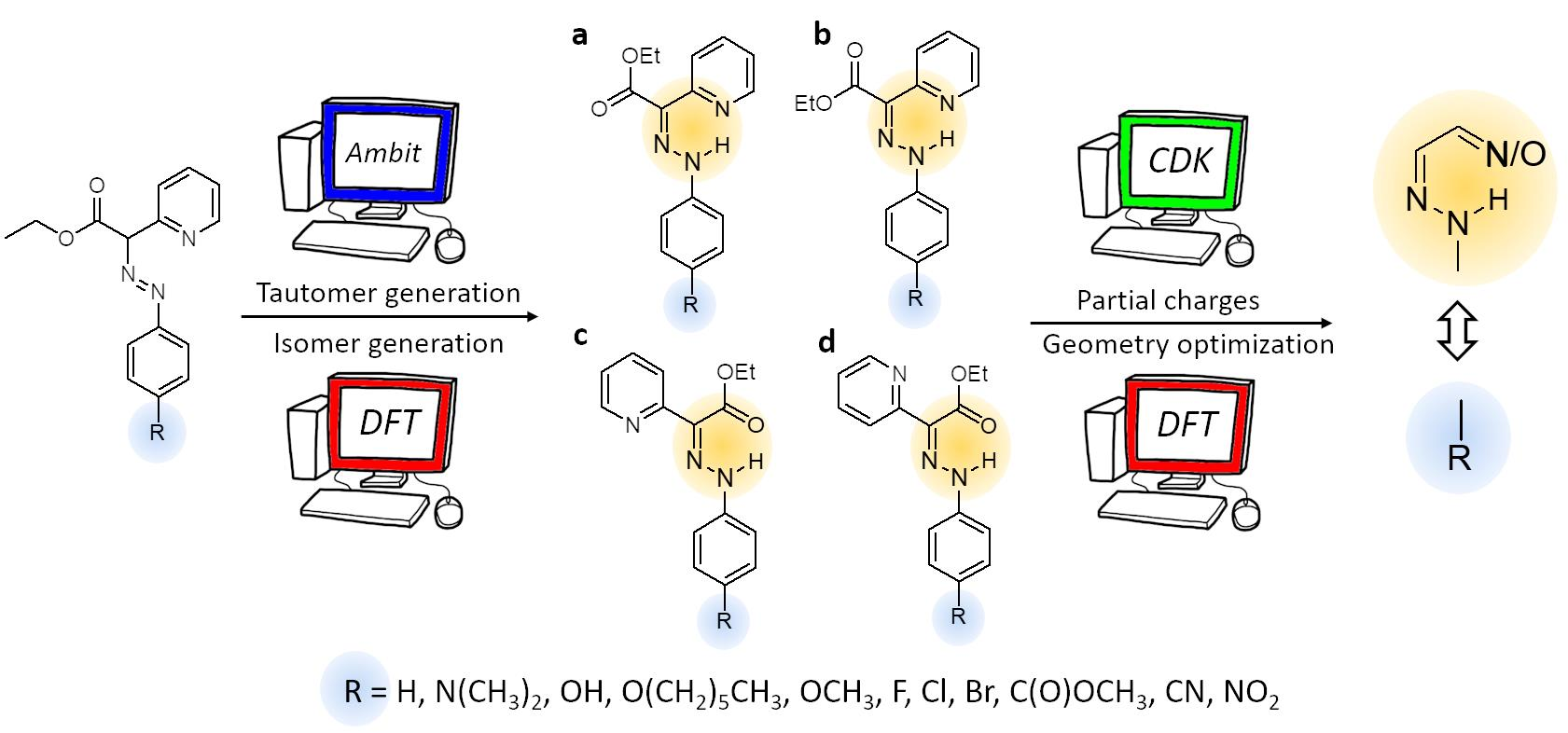
It is apparent that the adequacy of chemical property predictions strongly depends on the structure representation, including the proper treatment of the tautomeric forms. In a wider context of chemoinformatics activities, chemical structure representation tackles a variety of challenges concerning representation techniques on 1D, 2D and 3D structure levels, handling of implicit and explicit hydrogen atoms, aromaticity detection and representation and many more challenges that are out of the scope of the current paper. The open-source tool for automatic generation of tautomers of a given organic compound, Ambit-Tautomer, was used for preliminary screening of the possible constitutional isomers. The rough scoring of the tautomers generated by Ambit-Tautomer was performed with subsequent DFT calculations for selected structures (
Figure S1, Supporting Information), and high-energy nonconventional tautomers were excluded from further consideration. Selected keto tautomers of compounds
1–11 were subjected to quantum chemical calculations (full geometry optimization in the gas phase and in acetonitrile environment).
3.2. Para-Substituted Phenylhydrazones (Compounds 2–11)
The functional groups (atoms or groups of atoms) appended to the phenyl ring are shown in
Table 2. The isomers of compounds
2–
11 are identical to those for compound
1 (shown in
Figure 3). The substituents on the phenyl ring, selected by Su et al. [
8], range from typical electron-donating groups (EDGs) to typical electron-withdrawing ones (EWGs) (
Table 2).
For compounds
1,
2,
7 and
11 (unsubstituted system and –N(CH
3)
2, –Cl and –NO
2 substituted ones), the Gibbs energy differences ∆G
1 and ∆G
36 are given in
Table 1. The Gibbs energy differences in the gas phase (∆G
1) and in acetonitrile (∆G
36) calculated for the rest of the compounds in the series are given in
Table S1.
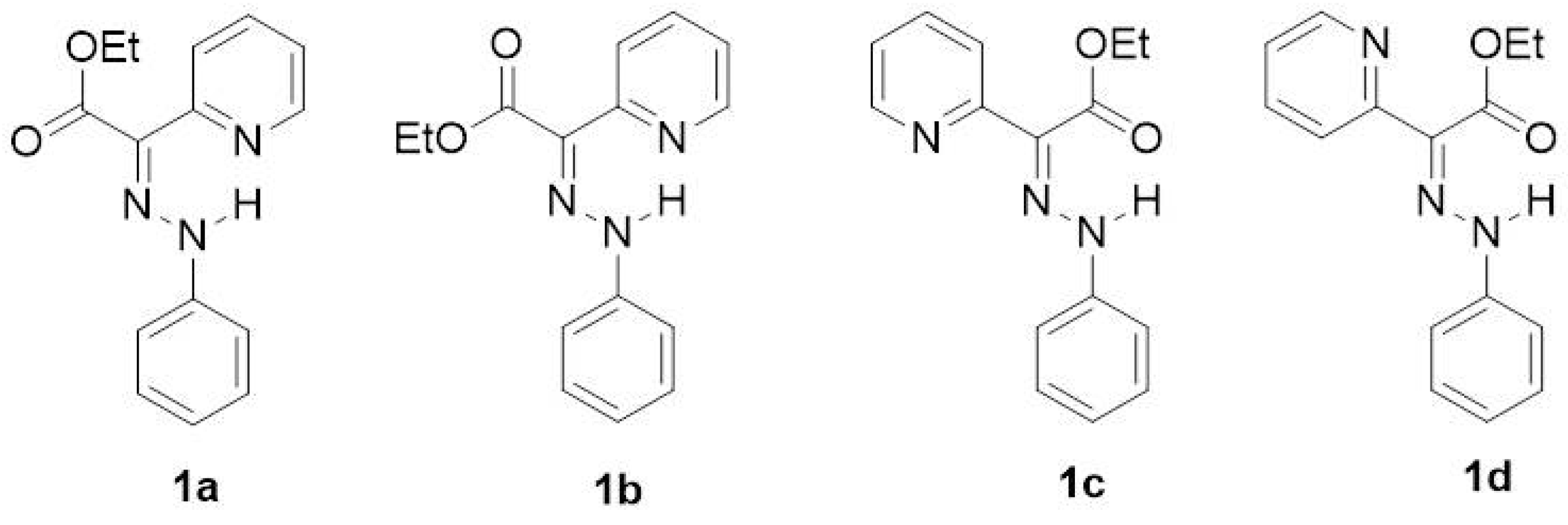
As expected, isomer
b is found to be energetically preferred over the isomers with Z-configuration (
c and
d) and over isomer
a (the second
E-isomer) for all studied compounds (
Table 1 and
Table S1). The results obtained at both computational levels slightly differ, as MN12-SX functional predicts higher Gibbs energy differences, but the energy order sequence is the same. The solvation has an important effect on the equilibrium of the isomers (
a–
d) considered. The acetonitrile solvent reduces the preference for the most-favored isomer
b over the
a and
c isomers.
The investigated isomers
a–
d of compounds
1–
11 contain intramolecular H-bonded phenyl and pyridine (pyridyl) rings. The presence of an EDG in the phenyl ring is expected to enhance the electron density and to increase the intramolecular hydrogen bond (IMHB) strength, whereas EWGs have the opposite effect. On the other hand, the acidity of the proton bound to the N1 atom should decrease in presence of EDGs. The dependencies between H-bond strength and the Hammett substituent constant σ experimentally observed by Su et al. [
8] are consistent with RAHB theory for the
Z-configurations (isomers with HN–N=C–C=O molecular fragment) of the different derivatives but not consistent for the
E-configurations (with HN–N=C–C=N fragment) of compounds
1–
11. We, therefore, decided to systematically investigate the structures of
a–
d isomers of the
1–
11 compound series. Selected bond lengths and N10⋯H distances calculated at M062X/6-31+G(d,p) level of theory for
a–
d isomers of compounds
1,
2,
7 and
11 are summarized in
Table 3. Derivatives with EDG (
2) and EWG (
7 and
11) are selected as representatives. Values calculated at ε = 1 and ε = 36 are compared.
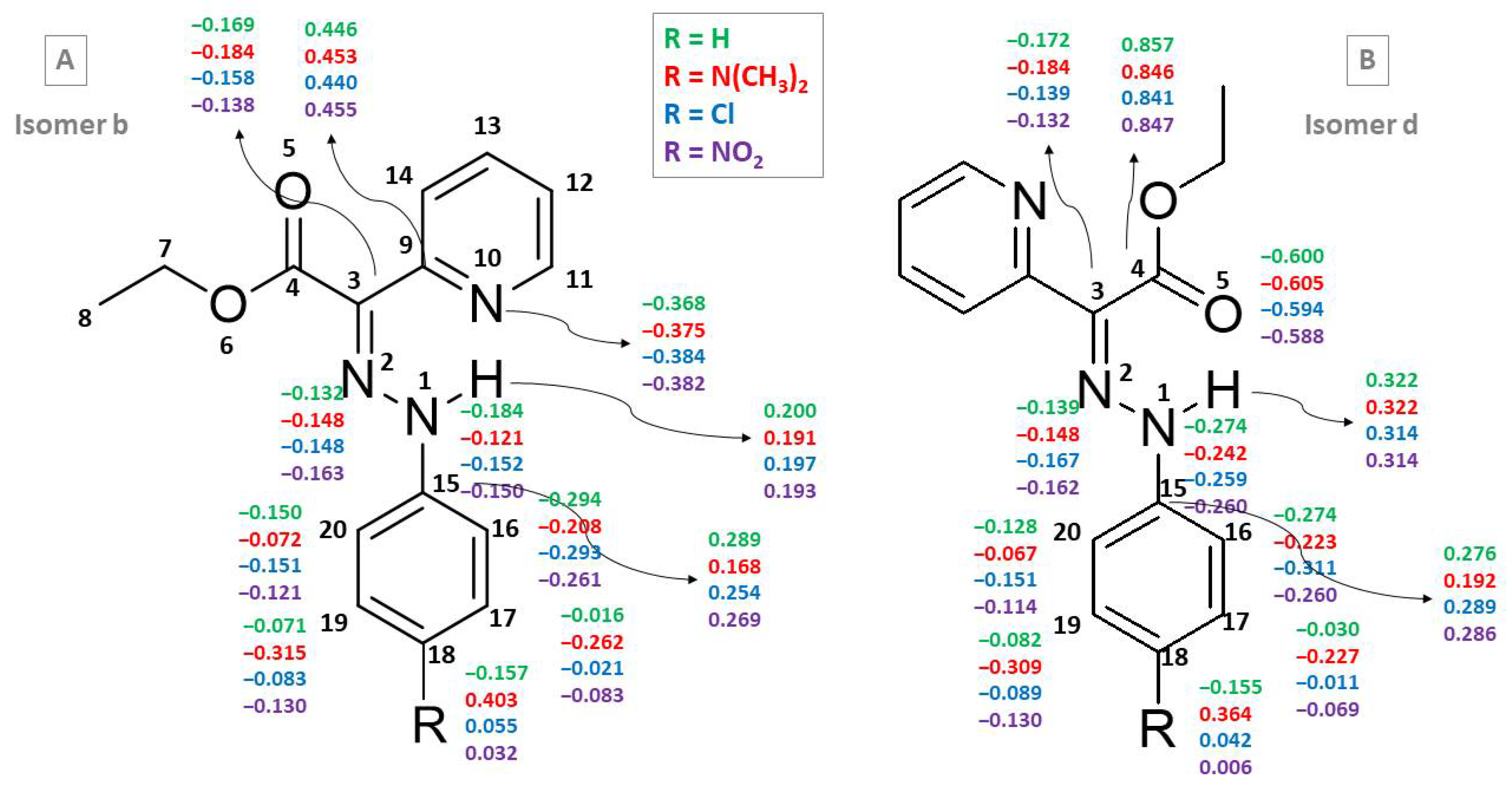
The IMHB results show that the NH⋯N unit of b isomers of compounds 1, 2, 7 and 11 is stronger than the corresponding NH⋯N or NH⋯O interactions of other forms and is consistent with the stability order. The d isomers are characterized by longer NH⋯O distances, while c isomers (with rotated pyridine ring, not involved in IMHB) have shorter NH⋯O distances, which does not support the stability order of the Z-isomers. It is interesting to differentiate trends in the NH⋯N and NH⋯O distances in presence of EDGs/EWGs. NH⋯N and NH⋯O distances calculated for 2a–d (–N(CH3)2 substituted in the phenyl ring) are slightly shorter (by ~0.02 Å) than those for the unsubstituted system. The EWG (–Cl or –NO2)-substituted compounds follow this same tendency found for EDG-substituted ones with two exceptions: 7d and 11d in acetonitrile have larger NH⋯O distances than 1d.
The single N1-N2 and double N2=C3 bonds have almost identical lengths of 1.30 Å in 1 and 2. In the EWG-substituted compounds 7 and 11, the double N2=C3 bond is slightly shorter (1.29 Å) than the single N1-N2 bond (1.31 Å–1.32 Å). The substitution effect is weaker for isomers b of compounds 1, 7 and 11; i.e., N1-N2 and N2=C3 bonds remain of almost identical length.
In this work, a popular model (CHELPG) of partial atomic charge was tested on the series of unsubstituted and
p-substituted phenylhydrazones. Partial atomic charges have application in the molecular modeling of different chemically relevant areas, including the investigation of the charge transfers within a single molecule. It should be noted that the model of partial atomic charges considered here is not used to predict the ordering of possible isomers of the compounds studied. We are turning our attention only to charges on atoms in the HN–N=C–C=N and HN–N=C–C=O molecular fragments (
Table 4).
As mentioned previously, the R substituents on the phenyl ring, selected by Su et al. [
8], are of two types: electron-donating (EDGs) and electron-withdrawing ones (EWGs). Usually, substituents with lone pairs (e.g., –OCH
3, –NH
2, –N(CH
3)
2) are electron-donating groups (EDG)—they activate the aromatic ring by increasing the electron density on the ring through a resonance-donating effect. Substituents with π bonds to electronegative atoms (e.g., –C=O, –NO
2) adjacent to the π-system are electron-withdrawing groups (EWG)—they deactivate the aromatic ring by decreasing the electron density on the ring through a resonance-withdrawing effect. Halogen substituents are not only inductive electron-withdrawing (due to their electronegativity) but also resonance-donating (lone pair donation). In summary, an EDG adds electron density to the π-system, making it more nucleophilic, and an EWG removes electron density from the π-system, making it less nucleophilic. How do these effects influence the
para-positioned HN–N=C–C=N and HN–N=C–C=O molecular fragments?
The
c and
d isomers of compounds
1,
2,
7 and
11 are characterized by larger partial charges of the prototropic H atom both in the gas phase and in acetonitrile (
Table 4 and
Figure 4). The sum of the partial charges of the atoms from the HN–N=C–C=N and HN–N=C–C=O fragments reveals different trends for compounds with these two fragments (
E- and
Z-isomers, respectively).
Figure 4 shows the CHELPG charges for the
b and
d isomers of compounds
1,
2,
7 and
11 in the gas phase.
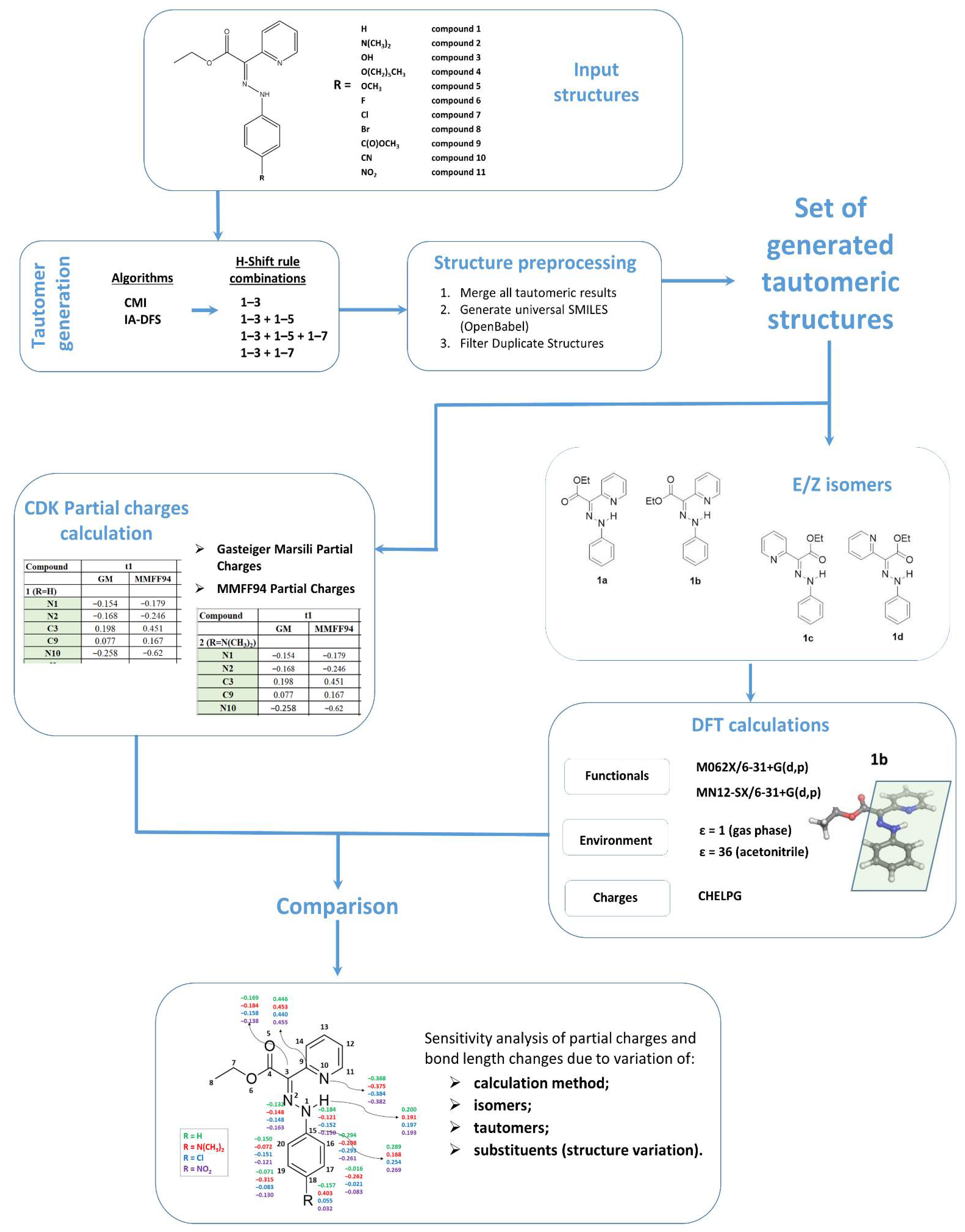
We performed sensitivity analysis of partial charges and bond length changes due to variation of different factors: calculation method, isomers, tautomers and substituents (structure variation). For this purpose, we calculated the root mean square deviation (
RMSD) of the studied parameter,
p (e.g., partial atomic charge or bond length), along the atoms/bonds (designated in the formula with index
) from the major structural fragment: N1-N2-C3-C9-N10⋯H21. For example, the
RMSD for comparing isomers
a and
b is calculated as follows:
or the comparison between Cl substituted and unsubstituted structures is performed by calculating
RMSD in the following manner:
When calculating
RMSD due to a variation of a particular factor (e.g., isomer
a vs.
b), all other factors were fixed (e.g., method, tautomer and substituent for the case of comparing isomers
a and
b). A variety of tables for different combinations of the other fixed factors are given in the
Supplementary Materials (see Tables S3–S18 and Raw Data Sheets 1–5).
Table 5 shows the deviations (
RMSD values) between partial charges of isomers
a and
b and between isomers
c and
d. RMSD values for the gas phase are roughly twice larger than the deviation obtained in a solution environment of acetonitrile. Charge redistribution due to isomerism is less influenced by the substituent type for the calculations in the gas phase.
Table 6 shows the RMSD changes of partial charges of the main structural fragment due to the substituent variation. Within the environment of acetonitrile solvent, RMSD of the charges due to substituents is about 8−10 times larger than the same RMSD in the gas phase for isomers
c and
d (e.g., 0.137 vs. 0.015), and it is about twice larger for isomers
a and
b. According to these calculations, the acetonitrile solvent is helpful for a larger charge redistribution.
The differences between DFT partial charges and those obtained with rapid methods for partial charge calculation (GM and MMFF94) show RMSD
DFT/GM values within the range 0.2–0.3 and RMSD
DFT/MMFF94 within the range 0.3–0.4 (see
Tables S8–S11 in the Supplementary Materials comparing tautomer 5 vs. isomer b). The sensitivity of the rapid methods of Gasteiger–Marsili and MMFF94 is not sufficient (RMSD values of about 0.05) to distinguish well the charge redistribution within tautomeric forms (see
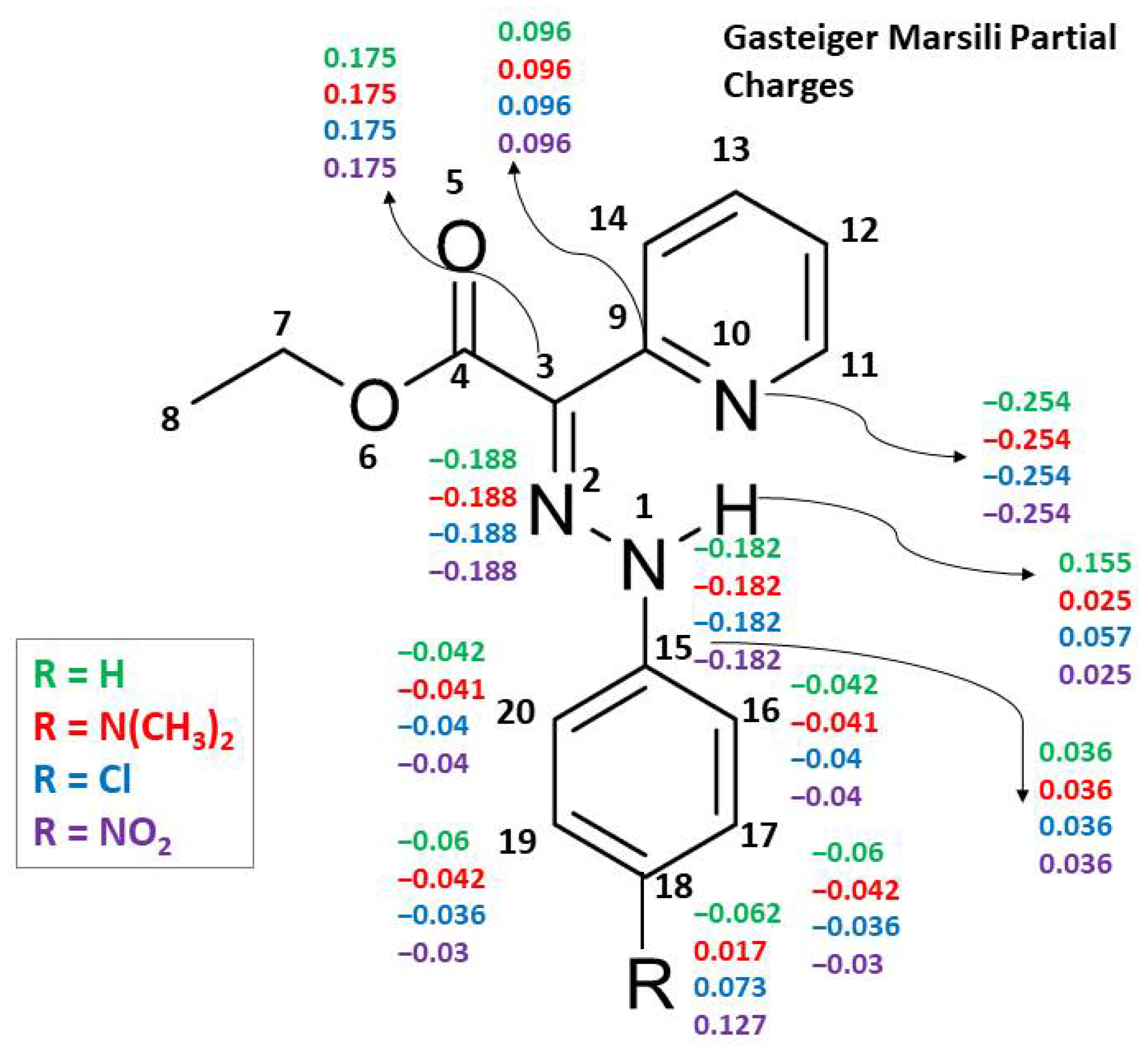
Supplementary Tables S12 and S13). The lower sensitivity of the rapid methods GM and MMFF94 is also seen by examining the partial charge changes, due to substituent variation, which are distributed up to topological distance 3 and hence not influencing the main structural fragment (see
Figure 5 and the tables with detailed structures and partial charges in the
Supplementary Materials). Such results are expected for rapid methods due to the application of the attenuation approach based only on topological information.
RMSD values for bond changes of the main structural fragment (i.e., atoms N1, N2, C3, C9, N10 and H21) show very small changes in the bond lengths (see
Supplementary Tables S14–S18). The major variation is for the hydrogen bonds (N10⋯H21, O5⋯H21), which vary around 0.01–0.05 Å.


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
