
A Computational Study of Molecular Mechanism of Chloroquine Resistance by Chloroquine Resistance Transporter Protein of Plasmodium falciparum via Molecular Modeling and Molecular Simulations


Abstract
:1. Introduction
2. Materials and Methods
2.1. Homology Model Building of PfCRT
2.2. Molecular Docking Calculations
2.3. Molecular Dynamics (MD) Simulations
3. Results
3.1. Homology Models of PfCRT
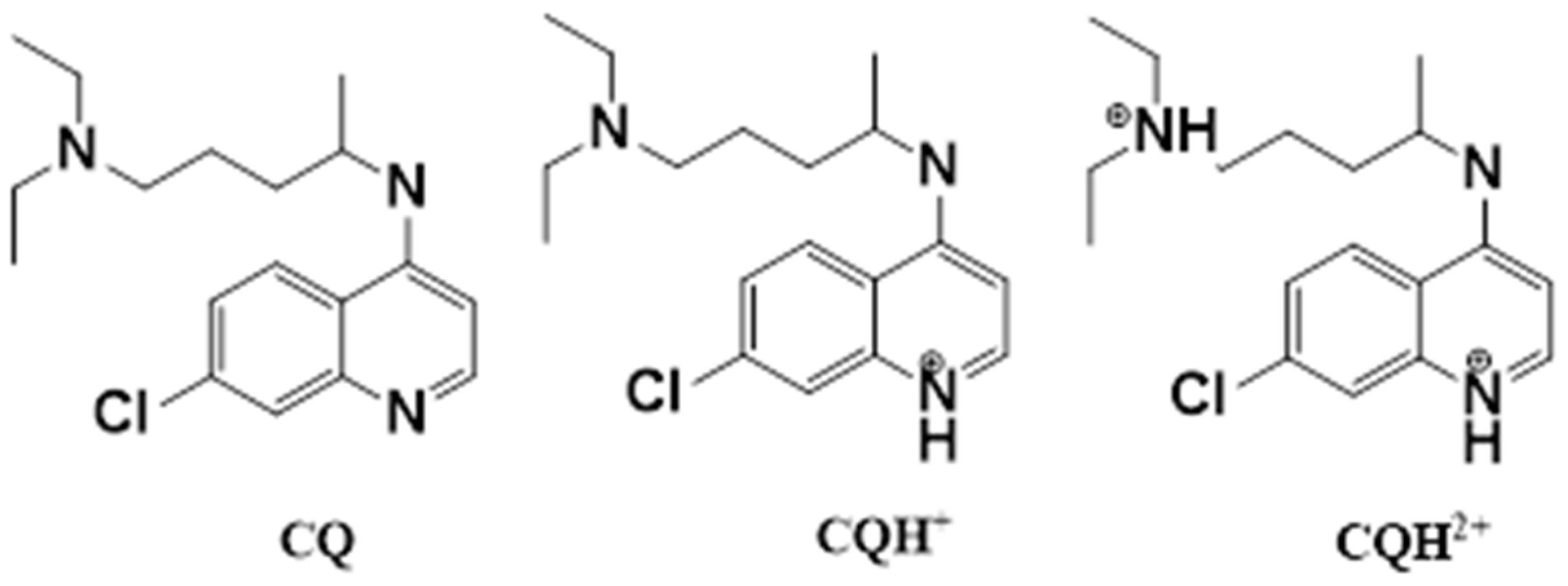
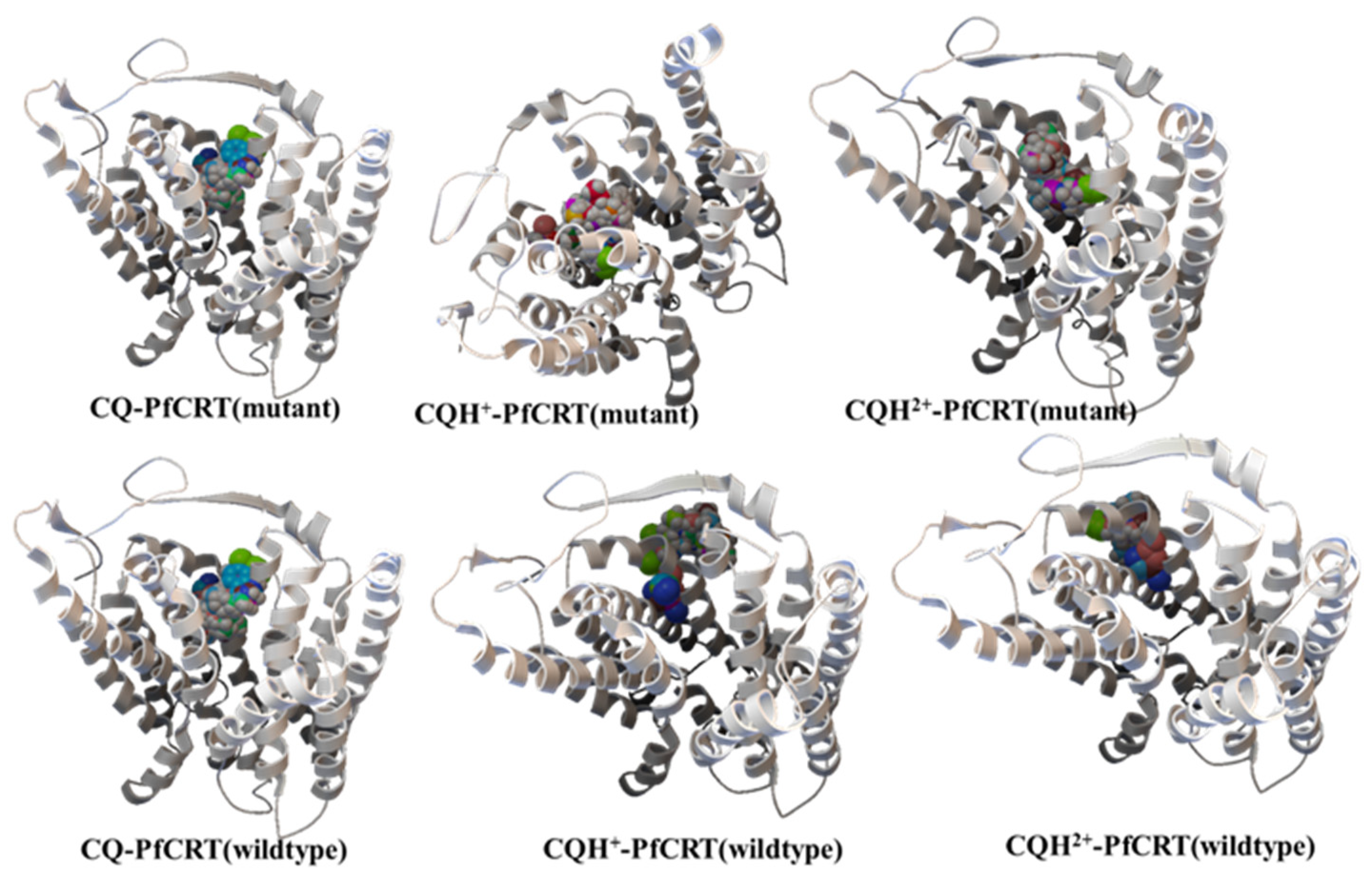
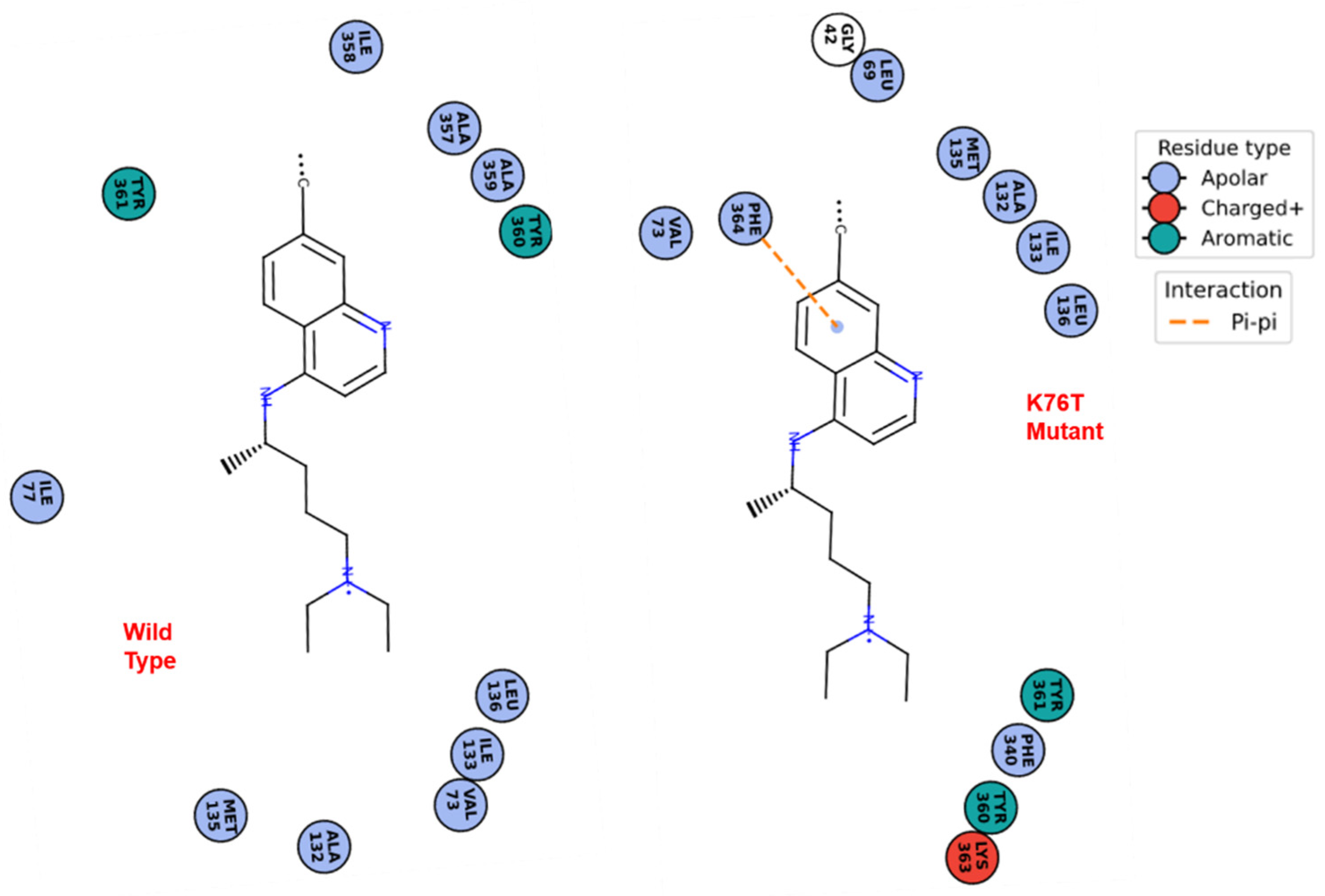
3.2. Molecular Docking Calculations
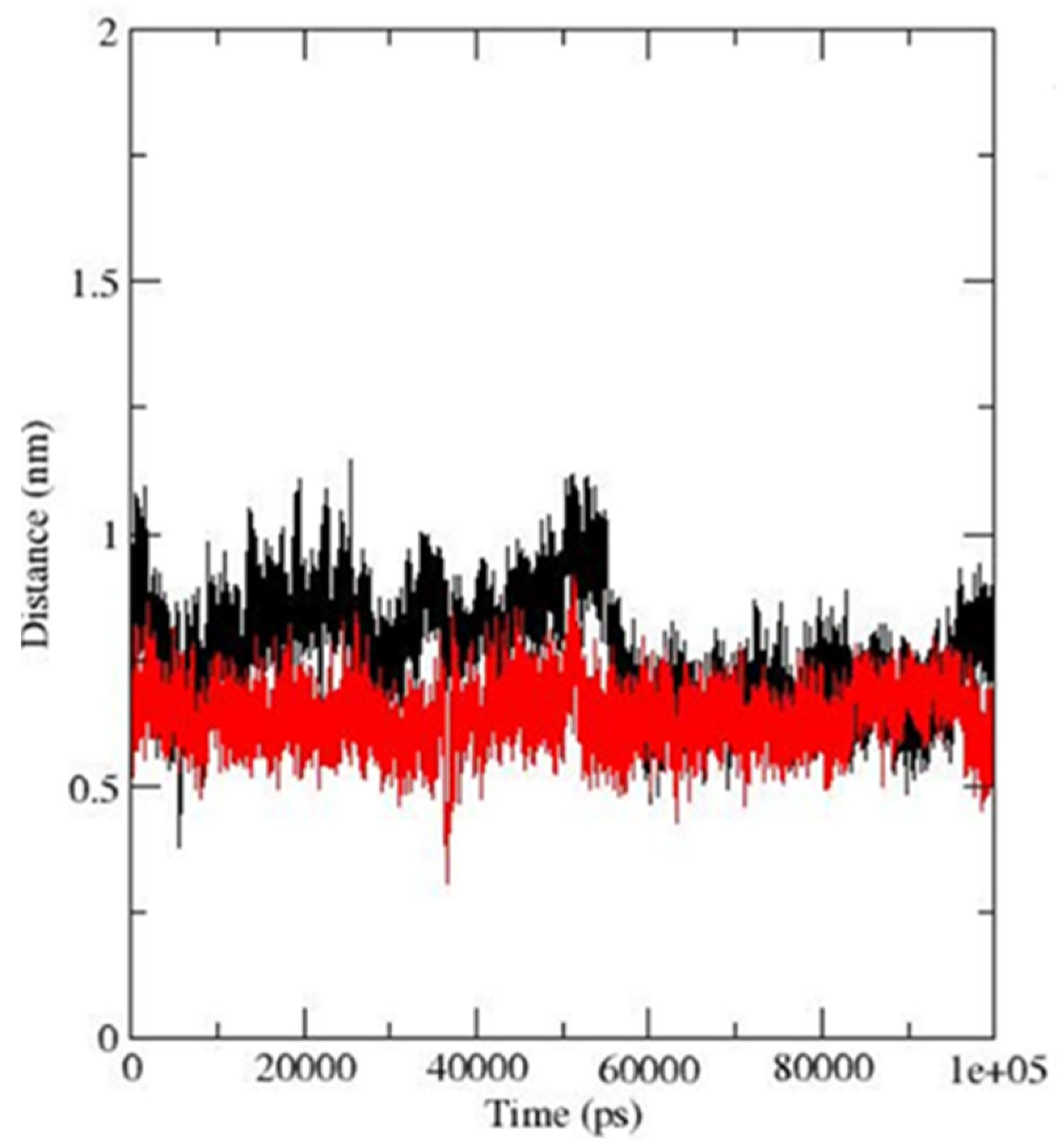
3.3. Molecular Dynamics Simulations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. World Malaria Report 2020: 20 Years of Global Progress and Challenges; World Health Organization: Geneva, Switzerland, 2020.
- Baird, J.J. 8-Aminoquinoline Therapy for Latent Malaria. Clin. Microbiol. Rev. 2019, 32, e00011-19. [Google Scholar] [CrossRef]
- World Health Organization. Guidelines for the Treatment of Malaria, 3rd ed.; WHO: Geneva, Switzerland, 2015.
- Sevene, E.; González, R.; Menéndez, C. Current knowledge and challenges of antimalarial drugs for treatment and prevention in pregnancy. Expert Opin. Pharmacother. 2010, 11, 1277–1293. [Google Scholar] [CrossRef] [PubMed]
- Dondorp, A.M.; Fanello, C.I.; Hendriksen, I.C.; Gomes, E.; Seni, A.; Chhaganlal, K.D.; Bojang, K.; Olaosebikan, R.; Anunobi, N.; Maitland, K.; et al. Artesunate versus quinine in the treatment of severe falciparum malaria in African children (AQUAMAT): An open-label, randomised trial. Lancet 2010, 376, 1647–1657. [Google Scholar] [CrossRef] [Green Version]
- Ashley, E.A.; Dhorda, M.; Fairhurst, R.M.; Amaratunga, C.; Lim, P.; Suon, S.S.; Anderson, J.M.; Mao, S.; Sam, B.; Sopha, C.; et al. Spread of artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2014, 371, 411–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medhi, B.; Patyar, S.; Rao, R.S.; Byrav, D.S.P.; Prakash, A. Pharmacokinetic and toxicological profile of artemisinin compounds: An update. Pharmacology 2009, 84, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Fairhurst, R.M. Understanding artemisinin-resistant malaria: What a difference a year makes. Curr. Opin. Infect. Dis. 2015, 28, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Fitch, C.D. Chloroquine resistance in malaria: A deficiency of chloroquine binding. Proc. Nat. Acad. Sci. USA 1969, 64, 1181–1187. [Google Scholar] [CrossRef] [Green Version]
- Leed, A.; DuBay, K.; Ursos, L.M.; Sears, D.; De Dios, A.C.; Roepe, P.D. Solution structures of antimalarial drug-heme complexes. Biochemisty 2002, 41, 10245–10255. [Google Scholar] [CrossRef]
- Cooper, R.A.; Ferdig, M.T.; Su, X.Z.; Ursos, L.M.; Mu, J.; Nomura, T.; Fujioka, H.; Fidock, D.A.; Roepe, P.D.; Wellems, T.E. Alternative mutations at position 76 of the vacuolar transmembrane protein PfCRT are associated with chloroquine resistance and unique stereospecific quinine and quinidine responses in Plasmodium falciparum. Mol. Pharma. 2002, 61, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Martin, R.E.; Marchetti, R.V.; Cowan, A.I.; Howitt, S.M.; Broer, S.; Kirk, K. Chloroquine transport via the malaria parasite’s chloroquine resistance transporter. Science 2009, 325, 1680–1682. [Google Scholar] [CrossRef] [Green Version]
- Cooper, R.A.; Lane, K.D.; Deng, B.; Mu, J.; Patel, J.J.; Wellems, T.E.; Su, X.; Ferdig, M.T. Mutations in transmembrane domains 1, 4 and 9 of the Plasmodium falciparum chloroquine resistance transporter alter susceptibility to chloroquine, quinine and quinidine. Mol. Microbiol. 2007, 63, 270–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidhu, A.B.; Valderramos, S.G.; Fidock, D.A. pfmdr1 mutations contribute to quinine resistance and enhance mefloquine and artemisinin sensitivity in Plasmodium falciparum. Mol. Microbiol. 2005, 57, 913–926. [Google Scholar] [CrossRef] [PubMed]
- Mu, J.; Ferdig, M.T.; Feng, X.; Joy, D.A.; Duan, J.; Furuya, T.; Subramanian, G.; Aravind, L.; Cooper, R.A.; Wootton, J.C.; et al. Multiple transporters associated with malaria parasite responses to chloroquine and quinine. Mol. Microbiol. 2003, 49, 977–989. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, C.P.; McLean, J.E.; Stein, J.E.; Lanzer, M. Evidence for a Substrate Specific and Inhibitable Drug Efflux System in Chloroquine Resistant Plasmodium falciparum Strains. Biochemisty 2004, 43, 16365–16373. [Google Scholar] [CrossRef] [PubMed]
- Krogstad, D.J.; Gluzman, I.Y.; Kyle, D.E.; Oduola, A.M.; Martin, S.K.; Milhous, W.K.; Schlesinger, P.H. Efflux of chloroquine from Plasmodium falciparum: Mechanism of chloroquine resistance. Science 1987, 238, 1283–1285. [Google Scholar] [CrossRef]
- Zhang, H.; Paguio, M.; Roepe, P.D. The antimalarial drug resistance protein Plasmodium falciparum chloroquine resistance transporter binds chloroquine. Biochemistry 2004, 43, 8290–8296. [Google Scholar] [CrossRef] [PubMed]
- Lehane, A.M.; Hayward, R.; Saliba, K.J.; Kirk, K. A verapamil-sensitive chloroquine-associated H+ leak from the digestive vacuole in chloroquine-resistant malaria parasites. J. Cell Sci. 2008, 121, 1624–1632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehane, A.M.; Kirk, K. Efflux of a range of antimalarial drugs and ‘chloroquine resistance reversers’ from the digestive vacuole in malaria parasites with mutant PfCRT. Mol. Microbiol. 2010, 77, 1039–1051. [Google Scholar] [CrossRef] [PubMed]
- Fidock, D.A.; Nomura, T.; Talley, A.K.; Cooper, R.A.; Dzekunov, S.M.; Ferdig, M.T.; Ursos, L.M.; Sidhu, A.B.; Naudé, B.; Deitsch, K.W.; et al. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol. Cell. 2000, 6, 861–871. [Google Scholar] [CrossRef]
- Sidhu, A.B.S.; Verdier-Pinard, D.; Fidock, D.A. Chloroquine Resistance in Plasmodium falciparum Malaria Parasites Conferred by pfcrt Mutations. Science 2002, 298, 210–213. [Google Scholar] [CrossRef] [Green Version]
- Nessler, S.; Friedrich, O.; Bakouh, N.; Fink, R.H.A.; Sanchez, C.P.; Planelles, G.; Lanzer, M. Evidence for activation of endogenous transporters in Xenopus laevis oocytes expressing the Plasmodium falciparum chloroquine resistance transporter, PfCRT. J. Biol. Chem. 2004, 279, 39438–39446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naude, B.; Brzostowski, J.A.; Kimmel, A.R.; Wellems, T.E. Dictyostelium discoideum expresses a malaria chloroquine resistance mechanism upon transfection with mutant, but not wild-type, Plasmodium falciparum transporter PfCRT. J. Biol. Chem. 2005, 280, 25596–25603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klonis, N.; Tan, O.; Jackson, K.; Goldberg, D.; Klemba, M.; Tilley, L. Evaluation of pH during cytostomal endocytosis and vacuolar catabolism of haemoglobin in Plasmodium falciparum. Biochem. J. 2007, 407, 343–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gligorijevic, B.; McAllister, R.; Urbach, J.S.; Roepe, P.D. Spinning disk confocal microscopy of live, intraerythrocytic malarial parasites. 2. Altered vacuolar volume regulation in drug resistant malaria. Biochemisty 2006, 45, 12411–12423. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.E.; Kirk, K. The Malaria Parasite′s Chloroquine Resistance Transporter is a Member of the Drug/Metabolite Transporter Superfamily. Mol. Biol. Evol. 2004, 21, 193801949. [Google Scholar] [CrossRef] [Green Version]
- Ecker, A.; Lehane, A.M.; Clain, J.; Fidock, D. PfCRT and its role in antimalarial drug resistance. Trends. Parasitol. 2012, 81, 504–514. [Google Scholar] [CrossRef] [Green Version]
- Lakshmanan, V.; Bray, P.G.; Verdier-Pinard, D.; Johnson, D.J.; Horrocks, P.; Muhle, R.A.; Alakpa, G.E.; Hughes, R.H.; Ward, S.A.; Krogstad, D. A critical role for PfCRT K76T in Plasmodium falciparum verapamil-reversible chloroquine resistance. EMBO J. 2005, 24, 2294–2305. [Google Scholar] [CrossRef]
- Tan, W.; Gou, D.M.; Tai, E.; Zhao, Y.Z.; Chow, L.M. Functional reconstitution of purified chloroquine resistance membrane transporter expressed in yeast. Arch. Biochem. Biophys. 2006, 452, 119–128. [Google Scholar] [CrossRef]
- Paguio, M.F.; Cabrera, M.; Reope, P.D. Chloroquine Transport in Plasmodium falciparum II: Analysis of PfCRT Mediated Drug Transport Using Proteoliposomes and a Fluorescent Chloroquine Probe. Biochemisty 2009, 48, 9482–9491. [Google Scholar] [CrossRef] [Green Version]
- Chinappi, M.; Via, A.; Marcatili, P.; Tramontano, A. On the Mechanism of Chloroquine Resistance in Plasmodium falciparum. PLoS ONE 2010, 5, e14064. [Google Scholar] [CrossRef] [Green Version]
- Bray, P.G.; Mungthin, M.; Hastings, I.M.; Biagini, G.A.; Saidu, D.K.; Lakshman, V.; Johnson, D.J.; Hughes, R.H.; Stocks, P.A.; O’Neill, P.M.; et al. PfCRT and the trans-vacuolar proton electrochemical gradient: Regulating the access of chloroquine to ferriprotoporphyrin IX. Mol. Microbiol. 2006, 62, 238–251. [Google Scholar] [CrossRef] [Green Version]
- The Universal Protein Resource (UniProt). Available online: https://www.uniport.org (accessed on 12 January 2021).
- Lobley, A.; Sadowski, M.I.; Jones, D.T. pGenTHREADER and pDomTHREADER: New Methods for Improved Protein Fold Recognition and Superfamily Discrimination. Bioinformatics 2009, 25, 1761–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGuffin, L.J.; Jones, D.T. Improvement of the GenTHREADER method for genomic fold recognition. Bioinformatics 2003, 19, 874–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchan, D.W.A.; Jones, D.T. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res. 2019, W1, W402–W407. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999, 292, 195–202. [Google Scholar] [CrossRef] [Green Version]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Fiser, A.; Do, R.K.; Sali, A. Modeling of loops in protein structures. Prot. Sci. 2000, 9, 1753–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using Modeller; Current Protocols in Bioinformatics 54; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; pp. 5.6.1–5.6.37. [Google Scholar]
- Fiser, A.; Šali, A. MODELLER: Generation and refinement of homology-based protein structure models. In Methods in Enzymology; Carter, C.W., Sweet, R.M., Eds.; Academic Press: San Diego, CA, USA, 2003; Volume 374, pp. 463–493. [Google Scholar]
- Lovell, S.C.; Word, J.M.; Richardson, J.S.; Richardson, D.C. The penultimate rotamer library. Proteins 2000, 40, 389–408. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Thornton, J.M. PROCHECK: Validation of protein structure coordinates. In International Tables of Crystallography, Volume F. Crystallography of Biological Macromolecules; Rossmann, M.G., Dordrecht, A.E., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2001; pp. 722–725. [Google Scholar]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexiblity. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Goodsell, D.S.; Morris, G.M.; Olson, A.J. Automated Docking of Flexible Ligands: Applications of AutoDock. J. Mol. Recog. 1996, 9, 1–5. [Google Scholar] [CrossRef]
- Abrahams, M.J.; Murtola, T.; Schulz, R.; Páss, S.; Smith, J.C.; Hess, B.; Lindhal, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general AMBER force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jakalian, A.; Bush, B.L.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J. Comput. Chem. 2000, 21, 132–146. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB ID 1 | Class | p-Value 2 | Alignment Score 3 |
|---|---|---|---|
| 1QGR (A) | Transporter–Receptor | 3 × 104 | 36.0 |
| 4ENE (A) | Transporter Protein | 8 × 104 | 57.0 |
| 3I1I (A) | Tranferase | 9 × 104 | 48.0 |
| 3RKO (B) | Oxidoreductase | 1 × 103 | 45.0 |
| 1WA5 (C) | Nuclear Transport | 1 × 103 | 45.0 |
| 4HEA (L) | Oxidoreductase | 0.001 | 53.0 |
| 1EHK | Oxidoreductase | 0.001 | 55.0 |
| 3ZKV (A) | Transport Protein | 0.001 | 40.0 |
| 3GJX (A) | Transport Protein | 0.001 | 60.0 |
| 2XWU (B) | Ligase | 0.002 | 56.0 |
| 1PW4 (A) | Membrane Protein | 0.002 | 46.0 |
| 3ORG (A) | Transport Protein | 0.002 | 40.0 |
| 4HAT (C) | Protein Transport/Antibiotic | 0.003 | 31.0 |
| 3NDO (A) | Lyase | 0.003 | 27.0 |
| Quality Assessment | Model 1 | Model 2 | Model 3 |
|---|---|---|---|
| QMean Score | 0.185 | 0.169 | 0.262 |
| Z-Score | −6.921 | −7.119 | −6.015 |
| Ramachandran Plot Summary | 88.8% core 0.5% disallowed | 90.1% core 0.5% disallowed | 90.1% core 1.0% disallowed |
| Bad Contacts | 17 | 16 | 14 |
| %-α-Helix | 70.3 | 69.1 | 66.5 |
| Modeller Z-Dope Score | 0.91 | 0.06 | 0.66 |
| Molecule | Binding Energy 1 | Ligand Efficiency 2 | Binding Energy 1 | Ligand Efficiency 2 |
|---|---|---|---|---|
| Wildtype PfCRT | K76T mutant of PfCRT | |||
| CQ | −3.7 | −0.17 | −4.08 | −0.19 |
| CQH+ | 59.6 | 27.13 | −4.15 | −0.19 |
| CQH2+ | 58.4 | 26.58 | −4.77 | −0.22 |
| Molecule | Coul-SR 1 | LJ-SR 2 |
|---|---|---|
| Wildtype | −14.74 (±0.53) | −140.71 (±3.0) |
| K64T Mutant | −10.96 (±2.4) | −160.75 (±1.8) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patel, C.; Roy, D. A Computational Study of Molecular Mechanism of Chloroquine Resistance by Chloroquine Resistance Transporter Protein of Plasmodium falciparum via Molecular Modeling and Molecular Simulations. Physchem 2021, 1, 232-242. https://0-doi-org.brum.beds.ac.uk/10.3390/physchem1030017
Patel C, Roy D. A Computational Study of Molecular Mechanism of Chloroquine Resistance by Chloroquine Resistance Transporter Protein of Plasmodium falciparum via Molecular Modeling and Molecular Simulations. Physchem. 2021; 1(3):232-242. https://0-doi-org.brum.beds.ac.uk/10.3390/physchem1030017
Chicago/Turabian StylePatel, Chandan, and Dipankar Roy. 2021. "A Computational Study of Molecular Mechanism of Chloroquine Resistance by Chloroquine Resistance Transporter Protein of Plasmodium falciparum via Molecular Modeling and Molecular Simulations" Physchem 1, no. 3: 232-242. https://0-doi-org.brum.beds.ac.uk/10.3390/physchem1030017