
Assessments of Bacterial Community Shifts in Sediments along the Headwaters of São Francisco River, Brazil

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Sites and Sampling
2.2. DNA Extraction, PCR Amplification, and High-Throughput Sequencing
2.3. Data Analyses
3. Results
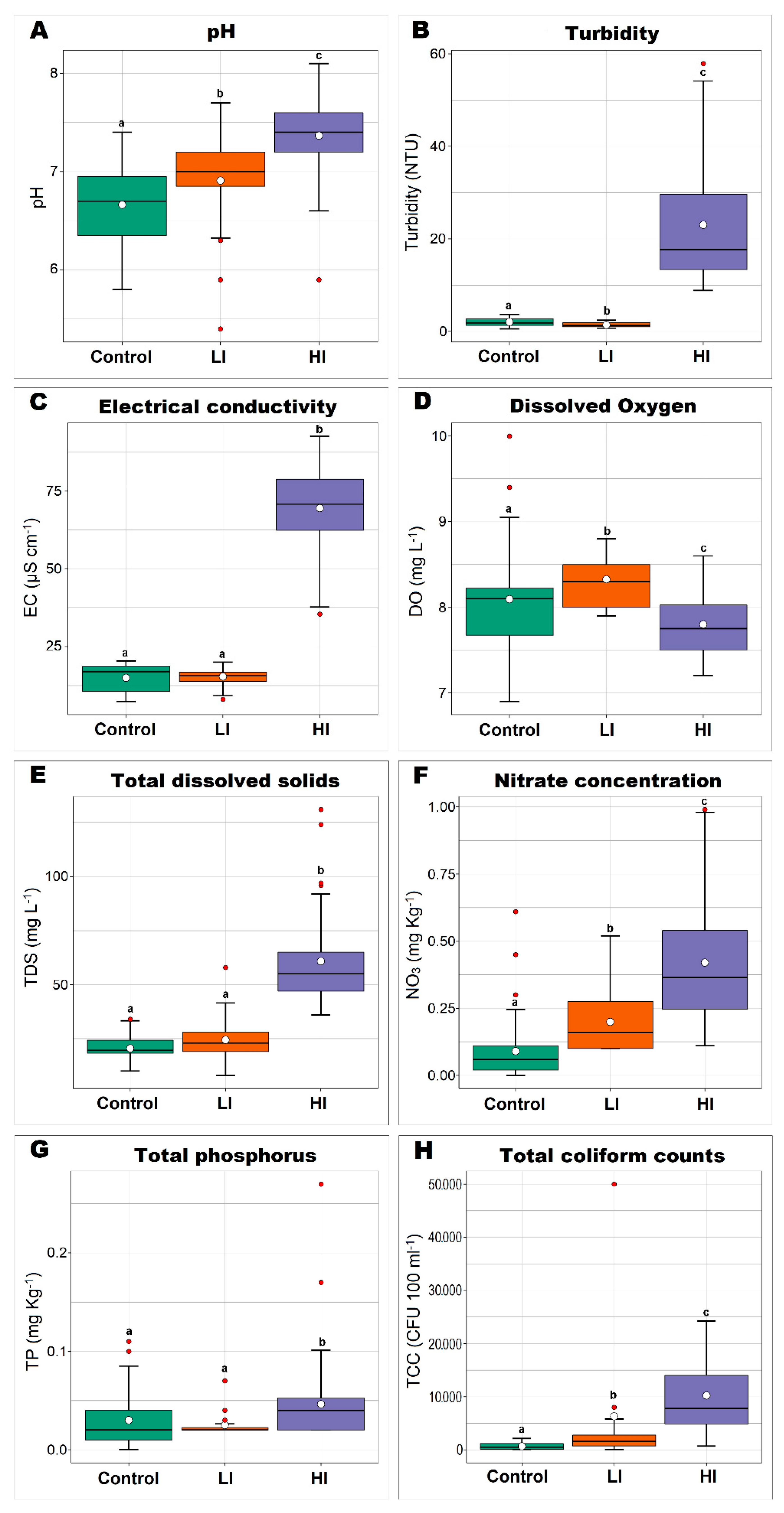
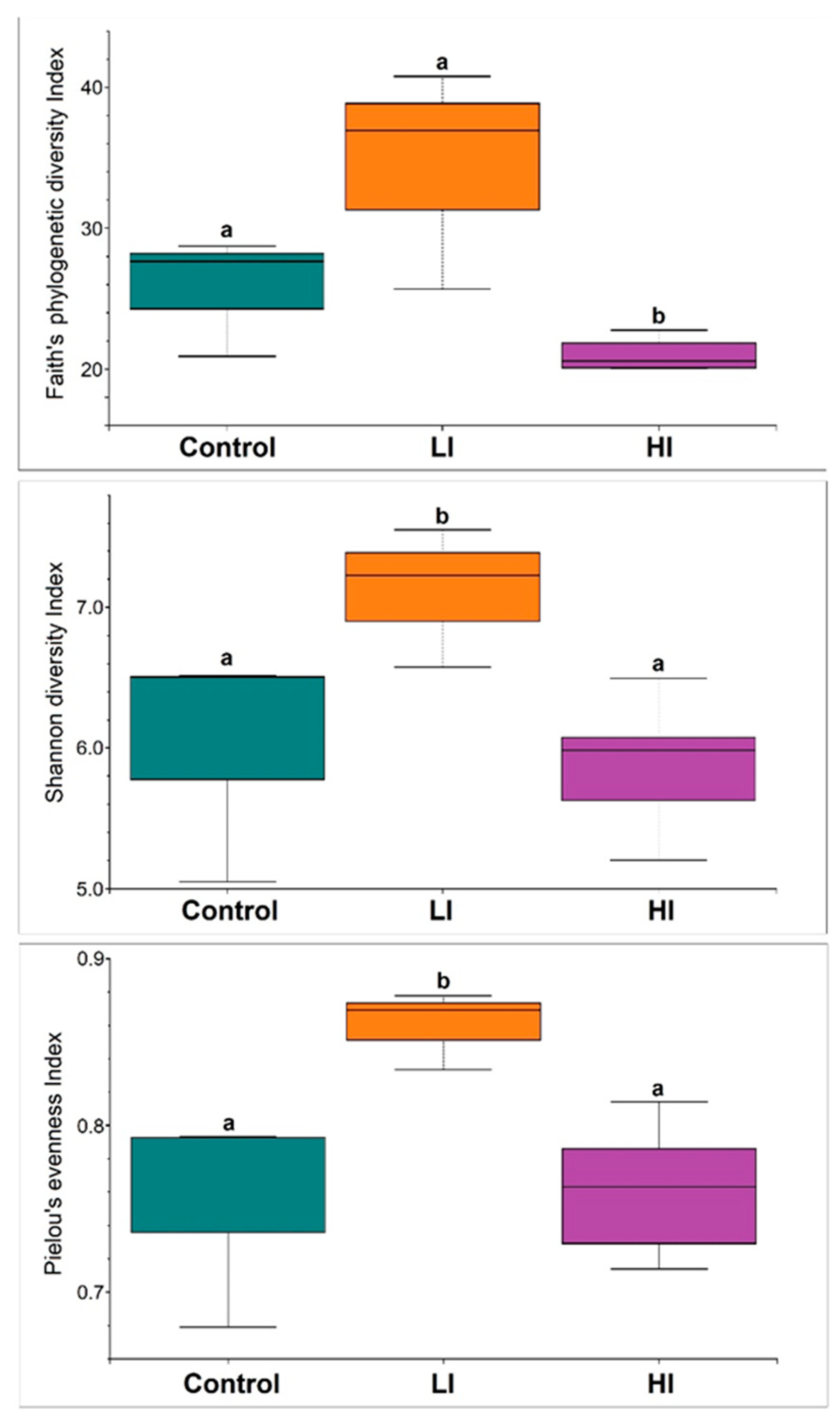
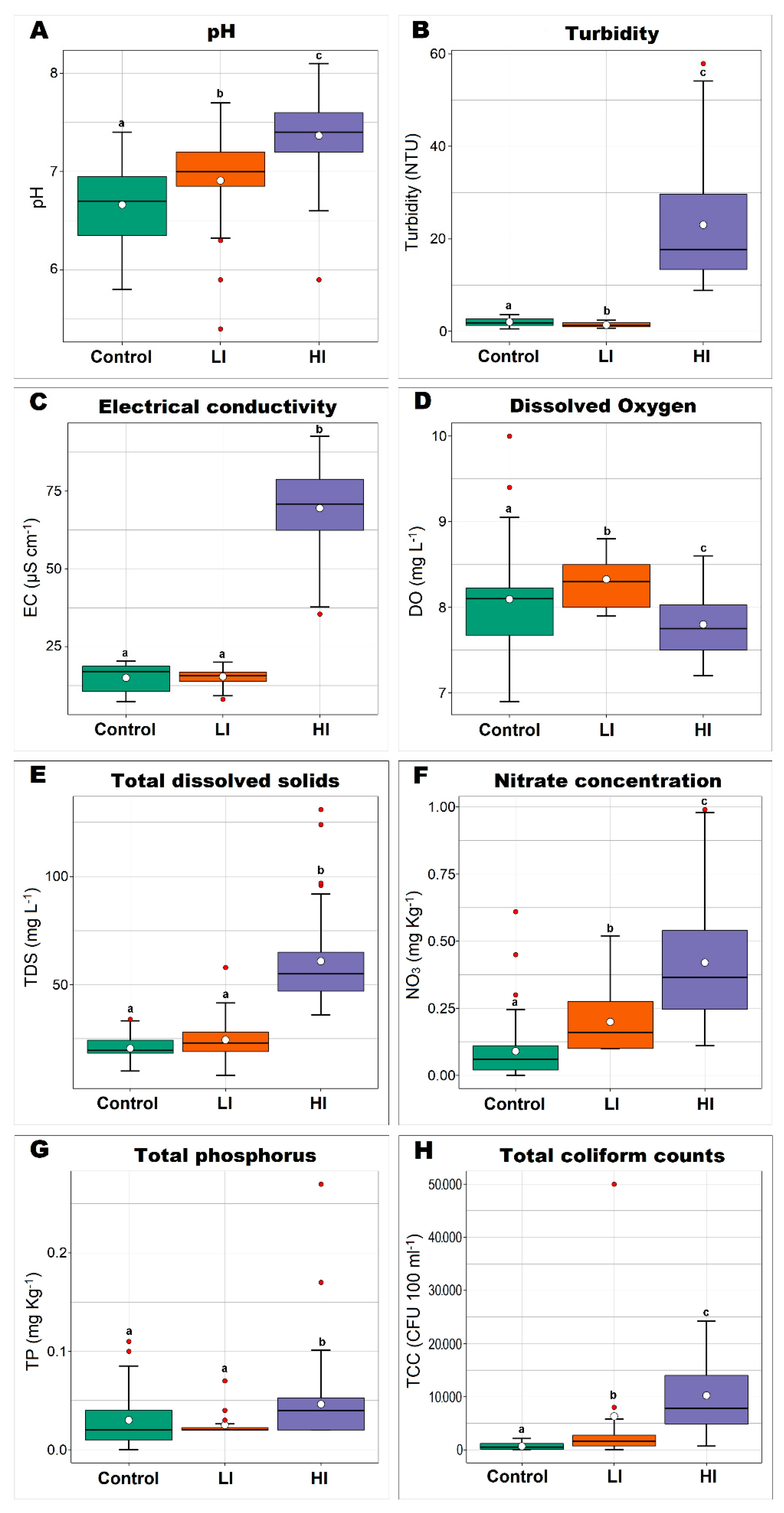
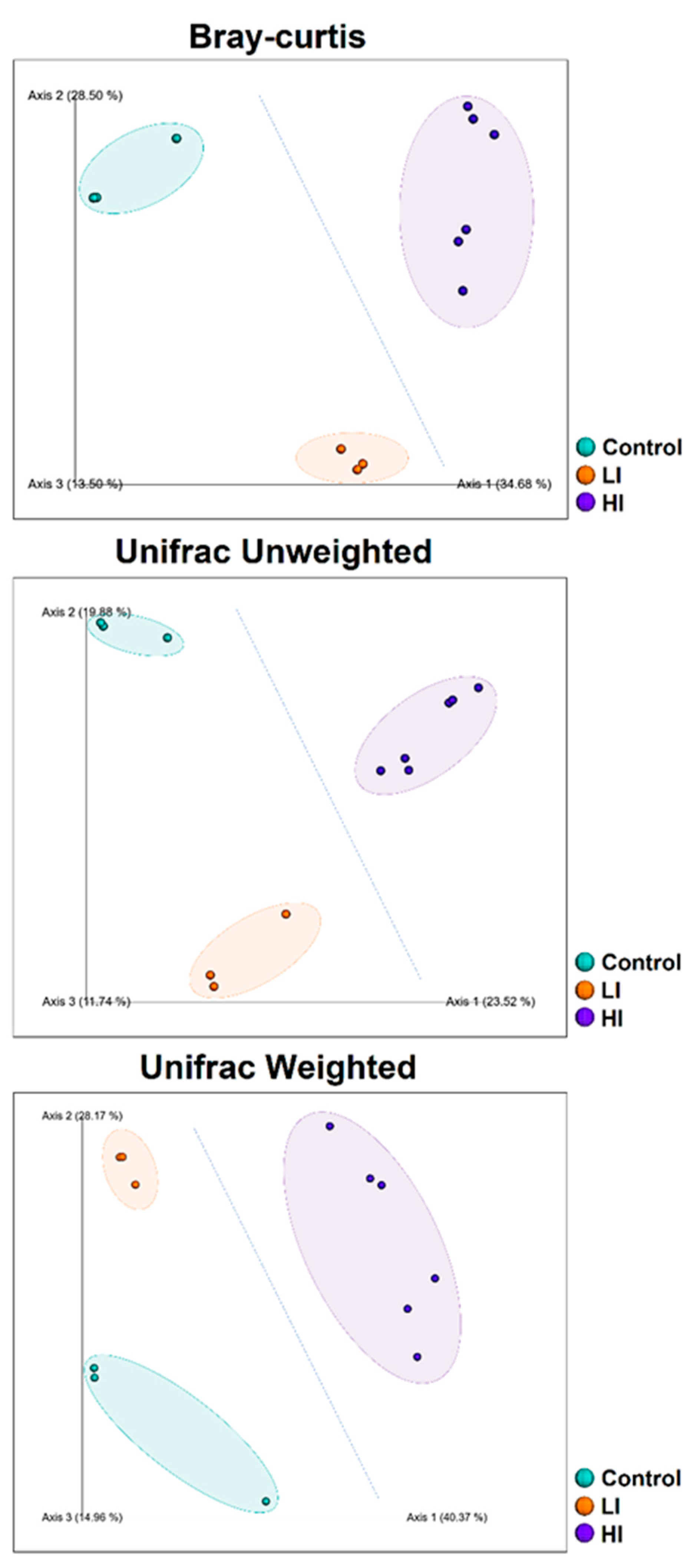
3.1. Diversity Indices of Sediment Bacterial Communities from Different Impacted Areas
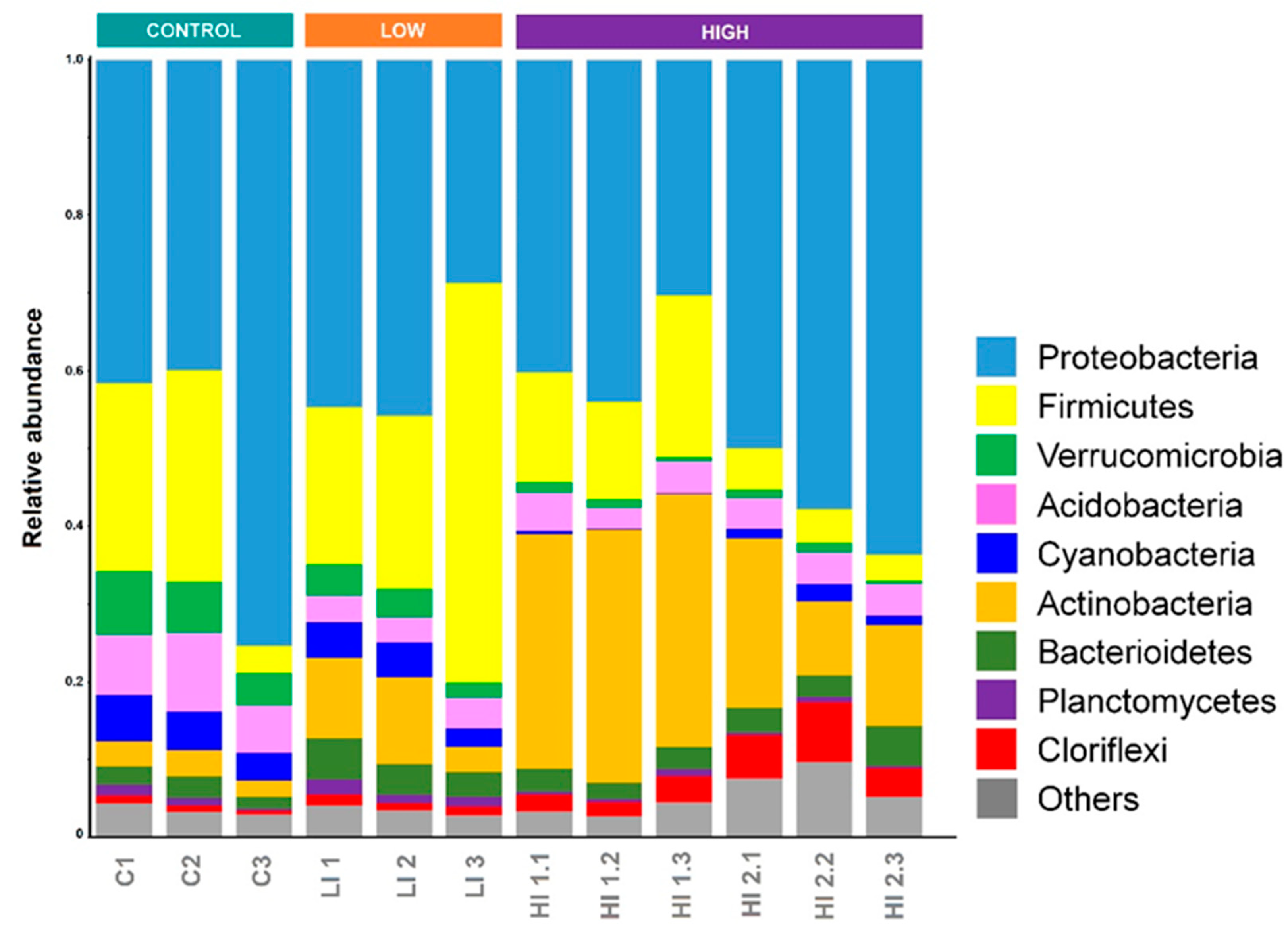
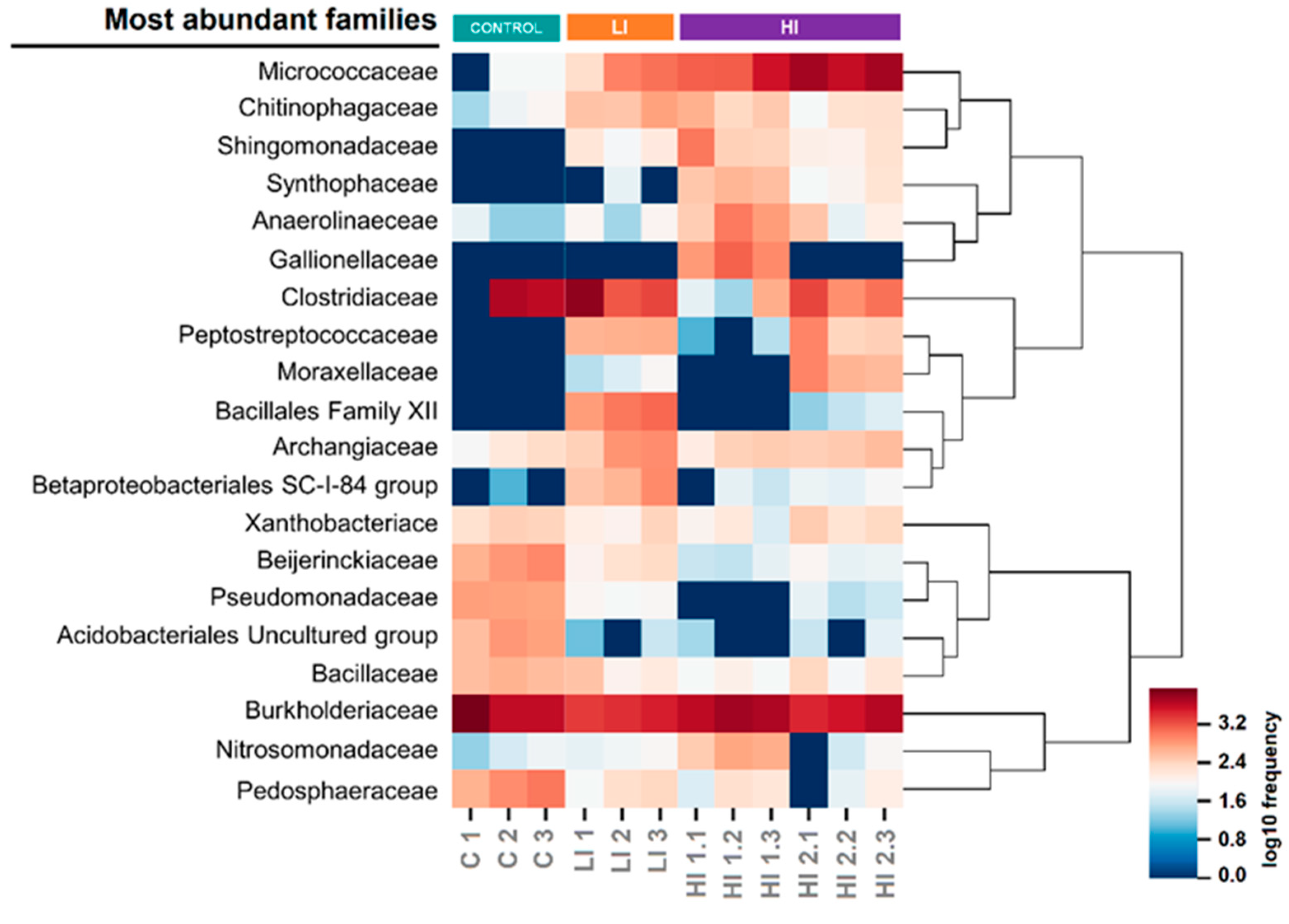
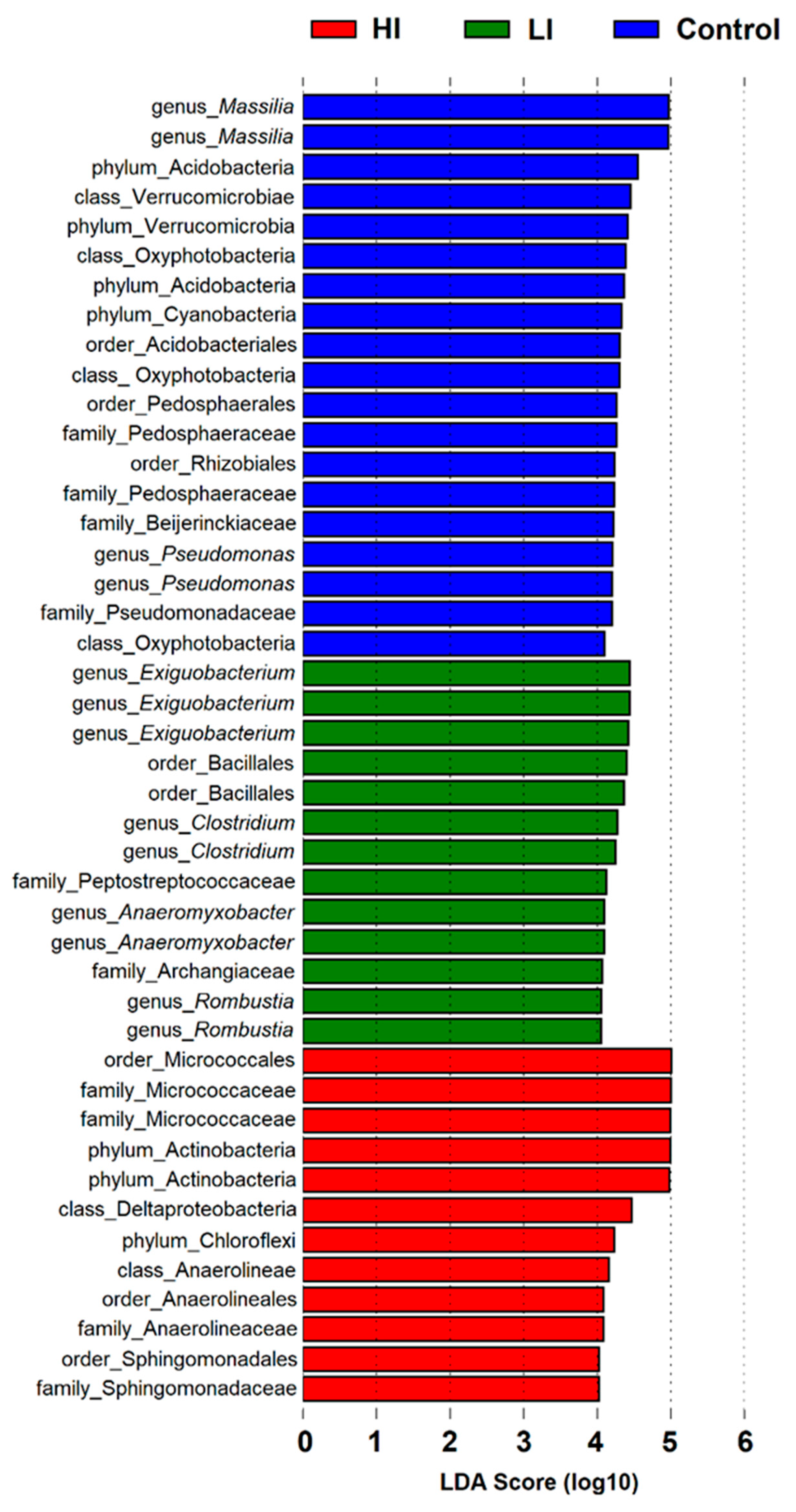
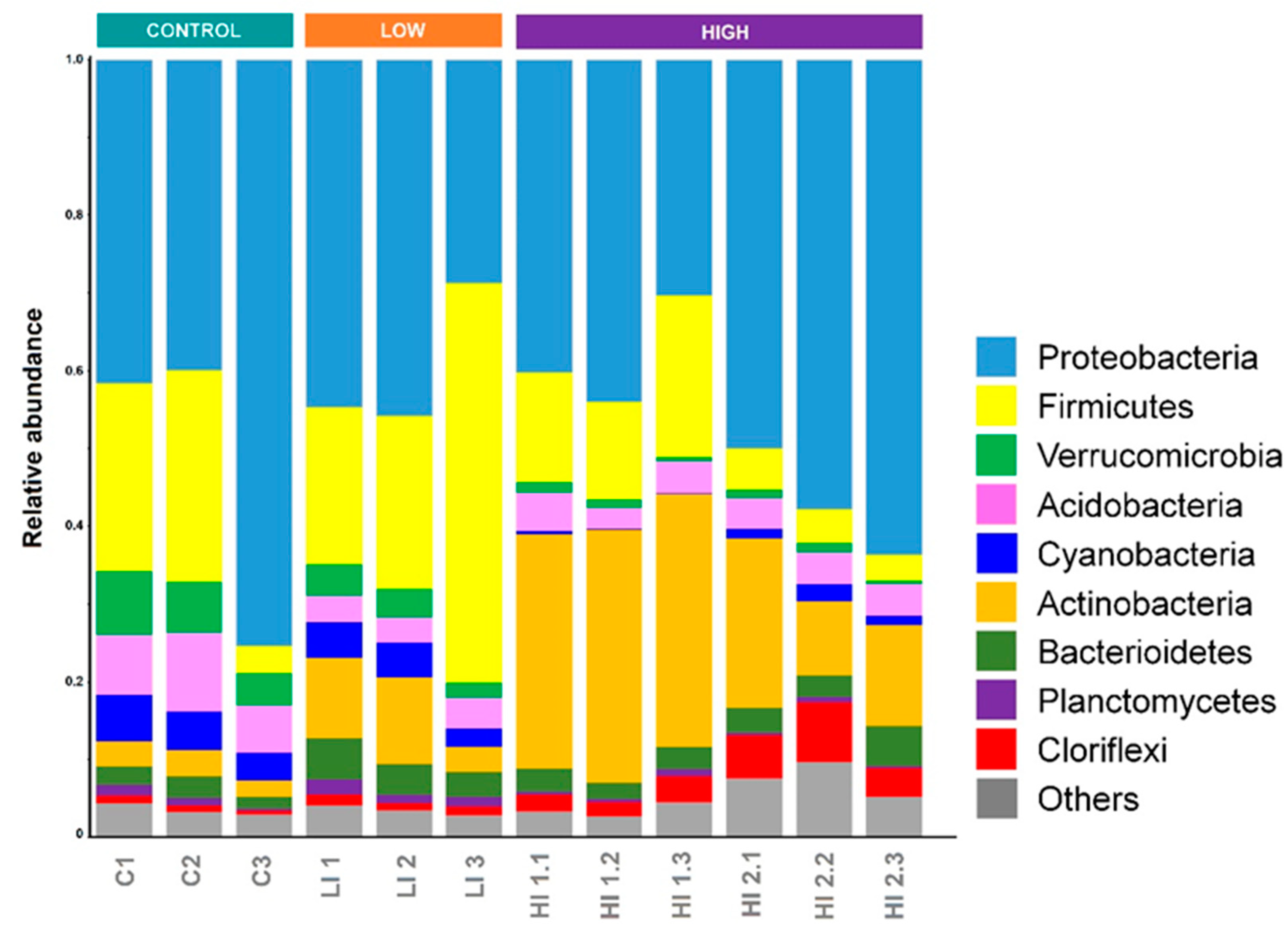
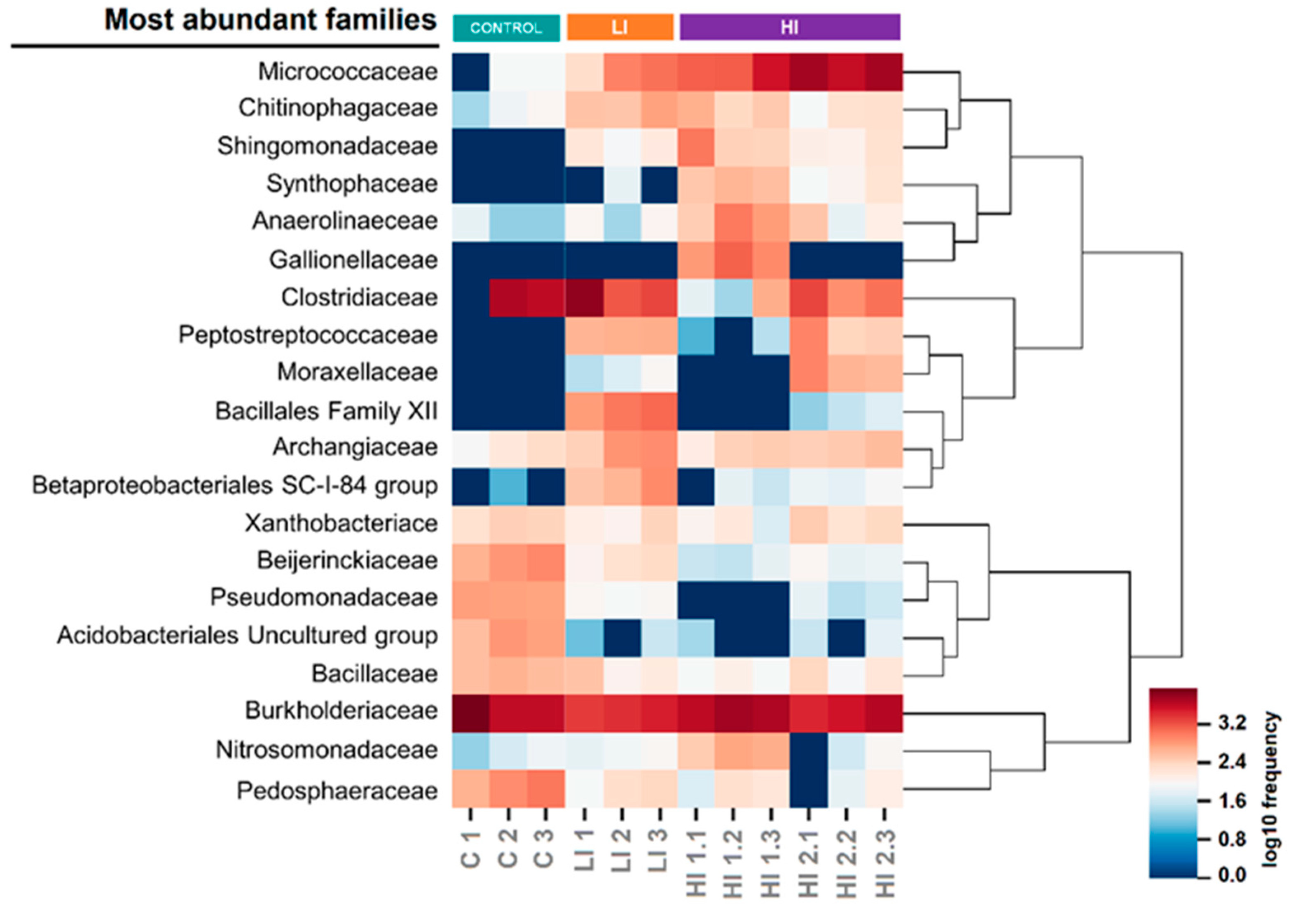
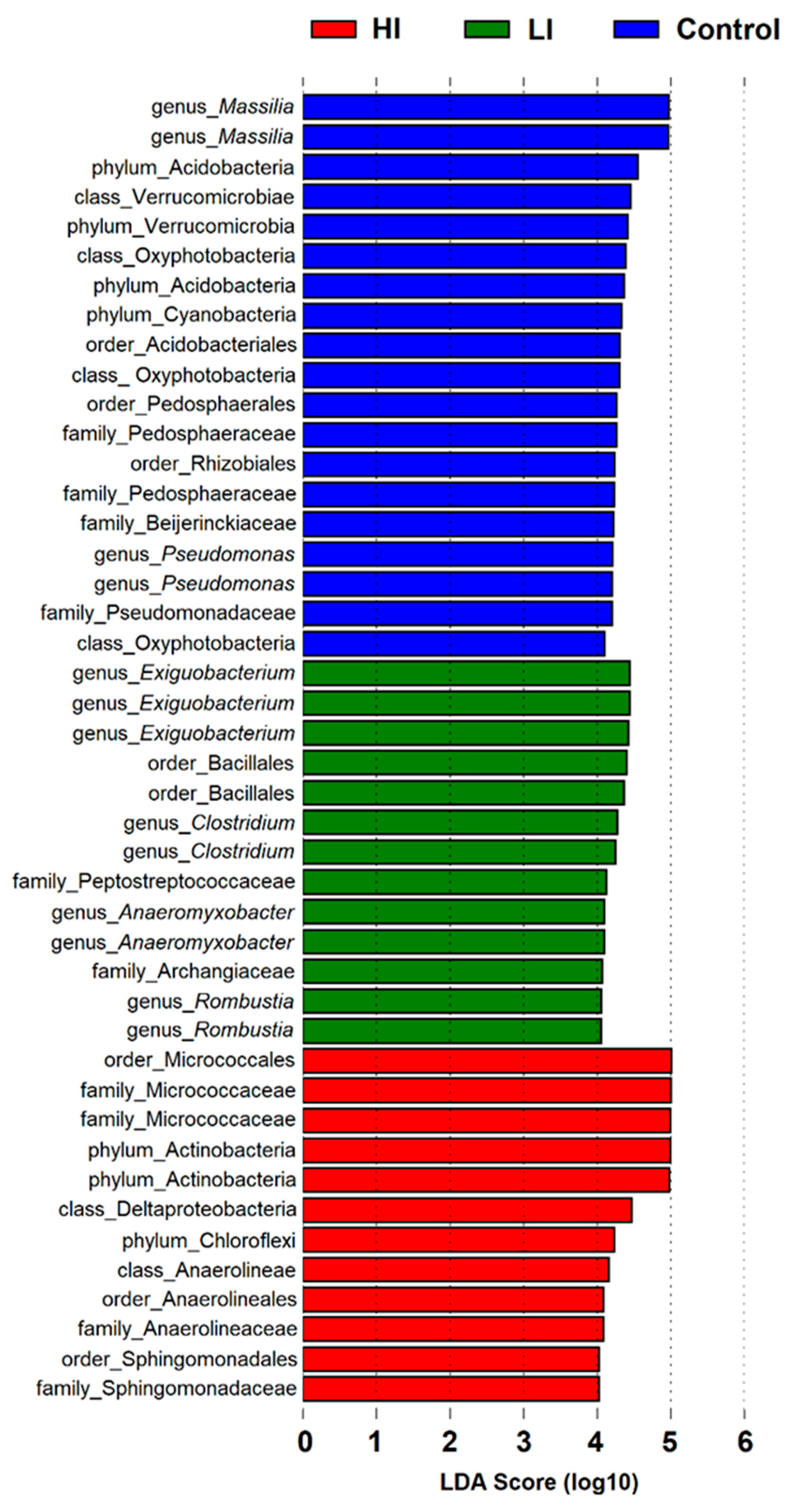
3.2. Distribution Pattern of Bacterial Taxa in Studied Areas
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Millennium Ecosystem Assessment. Ecosystems and Human Well-Being: Wetlands and Water; World Resources Institute: Washington, DC, USA, 2005; 80p. [Google Scholar]
- Nilsson, C.; Reidy, C.A.; Dynesius, M.; Revenga, C. Fragmentation and flow regulation of the world’s large river systems. Science 2005, 308, 405–408. [Google Scholar] [CrossRef] [Green Version]
- Vörösmarty, C.J.; McIntyre, P.B.; Gessner, M.O.; Dudgeon, D.; Prusevich, A.; Green, P.; Glidden, S.; Bunn, C.A.; Sullivan, C.A.; Liermann, C.R.; et al. Global threats to human water security and river biodiversity. Nature 2010, 467, 555–561. [Google Scholar] [CrossRef]
- Arthington, Á.H.; Naiman, R.J.; Mcclain, M.E.; Nilsson, C. Preserving the biodiversity and ecological services of rivers: New challenges and research opportunities. Freshw. Biol. 2010, 55, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Dale, V.H.; Beyeler, S.C. Challenges in the development and use of ecological indicators. Ecol. Indic. 2001, 1, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Dudgeon, D.; Arthington, A.H.; Gessner, M.O.; Kawabata, Z.I.; Knowler, D.J.; Lévêque, C.; Naiman, R.J.; Prieur-Richard, A.-H.; Soto, D.; Stiassny, M.L.J.; et al. Freshwater biodiversity: Importance, threats, status and conservation challenges. Biol. Rev. Biol. Proc. Camb. Philos. Soc. 2006, 681, 163–182. [Google Scholar] [CrossRef] [PubMed]
- Heink, U.; Kowarik, I. What are indicators? On the definition of indicators in ecology and environmental planning. Ecol. Indic. 2010, 10, 584–593. [Google Scholar] [CrossRef]
- Siddig, A.A.; Ellison, A.M.; Ochs, A.; Villar-Leeman, C.; Lau, M.K. How do ecologists select and use indicator species to monitor ecological change? Insights from 14 years of publication in Ecological Indicators. Ecol. Indic. 2016, 60, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Zeglin, L.H. Stream bacterial diversity in response to environmental changes: Review and synthesis of existing research. Front. Microbiol. 2015, 6, 454. [Google Scholar] [CrossRef] [Green Version]
- Bouchez, T.; Blieux, A.L.; Dequiedt, S.; Domaizon, I.; Dufresne, A.; Ferreira, S.; Godon, J.J.; Hellal, J.; Joulian, C.; Quaiser, A.; et al. Molecular microbiology methods for environmental diagnosis. Environ. Chem. Lett. 2016, 14, 423–441. [Google Scholar] [CrossRef]
- Grossart, H.-P.; Massana, R.; McMahon, K.D.; Walsh, D.A. Linking metagenomics to aquatic bacterial ecology and biogeochemical cycles. Limnol. Oceanogr. 2020, 65, 2–20. [Google Scholar] [CrossRef] [Green Version]
- Paerl, H.W.; Dyble, J.; Moisander, P.H.; Noble, R.T.; Piehler, M.F.; Pinckney, J.L.; Steppe, T.F.; Twomey, L.; Valdes, L.M. Bacterial indicators of aquatic ecosystem change: Current applications to eutrophication studies. FEMS Microbiol. Ecol. 2003, 46, 233–246. [Google Scholar] [CrossRef]
- Sims, A.; Zhang, Y.; Gajaraj, S.; Brown, P.B.; Hu, Z. Toward the development of bacterial indicators for wetland assessment. Water Res. 2013, 47, 1711–1725. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhao, T.; Wang, Q.; Li, L.; Shen, T.; Gao, G. Bacterial community composition in aquatic and sediment samples with spatiotemporal dynamics in large, shallow, eutrophic Lake Chaohu, China. J. Freshw. Ecol. 2019, 34, 575–589. [Google Scholar] [CrossRef]
- Yang, Y.; Li, S.; Gao, Y.; Chen, Y.; Zhan, A. Environment-driven geographical distribution of bacterial communities and identification of indicator taxa in Songhua River. Ecol. Indic. 2019, 101, 62–70. [Google Scholar] [CrossRef]
- Adhikari, N.P.; Liu, Y.; Liu, K.; Zhang, F.; Adhikari, S.; Chen, Y.; Liu, X. Bacterial community composition and diversity in Koshi River, the largest river of Nepal. Ecol. Indic. 2019, 104, 501–511. [Google Scholar] [CrossRef]
- Liu, J.; Chen, X.; Shu, H.Y.; Lin, X.R.; Zhou, Q.X.; Bramryd, T.; Shu, W.-S.; Huang, L.N. Bacterial community structure and function in sediments from e-waste contaminated rivers at Guiyu area of China. Environ. Pollut. 2018, 235, 171–179. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, J.; Li, H.; Yang, H.; Peng, C.; Peng, Z.; Lu, L. Shift in the bacterial community composition of surface water and sediment along an urban river. Sci. Total Environ. 2018, 627, 600–612. [Google Scholar] [CrossRef]
- Huang, W.; Chen, X.; Wang, K.; Chen, J.; Zheng, B.; Jiang, X. Comparison among the bacterial communities in the lake, lake wetland, and estuary sediments of a plain river network. Microbiologyopen 2017, 8, e644. [Google Scholar]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods. 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Sheng, H.F.; He, Y.; Wu, J.Y.; Jiang, Y.X.; Tam, N.F.; Zhou, H.W. Comparison of the levels of bacterial diversity in freshwater, intertidal wetland, and marine sediments by using millions of illumine tags. Appl. Environ. Microbiol. 2012, 78, 8264–8271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinger, L.; Gobet, A.; Pommier, T. Two decades of describing the unseen majority of aquatic bacterial diversity. Mol. Ecol. 2012, 21, 1878–1896. [Google Scholar] [CrossRef]
- Guo, J.; Zhao, L.; Lu, W.; Jia, H.; Sun, Y. Bacterial communities in water and sediment shaped by paper mill pollution and indicated bacterial taxa in sediment in Daling River. Ecol. Indic. 2016, 60, 766–773. [Google Scholar] [CrossRef]
- Niu, L.; Li, Y.; Wang, P.; Zhang, W.; Wang, C.; Li, J.; Wu, H. Development of a bacterial community-based index of biotic integrity (MC-IBI) for the assessment of ecological status of rivers in the Taihu Basin, China. Ecol. Indic. 2018, 85, 204–213. [Google Scholar] [CrossRef]
- Roberto, A.A.; Van Gray, J.B.; Leff, L.G. Sediment bacteria in an urban stream: Spatiotemporal patterns in community composition. Water Res. 2018, 134, 353–369. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Cheng, L.; Wang, L.; Zhu, T.; Cai, W.; Hua, Z.; Wang, Y.; Wang, W. Changes of Bacterial Communities in Response to Prolonged Hydrodynamic Disturbances in the Eutrophic Water-Sediment Systems. Int. J. Environ. Res. Public Health 2019, 16, 3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eggleton, J.; Thomas, K.V. A review of factors affecting the release and bioavailability of contaminants during sediment disturbance events. Environ. Int. 2004, 30, 973–980. [Google Scholar] [CrossRef]
- Saxena, G.; Marzinelli, E.M.; Naing, N.N.; He, Z.L.; Liang, Y.T.; Tom, L.; Mitra, S.; Ping, H.; Joshi, U.M.; Reuben, S.; et al. Ecogenomics reveals metals and land-use pressures on bacterial communities in the waterways of a megacity. Environ. Sci. Technol. 2015, 49, 1462–1471. [Google Scholar] [CrossRef]
- Xie, Y.; Wang, J.; Wu, Y.; Ren, C.; Song, C.; Yang, J.; Yu, H.; Giesy, J.P.; Zhang, X. Using in situ bacterial communities to monitor contaminants in river sediments. Environ. Pollut. 2016, 212, 348–357. [Google Scholar] [CrossRef]
- Xie, Y.; Wang, J.; Yang, J.; Giesy, J.P.; Yu, H.; Zhang, X. Environmental DNA metabarcoding reveals primary chemical contaminants in freshwater sediments from different land-use types. Chemosphere 2017, 172, 201–209. [Google Scholar] [CrossRef]
- Brasil MMA. Caderno da Região Hidrográfica do São Francisco; Ministério do Meio Ambiente, Secretaria de Recursos Hídricos: Brasília, Brasil, 2006; 148p.
- Lopes, J.E.G.; Barros, M.T.L.; Brandão, J.L.B. Projeto de Gerenciamento Integrado das Atividades Desenvolvidas em terra na bacia do São Francisco—Subprojeto 4.4, Determinação de Subsídios para Procedimentos Operacionais dos Principais Reservatórios da Bacia do São Francisco; ANA/GEF/PNUMA/OEA: Brasília, Brasil, 2002. [Google Scholar]
- Ministério do Desenvolvimento Urbano e Meio Ambiente. Conselho Nacional do Meio Ambiente—CONAMA. Resolução nº 357, de 17 de março de 2005. Dispõe sobre a classificação dos corpos de água e diretrizes ambientais para o seu enquadramento, bem como estabelece as condições e padrões de lançamento de efluentes, e dá outras providências. In Diário Oficial da República Federativa do Brasil; Ministério do Desenvolvimento Urbano e Meio Ambiente. Conselho Nacional do Meio Ambiente—CONAMA: Brasília, DF, Brasil, 2005. [Google Scholar]
- IGAM—Instituto Mineiro de Gestão de Águas. 2020; Portal InfoHidro. Available online: http://portalinfohidro.igam.mg.gov.br/ (accessed on 10 February 2020).
- Gilbert, J.A.; Jansson, J.K.; Knight, R. The Earth Microbiome project: Successes and aspirations. BMC Biol. 2014, 12, 69. [Google Scholar] [CrossRef] [Green Version]
- QIIME 2 Development Team. QIIME 2. Available online: https://docs.qiime2.org (accessed on 1 October 2017).
- Callahan, B.J.; McMurdie, P.J.; Holmes, S.P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017, 11, 2639. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2, high-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, T.M.; Hajibabaei, M. Scaling up: A guide to high-throughput genomic approaches for biodiversity analysis. Mol. Ecol. 2018, 27, 313–338. [Google Scholar] [CrossRef] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faust, K.; Raes, J. Bacterial interactions: From networks to models. Nat. Rev. Microbiol. 2012, 10, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wang, X.; Wang, C.; Miao, L.; Hou, J.; Yuan, Q. Shift in bacterioplankton diversity and structure: Influence of anthropogenic disturbances along the Yarlung Tsangpo River on the Tibetan Plateau, China. Sci. Rep. 2017, 7, 12529. [Google Scholar] [CrossRef] [PubMed]
- Tao, K.; Liu, Y.; Ke, T.; Zhang, Y.; Xiao, L.; Li, S.; Wei, S.; Chen, L.; Hu, T. Patterns of bacterial and archaeal communities in sediments in response to dam construction and sewage discharge in Lhasa River. Ecotoxicol. Environ. Saf. 2019, 178, 195–201. [Google Scholar] [CrossRef]
- Rasool, A.; Xiao, T. Response of bacterial communities to elevated thallium contamination in river sediments. Geomicrobiol. J. 2018, 35, 854–868. [Google Scholar] [CrossRef]
- Wei, J.; Gao, J.; Wang, N.; Liu, Y.; Wang, Y.; Bai, Z.; Zhuang, X.; Zhuang, G. Differences in soil bacterial response to anthropogenic disturbances in Sanjiang and Momoge Wetlands, China. FEMS Microbiol. Ecol. 2019, 95, fiz110. [Google Scholar] [CrossRef] [PubMed]
- Staebe, K.; Botes, M.; Madlala, T.; Oberholster, P.J.; Cloete, T.E. Bacterial community diversity as a potential bioindicator of AMD and steel plant effluent in a channelled valley bottom wetland. Water Air Soil Pollut. 2018, 229, 397. [Google Scholar] [CrossRef]
- Lv, X.; Yu, J.; Fu, Y.; Ma, B.; Qu, F.; Ning, K.; Wu, H. A meta-analysis of the bacterial and archaeal diversity observed in wetland soils. Sci. World J. 2014, 2014, 437684. [Google Scholar] [CrossRef] [Green Version]
- Marti, E.; Balcázar, J.L. Use of pyrosequencing to explore the benthic bacterial community structure in a river impacted by wastewater treatment plant discharges. Res. Microbiol. 2014, 165, 468–471. [Google Scholar] [CrossRef]
- Lu, X.M.; Lu, P.Z. Characterization of bacterial communities in sediments receiving various wastewater effluents with high-throughput sequencing analysis. Microb. Ecol. 2014, 67, 612–623. [Google Scholar] [CrossRef]
- Drury, B.; Rosi-Marshall, E.; Kelly, J.J. Wastewater treatment effluent reduces the abundance and diversity of benthic bacterial communities in urban and suburban rivers. Appl. Environ. Microbiol. 2013, 79, 1897–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalia, A.; Gosal, S.K. Effect of pesticide application on soil microorganisms. Arch. Agron. Soil Sci. 2011, 57, 569–596. [Google Scholar] [CrossRef]
- Wakelin, S.A.; Colloff, M.J.; Kookana, R.S. Effect of wastewater treatment plant effluent on microbial function and community structure in the sediment of a freshwater stream with variable seasonal flow. Appl. Environ. Microbiol. 2008, 74, 2659–2668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abia, A.L.K.; Alisoltani, A.; Keshri, J.; Ubomba-Jaswa, E. Metagenomic analysis of the bacterial communities and their functional profiles in water and sediments of the Apies River, South Africa, as a function of land use. Sci. Total Environ. 2018, 616, 326–334. [Google Scholar] [CrossRef]
- Wu, H.; Li, Y.; Zhang, J.; Niu, L.; Zhang, W.; Cai, W.; Zhu, X. Sediment bacterial communities in a eutrophic lake influenced by multiple inflow-rivers. Environ. Sci. Pollut. Res. 2017, 24, 19795–19806. [Google Scholar] [CrossRef]
- Hu, A.; Yang, X.; Chen, N.; Hou, L.; Ma, Y.; Yu, C.P. Response of bacterial communities to environmental changes in a mesoscale subtropical watershed, Southeast China. Sci. Total Environ. 2014, 472, 746–756. [Google Scholar] [CrossRef]
- Basta, T.; Buerger, S.; Stolz, A. Structural and replicative diversity of large plasmids, from sphingomonads that degrade polycyclic aromatic compounds and xenobiotics. Microbiology 2005, 151, 2025–2037. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, E.; Edward, E.-I.-C.; Delong, F.; Lory, S.; Stackebrandt, E.; Thompson, F. The Prokaryotes: Applied Bacteriology and Biotechnology; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- National Sanitation Foundation—NSF. Water Quality Index (WQI). Available online: http://www.nsf.org/consumer/earth_day/wqi.asp (accessed on 10 February 2020).
- García-Armisen, T.; İnceoğlu, Ö.; Ouattara, N.K.; Anzil, A.; Verbanck, M.A.; Brion, N.; Servais, P. Seasonal variations and resilience of bacterial communities in a sewage polluted urban river. PLoS ONE 2014, 9, e92579. [Google Scholar] [CrossRef]
- Van Rossum, T.; Peabody, M.A.; Uyaguari-Diaz, M.I.; Cronin, K.I.; Chan, M.; Slobodan, J.R.; Nesbitt, M.J.; Suttle, C.A.; Hsiao, W.W.L.; Tang, P.K.C.; et al. Year-long metagenomic study of river microbiomes across land use and water quality. Front Microbiol. 2015, 6, 1405. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Paula, M., Jr.; Costa Silva, T.A.; Araújo, A.S.; Lacorte, G.A. Assessments of Bacterial Community Shifts in Sediments along the Headwaters of São Francisco River, Brazil. Conservation 2021, 1, 91-105. https://0-doi-org.brum.beds.ac.uk/10.3390/conservation1020008
de Paula M Jr., Costa Silva TA, Araújo AS, Lacorte GA. Assessments of Bacterial Community Shifts in Sediments along the Headwaters of São Francisco River, Brazil. Conservation. 2021; 1(2):91-105. https://0-doi-org.brum.beds.ac.uk/10.3390/conservation1020008
Chicago/Turabian Stylede Paula, Marcos, Jr., Thiago Augusto Costa Silva, Amanda Soriano Araújo, and Gustavo Augusto Lacorte. 2021. "Assessments of Bacterial Community Shifts in Sediments along the Headwaters of São Francisco River, Brazil" Conservation 1, no. 2: 91-105. https://0-doi-org.brum.beds.ac.uk/10.3390/conservation1020008