger, M.; Zacharias, M.; Buchner, J. Molecular mechanism of amyloidogenic mutations in hypervariable regions of antibody light chains. J. Biol. Chem. 2021, 296, 100334. [Google Scholar] [CrossRef] [PubMed]
- Hurle, M.R.; Helms, L.R.; Li, L.; Chan, W.; Wetzel, R. A role for destabilizing amino acid replacements in light-chain amyloidosis. Proc. Natl. Acad. Sci. USA 1994, 91, 5446–5450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wall, J.; Schell, M.; Murphy, C.; Hrncic, R.; Stevens, F.J.; Solomon, A. Thermodynamic instability of human lambda 6 light chains: Correlation with fibrillogenicity. Biochemistry 1999, 38, 14101–14108. [Google Scholar] [CrossRef]
- Baden, E.M.; Randles, E.G.; Aboagye, A.K.; Thompson, J.R.; Ramirez-Alvarado, M. Structural insights into the role of mutations in amyloidogenesis. J. Biol. Chem. 2008, 283, 30950–30956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baden, E.M.; Owen, B.A.; Peterson, F.C.; Volkman, B.F.; Ramirez-Alvarado, M.; Thompson, J.R. Altered dimer interface decreases stability in an amyloidogenic protein. J. Biol. Chem. 2008, 283, 15853–15860. [Google Scholar] [CrossRef] [Green Version]
- Garofalo, M.; Piccoli, L.; Romeo, M.; Barzago, M.M.; Ravasio, S.; Foglierini, M.; Matkovic, M.; Sgrignani, J.; De Gasparo, R.; Prunotto, M.; et al. Machine learning analyses of antibody somatic mutations predict immunoglobulin light chain toxicity. Nat. Commun. 2021, 12, 3532. [Google Scholar] [CrossRef]
- Glenner, G.G. Amyloid deposits and amyloidosis. The beta-fibrilloses (first of two parts). N. Engl. J. Med. 1980, 302, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Sletten, K.; Natvig, J.B.; Husby, G.; Juul, J. The complete amino acid sequence of a prototype immunoglobulin-lambda light-chain-type amyloid-fibril protein AR. Biochem. J. 1981, 195, 561–572. [Google Scholar] [CrossRef] [Green Version]
- Solomon, A.; Frangione, B.; Franklin, E.C. Bence Jones proteins and light chains of immunoglobulins. Preferential association of the V lambda VI subgroup of human light chains with amyloidosis AL (lambda). J. Clin. Investig. 1982, 70, 453–460. [Google Scholar] [CrossRef]
- Annamalai, K.; Gührs, K.H.; Koehler, R.; Schmidt, M.; Michel, H.; Loos, C.; Gaffney, P.M.; Sigurdson, C.J.; Hegenbart, U.; Schönland, S.; et al. Polymorphism of Amyloid Fibrils In Vivo. Angew. Chem. Int. Ed. Engl. 2016, 55, 4822–4825. [Google Scholar] [CrossRef]
- Annamalai, K.; Liberta, F.; Vielberg, M.T.; Close, W.; Lilie, H.; Gührs, K.H.; Schierhorn, A.; Koehler, R.; Schmidt, A.; Haupt, C.; et al. Common Fibril Structures Imply Systemically Conserved Protein Misfolding Pathways In Vivo. Angew. Chem. Int. Ed. Engl. 2017, 56, 7510–7514. [Google Scholar] [CrossRef] [PubMed]
- Solomon, A.; Weiss, D.T.; Murphy, C.L.; Hrncic, R.; Wall, J.S.; Schell, M. Light chain-associated amyloid deposits comprised of a novel kappa constant domain. Proc. Natl. Acad. Sci. USA 1998, 95, 9547–9551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engvig, J.P.; Olsen, K.E.; Gislefoss, R.E.; Sletten, K.; Wahlström, O.; Westermark, P. Constant region of a kappa III immunoglobulin light chain as a major AL-amyloid protein. Scand. J. Immunol. 1998, 48, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Lavatelli, F.; Mazzini, G.; Ricagno, S.; Iavarone, F.; Rognoni, P.; Milani, P.; Nuvolone, M.; Swuec, P.; Caminito, S.; Tasaki, M.; et al. Mass spectrometry characterization of light chain fragmentation sites in cardiac AL amyloidosis: Insights into the timing of proteolysis. J. Biol. Chem. 2020, 295, 16572–16584. [Google Scholar] [CrossRef]
- Enqvist, S.; Sletten, K.; Westermark, P. Fibril protein fragmentation pattern in systemic AL-amyloidosis. J. Pathol. 2009, 219, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Morgan, G.J.; Kelly, J.W. The Kinetic Stability of a Full-Length Antibody Light Chain Dimer Determines whether Endoproteolysis Can Release Amyloidogenic Variable Domains. J. Mol. Biol. 2016, 428, 4280–4297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sternke-Hoffmann, R.; Pauly, T.; Norrild, R.K.; Hansen, J.; Dupré, M.; Tucholski, F.; Duchateau, M.; Rey, M.; Metzger, S.; Boquoi, M.; et al. Universal amyloidogenicity of patient-derived immunoglobulin light chains. BioRxiv 2021. [Google Scholar] [CrossRef]
- Bansal, A.; Schmidt, M.; Rennegarbe, M.; Haupt, C.; Liberta, F.; Stecher, S.; Puscalau-Girtu, I.; Biedermann, A.; Fändrich, M. AA amyloid fibrils from diseased tissue are structurally different from in vitro formed SAA fibrils. Nat. Commun. 2021, 12, 1013. [Google Scholar] [CrossRef]
- Schönfelder, J.; Pfeiffer, P.B.; Pradhan, T.; Bijzet, J.; Hazenberg, B.P.C.; Schönland, S.O.; Hegenbart, U.; Reif, B.; Haupt, C.; Fändrich, M. Protease Resistance of ex vivo Amyloid Fibrils implies the proteolytic Selection of disease-associated Fibril Morphologies. BioRxiv 2021. [Google Scholar] [CrossRef]
- Kumar, S.; Murray, D.; Dasari, S.; Milani, P.; Barnidge, D.; Madden, B.; Kourelis, T.; Arendt, B.; Merlini, G.; Ramirez-Alvarado, M.; et al. Assay to rapidly screen for immunoglobulin light chain glycosylation: A potential path to earlier AL diagnosis for a subset of patients. Leukemia 2019, 33, 254–257. [Google Scholar] [CrossRef]
- Foss, G.S.; Nilsen, R.; Cornwell, G.C., III; Husby, G.; Sletten, K. A glycosylated Bence Jones protein and its autologous amyloid light chain containing potentially amyloidogenic residues. Scand. J. Immunol. 1998, 47, 348–354. [Google Scholar] [CrossRef]
- Dispenzieri, A.; Larson, D.R.; Rajkumar, S.V.; Kyle, R.A.; Kumar, S.K.; Kourelis, T.; Arendt, B.; Willrich, M.; Dasari, S.; Murray, D. N-glycosylation of monoclonal light chains on routine MASS-FIX testing is a risk factor for MGUS progression. Leukemia 2020, 34, 2749–2753. [Google Scholar] [CrossRef]
- Mellors, P.W.; Dasari, S.; Kohlhagen, M.C.; Kourelis, T.; Go, R.S.; Muchtar, E.; Gertz, M.A.; Kumar, S.K.; Buadi, F.K.; Willrich, M.A.V.; et al. MASS-FIX for the detection of monoclonal proteins and light chain N-glycosylation in routine clinical practice: A cross-sectional study of 6315 patients. Blood Cancer J. 2021, 11, 50. [Google Scholar] [CrossRef]
- Prado, M.J.; Nicastri, A.L.; Costa, P.L.; Rockman, T.; Tersariol, I.L.; Nader, H.B.; Barros, R.T.; Prado, E.B. The renal and hepatic distribution of Bence Jones proteins depends on glycosylation: A scintigraphic study in rats. Braz. J. Med. Biol. Res. 1997, 30, 865–872. [Google Scholar] [CrossRef] [Green Version]
- Lim, A.; Wally, J.; Walsh, M.T.; Skinner, M.; Costello, C.E. Identification and location of a cysteinyl posttranslational modification in an amyloidogenic kappa1 light chain protein by electrospray ionization and matrix-assisted laser desorption/ionization mass spectrometry. Anal. Biochem. 2001, 295, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Klafki, H.W.; Kratzin, H.D.; Pick, A.I.; Eckart, K.; Karas, M.; Hilschmann, N. Complete amino acid sequence determinations demonstrate identity of the urinary Bence Jones protein (BJP-DIA) and the amyloid fibril protein (AL-DIA) in a case of AL-amyloidosis. Biochemistry 1992, 31, 3265–3272. [Google Scholar] [CrossRef]
- Omtvedt, L.A.; Husby, G.; Cornwell, G.G., 3rd; Kyle, R.A.; Sletten, K. The amino acid sequence of the glycosylated amyloid immunoglobulin light chain protein AL MS. Scand. J. Immunol. 1997, 45, 551–556. [Google Scholar] [CrossRef]
- Yazaki, M.; Liepnieks, J.J.; Callaghan, J.; Connolly, C.E.; Benson, M.D. Chemical characterization of a lambda I amyloid protein isolated from formalin-fixed and paraffin-embedded tissue sections. Amyloid 2004, 11, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Liu, H. Macro- and Micro-Heterogeneity of Natural and Recombinant IgG Antibodies. Antibodies 2019, 8, 18. [Google Scholar] [CrossRef] [Green Version]
- Klimtchuk, E.S.; Gursky, O.; Patel, R.S.; Laporte, K.L.; Connors, L.H.; Skinner, M.; Seldin, D.C. The critical role of the constant region in thermal stability and aggregation of amyloidogenic immunoglobulin light chain. Biochemistry 2010, 49, 9848–9857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, A.; Annamalai, K.; Schmidt, M.; Grigorieff, N.; Fändrich, M. Cryo-EM reveals the steric zipper structure of a light chain-derived amyloid fibril. Proc. Natl. Acad. Sci. USA 2016, 113, 6200–6205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piehl, D.W.; Blancas-Mejía, L.M.; Ramirez-Alvarado, M.; Rienstra, C.M. Solid-state NMR chemical shift assignments for AL-09 VL immunoglobulin light chain fibrils. Biomol. NMR Assign. 2017, 11, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Piehl, D.W.; Blancas-Mejía, L.M.; Wall, J.S.; Kennel, S.J.; Ramirez-Alvarado, M.; Rienstra, C.M. Immunoglobulin Light Chains Form an Extensive and Highly Ordered Fibril Involving the N- and C-Termini. ACS Omega 2017, 2, 712–720. [Google Scholar] [CrossRef]
- Close, W.; Neumann, M.; Schmidt, A.; Hora, M.; Annamalai, K.; Schmidt, M.; Reif, B.; Schmidt, V.; Grigorieff, N.; Fändrich, M. Physical basis of amyloid fibril polymorphism. Nat. Commun. 2018, 9, 699. [Google Scholar] [CrossRef] [PubMed]
- Hora, M.; Sarkar, R.; Morris, V.; Xue, K.; Prade, E.; Harding, E.; Buchner, J.; Reif, B. MAK33 antibody light chain amyloid fibrils are similar to oligomeric precursors. PLoS ONE 2017, 12, e0181799. [Google Scholar] [CrossRef]
- Lecoq, L.; Wiegand, T.; Rodriguez-Alvarez, F.J.; Cadalbert, R.; Herrera, G.A.; Del Pozo-Yauner, L.; Meier, B.H.; Böckmann, A. A Substantial Structural Conversion of the Native Monomer Leads to in-Register Parallel Amyloid Fibril Formation in Light-Chain Amyloidosis. Chembiochem 2019, 20, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Falcon, B.; Murzin, A.G.; Fan, J.; Crowther, R.A.; Goedert, M.; Scheres, S.H. Heparin-induced tau filaments are polymorphic and differ from those in Alzheimer’s and Pick’s diseases. Elife 2019, 8, e43584. [Google Scholar] [CrossRef]
- Kollmer, M.; Close, W.; Funk, L.; Rasmussen, J.; Bsoul, A.; Schierhorn, A.; Schmidt, M.; Sigurdson, C.J.; Jucker, M.; Fändrich, M. Cryo-EM structure and polymorphism of Aβ amyloid fibrils purified from Alzheimer’s brain tissue. Nat. Commun. 2019, 10, 4760. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.; Wiese, S.; Adak, V.; Engler, J.; Agarwal, S.; Fritz, G.; Westermark, P.; Zacharias, M.; Fändrich, M. Cryo-EM structure of a transthyretin-derived amyloid fibril from a patient with hereditary ATTR amyloidosis. Nat. Commun. 2019, 10, 5008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweighauser, M.; Shi, Y.; Tarutani, A.; Kametani, F.; Murzin, A.G.; Ghetti, B.; Matsubara, T.; Tomita, T.; Ando, T.; Hasegawa, K.; et al. Structures of α-synuclein filaments from multiple system atrophy. Nature 2020, 585, 464–469. [Google Scholar] [CrossRef]
- Kraus, A.; Hoyt, F.; Schwartz, C.L.; Hansen, B.; Hughson, A.G.; Artikis, E.; Race, B.; Caughey, B. Structure of an infectious mammalian prion. BioRxiv 2021. [Google Scholar] [CrossRef]
- Radamaker, L.; Lin, Y.H.; Annamalai, K.; Huhn, S.; Hegenbart, U.; Schönland, S.O.; Fritz, G.; Schmidt, M.; Fändrich, M. Cryo-EM structure of a light chain-derived amyloid fibril from a patient with systemic AL amyloidosis. Nat. Commun. 2019, 10, 1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radamaker, L.; Baur, J.; Huhn, S.; Haupt, C.; Hegenbart, U.; Schönland, S.; Bansal, A.; Schmidt, M.; Fändrich, M. Cryo-EM reveals structural breaks in a patient-derived amyloid fibril from systemic AL amyloidosis. Nat. Commun. 2021, 12, 875. [Google Scholar] [CrossRef]
- Swuec, P.; Lavatelli, F.; Tasaki, M.; Paissoni, C.; Rognoni, P.; Maritan, M.; Brambilla, F.; Milani, P.; Mauri, P.; Camilloni, C.; et al. Cryo-EM structure of cardiac amyloid fibrils from an immunoglobulin light chain AL amyloidosis patient. Nat. Commun. 2019, 10, 1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nokwe, C.N.; Hora, M.; Zacharias, M.; Yagi, H.; John, C.; Reif, B.; Goto, Y.; Buchner, J. The Antibody Light-Chain Linker Is Important for Domain Stability and Amyloid Formation. J. Mol. Biol. 2015, 427, 3572–3586. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Structure of a light chain V-domain. (A) Three-dimensional structure (PDB entry: 6MH2) and (B) two-dimensional topology diagram of a natively folded V-domain. The Greek-key β-sandwich is formed by two antiparallel beta sheets consisting of strands ABED (cyan) and CC′C″FG (brown). The three CDR loops (blue) cluster on the same side of the structure and form the antigen binding site. The conserved disulfide bridge (yellow) and tryptophan (orange) are highlighted. N: amino terminus; C: carboxy terminus.
Figure 1.
Structure of a light chain V-domain. (A) Three-dimensional structure (PDB entry: 6MH2) and (B) two-dimensional topology diagram of a natively folded V-domain. The Greek-key β-sandwich is formed by two antiparallel beta sheets consisting of strands ABED (cyan) and CC′C″FG (brown). The three CDR loops (blue) cluster on the same side of the structure and form the antigen binding site. The conserved disulfide bridge (yellow) and tryptophan (orange) are highlighted. N: amino terminus; C: carboxy terminus.

Figure 2.
Comparison of the structures of ex vivo AL fibrils. (A) Side view of the molecular model of the fibril. Polypeptide chains are shown as ribbon diagram. (B) Side view of one monomer layer as a ribbon diagram with surface representation. Numbers represent the height difference of the polypeptide chain. (C) Cross-sectional view of the protein conformation. Disordered segments are represented as dotted lines. Blue: CDRs. Yellow: canonical disulfide bridge. Orange: conserved tryptophan. AL λ1 (PDB entry: 6IC3), AL λ3 (PDB entry: 6Z1O), AL λ6 (PDB entry: 6HUD). N: amino terminus, C: carboxy terminus.
Figure 2.
Comparison of the structures of ex vivo AL fibrils. (A) Side view of the molecular model of the fibril. Polypeptide chains are shown as ribbon diagram. (B) Side view of one monomer layer as a ribbon diagram with surface representation. Numbers represent the height difference of the polypeptide chain. (C) Cross-sectional view of the protein conformation. Disordered segments are represented as dotted lines. Blue: CDRs. Yellow: canonical disulfide bridge. Orange: conserved tryptophan. AL λ1 (PDB entry: 6IC3), AL λ3 (PDB entry: 6Z1O), AL λ6 (PDB entry: 6HUD). N: amino terminus, C: carboxy terminus.

Figure 3.
Sequence alignment and location of the mutational positions in the fibril structure. (A) Sequence alignment of the AL proteins. Conserved residues (grey), disulfide bridge (yellow) and mutated residues in respect to the germline (red) are highlighted. Dot indicates identical residue. Dash indicates absence of a residue. (B) Cross-sectional view of the AL protein fold. The first and the last residue of the ordered segments, as well as the first and the last residue of the fibril protein are indicated. Asterisk: variable C terminal end reported (position 130, 135, 151). The numbering of the amino acids refers to the alignment of the 3 AL proteins. Red: mutated position. Blue: CDRs. Yellow: disulfide bridge. Dotted line: disordered segment. PDB entries: AL λ1 (6IC3), AL λ3 (6Z1O), AL λ6 (6HUD).
Figure 3.
Sequence alignment and location of the mutational positions in the fibril structure. (A) Sequence alignment of the AL proteins. Conserved residues (grey), disulfide bridge (yellow) and mutated residues in respect to the germline (red) are highlighted. Dot indicates identical residue. Dash indicates absence of a residue. (B) Cross-sectional view of the AL protein fold. The first and the last residue of the ordered segments, as well as the first and the last residue of the fibril protein are indicated. Asterisk: variable C terminal end reported (position 130, 135, 151). The numbering of the amino acids refers to the alignment of the 3 AL proteins. Red: mutated position. Blue: CDRs. Yellow: disulfide bridge. Dotted line: disordered segment. PDB entries: AL λ1 (6IC3), AL λ3 (6Z1O), AL λ6 (6HUD).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Haupt, C. The AL Amyloid Fibril: Looking for a Link between Fibril Formation and Structure. Hemato 2021, 2, 505-514. https://0-doi-org.brum.beds.ac.uk/10.3390/hemato2030032
AMA Style
Haupt C. The AL Amyloid Fibril: Looking for a Link between Fibril Formation and Structure. Hemato. 2021; 2(3):505-514. https://0-doi-org.brum.beds.ac.uk/10.3390/hemato2030032
Chicago/Turabian StyleHaupt, Christian. 2021. "The AL Amyloid Fibril: Looking for a Link between Fibril Formation and Structure" Hemato 2, no. 3: 505-514. https://0-doi-org.brum.beds.ac.uk/10.3390/hemato2030032